

Abstract
G protein-coupled receptors (GPCRs) are the largest family of membrane receptors in humans. They mediate nearly all aspects of human physiology and thus are of high therapeutic interest. GPCR signaling is regulated in space and time by receptor phosphorylation. It is believed that different phosphorylation states are possible for a single receptor, and each encodes for unique signaling outcomes. Methods to determine the phosphorylation status of GPCRs are critical for understanding receptor physiology and signaling properties of GPCR ligands and therapeutics. However, common proteomic techniques have provided limited quantitative information regarding total receptor phosphorylation stoichiometry, relative abundances of isomeric modification states, and temporal dynamics of these parameters. Here, we report a novel middle-down proteomic strategy and parallel reaction monitoring (PRM) to quantify the phosphorylation states of the C-terminal tail of metabotropic glutamate receptor 2 (mGluR2). By this approach, we found that mGluR2 is subject to both basal and agonist-induced phosphorylation at up to four simultaneous sites with varying probability. Using a PRM tandem mass spectrometry methodology, we localized the positions and quantified the relative abundance of phosphorylations following treatment with an agonist. Our analysis showed that phosphorylation within specific regions of the C-terminal tail of mGluR2 is sensitive to receptor activation, and subsequent site-directed mutagenesis of these sites identified key regions which tune receptor sensitivity. This study demonstrates that middle-down purification followed by label-free quantification is a powerful, quantitative, and accessible tool for characterizing phosphorylation states of GPCRs and other challenging proteins.
Introduction
G protein-coupled receptors (GPCRs) are the largest family of cell surface receptors in mammalian cells and are involved in the regulation of nearly every cellular process.1 Phosphorylation of a GPCR within the C-terminal tail and intracellular loops (ICLs) is a key posttranslational modification (PTM) which regulates and diversifies GPCR signaling by mediating interactions with multiple signaling partners, controlling receptor localization, and tuning receptor dynamics.2−5 Many GPCRs have multiple intracellular serine, threonine, or tyrosine residues which can potentially get phosphorylated. Additionally, several studies have found that different agonists6,7 or cellular conditions2,3,8 can produce unique phosphorylation patterns in a GPCR. These results prompted a model in which a specific GPCR phosphorylation state (defined by a set of phosphorylation at specific residues) is a mechanism to diversify potential signaling outcomes (dubbed the phosphorylation barcode)5,9−19 similar to the histone code hypothesis.20
Determining the phosphorylation status of GPCRs is not only critical for understanding receptor physiology but also necessary for mapping signaling properties of GPCR therapeutics and for identifying druggable targets within signal transduction pathways.21 The ability of a GPCR to adopt different phosphorylation states in the C-terminal domain (or phosphorylation barcode) was initially shown in β2-adrenergic receptor22 and M3-muscarinic acetylcholine receptor (M3-mAChR).9 These initial studies and subsequent studies of GPCR phosphorylation were performed using site-directed mutagenesis, bottom-up proteomics, phosphorylation-specific antibodies, or radiometric 32P labeling.16,23 While these techniques successfully overcome the technical challenges of analyzing large, hydrophobic proteins,24 they are limited in that they either require prior knowledge of phosphorylation sites or can only interrogate phosphorylation sites one at a time or in close proximity. Importantly, current methods cannot quantify the total phosphorylation stoichiometry (i.e., number of co-occurring phosphorylated residues on a single receptor), positional distribution of phosphorylation, and the heterogeneity and time evolution of these parameters. Thus, a general quantitative method for characterizing intact phosphorylation states of GPCRs is currently lacking.
Mass spectrometry (MS)-based proteomics is a powerful tool for characterizing PTMs and is capable of PTM quantification and localization by measuring the total mass and fragmentation spectra of a protein analyte.25 MS-based proteomics can be broadly divided into bottom-up, middle-down, and top-down workflows depending on the extent of peptide backbone cleavage prior to MS. Each approach has particular advantages and provides unique information coverage.26 Bottom-up entails extensive proteolysis to produce short peptide-level analytes prior to MS,26 while top-down entails direct analysis of whole proteins.27−29 Top-down is ideally suited to analyze heavily modified proteins, as co-occurring modifications are not separated by peptide cleavage and can be localized and quantified using tandem mass spectrometry (MS/MS or MSn).30−34 Despite the power of this method, large and hydrophobic proteins have remained difficult to characterize by top-down MS due to technical limitations. An intermediate between these methods, known as middle-down proteomics exists as a compromise in which partial proteolysis is performed, allowing the characterization of intact regions of interest while simplifying the analysis of large or hydrophobic proteins.35 Next, site-specific quantification of PTMs is generally performed using isotope-based methods or label-free methods via spectral counting of diagnostic ions. Conventional isotope-based methods (e.g., isobaric labeling, stable isotope labeling by amino acids in cell culture, and isotopically labeled internal standards) are generally applied to bottom-up workflows and have only recently been applied to intact protein workflows.36−38 As an alternative, quantification via spectral counting of diagnostic ions has been widely used for assessing both total protein modification and site-specific modification abundance.39−43 Spectral counting is generally performed by monitoring predefined precursor and fragment ions (selected reaction monitoring, SRM)44 or monitoring the assembly of fragment ions using high-resolution ion trap or Orbitrap instruments (parallel reaction monitoring, PRM).45 Ultimately, the development of a technically accessible sample preparation pipeline and complementary fragmentation analysis is necessary for advancing the study of GPCRs and other membrane proteins.
In this study, we developed a novel middle-down proteomic strategy coupled to PRM as a generalizable method to measure intact phosphorylation states of the C terminal tail of GPCRs. We used this method to characterize the phosphorylation of the C-terminal tail of human mGluR2 in a mammalian cell line. mGluR2 is a class C GPCR which mediates synaptic transmission and plasticity in the central nervous system and has emerged as a candidate therapeutic target in the treatment of anxiety, schizophrenia, pain disorders, and addiction.46,47 mGluR2 has 14 possible phosphorylation sites which are all located in the C-terminal tail. Since mGluR2 is known to not be internalized upon activation, we chose to investigate possible alternative roles for mGluR2 phosphorylation. Using our method, we identified novel basal and agonist-induced mGluR2 phosphorylation sites and quantified both phosphorylation stoichiometry and positional abundance of phosphorylations throughout the C-terminal tail. Next, we performed site-directed mutagenesis to explore the impact of receptor phosphorylation on mGluR2-dependent signaling. Importantly, we determined that phosphorylation of mGluR2 in regions proximal to transmembrane helix 7 (TM7) functionally tunes receptor sensitivity to an agonist. Overall, our results demonstrate the viability of middle-down proteomics as a characterization tool for GPCRs. As presented, our assay is ideal for detecting phosphorylation patterns within the C-terminal tail of receptors and cannot detect phosphorylations within the ICLs. However, for many receptors, phosphorylation is only possible in the C-terminus.
