

Abstract
Copper is an essential micronutrient that serves as a cofactor for enzymes involved in diverse physiological processes, including mitochondrial energy generation. Copper enters cells through a dedicated copper transporter and is distributed to intracellular cuproenzymes by copper chaperones. Mitochondria are critical copper utilizing organelle that harbors an essential cuproenzyme cytochrome c oxidase, which powers energy production. Mutations in copper transporters and chaperones that perturb mitochondrial copper homeostasis result in fatal genetic disorders. Recent studies have uncovered the therapeutic potential of elesclomol, a copper ionophore, for the treatment of copper deficiency disorders such as Menkes disease. Here we review the role of copper in mitochondrial energy metabolism in the context of human diseases and highlight the recent developments in copper therapeutics.
Keywords: Mitochondria, copper, Menkes disease, Wilson disease, mitochondrial disease, elesclomol
Copper as an ancient enzymatic cofactor
Copper is an essential trace element utilized by nearly all organisms from bacteria to humans. The evolution of copper-containing enzymes (cuproenzymes) likely occurred in response to the increasing need to utilize oxygen and oxygen-containing molecules in the primordial ocean [1]. This feature is still present today as many cuproenzymes use oxygen as a substrate to catalyze redox reactions. These enzymes participate in diverse physiological processes including mitochondrial energy generation and detoxification of free radicals [2]. Before describing different aspects of mitochondrial copper biology, we first present a brief overview of copper absorption, transport, and intracellular distribution in humans to introduce key players in copper homeostasis.
Copper absorption and transport in humans
Total body copper in an average adult human is approximately 100 mg. The recommended daily intake of copper for adults is ~1.5 mg, which is mostly acquired through diet [3]. Dietary copper is imported as cuprous (Cu1+) ions via copper transport protein 1, CTR1 (Figure 1) [4]. Various cupric (Cu2+) reductases facilitate this process, including cytochrome b ferric/cupric reductase, STEAP metalloreductases, and cytochrome B reductase 1 [5, 6]. Copper could also be imported as cupric (Cu2+) ions via divalent metal transporter, DMT1 [7, 8]. Dietary copper is then exported from enterocytes into the portal blood circulation via ATP7A, a copper-transporting P-type ATPase, which translocate to the basolateral membrane from its trans-Golgi location in response to excess cellular copper in enterocytes (Figure 1) [9].
Figure 1.
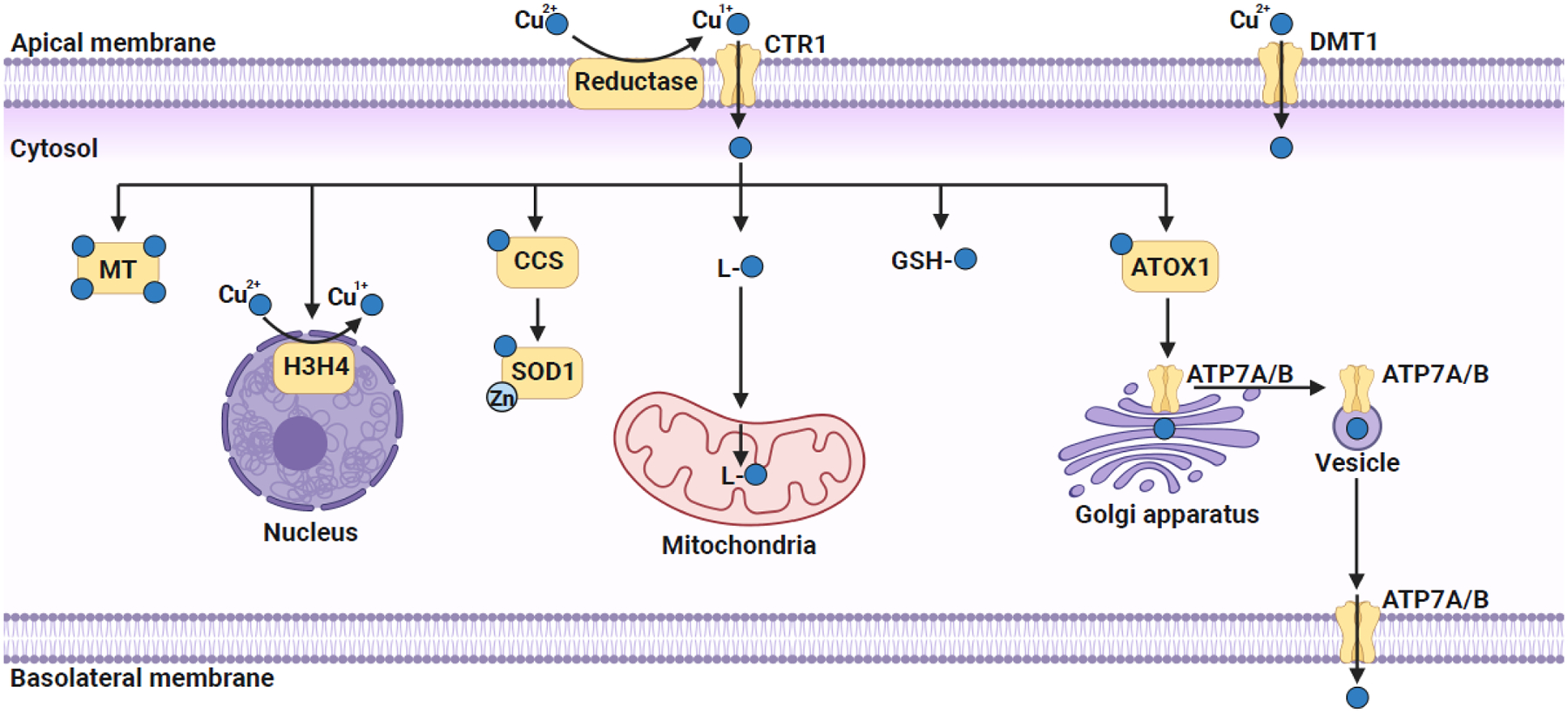
Copper trafficking in a polarized mammalian cell.
Extracellular Cu2+ is reduced to Cu1+ by plasma membrane-localized reductases that allows its import via the high-affinity plasma membrane copper transporter CTR1. Extracellular Cu2+ could also be imported by a non-specific divalent metal transporter DMT1. Once in the cytosol, copper is immediately bound to either a non-proteinaceous ligand (GSH or CuL) or a metallochaperone for subsequent trafficking to various organelles. CuL is proposed to transport copper to mitochondria, where it is stored within the mitochondrial matrix and acts as a feedstock for the metallation of mitochondrial cuproenzymes. In the cytosol, CCS, a metallochaperone, binds and transfers copper to Cu/Zn SOD1. Copper is delivered to the Golgi lumen, via the combined action of cytosolic metallochaperone ATOX1 and the Golgi membrane localized copper pumps ATP7A and ATP7B. Recently, histones H3–H4 tetramer have been shown to act as copper reductases that increase the bioavailable copper pool by catalyzing the reduction of Cu2+ to Cu1+. Excess copper is either bound to metallothionein (MT) or pumped out of the cell via the action of ATP7A/B, which translocates to the plasma membrane via vesicular trafficking. The figure was created with Biorender.com.
Once in the blood, copper is bound to α2-macroglobulin, albumin, and other ligand(s) and is delivered to the liver [10]. As in enterocytes, copper transport into hepatocytes is mediated by cupric reductases and CTR1. In these cells, ATP7B, a homolog of ATP7A, is expressed which facilitates copper loading into secretory cuproenzymes in the trans-Golgi network. One critical secretory cuproenzyme specifically expressed in the liver is ceruloplasmin, which is released into the blood and facilitates cellular absorption of serum iron [10]. When liver copper storage exceeds body requirements, ATP7B translocates to the canalicular membrane, where it mediates cellular copper export into the bile which is eliminated through feces [11].
