

Abstract
Biopharmaceutical classification system (BCS) is an advanced tool used for classifying medicines based on dissolution, water solubility, and intestinal permeability, which affect the absorption of active pharmaceutical ingredients (API) from immediate-release solid oral forms. It is useful to the formulation researchers to develop novel dosage forms based on modernistic rather than experimental approaches. The current review focuses on the fundamentals, objectives, guidance of BCS, characteristics of BCS drugs, their importance and applications of BCS. This review explains the challenges in drug development in terms of solubility and in vivo disposition. In the current review, new strategies for improving BCS II drug solubility as well as biopharmaceutical drug disposition properties which are utilized throughout the early stages of drug development and commercialization are mainly discussed.
Keywords: Bioavailability, biopharmaceutical classification system, drug solubility, dissolution, drug disposition, bioequivalence, new drug application
INTRODUCTION
Biopharmaceutical classification system (BCS) is an advanced tool used for classifying drug substances on dissolution, intestinal permeability and water solubility.1 In 1995, a theoretical approach for comparing in vitro drug dissolution with in vivo bioavailability was first conducted by Amidon et al.1 BCS is a pharmaceutical development tool that is used for basic management in the drug discovery and early development of novel medications.1,2
The criteria for BCS direction for biowaiver are given by the United States Food and Drug Administration (FDA or USFDA), World Health Organization (WHO) and European Medicines Agency.3,4 The BCS data assist the particular researcher in constructing a dosage form based on intuition rather than experimental approaches (FDA rules, 2000).5 The BCS conceptual structure requirements can be linked to New Drug Application and Abbreviated New Drug Application approvals as well as scale up and post-approval alterations in medication manufacturing.
BCS is a conceptual structure that discusses three rate-limiting phases in oral retention.
• Release of drugs from dosage form
• Gastrointestinal (GI) tract arrangement of disintegrated form
• Saturation through GI membrane into hepatic circulation6
The intestinal permeability arrangement is determined by a correlation with intravenous infusion and solubility characterization is determined according to United States Pharmacopeia (USP).7
BCS follows fick’s first law utilized for membrane permeability
Jf= Pm Ci (equation 1)
Where,
Jf = Drug flux rate (mass/area/time)
Pm = Membrane permeability
Ci = Concentration of the drug at the intestinal membrane surface
BCS acts as a regulatory tool and replaces certain bioequivalent studies, which are have been accurate in vitro dissolution tests and ensure avoiding unnecessary drug exposure to healthy volunteers.8
BCS classification
According to BCS system, drugs are classified into four types based on their intestinal permeability and solubility. BCS classification is based on key parameters like solubility, dissolution rate and permeability, which control absorption. In case of class I drugs, absorption is maximum, class II drugs are showing solubility limited, class III drugs have permeability limited, class IV drugs have poorly absorbed mentioned in Table 1.6,7,8 Apparent permeability index (Papp) is the index used to assess the degree of permeability of drug substances. The permeability coefficient, which is a measure of flow to the drug concentration in the donor compartment. Papp of any drug substance can be calculated using in vitro, ex vivo, in situ, and in vivo techniques.9
Table 1. BCS classification system6,7,8.
