

Abstract
In cap‐dependent translation, the eukaryotic translation initiation factor 4A (eIF4A1) is an mRNA helicase is involved in unwinding of the secondary structure, such as the stem‐loops, at the 5′‐leader regions of the key oncogenic mRNAs. This facilitates ribosomal scanning and translation of the oncogenic mRNAs. eIF4A1 has a regulatory role in translating many oncoproteins that have vital roles in several steps of metastases. Sridharan et. al. have discovered and provide a novel insight into how eIF4A1 can play a regulatory role in drug resistance by influencing the levels of pluripotent Yamanaka transcription factors and ATP‐binding cassette (ABC) transporters in triple‐negative breast cancer (TNBC) stem‐like cells. These findings may help us understand the molecular underpinnings of chemoresistance, especially in established metastases in TNBC. Importantly, eIF4A1 may form a novel clinical target in metastatic TNBC and the drug eFT226 from Effector Therapeutics targeting eIF4A1 is already in phase1‐2 clinical trial.
Keywords: ABC transporters, breast cancer stemness, chemoresistance, eIF4A1, triple‐negative breast cancer
Triple‐negative breast cancer (TNBC), defined by the lack of estrogen (ER) and progesterone receptors (PR), and the absence of human epidermal growth factor receptor 2 (HER2) overexpression, often leads to high‐grade invasive ductal carcinoma (IDC) in the patients accounting for one‐fourth of all breast cancer deaths. 1 , 2 Although the prevalence of TNBC is only around 15%, the metastatic nature of TNBC results in poor clinical outcome in majority of TNBC patients. Furthermore, the median overall survival (OS) in metastatic TNBC (mTNBC) is around 18 months, whereas in the luminal breast cancer cases, expressing ER/PR or HER2, it exceed 5 years. It affects more of the premenopausal and young African American women. Following standard platinum/taxane/anthracycline neoadjuvant chemotherapy (NACT) in TNBC patients, there is an initial response but followed by an increased rate of relapse frequently accompanied by distant metastases (TNBC paradox). The pathological complete response (pCR) to FDA‐approved targeted therapy against poly ADP ribose polymerase (PARP) in TNBC patients is unsatisfactory with the development of resistance. Relapse due to drug resistance or chemoresistance is a serious clinical problem frequently encountered in the TNBC patients. One of the main reasons for the relapse is attributed to the presence of a small subset of cells in the tumor called breast cancer stem‐like cells (BCSCs) or tumor‐initiating cells (TICs). BCSCs play a paramount role in tumor initiation, progression, and metastasis. 1 , 2 , 3 In terms of resistance to therapy, BCSCs impart either constitutive or acquired resistance to chemotherapeutics or radiotherapy, which leads to poor prognosis. 4 After the administration of the standard‐of‐care NACT against TNBC, which relies on actively dividing cells, the BCSCs survive the therapy along with some stromal cells that constitutes the minimal residual disease (MRD). 4 , 5 MRD is usually not detected by routine, clinical imaging techniques as those rely on certain minimum number of cells to be detected. After cessation of the cytotoxic or radiotherapy, the BCSCs from MRD, under appropriate stimulatory conditions, proliferate and undergo multi‐lineage differentiation program, replenishing the whole heterogeneous tumor. Such relapsed tumors are highly aggressive and are usually less responsive to the previously employed chemotherapy. They also likely become cross‐resistant to structurally and functionally unrelated chemotherapeutics, resulting in multidrug‐resistant (MDR) tumor cells. The unresponsive or chemoresistant MDR tumors present a grave prognosis as they are highly metastatic in nature. The intrinsic or acquired drug resistance and tolerance by BCSCs may arise due to many virtues in BCSCs. One of them is their ability to express a family of enzymes, such as aldehyde dehydrogenases (ALDHs). ALDHs can detoxify the drugs through metabolic conversion to less harmful or harmless products. Energetic BCSCs, which are a subset of BCSCs, display a high ALDH expression and demonstrate an upregulated capacity to proliferate and grow in an anchorage‐independent manner. 6 , 7 The other virtue by which the BCSCs display chemoresistance is through their expression of the ATP‐binding cassette (ABC) drug transporters on their plasma membrane. ABC transporters are integral membrane proteins that bind to the chemotherapeutic drugs in the cytoplasm of the tumor cells and pump them out to the cell exterior. Indeed, the BCSCs constitutively express ABCG2 or breast cancer resistance protein (BCRP) and serve as one of the key markers that are employed to identify BCSCs. 8 The other two frequently encountered drug transporters in BCSCs are ABCB1 or P‐glycoprotein and ABCC1. These ABC transporters and possibly other influx and efflux transporters that are co‐expressed, co‐localized, and have a substantial overlap in their functions may impart the ability to BCSCs to withstand inimical exposure to xenobiotics including chemotherapeutic drugs. This points a compelling need to develop more effective treatments for mTNBC patients and provides a strong rationale to target the BCSC compartment of the tumor or co‐target BCSCs and bulk tumor cells (non‐BCSCs) to overcome drug resistance in mTNBC.