Copper abundance varies across organs with tissues such as the skeletal muscle, brain, and heart, containing approximately 23, 8, and 1% of total body copper, respectively [12]. Copper entry into the brain through the blood-brain barrier requires the combined action of CTR1 and ATP7A [13]. In the brain, copper functions as an essential cofactor for several cuproenzymes including dopamine-β-hydroxylase and peptidylglycine α-amidating monooxygenase which are required for the biosynthesis of neurotransmitter (norepinephrine) and amidated neuropeptides, respectively. The clearance of copper from brain cells occurs via ATP7A, which promotes the excretion of copper through blood cerebrospinal fluid barrier [13].
Intracellular copper distribution
Upon cellular import, copper is immediately bound to either a copper chaperone(s) or to a non-proteinaceous ligand such as glutathione, that shuttles copper to various cuproenzymes present in different subcellular compartments [14, 15]. CCS is one such metallochaperone (see Glossary) that escorts copper from the plasma membrane localized CTR1 to copper-zinc superoxide dismutase, SOD1, which is mainly found in the cytoplasm [16]. Similarly, the cytosolic copper metallochaperone ATOX1 delivers copper to ATP7A and ATP7B, which are present on the Golgi membrane in the trans-Golgi network, where they pump copper into the Golgi lumen for its incorporation into several secretory cuproenzymes [1, 2]. Excess cytoplasmic copper can be sequestered by metallothioneins, cysteine-rich proteins that provide resistance to cellular copper toxicity (Figure 1) [17]. A recent study has uncovered an unexpected role of the nuclear protein complex, histone H3–H4 tetramer, in reducing Cu2+ to Cu1+ and in maintaining mitochondrial and cytosolic cuproenzymes activity [18].
Mitochondrial copper homeostasis
Mitochondria serve as a major copper storage site, containing a pool of copper whose molecular identity is unknown [19, 20]. Typically, mammalian mitochondria contain 30–50 nanograms of copper per milligram of mitochondrial protein [21]. Under copper deficiency, cells prioritize mitochondrial copper homeostasis suggesting its critical requirement for this organelle [22]. A significant fraction (~25%) of the mitochondrial copper pool is utilized by cytochrome c oxidase (CcO), the most critical cuproenzyme of mitochondria [23]. As the terminal enzyme of the mitochondrial respiratory chain, CcO catalyzes the electron transfer from reduced cytochrome c to molecular oxygen, while simultaneously pumping protons from the matrix to the intermembrane space (IMS) to contribute to the proton gradient that powers mitochondrial ATP synthesis. This multi-subunit complex is localized to the inner mitochondrial membrane (IMM) and contains copper and heme as cofactors [24]. Three copper ions are bound within the catalytic core of CcO as CuA and CuB sites, which are present in COX2 and COX1 subunits, respectively [24]. Another important cuproenzyme present in mitochondria is SOD1, which is localized to the IMS, where it neutralizes superoxides produced by the mitochondrial respiratory chain.
For a long time, the evolutionarily conserved protein, COX17, was thought to be the mitochondrial copper shuttle due to its dual localization in the cytoplasm and mitochondria [14]. However, COX17 enters mitochondria in an unfolded state which is unlikely to carry copper into mitochondria [25]. Another piece of evidence that questioned the copper-shuttling role of COX17 was a creatively designed experiment where COX17 was tethered to the IMM using the transmembrane domain of SCO2, a known IMM localized protein [26]. The IMM-tethered COX17 was exclusively localized to mitochondrial IMS and was fully functional, a finding inconsistent with the copper-shuttling role of COX17 [26]. The current hypothesis is that copper is transported into mitochondria via a non-proteinaceous anionic ligand (CuL) [19, 20]. This non-proteinaceous anionic ligand has been identified in the cytosol in both the copper-bound and unbound forms, however, the molecular identity of this ligand has remained unknown to date [20]. Recent advances in chromatographic and mass spectrometry techniques which were used to identify low molecular mass labile metal pools in E. coli [27], could prove to be extremely useful in addressing this long-standing question.
CuL likely enters mitochondria by passive diffusion through porin, a large outer mitochondrial membrane channel (Figure 2). Once in the IMS, copper could be used for the metallation of CcO and SOD1 because the molecular machinery for copper insertion into these proteins resides in the IMS (Figure 2). Curiously, the cytosolic copper is first transported into the mitochondrial matrix via the IMM localized transporter SLC25A3 in mammals and Pic2 in yeast [28, 29]. Pic2-deleted yeast cells do not display a robust mitochondrial copper-deficient phenotype [29]. Moreover, copper supplementation in the growth media can overcome CcO deficiency in SLC25A3 knockout cells [28]. Together, these results suggest an alternate mechanism of copper import into mitochondria. Consistent with this idea, mitochondrial iron transporters from yeast (Mrs3) and fish (MFRN1) have been shown to transport copper as well [30, 31]. A more recent phylogenetic analysis of Pic2 and SLC25A3 identified specific amino acid requirements for copper transport establishing their important role in mitochondrial copper import [32]. Within the mitochondrial matrix, copper is stored in its ligand-bound form and is made bioavailable for the metallation of CcO when required [20]. Interestingly, the matrix copper must be exported back via an unidentified transporter into the IMS for its delivery to CcO as well as SOD1 (Figure 2) [33]. Apart from serving as a feedstock for CcO and SOD1, the biochemical role of this matrix copper pool remains to be elucidated (Figure 2).
Figure 2.
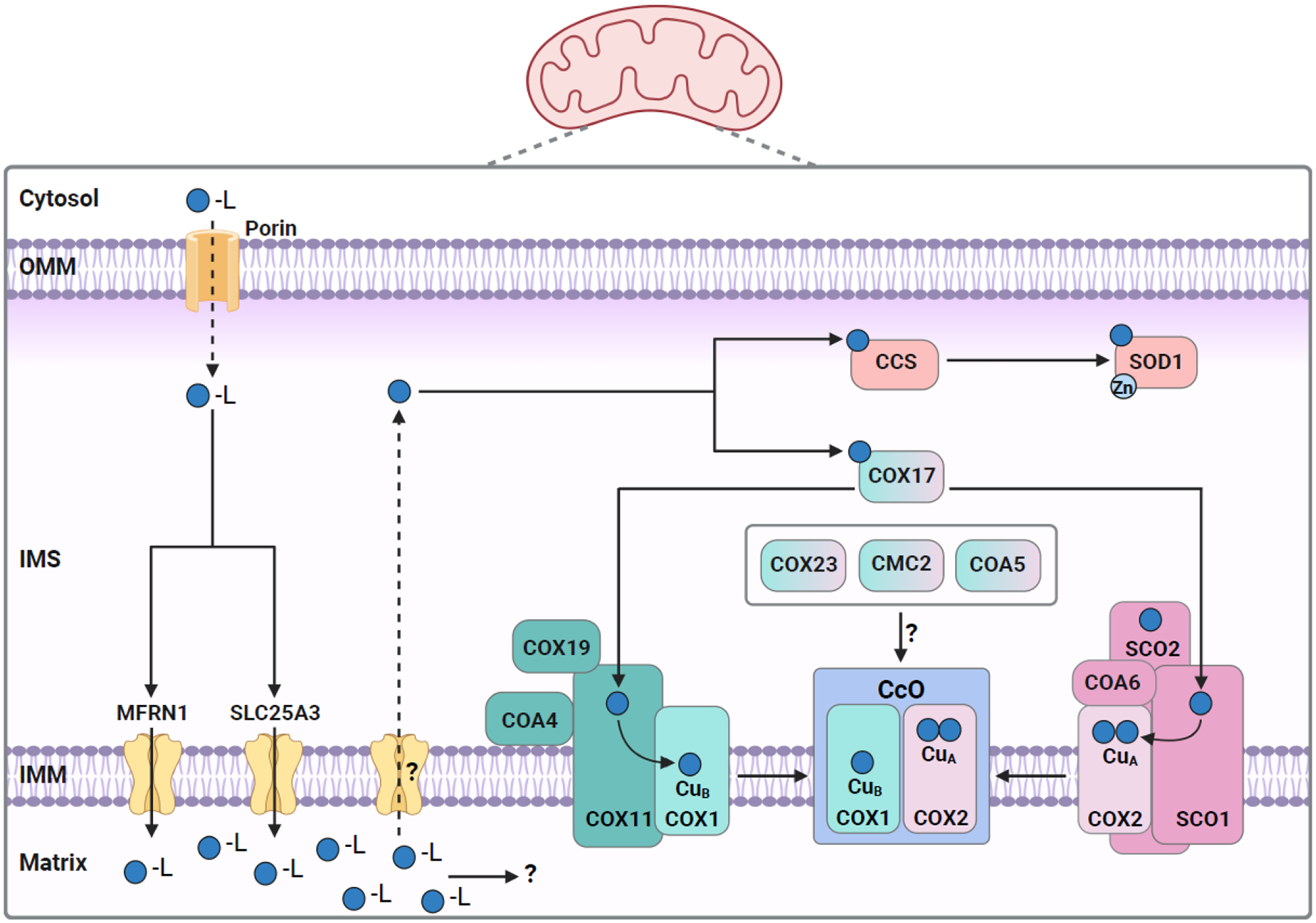
Copper trafficking within mammalian mitochondria.