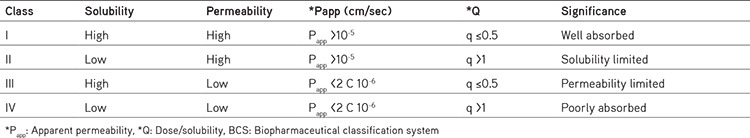
Solubility
The amount of a substance that can be dissolved in a given amount of solvent is called solubility. A medicine that can be dissolved in 250 mL or less of water throughout a pH range of 1-8 is deemed an excellent dissolved pharmaceutical.10
Permeability
Permeability is the quality or state of being permeable. When a medicine has an absorption rate of more than 90% of the prescribed dosage and is stable in the stomach, it is termed an exception penetrable pharmaceutical.11
Dissolution rate
The process by which a solute dissolves into a solvent and produces a solution is known as dissolution. When 85% of the labeled quantity of drug substance dissolved in 30 min using USP equipment 1 at 100 rpm or apparatus 2 at 50 rpm in a volume of 900 mL buffer solutions (0.1 N HCl/pH 4.5 buffer/pH 6.8 buffer without enzymes), the drug product is regarded to have fast dissolution.12,13,14
Dimensionless parameter
BCS characterization is connected to medication dissolution and absorption display, which are essential factors for regulating medication absorption as a set of dimensionless numbers.15,16 Drug properties and their corresponding dimensionless parameters and their significance related to them are highlighted in Table 2.
Table 2. Drug properties influencing absorption6,7,8.
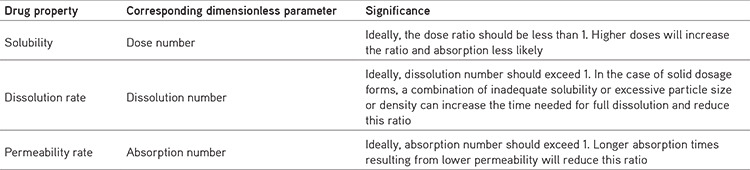
Absorption number (An) = Average residence time/average absorption time (equation 2)
The dissolution number (DSn) = Average residence time/average dissolution time (equation 3)
Dose number (Dn) = Mass of drug/uptake volume of 250 mL × drug solubility (equation 4)
Objectives
• The goal of BCS is to evaluate in vivo performance of medicinal products based on in vitro permeability and solubility data.17
• To provide techniques for categorizing medicinal products based on solubility and permeability properties as well as dosage form dissolution.
• We improved the efficiency of drug development and review processes by proposing a mechanism to perform clinical bioequivalence tests expandable.
Importance
To replace certain bioequivalent studies, BCS acts as a regulatory tool. It is applicable in both preclinical and clinical examinations. BCS can reduce the time and money for the immediate release orally administered drugs, which meet particular criteria; the FDA will allow a waiver for costly and tedious bioequivalence studies. It acts as a guiding tool for selecting the formulation of new dosage forms, development of various oral drug delivery systems.18
Class II drugs
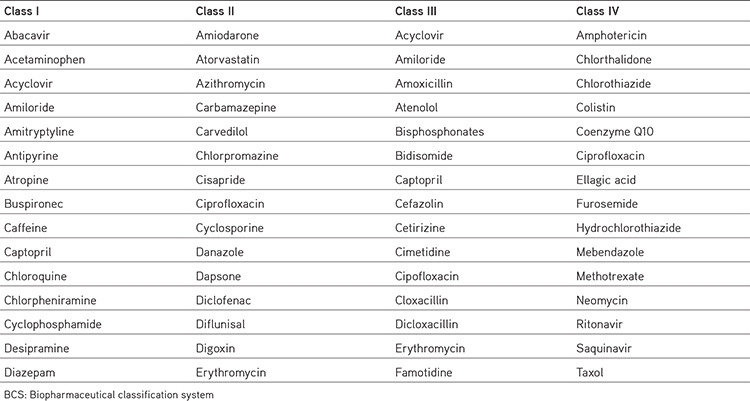
BCS class II drugs have high permeability and low solubility. These medications have a high absorption number, but a small disintegration number. In vivo drug dissolution is then a rate limiting advanced step for absorption, except in very high dose numbers. These drugs have varied bioavailability and require improved solubility or dissolution to increase bioavailability. These compounds are suitable to outline the sustained release and controlled release formulations. In vitro-in vivo correlation applies normally to class II drugs. Based on solubility and permeability, drugs are classified into four types (class I to class IV) examples mentioned in Table 3.18
Table 3. Examples of some model drugs as per BCS10,11,12,13,14.