A recent comprehensive review discusses various molecular targets in BCSCs that could be potentially targeted in combination with standard NACT. In particular, targeting various signaling receptors and their downstream mediators or effectors that would reduce stemness and overcome chemoresistance of BCSCs were described. 1 Ideally, an effective strategy would be to target the BCSCs that reduce their cancer stemness or plasticity. Targeting such mechanisms underlying cancer stemness or plasticity may lead to a durable therapy response. Sridharan et al, have discovered a key vulnerable node in triple‐negative BCSCs; in that, they are dependent on eukaryotic translation initiation factors (eIFs), especially eIF4A1. 9 They implicated a possible role for eIF4A1 in mediating drug resistance in their paclitaxel‐resistant TNBC model in vitro. eIF4A1 is an mRNA helicase that unwinds the classical secondary structures located at the 5′‐leader sequence of selected, vital oncogenic mRNAs 10 (Figure 1). The eIF4A1‐facilitated translation of oncogenic mRNA repertoire leads to the synthesis of many oncoproteins, such as survivin or BIRC5, myeloid cell leukemia 1(MCL1), cyclin D1, cyclin D3, mucin‐1C (MUC‐1C), Rho kinase 1 (ROCK1), ADP ribosylation factor 6 (ARF6), and murine double minute 2/human double minute 2 (MDM2/HDM2) and ADP ribosylation factor 6 (AFR6), which are vital for tumor cell survival both at primary and metastatic sites, proliferation, migration, local invasion, metastasis, and chemoresistance. 11 , 12 , 13 , 14 , 15 , 16
FIGURE 1.
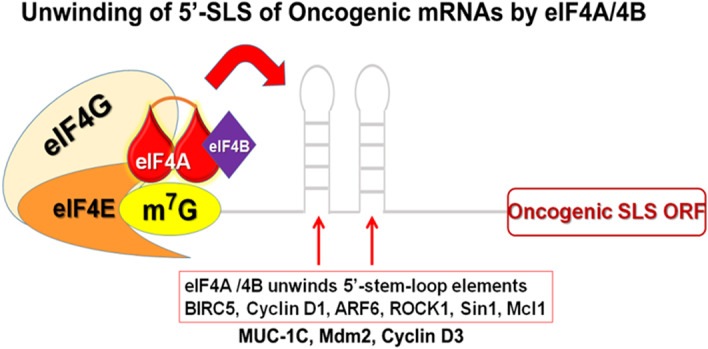
Unwinding of 5′‐leader sequence of oncogenic mRNAs by eIF4A1. eIF4A1 bound to the cap structure of the oncogenic mRNAs will unwind the classical secondary stem‐loop structures at the 5′‐leader sequence of oncogenic mRNAs. This will facilitate the facile scanning of the ribosome for the first AUG codon
Survivin plays a key role as a functional checkpoint for both mitosis and apoptosis in cancer cells; survivin and MCL1 are involved in chemoresistance as well. 17 Cyclin D1 and cyclin D3 are also vital for clonogenicity and chemoresistance of the BCSCs.. 10 , 18 Although nuclear cyclin D1 is known for its role in cell proliferation, 19 the cytoplasmic cyclin D1 has a novel, non‐canonical role in cell migration. 20 , 21 Cyclin D1 activates CDK4/6, a current target in the clinics with palbociclib for chemoresistant forms of BC. 22 ARF6 is one of the key proteins required for cell adhesion, migration, and invasion of cancer cells. 23 ROCK1 promotes cell polarization and persistent directional migration (chemotaxis). 24 , 25 MDM2/HDM2, being an E3 ligase, can ubiquitinate wild‐type p53 and target it for degradation. 