Copper bound to a ligand (CuL) likely enters mitochondria via OMM localized porin and is then transported into the matrix by the IMM localized transporters SLC25A3 and MFRN1. The function of copper within the matrix is not known but this copper is transported back across the IMM by an unknown transporter. Once in the IMS, copper binds metallochaperones CCS and COX17, for its delivery to SOD1 and CcO, respectively. COX17 transfers copper to metallochaperones, COX11 and SCO1, which in turn inserts it into CcO subunits COX1 and COX2, respectively. COX11, COX19, and COA4 are components of the copper delivery pathway to the CuB site within the COX1 subunit. SCO1, SCO2, and COA6 are components of the copper delivery pathway to the CuA site within the COX2 subunit. The IMS-localized proteins COX23, CMC2, and COA5 are essential for CcO assembly but their involvement in the copper delivery pathway remains unknown. Abbreviations: OMM, outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane. The figure was created with Biorender.com.
The matrix copper is mobilized back into the IMS, where it becomes available to metallochaperones involved in the copper delivery to SOD1 and the copper-containing subunits of CcO (Figure 2). SOD1 receives its copper cofactor from CCS, which is also found in the IMS, though the immediate donor of copper to mitochondrial CCS remains to be determined. Mobilization of copper from the mitochondrial matrix to the IMS for its delivery to copper sites in CcO subunits requires several evolutionarily conserved proteins (Table 1). The delivery of copper to CcO is a very complex process, driven by copper affinity gradients and dedicated copper metallochaperones that facilitate the sequential transfer of copper [34, 35]. The function of these CcO assembly factors can be broadly classified into two types: 1) copper metallochaperones such as COX17 and SCO1, that directly bind and deliver copper through specific protein-protein interactions, or 2) accessory factors such as COA6 and SCO2, that do not transfer copper themselves but rather facilitate this process (Figure 2). Copper delivery to COX1 and COX2 occurs as a modular process, requiring distinct sets of proteins for copper insertion into each subunit [35] (Figure 2). The IMS-localized CX9C copper metallochaperone COX17 receives copper from the matrix and transfers it to metallochaperones SCO1, SCO2, and COX11 [36]. SCO1 functions as the COX2-specific metallochaperone that inserts copper into the CuA site of CcO [37]. COX11 functions as the COX1-specific metallochaperone that delivers copper to the CuB site of CcO [38, 39]. A prerequisite for copper binding by these cysteine-based copper metallochaperones is the presence of their cysteines in a reduced form to enable copper binding. This function is provided by SCO2 and COA6, which have been shown to function as disulfide reductases of SCO1 and COX2 proteins [37, 40–42].
Table 1.
Genes involved in mitochondrial copper biology.
| Yeast Gene | Human Gene | Function | Refs |
|---|---|---|---|
| COX1 | COX1 | Subunit of the catalytic core of CcO, contains the CuB site, catalyzes electron transport to molecular oxygen | [24] |
| COX2 | COX2 | Subunit of the catalytic core of CcO, contains the CuA site, catalyzes electron transport to molecular oxygen | [24] |
| COX11 | COX11 | Metallochaperone, a component of the copper delivery pathway to the CuB site | [38, 39] |
| COX17 | COX17 | Metallochaperone, transfers copper to metallochaperones SCO1 and COX11 | [36] |
| COX19 | COX19 | Precise function unknown, a component of the copper delivery pathway to the CuB site | [46] |
| COA4 | COA4 | Precise function unknown, a component of the copper delivery pathway to the CuB site | [45] |
| COA6 | COA6 | Disulfide reductase of COX2 and SCO1, a component of the copper delivery pathway to the CuA site | [41, 42, 97] |
| SCO1 | SCO1 | Metallochaperone, required for copper delivery to the CuA site | [37] |
| SCO2 | SCO2 | Copper-binding thiol-disulfide oxidoreductase of COX2, a component of the copper delivery pathway to the CuA site | [37, 40] |
| PIC2 | SLC25A3 | Mitochondrial copper importer | [28, 29] |
| CCS1 | CCS | Metallochaperone, transfers copper to SOD1 | [16] |
| SOD1 | SOD1 | Cytosol and mitochondria localized copper-zinc superoxide dismutase, detoxifies superoxides | [16, 98] |
In addition to these proteins, several evolutionarily conserved twin CX9C motif-containing IMS proteins including, Cox19, Cox23, Cmc1, Cmc2, Coa4, and Pet191 and have been implicated in the assembly of CcO [43]. Despite sharing a similar fold, these proteins lack the N-terminal cysteine-rich (CCXC) copper-binding motif found in Cox17, suggesting that they may not function as copper metallochaperones. Genetic suppression screens in yeast have linked some of these genes to the CcO copper delivery pathway, suggesting that these proteins may play a non-metallochaperone role in the delivery of copper to CcO. For example, suppression of the respiratory growth defect of yeast cox23 null mutant by COX17 overexpression links it to the mitochondrial copper delivery pathway to CcO [44]. A recent yeast genetic suppressor study has placed Coa4 upstream of Cox11 in the copper delivery pathway to the CuB site [45]. Along with genetic interaction studies, protein:protein interaction studies have also proved valuable in placing CX9C proteins to the copper delivery pathway to CcO. For example, a SILAC-based quantitative proteomic study uncovered a dynamic physical interaction between Cox19 and Cox11, placing Cox19 in the copper delivery to the CuB site [46]. Another recent study characterizing human cell lines lacking COX11, COX19, and PET191 (COA5) showed that CcO copper chaperones form macromolecular assemblies and cooperate with several twin CX9C proteins to coordinate copper transfer sequentially to the CuA and CuB sites [47]. Despite this progress, the precise molecular function of these proteins in copper delivery to CcO remains unknown.
