

Abstract
Nanoparticles are widely studied as theranostic vehicles for cancer, however, clinical translation has been limited due to poor tumor-specificity. Features that maximize tumor uptake remain controversial, particularly using clinically relevant, orthotopic models. Herein, we report a systematic study that assesses two major features for impact on tumor specificity, i.e., active vs. passive targeting and nanoparticle size, to evaluate relative influences for a pancreatic cancer model, in vivo. Active targeting via V7 peptide is superior to passive targeting for uptake by orthotopic pancreatic tumors, irrespective of nanoparticle size, observed through cell internalization and release mechanisms. However, size has a secondary effect on uptake for actively-targeted nanoparticles. Nanoparticle size had no significant effect on uptake of passively-targeted nanoparticles. Results highlight the superiority of active targeting over nanoparticle size for pancreatic tumor uptake. These findings suggest a framework for optimizing similar non-aggregate nanoparticles for diagnostic and therapeutic treatment of pancreatic and recalcitrant cancers.
Keywords: actively targeted nanoparticles, nanoparticle size, mesoporous silica nanoparticles, tumor specificity, pancreatic cancer
Graphical Abstract
Introduction
While many nanoparticles have been evaluated, both pre-clinically and clinically, for cancer, there are many barriers and few guiding principles leading to an overall particle design that have been validated in a clinically relevant in vivo setting1. In terms of tumor specificity and uptake, nanoparticles consistently underperform in a clinical setting, leading to inadequate agent delivery to the lesion, thus reducing diagnostic and therapeutic efficacy. Prior studies examining the tumor-specific features of nanoparticles are largely limited to computational and in vitro models. The failure to use clinically relevant in vivo models has led to controversy in the context of improving tumor specificity of nanoparticles; in vitro and computational models do not fully recapitulate the in vivo situation due to the complexities of the cancer microenvironment.2, 3 While active targeting and nanoparticle size, two of the most critical features known to affect tumor uptake2, have been assessed, their relative influences on tumor specificity have not been rigorously studied using in vivo models. Determination of relative influences of critical nanoparticle features on tumor specificity will accelerate the translation of nanoparticles into clinically viable theranostic tools for cancer.
Active targeting options, such as with antibodies (e.g. Her24) or peptides (e.g. RGD5, folate targeting6, tumor necrosis factor7), represent techniques to actively target cancers specifically through extracellular expression of receptors or other proteins. However, many cancers, including pancreatic cancer, have few, if any, identified receptors to facilitate active targeting of the tumor cells. The heterogeneity of pancreatic cancer hinders the development of specific and efficacious extracellular protein targeting.8 However, acidosis resulting from tissue hypoxia and the Warburg effect is a microenvironmental hallmark of aggressive cancers, which can be exploited for active targeting.9, 10 Utilization of the pH-low insertion peptide variant 7 (V7) allows for tumor cell specific targeting in acidic pH (6.8–6.6).9, 11, 12 The amino acid sequence and physical properties of V7 cause the peptide to undergo a conformational change into an active alpha helix state under acidic conditions, i.e. pH 6.8–6.6, potentiating its insertion into the target cell membrane.11 Specifically, V7 responsiveness to the transformation and internalization is facilitated directly by acidity in the pancreatic cancer microenvironment.9, 12 In addition to the ability of V7 to actively target the acidic microenvironment of pancreatic cancer, the “brush” conformation and high density of V7 reduces protein corona formation, similarly to PEG but without the deleterious PEG-associated toxicity concerns13. Limiting protein corona formation is beneficial since the protein corona is associated with reduced tumor specificity and accumulation.13
Core nanovehicle size is an additional critically important feature of nanoparticles for tumor uptake. Previous in vitro and computational modeling studies suggest that a nanovehicle size range centered on 50 nm is optimal for enhanced drug delivery and tumor specificity2, 14, 15. However, these studies failed to incorporate either active targeting or appropriate clinically-relevant testing in in vivo models. For pancreatic cancer, a nanoparticle size range centered on 30 nm has been suggested. This is based on the vasculature collapse that occurs in pancreatic cancer and in turn reduces the importance of the enhanced permeability and retention (EPR) effect.16 These studies suggest that nanoparticle uptake would be optimal with nanovehicles in a size range centered near 30–50 nm. However, there are currently too few studies evaluating this idea using viable translational nanoparticles in a clinically relevant model, especially when evaluated in combination with active targeting, when has emerged as a necessary feature for maximum delivery efficacy.3 A rigorous examination of the relative influences of active targeting vs. passive targeting and nanoparticle size on tumor uptake in a clinically relevant pancreatic cancer model is critically needed.
Mesoporous silica nanoparticles (MSNs) are tailorable nanoparticles that can enhance delivery of therapeutic drugs or diagnostic contrast agents and have been used with pre-clinical and clinical imaging platforms.17, 18 Applications include anticancer therapies19–21 as well as non-cancer treatments such as antibiotic delivery22 – each application exploits the enhanced delivery capabilities of MSNs. Changes in the core physical parameters of MSNs such as size23, shape24, pore topology25, and surface chemistry26 affect the properties of the MSNs, including payload retention, targeted delivery, off-target delivery, and toxicity.27–30 Facile alterations in MSN synthesis allow for fine-tuning of physical properties which determine clinical potential (Scheme 1). MSNs generally have a longer circulation time in the blood, have a higher capacity of cargo per unit volume, and have more favorable cargo release kinetics than other types of nanoparticles such as metal nanoparticles.31 The rigorous and varied tunability of MSNs makes agent encapsulation, retention, and release highly controllable and suitable as theranostic tools for cancer.
Scheme 1.
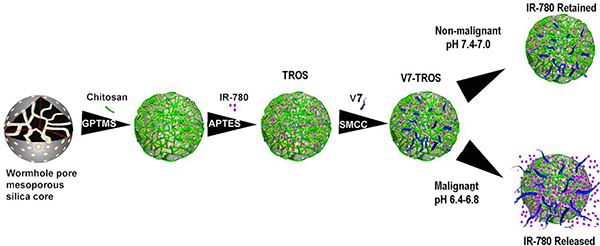
Schematic illustration of the components and formation of V7-TROS nanoparticles.
Nanoparticle translation is often hindered by an inability to accurately track the biodistribution in vivo. The newly emerging imaging modality, Multispectral optoacoustic tomography (MSOT), has demonstrated potential to track particle tumor specificity and uptake in vivo and in real-time.32–35 MSOT uses multi-wavelengths of near infrared (NIR) laser light to excite thermoelastic expansion of specifically selected chromophores. Synchronized high energy lasers allow for high resolving power on the order of less than 10 cells for detection of small tumor masses, exceeding current resolution limits of 0.1–1mm with conventional modalities such as PET/SPECT, and MRI.36, 37 The chromophores can be endogenous (e.g., deoxy- or oxy-hemoglobin) or exogenous contrast agents (e.g., indocyanine green (ICG)) and can be targeted simultaneously for multi-agent imaging and multi-variable biodistribution analysis.9, 38, 39 This allows for high-resolution imaging with contrast levels unachieved with ultrasound, and CT, for high-sensitivity detection of cancer35. The multimodal NIR and optoacoustic approach allows for signal penetration at depths of up to 5 centimeters33 for examination of tumors positioned orthotopically, such as pancreatic tumors within mice, which far bypasses the penetration limit of optical imaging methods40. The high concordance of MSOT with existing gold-standard imaging (such as radiography), the rapid speed and efficiency of MSOT, and its increasing usage all point to MSOT imaging emerging as a reliable and clinically-relevant metric of biodistribution.41–43 MSOT has the potential to synergize well with nanoparticles equipped with high contrast per particle32. MSOT has shown capabilities for sub millimeter detection of contrast agents previously44; far more sensitive than current gold-standard imaging modalities, which are largely limited to a minimum 1-centimeter detection.
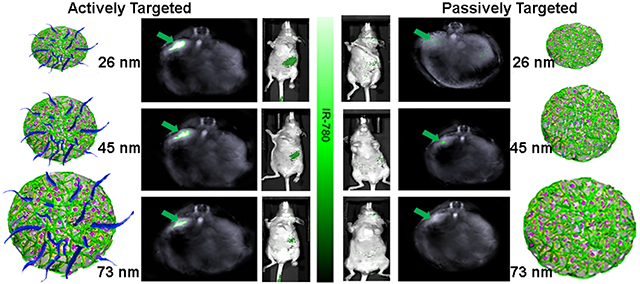
The present study focuses on using MSOT to determine the relative influences of active targeting vs. passive targeting and nanoparticle size on tumor specific delivery and uptake of MSN nanoparticles in a clinically-relevant in vivo model of pancreatic cancer. While many characteristics of nanoparticles can influence tumor specificity and uptake, evaluation of all such features simultaneously is not feasible. Therefore, herein focus is placed on two of the most critical components: active targeting and size. Individual MSNs of 26 nm, 45 nm, and 73 nm, each one in combination with either active or passive targeting were examined. Our data show that active targeting is the primary influence on tumor specific nanoparticle uptake with size as a secondary factor. The greatest tumor-specific nanoparticle uptake was seen with actively targeted V7-TMOS (26 nm). A unique finding was that even the largest actively targeted particle, V7-TPOS, had greater tumor specific uptake than the smallest passively targeted particle (TMOS), underlining active targeting via V7 as the predominant determinant of tumor specific uptake. Actively targeted particles showed decreasing uptake with increasing particle size, i.e., specificity was greatest in V7-TMOS and lowest in V7-TPOS. Passively targeted MSNs did not exhibit significant changes in uptake as a result of size. These data clearly support the greater importance of active targeting over size in the context of promoting tumor specific uptake. In addition, V7-TMOS coupled with MSOT imaging was able to achieve sub-millimeter detection of small tumor presence in vivo in contrast to NIR fluorescence. In pursuit of MSN translation to the clinic, it is imperative to incorporate active targeting to realize the critical need of theranostics for pancreatic and other recalcitrant cancers. Results are expected to readily translate to similar non-aggregate nanoparticle systems for relevant tumor types.
Results
The future development of nanoparticles as theranostic tools for cancer requires the design of nanoparticles with distinct features that facilitate improved tumor-specific uptake in clinically relevant orthotopic in vivo models. Active targeting and optimizing nanoparticle size will not only increase tumor-specific uptake, but also have the potential to improve biocompatibility and reduce the toxicity that result from deleterious off-target accumulation. These latter two in vivo-relevant variables correspond to the current clinical safety concerns associated with nanoparticle-based delivery systems. Thus, the focus of this study is to evaluate the effects of active targeting and nanoparticle size, and determine their relative influences, on tumor specificity and uptake of MSNs. This study evaluated both V7 actively targeted and passively targeted MSNs in a narrow size range (~25nm-75 nm) using optoacoustic imaging in a clinically relevant, orthotopic in vivo mouse model. MSNs with wormhole-like pores of sizes 26 nm, 45nm, and 73 nm were prepared loaded with the optoacoustic contrast agent IR780, coated with a pH-sensitive polysaccharide gatekeeper, and actively targeted for pancreatic cancer with V7. MSNs are specifically referred to by silica precursor and resulting size as follows: 26 nm- tetramethyl orthosilicate (TMOS); 45 nm- tetraethyl orthosilicate (TEOS); and 73 nm- tetrapropyl orthosilicate (TPOS). Collectively, the MSNs will be referred to as TROS, where “R” corresponds to the length of the four hydrocarbon chains extending from the silicate core. Each different sized MSN, both with and without V7 active targeting, was intravenously injected into mice with orthotopic pancreatic tumors. Comparisons were made between targeted particles of different sizes and between each of the targeted experimental groups and passively targeted (negative) controls. Physical characterizations, in vitro experimental results, and ex vivo secondary confirmations corroborate the efficacy of our in vivo MSOT imaging-based methodology. Following, the limit of MSOT sensitivity was compared to NIR fluorescence by recording sub-millimeter tumors in vivo. A focus herein on comparison of targeting and size of MSNs in an in vivo model frames the discussion for improving the efficacy of a nanomedicine-based treatment delivery system in pancreatic cancer.
Physical Characteristics of TROS Nanoparticles
Defining the optimum size range of core nanoparticles is necessary for their successful theranostic use in cancer. This study evaluated the combined effect of nanoparticle active targeting and size on tumor specificity in an orthotopic in vivo mouse model. Particle size was controlled by a modified Stober synthesis approach.45 The synthesis procedure is described in the SI document and graphically outlined in Scheme 1. Three size distinct core MSNs, TMOS, TEOS, and TPOS, were successfully synthesized in a narrow size range with low batch-to-batch variability.
Average core particle diameters were 26 ± 3.1 nm, 45 ± 3.8 nm, and 73 ± 4.2 nm (TMOS, TEOS, and TPOS) with corresponding average pore sizes of 1.3 ± 0.2 nm, 1.5 ± 0.2 nm, and 1.7 ± 0.2 nm for TMOS, TEOS, and TPOS, respectively, as measured by transmission electron microscopy (TEM) (n=25 particles/pores) (Figure 1A–C). The hydrodynamic diameter for the particles were slightly larger as measured with dynamic light scattering (DLS) (Figure 1D). Full DLS distribution are included (Figure 1H) DLS measurements of hydrodynamic diameter have been reported to over-estimate core MSN diameter.45, 46 Polydispersity index (PDI) showed that MSNs were prepared as a monodisperse distribution (Figure 1E). The mean PDI of core TMOS, TEOS, and TPOS, was low and stable over several months. BJH analysis was used to interpret BET results47 to determine surface area and pore volume (Figure 1F–G). Decreases in surface area dependent upon size is a result of differences in hydrolysis rates.45 Precursor selection determines preferential nucleation of new particles and outward growth of existing particles in the synthesis mixture.45
Figure 1.
