

Abstract
The RNA‐binding protein Quaking (QKI) has emerged as a potent regulator of cellular differentiation in developmental and pathological processes. The QKI gene is itself alternatively spliced to produce three major isoforms, QKI‐5, QKI‐6, and QKI‐7, that possess very distinct functions. Here, we highlight roles of the different QKI isoforms in neuronal, vascular, muscle, and monocyte cell differentiation, and during epithelial–mesenchymal transition in cancer progression. QKI isoforms control cell differentiation through regulating alternative splicing, mRNA stability and translation, with activities in gene transcription now also becoming evident. These diverse functions of the QKI isoforms contribute to their broad influences on RNA metabolism and cellular differentiation.
This article is categorized under:
RNA Interactions with Proteins and Other Molecules > Protein‐RNA Interactions: Functional Implications
RNA Processing > Splicing Regulation/Alternative Splicing
RNA in Disease and Development > RNA in Development
Keywords: RNA, RNA‐binding protein
The RNA‐binding protein Quaking potently regulates cell differentiation via diverse impacts on gene expression and RNA metabolism.
1. INTRODUCTION
The Quaking RNA‐binding proteins (RBPs) are composed of three major isoforms (QKI‐5, QKI‐6, and QKI‐7) that play diverse functions in RNA metabolism with widespread impacts on cellular differentiation. The Quaking gene (Ebersole et al., 1996) derives its name from a “Quaking” phenotype observed in the Quaking viable (Qk v ) mutant mouse (Ebersole et al., 1996; Sidman et al., 1964), resulting from severe demyelination of the central and peripheral nervous system. Close examination of the Qk v mouse revealed a mosaic pattern of QKI isoform expression, where QKI‐5 is retained in less affected regions of the brain while loss of all three major isoforms correlates with severe dysmyelination (Hardy et al., 1996). In addition to neural tissues, QKI proteins are prominently expressed in developing heart and vascular tissues, with Qki mutant mice displaying cardiovascular defects (review in Justice & Hirschi, 2010). These pioneering studies pointed to important roles of QKI in cellular differentiation and homeostasis.
More recently, QKI proteins have been found to contribute to neural stem cell (NSC; Hayakawa‐Yano et al., 2017), vascular (van der Veer et al., 2013), muscle (Chen et al., 2021), and monocyte (De Bruin, Shiue, et al., 2016) cell differentiation through an array of mechanisms including regulating alternative splicing, RNA stability and gene transcription. In addition, QKI also plays diverse roles during cancer progression, influencing epithelial–mesenchymal transition (EMT) and associated alternative splicing patterns (Pillman et al., 2018). This review provides a background on the discovery of QKI, function of its major isoforms and focuses on recent discoveries of QKIs functions in cellular differentiation.
2. IDENTIFICATION OF THE QUAKING GENE
The Quaking protein (QKI) was named after the phenotype observed in the Qk v mouse from which the QkI gene was discovered (Sidman et al., 1964). These mice possess a spontaneous recessive mutation which at 10 days after birth causes severe demyelination of the central nervous system leading to the characteristic “Quaking” (meaning shaking or shuddering) phenotype (Sidman et al., 1964). Homozygous male mice were also found to be infertile due to a defect in spermatid differentiation (Bennett et al., 1971).
Characterization of the mutation responsible for the Quaking phenotype revealed a 1 Mb deletion in the mouse Chromosome 17 (Ebersole et al., 1992; Ebersole et al., 1996). This deletion was found to impact four distinct genes, parkin, Parkin co‐regulated gene (pacrg) and two previously uncharacterized genes initially named QkI and QkII (Ebersole et al., 1996). The genes parkin and pacrg are both associated with juvenile familial Parkinson's disease and their disruption has no influence on the Quaking phenotype, except for pacrg which was found to be responsible for the homozygous male infertility (Itier et al., 2003; Lorenzetti et al., 2004). The QkII gene transcribed a small single exon gene, which has since been found to code for a long noncoding RNA, now identified as CAHM (Pedersen et al., 2014). Mapping of the Qk v mice deletion break point was localized to 913 bp upstream of the transcription initiation site for QkI, leaving the coding region intact, but likely disrupting important promotor and enhancer sequences and consequently reducing transcription of the coded protein (Hardy et al., 1996; Kondo et al., 1999).