Another example of the importance of maintaining mitochondrial copper homeostasis comes from a recent study investigating the role of copper ionophore-induced cell death. This study identified lipoylated proteins of the tricarboxylic acid cycle as the target for copper toxicity [48]. Utilizing genome-wide CRISPR screens, the authors showed that the deletion of components of the lipoic acid pathway or lipoylation target, the pyruvate dehydrogenase complex, confers resistance to the two copper ionophores tested [48]. Interestingly, cell death caused by copper overload occurred via a mechanism distinct from other forms of cell death including, apoptosis, ferroptosis, pyroptosis, and necroptosis. Therefore, the authors named this unique form of copper-dependent cell death “cuproptosis” [48]. These findings have important implications in conditions of mitochondrial copper overload, such as in Wilson disease where copper accumulation is observed in certain organs such as the liver.
Considering this finding, it is imperative to identify mitochondrial copper detoxification mechanisms. These would include mitochondrial copper export machinery, copper sequestering proteins, or antioxidant defense mechanisms that would counter copper-induced ROS production. As mentioned previously, mitochondrial matrix copper is exported for the metallation of CcO and SOD1 and it is expected that the same mitochondrial copper exporter plays a role in maintaining copper homeostasis under conditions of mitochondrial copper overload. Alternatively, copper-binding proteins or other ligands may be trafficked to mitochondria to sequester copper and counter its toxicity.
Mitochondrial dysfunction in human genetic diseases of copper metabolism
Mitochondrial and non-mitochondrial cuproenzymes have many different biochemical functions and owing to their diverse roles in cellular and organismal physiology, loss-of-function mutations in genes required for copper absorption, elimination, or transport often result in fatal or severely debilitating disorders (Figure 3). For example, mutations in CcO assembly factors that are involved in copper delivery to CcO including SCO1, SCO2, and COA6, have been shown to cause fatal infantile mitochondrial disorders. These disorders are typically characterized by cardioencephalomyopathy, though significant genetic and clinical heterogeneity is observed in these patients. Mutations and clinical presentations for each of these genes are described in Box 1. Mutations in copper transporters – ATP7A and ATP7B – that lead to whole body, organ, and intracellular copper imbalance cause Menkes disease and Wilson disease, respectively [49, 50]. Although not prototypical mitochondrial disorders, perturbations in mitochondrial copper content in these disorders significantly contribute to the disease pathology. Genetics, epidemiology, and clinical pathology associated with these diseases are described below.
Figure 3.
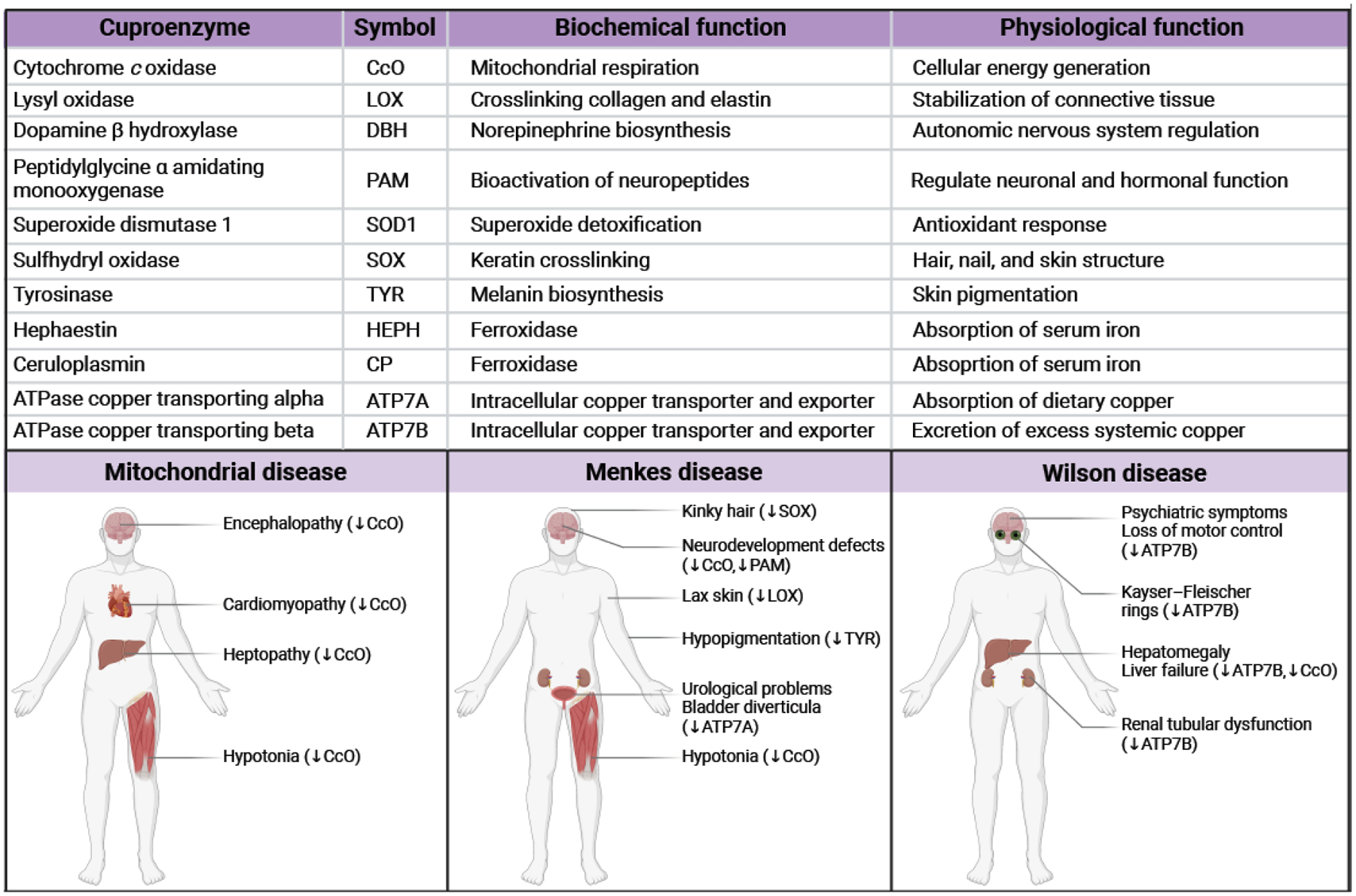
Cuproenzymes and their role in human physiology and pathology.
The formal and abbreviated names of cuproenzymes as well as their enzymatic and physiological role(s) are listed. Typical clinical presentations of mitochondrial disorders, Menkes disease, and Wilson disease are depicted below, and causative enzyme(s) are indicated. Downward-facing arrows indicate reduced enzymatic activity. The figure was created with Biorender.com.
Box 1. Disorders of the mitochondrial copper delivery pathway.
SCO1:
Mutations in SCO1 have been identified in four infants [100–102]. These patients presented with a variety of cardiac, liver, and neurological ailments including ventricular hypertrophy, hepatomegaly, and brain atrophy. Metabolic acidosis with increased excretion of lactate, fumarate, succinate, and malate in urine has also been reported. All patients died before the age of 6 months. Molecular studies on patient DNA have revealed a variety of loss-of-function mutations including frameshift and point mutations (P174L, G132S, M294V, V93X) some of which introduce a premature stop codon. Biochemical studies on patient samples showed decreased CcO activity in the liver and skeletal muscles, and a reduction in CcO containing supercomplexes in skeletal muscles [100, 101].
