

Abstract
To investigate the function of miR‐454 in ischemic stroke, this study was carried out. Cerebral ischemia/reperfusion (I/R) injury animal model and a SHSY5Y cell culture model of oxygen‐glucose deprivation/reoxygenation (OGD/R) were constructed. The effects of miR‐454 were detected by evaluating the levels of biochemical markers, gene expression, and pathophysiological markers. The results showed that NOX4 level was elevated, while miR‐454 expression was reduced in I/R brain samples and in OGD/R‐treated cells. The miR‐454 agomir declined NOX4 level and reactive oxygen species (ROS) production in rats suffering from I/R. Furthermore, microRNA‐145 (miR‐454) overexpression inhibited NOX4 level and ROS production in cells treated by OGD/R and decreased luciferase activity in cells transfected with NOX4‐wild type (WT) reporter plasmid. Meanwhile, our results proved that the protected effects of miR‐454 on SH‐SY5Y cells treated by OGD/R were reversed by pcDNA‐NOX4 transfection. MiR‐454 protected animals from brain injury induced by cerebral I/R via directly regulating its target gene NOX4, illustrating a curatively potential target for treating ischemic stroke.
Keywords: cerebral ischemia/reperfusion, miR‐454, NOX4, oxidative stress
MiR‐454 protected brain injury via directly regulating NOX4
1. BACKGROUND
Ischemic stroke, one of the cerebrovascular diseases, is triggered by stenosis or occlusion of cerebral blood vessels and could result in cerebral ischemia, hypoxia, necrosis, and even brain dysfunction.[ 1 , 2 ] However, due to delayed treatment acceptance and rapid neuronal damage or necrosis, numerous patients still have poor prognoses with certain adverse sequelae.[ 3 , 4 ] Therefore, exploring the molecular and pathophysiological mechanisms of ischemic stroke is important for finding more critical therapeutic strategies.
Abnormal microRNA (miRNAs) expression has been observed in different types of brain diseases, such as neurodegenerative diseases, ischemic stroke, and brain tumors.[ 5 , 6 , 7 ] Studies have revealed that miR‐181 suppression is presented to diminish brain injury and improve behavioral recovery following ischemia.[ 8 ] From this perspective, identification and intervention of specific miRNA might alleviate or reverse the structural and functional abnormalities of brain tissues and neuron cells caused by ischemic stroke, stimulating brain recovery and enhancing the prognosis of patients with stroke.
ROS are important molecules regulating neuron cell growth and death[ 9 , 10 ] and key factors involving the pathogenesis of oxidative stress during ischemic stroke. As a member of the 5,10‐methylenetetrahydrofolate reductase (NADPH) oxidase family, NOX4 could promote ROS production,[ 11 ] and NOX4 knockout protected mice from oxidative stress and neuronal apoptosis induced by transient or permanent cerebral ischemia.[ 12 ] Some miRNAs, like microRNA‐132, microRNA‐126a, microRNA‐29c, microRNA‐29a, and exhibited altered expressions and possibly targeted NOX4 and NADPH oxidase 2(NOX2) genes in hyperglycemic animals treated with acute ischemic stroke.[ 13 ] However, miR‐454 expression and its underlying pathophysiological mechanism in cerebrovascular diseases remain unclear.
Here, elevating miR‐454 expression was found to alleviate I/R‐induced NOX4 upregulation and brain damage, suggesting that miR‐454 might be vital to protect neurons and reduce brain damage and would be an ischemic stroke therapy target.
2. MATERIALS AND METHODS
2.1. Animals
The study was approved by the Animal Care Committee of Ningxia Hui Autonomous Region People's Hospital. Sprague Dawley rats (n = 40, male 250−300 g) were used in the study. In short, after anesthetizing by 40 mg/kg pentobarbital sodium (3%, w/v) per rat, the left internal carotid artery of rats was isolated. To block the blood flow from the anterior cerebral artery, posterior cerebral artery, middle cerebral artery, internal carotid artery, and a surgical nylon monofilament with a 4/0 rounded tip was utilized to be added to the anterior cerebral artery. Nylon monofilament was taken out after 2 h occlusion, blood reperfusion was restored for 24 h. Animals in the sham group experienced the same procedure without inserting the nylon monofilament into the internal carotid artery. Neurological deficit scores were analyzed after 24 h of reperfusion, and the brain infarct was observed by triphenyltetrazolium chloride (TTC) staining, mRNA level, and protein expression.