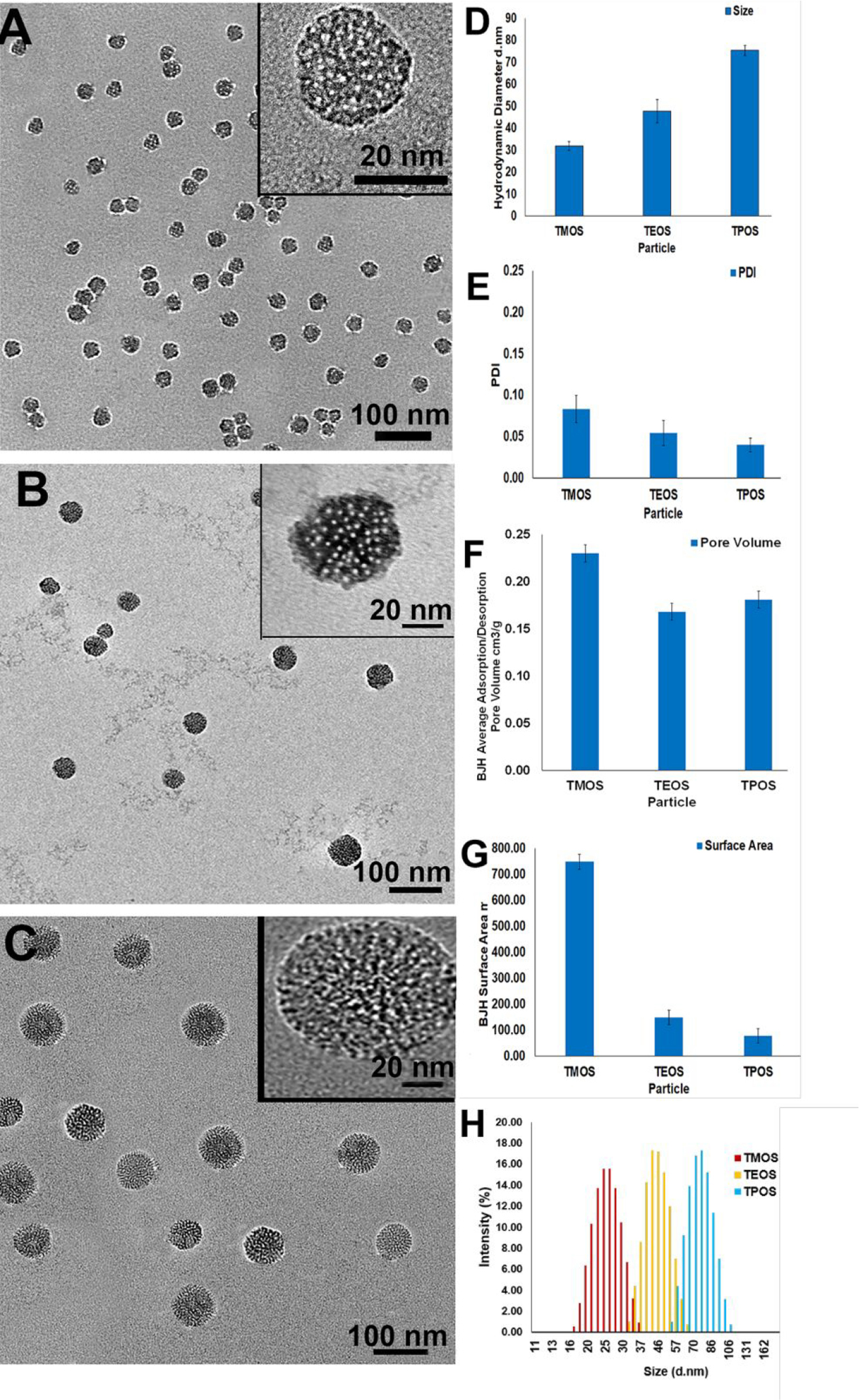
Characterization of TROS particles. Low magnification transmission electron micrographs (TEM) images of (A) TMOS, (B) TEOS, and (C) TPOS demonstrate an average particle diameter of 26 ± 3.1 nm, 45 ± 3.8 nm, and 73 ± 4.2 nm. Each TEM image also contains a high magnification image in the inset. (D) Corresponding DLS data of 32±3 d. nm for TMOS, 48±5 d. nm for TEOS, and 75±2 d. nm for TPOS closely resemble the TEM data. Additional characterization charts depict the (E) polydispersity index, PDI, of the TROS particles, (F) BJH method-computed adsorption/desorption pore volume and (G) surface area of the TROS particles following BET analysis. Full DLS distribution of TMOS, TEOS, and TPOS (H).
TROS synthesis was followed by conjugation with a natural polysaccharide, chitosan, as a gatekeeping molecule for encapsulation of the contrast agent, IR780, within MSN pores. IR780 is a hydrophobic non-aggregate cargo, and was selected in this study due to its ability to generate an optoacoustic signal.27, 39, 48 Chitosan conjugation was confirmed by increased zeta potential (Figure 2A). IR780 encapsulation increased with increases in particle size: 1.01×108 dye molecules/TMOS particle, 3.38×108 dye molecules/TEOS particle, and 5.62×108 dye molecules/TPOS particle (Figure 2B). Following dye encapsulation, TROS particles were actively targeted by functionalization with Variant 7 (V7) tumor-targeting ligand, again confirmed by zeta potential measurement (Figure 2A). Differences in the conjugation density of V7 peptide was observed according to TROS size: 2.0×103 V7 molecules/particle for TMOS, 2.0×103 V7 molecules/particle for TEOS, and 2.0×103 V7 molecules/particle for TPOS.
Figure 2.
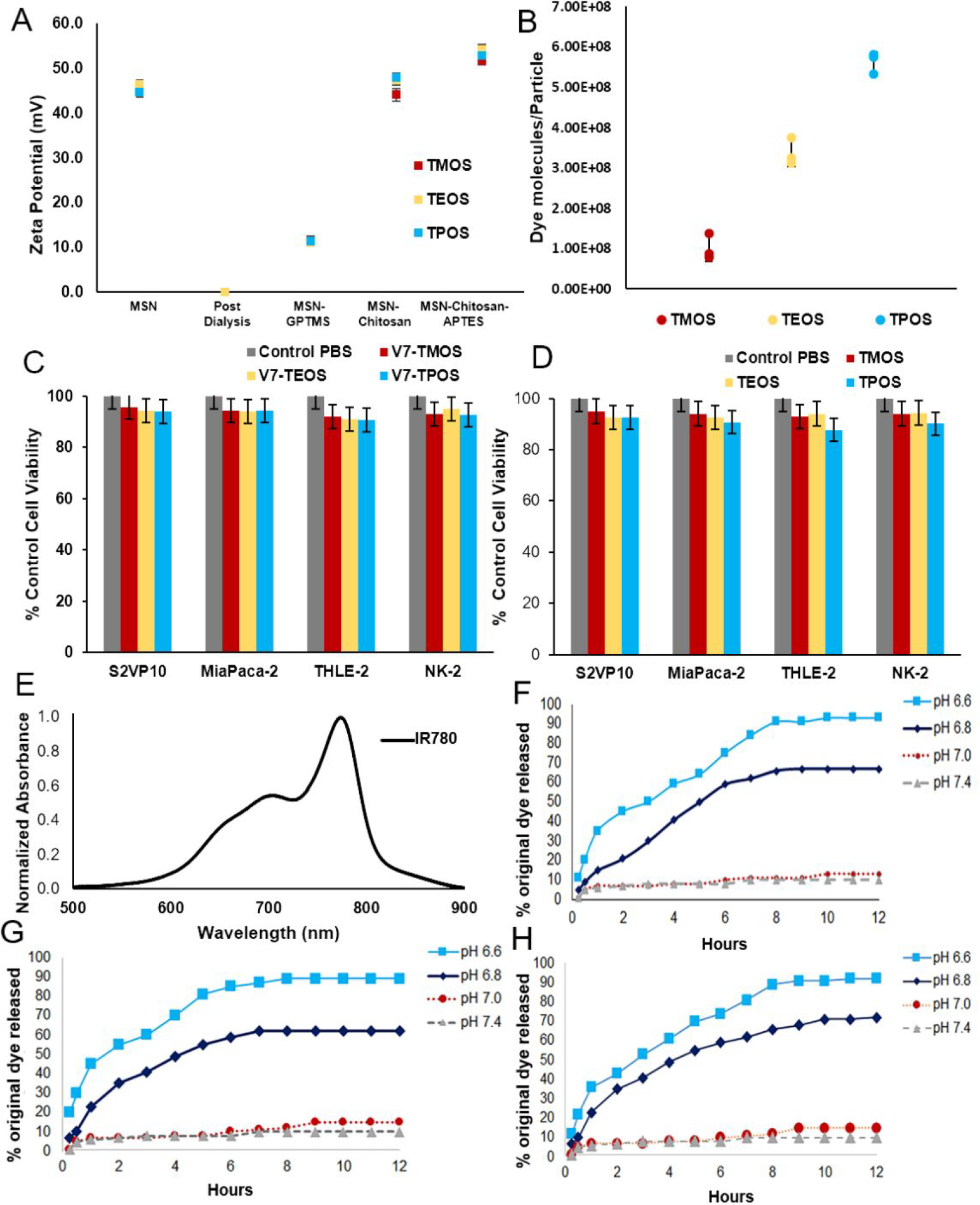
Characteristics of TROS (A) Zeta potential measurements confirming cleaning and conjugation of TROS particles. (B) Dye molecules per particle for three sizes of TROS particles. (C) No significant toxicity was observed in malignant (S2VP10 and MiaPaca-2) or non-malignant (THLE-2 and NK-2) cells at any particle size for V7 targeted TROS p>0.05. (D) No significant toxicity was observed in malignant (S2VP10 and MiaPaca-2) or non-malignant (THLE-2 and NK-2) cells at any particle size for passively targeted TROS p>0.05. (E) Absorbance spectra of optoacoustic contrast agent IR-780. (F) IR-780 release from TMOS particles at pH 7.4–6.6. (G) IR-780 release from TEOS particles at pH 7.4–6.6. (H) IR-780 release from TPOS particles at pH 7.4–6.6.
V7-TROS toxicity evaluated in pancreatic tumor cells and non-malignant cells.
Due to limited data on toxicity of chitosan capped MSNs, S2VP10 and MiaPaca-2 pancreatic cancer cells and non-malignant liver (THLE-2) and kidney (NK-2) cells were treated with V7-TROS and passively targeted TROS for 24 h and evaluated cell viability. No statistically significant differences in cell viability were observed in any cell line as a result of active targeting and size. (p>0.05) (Figure 2C–D).
pH-specific release of TROS
IR-780 (Figure 2E) was loaded into nanoparticles as described. The efficacy of chitosan for contrast agent release was assessed by a dye release assay that simulates acidic conditions.49 (Figure 2F–H) The release characteristics of TMOS, TEOS, and TPOS nanoparticles were all similar for in vitro pH-controlled cargo release. TMOS, TEOS, and TPOS particles released the most, 90%–94%, of the encapsulated IR780 dye at pH 6.6, with lower release, 62–72%, at pH 6.8 (TMOS p=0.0043, TEOS p=0.0490, TPOS=p=0.1271), and 10%–17% at pH 7.4 (TMOS p=0.0010, TEOS p=0.0010, TPOS p=0.0010).
pH-specific cell uptake of V7-TROS evaluated in vitro
S2VP10 cells were treated with passively targeted TROS and V7-TROS particles, each with IR780 cargo, for 2 h under pH 7.4, 6.8, or 6.6 conditions. After washing of the cells, the NIR fluorescence data showed that uptake of V7-TROS with IR780 cargo was pH specific (Figure 3A–B); passively-targeted TROS had minimal cell uptake, due to the lack of tumor targeting peptide, V7. No passively-targeted MSNs, irrespective of size, exhibited any significant pH-sensitivity. V7-TROS had significantly greater specificity at acidic pH than did passively targeted-particles at any pH (Figure 3A–B). Specifically, V7-TMOS exhibited the greatest enhancement of in vitro uptake in response to pH: uptake increased by 8.5-fold at pH 6.8 and 19.2-fold at pH 6.6 (p=0.002, p=0.0001 respectively), compared to V7-TMOS at pH 7.4. V7-TEOS and V7-TPOS experienced similar, but less dramatic, trends; V7-TEOS increased 7.8-fold at pH 6.8 and 16.5-fold at pH 6.6 and V7-TPOS 6.2-fold at pH 6.8 and 15.8-fold at pH 6.6 (p<0.005).
Figure 3.