The QkI gene was found to produce three mRNA transcripts of five, six, and seven kilobases in length, which were named QkI‐5, QkI‐6, and QkI‐7, respectively. All three isoforms share a coding sequence up until a carboxy‐terminal region that is unique for each of them (Ebersole et al., 1996) (Figure 1). Three major domains were identified in the coding sequence of QkI, based on their conservation in other species (Ebersole et al., 1996). The hnRNP K homology (KH) domain, an RNA‐binding domain, is flanked by the upstream QUA1 domain and downstream QUA2 (Ebersole et al., 1996). The QUA1 domain shares strong homology with a sequence in the Xenopus version of Quaking, pXqua, which was found to be responsible for homodimerization and is necessary for RNA binding (Chen et al., 1997; Chen & Richard, 1998; Ebersole et al., 1996; Zorn & Krieg, 1997). Studies of the QUA2 domain in the QkI homologs, GLD‐1 and Splicing factor 1 (SF1), found it to support the KH domain in RNA binding (Daubner et al., 2014; Liu et al., 2001). These three domains form a larger tripartite domain known as the signal transduction and activation of RNA (STAR) domain (Figure 1) and proteins which contain this domain are classified as members of the STAR family (Vernet & Artzt, 1997).
FIGURE 1.
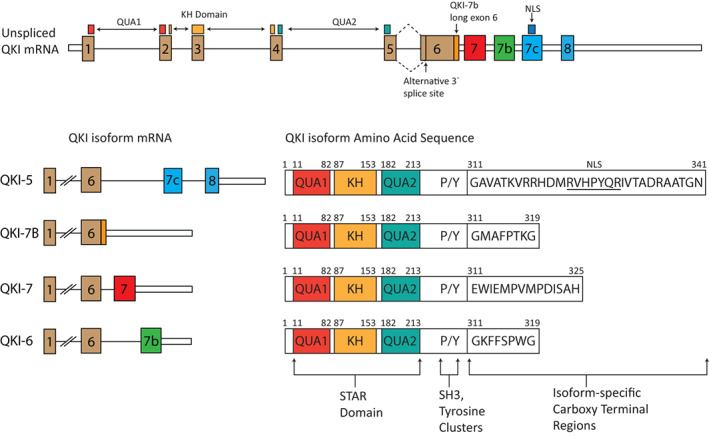
The human QKI gene, major isoforms and protein products. The QUA1 dimerization domain, KH RNA binding domain and QUA2 domains are present in all isoforms and are called the STAR domain. Alternative splicing and alternative polyadenylation events generate the major splice isoforms of QKI, while an N‐terminal truncation in Exon 6 has also been detected. Post‐translational phosphorylation can occur in SH3 and tyrosine clusters. QKI‐5 has a distinct 3'UTR and has a NLS in its C‐terminal region restricting its activity to the nucleus. NLS, nuclear localization signal; QKI, RNA‐binding protein Quaking; SRC, SRC homology 3 domain (SH3); STAR, signal transduction and activation of RNA
3. STRUCTURE OF QKI PROTEINS AND FUNCTIONS IN RNA REGULATION
3.1. The structure of the QKI pre‐mRNA leads to the generation of alternative isoforms
The three major protein isoforms produced from the human QKI locus, QKI‐5, QKI‐6, and QKI‐7, as well as the less studied isoform known as QKI‐7B, are generated through complex patterns of alternative splicing (Kondo et al., 1999). Although Exons 1–6 are included in each transcript, the remaining exons are isoform specific with QKI‐7 containing Exon 7, QKI‐6 containing Exon 7b and QKI‐5 containing Exons 7c and 8 (Figure 1). QKI‐7B ends at Exon 6, where the 5′ splice is not utilized, and a downstream stop codon defines the end of the coding region. This mechanism of splicing is known as alternative last exons (ALE), and is caused by different definitions of the terminal exon through alternative poly(A) site (PAS) selection (Dutertre et al., 2014). Definition of an internal exon requires the recognition of a downstream 5′ splice site before the splicing machinery commits to intron excision (Robberson et al., 1990). The definition of the terminal exon, and excision of the terminal intron, relies on the presence of a 5′ splice site and 3′ splice site in the intron and a downstream PAS (Niwa et al., 1990; Niwa & Berget, 1991; Rigo & Martinson, 2008). This phenomenon is likely exploited to produce the QKI isoforms where competing PAS sites determine the end of the transcript and a switch between production of QKI‐5 or QKI‐6 and QKI‐7.