SCO2:
As compared to SCO1, a much larger number of patients have been reported with pathogenic mutations in SCO2 [103–106]. These patients presented with hypertrophic cardiomyopathy, hepatomegaly, muscle hypotonia, spinal muscular atrophy, neuropathy, and increased lactate in blood and cerebrospinal fluid. The mutational spectrum in SCO2 patients is broad with some mutations leading to death within six months of age whereas other patients survive to adulthood. For example, patients that carry compound heterozygous mutations with E140K (the most common mutation reported in SCO2 patients) and one of the following mutations W36X, Q53X, R90X, C133S, L151P, M177T, S225F, R171W die within six months of birth [103–108]. There have been cases of compound heterozygous mutations (E140K/P169T and D135G/R171Q) in patients with axonal Charcot-Marie-Tooth disease where patients survived through infancy and developed neuropathy in early adolescence [109]. Biochemical studies on patient tissues revealed decreased CcO activity in skeletal muscles, heart, liver, and fibroblasts [103, 104].
COA6:
Mutations in COA6 have been identified in two patients with hypertrophic cardiomyopathy [110, 111]. A W59C mutation and an E87X mutation were identified in one patient, and a homozygous W66R mutation in another patient. Biochemical studies revealed decreased COX activity in patient heart and skin fibroblasts [110, 111]. Interestingly, copper supplementation was able to rescue the COX2 and CcO levels in cultured patient skin fibroblasts [111].
Menkes disease
Menkes disease is an X-linked multisystemic disorder caused by pathogenic mutations in ATP7A and typically results in death during early childhood [49]. Menkes is a relatively rare disease with incidence estimates ranging from 1/50,000 to 1/360,000 live births depending on the population surveyed, though these numbers may be an underestimate [49]. Clinical presentations for Menkes disease include failure to thrive, connective tissue defects, kinky hair, hypotonia, seizures, and progressive neurodegeneration [51] (Figure 3). As ATP7A is required for directional transport of copper across polarized epithelial cells of the intestine, kidney, and blood-brain barrier, complete or partial loss-of-function of this protein results in systemic copper deficiency in major organs of the body, including the brain, with copper accumulation observed in the polarized epithelial cells. Menkes patients exhibit decreased activity of multiple cuproenzymes and was historically connected to mitochondrial dysfunction due to reduced CcO activity [51, 52]. Ragged-red fibers, a typical feature of classical mitochondrial disorders, were also reported in the skeletal muscle of a Menkes disease patient [53]. Some of the defects observed in the brain of Menkes disease patients including neuronal cell loss in the cerebellum and severe demyelination are partly similar to those in patients with Leigh syndrome, a mitochondrial disorder characterized by biochemical defects in mitochondrial respiratory chain complexes including CcO [54]. The hypoactivity of brain CcO is the primary driver for neurodevelopmental defects observed in these patients. Mitochondrial redox imbalance could also significantly contribute to pathologies caused by the accumulation of copper [55]. Diminished activity of other brain cuproenzymes including dopamine b-hydroxylase and peptide α -amidating monooxygenase, also contributes to clinical presentations. The substrate and product of dopamine β-hydroxylase – dopamine and norepinephrine – are used as reliable diagnostic markers for this disease [56].
Wilson disease
Wilson disease is a rare autosomal recessive genetic disorder caused by mutations in ATP7B [50]. Unlike Menkes disease, which manifests in infancy, clinical presentations of Wilson disease typically become apparent during the teenage years [50]. The prevalence of Wilson disease is also higher than Menkes disease, at 1:30,000 live births [50]. As ATP7B is primarily tasked with eliminating surplus copper from the body, this disorder is characterized by copper accumulation in the liver, brain, kidney, and other tissues dependent on ATP7B function [57]. This disease is progressive and if left untreated results in liver failure, as well as neurological and psychiatric symptoms in a subset of patients [50] (Figure 3). Mitochondrial structural abnormalities have been reported in Wilson disease pathology [58]. As copper accumulates in Wilson disease patient cells and Wilson animal models, mitochondrial membrane integrity is compromised [59]. Critically, the disease status correlated with mitochondrial but not whole-liver copper content [59]. Interestingly, even in pre-symptomatic patients and animal models of Wilson disease, ultrastructural alterations of mitochondria in hepatocytes have been observed [59, 60]. The liver of Wilson disease patients exhibits increased levels of mitochondrial ROS and apoptotic markers [61]. This hepatic injury results in liver damage in the form of cirrhosis and chronic hepatitis, culminating in progressive liver failure [50]. Wilson disease patients with acute hepatic failure present with mitochondrial respiratory chain deficits [62]. In the Wilson disease mouse model tx-j, CcO activity progressively decreased as normalized to mitochondrial abundance [63]. A recent study reported mitochondrial DNA-depletion like syndrome in Wilson disease models and patients. Specifically, mitochondrial DNA copy number was found to be decreased in the blood of Wilson disease patients as well as in the liver of the tx-j mouse model. This finding was consistent with a metabolomic signature that highlighted enriched DNA synthesis/replication pathways in serum metabolomics of Wilson disease patients [64]. Thus, mitochondrial copper burden appears to be one of the key causes of Wilson disease pathology.
The expression of metallothioneins, MT1 and MT2, are highly upregulated in models of Wilson disease and provide protection against non-sequestered copper [65, 66]. Additionally, metallothioneins have been suggested to serve as highly sensitive and specific markers for the detection of Wilson disease in human patients [67]. Interestingly, a previous study had shown that MT1 can also localize to the IMS of mitochondria [68]. This finding raises the question of whether metallothioniens play a protective role in mitochondria of Wilson disease patients.
Additional human pathologies associated with disrupted mitochondrial copper homeostasis
Several neurodegenerative disorders have been linked to either the aberrant function of mitochondrial copper-binding proteins or perturbation in mitochondrial copper homeostasis. Specifically, a subset of amyotrophic lateral sclerosis (ALS) cases are caused by mutations in SOD1, and mitochondrial accumulation of mutant SOD1 is an important contributor to the pathology of this disorder [69]. There is also a large body of literature suggesting that other common neurodegenerative disorders including Parkinson’s disease and Alzheimer’s disease are also characterized by a disruption in copper homeostasis [reviewed in 70]. MEDNIK syndrome and Huppke-Brendel syndrome are rare disorders of copper metabolism that exhibit reduced serum copper and ceruloplasmin levels [71, 72]. MEDNIK syndrome is caused by pathogenic mutations in AP1S1. These patients exhibit symptoms and biochemical markers that are similar to both Menkes (hypotonia, developmental delay, cerebral atrophy, reduced CcO abundance) and Wilson disease (liver copper overload) [71, 73]. This is likely because AP1S1 is required for the proper intracellular trafficking of ATP7A and ATP7B proteins. Huppke-Brendel syndrome is caused by pathogenic mutations in SLC33A1, which encodes an acetyl-coA transporter. These patients exhibit hypotonia, developmental delay, seizures, and neurodevelopmental defects including cerebellar hypoplasia and severe demyelination, clinical presentations that are reminiscent of Menkes disease [72]. Reduced CcO activity has also been reported in one patient [74]. The molecular mechanism by which defective acetyl-coA transporter cause copper imbalance remains to be determined. Additionally, pathogenic mutations in the mitochondrial copper importer SLC25A3 have been reported and shown to cause muscular hypotonia and progressive hypertrophic cardiomyopathy [75].