2.2. Cell culture and OGD/R cell model
SH‐SY5Y and HEK 293 cell lines were cultured in Gibco Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS), 1% nonessential amino acids, 1% glutamax, and (1%) and 1 mM sodium pyruvate in an incubator at 37°C with 5% CO2. Then, SHSY5Y cells were moved to a hypoxic incubator chamber to diminish oxygen content for 10 min. Subsequently, glucose‐free DMEM was added to each well to culture cells at OGD condition at 37°C for 2 h. Cells were subjected to normal Gibco DMEM:F‐12 (DMEM/F12) containing 10% FBS and to a reoxygenation period in normoxic conditions with 5% CO2. Cells treated similarly without OGD/R were used as a control group.
2.3. Cerebral blood flow measurement
In this experiment, laser doppler flowmetry was used. In brief, the rat's head was held by a stereotaxic device, and the skin over the calvarium was incised to uncover the bregma. The cerebral blood flow was analyzed dynamically by the laser Doppler flow probe, which had been placed after burr holes were drilled at the bregma.
2.4. Enzymatic activity measurement and ROS production
Total NOX activity was determined, and Caspase‐3 activity was measured. ROS production was measured using a ROS Kit. Malondialdehyde (MDA) and superoxide dismutase (SOD) in cells and tissues were measured using commercial kits. With a spectrophotometer, absorbance at 450 nm was measured.
2.5. Cell transfection and apoptosis
MiR‐454 agomir was from Ribo, and pcDNA‐NC and pcDNA‐NOX4 were from Hanbio Co., Ltd. For transfection, Lipofectamine 2000 was used. In brain tissues, cellular apoptosis was measured by TUNEL assay using commercial kits (Beyotime). TdT‐mediated dUTP nick‐end labeling (TUNEL)‐positive cells were presented in percentage. SHSY5Y cell apoptosis was assessed by Hoechst 33258 staining kit (Beyotime) and flow cytometry.
2.6. Hematoxylin−eosin (H&E) staining
Rat brains were removed instantly at 24 h after I/R injury by cutting at 4 mm behind chiasma along the coronal plane. After that, half of the forebrains were fixed, embedded, and prepared as 6‐μm‐thick coronal sections with the dorsal hippocampus (bregma −3.8 mm). H&E staining was carried out.
2.7. Bioinformatic analysis and luciferase reporter gene experiment
The potential target miRNAs of NOX4 were predicted using the website of TargetScan.org and microrna.org. NOX4 3′‐UTR fragments containing wild‐type or mutated miR‐454 binding site were inserted into pmirGLO. These vectors were cotransfected with NC miRNA or miR‐454 agomir into HEK293T cells. Then, following the manufacturer's protocols, luciferase activity was measured with a luminometer.
2.8. Intracerebroventricular infusion of miR‐454 agomir
miR‐454 overexpression was managed following the guideline. In brief, miR‐454 agomir was subjected to intracerebroventricular infusion via the right lateral ventricle. Two days after injection, rats were feasible for I/R treatment.
2.9. Cell viability assay
MTT assay was used here, and 570 nm absorbance (A) was measured using a microplate reader and converted into viability in percentage.
2.10. Real‐time polymerase chain reaction (PCR)
From each sample, total RNA was extracted by Trizol reagent. After RNA (100 ng) was reverse‐transcribed into cDNA, NOX4 and miRNA levels were determined on an ABI 7300 Real‐Time PCR System and normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and U6 snRNA, respectively. NOX4 and miRNA relative expression was calculated by ABI software with the comparative C t method. Sequences of primers used in real‐time quantitative PCR (qRT‐PCR) were shown in Table 1.
Table 1.
Sequences of primers used in qRT‐PCR
| Gene | Forward primer (5′‐3′) | Reversed primer (5′‐3′) |
|---|---|---|
| miR‐129 | GGGGGCTTTTTGCGGTCTGG | AGTGCGTGTCGTGGAGTC |
| miR‐32 | GCGGCGTATTGCACATTACT | TCGTATCCAGTGCAGGGTC |
| miR‐330‐5p | TCTCTGGGCCTGTGTCTTAG | CAGTGCGTGTCGTGGAGT |
| miR‐363 | GCCGAGAATTGCACGGTAT | CTCAACTGGTGTCGTGGA |
| miR‐375 | CCCCAAGGCTGATGCTGAGAAG | GCCGCCCGGCCCCGGGTCTTC |
| miR‐96 | GCCCGCTTTGGCACTAGCACATT | GTGCAGGGTCCGAGGT |
| miR‐454 | GGGACCCTATCAATATTGT | CAGTGCGTGTCGTGGAGT |
| U6 | CTTCGGCAGCACATATACT | AAAATATGGAACGCTTCACG |
| NOX4 | CTGACAGGTGTCTGCATGGT | ACTTCAACAAGCCACCCGAA |
| GAPDH | TCATGCATGCTGACGCTAC | TTGTACTGCCTGCACTGC |
2.11. Western blot
Proteins were quantified and subjected to Western blot. The primary antibodies against GAPDH (1:1000), cleaved‐Caspase3 (1:500), or NOX4 (1:500) were used to incubate the membrane at 4°C for 16 h, and then secondary antibodies were used for 1 h. Their intensity was analyzed using ImageJ 1.43 and normalized to GAPDH (Millipore).