Biowaiver
Biowaiver is most commonly used in the administrative drug approval procedure, when the drug application is confirmed based on the proof of proportionality other than in vivo comparison testing. This waiver applies to both the pre- and post-approval stages. BCS-based biowaiver is applicable for immediate-release solid oral formulations containing the API approved by WHO.19
Biowaiver extension potential
BCS class II medicines are effective and completely absorbed, when taken orally. Class II drugs are weak acids with pKa values of ≤4.5 and intrinsic solubility (dissolvability of the unionized form) of ≥0.01 mg/mL, which are ineffectively dissolvable. At pH values typical of the fasted state in the jejunum (about pH 6.5), these medications will have a solubility of >1 mg/mL, produce about rapid and steady dissolution of the medication. Class II drugs are inadequately dissolvable at gastric pH, in which pH is considerably less than pKa because the small intestinal transit time is more consistent and when fasting longer than the gastric residence time (3 hr), drugs these physical characteristics will have enough time to dissolve. Class II drugs meet the permeability measure, biowaiver for products that break down quickly at the pH levels regularly in the small digestive tract, it has been suggested that BCS class II drugs have a biowaiver enhancement potential.19
Applications
Dissolution or solubility is the rate limiting factor in BCS II and it has a substantial impact on absorption and bioavailability. Lyophilization, micronization, microemulsion, inclusion of surfactants, solid dispersion, and use of complexing agents such as cyclodextrins; these are the methods used to improve solubility.19,20,21,22 Zer-Os tablet innovation, soft gel, triglas, and nano-sized formulations are enhancement techniques, for example; nanocrystals, nanosuspension, and nanoemulsions are useful methods for increasing the solubility and bioavailability of low water soluble drugs23,24,25 mentioned in Table 4.
Table 4. Techniques employed for BCS II drugs.
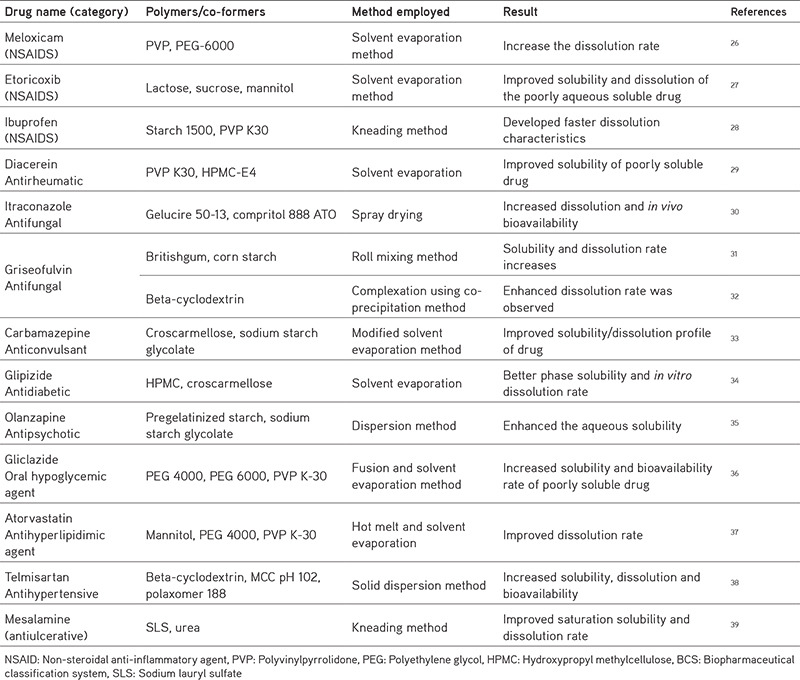
Techniques to enhance the solubility of BCS II drugs
Physical modifications
Micronization: Spray drying or use fluid energy or a jet mill to reduce the particle size to 1-10 microns. A reduced particle size will increase the surface area and improves bioavailability. Examples: Griseofulvin, sulfa, and certain steroidal drugs.26
Nanoionization: Powdered drug is converted to nanocrystals of size 200-600 nm using technologies such as pearl processing, homogenization in water, and homogenization using non-aqueous medium. Examples: estradiol, doxorubicin, cyclosporin, and paclitaxel.27
Sonocrystallization: Ultrasound in the range of 20 KHz-5 KHz is used to induce crystallization in sonocrystallization. Examples: This method increased the solubility of ketoconazole by 5.517 folds.28
Use of polymorphs, amorphous, solvates, and metastable form: Because the vitality required to transfer the crystal lattice is more than that necessary for amorphous solid, amorphous forms are more soluble than crystal structures. Metastable forms are more soluble than stable ones. Because hydrates are already associated with water, anhydrates are more soluble, so require less energy for crystal separation. Thus, the order of solubility of different solid forms of drugs is
Amorphous > Metastable > Stable > Anhydrates > Hydrates > Solvates > Non-solvates