Copper therapeutics
The therapeutic value of pharmacological agents that can restore cellular and tissue copper homeostasis is increasingly being realized [76, 77]. Many copper-transporting pharmacological agents were initially developed as anti-cancer drugs because cancer cells have a low threshold for tolerating oxidative agents like copper [78, 79]. Owing to their high efficiency in transporting copper across biological membranes, these chemotherapeutic drugs are attractive copper delivery agents in diseases of genetic copper deficiencies (Table 2). For example, our recent studies on elesclomol, a copper ionophore that was originally developed as a chemotherapeutic agent [80], have shown that the administration of low doses of elesclomol ameliorates disease phenotypes in yeast, zebrafish, and mouse models of copper deficiency, including a mouse model of Menkes disease [81, 82]. Elesclomol-copper complex (ES-Cu) restores mitochondrial function in copper deficient cells by making copper bioavailable to CcO [81, 82]. The unique ability of ES-Cu to deliver copper to mitochondria is likely dependent on mitochondrial reductase FDX1, which was recently shown to bind ES-Cu [83]. More recently, our lab has demonstrated that ES-Cu can also make copper bioavailable to the Golgi compartment in a Ccc2 (a yeast ortholog of ATP7A/ATP7B)-dependent manner [84]. This is an important observation as many cuproenzymes are metallated in the Golgi lumen [2]. Notably, other copper-binding drugs such as disulfiram or copper salts such as Cu-histidinate have shown limited efficacy in the treatment of Menkes disease patients [56, 76, 85]. Therefore, there is a dire need for better copper delivery agents. Our recent pre-clinical studies provide a strong motivation for repurposing ES-Cu for the treatment of copper deficiency disorders, including Menkes disease [81, 82, 86].
Table 2.
Copper therapeutics
| Compound | Potential therapeutic application | Chemical structure | Refs |
|---|---|---|---|
| Copper ionophores | |||
| Elesclomol | Menkes disease, Mitochondrial disorders, Cancer |

|
[80, 86] |
| CuATSM | ALS, Parkinson’s disease, Cancer |

|
[87, 88, 99] |
| CuGTSM | Alzheimer’s disease, Cancer |
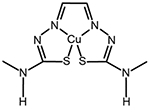
|
[89, 99] |
| Disulfiram | Cancer, Alcohol abuse drug |
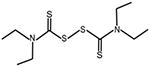
|
[79] |
| Copper chelators | |||
| D-penicillamine | Wilson disease |
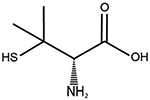
|
[90, 92] |
| Trientine | Wilson disease |
|
[90, 92] |
| Tetrathiomolybdate | Wilson disease |
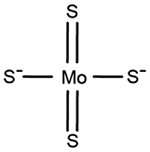
|
[90, 92, 93] |
Other copper transporting complexes such as CuATSM and CuGTSM have also been investigated for their use in treating neurodegenerative diseases. For example, CuATSM has been shown to increase the life span of a mouse model of ALS [87] and improve disease phenotypes in animal models of Parkinson’s disease [88]. CuGTSM was reported to be neuroprotective in an animal model of Alzheimer’s disease [89]. While these copper-ionophores have shown early promise in the treatment of animal studies, they likely have a narrow therapeutic window. Therefore, the next challenge lies in expanding their therapeutic index.
In contrast to copper delivery agents, copper chelation therapy aims to remove excess copper accumulation in tissues. The development of multiple copper chelators has allowed for the reduction of copper burden in Wilson disease patients, significantly improving the survival and quality of life (Table 2). For example, D-penicillamine has classically been utilized as a Wilson disease therapeutic as it binds excess free copper forming a copper complex that is excreted through the urine, thereby lowering blood copper levels and preventing hepatic copper accumulation when administered chronically [90]. Though D-penicillamine improves liver symptoms, it has been linked to the worsening of neurologic symptoms in a subset of Wilson disease patients [91]. The subsequent copper chelator developed was trientine, which has an improved safety profile [92]. Tetrathiomolybdate, a more recently developed therapeutic for Wilson disease, is deemed the safest for preserving neurological function and is an effective agent for reducing hepatic copper accumulation [92, 93].
Concluding remarks
Studies from yeast genetics and rare human genetic disorders have highlighted the essential role of mitochondrial copper in cellular and organismal health. Mitochondrial copper deficiency or disruption in copper delivery to CcO underlies a variety of clinical presentations typically affecting neurodevelopment and often resulting in fatal infantile disorders with no Food and Drug Administration (FDA)-approved treatment. Though, exciting studies with elesclomol have opened new possibilities for repurposing this cancer drug for the treatment of copper deficiency disorders such as Menkes disease and associated syndromes [86]. It is also becoming increasingly apparent that the mitochondrial copper pool influences mitochondrial redox homeostasis, iron-sulfur cluster biogenesis, and TCA cycle flux via lipoylation [48, 55, 94]. However, whether matrix copper engages with these pathways under normal physiological conditions remains to be seen. The presence of a copper pool within the mitochondrial matrix raises an important question - Are there yet to be discovered mitochondrial cuproproteins or copper-regulated enzymes that function in the matrix of mitochondria? (See Outstanding Questions). With the ability to make precise genetic lesions through CRISPR-Cas9, the availability of targeted copper delivery through elesclomol, advances in analytical methods such as mass spectrometry-based proteomics and metabolomics, and the emergence of powerful in vivo imaging techniques including nanoscale secondary ion mass spectrometry (nanoSIMS) and X-ray fluorescence microscopy (XFM) [95, 96], we are poised to find answers to some of these outstanding questions and advance our knowledge of mitochondrial copper biology.
Outstanding questions.
What are the cellular and organismal biomarkers of copper deficiency and excess?
What is the chemical identity of copper-ligand that transports copper to the mitochondria?
What is the role of the mitochondrial copper pool?
What are the molecular players involved in mitochondrial copper export?
How sensitive are currently available methods for in vivo copper detection?
Highlights.
Copper is an essential trace element that is crucial for the activity of several cuproenzymes.
Mitochondria play a key role in cellular copper homeostasis and house critical cuproenzymes including, cytochrome c oxidase and superoxide dismutase.
Perturbations in mitochondrial copper homeostasis cause human genetic disorders.
Copper therapeutics targeting mitochondria are gaining interest in the treatment of rare genetic disorders.