26 In addition, perturbation of the chemokine GPCR, CXCR4, signaling promotes BC cell migration by regulating tumor cell adhesion events through provision of an optimal level of ROCK1 activity for effective cell migration. 27 The chemokine receptor, CXCR4, has been demonstrated to activate Gαi/mTORC1 axis, which is upstream of eIF4A to promote spontaneous metastasis. 28 Signaling from CXCR4 can also activate ribosomal S6 kinases—p90 ribosomal S6 kinase (p90rsk ‐ via ERK pathway) 29 and p70‐S6 kinase (p70rsk ‐ via mTORC1 pathway) (Figure 2). 28 These two major kinases feed into eFF4A by phosphorylating its endogenous inhibitor programmed cell death 4 (PDCD4) and targets it for degradation. This frees up some eIF4A from the PDCD4‐bound pool, which now can be incorporated into the eIF4F complex to initiate the cap‐dependent translation of oncogenic mRNAs. 30 Interestingly, high level of expression of the chemokine receptor, CXCR4, in TNBC specimens predicts poor clinical outcome. 31
FIGURE 2.
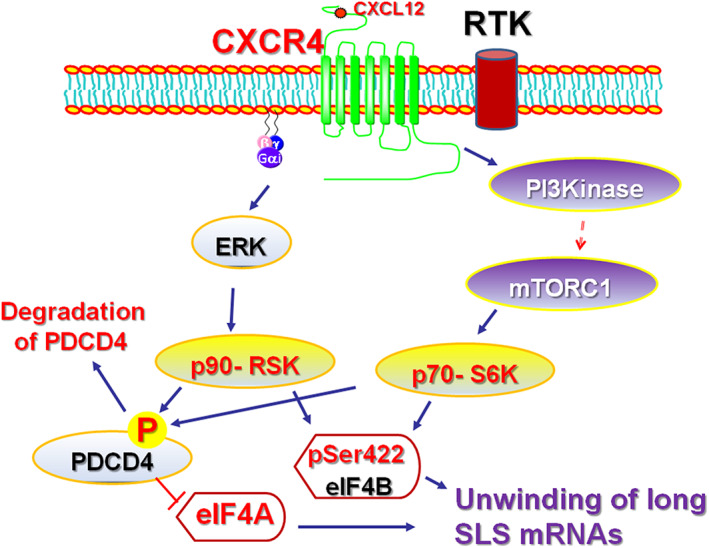
Signaling from G‐protein coupled receptors and receptor tyrosine kinases feed into activation of eIf4A1. The main signaling nodes, the phosphatidylinositol 3‐kinase (PI3K) and extracellular signal‐regulated (ERK) kinases, are activated by GPCRs, such as CXCR4, and receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2). PI3K and ERK subsequently activate ribosomal S6 kinases that phosphorylate the programmed cell death 4 (PDCD4) and mark it for degradation. This frees up the eIF4A1 to participate in the cap‐dependent protein translation. These pathways phosphorylate eIF4B, an ancillary protein that enhances the activity of eIF4A1, which speeds up the protein translation process
Through targeting this single mRNA helicase molecule, eIF4A1, it seems, the translation of a whole gamut of aforementioned oncogenic mRNAs can be inhibited. Sridharan et al, have demonstrated that some of the Yamanaka factors or transcription factors that regulate pluripotency or stemness or plasticity, such as OCT4, SOX2, and NANOG, were significantly downregulated when eIF4A1 was genetically ablated. These pluripotent transcription factors also cause drug resisatnce. A similar outcome was obtained when eIF4A1 was pharmacologically targeted with the natural, small molecule inhibitor, Rocaglamide A. This was the first report that highlights that targeting of eIF4A could downregulate all three pluripotency transcription factors that regulate BC stemness. 