Eutectic mixtures: The soluble carrier in the eutectic mixtures dissolves when exposed to water, leaving the drug in a microcrystalline state that solubilize rapidly. They are inexpensive and easily prepared. Examples: paracetamol with urea, griseofulvin with urea, griseofulvin with succinic acid.29
Solid dispersions: A hydrophilic matrix (polyvinylpyrrolidone, povidone, polyethylene glycol, surfactant such as sodium lauryl sulfate, tween 80, pluronic F-68) and hydrophobic drug (fats, oils, waxes, alkanes, and other greasy substances) are used in preparing solid dispersions. Methods for preparing solid dispersions including.
Hot-melt method (fusion method): Drug and the carrier are heated directly until they melt and then rapidly cooled with ice by continuous stirring to solidify. After that, it is crushed, pulverized, sieved and compressed into tablets.30
Solvent evaporation method: Medication and the carrier were dissolved in a common solvent and the dissolved content was evaporated under vacuum to form an amorphous precipitate.30 Examples: Meloxicam, naproxen, nimesulide.31
Hot melt extrusion: It is the same as the combination technique, except the extruder does the extreme mix. It is appropriate for large-scale preparations.31 Examples: Ritonavir.32
Chemical modifications
Change in pH: The easiest approach to enhance solubility of organic ionized solutions is to change the pH of the formulation. A change in pH can be done by;
• Use of buffers
• In situ salt formation
Salt formation: When compared with pure API drugs, salt forms have better solubility. Example: Antacid metal salts of acidic medicines, such as penicillin, solid corrosive salts of vital pharmaceuticals, such as atropine.33
Prodrug: Solubility of the drugs can be increased by converting a pharmacologically inactive substance into a pharmacologically active drug. Examples: acyclovir, fluorouracil, cyclophosphamide, carbamazepine, captopril, and carisoprodol.
Atomic elucidation with cyclodextrins: The beta and gamma ray cyclodextrins can form sub-atomic consideration structures since they have a cavity to accommodate lipophilic medicines as guests and the exterior of the transporter is hydrophilic. As a result, there is a significant increase in dissolving rate and solubility. Thiazide diuretics, barbiturates, and benzodiazepines are examples of drugs with enhanced bioavailability due to this method.34
Derivatization: Conversion of a chemical compound into a product, which shows a similar chemical structure called derivative with different solubilities’s that of the adduct.35
Miscellaneous modifications
Super critical fluid (SCF) recrystallization: These fluids have temperatures and pressures that are higher than their critical temperature and exhibit the characteristics of both gases and liquids. SCFs are profoundly compressible at close fundamental temperatures, modifying thickness and mass power by allowing weight modification. When the drug particles were dissolved in SCF, they crystallized with smaller molecule sizes.36
Use of surfactants: Surfactants increase the disintegration rate by advancing wetting and infiltration of disintegration liquid into the medication particles, when used in the focus beneath their basic micelle fixation because drug captured in the micelle structure failed to partition in the dissolution fluid above the critical micelle concentration. Example: a steroid-like spironolactone bioavailability has been enhanced by this technique.36,37,38
Solvent deposition: Poorly soluble medicines are dissolved and deposited on an inert, hydrophilic, and solid matrix by evaporation of the solvent using organic solvents such as alcohol. Example: Nifedipine.