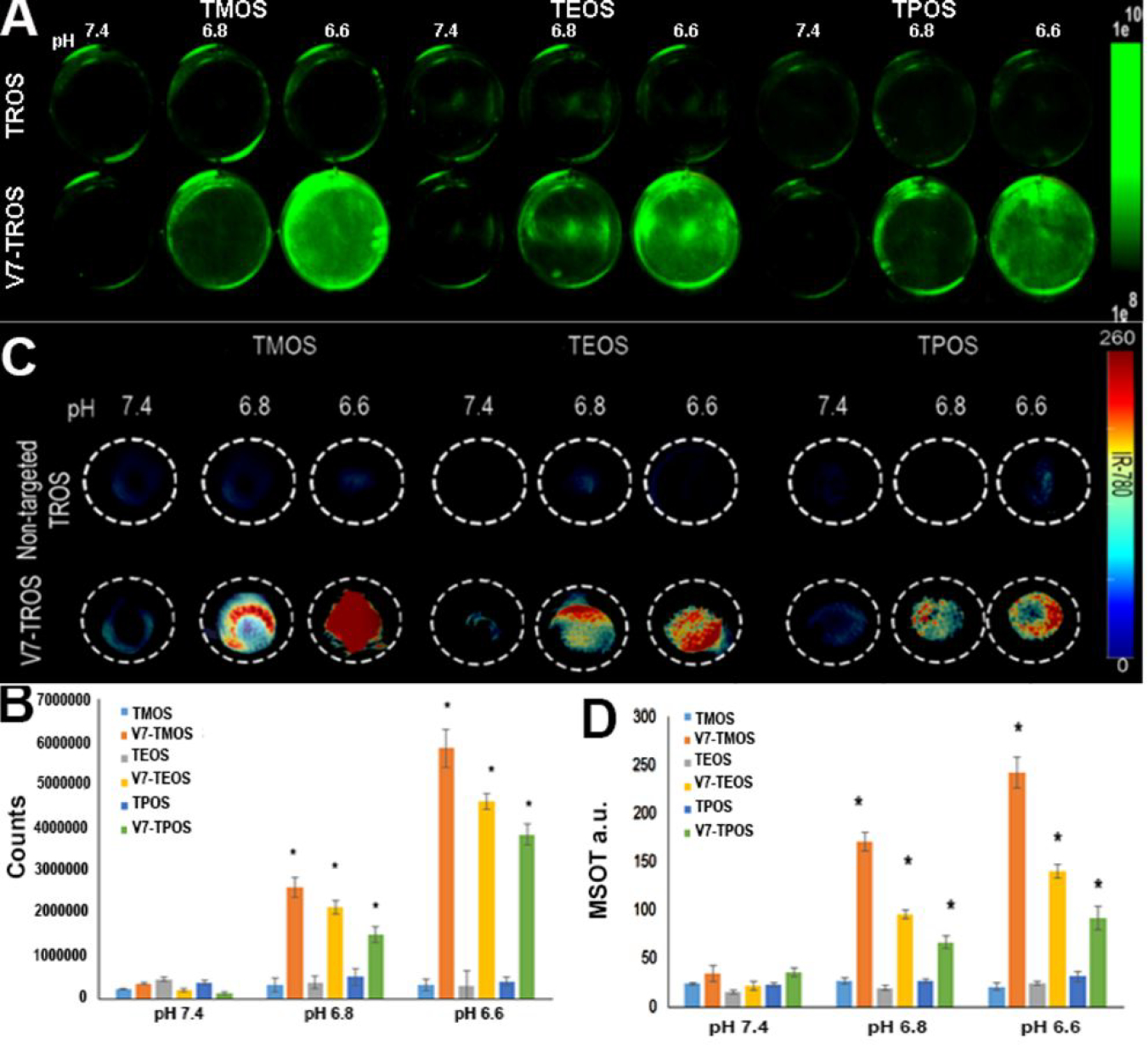
NIR and MSOT confirmation of TROS particle pH-specific cell uptake and cargo release in vitro. Tumor cells were plated and treated with either passively targeted TROS or V7-TROS particles followed by evaluation using (A-B) NIR fluorescent imaging or (C-D) MSOT imaging. (A) The uptake of the TROS nanoparticles with IR780 cargo is shown in the green color bar and (B) quantitatively depicted. Tissue mimicking phantoms containing cells treated with passively targeted TROS or V7-TROS were imaged using MSOT (C) with (D) corresponding quantitative measurements.
Using tissue-mimicking phantoms, cell uptake of TROS particles was confirmed using MSOT (Figure 3C–D). The optoacoustic signal observed in the tissue phantoms using MSOT confirmed increasing cellular uptake of the particles corresponding to V7 targeting and decreasing particle size. The results were consistent with NIR fluorescence data reported above; V7 targeting was the strongest influence on uptake, with smaller particles having a significant, but secondary, impact (Figure 3A–D). Passively-targeted TROS particles were detected at levels <35 a.u. in all samples. V7-TMOS samples contained 35 a.u., 171 a.u., and 243 a.u. for the pH 7.4, 6.8, and 6.6 samples respectively. V7-TEOS and V7-TPOS exhibited similar trends, but attained lower maximal values: 141 a.u. and 92 a.u respectively, at pH 6.6. Overall, for each V7-TROS, the signals at pH 6.6 were significantly higher than at either pH 7.4 or 6.8 (p<0.005). (Figures 3B–D). The increased signal in TMOS is even more significant when considering that a single TPOS particle has substantially greater cargo capacity than does the smaller TMOS particle (Figure 1). Comparison of the proportion of fluorescent signal in Figures 3A and 3B, for V7-TROS from pH 7.4 to 6.6, to the analogous signal measured by MSOT in Figures 3C and 3D shows that V7-TEOS and V7-TPOS produce a higher magnitude (V7-TEOS<V7-TPOS) of fluorescent signal per unit of MSOT signal than does V7-TMOS (p<0.00127).
Cell internalization of TROS with propidium iodide
To further substantiate the internalization mechanism of TROS nanoparticles, propidium iodide was encapsulated within TROS in place of IR780. Propidium iodide (PI) is a cell membrane impermeable dye that exhibits differential fluorescence depending on intracellular or extracellular presence.50 Most often propidium iodide binds to nucleic acid in dead cells and shows red fluorescence. However, when propidium iodide is not bound to nucleic acid, fluorescence is green-shifted by approximately 15 nm.50 When encapsulated within TROS, mechanism of entry into live cells can be confirmed through intracellular red fluorescence, while extracellular propidium iodide is impermeable and is removed via post-treatment washes, or, if remaining present, detected via green-shifted fluorescence.50, 51 Figure 4 indicates red fluorescence is observed significantly in actively targeted, pH 6.6 samples only (TMOS p=5*10−14, TEOS p=5*10−14, TMOS p=4*10−10). Specifically, at pH 6.6, V7-TMOS images showed an average red fluorescence count of 8943, V7-TEOS at 9072, and V7-TPOS at 2310 (Figure 4B). Small amounts (<60 counts) of red fluorescence were observed in V7-TROS at pH 7.4. Negligible red and green fluorescence was observed in all other treatments (Figure 4B). This implies that not only is V7 crucial for internalization of MSNs, but also that V7 is active in a cancerous pH of 6.6, but minimally active in a physiological pH of 7.4. Low levels of green fluorescence in passively targeted treatments and pH 7.4 media are suspectedly a result of washing following treatment; because TROS and PI are not bound to cells due to inactive V7, PI is removed due to PBS washing. In addition, S2VP10 cells in the presence of acidic media at pH 6.6 did not undergo cell death, evident through the absence of intracellular propidium iodide and red fluorescence.
Figure 4.
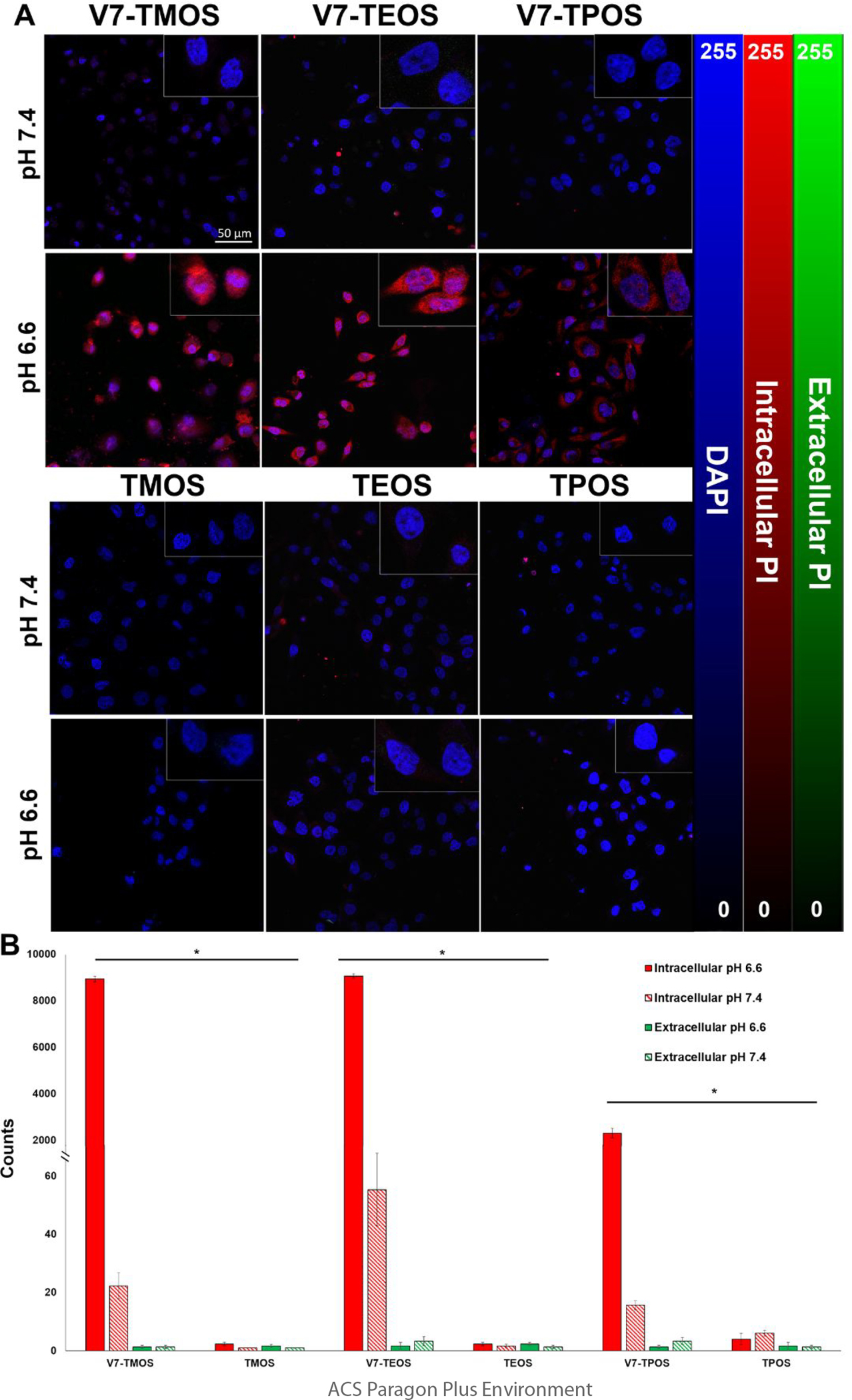
Cell internalization of V7-TROS and passively targeted TROS in acidic, cancerous environments through detection of Propidium iodide in PDAC cell line S2VP10. (A) Fluorescence confocal microscopy images displaying cell internalization. Rows are characterized by the pH of the media (pH 7.4 or pH 6.6). Columns are characterized by treatment type (actively targeted MSNs of all sizes and passively targeted MSNs of all sizes). Quantification shows red vs. green fluorescence observed for each treatment for pH 7.4 and pH 6.6 (B). *p<3×10−10
In vivo kinetics of TROS
In our study, effects of active targeting vs. passive targeting and size on tumor-specific uptake were compared following intravenous injection of TROS into mice orthotopically implanted with pancreatic cancer cells.38 Mice were iv injected with 100 μl of solution containing 8*106 V7-TROS or passively-targeted TROS particles, each loaded with IR780 cargo at 2.7mM (0.1 OD600/100 μl). Concentrations were selected based upon previous evaluations to heighten efficacy52. To evaluate pharmacokinetic properties of V7-TROS and TROS, mice were imaged at 0h, 4h, 8h, and 24h post injection (Figure 5A). While some accumulation of V7-TROS in tumors was observed 4 h post injection for V7-TMOS, V7TEOS, and V7-TPOS (29.2 a.u., 8.2 a.u., and 5.7 a.u.), respectively, peak tumor signals were attained at 8h post injection (65.3 a.u., 29.1 a.u., and 14.5 a.u.) p=0.0008, p=0.0007, and p=0.005. The signal of the V7-TROS was significantly lower after 24 h (17.6 a.u., 3.6 a.u., 1.6 a.u.) p<0.00001 which indicates 8h as the optimal timepoint for evaluation of biodistribution (Figure 5A). Among the V7 TROS treatments, the smaller V7-TMOS particles had significantly greater tumor accumulation than either the V7-TEOS (p=0.0003) or V7-TPOS (p=0.0001) at the maximum signal timepoint of 8h. In contrast, no significant differences of signal in tumors treated with any passively targeted TROS were seen at any timepoint: 2.9 a.u.-1.0 a.u. p=0.672.
Figure 5.