2.12. Statistical analysis
The quantitative data were analyzed by statistical software SPSS 20.0 and presented as means ± standard deviation (SD). The nonparametric Mann−Whitney test or Student's t test was utilized for comparison between the two groups. For multiple groups, one‐way analysis of variance (ANOVA) followed by the Newman−Keuls test or Tukey's HSD was utilized. A p value < 0.05 was considered statistically significant.
3. RESULTS
3.1. NOX4 and miR‐454 expression in rat's brain
After cerebral I/R was constructed, we found that neurological function scores and infarct volume of rats were raised after 24 h of I/R operation reperfusion (p < 0.01) (Figure 1A,B). Moreover, cerebral I/R operation reduced SOD activity (Figure 1C), increased MDA content and ROS production (p < 0.01) (Figure 1D,E), and enhanced NOX4 expression in rat brains at both protein and mRNA levels (p < 0.01) (Figure 1F,G). Bioinformatics analysis showed that miR‐363, miR‐32, miR‐454, miR‐129, miR‐330‐5p, miR‐96, and miR‐375 might be the target of NOX4. We found that in the brain tissues and serum, the miR‐454 level was significantly reduced in I/R rats (p < 0.01) (Figure 1H,I) Therefore, miR‐454 might be a critical target of NOX4 (Figure 1G).
Figure 1.
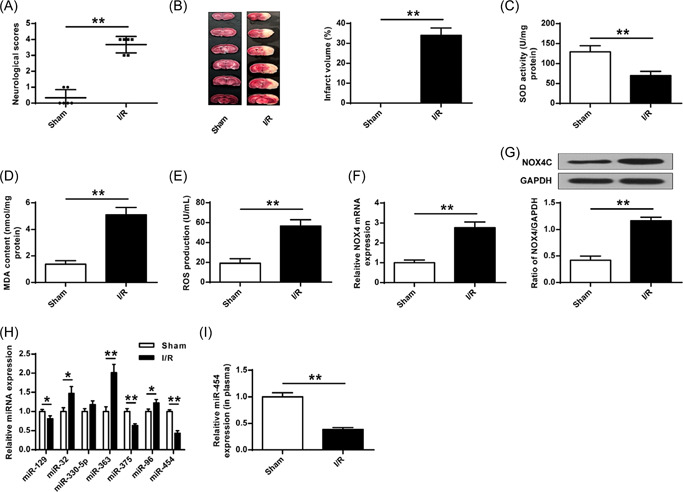
NOX4 and miR‐454 expression after cerebral I/R treatment. (A) Neurobehavioral outcomes. (B) Infarct volume. (C) SOD activity determined by ELISA. (D) MDA content determined by ELISA. (E) ROS production. (F, G) NOX4 expression. (H) miRNAs expression. (I) miR‐454 in plasma. I/R, ischemia/reperfusion; ROS, reactive oxygen species; SOD, superoxide dismutase. *p < 0.05, **p < 0.01. n = 6.
3.2. MiR‐454 agomir injection leads to reduction of brain injury
To test our hypothesis, miR‐454 agomir was used and it was found that miR‐454 agomir increased miR‐454 level in rat brain tissues (Figure 2A, p < 0.01). Moreover, cerebral blood flow was reduced in rats suffering from filament insertion and recovered to the baseline level when the filament was removed (Figure 2B). However, miR‐454 agomir significantly attenuated cerebral I/R‐increased neurological score (Figure 2C), infarct volume (Figure 2D), and brain water content (Figure 2E) (p < 0.01 for all). Moreover, H&E staining showed that miR‐454 agomir significantly reduced neuron damages induced by cerebral I/R (p < 0.01) (Figure 2F).
Figure 2.