Precipitation: Medication that is poorly water-soluble is first dissolved in a suitable organic solvent, then quickly mixed with a non-dissolvable to precipitate the medication in nanosize particles, and this result is known as a hydrosol.39 Example: cyclosporine.
Co-solvents: Solubility is low for weak electrolytes and non-polar compounds. Solubility can be increased by altering the polarity of those molecules by adding organic co-solvents (mixing miscible or partially miscible solvents) to water, which drastically affects medication solubility.40 Example: Etoricoxib, glipizide, glyburide, glimepiride and pioglitazone.
Hydrotrophy: The addition of a significant number of additives (hydrotropic agent) to the drug solution increases the medication’s water solubility.41 Example: Ethanol, resorcinol, pyrogallol, catechol and procaine hydrochloride.
Selective adsorption on insoluble carriers: Adsorbents can enhance solubility by forming weak physical bonding between the drug and adsorbent and can also by hydration and swelling of clay in aqueous media. Example: inorganic clay bentonite can improve the dissolution of drugs like griseofulvin, prednisone, and indomethacin.42
Drug disposition
The significant route of elimination of drugs showing high intestinal permeability in humans is mainly by metabolism and the drugs having weak intestinal permeability rates are mainly excreted as unchanged drugs in the urine and bile in humans. In 2005 drug disposition was first observed by Wu and Benet43, who proposed a system called Biopharmaceutics Drug Disposition Classification System (BDDCS): in case of class 1 and 2 drugs showing extensive metabolism, class 3 and 4 drugs showing a poor metabolism rate shown in Table 5.43,44,45
Table 5. Biopharmaceutics drug disposition classification system43,44,45.
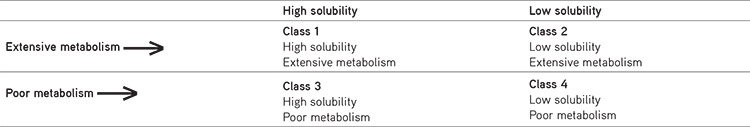
BDDCS system estimates the effect of food, absorption as well as efflux transporters, route of excretion on overall drug absorption and the permeability of immediate-release oral dose forms is less than bioavailability. BDDCS system as an extension of BCS.44
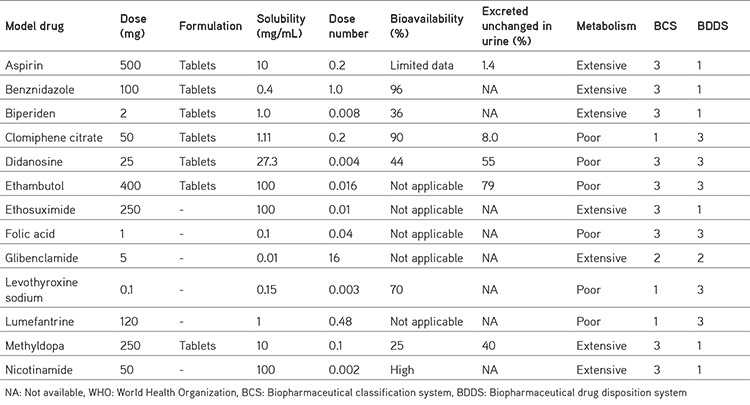
Because BDDCS is a replacement for permeability, they proposed that medications that demonstrate metabolism as a main route of elimination be deemed highly permeable. Low permeable drugs are those, whose primary route of excretion is renal and biliary excretion of unmodified medicine.45 Data on medication disposition for a few medicines from the WHO essential drug list are shown in Table 6.46
Table 6. Drug disposition data from WHO essential medicines list46.