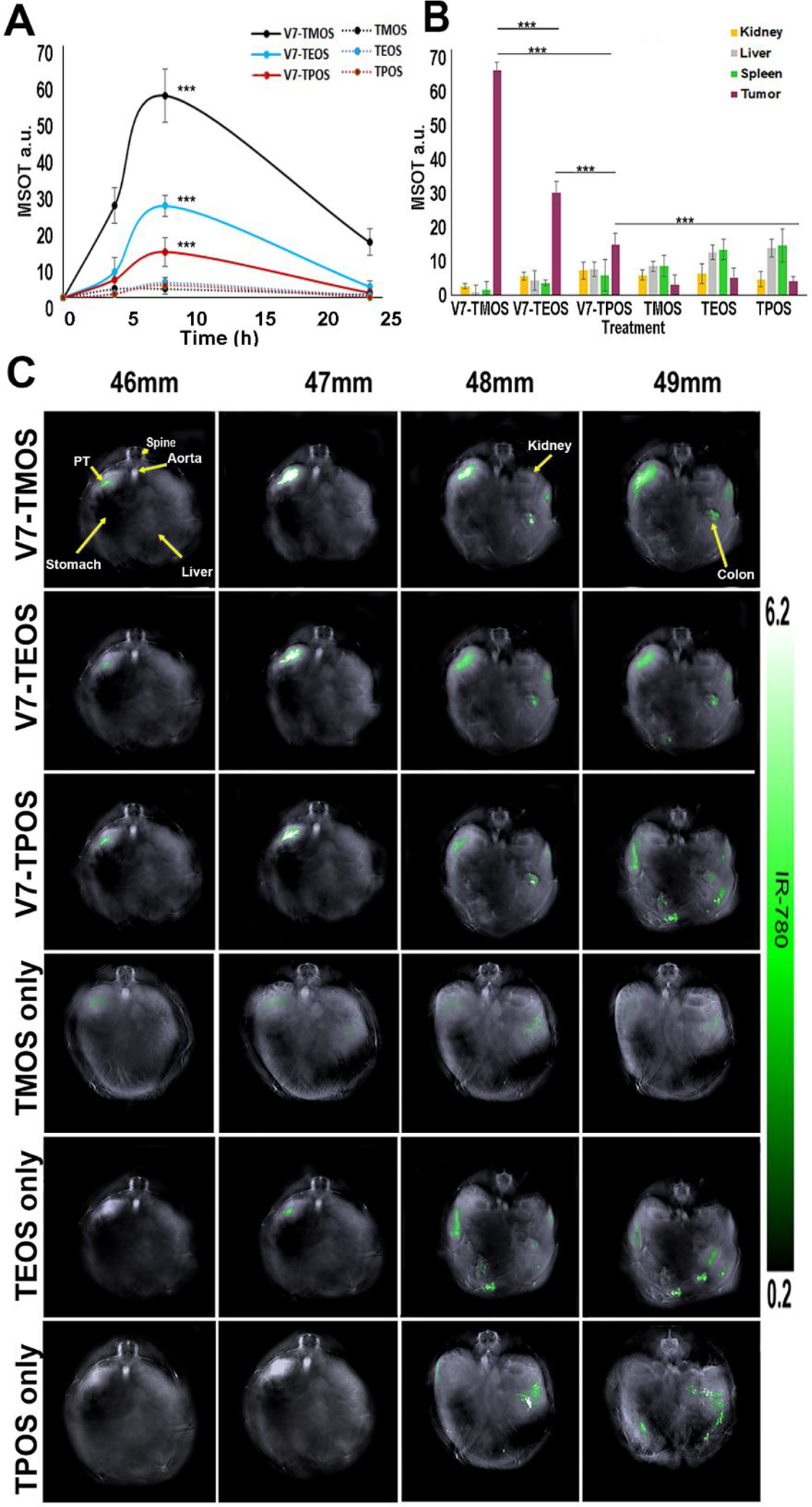
In vivo biodistribution of V7-TROS and passively targeted TROS particles injected into mice bearing orthotopic pancreatic tumors imaged via MSOT (A) Pharmacokinetic assessment of TROS signal within the tumor at timepoints of 0, 4 h, 8 h, and 24 h post intravenous injection (B) After 8h, Biodistribution of TROS was quantified within the tumor, kidney, liver, and spleen(C) Biodistribution of TROS in axial slices showing accumulation within the tumor, kidney, liver, and spleen. *p<0.05, **p<0.01, ***p<0.0001
In vivo biodistribution of TROS
Figure 5B–C shows MSOT data comparing pancreatic tumor accumulation of both actively- and passively- targeted TROS particles loaded with IR780 dye 8 h post injection. Active targeting of TROS caused significantly greater tumor accumulation than passively targeted TROS. Specifically, MSOT recorded 65.3 a.u. for V7-TMOS and only 2.9 a.u. for TMOS (p=0.00001). Similarly, for the actively- and passively-targeted TEOS and TPOS the MOST signals were 29.2 a.u. vs. 2.8 a.u. (p=0.00002) and 14.5 a.u. vs. 2.8 a.u. (p=0.00023) respectively. These results show that active targeting leads to greater accumulation of nanoparticles in the tumor than passive targeting. In addition, TROS size has a secondary effect on tumor accumulation, but only in the context of actively targeted TROS and not in the case of passively targeted TROS. Specifically, the V7-TMOS (26 nm) signal (65.3 a.u) within the tumor was significantly higher than either V7-TEOS (45 nm) at 29.2 a.u. (p=0.000002) or V7-TPOS (73 nm) at 14.5 a.u. (p=0.000001). As the amount of signal/particle is proportional to particle size, i.e., 1.01×108 dye molecules/TMOS particle, 3.38×108 dye molecules/TEOS particle, and 5.62×108 dye molecules/TPOS particle (Figure 1D), substantially more V7-TMOS particles accumulated within the tumor than direct comparison of the a.u. values would suggest. In contrast to the actively-targeted TROS, differences in tumor accumulation for passively targeted TMOS (26 nm), TEOS (45 nm), and TPOS (73 nm) were not significant (p>0.05). These results reinforce the notion that while nanoparticle size can influence tumor accumulation, utilization of active targeting over passive targeting should be prioritized.
Figures 5B also shows off-target accumulation and delivery of actively- and passively-targeted TROS to vital organs at the same time point (8 h) as tumor accumulation. Actively- targeted TROS accumulated in off-target organs at lower levels than the passively-targeted TROS. V7-TMOS off-target delivery was recorded at 2.8 a.u., 1.2 a.u., and 1.3 a.u. for the kidney, liver, and spleen, respectively, while passively-targeted TMOS were recorded at values of 6.1 a.u., 8.2. a.u., and 8.3 a.u. for the same organs. The values for passively-targeted TROS were significantly higher than those for V7-TMOS in the case of both liver (p=0.0001) and spleen (p=0.0131). The data for V7-TEOS vs. TEOS and V7-TPOS vs. TPOS followed similar trends, i.e., for both accumulation of the passively-targeted TROS was higher in the liver and spleen. Overall, actively-targeted TROS not only showed significantly higher accumulation within the tumor, but also significantly lower off-target delivery than passively targeted TROS. The impact of TROS size on tumor accumulation and off-target delivery are shown, although the effect of size is clearly minor compared to active vs. passive targeting.
Secondary confirmation of TROS biodistribution
The in vivo MSOT biodistribution results were confirmed in vivo using NIR fluorescence (Figure 6A–B). While 2D fluorescence has benefits relating to topical applications in real-time, limited tissue penetration hinders whole-body imaging. Therefore, MSOT was primarily used in this study for real-time imaging without organ removal. NIR fluorescence is a strong secondary confirmation option. The pancreas region of the mice treated with V7-TROS showed the highest accumulation (Figure 6A). The V7-TROS particles had significantly higher accumulation compared to passively targeted TROS p=3×10−7. V7-TMOS had the highest tumor accumulation with 4.3×106 photons/s followed by V7-TEOS 4.5×105 photons/s and V7-TPOS 5.4×104 photons/s, (V7-TMOS vs V7-TEOS p=0.00001, V7-TMOS vs V7-TPOS p=0.000001, V7-TEOS vs V7-TPOS p=0.000002). The comparable passively-targeted TMOS, TEOS, and TPOS contained 1.7×103 photons/s, 1.5 ×103 photons/s, and 1.1×103 photons/s, respectively, and did not significantly vary based on size p=0.43. NIR fluorescence imaging confirmed the trends of accumulation observed with MSOT in our in vivo orthotopic mouse model. After euthanasia, mouse organs were excised and MSOT signals were confirmed ex vivo using NIR fluorescence (Figure 6C). The results confirmed the measurements recorded with MSOT in terms of biodistribution and tumor specificity. Maximum signal and accumulation within the tumor of actively targeted V7-TMOS (2.8×105 photons/s), V7-TEOS (9.1×104 photons/s), and V7-TPOS (7.7×103 photons/s), compared to their passively targeted counterparts TMOS (8.8×102 photons/s), TEOS (7.2×102 photons/s), and TPOS (4.8×102 E2 photons/s) was significant p=0.000004. Further post hoc analysis also demonstrated significant differences between V7-TMOS vs TMOS (p=0.0080), V7-TEOS vs TEOS (p=0.00035), and V7-TPOS vs TPOS (p=0.0088). However, accumulation solely based upon nanoparticle size demonstrated significant differences between the smallest and largest sizes tested of TMOS vs TPOS (p=0.0432), but not significantly different between TMOS vs TEOS p=0.3111. These results confirm and emphasize the critical importance of active targeting for tumor accumulation, with size, < 50 nm, as a secondary influencing factor in particle accumulation within tumors, particularly in the case of actively-targeted particles.
Figure 6.
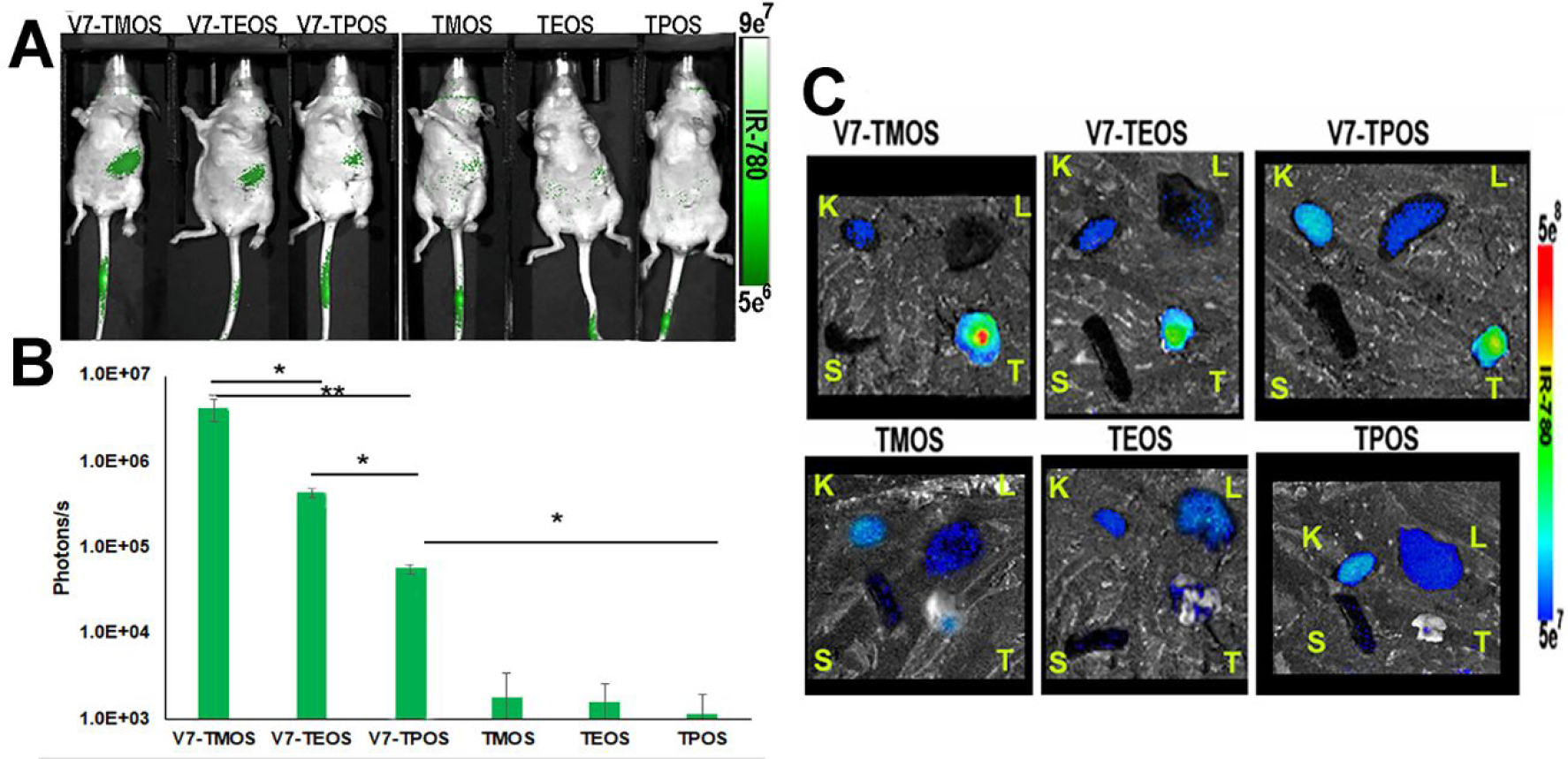
(A) In vivo NIR fluorescence imaging for IR780 dye served as a secondary in vivo imaging method where tumor signals were quantified (B). (C) Secondary confirmation of IR780 signal within ex vivo organs 8h post injection via NIR fluorescence. Kidney (K), liver (L), spleen (S), and pancreas tumor (T) were imaged using NIR fluorescence imaging. Signal uptake observed with NIR imaging corresponded to MSOT in vivo signals. *p<0.05, **p<0.01, ***p<0.0001
Detection of sub-millimeter tumors using V7-TROS
Following determination of V7-TMOS as the optimal treatment for detection in this study, small, sub-millimeter tumors imaged with the same in vivo model to investigate the sensitivity of MSOT imaging, both with and without V7-TMOS, compared to fluorescence (Figure 7). Figure 7A and 7C show that while V7-TMOS and oxy/deoxyhemoglobin where capable of detecting a 0.72mm PDAC tumor, V7-TMOS was able to detect a 0.23 mm PDAC tumor Figure 7B while oxy/deoxyhemoglobin was not 7D. Whole-body NIR fluorescence Figure 7E–F shows that detection was achieved with the 0.72mm tumor, signal was insufficient to confirm present of the smaller, 0.23mm tumor. Figure 7G confirms that excised tumor shows NIR fluorescence signal ex vivo. These results show that MSOT has capabilities to synergize with contrast-carrying nanoparticles for the detection of small tumors.