Acknowledgments
We thank Prof. Hans Zischka and Miriam C. Stein for their help in the preparation of this article. The research in the author’s laboratory is supported by the National Institute of General Medical Sciences, National Institutes of Health, United States awards R01GM143630 and R01GM111672 to V.M.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- CcO assembly factor
A broad term used to describe any proteins that are required for the assembly of mitochondrial cytochrome c oxidase
- Chelator
Small molecule that binds metal ions, making it non-bioavailable
- Ionophore
Small molecule that reversibly binds and transports ions through biological membranes
- Metallochaperone
Metal-binding protein that inserts metal ions into target protein(s) via specific protein-protein interactions
- Mitochondrial disorders
Mitochondrial disorders are defined by a biochemical defect in the mitochondrial respiratory chain and are caused by mutations in proteins that localize to mitochondria
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gupta A and Lutsenko S (2012) Evolution of copper transporting ATPases in eukaryotic organisms. Curr. Genomics 13, 124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim BE et al. (2008) Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol 4, 176–185 [DOI] [PubMed] [Google Scholar]
- 3.Chambers A et al. (2010) An exposure-response curve for copper excess and deficiency. J. Toxicol. Environ. Health B Crit. Rev 13, 546–578 [DOI] [PubMed] [Google Scholar]
- 4.Nose Y et al. (2006) Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 4, 235–244 [DOI] [PubMed] [Google Scholar]
- 5.Knöpfel M and Solioz M (2002) Characterization of a cytochrome b558 ferric/cupric reductase from rabbit duodenal brush border membranes. Biochem. Biophys. Res. Commun 291, 220–225 [DOI] [PubMed] [Google Scholar]
- 6.Wyman S et al. (2008) Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 582, 1901–1906 [DOI] [PubMed] [Google Scholar]
- 7.Arredondo M et al. (2003) DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am. J. Physiol. Cell Physiol 284, C1525–C1530 [DOI] [PubMed] [Google Scholar]
- 8.Pezacki AT et al. (2022) Oxidation state-specific fluorescent copper sensors reveal oncogene-driven redox changes that regulate labile copper(II) pools. Proc. Natl. Acad. Sci. U. S. A 119, e2202736119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linz R and Lutsenko S (2007) Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J. Bioenerg. Biomembr 39, 403–407 [DOI] [PubMed] [Google Scholar]
- 10.Linder MC (2016) Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics 8, 887–905 [DOI] [PubMed] [Google Scholar]
- 11.Roelofsen H et al. (2000) Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119, 782–793 [DOI] [PubMed] [Google Scholar]
- 12.Collins JF (2021) Copper nutrition and biochemistry and human (patho) physiology. In Adv. Food Nutr. Res 96, 311–364 [DOI] [PubMed] [Google Scholar]
- 13.Lutsenko S et al. (2010) Copper handling machinery of the brain. Metallomics 2, 596–608 [DOI] [PubMed] [Google Scholar]
- 14.Robinson NJ and Winge DR (2010) Copper metallochaperones. Annu. Rev Biochem 79, 537–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maryon EB et al. (2013) Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol 304, C768–C779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Culotta VC et al. (1997) The copper chaperone for superoxide dismutase. J. Biol. Chem 272, 23469–23472 [DOI] [PubMed] [Google Scholar]
- 17.Gudekar N et al. (2020) Metallothioneins regulate ATP7A trafficking and control cell viability during copper deficiency and excess. Sci. Rep 10, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attar N et al. (2020) The histone H3–H4 tetramer is a copper reductase enzyme. Science 369, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cobine PA et al. (2004) Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J. Biol. Chem 279, 14447–14455 [DOI] [PubMed] [Google Scholar]
- 20.Cobine PA et al. (2006) Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J. Biol. Chem 281, 36552–36559 [DOI] [PubMed] [Google Scholar]
- 21.Zischka H and Einer C (2018) Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol 102, 71–75 [DOI] [PubMed] [Google Scholar]
- 22.Dodani SC et al. (2011) A targetable fluorescent sensor reveals that copper-deficient SCO1 and SCO2 patient cells prioritize mitochondrial copper homeostasis. J. Am. Chem. Soc 133, 8606–8616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medlock AE et al. (2022) Prime Real Estate: Metals, Cofactors and MICOS. Front. Cell Dev. Biol 10, 892325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukihara T et al. (1995) Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å. Science 269, 1069–1074 [DOI] [PubMed] [Google Scholar]
- 25.Koch JR and Schmid FX (2014) Mia40 targets cysteines in a hydrophobic environment to direct oxidative protein folding in the mitochondria. Nat. Commun 5, 3041. [DOI] [PubMed] [Google Scholar]
- 26.Maxfield AB et al. (2004) Cox17 is functional when tethered to the mitochondrial inner membrane. J. Biol. Chem 279, 5072–5080 [DOI] [PubMed] [Google Scholar]
- 27.Brawley HN and Lindahl PA (2021) Low-molecular-mass labile metal pools in Escherichia coli: advances using chromatography and mass spectrometry. J. Biol. Inorg. Chem 26, 479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulet A et al. (2018) The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem 293, 1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vest KE et al. (2013) Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem 288, 23884–23892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vest KE et al. (2016) Overlap of copper and iron uptake systems in mitochondria in Saccharomyces cerevisiae. Open Biol. 6, 150223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christenson ET et al. (2018) In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J. Biol. Chem 293, 3819–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X et al. (2021) Mitochondrial copper and phosphate transporter specificity was defined early in the evolution of eukaryotes. Elife 10, e64690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker ZN et al. (2017) The mitochondrion: a central architect of copper homeostasis. Metallomics 9, 1501–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banci L et al. (2010) Affinity gradients drive copper to cellular destinations. Nature 465, 645–648 [DOI] [PubMed] [Google Scholar]
- 35.Timón-Gómez A et al. (2018) Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol, 76, 163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horng YC et al. (2004) Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem 279, 35334–35340 [DOI] [PubMed] [Google Scholar]
- 37.Morgada MN et al. (2015) Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. U. S. A 112, 11771–11776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carr HS et al. (2002) Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu (I)-binding protein. J. Biol. Chem 277, 31237–31242 [DOI] [PubMed] [Google Scholar]
- 39.Hiser L et al. (2000) Cox11p is required for stable formation of the CuBand magnesium centers of cytochrome c oxidase. J. Biol. Chem 275, 619–623 [DOI] [PubMed] [Google Scholar]
- 40.Leary SC et al. (2009) Human SCO2 is required for the synthesis of COII and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet 18, 2230–2240 [DOI] [PubMed] [Google Scholar]
- 41.Soma S et al. (2019) COA6 is structurally tuned to function as a thiol-disulfide oxidoreductase in copper delivery to mitochondrial cytochrome c oxidase. Cell Rep. 29, 4114–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pacheu-Grau D et al. (2020) COA6 facilitates cytochrome c oxidase biogenesis as thiol-reductase for copper metallochaperones in mitochondria. J. Mol. Biol 432, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longen S et al. (2009) Systematic analysis of the twin Cx9C protein family. J. Mol. Biol 393, 356–368 [DOI] [PubMed] [Google Scholar]
- 44.Barros MH et al. (2004) COX23, a homologue of COX17, is required for cytochrome oxidase assembly. J. Biol. Chem 279, 31943–31947 [DOI] [PubMed] [Google Scholar]
- 45.Swaminathan AB et al. (2022) A yeast suppressor screen links Coa4 to the mitochondrial copper delivery pathway for cytochrome c oxidase. Genetics 221, iyac090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bode M et al. (2015) Redox-regulated dynamic interplay between Cox19 and the copper-binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol. Biol. Cell 26, 2385–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nývltová E et al. (2022) Coordination of metal center biogenesis in human cytochrome c oxidase. Nat. Commun 13, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsvetkov P et al. (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tümer Z and Møller LB (2010) Menkes disease. Eur. J. Hum. Genet 18, 511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Członkowska A et al. (2018) Wilson disease. Nat. Rev. Dis. Primers 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaler SG (2013) Inborn errors of copper metabolism. Handb. Clin. Neurol 113, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedespan JM et al. (1999) Menkes disease: study of the mitochondrial respiratory chain in three cases. Eur. J. Paediatr. Neurol 3, 167–170 [DOI] [PubMed] [Google Scholar]
- 53.Morgello S et al. (1998). Menkes kinky hair disease with ragged red fibers. Dev. Med. Child Neurol 30, 812–816 [PubMed] [Google Scholar]