Results and Discussion
Development of a Middle-Down Proteomic Assay
To characterize complete phosphorylation states, the intracellular C-terminus of mGluR2 must remain intact. To selectively isolate and analyze the mGluR2 C-terminus, we generated a stable cell line expressing human mGluR2 with an EGFP at the N-terminus and a TEV protease site following TM7 between residues A830 and P831, in HEK293T cells (Figures 1, S1A). We verified that this receptor traffics to the plasma membrane very efficiently and is functional (Figure S1). This specific design allowed for the isolation of the intact C-terminal tail with high yield and purity via anti-GFP immunoprecipitation followed by TEV digestion (Figure 1A, Table S1, Figure S2). After purification, we used reverse-phase liquid chromatography–mass spectrometry (LC–MS) to quantify total phosphorylation and positional distribution of phosphorylation via PRM.48 We isolated individual phosphorylation stoichiometries (e.g., monophosphorylated, diphosphorylated, etc.), followed by higher energy collisional dissociation (HCD), to produce peptide-level fragment ions holding the modification (Figure 1B). Given the pool of fragment ions we detected, the C-terminal tail was divided into four sections with phosphorylation localized and relatively quantified within these defined regions. Relative abundances of isomers were then quantified by comparing the intensities of fragment ions (Figure S3).
Figure 1.

Development of a novel middle-down proteomic assay. (A) Overview of middle-down MS assay. (B) Overview of isolation, fragmentation, and PRM of phosphorylated precursors for relative quantification of isomers, an example is given for a diphosphorylated tail. (C) Sequence of human mGluR2 (accession #Q14416) cleavage product and potential phosphorylation sites (red) used for PRM studies. Residue numbering in (C) is based on the endogenous sequence reported in accession #Q14416-01.
Gas collision-based fragmentation can eject labile PTMs as neutral losses (Figures S4 and S5), which would interfere with localization and quantification; however, the propensity to eject is highly dependent on the size and charge of the starting analyte (Supporting Discussion 1, Figures S4–S6).49−51 To determine MS2 parameters which minimized neutral loss of phosphorylation from the mGluR2 tail, we performed a PRM assay on a synthetic diphosphorylated mGluR2 tail with phosphorylation at defined sites. We found that for normalized collision energies (NCEs) ≤ 20 neutral loss of metaphosphoric (HPO3) or phosphoric acid (H3PO4) occurred in a minority of fragment ions and accounted for less than 5% of fragment ion intensity for any given fragment ion species (Tables S2 and S3, Figure S7). Quantifying the extent of phosphorylation ejection for proteins with several unknown positional isomers is more challenging as neutral loss of phosphoric acid or water can lead to isobaric fragment ions (Supporting Discussion 1, Figure S6). However, we performed a statistical comparison of −18.01 Da losses from fragment ions originating from unmodified mGluR2 tails versus monophosphorylated mGluR2 tails derived from the stable cell line and found no statistical increase, indicating there is no notable loss of phosphoric acid (Figure S8).
mGluR2 Phosphorylation States Are Heterogeneous and Exist as Multiple Isomers
First, we analyzed the intact, isolated C-terminal tails of mGluR2 and found that each tail can have between 0 and 4 total phosphorylations simultaneously (Figure 2A). These modification states were identified with high confidence, reproducibility, and purity (intact mass error <1 ppm and assigned fragment P-score of 4.2 × 10–21 for the unmodified form, Table S1 and Figure S2). Isotopic envelopes for each phosphoform were identified based on a Pearson correlation and summed across the LC timescale (Figure S9) revealing a large change in relative intensity between phosphoforms, with the unmodified tail representing 75.5 ± 3.1% of the summed signal intensity (Figure 2B). Subsequent monophosphorylated, diphosphorylated, and triphosphorylated forms accounted for 21.0 ± 2.3, 3.5 ± 1.1, and 0.1 ± 0.1% relative intensity, respectively (Figure 2B, Table S4). It is important to note that electrospray ionization efficiencies of peptides and small proteins could be impacted by the total number and position of phosphorylations and thus the measured amplitude of phosphorylated species could be an underestimation of the true concentrations in the sample.
Figure 2.

mGluR2 is subject to co-occurring phosphorylations. (A) Intact mass spectra of the mGluR2 C-terminus (4+ charge state). (B) Relative summed intensity across the LC-timescale (%) of intact phosphorylated C-terminal tails before l-glutamic acid treatment. Unmodified mGluR2 is denoted in gray, monophosphorylated mGluR2 in light blue, diphosphorylated mGluR2 in dark blue, triphosphorylated mGluR2 in green, and tetraphosphorylated mGluR2 in gold.
Next, we observed that all phosphorylation stoichiometries eluted as multiple peaks, as measured by reverse-phase chromatography (Figure S2C), suggesting that these phosphorylation states existed as multiple isomers. In total, we recorded over 500 unique fragment ions per replicate and observed high sequence coverage between ranges b16–b20, b23–b27, b28–b37, y23–y27, and y6–y12 consistently across all samples (assuming the N-terminal glycine of the cleavage product is the first residue as shown in Figure 1C). Based on this distribution, we segmented the C-terminal tail into four regions to further quantify the isomeric distributions (Figure 3A).32−34,52 In this analysis, the phosphorylation content was localized within regions A (T832, S833, S837), B (S843, S844, S845), C (S850, S852, T857), and D (S867, T868, T869, S870, S871) for the monophosphorylated, diphosphorylated, and triphosphorylated precursors. For all precursors, we found that phosphorylation is not uniformly distributed among the four regions. Specifically, regions A and D were highly modified (Figure 3B–D, values provided in Table S5). For the monophosphorylated population, regions A and D contained 26.0 ± 2.5 and 53.6 ± 1.2% relative phosphorylation content, respectively. Similarly, for the higher stoichiometries (Figure 3C, D), regions A + B and region D carried most of the phosphorylation content. These results are consistent with previously reported phosphorylation of mGluR2 found in high-throughput studies.53−56 Importantly, for all precursors, each region is partially modified with a varying probability that is not proportional to the number of possible phosphorylated residues. The nonuniform distribution of phosphorylation suggests a specific mechanism that controls these modifications, as opposed to stochastic modification by kinases. Importantly, these data validate that multiple phosphorylations can occur on a single receptor, which was previously unknown. Overall, these data demonstrate that mGluR2 phosphorylation states are very heterogeneous among the population and are indeed combinatorial.
Figure 3.