Figure 7.
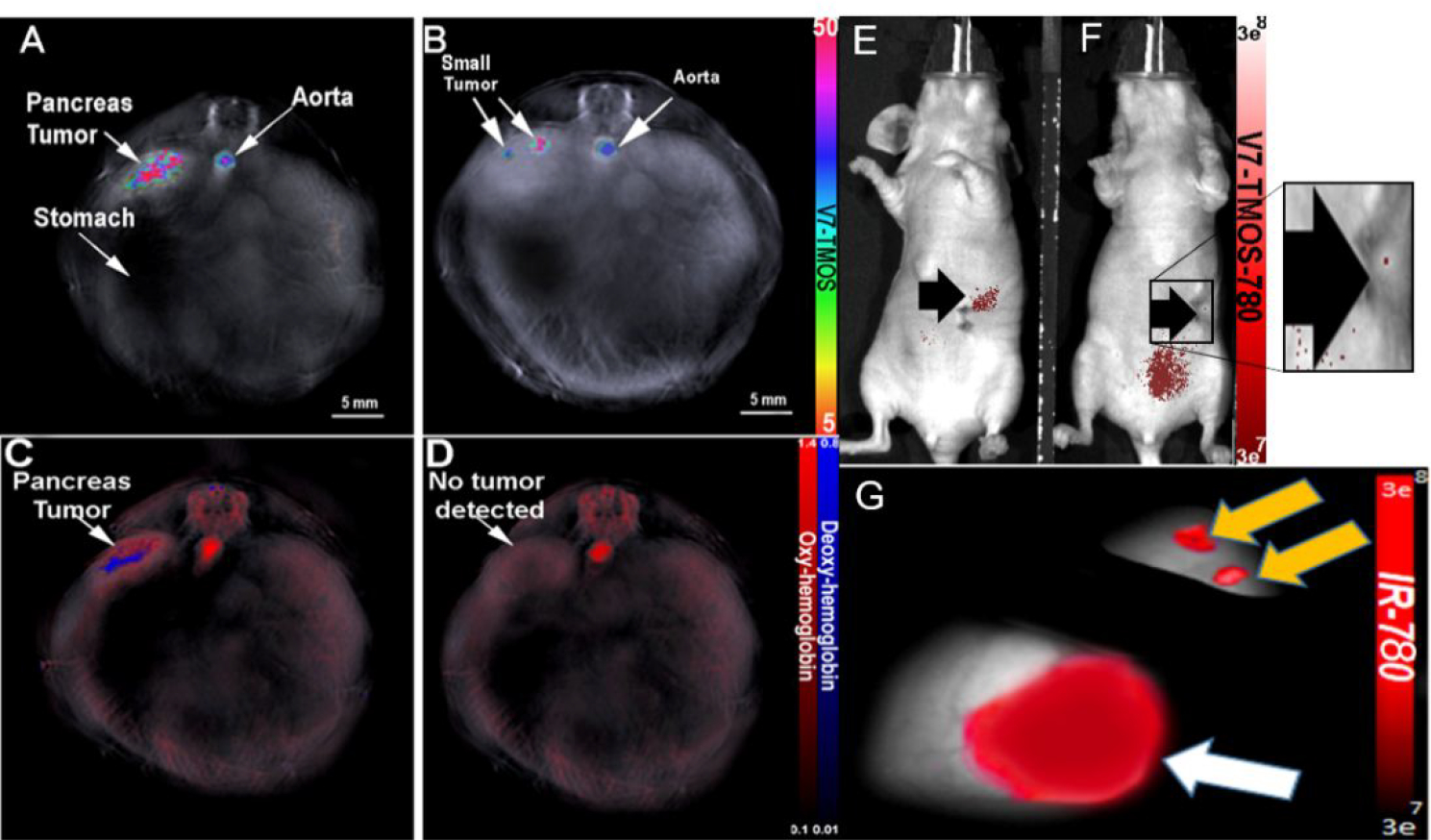
MSOT detection of sub-millimeter PDAC tumor via V7-TMOS in vivo. Orthotopic sub-millimeter (0.72mm) PDAC tumor detected using V7-TMOS (A) or oxy/deoxyhemoglobin (C). Orthotopic sub-millimeter (0.23mm) PDAC tumor detected with V7-TMOS (B). The 0.23mm tumor was not detected with oxy/deoxyhemoglobin only (D). NIR fluorescence was used to detect sub-millimeter tumors of size 0.72mm (E). NIR fluorescence was unable to detect PDAC tumors of 0.23mm (F). Confirmation of sub-millimeter tumor using NIR fluorescence(G).
Evaluation of TROS toxicity
Histological evaluation of the liver and kidney was performed by a Board-certified pathologist (WEG) and showed no change in the tissue outside of the range of normal (Figure 8A–G). These results support cellular viability conclusions in Figure 2C–D that TROS and V7-TROS are non-toxic.
Figure 8.
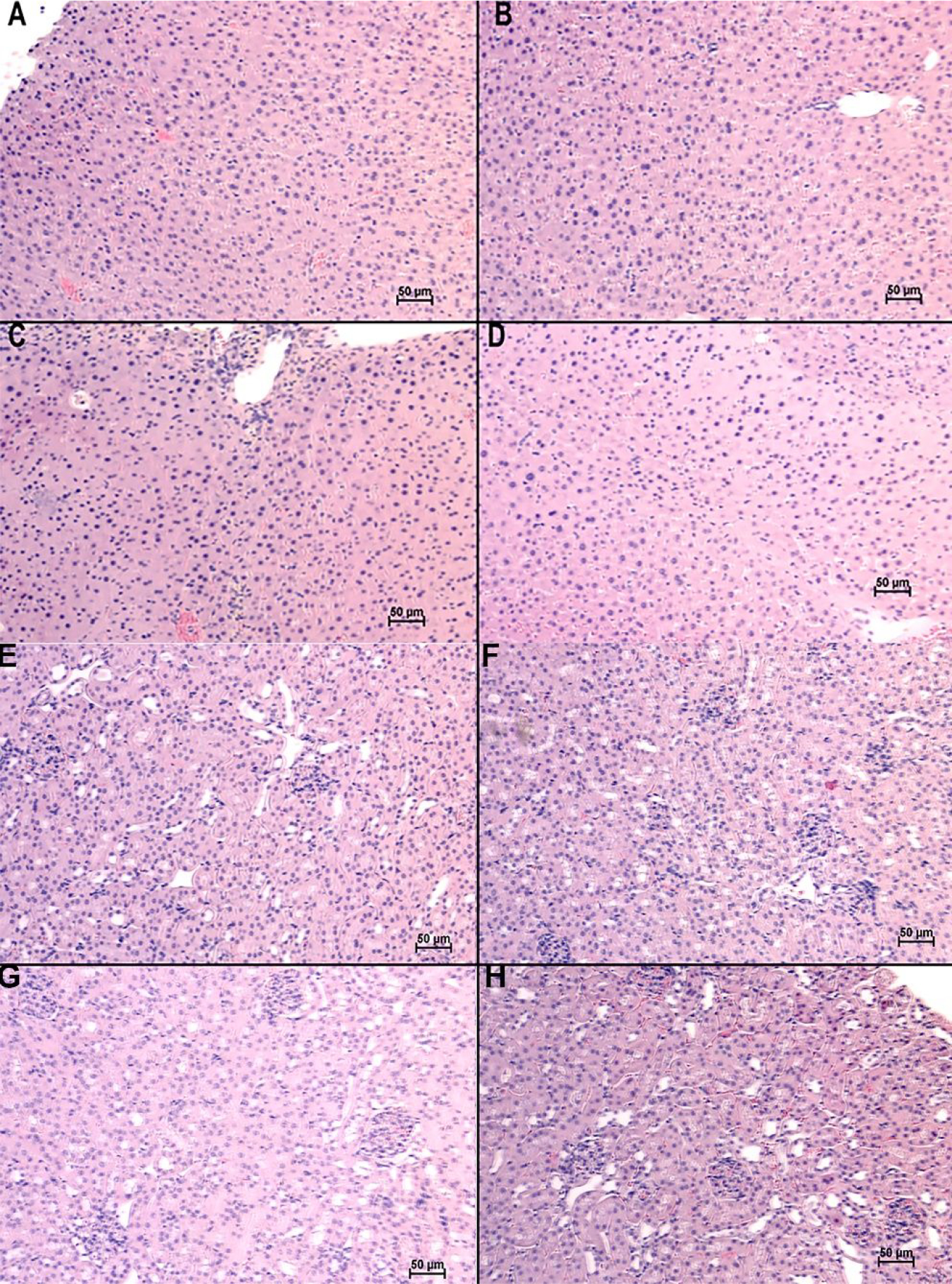
Histological evaluation of Liver and Kidney. (A-D) Livers and (E-H) Kidneys of (A, E) untreated mice, and mice treated with (B, F) V7-TMOS, (C, G) V7-TEOS, (D, H) V7-TPOS and evaluated for potential toxicity. At the time of euthanasia, all livers and kidneys appeared to be within the range of normal/non-diseased.
Discussion
Inspired by previous research on the individual effects of nanoparticle size and targeting using in vitro and computational models53–55, our study investigates both active-targeting and nanoparticle size to assess the optimal particle characteristics for in vivo theranostic applications in clinically-relevant model. Active targeting, through V7, was evaluated against passive targeting in three analogously prepared mesoporous silica nanoparticles, varying only in nanoparticle size. The range of sizes (~25–75 nm) were chosen in this study to avoid potential glomerular filtration by the kidney, but also to test the limits of the efficacy of sequestration by Kupffer cells in the liver56, 57, as well as utilizing the effective range of the EPR effect in a poorly vascularized cancer model.16
Active targeting was introduced to the MSNs through conjugation of V7 pH Low Insertion peptide. Pancreatic cancer is characterized as a highly acidic microenvironment due to rapid metabolism and resulting acidosis. Acidosis is strongly correlated with tumor progression, but highly present surrounding the microenvironment and tumor cells in early stages as well, thus serving as an intriguing feature PDAC marker58–60. V7 has been described previously within the context of in vivo tumor targeting.38 The acidic targeting properties of the pH-responsive V7 targeting peptide is mediated by the acidosis inherent to solid tumors, and exacerbated by the increased Warburg effect in pancreatic cancer.61, 62 During the transition from physiologic pH to extracellular tumor pH, V7 undergoes a conformational change into a transmembrane alpha helical structure, leading to binding to and uptake by tumor cells.63–65 Actively targeting for the acidity of pancreatic cancer may represent a more efficient option than targeting for an extracellular protein or receptor due to the high heterogeneity of pancreatic cancer.9 While these tumors do not uniformly express receptors or other proteins, the entire pancreatic tumor is highly acidic.8, 10
Active targeting had the greatest impact on tumor-specificity both in vitro and in vivo (Figures 3–6). Size showed a secondary influence on MSNs that were actively targeted, with the smallest (26 nm) particles showing the highest uptake. The large increase in of uptake of V7-TROS compared to passively-targeted TROS was due to V7 peptide and the insertion into the membrane at acidic pH in vitro (Figure 3). Our results show that at pH 6.6 V7-TMOS had the highest uptake compared to V7-TEOS (p=0.0011) or V7-TPOS (p=0.000058). Using propidium iodide as an intracellularly specific reporter dye, Figure 4 depicts the improved internalization mechanism of V7-TROS as compared to passively targeted TROS. PI is a cell-impermeable dye that expresses differential fluorescence depending on intra/extracellular presence.50 Namely, extracellular PI (unbound to nucleic acid) exhibits 15 nm green-shifted fluorescence compared to intracellular (nucleic acid bound) PI.50 As such, TROS loaded with PI can be used to confirm V7-TROS are internalized by the cell. Significant counts of red fluorescing PI were observed in cells treated with V7-TROS at pH 6.6 (TMOS p=5*10−14, TEOS p=5*10−14, TMOS p=4*10−10), but minimal counts were observed in V7-TROS at pH 7.4 as well as passively-targeted TROS. This data suggests that V7 is the key component for facilitating the cell internalization mechanism at pH levels of 6.6, but is largely unactive at pH levels of 7.4. As the particle size increased, tumor uptake, observed by IR780 detection with MSOT, decreased while there was increasing off-target uptake, despite larger particles carrying significantly more contrast agent in vivo (Figure 5B). Decreased targeting efficacy with larger particles is conceivably attributable to the increased uptake of nanoparticles by macrophages and Kupffer cells.56 The upper and lower boundaries of particle size exclusion in the circulatory system have been examined extensively. In particular, spherical MSNs greater than 150 nm are known to be rapidly sequestered by Kupfer cells within the liver.66, 67 Furthermore, an enhanced-EPR effect evidently contributes to the less than 50 nm hypothesis for hypovascular tumors.16 The incidence of lower tumor-specific delivery for V7-TPOS than for V7-TMOS or V7-TEOS is suggestive of a limitation for targeted delivery of larger particles on account of the influence of the aforementioned factors. In vitro, MSN cellular membrane-mediated endocytosis is known to be size-dependent68, but onset of size-mediated exclusion from targeted delivery may be closer to 75 nm than the >150 nm which one might expect on the basis of Kupffer cells, the EPR effect, or size effect individually. This study provides further strong evidence that active-targeting should be addressed as a priority before optimizing for particle size. Once active-targeting is achieved, a size range of 15–50 nm in core particle diameter23, 69–71 should be incorporated for optimal cellular uptake of nanoparticles. From a clinical standpoint, improving tumor-specificity of a contrast agent could prove valuable for margin assessment, as any improvement in a disease with outlooks as dismal as PDAC would be a major improvement.