- 54.Walker M et al. (2022) On the dynamic and even reversible nature of Leigh syndrome: Lessons from human imaging and mouse models. Curr. Opin. Neurobiol 72, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhattacharjee A, et al. (2016) The activity of Menkes disease protein ATP7A is essential for redox balance in mitochondria. J. Biol. Chem 291, 16644–16658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaler SG et al. (2008) Neonatal diagnosis and treatment of Menkes disease. N. Eng. J. Med 358, 605–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dzieżyc-Jaworska K et al. (2019) Clinical manifestations of Wilson disease in organs other than the liver and brain. Ann. Transl. Med 7, S62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zischka H and Lichtmannegger J (2014) Pathological mitochondrial copper overload in livers of Wilson’s disease patients and related animal models. Ann. N Y Acad. Sci 1315, 6–15 [DOI] [PubMed] [Google Scholar]
- 59.Lichtmannegger J et al. (2016) Methanobactin reverses acute liver failure in a rat model of Wilson disease. J. Clin. Invest 126, 2721–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sternlieb I (1992) Fraternal concordance of types of abnormal hepatocellular mitochondria in Wilson’s disease. Hepatology 16, 728–732 [DOI] [PubMed] [Google Scholar]
- 61.Polishchuk EV et al. (2019) Activation of autophagy, observed in liver tissues from patients with Wilson disease and from ATP7B-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology 156, 1173–1189 [DOI] [PubMed] [Google Scholar]
- 62.Gu M et al. (2000) Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. Lancet 356, 469–474 [DOI] [PubMed] [Google Scholar]
- 63.Roberts EA et al. (2008) Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol. Genet. Metab 93, 54–65 [DOI] [PubMed] [Google Scholar]
- 64.Medici V et al. (2020) mtDNA depletion-like syndrome in Wilson disease. Liver Int. 40, 2776–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muchenditsi A et al. (2021) Systemic deletion of Atp7b modifies the hepatocytes’ response to copper overload in the mouse models of Wilson disease. Sci. Rep 11(1), 5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang CC et al. (2018) Metallothionein is elevated in liver and duodenum of Atp7b (−/−) mice. Biometals 31, 617–625 [DOI] [PubMed] [Google Scholar]
- 67.Rowan DJ et al. (2022) Metallothionein immunohistochemistry has high sensitivity and specificity for detection of Wilson disease. Mod. Pathol 35, 946–955 [DOI] [PubMed] [Google Scholar]
- 68.Ye B et al. (2001) Zinc metallothionein imported into liver mitochondria modulates respiration. Proc. Natl. Acad. Sci. U. S. A 98, 2317–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tafuri F et al. (2015) SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci 9, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Acevedo K et al. , (2019) Redox active metals in neurodegenerative diseases. J. Biol. Inorg. Chem 24, 1141–1157 [DOI] [PubMed] [Google Scholar]
- 71.Incecik F, et al. (2018) MEDNIK syndrome with a frame shift causing mutation in AP1S1 gene and literature review of the clinical features. Metab. Brain Dis 33, 2065–2068 [DOI] [PubMed] [Google Scholar]
- 72.Huppke P, et al. (2012) Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am. J. Hum. Genet 90, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinelli D et al. (2013) MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain 136, 872–881 [DOI] [PubMed] [Google Scholar]
- 74.Chiplunkar S et al. (2016) Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1. Metab. Brain Dis 31, 1195–1198 [DOI] [PubMed] [Google Scholar]
- 75.Bhoj E et al. (2014) Pathologic variants of the mitochondrial phosphate carrier SLC25A3: two new patients and expansion of the cardiomyopathy/skeletal myopathy phenotype with and without lactic acidosis. JIMD Rep. 19, 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horn N et al. (2019) Chelating principles in Menkes and Wilson diseases: Choosing the right compounds in the right combinations at the right time. J. Inorg. Biochem 190, 98–112 [DOI] [PubMed] [Google Scholar]
- 77.Hunsaker EW and Franz KJ (2019) Emerging opportunities to manipulate metal trafficking for therapeutic benefit. Inorg. Chem 58, 13528–13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duncan C and White AR (2012) Copper complexes as therapeutic agents. Metallomics 4, 127–138 [DOI] [PubMed] [Google Scholar]
- 79.Chen D et al. (2006) Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 66, 10425–10433 [DOI] [PubMed] [Google Scholar]
- 80.Chen S et al. (2013) Syntheses and antitumor activities of N’1,N’3-dialkyl-N’1,N’3-di-(alkylcarbonothioyl) malonohydrazide: the discovery of elesclomol. Bioorg. Med. Chem. Lett 23, 5070–5076 [DOI] [PubMed] [Google Scholar]
- 81.Soma S et al. (2018) Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. U. S. A 115, 8161–8166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guthrie LM et al. (2020) Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 368, 620–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsvetkov P et al. (2019) Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol 15, 681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garza NM et al. (2022) Elesclomol elevates cellular and mitochondrial iron levels by delivering copper to the iron import machinery. J. Biol. Chem 298, 102139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ogawa E and Kodama H (2012) Effects of disulfiram treatment in patients with Menkes disease and occipital horn syndrome. J. Trace Elem. Med. Biol 26, 102–104 [DOI] [PubMed] [Google Scholar]
- 86.Gohil VM (2021) Repurposing elesclomol, an investigational drug for the treatment of copper metabolism disorders. Expert Opin. Investig. Drugs 30, 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Williams JR et al. (2016) Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SODG93A mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis 89, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hung LW et al. (2012) The hypoxia imaging agent CuII (atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med 209, 837–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crouch PJ et al. (2009) Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. U. S. A 106, 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yuan X-Z et al. (2021) Management perspective of Wilson’s disease: early diagnosis and individualized therapy. Curr. Neuropharmacol 19, 465–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brewer GJ et al. (1987) Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch. Neurol 44, 490–493 [DOI] [PubMed] [Google Scholar]
- 92.Baldari S et al. (2020) Current biomedical use of copper chelation therapy. Int. J. Mol. Sci 21, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brewer GJ et al. (2006) Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch. Neurol 63, 521–527 [DOI] [PubMed] [Google Scholar]
- 94.Vallières C et al. (2017) Mitochondrial ferredoxin determines vulnerability of cells to copper excess. Cell Chem. Biol 24, 1228–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hong-Hermesdorf A et al. (2014) Subcellular metal imaging identifies dynamic sites of Cu accumulation in Chlamydomonas. Nat. Chem. Biol 10, 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leary SC and Ralle M (2020) Advances in visualization of copper in mammalian systems using X-ray fluorescence microscopy. Curr. Opin. Chem. Biol 55, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stroud DA et al. (2015) COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet 24, 5404–5415 [DOI] [PubMed] [Google Scholar]
- 98.Robinett NG et al. (2018) Eukaryotic copper-only superoxide dismutases (SODs): A new class of SOD enzymes and SOD-like protein domains. J. Biol. Chem 293, 4636–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cater MA et al. (2013) Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem. Biol 8, 1621–1631 [DOI] [PubMed] [Google Scholar]
- 100.Valnot I et al. (2000) Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am. J. Hum. Genet 67, 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stiburek L et al. (2009) Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol 296, C1218–C1226 [DOI] [PubMed] [Google Scholar]
- 102.Leary SC, et al. (2013) Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat 34, 1366–1370 [DOI] [PubMed] [Google Scholar]
- 103.Papadopoulou LC et al. (1999) Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet 23, 333–337 [DOI] [PubMed] [Google Scholar]
- 104.Jaksch M et al. (2000) Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet 9, 795–801 [DOI] [PubMed] [Google Scholar]
- 105.Verdijk RM et al. (2008) Phenotypic consequences of a novel SCO2 gene mutation. Am. J. Med. Genet. A 146A, 2822–2827 [DOI] [PubMed] [Google Scholar]
- 106.Tarnopolsky MA et al. (2004) Novel SCO2 mutation (G1521A) presenting as a spinal muscular atrophy type I phenotype. Am. J. Med. Genet. A 125A, 310–314 [DOI] [PubMed] [Google Scholar]
- 107.Pronicka E et al. (2013) The natural history of SCO2 deficiency in 36 Polish children confirmed the genotype-phenotype correlation. Mitochondrion 13, 810–816 [DOI] [PubMed] [Google Scholar]
- 108.Sacconi S et al. (2003) Mutation screening in patients with isolated cytochrome c oxidase deficiency. Pediatr. Res 53, 224–230 [DOI] [PubMed] [Google Scholar]
- 109.Rebelo AP et al. (2018) SCO2 mutations cause early-onset axonal Charcot-Marie-Tooth disease associated with cellular copper deficiency. Brain 141, 662–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Calvo SE et al. (2012) Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med 4, 118ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baertling F et al. (2015) Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum. Mutat 36, 34–38 [DOI] [PubMed] [Google Scholar]