mGluR2 is subject to basal isomeric phosphorylation states. (A) Sequence of mGluR2 cleavage product and potential phosphorylation sites (red) used for PRM studies, b-type and y-type fragment ions utilized for PRM studies are denoted as black flags. Fragment ion numbering is based on the number of residues in the cleavage product (P2 = PA830 in accession #Q14416-1). Phosphorylation content was localized between regions A, B, C, and D for the (B) mono-, (C) di-, (D) and triphosphorylated populations. The intact precursor corresponding to each set of plots is shown pictorially on the left. Color fill denotes the number of phosphorylations localized within a given region using fragment ions from the PRM assay. No phosphorylation is denoted with gray, monophosphorylation is denoted with light blue, diphosphorylation is denoted with dark blue, and triphosphorylation is denoted with green. For the higher stoichiometries, regions A and B were combined based on fragment ion coverage. *For the triphosphorylated population, y19 to y16 were used to calculate occupancy in region D + T857. Error bars represent the standard error of the mean (N = 3 biological replicates and 3 technical replicates).
mGluR2 Activation Increases Total Phosphorylation Abundance
After quantifying the basal phosphorylation state of mGluR2, we then investigated the effect of agonist treatment on mGluR2 phosphorylation. For many GPCRs, agonist-induced activation results in receptor phosphorylation by GPCR kinases (GRKs). Additionally, the location and abundance of phosphorylations can change over time post-agonist exposure and, in some cases, the agonist type. For example, recent temporal studies of angiotensin II type 1 and β2-adrenergic receptors show that agonist-induced GRK activity can persist anywhere from <30 s to 30–60 min following ligand exposure.57 To investigate and quantify this dynamic aspect in our system, we treated mGluR2-expressing cells with 1 mM l-glutamic acid and collected the cells at 0-, 10-, 30-, or 60-minutes post-activation for analysis. We found that after the addition of l-glutamic acid, the C-terminal tail is progressively phosphorylated with all phosphorylation stoichiometries increasing relative to the unmodified tail throughout the 60 minute treatment window (Figures 4A, B, S10, Table S4). Overall, these findings demonstrate that the mGluR2 C-terminus is basally phosphorylated in our model system and these phosphorylation states rapidly and consistently increase upon agonist stimulation. These findings are consistent with the model that receptor activation is a regulator of overall phosphorylation abundance on the GPCR C-terminal tail.
Figure 4.

mGluR2 activation enhances intracellular phosphorylation abundance. (A) Normalized intensity of intact phosphorylated C-terminal tails over 1 mM l-glutamic acid treatment (0–60 min). The intensity for a given phosphorylated stoichiometry is normalized to the intensity of the unmodified mGluR2 tail in the corresponding LC–MS run. Values for each phosphoform are denoted by color. Values for the monophosphorylated population are shown in light blue, the diphosphorylated population is shown in dark blue, the triphosphorylated population is shown in green, and the tetraphosphorylated population is shown in gold. (B) Each phosphorylated stoichiometry is provided as an individual inset. Error bars represent the standard error of the mean (N = 3 biological replicates and 3 technical replicates). Brackets indicate a statistical comparison between two data points using a Wilcoxon–Mann–Whitney test. Significant differences are denoted as follows: * = p < 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
mGluR2 Activation Dynamically Shifts Positional Phosphorylation Abundance Over Time
Next, we quantified how agonist treatment shifts the relative phosphorylation abundance within the defined regions presented in Figure 3 over time. We did this by calculating the difference in log2(fragment ion intensity) for differentially modified fragment ions. For example, comparing unmodified versus monophosphorylated fragment ions derived from the monophosphorylated population showed statistically significant changes in phosphorylation position over l-glutamic acid treatment (Figures 5A, S11–S13). Specifically, all sites N-terminal to S867 decreased in phosphorylation content after l-glutamic acid treatment while the distal sites from S867 to S871 increased in phosphorylation content. We then converted ratios of fragment ion intensities into relative percentages, showing phosphorylation content within a defined region (Figure 5B, C). For the monophosphorylated population region D, which was highly basally modified, the relative abundance of phosphorylation increased from 53.5 ± 7.8% (t = 0 min) to 70.5 ± 11.1% (t = 60 min) after l-glutamic acid treatment (Figure 5B). Inversely, region A underwent a decrease in phosphorylation abundance from 26.6 ± 8.7% (t = 0 min) to 18.6 ± 8.3% (t = 60 min). We saw a similar l-glutamic acid dependence for the diphosphorylated and triphosphorylated populations (Figures 5C, S12, and S13). For the diphosphorylated population, region D increased in both monophosphorylation and diphosphorylation, as relative abundances shifted from 4.8 ± 0.7% (t = 0 min) to 13.4 ± 3.6% (t = 60 min) and 5.9 ± 2.2% (t = 0 min) to 21.5 ± 7.8% (t = 60 min), respectively (Table S5). Regions A + B proportionally decreased in phosphorylation for both monophosphorylated and diphosphorylated populations, as relative abundances shifted from 22.4 ± 3.8% (t = 0 min) to 20.6 ± 6.7% (t = 60 min) and 46.5 ± 8.4% (t = 0 min) to 20.6 ± 11.9% (t = 60 min), respectively. Together, our results showed that generally, regions A, B, and D undergo statistically significant changes in phosphorylation amplitude after l-glutamic acid treatment. Specifically, while agonist treatment overall increased the density of phosphorylation on the C-terminal tail, the increase is not uniformly distributed across the tail. In fact, locally, the region closest to TM7 showed a reduction in monophosphorylation, while the region at the very C-terminus increased in monophosphorylation.
Figure 5.

mGluR2 activation dynamically shifts positional phosphorylation abundance over time. (A) The difference in log 2 (fragment ion intensity) for unmodified and monophosphorylated fragment ions derived from the monophosphorylated precursor population shows statistically significant changes in phosphorylation position over 1 mM l-glutamic acid treatment (0 to 60 min). The population of averaged fragment ions is listed above each box plot. These ratios of fragment ions can be converted into relative percentages showing phosphorylation content within a defined region. Relative phosphorylation content was calculated for the (B) mono- and (C) diphosphorylated precursor populations. Color fill denotes the number of phosphorylations localized within a given region using fragment ions from the PRM assay. No phosphorylation is denoted with gray, monophosphorylation is denoted with light blue, and diphosphorylation is denoted with dark blue. Error bars represent the standard error of the mean (N = 3 biological replicates and 3 technical replicates). Brackets indicate a statistical comparison between two data points using a Wilcoxon–Mann–Whitney test. Significant differences are denoted as follows: * = p < 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p ≤ 0.0001.