Data of off-target delivery reinforces our findings that suggest active-targeting is absolutely critical to achieving high tumor specificity. The actively targeted TROS exhibit maximal tumor accumulation as well as minimal off-target delivery. In addition, the size of the actively- and passively-targeted TROS had relatively minor impact on off-target delivery. Increase in size did lead to higher accumulation in the liver, V7-TMOS compared to V7-TEOS (p=0.0166) and V7-TPOS (p=0.0006). Signal from the liver trended to be higher in passively-targeted TEOS and was significantly higher in TPOS (p=0.008) compared to TMOS. This increased liver uptake may be attributed to increased Kupffer cell sequestration which has been previously reported as a result of larger nanoparticle size.56 While size increase did not significantly affect splenic uptake of actively targeted TROS (p=0.219), passively targeted TEOS and TPOS had increased splenic uptake compared to TMOS (p=0.042 and p=0.015 respectively). No statistical differences were observed in kidney uptake of either actively- or passively-targeted TROS, likely due to the nanoparticles having a size larger than the kidney filtration boundary.57
In addition to size-based toxicity concerns, the nanoparticle system used avoids the use of poly (ethylene glycol (PEG) as a nanoparticle coating. PEGylated nanoparticles give rise to toxicity concerns based on inhibition of cellular uptake and extended circulation time72–76; in contrast our data indicates no toxicity associated with the use of V7-TROS for V7 targeting. The lack of cytotoxicity in V7-TROS nanoparticles was likely due to the near complete removal of surfactant used in TROS synthesis, visually evident in TEM images. While the toxicity of each of the TROS particle types was insignificant, it should be noted that larger TPOS particles tended towards greater toxicity than TMOS or TEOS particles. While each TROS particle is relatively small (<75 nm), there are subtle changes amongst the particle in viability that may influence particle behavior in vivo. Chitosan has previously been shown to encourage protein corona formation, potentially leading to toxicity and hindered targeting efficacy77, the addition of V7 in this system largely counteracts that effect. The dense conjugation of V7 on the MSN surface creates a “brush” conformation similar to PEG that restricts surface interactions with proteins.13 This not only increases efficacy of specific targeting, but decreases off-target accumulation and toxicity.78 This obviates any need to PEGylate the MSNs, which itself raises concerns of toxicity, especially potential anaphylactic shock or hypersensitivity.72–75, 79
Defining the characteristics of nanoparticles is critical in its successful translation to the clinic. Active targeting and size have been identified as potential influencers on tumor-specificity, but to date, studies evaluating these features rarely used clinically relevant, in vivo models. Here, an orthotopic in vivo mouse model is used to evaluate the effects of active targeting and particle size on tumor specificity using MSOT imaging. TROS exhibit the potential to carry and deliver high amounts of contrast agent to tumors compared to conventional techniques such as antibody targeting. MSOT was chosen to measure TROS kinetics and biodistribution in this study due to its high resolution, its sensitivity, and deeper penetration into tissue44. The use of MSOT also has the benefit of its prior application in clinical trials.33, 34 MSOT and active-targeting of nanoparticles shows excellent synergy, as rapid tumor accumulation is visualized, without the a longer exposure, which is often required for significant tumor accumulation.80, 81 This is attributed to the high specificity of V7 for the tumor microenvironment, rather than conventional passive diffusion of nanoparticles throughout the body. Appropriate circulation time in the blood prior to clearance is determined to be long enough to allow for tumor accumulation all the while circumventing toxicity (Figure 5B). Our data shows that V7-TROS tumor accumulation peaked at approximately 8 h following injection, while passively targeted TROS remained at a low-level within the tumor for the duration of the experiment (Figure 5A). Following determination of V7-TMOS as the superior nanoparticle for tumor targeting and detection, MSOT sensitivity limits with V7-TMOS were explored (Figure 7). While oxy/deoxyhemoglobin differences were capable of detecting the 0.72mm PDAC tumors used in Figure 5–6 using MSOT, each were not sufficient to detect the 0.23mm PDAC tumor. In contrast, V7-TMOS allowed for clear distinction of sub-millimeter PDAC tissue from benign tissue using MSOT, heightening potential for MSOT utilization for small tumor identification. In comparison to NIR fluorescence, which was not capable of distinguishing the 0.23mm PDAC tumor from off-target V7-TMOS accumulation.
In summary, nanobiotechnologies have universally seen suboptimal clinical outcomes in cancer-related applications due to a poor understanding of how to reliably improve tumor specificity. Numerous structural features of nanoparticles have been previously manipulated to achieve high tumor uptake paired with low off-target accumulation. However, the resulting data remains controversial due to the lack of clinically appropriate in vivo models. This study examines the influence of active targeting vs. passive targeting simultaneously with changes in particle size to determine combinations that produce high tumor-specific uptake in a clinically-relevant orthotopic model of pancreatic cancer in vivo. Nanoparticles were prepared as TMOS (26 nm), TEOS (45 nm), and TPOS (73 nm) and either actively targeted with V7 peptide or used passive targeting. Our results show a strong, primary influence of active targeting, with a secondary influence of particle size, on tumor uptake. Ultimately, V7 targeted 26 nm TMOS particles resulted in the greatest tumor specificity in vivo. The superior influence of active targeting was further shown as the actively targeted largest particle (V7-TPOS-73 nm), had higher uptake than the passively targeted smallest particle (TMOS-26 nm). Particle size had a secondary, but important, influence, with particles < 50 nm having the best uptake profile among the tested particles. In addition, MSOT showed the capability to out-perform many clinical imaging modalities through detection of 0.23mm PDAC tumors with V7-TMOS, and 0.72mm PDAC tumors without any exogenous contrast. Our study provides valuable insight for the future development of similar non-aggregate nanoparticles for cancer theranostics using powerful imaging modalities such as MSOT.
Materials and Methods
Chemicals
Reagents purchased from Sigma Aldrich include: methanol (ACS reagent, ≥99.8%, 179337), CTAB hexadecyltrimethylammonium bromide (molecular biology, ≥99%, H6269), ammonium nitrate (≥99.0%, A9642), low molecular weight chitosan (448869), 1-propanol (anhydrous, ≥99.7%, 279544), GPTMS (3-glycidyloxypropyl) trimethoxysilane (≥98%, 440167), TEOS tetraethyl orthosilicate (gas chromatography, ≥99.0%, 86578), TMOS tetramethyl orthosilicate (≥99%, 341436), TPOS tetrapropyl orthosilicate (deposition grade, ≥98%, 679240), benzoylated dialysis tubing (D7884), TEA triethanolamine (gas chromatography, ≥99.0%, 90279), IR780 dye (≥98%, 425311), SMCC (succinimidyl 4-(N-maleimidomethyl)cuclohexane-1-carboxylate) (Powder, ≥98%, M5525), propidium iodide solution (1mg/mL, P4864). MQ water dispensed through MilliPore, Sigma. Glacial Acetic Acid (certified ACS, ≥99.7%, A38C-212) obtained through Fisher. Ethyl alcohol (anhydrous, 200 proof) sourced from Warner Graham Company. pHLIP ligand V7 obtained from CSBio.
Cell Lines
The two malignant human cancer cell lines utilized herein were S2VP10L and MiaPaca-2. S2PV10 originates from University of Nebraska, under care of M. Hollingsworth, and a variant, S2VP10L, which constitutively expresses firefly luciferase for in vivo tumor growth tracking, was generated. MiaPaca-2, THLE-2 (normal liver), and NK-2 (normal kidney) were purchased from ATCC. All cancer cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% L-glutamine. For evaluation of potential toxicity of TROS and V7-TROS on non-malignant cells, THLE-2 cells and NK-2 cells were grown in standard DMEM with 10% FBS was supplemented with the Lonza Bullet Kit (CC3170) without the addition of gentamycin, amphotericin, and epinephrine. Cells were grown and maintained in a humified incubator at 37°C and 5% CO2 until treated with pH-specific media.
pH-specific Media preparation for the in vitro nanoparticle assays
For evaluation of pH-specificity of particle uptake and cell viability of tumor cells, pH specific media was utilized. Briefly, phosphate buffer (25 mM) at the desired pH (7.4, 6.6) was prepared by dissolving sodium phosphate monobasic and dibasic solution dissolved in distilled water (Sigma-Aldrich, St. Louis, MO, USA). 13.6 g of DMEM powder was dissolved in the autoclaved phosphate buffer (25mM) at the desired pH (7.4 or 6.6). The solution was enriched with 10% FBS and 1% L-glutamine and filtered through grade-1 Whatman qualitative filter paper (Sigma-Aldrich, St. Louis, MO, USA). The pH of the solution was determined by pH meter (Denver Instrument Ultrabasic, Bohemia, NY, USA). The acidity and basicity adjustments of the pH solutions were performed with sterilized sodium hydroxide (1 M) or hydrochloric acid (1 M).
Nanoparticle Synthesis and Characterizations
Core Particle Synthesis
The three prepared colloidal mesoporous silica core particles were prepared by identical methods with the only exception being substitution of analogous TROS and corresponding R(OH). Briefly, 2.00 g CTAB, 240 mL MQ water, 0.420 g TEA, and 88.0mmol ROH are combined into a flask, with moderate to vigorous stirring, under reflux and slowly heated to 80°C (approx.30min). At temperature, 11.0mmol of TROS is added dropwise at a rate of approximately 0.5 mL/min and reflux is continued at 80 °C for 12–16 h. At completion, the reaction vessel is removed from heat and cooled to room temperature. A 0.5–1.0 mL aliquot of the resultant solution is reserved for dynamic light scattering (DLS) analysis of polydispersity (PDI) and hydrodynamic diameter.
Dynamic Light Scattering Analysis
A Zetasizer Nano ZS was used for analysis of colloid particle diameter and PDI following synthesis and dialysis processes. The instrument is equipped with a 5mW HeNe laser exciting at 633 nm wavelength and detects scattered light at 1730nm. Software settings indicate a solvent refractive index and viscosity of 1.33 and 0.89 cP, respectively. Measurements were collected in single angle backscatter (173°).
A Zetasizer Ultra was used for analyzing chitosan coated TROS. Measurements for the size parameter were collected in triplicate in multi-angle DLS (MADLS) mode (173°, 90°, 7°). Particle concentration measurements were collected in triplicate and preceded by a single MADLS size measurement for comparison between reported size distributions in both measurement types. General purpose data analysis settings, a dispersant RI and viscosity of 1.43 and 16.1118 (ethylene glycol), and material RI and abs of 1.33 and 0.01 were used for all particle concentration and MADLS measurements to optimize collected correlograms. An input of 145kcps for dispersant mean count rate was used exclusively for particle concentration measurements.
Removal of Core MSN Surfactant Scaffold
A 1:1 (v/v) solution of 2 M Glacial Acetic Acid and Ethyl Alcohol was prepared, in quantity 1 L, into which was placed dialysis tubing containing one of the three synthesized particles. The dialysis vessel was sealed to prevent evaporation and stirring of the external solution was set to between 300–500 rpm with a 39 mm or 50 mm stir bar. The external solution was replaced every 12 h for a period of 3–4 days. Particle solutions were then removed from the acid and alcohol solution, rinsed (externally) with MQ water, and submerged in 1 L of MQ water for two periods of 12 h with continued stirring. Particle solutions were then removed from their tubing and stored in 1 L glass media bottles.
Briefly, 50 mL of each particle solution was separately heated to 60 °C in a 500 mL flask under reflux to which was added, dropwise, an equivalent volume of 74.95 mM Ammonium Nitrate in MQ water. The reaction was run for 1h with moderate stirring and then allowed to cool to room temperature without stirring. These particle solutions were removed again to dialysis tubing, dialyzed twice for 12 h in MQ water, and again stored in 1 L glass vessels.