9 Furthermore, the landmark finding is that when eIF4A1 is targeted by Rocaglamide A, the protein level of ABCB1 or P‐glycoprotein was significantly reduced. This was without any direct targeting of any of the ABC drug transporters. The mechanistic details as to how the targeting of eIF4A1 would reduce the BC stemness or diminish the protein levels of drug transporters remains to be elucidated. The interesting feature with targeting of eIF4A1 was equally effective in both therapy‐naïve and paclitaxel‐resistant TNBC cells. Furthermore, knocking out of the eIF4A1 in paclitaxel‐resistant TNBC cells reduced the pre‐existing resistance to paclitaxel dramatically. Overall, this brings a salient feature in that targeting eIF4A1 controls both BC stemness as well as drug resistance. Moreover, the stemness and chemoresistance are highly related to each other. 8 , 9 Importantly, the protein level of eIF4A1 is present in similar amounts between BCSCs and non‐BCSCs (bulk tumor cells). So, when eIF4A1 is targeted, both cellular populations will perish at the same time with less chance for MRD and tumor relapse. Thus, eIF4A1 is an actionable novel molecular target in the BCSC compartment, and controlling the helicase activity of eIF4A1 may lead to a favorable outcome in clinical chemoresistant cases of TNBC. Currently, Effector Therapeutics (NCT04092673) is recruiting patients for a phase I‐II clinical trial for targeting eIF4A1 with their synthetic small‐molecule inhibitor eFT226, which is somewhat analogous to Rocaglamide A.
As BCSCs play a role in drug tolerance and resistance, targeting the plasticity may lead to a profound and more durable clinical response. Targeting eIF4A1 seems a promising strategy to overcome TNBC stemness and chemoresistance in in vitro systems. A combinatorial treatment approach comprehensively targeting stem‐like cells may overcome the MDR encountered in the clinic and may result in a better objective treatment response in mTNBC.
ETHICAL STATEMENT
Not applicable
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
AUTHOR CONTRIBUITIONS
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, D.R., A.K.T.; Data curation, D.R.; Funding acquisition, D.R., A.K..; Supervision, D.R.; Visualization, D.R.; Writing original draft, D.R., A.K.T.; Writing review editing, D.R., A.K.T.; Project administration, D.R., A.K.T.
ACKNOWLEDGEMENTS
This manuscript has been supported, in part, by National Institute of Health (NIH)/National Cancer Institute (NCI) grant R21CA202176, Ohio Cancer Research foundation (OCR), and University of Toledo startup grant (F110796) (DR); University of Toledo startup grant and Susan G. Komen Breast Cancer Foundation (CCR18548498) (AT).
Funding information NCI‐NIH, Grant/Award Number: R21CA202176; Susan G. Komen for the Cure, Grant/Award Number: CCR18548498; Ohio Cancer Research foundation (OCR), and University of Toledo, Grant/Award Number: F110796
Contributor Information
Dayanidhi Raman, Email: dayanidhi.raman@utoledo.edu.