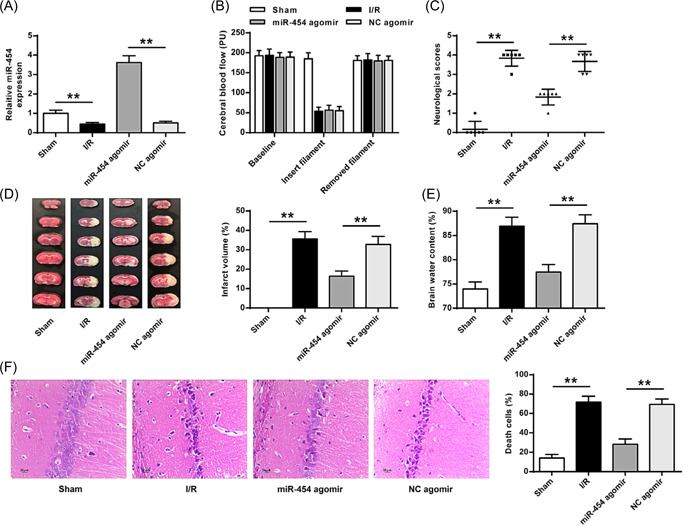
MiR‐454 agomir reduced brain injury. (A) miR‐454 agomir transfection efficiency was evaluated. (B) Alterations of cerebral blood flow (PU). (C) Neurobehavioral outcomes. (D) TTC staining. (E) Brain water content. (F) H&E staining (×200). H&E, hematoxylin‐eosin; TTE, triphenyltetrazolium chloride.**p < 0.01. n = 6.
3.3. MiR‐454 agomir reduced oxidative stress and apoptosis
We further evaluated the miR‐454 agomir effect on oxidative stress and apoptosis in rat brain tissues. MiR‐454 overexpression reduced NOX4 mRNA (Figure 3A) and proteins (Figure 3B) levels, NOX activity (Figure 3C), MDA (Figure 3E), and ROS (Figure 3F) contents but increased SOD activity (Figure 3D) in I/R group rat brain tissues. To explore miR‐454 agomir effect on brain tissue apoptosis in I/R rats. miR‐454 overexpression was found to reverse I/R‐enhanced caspase‐3 level (Figure 3G, p < 0.01) and activity (Figure 3H, p < 0.05) and tissue apoptosis (Figure 3I, p < 0.01), while injecting NC agomir had no significant effects on I/R‐induced all changes in I/R rat brain tissues. These results proved that the change in miR‐454 expression affected the brain tissue oxidative stress and apoptosis.
Figure 3.
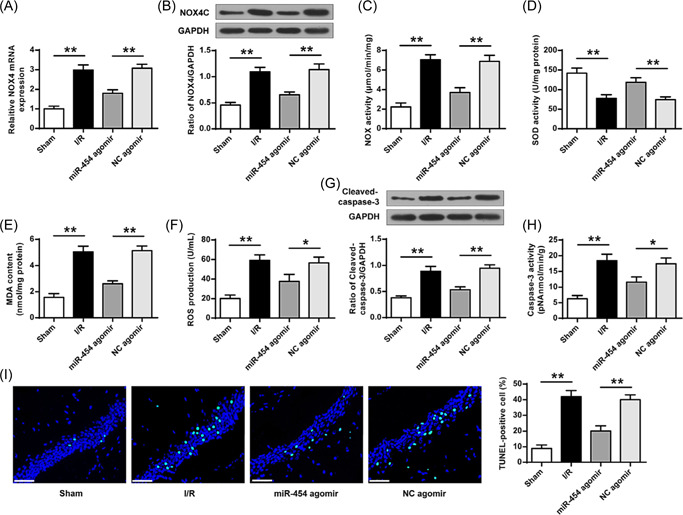
MiR‐454 agomir reduced cell apoptosis and oxidative stress. (A, B) NOX4 expression. (C) Total NOX enzyme activity. (D, E) MDA content and SOD activity determined by ELISA. (F) ROS production. (G) The level of Caspase‐3 protein. (H) The enzyme activity of Caspase‐3. (I) TUNEL staining (×400). Scale bar = 50 μm. MDA, malondialdehyde; ROS, reactive oxygen species; SOD, superoxide dismutase; TUNEL, TdT‐mediated dUTP nick‐end labeling. *p < 0.05, **p < 0.01. n = 6.
3.4. NOX4 is a direct target gene of miR‐454
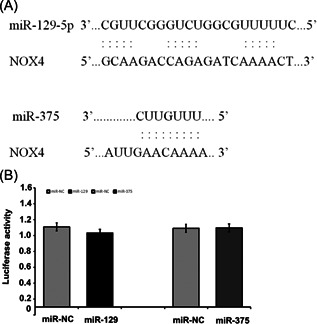
Bioinformatics analysis found that NOX4 could be the target gene of miR‐454 (Figure 4A). The data showed that miR‐454 overexpression reduced the luciferase activity of the reporter vector containing the NOX4 3′‐UTR WT (Figure 4B, p < 0.01) but not NOX4 3′‐UTR MT (Figure 4B, p > 0.05). Furthermore, miR‐454 overexpression decreased NOX4 expression in SH‐SY5Y cells (Figure 4C,D, p < 0.01), proving that miR‐454 targets NOX4.