Particle Lyophilizing, Brunauer-Emmett-Teller (BET) and Nitrogen Adsorption/Desorption Analyses
Fully processed TROS colloids were vacuum centrifuged to lyophilized powders using an Eppendorf Vacufuge 5301. 1.6–1.8 mL of fully processed TROS colloid was pipetted into each of 48 2 mL Eppendorf micro centrifuge tubes all of which were placed, open capped, into an F-45–48-11 rotor. Internal chamber temperature was set to ambient (i.e., no temperature selected). Rate of sample evaporation was in the range 0.15 to 0.22 mL/h. Maximum vacuum achieved was 9 mbar and maximum rotor speed was 1400 rpm.
Using a TriStar analyzer, powders were vacuum dried at 120 °C overnight, were submerged in a liquid nitrogen bath using pure nitrogen gas, and a subsequent seven-point BET isotherm and 50-point adsorption/desorption isotherm were collected. For specific surface area, pore diameter and pore volume measurements, the BET and BJH methods, respectively, of calculation were used.
Sample Preparation and Transmission Electron Microscopy (TEM) Analysis
To prepare samples for TEM analysis, fully processed TROS colloids were diluted to 1:100 in MQ water. 20 μL of this solution was then pipetted onto 200 mesh copper formvar and, separately, lacey carbon electron micrograph grids. Grids were incubated for 30 minutes, had liquid wicked off with filter paper, and were allowed to air dry for 30 min. Grids were stored in stock grid holders. TEM analysis was performed on a Tecnai-F20 transmission electron microscope.
Surface Cross-Linking Functionalization and Chitosan Capping of Core MSNs
To apply GPTMS surface functionalization and attach chitosan polymer, the following steps were taken. Post-processed MSN colloids were diluted 4:1 in 100% Ethyl Alcohol and ultrasonicated for 3minutes at room temperature. pH of the colloidal solution at this stage was between 4 and 4.5. 0.1 mL GPTMS was added to the colloidal solution which was then shaken vigorously (2500 rpm) for 3 h on a vortex.
To derive the capping solution of chitosan, a 1% w/v chitosan solution in 5% nitric acid was prepared and stirred w/ 33 mm stir bar at room temp for 2h. 90 mL of total chitosan solution was homogenized by swirling and quantitatively transferred into 6 15 mL falcon tubes in 15 mL aliquots. Tubes were centrifuged at room temp at 200 rpm (8×g) for 30 min. Light gold-colored pellet was apparent, with transparent solution above, in each tube. The top 3.33 mL of each of the six tubes (cumulatively, top 22% of total volume) was drawn off and combined 6:5 with the TROS fully processed colloid. This MSN-Chitosan mixture was shaken vigorously (2500 rpm) for 12–16 h (i.e., overnight). MSN-Chitosan was isolated by centrifuging a 50 mL volume of solution at 24 °C and 17,000 rpm for 30 min. Pellet volume of approximately 50–100 μL, was not gelled or solid and not adherent to tube.
Loading of IR780 Contrast Agent, Retention and Release Assay, Dye per Particle analyzed by UV-VIS
To load chitosan capped TROS with IR780, the following steps were performed. Total volume of MSN-Chitosan pellet was combined with 80 μL ATPES and 100 μL 1mM IR780 contrast agent, pH of solution was lowered to 4–4.5 using 1M HCl, and the solution was shaken for 5–6 h. Solution pH was then increased to 7.4. Dye loaded particles were isolated by centrifugation at 10,000 rpm for 5 min, dispersed in 500 μL pH 7.4 PBS, and shaken for 30 min for a total of 3 iterations. Maximum solution absorbance was measured by UV-vis for dispersed dye-loaded particles which were then diluted to 1 OD/mL of IR780 signal measured at 653 nm wavelength. For retention and release assays, dye loaded particle solutions were pH adjusted to 7.4 or 6.6 and absorbance of the supernatant at 653 nm was reported for various time intervals following pH change.
Fluorescence OD per particle was determined by centrifugal isolation of dye-loaded MSNs followed by measurement of fluorescent signal in the solution supernatant and calculation of dye concentration in the isolated particles. Dye molecules per particle are reported as 101 million for TMOS, 338 million for TEOS, and 562 million for TPOS. Measurements taken for a particle concentration in sample solution of 8 million particles/mL.
Preparation of pHLIP V7 Ligand and Conjugation to Particle
V7 pHLIP peptide (V7 pHLIP sequence ACEEQNPWARY LEWLFPTETLLLEL, CS Bio, Menlo Park, CA, USA) was added as an active targeting ligand. To prepare a pHLIP V7 solution for particle attachment, 5mg of lyophilized pHLIP V7 powder was thawed from −80°C and combined with 2.44 mL of pH 7.4 PBS and swirled by hand for 15 min until reasonably dissolved. This solution was aliquoted and stored at −80°C until it was used. Briefly, 75 μL of SMCC NH2-malemide linker and 100 μL of pHLIP V7 solution was added to the previous dye-loaded and chitosan capped TROS solution. Total solution was then shaken at room temp in the dark for 3–5 h and subsequently stored at −4°C for use in targeted particle applications. V7-TROS was washed in in PBS and centrifuged at 10,000 g for 10 min. The supernatant was collected to determine the binding efficiency of V7 to TROS with a protein assay. Results corresponded to an approximate conjugation density of 2000 V7 molecules/particle for TMOS, 2000 V7 molecules/particle for TEOS, and 200 million V7 molecules/particle for TPOS.
In Vitro Experiments
Cell viability
Pancreatic tumor cells, S2VP10L and MiaPaca-2, along with non-malignant hepatocytes THLE-2 and non-malignant proximal tubule kidney cells were plated into 96 well plates at 5 × 103 cells per well for 24 h in standard DMEM media with 10% FBS and 1% L-glutamine. Both THLE-2 and NK-2 cells received extra supplements from the Lonza Bullet kit as indicated above. The cells were treated with 2.5 μL of TROS or V7-TROS particles in a 96 well plate for 24 and 48 h at 37°C with 5% CO2. Cell viability was measured using ATPlite™ according to the manufacturer’s instructions (Perkin Elmer, Waltham, MA, USA). Adenosine triphosphate (ATP) levels were measured using a plate reader (Packard TopCount NXT, Meriden, CT, USA) and normalized to phosphate-buffered saline (PBS) treated control.
In vitro evaluation of TROS particles
Evaluation of pancreatic cancer cell uptake of TROS and V7-TROS containing IR780 was conducted using NIR fluorescent imaging and tissue mimicking phantoms. 5 × 105 pancreatic cancer cells (S2VP10 and MiaPaca-2) were grown in 6 well plates with the pH media 7.4 or 6.6 respectively. The cells were incubated for 2h with TMOS, TEOS, and TPOS with and without V7 peptide, respectively. Particles contained encapsulated IR780 at a concentration of 2.7mM (0.1 OD/mL) and a particle concentration in particle sample of 8×106 particles/mL. The cells were washed (3 ×) with the pH specific PBS containing 10% FBS. The uptake of IR780 dye from the particles was measured in two ways: 1) by analyzing the fluorescence intensity using an Odyssey Infrared System (LI-COR) in both the cell lines with dosimetry performed using LI-COR software; 2) Tissue mimicking phantoms were prepared for multispectral optoacoustic tomography (MSOT) analysis. Cell pellets were added to the agar gel-based tissue mimicking phantoms. Tissue mimicking phantoms were prepared from 1.3 %w/w of agar and 6 % v/v intralipid was added to DI water which results in creating a gel with a reduced light scattering coefficient of μ = 10 cm−1. S2VP10 cells treated with TMOS, TEOS, and TPOS with and without V7 peptide at pH 7.4 and 6.6 containing IR780 were added to the 3 mm diameter cylindrical opening in the tissue phantoms. All samples were evaluated at multiple wavelengths (680, 710, 730, 740, 760, 760, 770, 780, 800, 850 and 900 nm). At least 20 average readings were taken during the analysis at each wavelength.
Cell internalization of TROS with Propidium iodide
Propidium iodide was used in place of IR780 to evaluate cell internalization prior to dye release. Propidium iodide usually emits green fluorescence, but when bound to nucleic acid, red fluorescence is instead emitted. Propidium iodide is usually impermeable to the cell membrane, but can be allowed entry into the cell by encapsulation within TROS. S2VP10 cells were grown to confluency in a 6-well plate containing 12mm cover glasses in wells. Cells were grown overnight in DMEM with 10% FBS and 1% L-glutamine. pH-specific DMEM (pH 7.4 or 6.6) was used to mimic benign and malignant conditions for pH-specific internalization and release. 80 μL of PBS (control) or V7-TROS with propidium iodide was added to the plate for 1.5 hr. Plates with then fixed with 4% formaldehyde for 10 min at room temperature. Cells were washed with PBS to remove free TROS and propidium iodide. The cover glasses were removed and mounted with VectaShield Vibrance Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). The samples were dried in the darkness for 2 hrs. Images were taken at 400X magnification using a Leica SP8 Confocal Microscope. Fluorescence counts of images were determined through channel splitting and thresholding via ImageJ.
In vivo Experiments
Human pancreatic cancer xenograft mouse models
Athymic female mice aged 4 wk were used for this study in strict adherence to the Wake Forest University Institutional Animal Care and Use Committee approved protocol. Mice were placed on a 2920X alfalfa free-rodent diet (Harlan Laboratories, Indianapolis, IN) to reduce background signal during imaging. An established model for orthotopic cell implantation into the mouse pancreas was followed as previously described.38 Briefly, 10 mice were anesthetized with isoflurane, and the abdomen was then prepped with betadine. A 1 cm incision was made in the left upper quadrant, with the pancreas exposed by retraction of the spleen. Luciferase-cloned S2VP10 cells (S2VP10L) were suspended in serum-free RPMI medium at 4°C in a sterile tube. A solution of 1.5 × 105 cells per 30 μL was drawn up using a 28-gauge needle and injected into the pancreas. A sterile cotton-tipped applicator was held over the injection site for 30 s to prevent peritoneal leakage. The organs were returned to the abdomen with the skin, and peritoneum closed in a single layer using 5–0 prolene sutures. Mice recovered underneath a warming blanket and were returned to their cages with food and water ad libitum after regaining full mobility.
The raw data of the multispectral analyzed samples were reconstructed with the ViewMSOT software version 3.8 (Ithera Medical, Munich, Germany). Using ViewMSOT software wavelengths corresponding to the IR780 dye were reconstructed at a resolution of 75 μm. The Multispectral Processing was conducted using linear regression with ViewMSOT 3.8 (Ithera Medical, Munich, Germany). Image stacks were imported into orthogonal views. The region of interest (ROI) was plotted over the tumor site, and the settings were kept constant for all the image slices obtained throughout the experiment. The signal intensity obtained from TROS nanoparticles containing IR780 at the tumor site was represented in MSOT a.u units. The MSOT a.u. values for the TROS nanoparticles were compared using SAS 9.3 Cary, NC, USA.
Evaluation of Nanoparticles using MSOT
Mice were intravenously injected, at the tail vein, with actively, V7, targeted and passively, no peptide, targeted TMOS, TEOS, and TPOS. Injected MSN probe contained encapsulated IR780 at a concentration of 2.7mM (0.1 OD/mL) and an MSN concentration in solution of 8 × 106 particles/mL. After intravenous injection of probe, mice were imaged at 8 h using InVision 512 TF MSOT (iThera Medical, Munich, Germany). Mice were imaged ventral side up within the animal holder and positioned in a nose cone for anesthesia delivery. Anesthesia was maintained at 1.5% isoflurane in 0.8 L medical air and 0.1 L O2 throughout imaging. Imaging was performed using axial slices with a 0.2-mm step through the liver-tumor-kidney region, at wavelengths of 680, 710, 730, 740, 750, 760, 770, 780, 800, 850, and 900 nm for each position. For each wavelength, 25 frames were obtained and averaged. To minimize the influence of animal movement in the images, an acquisition time of 10 μs per frame was used.9 Respiration rate and signs of distress were monitored through all stages of the imaging procedure. After the last imaging time point of 24 h, animals were euthanized via carbon dioxide overdose and cervical dislocation. Pancreas tumor, liver, spleen, and kidney were removed and imaged using the AMI (Spectral Imaging Instruments, Tucson, AZ, USA).
Pharmacokinetics and biodistribution of TROS with MSOT
Pharmacokinetics of TROS particles were evaluated using orthotopically implanted xenograft mice. Mice were tail-vein injected with V7-TROS or passively targeted TROS particles loaded with IR780 cargo at 2.7mM (0.1 OD/mL). Mice were imaged ventral side up while under a 1.5% isoflurane anesthesia using MSOT at 0 h, 4h, 8 h, and 24 h post injection. Axial slices were recorded at 0.2 mm-steps throughout regions that contain the liver, tumor, kidney, and spleen. Multiwavelength illumination between 680 nm and 900 nm was utilized for spectral enrichment. 10 microsecond acquisition time and 25 acquisitions per wavelength were used for image capture. Mice were continuously monitored to ensure distress minimization.
Ex vivo organ analysis
After the 8 h imaging time point, several control and V7-TROS treated mice were euthanized, and evaluation of signal accumulation within the tumor was compared with off-target organs using fluorescence imaging by Advanced Molecular Imager (AMI). ROI analysis of IR780 cargo accumulation was used to verify MSOT data in the liver, kidney, and pancreas tumor. Liver and kidney were also evaluated for off-target toxicity using H&E staining.
Statistical analysis.