Phosphorylation States Fine-Tune mGluR2 Signaling
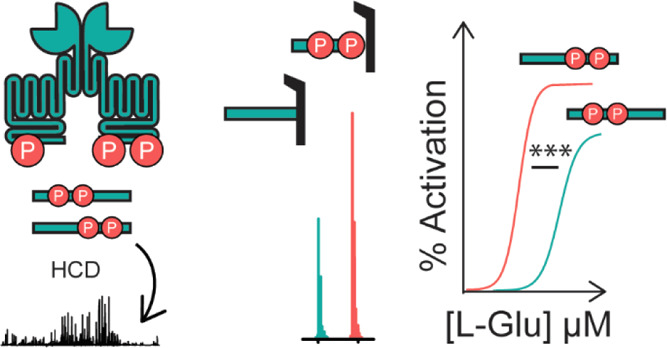
Given the identification of novel and l-glutamic acid-sensitive phosphosites, we sought to explore how mimicking phosphorylation at these newly identified modified regions would impact G protein-dependent signaling. To explore the impact of phosphorylation on G protein-dependent signaling, we performed site-directed mutagenesis of regions A, and D individually. To simulate constant phosphorylation, we constructed mutants in which all candidate phosphorylation sites (serine and threonine, S/T) were mutated to phosphomimetic glutamic acid (E).58,59 Equivalent phosphosilencing mutants were made by mutating candidate phosphorylation sites to alanine (S/T to A). mGluR2 signaling was measured using the −22F cAMP pGloSensor to quantify Gαi-dependent suppression of cAMP, as described previously.60 mGluR2 maximum efficacy (EMAX) and half maximal effective concentration (EC50) were calculated from the inhibition of forskolin (FSK)-dependent cAMP response with increasing concentrations of l-glutamic acid.60 Strikingly, we observed that substitution of phosphomimetic glutamic acid in phosphosites proximal to TM7 (region A) significantly decreased the maximum efficacy and increased the half-maximal effective concentration of l-glutamic acid, as demonstrated by the rightward shift in the concentration–response curve (Figure 6A–C). However, substitution to glutamic acid in the distal region (region D) did not impact the Gi-dependent response of mGluR2 to l-glutamic acid in comparison to wild-type receptors. This newly identified region of glutamate-sensitive phosphorylation sites could contribute to Gi-independent functions of mGluR2. Together these results show that phosphorylation in the C-terminal domain of mGluR2 affects receptor signaling in a region-specific manner. Specifically, phosphorylation proximal to TM7 critically modulates receptor sensitivity to l-glutamic acid in contrast to distal phosphorylation and supports a model of C-terminal phosphorylation “barcodes” diversifying and fine-tuning GPCR signaling events. GPCR phosphorylation is critical for almost all aspects of receptor signaling including trafficking, localization, activation, desensitization, and internalization.4 Despite this importance, the extent and heterogeneity of phosphorylation for individual receptors are not well characterized because of technical limitations. Despite the number of mGluR2 phosphorylation sites identified previously and herein, only phosphorylation at S843 via cAMP-dependent protein kinase (PKA) was previously reported.53 Indirect biochemical data demonstrate that mGluR2 desensitization is also impacted by active levels of protein kinase C (PKC) and GPCR kinase 2 (GRK2),61 suggestive of a greater, unexplored significance for mGluR2 phosphorylation. Using our approach, we ultimately determined that phosphorylation proximal to TM7 acts to tune the sensitivity of mGluR2 to l-glutamic acid, including at several sites N-terminal to S843. Interestingly, two recent structures of Gi-bound mGluR2 (PDB: 7MTS(62) and 7E9G(63)) support this observation and suggest that C-terminal residues interact with the Gα Ras domain. According to these structures, instead of inserting the Gi α5 helix into the transmembrane binding cavity similar to class A GPCRs, in mGluR2, Gi inserts into a shallow pocket formed between TM3, ICL2, ICL3, and the C-terminus.62 Although the structure of residues 831–872 is not resolved, the deletion of residues 826–872 significantly impacted G protein-dependent signaling, while the reintroduction of residues 826–833 partially recovered G protein activity.62 Coulombic electrostatic potential maps of the mGluR2–Gi complex show that the mGluR2 G protein pocket is positively charged (Figure 6D), and the complementary Gi α5 helix is correspondingly negatively charged. Therefore, it is possible that the introduction of negative charge via phosphorylation (or phosphomimetic mutation) could disrupt the mGluR2-Gi binding via interactions with the intracellular binding pocket or repulsion of the Gi α5 helix. This could explain our observation that only phosphorylation proximal to TM7 (regions A and B) can reduce receptor sensitivity. Moreover, this tuning could serve as a mechanism for re-sensitization of mGluR2 following prolonged agonist activation given that regions A and B also experienced a statistically significant decrease in phosphorylation abundance after agonist treatment. Given that mGluR2 is uniquely resistant to agonist-induced internalization in comparison to other mGluRs which are reported to internalize and recycle in a phosphatase-dependent process upon agonist stimulation,64,65 this phosphorylation-dependent tuning could serve as a novel re-sensitization mechanism following prolonged activation. Curiously, distal C-terminal phosphorylation states did not contribute to Gi-dependent signaling in our assay. Instead, these phosphorylation states may contribute to processes downstream or independent of G protein-coupling, such as receptor trafficking or tuning protein–protein interactions such as interactions between mGluR2 and PDZ-domain-containing proteins.66,67 In this model, different functionalities are encoded in the C-terminal tail in a modular form. Undoubtedly, the identification of these novel glutamate-sensitive phosphorylation sites on the distal C-terminus warrants future study into their role in G protein-independent functions of mGluR2.
Figure 6.

Intracellular phosphorylation states fine-tune mGluR2 signaling. (A) Dose–response curves for wild-type mGluR2 (blue); T832E, S833E, and S837E mGluR2 (orange); T832A, S833A, and S837A mGluR2 (purple); S867E, T868E, T869E, S870E, and S871E mGluR2 (pink); and S867A, T868A, T869A, S870A, and S871A mGluR2 (green). Quantification of (B) half-maximal effective concentration (EC50) and (C) maximal efficacy (EMAX) for l-glutamic acid. (D) Coulombic electrostatic potential map for CryoEM structure 7MTS; inset shows the intracellular pocket of mGluR2 complexed with Gi. Error bars represent the standard error of the mean (N = 3 biological replicates and 2 technical replicates). Asterisks indicate a statistical comparison using a one-way ANOVA with multiple comparisons to WT. Significant differences are denoted as follows: *** = p ≤ 0.001, **** = p ≤ 0.0001.
Conclusions
Developing platforms to study phosphorylation states of GPCRs is necessary for understanding receptor physiology and mapping signaling properties of GPCR therapeutics. In this work, we developed a quantitative strategy based on middle-down MS and spectral counting via PRM to profile the phosphorylation states of GPCRs in human cells. Using this approach, we quantified a previously unknown level of heterogeneity in both the degree of phosphorylation stoichiometry and positional distribution in mGluR2. Importantly, we found that phosphorylation is distributed nonuniformly throughout the tail, suggesting a nonrandom process controlling the addition and maintenance of phosphorylation modifications. Moving forward, we foresee this technique being adapted and applied to other receptors to illuminate the vast landscape of intracellular GPCR phosphorylation. Moreover, this powerful approach can be readily used to quantify other important PTMs such as ubiquitination and palmitoylation. Finally, we expect that combining this strategy with purification procedures for different organelles will further enable the exploration and decoding of the phosphorylation barcode and its essential role in receptor trafficking, desensitization, recycling, and degradation.