Figure 4.
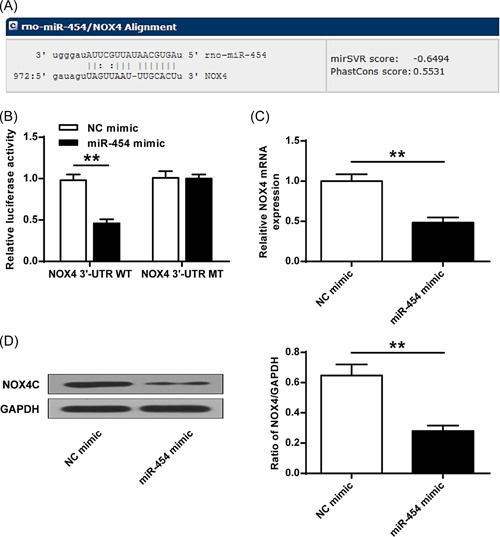
miR‐454 directly suppresses NOX4 expression. (A) The predicted binding site of miR‐454 and 3′‐UTR of NOX4. (B) Luciferase activity detection. (C) miR‐454 effect on mRNA level of NOX4 in cells. (D) miR‐454 effect on NOX4 protein expression in cells. **p < 0.01. n = 3.
3.5. Exogenous miR‐454 agomir suppressed oxidative stress in cells
MiR‐454 expression was reduced in SH‐SY5Y cells suffering from OGD/R (Figure 5A, p < 0.01). Additionally, OGD/R reduced cell viability, which could be reversed by miR‐454 agomir, but not NC agomir (p < 0.05) (Figure 5B). Moreover, OGD/R increased NOX4 expression, which was suppressed by miR‐454 agomir, but not NC agomir (Figure 5C,D, p < 0.01). Consistently, OGD/R increased SOD activity, ROS production, MDA content, NOX activity and which was also reduced by miR‐454 agomir (Figure 5E,F, p < 0.01). These proved that miR‐454 overexpression is related to NOX4 upregulation and suppressed oxidative stress in cells.
Figure 5.
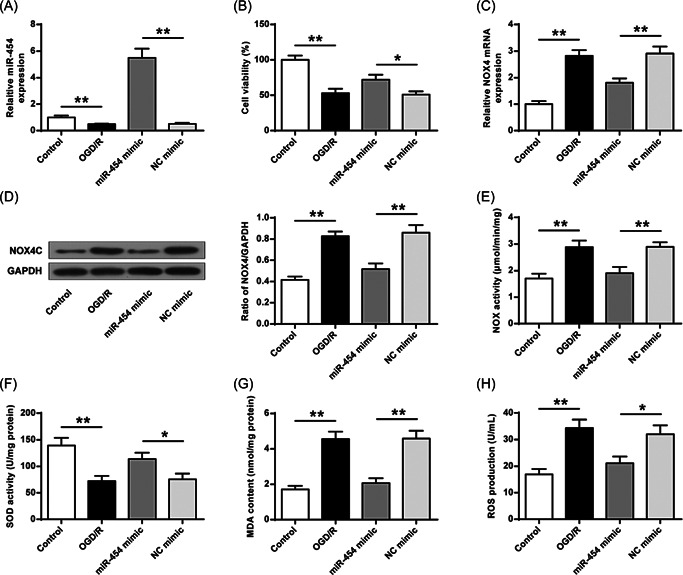
Exogenous miR‐454 agomir suppresses oxidative stress. (A) Transfection efficiency of miR‐454 agomir was evaluated. (B) Cell viability determined. (C, D) The mRNA and protein expression of NOX4. (E) Total enzyme activity of NOX. (F, G) SOD activity and MDA content determined by ELISA. (H) ROS production. MDA, malondialdehyde; ROS, reactive oxygen species; SOD, superoxide dismutase. *p < 0.05, **p < 0.01. n = 3.
3.6. Exogenous miR‐454 agomir decreased cell apoptosis
The caspase‐3 expression and activity were detected. The results showed that OGD/R treatment upregulated caspase‐3 expression and increased its activity, while these increases were partly reversed by miR‐454 agomir (Figure 6A,B, p < 0.05, p < 0.01). Moreover, SH‐SY5Y cell apoptosis was elevated compared with the control by OGD/R treatment, while was attenuated by miR‐454 agomir, but not NC agomir (Figure 6C, p < 0.01). Furthermore, miR‐454 agomir reduced OGD/R‐induced cell death (Figure 6D). These data together indicated that miR‐454 overexpression protected SH‐SY5Y cells and alleviated cell apoptosis or death.