Statistical evaluation of the in vitro studies was performed using analysis of variance (ANOVA) and Tukey multiple comparison test with significance defined at p<0.05. In vivo pharmacokinetics were analyzed with repeated measures ANOVA at significance of p < 0.05. The in vivo biodistribution and accumulation of the IR780 loaded V7-TROS and TROS nanoparticles containing IR780 in the pancreatic tumor, liver, and kidney were statistically analyzed using ANOVA with significance at p < 0.05.
Supplementary Material
Acknowledgements
The authors acknowledge funding sources:
National Institute of Health grant R01CA205941
National Institute of Health grant R01CA212350
National Institute of Health grant R01EB020125
National Institute of Health grant P30CA225520
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Poon W, Kingston BR, Ouyang B, Ngo W and Chan WC, Nature Nanotechnology, 2020, 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Sykes EA, Chen J, Zheng G and Chan WC, ACS nano, 2014, 8, 5696–5706. [DOI] [PubMed] [Google Scholar]
- 3.Frieboes HB, Wu M, Lowengrub J, Decuzzi P and Cristini V, PloS one, 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korangath P, Barnett JD, Sharma A, Henderson ET, Stewart J, Yu S-H, Kandala SK, Yang C-T, Caserto JS and Hedayati M, Science Advances, 2020, 6, eaay1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haubner R, Wester H-J, Burkhart F, Senekowitsch-Schmidtke R, Weber W, Goodman SL, Kessler H and Schwaiger M, Journal of Nuclear Medicine, 2001, 42, 326–336. [PubMed] [Google Scholar]
- 6.Gao W, Xiang B, Meng T-T, Liu F and Qi X-R, Biomaterials, 2013, 34, 4137–4149. [DOI] [PubMed] [Google Scholar]
- 7.Dancy JG, Wadajkar AS, Connolly NP, Galisteo R, Ames HM, Peng S, Tran NL, Goloubeva OG, Woodworth GF and Winkles JA, Science advances, 2020, 6, eaax3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du W and Elemento O, Oncogene, 2015, 34, 3215–3225. [DOI] [PubMed] [Google Scholar]
- 9.Kimbrough CW, Khanal A, Zeiderman M, Khanal BR, Burton NC, McMasters KM, Vickers SM, Grizzle WE and McNally LR, Clinical cancer research, 2015, 21, 4576–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillies RJ, Verduzco D and Gatenby RA, Nature Reviews Cancer, 2012, 12, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weerakkody D, Moshnikova A, Thakur MS, Moshnikova V, Daniels J, Engelman DM, Andreev OA and Reshetnyak YK, Proceedings of the National Academy of Sciences, 2013, 110, 5834–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreev OA, Engelman DM and Reshetnyak YK, Frontiers in physiology, 2014, 5, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao W and Gao H, International Journal of Pharmaceutics, 2018, 552, 328–339. [DOI] [PubMed] [Google Scholar]
- 14.Blanco E, Shen H and Ferrari M, Nature biotechnology, 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh N and Park J-H, International journal of nanomedicine, 2014, 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano M, Miyazono K and Uesaka M, Nature nanotechnology, 2011, 6, 815. [DOI] [PubMed] [Google Scholar]
- 17.Bharti C, Nagaich U, Pal AK and Gulati N, International journal of pharmaceutical investigation, 2015, 5, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayan R, Nayak UY, Raichur AM and Garg S, Pharmaceutics, 2018, 10, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X and Ridgway J, Nature, 2010, 464, 1052–1057. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, Chai F, Fu Q, Zhang L, Liu H, Li L, Liao Y, Su Z, Wang C and Duan B, Journal of Materials Chemistry, 2011, 21, 5299–5306. [Google Scholar]
- 21.Gary-Bobo M, Mir Y, Rouxel C, Brevet D, Basile I, Maynadier M, Vaillant O, Mongin O, Blanchard-Desce M and Morère A, Angewandte Chemie International Edition, 2011, 50, 11425–11429. [DOI] [PubMed] [Google Scholar]
- 22.Shen S-C, Ng WK, Shi Z, Chia L, Neoh KG and Tan RBH, Journal of Materials Science: Materials in Medicine, 2011, 22, 2283. [DOI] [PubMed] [Google Scholar]
- 23.Shang L, Nienhaus K and Nienhaus GU, Journal of nanobiotechnology, 2014, 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daum N, Tscheka C, Neumeyer A and Schneider M, Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology, 2012, 4, 52–65. [DOI] [PubMed] [Google Scholar]
- 25.Yanes RE and Tamanoi F, Therapeutic delivery, 2012, 3, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreira AF, Dias DR and Correia IJ, Microporous and Mesoporous Materials, 2016, 236, 141–157. [Google Scholar]
- 27.Gurka MK, Pender D, Chuong P, Fouts BL, Sobelov A, McNally MW, Mezera M, Woo SY and McNally LR, Journal of controlled release, 2016, 231, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamaeva V, Sahlgren C and Lindén M, Advanced drug delivery reviews, 2013, 65, 689–702. [DOI] [PubMed] [Google Scholar]
- 29.Wen J, Yang K, Liu F, Li H, Xu Y and Sun S, Chemical Society Reviews, 2017, 46, 6024–6045. [DOI] [PubMed] [Google Scholar]
- 30.Kaasalainen M, Aseyev V, von Haartman E, Karaman DŞ, Mäkilä E, Tenhu H, Rosenholm J and Salonen J, Nanoscale research letters, 2017, 12, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christian DA, Cai S, Garbuzenko OB, Harada T, Zajac AL, Minko T and Discher DE, Molecular pharmaceutics, 2009, 6, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNally LR, Mezera M, Morgan DE, Frederick PJ, Yang ES, Eltoum I-E and Grizzle WE, Clinical cancer research, 2016, 22, 3432–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoffels I, Morscher S, Helfrich I, Hillen U, Leyh J, Burton NC, Sardella TC, Claussen J, Poeppel TD and Bachmann HS, Science translational medicine, 2015, 7, 317ra199–317ra199. [DOI] [PubMed] [Google Scholar]
- 34.Knieling F, Neufert C, Hartmann A, Claussen J, Urich A, Egger C, Vetter M, Fischer S, Pfeifer L and Hagel A, The New England journal of medicine, 2017, 376, 1292. [DOI] [PubMed] [Google Scholar]
- 35.MacCuaig WM, Jones MA, Abeyakoon O and McNally LR, Radiology: Imaging Cancer, 2020, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhutiani N, Samykutty A, McMasters KM, Egilmez NK and McNally LR, Photoacoustics, 2019, 13, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalyane D, Raval N, Maheshwari R, Tambe V, Kalia K and Tekade RK, Materials Science and Engineering: C, 2019. [DOI] [PubMed] [Google Scholar]
- 38.Kimbrough CW, Hudson S, Khanal A, Egger ME and McNally LR, Journal of surgical research, 2015, 193, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeiderman MR, Morgan DE, Christein JD, Grizzle WE, McMasters KM, L. R. J. A. b. s. McNally and engineering, 2016, 2, 1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tallury P, Payton K and Santra S, 2008, 3, 579–592. [DOI] [PubMed] [Google Scholar]
- 41.Stoffels I, Petri M, Morscher S, Burton N, Schadendorf D, Gunzer M and Klode J, Journal of Nuclear Medicine, 2016, 57, 412–412. [Google Scholar]
- 42.Lutzweiler C, Meier R, Rummeny E, Ntziachristos V and Razansky D, Optics letters, 2014, 39, 4061–4064. [DOI] [PubMed] [Google Scholar]
- 43.Razansky D, Buehler A and Ntziachristos V, Nature protocols, 2011, 6, 1121. [DOI] [PubMed] [Google Scholar]
- 44.Bhutiani N, Kimbrough C, Burton N, Morscher S, Egger M, McMasters K, Woloszynska-Read A, El-Baz A and McNally L, Biotechnic & histochemistry, 2017, 92, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada H, Urata C, Aoyama Y, Osada S, Yamauchi Y and Kuroda K, Chemistry of Materials, 2012, 24, 1462–1471. [Google Scholar]
- 46.Möller K, Kobler J and Bein T, Adv. Funct. Mater, 2007, 17, 605. [Google Scholar]
- 47.Lehman SE, Morris AS, Mueller PS, Salem AK, Grassian VH and Larsen SC, Environmental Science: Nano, 2016, 3, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laramie M, Smith M, Marmarchi F, McNally L and Henary M, Molecules, 2018, 23, 2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeiderman MR, Morgan DE, Christein JD, Grizzle WE, McMasters KM and McNally LR, ACS biomaterials science & engineering, 2016, 2, 1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin W, Kimbrough CW, Gomez-Gutierrez JG, Burns CT, Chuong P, Grizzle WE and McNally LR, Journal of nanobiotechnology, 2015, 13, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samykutty A, Grizzle WE, Fouts BL, McNally MW, Chuong P, Thomas A, Chiba A, Otali D, Woloszynska A, Said N, Frederick P, Jasinski J, Liu J and McNally LR, Biomaterials, 2018, 182, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ouyang B, Poon W, Zhang Y-N, Lin ZP, Kingston BR, Tavares AJ, Zhang Y, Chen J, Valic MS and Syed AM, Nature Materials, 2020, 1–10. [DOI] [PubMed] [Google Scholar]
- 53.Lu F, Wu SH, Hung Y and Mou CY, small, 2009, 5, 1408–1413. [DOI] [PubMed] [Google Scholar]
- 54.Chauhan VP, Stylianopoulos T, Martin JD, Popović Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D and Jain RK, Nature nanotechnology, 2012, 7, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stylianopoulos T, Soteriou K, Fukumura D and Jain RK, Annals of biomedical engineering, 2013, 41, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadauskas E, Wallin H, Stoltenberg M, Vogel U, Doering P, Larsen A and Danscher G, Particle and fibre toxicology, 2007, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan KW-Y and Wong W-T, Coordination Chemistry Reviews, 2007, 251, 2428–2451. [Google Scholar]
- 58.Swietach P, Vaughan-Jones RD and Harris AL, Cancer and Metastasis Reviews, 2007, 26, 299–310. [DOI] [PubMed] [Google Scholar]
- 59.Cruz-Monserrate Z, Roland CL, Deng D, Arumugam T, Moshnikova A, Andreev OA, Reshetnyak YK and Logsdon CD, Scientific reports, 2014, 4, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C and Rivoltini L, 2017. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Lin Y and Gillies RJ, Journal of Nuclear Medicine, 2010, 51, 1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vander Heiden MG, Cantley LC and Thompson CB, science, 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reshetnyak YK, Andreev OA, Segala M, Markin VS and Engelman DM, Proceedings of the National Academy of Sciences, 2008, 105, 15340–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andreev OA, Dupuy AD, Segala M, Sandugu S, Serra DA, Chichester CO, Engelman DM and Reshetnyak YK, Proceedings of the National Academy of Sciences, 2007, 104, 7893–7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reshetnyak YK, Andreev OA, Lehnert U and Engelman DM, Proceedings of the National Academy of Sciences, 2006, 103, 6460–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaumet M, Vargas A, Gurny R and Delie F, European journal of pharmaceutics and biopharmaceutics, 2008, 69, 1–9. [DOI] [PubMed] [Google Scholar]
- 67.Decuzzi P, Godin B, Tanaka T, Lee S-Y, Chiappini C, Liu X and Ferrari M, Journal of Controlled Release, 2010, 141, 320–327. [DOI] [PubMed] [Google Scholar]
- 68.Zhang S, Li J, Lykotrafitis G, Bao G and Suresh S, Advanced materials, 2009, 21, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma N, Ma C, Li C, Wang T, Tang Y, Wang H, Mou X, Chen Z and He N, Journal of nanoscience and nanotechnology, 2013, 13, 6485–6498. [DOI] [PubMed] [Google Scholar]
- 70.He X, Nie H, Wang K, Tan W, Wu X and Zhang P, Anal. Chem, 2008, 80, 9597–9603. [DOI] [PubMed] [Google Scholar]
- 71.Choi CHJ, Zuckerman JE, Webster P and Davis ME, Proceedings of the National Academy of Sciences, 2011, 108, 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang X-D, Di Wu XS, Liu P-X, Yang N, Zhao B, Zhang H, Sun Y-M, Zhang L-A and Fan F-Y, International journal of nanomedicine, 2011, 6, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerdá VJ, Pacheco RR, Witek JD, de la Calle FMM and de la Sen Fernández ML, Allergy, Asthma & Clinical Immunology, 2019, 15, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gachoka D, ACG Case Reports Journal, 2015, 2, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wylon K, Dölle S and Worm M, Allergy, Asthma & Clinical Immunology, 2016, 12, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hatakeyama H, Akita H and Harashima H, Advanced drug delivery reviews, 2011, 63, 152–160. [DOI] [PubMed] [Google Scholar]
- 77.Almalik A, Benabdelkamel H, Masood A, Alanazi IO, Alradwan I, Majrashi MA, Alfadda AA, Alghamdi WM, Alrabiah H and Tirelli N, Scientific reports, 2017, 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lynch I and Dawson KA, Nano today, 2008, 3, 40–47. [Google Scholar]
- 79.Liu J, Yu M, Ning X, Zhou C, Yang S and Zheng JJACIE, 2013, 52, 12572–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ernsting MJ, Murakami M, Roy A and Li S-D, Journal of controlled release, 2013, 172, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li S-D and Huang L. J. M. p., 2008, 5, 496–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.