Amit K. Tiwari, Email: amit.tiwari@utoledo.edu.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Sridharan S, Howard CM, Tilley AMC, et al. Novel and alternative targets against breast cancer stemness to combat chemoresistance. Front Oncol. 2019;9:1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sampieri K, Fodde R. Cancer stem cells and metastasis. Semin Cancer Biol. 2012;22:187‐193. [DOI] [PubMed] [Google Scholar]
- 3. Dittmer J. Breast cancer stem cells: features, key drivers and treatment options. Semin Cancer Biol. 2018;53:59‐74. [DOI] [PubMed] [Google Scholar]
- 4. Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther. 2016;160:145‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross AA. Minimal residual disease in solid tumor malignancies: a review. J Hematother. 1998;7:9‐18. [DOI] [PubMed] [Google Scholar]
- 6. Sotgia F, Fiorillo M, Lisanti MP. Hallmarks of the cancer cell of origin: comparisons with “energetic” cancer stem cells (e‐CSCs). Aging. 2019;11:1065‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fiorillo M, Sotgia F, Lisanti MP. “Energetic” cancer stem cells (e‐CSCs): a new hyper‐metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front. Oncologia. 2018;8:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor‐initiating cells. Expert Opin Drug Metab Toxicol. 2016;12:633‐644. [DOI] [PubMed] [Google Scholar]
- 9. Sridharan S, Robeson M, Bastihalli‐Tukaramrao D, et al. Targeting of the eukaryotic translation initiation factor 4A against breast cancer Stemness. Front Oncol. 2019;9:1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Waldron JA, Raza F, Le Quesne J. eIF4A alleviates the translational repression mediated by classical secondary structures more than by G‐quadruplexes. Nucleic Acids Res. 2018;46:3075‐3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malka‐Mahieu H, Newman M, Desaubry L, Robert C, Vagner S. Molecular pathways: the eIF4F translation initiation complex‐new opportunities for cancer treatment. Clin Cancer Res. 2017;23:21‐25. [DOI] [PubMed] [Google Scholar]
- 12. Leppek K, Das R, Barna M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2017;19:158‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steinberger J, Chu J, Maiga RI, Sleiman K, Pelletier J. Developing anti‐neoplastic biotherapeutics against eIF4F. Cell Mol Life Sci. 2017;74:1681‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gandin V, Masvidal L, Hulea L, et al. nanoCAGE reveals 5′ UTR features that define specific modes of translation of functionally related MTOR‐sensitive mRNAs. Genome Res. 2016;26:636‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rubio CA, Weisburd B, Holderfield M, et al. Transcriptome‐wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 2014;15:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin C, Rajabi H, Rodrigo CM, Porco JA Jr, Kufe D. Targeting the eIF4A RNA helicase blocks translation of the MUC1‐C oncoprotein. Oncogene. 2013;32:2179‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee KM, Giltnane JM, Balko JM, et al. MYC and MCL1 cooperatively promote chemotherapy‐resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26:633–647.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petty WJ, Voelzke WR, Urbanic JJ, et al. High cyclin D3 expression confers erlotinib resistance in aerodigestive tract cancer. Lung Cancer. 2011;74:384‐391. [DOI] [PubMed] [Google Scholar]
- 19. Lee RJ, Albanese C, Fu M, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F‐dependent signaling pathway. Mol Cell Biol. 2000;20:672‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Z, Jiao X, Wang C, et al. Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res. 2006;66:9986‐9994. [DOI] [PubMed] [Google Scholar]
- 21. Li Z, Wang C, Jiao X, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol. 2006;26:4240‐4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lamb R, Lehn S, Rogerson L, Clarke RB, Landberg G. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor‐dependent divergent functions in breast cancer migration and stem cell‐like activity. Cell Cycle. 2013;12:2384‐2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sabe H. Requirement for Arf6 in cell adhesion, migration, and cancer cell invasion. J Biochem. 2003;134:485‐489. [DOI] [PubMed] [Google Scholar]
- 24. Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lammermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol. 2009;21:636‐644. [DOI] [PubMed] [Google Scholar]
- 26. Cunningham TA, Chapman E, Schatz JH. eIF4A inhibition: ready for primetime? Oncotarget. 2018;9:35515–35516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Struckhoff AP, Vitko JR, Rana MK, et al. Dynamic regulation of ROCK in tumor cells controls CXCR4‐driven adhesion events. J Cell Sci. 2010;123:401‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dillenburg‐Pilla P, Patel V, Mikelis CM, et al. SDF‐1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/Galphai/mTORC1 axis. FASEB J. 2015;29:1056‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saini V, Staren DM, Ziarek JJ, et al. The CXC chemokine receptor 4 ligands ubiquitin and stromal cell‐derived factor‐1alpha function through distinct receptor interactions. J Biol Chem. 2011;286:33466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Not applicableShahbazian D, Parsyan A, Petroulakis E, Hershey J, Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto‐oncogenic signaling pathways. Cell Cycle. 2010;9:4106‐4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chu QD, Panu L, Holm NT, Li BD, Johnson LW, Zhang S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J Surg Res. 2010;159:689‐695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.