Figure 6.
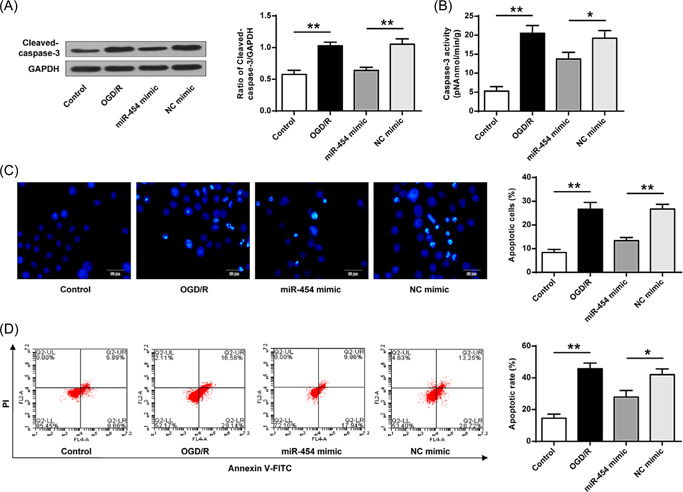
Exogenous miR‐454 agomir decreases apoptosis in cultured cells. (A) Caspase‐3 protein expression. (B) Caspase‐3 enzyme activity. (C) Hoechst 33258 staining (×200). (D) Flow cytometry detection. *p < 0.05, **p < 0.01. n = 3.
3.7. Exogenous miR‐454 agomir suppresses oxidative stress and apoptosis
At last, we explored whether NOX4 is directly implicated in the protective effect of miR‐454 following ischemic injury. pcDNA‐NOX4 and miR‐454 agomir were cotransfected into SH‐SY5Y cells treated by OGD/R. The transfection efficiency of pcDNA‐NOX4 was confirmed (Figure 7A, p < 0.01). MiR‐454p overexpression significantly elevated SH‐SY5Y cell proliferation after OGD/R treatment, but NOX4 overexpression diminished this elevation (Figure 7B, p < 0.05). Furthermore, NOX4 overexpression reversed the neuroprotective effects of miR‐454 on SH‐SY5Y cells treated by OGD/R (Figure 7C−I). These data revealed that miR‐454 overexpression suppressed apoptosis and oxidative stress by directly targeting NOX4 in SH‐SY5Y cells.
Figure 7.
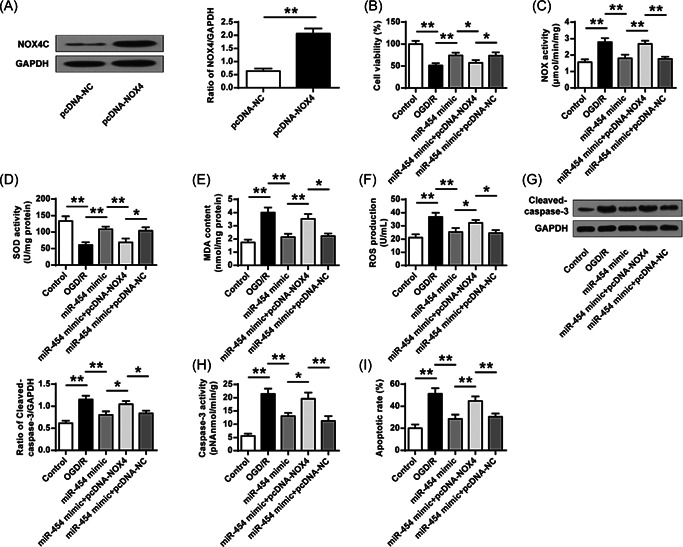
Exogenous miR‐454 agomir suppresses oxidative stress and apoptosis. (A) Western blot detected the transfection efficiency of pcDNA‐NOX4. (B) MTT assay determined the cell viability. (C) Total NOX enzyme activity. (D, E) SOD activity and MDA content determined by ELISA. (F) ROS production. (G) Caspase‐3 protein expression. (H) Caspase‐3 enzyme activity. (I) Cell death calculation using flow cytometry detection. ROS, reactive oxygen species; SOD, superoxide dismutase. *p < 0.05, **p < 0.01. n = 3.