CONCLUSION
BCS serves as a regulatory tool for the progress of various oral drug transport advancements. The BCS considers three major factors, dissolution, solubility and intestinal permeability, which govern the rate and degree of medication absorption from immediate solid dosage forms. It is a controlling device for anticipating in vivo execution of the medicinal substance and the improvement of the medication delivery system. The data generated from the solubility and permeability in pipeline drug discovery or development can be used for early pipeline compound categorization. The BCS’s advantageous circumstances include reduced medication exposure to a large panel of human participants and in some cases shorter drug product development time, in addition to significant cost savings.
Acknowledgments
The authors are thankful to the management of Vignan’s Foundation for Science, Technology and Research (VFSTR) for providing the necessary facilities and infrastructure to complete this work.
Footnotes
Ethics
Peer-review: Externally peer-reviewed.
Authorship Contributions
Concept: R.S., Design: R.S., J.C., Data Collection or Processing: R.S., S.K., Analysis or Interpretation: R.S., J.C., Literature Search: R.S., S.K., Writing: R.S., J.C., S.K.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.Reddy BBK, Karunakar A. Biopharmaceutics classification system: a regulatory approach. Dissolution Technol. 2011;18:31–37. [Google Scholar]
- 3.Guidance for Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Rockville, MD, Department of Health and Human Services, US Food and Drug Administration, 2000. Available from: [Internet] https://www.gmp-compliance.org/files/guidemgr/UCM070246.pdf.
- 4.No Authors. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations, Forty-ninth report. Geneva, World Health Organization. 2015;992:134–184. [PubMed] [Google Scholar]
- 5.Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. European Medicines Agency. 2008. (CPMP/EWP/QWP/1401/98 Rev.1). Available from: [Internet] https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf.
- 6.Siya D, Kunde S, Bhilegaonkar S, Gajre P. Biopharmaceutical classification system: a brief account. IJRM Human. 2015;1:20–46. [Google Scholar]
- 7.Wagh MP, Patel JS. Biopharmaceutical classification system: scientific basis for biowaiver extensions. Int J Pharm Pharm Sci. 2010;2:12–19. [Google Scholar]
- 8.Chavda HV, Patel CN, Anand IS. Biopharmaceutics classification system. Sys Rev Pharm. 2010;1:62–69. [Google Scholar]
- 9.Dokoumetzidis A, Valsami G, Macheras P. Modeling and simulation in drug absorption processes. Xenobiotica. 2007;37:1052–1065. doi: 10.1080/00498250701502114. [DOI] [PubMed] [Google Scholar]
- 10.Verbeeck RK, Junginger HE, Midha KK, Shah VP, Barends DM. Biowaiver monographs for immediate-release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: chloroquine phosphate, chloroquine sulfate, and chloroquine hydrochloride. J Pharm Sci. 2005;94:1389–1395. doi: 10.1002/jps.20343. [DOI] [PubMed] [Google Scholar]
- 11.Nagesh XB, Murti V. BCS Class IV Drugs: Develop or Discard? AAPS annual meeting and exposition. San Diego Convention Centres, San Diego, CA. 2007:11–15. [Google Scholar]
- 12.Bala I, Bhardwaj V, Hariharan S, Sitterberg J, Bakowsky U, Ravi Kumar MN. The design of biodegradable nanoparticles: a novel approach to encapsulating poorly soluble phytochemical ellagic acid. Nanotechnology. 2005;16:2819–2822. [Google Scholar]