Experimental Section
Molecular Cloning
For the calcium mobilization assay68 and initial screenings, human mGluR2 was cloned from the GRM2-Tango (Addgene #66388) into a pcDNA3.1 + expression vector with an N-terminal GFP-tag via Gibson assembly. A Tobacco Etch Virus nuclear-inclusion-a endopeptidase (TEV) protease site (ENLYFQ|G) was inserted between residues A830 and P831 via mutagenesis. This GFP-mGluR2 (TEV) construct was then moved into a lentiviral plasmid driven by the SFFV promoter for lentiviral transduction, which is described in detail below. For the −22F pGloSensor camp assay, human mGluR2 in a pcDNA3.1 + expression vector was purchased from GenScript (ORF Clone: OHu32034C). Point mutations at phosphorylated regions were made to this plasmid using PfuUltra High-Fidelity DNA Polymerase (Agilent) and using a QuikChange II Site-Directed Mutagenesis kit (Agilent). All other DNA restriction enzymes, DNA polymerases, and DNA ligases were from New England Biolabs. All recombinant DNA work was verified by sequencing (ACGT Inc). Plasmid preparation kits were obtained from Macherey-Nagel.
Cell Culture Conditions
HEK293T cells were purchased from Sigma and maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM) (Corning), 10% (v/v) fetal bovine serum (GE Healthcare), 100 unit/mL penicillin–streptomycin (Gibco), and 10 mM HEPES (pH 7.4, Gibco). Cells were grown adherent at 37 °C under 5% CO2. Cells were passaged using 0.05% trypsin–EDTA solution (Gibco) and maintained below a passage number of 30. For all experiments, cells were transiently transfected using the Lipofectamine 3000 reagent kit (Thermo Scientific). The empty vector pcDNA3 was used to normalize DNA amounts when needed.
mGluR2 Purification and MS Sample Preparation
The GFP-mGluR2 (TEV) construct described above was transduced into HEK293T cells, sorted by GFP signal, and validated for mGluR2 functionality using fluorometric calcium imaging68 as described in the Supporting Information. This stable cell line was treated with 1 mM l-glutamic acid and returned to the incubator at 37 °C and 5% CO2 for either 0, 10, 30, or 60 min. After treatment, the mGluR2 C-terminus was purified using a custom anti-GFP resin and TEV protease. This purification and resin production are described in the Supporting Information.
LC–MS Analysis
mGluR2 samples in buffer A (5% ACN, 0.2% formic acid, aqueous) were subjected to reverse-phase LC using an Ultimate 3000 LC system (Thermo Scientific, San Jose, CA). Six microliters of the sample was loaded onto a trap column (20 mm, 150 μM inner diameter) packed in-house with PLRP-S resin (Agilent, Santa Clara, CA, USA) and washed prior to analytical separation. Analytical separation was performed using an in-house packed capillary also containing PLRP-S resin (150 mM, 75 μM inner diameter). Both columns were maintained at 55 °C. The gradient consisted of a ramp of buffer B (5% water and 0.2% formic acid in acetonitrile) from 15 to 30% over 30 min, with a total run time of 60 min including column wash (at 90% buffer B) and re-equilibration into buffer A. The outlet of the column was coupled to 15 ± 1 μM uncoated spray tip (PicoTip EMITTER Silica Tip) packed with <1 mm PLRP-S resin and sprayed into a nanoelectrospray ionization source at 2 kV potential for protein ionization.
All MS was performed on an Orbitrap Eclipse Tribrid (Thermo Scientific, San Jose, CA) operating in intact protein, low-pressure mode. Intact protein scans (MS1) were performed with a source activation of 15 V, resolving power of 120,000 (at 200 m/z), maximum injection time of 100 ms, and normalized ACG target of 300% using a scan window from 700–1400 m/z. Intact scans (MS1) alternated with fragmentation scans. For fragmentation (MS2), a single phosphorylated state within the 4+ precursor charge envelope was quadrupole isolated using a 2.2 m/z window and subjected to an HCD collision energy of 15–20%. All MS2 scans were performed using a source activation of 15 V, a resolving power of 60,000 (at 200 m/z), and a maximum injection time of 500 ms. All MS scans were obtained by averaging 10 microscans.
MS Data Analysis and Statistical Analysis
To compare abundances of intact phosphorylation states, the MS1 scans were searched for isotopic patterns of target phosphorylation states in the 4+ charge state based on their chemical formula as previously described.69,70 A window of 10 ppm mass tolerance was allowed to account for mass accuracy drift. The relative intensities of the experimental and theoretical isotopic distribution were used to produce a fitting score ranging from 0 to 1 based on Pearson correlation coefficient calculation. Phosphorylation state intensities were calculated by summing all isotopomers for scans with fitting scores ≥0.5. The total intensity per LC–MS run was calculated for each phosphorylation state individually, and the average intensity and standard error of the mean were calculated using a custom R script; statistical comparisons between intensities were done using the Wilcoxon–Mann–Whitney test.
The sequence of the TEV cleavage product was used to produce a list of all theoretical b- and y-type fragment ions with a PTM set including 0–4 phosphorylations or 0–4–18 Da neutral losses, and charge states from 1 to 4+. An isotope fitting algorithm71,72 developed in-house was used for matching theoretical and experimental fragment ions using a maximum error tolerance of 10 ppm, sub-error tolerance of 5 ppm, a minimum assigned score of 0.5, and a minimum S/N cutoff of 3. Total fragment ion intensity was calculated by summing the intensity of all matched fragment ion isotopomers. Initial results were validated in TDValidator 1.0 (Proteinaceous Inc., Evanston, IL, USA) using identical parameters, and all fragmentation maps were generated in TDValidator 1.0. All identified fragment ions and intensities were imported into R, and a custom script was used to perform the fragment ion relative ratios analysis as previously described.32,33 Fragment ions between defined regions A, B, C, and D were averaged to produce the ratio and standard deviation of the ratio. Relative percentages were calculated using these ratios, and calculations were performed for both the upper and lower bounds of the ratio to define error bars for the percentage calculations. Statistical comparisons were done using the Wilcoxon–Mann–Whitney test.