A less studied variation on QKI alternative splicing is the presence of an alternative 3′ splice site in Exon 6, 24 nucleotides downstream from the major 3′ splice site. When used, this splice site causes the loss of eight amino acids encoded within Exon 6. Recently, Polypyrimidine tract‐binding protein 1 (PTBP1) was found to regulate the utilization of this splicing site (Mironov et al., 2021); however, further studies are required to determine the consequences of these eight amino acids for QKI activity.
As alternative splicing between the QKI isoforms is predominantly limited to the 3′ end of the transcript, the protein products all contain the STAR domain, and only differ in their carboxy‐terminal (C‐terminal) tails, which are distinct for each isoform (Ebersole et al., 1996). To date, little is known about the functions of these regions, except for QKI‐5, which contains a nuclear localization signal (NLS) that directs its localization to the nucleus (Wu et al., 1999). This highlights the importance of performing studies that focus on identifying the functions of these regions.
A striking feature of the QKI isoform mRNAs is the lengths of their 3′ untranslated regions (UTRs), spanning 4173 bp for QKI‐6, 5434 bp for QKI‐7, and 6399 bp for QKI‐7B, while the QKI‐5 3′ UTR is completely distinct from the other isoforms and predominantly ends 3086 bp downstream from its stop codon (Pillman et al., 2018). Another interesting observation is that these 3′ UTRs are highly conserved between vertebrates (Artzt & Wu, 2010). The lengths of these 3′ UTRs, their conservation, and the fact that QKI‐5 contains a distinct 3′ UTR from QKI‐6, QKI‐7, and QKI‐7B allows for isoform specific regulation by miRNAs and by the QKI isoforms themselves (Fagg et al., 2017; Pillman et al., 2018).
3.2. The QUA1 domain mediates the formation of QKI isoform heterodimers
The QUA1 domain is required for the dimerization of the QKI isoforms, which allows them to recognize and bind a bipartite consensus sequence (Chen & Richard, 1998; Galarneau & Richard, 2005). As most KH domain‐containing proteins bind using multiple KH domains (Valverde et al., 2008), the ability of QKI and other STAR‐domain proteins to dimerize likely overcomes the limitation of having a single KH domain. Interestingly, the QKI homolog, Src‐associated substrate in mitosis of 68 kDa (SAM68), requires RNA‐binding activity to form a stable homodimer, whereas QKI was found to dimerize in the presence and the absence of RNA (Chen et al., 1997; Chen & Richard, 1998). Another STAR protein, SF1, lacks a QUA1 domain while still retaining the ability to bind RNA as a splicing factor. However, as SF1 recognizes the branchpoint sequence to facilitate intron splicing, it is likely that cooperative binding with the splicing machinery enhances its binding strength and specificity (Berglund et al., 1998).
As all QKI isoforms contain the same QUA1 domain, heterodimerization between different isoforms can occur (Pilotte et al., 2001). This allows the cytoplasmic isoform QKI‐7 to trans‐localize to the nucleus by shuttling with QKI‐5 (Pilotte et al., 2001). Furthermore, hetero‐dimerization has been suggested as a mechanism to allow QKI‐6 and QKI‐7 access to the nucleus to promote the export of nuclear retained mRNAs (Larocque et al., 2002). Some small proteins (<60 kDa) can diffuse into the nucleus freely without a NLS, whereas larger proteins diffuse at a reduced rate (Timney et al., 2016; Wang & Brattain, 2007). Monomeric QKI isoforms are approximately 37 kDa, so it is possible that QKI‐6 and QKI‐7 can access the nucleus freely as a monomer. However, they may have a reduced ability to enter as dimer, unless heterodimerized with QKI‐5. To gain a better understanding of how the QKI isoforms cooperate, more work is required to determine if nuclear localization of QKI‐6 and QKI‐7 requires heterodimerization with QKI‐5.
3.3. Posttranslational modification of QKI and its regulation by signal transduction pathways
A characteristic of STAR proteins is their regulation in response to signal transduction pathways (Vernet & Artzt, 1997). Proteins of this classification are found to contain various sites that permit posttranslational modification, including tyrosine residues, proline‐rich SH3‐binding domains, WW‐binding sites, and arginine‐glycine methylation sites. STAR proteins have been found to be directed to regulate mRNA metabolism as downstream targets of cell signaling and, in the case of SAM68, as a mediator of cell signals (Lock et al., 1996; Richard et al., 1995). However, little is known about the role that QKI plays in cell signaling.