- 13.Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–246. doi: 10.1016/s0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- 14.Sharma P, Varma MV, Chawla HP, Panchagnula R. In situ and in vivo efficacy of peroral absorption enhancers in rats and correlation to in vitro mechanistic studies. Farmaco. 2005;60:874–883. doi: 10.1016/j.farmac.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Kumar S, Bhargava D, Thakkar A, Arora S. Drug carrier systems for solubility enhancement of BCS class II drugs: a critical review. Crit Rev Ther Drug Carrier Syst. 2013;30:217–256. doi: 10.1615/critrevtherdrugcarriersyst.2013005964. [DOI] [PubMed] [Google Scholar]
- 16.Jaiswal SB. Biopharmaceutics and pharmacokinetics-a treatise. Vallabh Prakashan, (1st eds). 1995:347–352. [Google Scholar]
- 17.Yasir M, Asif M, Kumar A, Aggarval A. Biopharmaceutical classification system: an account. Int J PharmTech Res. 2010;2:1681–1690. [Google Scholar]
- 18.Ramesh V, Meenakshi S, Jyothirmayee N, Bullebbai M, Noorjahan SK, Rajeswari G, Nagesh Babu G, Madhavi D. Enhancement in solubility, dissolution rate and bioavailability of BCS class II drugs. Int J Pharm Chem. 2016;2:80–95. [Google Scholar]
- 19.Devane J. Oral drug delivery technology: addressing the solubility/permeability paradigm. Pharm Technol. 1998;22:68–80. [Google Scholar]
- 20.Sunder S, Nair R. Methods of canonization of drugs for enhancing their dissolution. Euro J Adv Engg Tech. 2016;3:101–110. [Google Scholar]
- 21.Reddy MS, Narendra Y, Fazal-Ul-Haq S. Md. Solubility enhancement of the poorly soluble drug ketoconazole by self-emulsifying drug delivery system. Int J Pharm Biol Sci. 2018;8:111–127. [Google Scholar]
- 22.Shah K, Borhade S, Londhe V. The utilization of co-crystallization for solubility enhancement of a poorly soluble antiretroviral drug – ritonavir. Int J Pharm Pharm Sci. 2014;6:556–558. [Google Scholar]
- 23.Nayak AK, Panigrahi PP. Solubility enhancement of etoricoxib by cosolvency approach. ISRN Physical Chemistry. 2012:1–5. [Google Scholar]
- 24.Seeder N, Kanojia M. Co-solvent solubilization of some poorly-soluble antidiabetic drugs. Pharm Dev Technol. 2009;14:185–192. doi: 10.1080/10837450802498894. [DOI] [PubMed] [Google Scholar]
- 25.Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012;2012:195727. doi: 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jafar M, Mhg D, Shareef A. Enhancement of dissolution and anti-inflammatory effect of meloxicam using solid dispersions. Int J Appl Pharm. 2010;2:22–27. [Google Scholar]
- 27.Das A, Nayak AK, Mohanty B, Panda S. solubility and dissolution enhancement of etoricoxib by the solid dispersion technique using sugar carriers. ISRN Pharm. 2011;2011:819765. doi: 10.5402/2011/819765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdary KPR, Chandra DU, Parimala V, Indira M. A factorial study on the formulation development of ibuprofen tablets employing starch 1500 and PVP K 30. Int J Pharm Sci Res. 2012;3:189–193. [Google Scholar]
- 29.Deshmukh DB, Gaikwad PD, Bankar VH, Pawar SP. Dissolution enhancement of poorly water soluble diacerein by solid dispersion technique. J Pharm Sci Res. 2010;2:734–739. [Google Scholar]
- 30.Prasad RS, Yandrapu SK, Manavalan R. Preparation and characterization of itraconazole solid dispersions for improved oral bioavailability. Int J ChemTech Res. 2010;2:133–142. [Google Scholar]