cAMP Assay
HEK 293T/17 cells were cultured in DMEM supplemented with 10% fetal bovine serum, minimum essential medium nonessential amino acids (Life Technologies), 1 mM sodium pyruvate, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified incubator containing 5% CO2. For each experiment, cells were seeded in 6 cm dishes without penicillin and streptomycin and transfected the following day at ∼70% confluency. Cells were transiently transfected using Lipofectamine LTX with Plus Reagent with 2.52 μg of Promega -22F cAMP pGloSensor plasmid and 0.42 μg mGluR2 constructs. Cells were washed with HEPES-buffered Tyrode’s solution (Alfa Aesar J67607) using centrifugation and incubated with Promega GLO reagent in this buffer for ∼2 h in 96-well format. Following incubation, cells were pretreated with various concentrations of l-glutamic acid before baseline luminescence was read on a BMG LabTech PHERAstar FSX. After 5 min, wells were injected with 1 μM of FSK, and readings were continued for up to 20 min. mGluR2 activation was calculated as the decrease in FSK-mediated luminescence amplitude.60
Acknowledgments
This work was supported by NIH grants T32GM105538 and F31MH129114-01 (to A.N.I.), P41GM108569 and RF1AG063903-01 (to N.L.K.), R01GM140272-02 (to R.V.), and MH105482 (to K.A.M.). This work was supported by the Northwestern University—Flow Cytometry Core Facility through NIH grants NCI CA060553, 1S10OD011996-01, and 1S10OD026814-01. Peptide Synthesis was performed at the Peptide Synthesis Core Facility of the Simpson Querrey Institute for BioNanotechnology at Northwestern University. This facility has current support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
Abbreviations
- GPCR
g protein-coupled receptor
- LC–MS
liquid chromatography–mass spectrometry
- HCD
higher-energy collisional dissociation
- mGluR2
metabotropic glutamate receptor 2
- PRM
parallel reaction monitoring
- TM7
transmembrane helix 7
- GFP
green fluorescent protein
- IP
immunoprecipitation
- TEV
tobacco etch virus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10697.
Additional experimental details for preparing the nanobody resin, generating the stable cell line, and purifying the mGluR2 C-terminus; sample MS1 and MS2 spectra; theoretical and assigned intact monoisotopic masses of the purified mGluR2 C-terminus, and considerations for using collision-based fragmentation for phosphoprotein fragmentation and site localization (PDF)
The authors declare the following competing financial interest(s): N.L.K. and R.T.F. are involved in commercialization of proteomics software.
Notes
All MS data is available at the MassIVE repository (http://massive.ucsd.edu/MSV000090376/).
Supplementary Material
References
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin A. B. G-protein-coupled receptor phosphorylation: where, when and by whom. Br. J. Pharmacol. 2008, 153, S167–S176. 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin A. B.; Butcher A. J.; Kong K. C. Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 2008, 29, 413–420. 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan A.; Cheng N.; Trejo J. Post-Translational Modifications of G Protein-Coupled Receptors Control Cellular Signaling Dynamics in Space and Time. Pharmalcol. Rev. 2021, 73, 120–151. 10.1124/pharmrev.120.000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett S. B. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 2011, 4, pe36. 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Wang H.; Wang M.; Wang D.; Zhang Z.; Wei R.; Gao X.; Zhang R.; Wang C.; Chen J. A novel phosphorylation site on orexin receptor 1 regulating orexinA-induced GRK2-biased signaling. Cell Signal. 2020, 75, 109743. 10.1016/j.cellsig.2020.109743. [DOI] [PubMed] [Google Scholar]
- Pitcher J.; Lohse M. J.; Codina J.; Caron M. G.; Lefkowitz R. J. Desensitization of the isolated .beta.2-adrenergic receptor by .beta.-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry 1992, 31, 3193–3197. 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- Butcher A. J.; Prihandoko R.; Kong K. C.; McWilliams P.; Edwards J. M.; Bottrill A.; Mistry S.; Tobin A. B. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 2011, 286, 11506–11518. 10.1074/jbc.m110.154526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara E.; Crépieux P.; Gauthier C.; Martinat N.; Piketty V.; Guillou F.; Reiter E. A Phosphorylation Cluster of Five Serine and Threonine Residues in the C-Terminus of the Follicle-Stimulating Hormone Receptor Is Important for Desensitization But Not for β-Arrestin-Mediated ERK Activation. Mol. Endocrinol. 2006, 20, 3014–3026. 10.1210/me.2006-0098. [DOI] [PubMed] [Google Scholar]
- Dwivedi-Agnihotri H.; Chaturvedi M.; Baidya M.; Stepniewski T. M.; Pandey S.; Maharana J.; Srivastava A.; Caengprasath N.; Hanyaloglu A. C.; Selent J.; Shukla A. K. Distinct phosphorylation sites in a prototypical GPCR differently orchestrate β-arrestin interaction, trafficking, and signaling. Sci. Adv. 2020, 6, eabb8368 10.1126/sciadv.abb8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A. I.; Perry N. A.; Gurevich V. V.; Iverson T. M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 14139–14149. 10.1073/pnas.1918736117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S. M.; Pack T. F.; Wilkins A. D.; Urs N. M.; Urban D. J.; Bass C. E.; Lichtarge O.; Caron M. G. Elucidation of G-protein and β-arrestin functional selectivity at the dopamine D2 receptor. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 7097–7102. 10.1073/pnas.1502742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prihandoko R.; Bradley S. J.; Tobin A. B.; Butcher A. J. Determination of GPCR Phosphorylation Status: Establishing a Phosphorylation Barcode. Curr. Protoc. Pharmacol. 2015, 69, 2.13.1–2.13.26. 10.1002/0471141755.ph0213s69. [DOI] [PubMed] [Google Scholar]
- Latorraca N. R.; Masureel M.; Hollingsworth S. A.; Heydenreich F. M.; Suomivuori C. M.; Brinton C.; Townshend R. J. L.; Bouvier M.; Kobilka B. K.; Dror R. O. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825. 10.1016/j.cell.2020.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Yang F.; Zhang D.; Liu Z.; Lin A.; Liu C.; Xiao P.; Yu X.; Sun J. P. Phosphorylation of G Protein-Coupled Receptors: From the Barcode Hypothesis to the Flute Model. Mol. Pharmacol. 2017, 92, 201–210. 10.1124/mol.116.107839. [DOI] [PubMed] [Google Scholar]