4. DISCUSSION
Ischemic stroke triggers a series of reactions that lead to rapid neuronal damage and death.[ 1 ] Studies have shown that miRNA expression is altered in ischemic stroke and affects the outcome of stroke.[ 5 , 14 , 15 ] For instance, miR‐424, miR‐99a, and miR‐23a‐3p upregulation attenuates oxidative stress and consequently protect the brain following an ischemic injury.[ 16 , 17 , 18 ] Our study found that the miR‐454 level is decreased in I/R‐treated rats. NOX4 expression is increased in I/R‐treated rats, which is inhibited by miR‐454 overexpression. NOX4 is a target of miR‐454. Beyond that, the elevation of NOX4 partially reduced the protective effect of miR‐454 on neuronal cells, suggesting that miR‐454 acts as a neuronal protection function in ischemic stroke via regulating NOX4 expression and activity.
Studies have shown that restored miR‐454‐3p level inhibits glioma cell function by directly targeting ATG12,[ 19 ] and miR‐454 is downregulated in glioblastoma tumor cells to alter cell proliferation and cycle by binding to its target gene PDK1.[ 20 ] Our study showed that miR‐454 treatment alone significantly alleviates I/R‐induced neurological function decline, diminishes cerebral infarction and cerebral edema, and decreases cell death after cerebral ischemia/reperfusion. Thereafter, we verified that miR‐454 overexpression increases SOD activity and decreases ROS production and MDA content via inhibiting NOX4 expression and enzymatic activity. These data suggest that miR‐454 function in ischemic stroke disease could be realized by regulating its downstream target NOX4.
NOX4, a member of the NOX family, promotes ROS production[ 11 ] and is recognized as the major source of neurodegeneration in ischemic stroke disease.[ 21 ] We verified that the protective effects of miR‐454 agomir, including increased cell activity, decreased cleaved caspase 3 levels, and reduced neuronal apoptosis in OGD/R‐treated cells, are reversed by pcDNA‐NOX4. Moreover, NOX4 expression regulated by miR‐454 may ultimately alter ROS production, thereby mediating oxidative stress in ischemic stroke.
Following decades of research, tissue plasminogen activator (tPA) is still the main treatment for clinical stroke.[ 4 , 22 ] At present, many preclinical trials have shown that effective neuroprotective drugs have not been successfully translated into clinical medications, and new therapies for ischemic stroke are urgently needed.[ 23 ] More and more attention has been paid to the neuroprotective strategies to alleviate or reverse brain injury to overcome the obstacles encountered in the administration of stroke.[ 24 , 25 ] Therefore, the oxidative stress protection‐related pathway is becoming a new hotspot. Our study illustrates that the ROS production and the expression of related molecules, including SOD and MDA, are also regulated by the levels of miR‐454 and NOX4. This study revealed a functional transcription regulation molecular. The miR‐454 may become a new biomarker related to ischemic stroke. Its clinical value needs to be evaluated in clinical cohorts in the future.
5. CONCLUSION
Our study demonstrated that miR‐454 plays a crucial role in protecting against neuron damages following ischemic stroke by regulating NOX4, suggesting that miR‐454 might be an attractive neuroprotective agent for ischemic stroke treatment by regulating NOX4.
AUTHOR CONTRIBUTIONS
Tao Zhang, Xuqing Cao: Concept, manuscript writing, editing, and review. Haiping Han, Yan Zhou, Zhimei Liu, Tingjie Ma: Data collection and analysis, manuscript preparation. All authors have read and approved the submission of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
All procedures were approved by the Animal Care Committee of Ningxia Hui Autonomous Region People's Hospital. Procedures operated in this researchstudy were completed in keeping with the standards set out in the principles of ethical animal research outlined in the National Institutes of Health Laboratory Animal Care and Use Guidelines.