- 31.Saito M, Ugajin T, Nozawa Y, Sadzuka Y, Miyagishima A, Sonobe T. Preparation and dissolution characteristics of griseofulvin solid dispersions with saccharides. Int J Pharm. 2002;249:71–79. doi: 10.1016/s0378-5173(02)00455-6. [DOI] [PubMed] [Google Scholar]
- 32.Omar SM, Ibrahim F, Ismail A. Formulation and evaluation of cyclodextrin-based nanosponges of griseofulvin as a pediatric oral liquid dosage form for enhancing bioavailability and masking bitter taste. Saudi Pharm J. 2020;28:349–361. doi: 10.1016/j.jsps.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalyanwat R, Gupta S, Songara RKr, Jain D, Sushma Patel. Study on the enhancement of the dissolution rate of carbamazepine by solid dispersion. Int J Compr Adv Pharm. 2011;5:1–4. [Google Scholar]
- 34.Shivalingam MR, Jyothibasu T, Reddy YVK, AppaRao B, Tejaswi V, Nagaanusha D. Formulation and evaluation of solid dispersions of glipizide for dissolution rate enhancement. Int J Pharm Res Dev. 2011;3:231–239. [Google Scholar]
- 35.Krishnamoorthy V, Nagalingam A, Priya Ranjan Prasad V, Parameshwaran S, George N, Kaliyan P. Characterization of olanzapine-solid dispersions. Iran J Pharm Res. 2011;10:13–24. [PMC free article] [PubMed] [Google Scholar]
- 36.Shavi GV, Kumar AR, Usha YN, Armugam K, Ranjan OP, Ginjupalli K, Pandey S, Udupa N. Enhanced dissolution and bioavailability of gliclazide using solid dispersion techniques. Int J Drug Deliv. 2010;2:49–57. [Google Scholar]
- 37.Bobe KR, Subrahmanyam CR, Suresh S, Gaikwad DT, Patil MD, Khade TS, Gavitre BB, Kulkarni VS, Gaikwad UT. Formulation and evaluation of the solid dispersion of atorvastatin with various carriers. Int J Compr Pharm. 2011;1:1–6. [Google Scholar]
- 38.Ramesh V, Jat RK, Chowdary KPR. The formulation of telmisartan tablets employing solid dispersions in MCC PH102 and Poloxamer188 as per 22 factorial design. World J Pharm Res. 2015;4:1397–1405. [Google Scholar]
- 39.Jejurkar L, Tapar KK. Preparation and characterization of mesalamine solid dispersions by kneading method. Int J Pharm Sci Res. 2011;2:2623–2628. [Google Scholar]
- 40.Liu j, Cao F, Zhang C, Ping Q. Use of polymer combinations in the preparation of solid dispersion of a thermally unstable drug by hot melt extrusion. Acta Pharm Sin B. 2013;3:263–272. [Google Scholar]
- 41.Akshay M, Ashwini D. Griseofulvin: BCS classification and solubility enhancement techniques. Int J Res Dev Pharm L Sci. 2012;1:112–121. [Google Scholar]
- 42.Ramu S, Jithendra CH, Sumalatha K, Dharani G, Rachana J, Manasa K, Anitha P. Co-crystals: a review of recent trends in co crystallization of BCS class II drugs. Res J Pharm Tech. 2019;12:3117–3124. [Google Scholar]
- 43.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 44.Benet LZ, Amidon GL, Barends DM, Lennernäs H, Polli JE, Shah VP, Stavchansky SA, Yu LX. The use of BDDCS in classifying the permeability of marketed drugs. Pharm Res. 2008;25:483–488. doi: 10.1007/s11095-007-9523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benet LZ. Role of (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development. J Pharm Sci. 2013;102:34–42. doi: 10.1002/jps.23359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khandelwal A, Bahadduri PM, Chang C, Polli JE, Swaan PW, Ekins S. Computational models to assign biopharmaceutics drug disposition classification from molecular structure. Pharm Res. 2007;24:2249–2262. doi: 10.1007/s11095-007-9435-9. [DOI] [PubMed] [Google Scholar]