- Sente A.; Peer R.; Srivastava A.; Baidya M.; Lesk A. M.; Balaji S.; Shukla A. K.; Babu M. M.; Flock T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 538–545. 10.1038/s41594-018-0071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont R. T. Keys to the Kingdom: GPCR phosphorylation patterns direct β-arrestin. EMBO Rep. 2020, 21, e51249 10.15252/embr.202051249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley S. J.; Wiegman C. H.; Iglesias M. M.; Kong K. C.; Butcher A. J.; Plouffe B.; Goupil E.; Bourgognon J.-M.; Macedo-Hatch T.; LeGouill C.; Russell K.; Laporte S. A.; König G. M.; Kostenis E.; Bouvier M.; Chung K. F.; Amrani Y.; Tobin A. B. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 4524–4529. 10.1073/pnas.1521706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Coradin M.; Porter E. G.; Garcia B. A. Accelerating the Field of Epigenetic Histone Modification Through Mass Spectrometry-Based Approaches. Mol. Cell. Proteomics 2021, 20, 100006. 10.1074/mcp.R120.002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Zhang S.; Zhang X.; Liu H. QR code model: a new possibility for GPCR phosphorylation recognition. Cell Commun. Signal. 2022, 20, 23. 10.1186/s12964-022-00832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles K. N.; Xiao K.; Ahn S.; Shukla A. K.; Lam C. M.; Rajagopal S.; Strachan R. T.; Huang T.-Y.; Bressler E. A.; Hara M. R.; Shenoy S. K.; Gygi S. P.; Lefkowitz R. J. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 2011, 4, ra51. 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher A. J.; Hudson B. D.; Shimpukade B.; Alvarez-Curto E.; Prihandoko R.; Ulven T.; Milligan G.; Tobin A. B. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J. Biol. Chem. 2014, 289, 18451–18465. 10.1074/jbc.m114.568816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vit O.; Petrak J. Integral membrane proteins in proteomics. How to break open the black box?. J. Proteonomics 2017, 153, 8–20. 10.1016/j.jprot.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Chait B. T. Mass Spectrometry: Bottom-Up or Top-Down?. Mol. Cell. Proteomics 2006, 314, 65–66. 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- Durbin K. R.; Fornelli L.; Fellers R. T.; Doubleday P. F.; Narita M.; Kelleher N. L. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J. Proteome Res. 2016, 15, 976–982. 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby T. K.; Fornelli L.; Kelleher N. L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Brown K. A.; Lin Z.; Ge Y. Top-Down Proteomics: Ready for Prime Time?. Anal. Chem. 2018, 90, 110–127. 10.1021/acs.analchem.7b04747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.; Wei H.; Garrison T. R.; Lefkowitz R. J. Reciprocal Regulation of Angiotensin Receptor-activated Extracellular Signal-regulated Kinases by β-Arrestins 1 and 2. J. Biol. Chem. 2004, 279, 7807–7811. 10.1074/jbc.c300443200. [DOI] [PubMed] [Google Scholar]
- Escobar E. E.; Venkat Ramani M. K.; Zhang Y.; Brodbelt J. S. Evaluating Spatiotemporal Dynamics of Phosphorylation of RNA Polymerase II Carboxy-Terminal Domain by Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 8488–8498. 10.1021/jacs.1c03321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento J. J.; Bullock C. R.; LeDuc R. D.; Mizzen C. A.; Kelleher N. L. Combinatorial Modification of Human Histone H4 Quantitated by Two-dimensional Liquid Chromatography Coupled with Top Down Mass Spectrometry. J. Biol. Chem. 2008, 283, 14927–14937. 10.1074/jbc.m709796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento J. J.; Mizzen C. A.; Kelleher N. L. Quantitative Analysis of Modified Proteins and Their Positional Isomers by Tandem Mass Spectrometry: Human Histone H4. Anal. Chem. 2006, 78, 4271–4280. 10.1021/ac0600050. [DOI] [PubMed] [Google Scholar]
- Siuti N.; Roth M. J.; Mizzen C. A.; Kelleher N. L.; Pesavento J. J. Gene-Specific Characterization of Human Histone H2B by Electron Capture Dissociation. J. Proteome Res. 2006, 5, 233–239. 10.1021/pr050268v. [DOI] [PubMed] [Google Scholar]
- Cristobal A.; Marino F.; Post H.; van den Toorn H. W. P.; Mohammed S.; Heck A. J. R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. 10.1021/acs.analchem.6b03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quijada J. V.; Schmitt N. D.; Salisbury J. P.; Auclair J. R.; Agar J. N. Heavy Sugar and Heavy Water Create Tunable Intact Protein Mass Increases for Quantitative Mass Spectrometry in Any Feed and Organism. Anal. Chem. 2016, 88, 11139–11146. 10.1021/acs.analchem.6b03234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Wang Z.; Cupp-Sutton K. A.; Guo Y.; Kou Q.; Smith K.; Liu X.; Wu S. Quantitative Top-Down Proteomics in Complex Samples Using Protein-Level Tandem Mass Tag Labeling. J. Am. Soc. Mass Spectrom. 2021, 32, 1336–1344. 10.1021/jasms.0c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Sutton K. A.; Wu S. High-throughput quantitative top-down proteomics. Mol. Omics 2020, 16, 91–99. 10.1039/c9mo00154a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilson K. A.; Ali N. A.; Muralidharan S.; Mirzaei M.; Mariani M.; Assadourian G.; Lee A.; van Sluyter S. C.; Haynes P. A. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- Clough T.; Key M.; Ott I.; Ragg S.; Schadow G.; Vitek O. Protein Quantification in Label-Free LC-MS Experiments. J. Proteome Res. 2009, 8, 5275–5284. 10.1021/pr900610q. [DOI] [PubMed] [Google Scholar]
- Escobar E. E.; Venkat Ramani M. K.; Zhang Y.; Brodbelt J. S. Evaluating Spatiotemporal Dynamics of Phosphorylation of RNA Polymerase II Carboxy-Terminal Domain by Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 8488–8498. 10.1021/jacs.1c03321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayel Ö.; Tonelli F.; Virreira Winter S. V.; Geyer P. E.; Fan Y.; Sammler E. M.; Alessi D. R.; Steger M.; Mann M. J. M.; Proteomics C. Accurate MS-based Rab10 phosphorylation stoichiometry determination as readout for LRRK2 activity in Parkinson’s disease. Mol. Cell. Proteomics 2020, 19, 1546–1560. 10.1074/mcp.ra120.002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweredoski M. J.; Moradian A.; Raedle M.; Franco C.; Hess S. High Resolution Parallel Reaction Monitoring with Electron Transfer Dissociation for Middle-Down Proteomics. Anal. Chem. 2015, 87, 8360–8366. 10.1021/acs.analchem.5b01542. [DOI] [PubMed] [Google Scholar]
- Lange V.; Picotti P.; Domon B.; Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2008, 4, 222. 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Oh H. J.; Han D.; Wang J. I.; Park I. A.; Ryu H. S.; Kim Y. Parallel Reaction Monitoring-Mass Spectrometry (PRM-MS)-Based Targeted Proteomic Surrogates for Intrinsic Subtypes in Breast Cancer: Comparative Analysis with Immunohistochemical Phenotypes. J. Proteome Res. 2020, 19, 2643–2653. 10.1021/acs.jproteome.9b00490. [DOI] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzitelli M.; Palazzo E.; Maione S.; Neugebauer V. Group II Metabotropic Glutamate Receptors: Role in Pain Mechanisms and Pain Modulation. Front. Mol. Neurosci. 2018, 11, 383. 10.3389/fnmol.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. C.; Russell J. D.; Bailey D. J.; Westphall M. S.; Coon J. J. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 2012, 11, 1475–1488. 10.1074/mcp.o112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil S. A.; Jedrychowski M.; Schwartz D.; Elias J. E.; Villén J.; Li J.; Cohn M. A.; Cantley L. C.; Gygi S. P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12130–12135. 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert M.; Yates L. M.; Schümann M.; Perlman D.; Fiedler D.; Krause E. Unambiguous Identification of Serine and Threonine Pyrophosphorylation Using Neutral-Loss-Triggered Electron-Transfer/Higher-Energy Collision Dissociation. Anal. Chem. 2017, 89, 3672–3680. 10.1021/acs.analchem.6b05095. [DOI] [PubMed] [Google Scholar]