Zhang T., Han H., Zhou Y., Liu Z., Ma T., Cao X., J. Biochem. Mol. Toxicol. 2022;36:e23153. 10.1002/jbt.23153
DATA AVAILABILITY STATEMENT
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Hankey G. J., Lancet 2017, 389(10069), 641. [DOI] [PubMed] [Google Scholar]
- 2. Phipps M. S., Cronin C. A., BMJ (Clinical Research Ed.) 2020, 368, l6983. [DOI] [PubMed] [Google Scholar]
- 3. Rodrigues F. B., Neves J. B., Caldeira D., Ferro J. M., Ferreira J. J., Costa J., BMJ (Clinical Research Ed.) 2016, 353, i1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saver J. L., Goyal M., van der Lugt A., Menon B. K., Majoie C. B., Dippel D. W., Campbell B. C., Nogueira R. G., Demchuk A. M., Tomasello A., Cardona P., Devlin T. G., Frei D. F., du Mesnil de Rochemont R., Berkhemer O. A., Jovin T. G., Siddiqui A. H., van Zwam W. H., Davis S. M., Castaño C., Sapkota B. L., Fransen P. S., Molina C., van Oostenbrugge R. J., Chamorro Á., Lingsma H., Silver F. L., Donnan G. A., Shuaib A., Brown S., Stouch B., Mitchell P. J., Davalos A., Roos Y. B., Hill M. D., Hermes C., JAMA 2016, 316(12), 1279. [DOI] [PubMed] [Google Scholar]
- 5. Khoshnam S. E., Winlow W., Farbood Y., Moghaddam H. F., Farzaneh M., J. Stroke 2017, 19(2), 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu H., Guo J., Shen Y., Dong W., Gao H., Miao Y., Li C., Zhang Y., Clin. Cancer Res. 2018, 24(22), 5757. [DOI] [PubMed] [Google Scholar]
- 7. Juźwik C.A., S.S.D., Zhang Y., et al., Prog. Neurobiol. 2019, 182, 101664. [DOI] [PubMed] [Google Scholar]
- 8. Xu L. J., Ouyang Y. B., Xiong X., Stary C. M., Giffard R. G., Exp. Neurol. 2015, 264, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma M. W., Wang J., Zhang Q., Wang R., Dhandapani K. M., Vadlamudi R. K., Brann D. W., Mol. Neurodegener. 2017, 12(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma M. W., Wang J., Dhandapani K. M., Wang R., Brann D. W., Redox Biol. 2018, 16, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yao H., Ago T., Kitazono T., Nabika T., Int. J. Mol. Sci. 2017, 18, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleinschnitz C., Grund H., Wingler K., Armitage M. E., Jones E., Mittal M., Barit D., Schwarz T., Geis C., Kraft P., Barthel K., Schuhmann M. K., Herrmann A. M., Meuth S. G., Stoll G., Meurer S., Schrewe A., Becker L., Gailus‐Durner V., Fuchs H., Klopstock T., de Angelis M. H., Jandeleit‐Dahm K., Shah A. M., Weissmann N., Schmidt H. H., PLoS Biol. 2010, 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z., Tuo Y. H., Chen J. W., Wang Q. Y., Li S., Li M. C., Dai G., Wang J. S., Zhang Y. L., Feng L., Shi Z. S., J. Neurointerv. Surg. 2017, 9(7), 702. [DOI] [PubMed] [Google Scholar]
- 14. Koutsis G., Siasos G., Spengos K., Curr. Top. Med. Chem. 2013, 13(13), 1573. [DOI] [PubMed] [Google Scholar]
- 15. Li G., Morris‐Blanco K. C., Lopez M. S., Yang T., Zhao H., Vemuganti R., Luo Y., Prog. Neurobiol. 2018, 163‐164, 59. [DOI] [PubMed] [Google Scholar]
- 16. Zhao H., Tao Z., Wang R., Liu P., Yan F., Li J., Zhang C., Ji X., Luo Y., Brain Res. 2014, 1592, 65. [DOI] [PubMed] [Google Scholar]
- 17. Liu P., Zhao H., Wang R., Wang P., Tao Z., Gao L., Yan F., Liu X., Yu S., Ji X., Luo Y., Stroke 2015, 46(2), 513. [DOI] [PubMed] [Google Scholar]
- 18. Tao Z., Zhao H., Wang R., Liu P., Yan F., Zhang C., Ji X., Luo Y., J. Neurol. Sci. 2015, 355(1‐2), 113. [DOI] [PubMed] [Google Scholar]
- 19. Shao N., Xue L., Wang R., Luo K., Zhi F., Mol. Cancer Ther. 2019, 18(2), 459. [DOI] [PubMed] [Google Scholar]
- 20. Fang B., Zhu J., Wang Y., Geng F., Li G., Biomed. Pharmacother. 2015, 75, 148. [DOI] [PubMed] [Google Scholar]
- 21. Radermacher K. A., Wingler K., Langhauser F., Altenhöfer S., Kleikers P., Hermans J. J., Hrabě de Angelis M., Kleinschnitz C., Schmidt H. H., Antioxid. Redox Signal. 2013, 18(12), 1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z. G., Chopp M., Lancet Neurol. 2009, 8, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kikuchi K., Tanaka E., Murai Y., Tancharoen S., CNS Drugs 2014, 28, 929. [DOI] [PubMed] [Google Scholar]
- 24. Sekerdag E., Solaroglu I., Gursoy‐Ozdemir Y., Curr. Neuropharmacol. 2018, 16, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Venkat P., Shen Y., Chopp M., Chen J., Neuropharmacology 2018, 134, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.