- Potel C. M.; Lemeer S.; Heck A. J. R. Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Anal. Chem. 2019, 91, 126–141. 10.1021/acs.analchem.8b04746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer S. M.; Sidoli S.; Coradin M.; Schack Jespersen M.; Schwämmle V.; Jensen O. N.; Garcia B. A.; Brodbelt J. S. Extensive Characterization of Heavily Modified Histone Tails by 193 nm Ultraviolet Photodissociation Mass Spectrometry via a Middle-Down Strategy. Anal. Chem. 2018, 90, 10425–10433. 10.1021/acs.analchem.8b02320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffhauser H.; Cai Z.; Hubalek F.; Macek T. A.; Pohl J.; Murphy T. J.; Conn P. J. cAMP-Dependent Protein Kinase Inhibits mGluR2 Coupling to G-Proteins by Direct Receptor Phosphorylation. J. Neurosci. 2000, 20, 5663. 10.1523/jneurosci.20-15-05663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad J. C.; Barkan D. T.; Gulledge B. F.; Thalhammer A.; Sali A.; Schoepfer R.; Burlingame A. L. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 2012, 11, 215–229. 10.1074/mcp.o112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Nagaraj N.; Zougman A.; Gnad F.; Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 2010, 9, 3280–9. 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- Tweedie-Cullen R. Y.; Reck J. M.; Mansuy I. M. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J. Proteome Res. 2009, 8, 4966–4982. 10.1021/pr9003739. [DOI] [PubMed] [Google Scholar]
- Paek J.; Kalocsay M.; Staus D. P.; Wingler L.; Pascolutti R.; Paulo J. A.; Gygi S. P.; Kruse A. C. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell 2017, 169, 338–349. 10.1016/j.cell.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman S. M.; Serber Z.; Ferrell J. E. Jr. A mechanism for the evolution of phosphorylation sites. Cell 2011, 147, 934–946. 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissmeyer N.; Schnittger A. Use of phospho-site substitutions to analyze the biological relevance of phosphorylation events in regulatory networks. Methods Mol. Biol. 2011, 779, 93–138. 10.1007/978-1-61779-264-9_6. [DOI] [PubMed] [Google Scholar]
- Dunn H. A.; Patil D. N.; Cao Y.; Orlandi C.; Martemyanov K. A. Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 5022–5027. 10.1073/pnas.1722498115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli L.; Nicoletti F.; De Blasi A. Molecular mechanisms that desensitize metabotropic glutamate receptor signaling: An overview. Neuropharmacology 2013, 66, 24–30. 10.1016/j.neuropharm.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Seven A. B.; Barros-Álvarez X.; de Lapeyrière M.; Papasergi-Scott M. M.; Robertson M. J.; Zhang C.; Nwokonko R. M.; Gao Y.; Meyerowitz J. G.; Rocher J.-P.; Schelshorn D.; Kobilka B. K.; Mathiesen J. M.; Skiniotis G. G-protein activation by a metabotropic glutamate receptor. Nature 2021, 595, 450–454. 10.1038/s41586-021-03680-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Han S.; Cai X.; Tan Q.; Zhou K.; Wang D.; Wang X.; Du J.; Yi C.; Chu X.; Dai A.; Zhou Y.; Chen Y.; Zhou Y.; Liu H.; Liu J.; Yang D.; Wang M.-W.; Zhao Q.; Wu B. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 2021, 594, 583–588. 10.1038/s41586-021-03495-2. [DOI] [PubMed] [Google Scholar]
- Mahato P. K.; Pandey S.; Bhattacharyya S. Differential effects of protein phosphatases in the recycling of metabotropic glutamate receptor 5. Neurosci 2015, 306, 138–150. 10.1016/j.neuroscience.2015.08.031. [DOI] [PubMed] [Google Scholar]
- Iacovelli L.; Molinaro G.; Battaglia G.; Motolese M.; Di Menna L.; Alfiero M.; Blahos J.; Matrisciano F.; Corsi M.; Corti C.; Bruno V.; De Blasi A.; Nicoletti F. Regulation of group II metabotropic glutamate receptors by G protein-coupled receptor kinases: mGlu2 receptors are resistant to homologous desensitization. Mol. Pharmacol. 2009, 75, 991–1003. 10.1124/mol.108.052316. [DOI] [PubMed] [Google Scholar]
- Ritter-Makinson S. L.; Paquet M.; Bogenpohl J. W.; Rodin R. E.; Chris Yun C.; Weinman E. J.; Smith Y.; Hall R. A. Group II metabotropic glutamate receptor interactions with NHERF scaffold proteins: Implications for receptor localization in brain. Neuroscience 2017, 353, 58–75. 10.1016/j.neuroscience.2017.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R. Structure of metabotropic glutamate receptor C-terminal domains in contact with interacting proteins. Front. Mol. Neurosci. 2012, 5, 52. 10.3389/fnmol.2012.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarda V.; Calo’ G. Chimeric G proteins in fluorimetric calcium assays: experience with opioid receptors. Methods Mol. Biol. 2013, 937, 293–306. 10.1007/978-1-62703-086-1_18. [DOI] [PubMed] [Google Scholar]
- Ntai I.; Kim K.; Fellers R. T.; Skinner O. S.; Smith A. D.; Early B. P.; Savaryn J. P.; LeDuc R. D.; Thomas P. M.; Kelleher N. L. Applying Label-Free Quantitation to Top Down Proteomics. Anal. Chem. 2014, 86, 4961–4968. 10.1021/ac500395k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckler H. d. S.; Fornelli L.; Mutharasan R. K.; Thaxton C. S.; Fellers R.; Daviglus M.; Sniderman A.; Rader D.; Kelleher N. L.; Lloyd-Jones D. M.; Compton P. D.; Wilkins J. T. A Targeted, Differential Top-Down Proteomic Methodology for Comparison of ApoA-I Proteoforms in Individuals with High and Low HDL Efflux Capacity. J. Proteome Res. 2018, 17, 2156–2164. 10.1021/acs.jproteome.8b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin K. R.; Skinner O. S.; Fellers R. T.; Kelleher N. L. Analyzing internal fragmentation of electrosprayed ubiquitin ions during beam-type collisional dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 782–787. 10.1007/s13361-015-1078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Fornelli L.; Compton P. D.; Sharma S.; Canterbury J.; Mullen C.; Zabrouskov V.; Fellers R. T.; Thomas P. M.; Licht J. D.; Senko M. W.; Kelleher N. L. Unabridged Analysis of Human Histone H3 by Differential Top-Down Mass Spectrometry Reveals Hypermethylated Proteoforms from MMSET/NSD2 Overexpression. Mol. Cell. Proteomics 2016, 15, 776–790. 10.1074/mcp.m115.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.