The QKI amino acid sequence was found to contain RG boxes, tyrosine clusters, and putative SH3‐binding sites (Ebersole et al., 1996). Subsequently, QKI has been shown to be phosphorylated by Src‐protein tyrosine kinases and arginine methylated (Côté et al., 2003; Zhang et al., 2003). Phosphorylation of QKI by Src was discovered to decrease its ability to bind RNA, repressing its ability to regulate mRNAs (Zhang et al., 2003). During myelination, decreased phosphorylation of QKI coincides with myelin basic protein (MBP) mRNA accumulation (Zhang et al., 2003) and the Src family member, Fyn, was shown to be required for the upregulation of important myelin genes, with loss of Fyn leading to reduction of the Exon 2 containing MBP isoform (Lu et al., 2005). As QKI was shown to promote the expression of this isoform, it is suggested that Fyn might phosphorylate QKI to indirectly repress this isoform (Lu et al., 2005).
3.4. The QKI RNA‐binding interface and discovery of the QKI regulatory element
The QKI‐binding motif was initially identified using the systematic evolution of ligands by exponential enrichment method, which uncovered a motif, NACUAAY, followed by a half site, UAAY, within 20 nucleotides away (Galarneau & Richard, 2005). Since then, various cross‐linked immunoprecipitation (CLIP)‐Seq experiments have been performed that have also confirmed the QKI motif to be ACUAAY (Hafner et al., 2010; Pillman et al., 2018; Teplova et al., 2013; Van Nostrand et al., 2016). The tendency of QKI to bind a full site and half site in combination provides strong evidence that QKI preferentially recognizes two sites as a dimer (Galarneau & Richard, 2005; Teplova et al., 2013).
The QKI isoforms, like other STAR proteins, all have a QUA2 domain adjacent to the KH domain that strengthens their association with RNA. In the case of the QKI homolog SF1, the QUA2 domain was found to specifically recognize the 5′ bases of the QKI motif (ACU), while the KH domain was found to recognize the 3′ bases (UAAC) (Daubner et al., 2014; Liu et al., 2001). The QUA2 domain is required for successful binding of SF1 to the QKI motif and deletion of the QUA2 domain from mouse Qk has been shown to severely disrupt RNA binding (Chen & Richard, 1998; Liu et al., 2001).
3.5. Distinct mechanisms of RNA regulation by the QKI isoforms
The QKI isoforms engage in many different mechanisms of mRNA regulation including pre‐mRNA splicing, nuclear export, differential localization, translation, and mRNA stability (Figure 2). Since each isoform shares the same RNA‐binding interface, it likely that the differences in function of the QKI isoforms are due to their unique C‐terminal regions. The only established function of any of the C‐terminal regions is for QKI‐5, which contains a NLS that permits its nuclear function in splicing (Wu et al., 1999). Most of the differences in function between the nuclear QKI‐5 and cytoplasmic QKI‐6 and QKI‐7 are likely due to localization. However, the C‐terminal regions of QKI‐6 and QKI‐7 are distinct from one another and so it is likely that these isoforms have unique functions.
FIGURE 2.
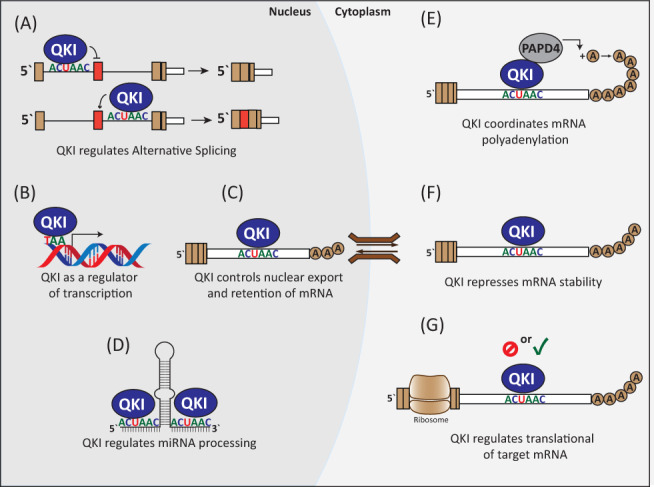
QKI coordination of gene expression and RNA metabolism. (a) Nuclear expression of QKI controls alternative splicing via binding upstream (exon inclusion) or downstream (exon exclusion) to specific motifs. (b) QKI can act as a co‐factor to influence gene transcription. (c) Binding of QKI to mRNAs can facilitate nuclear export or retention. (d) Processing of miRNAs can be enhanced through QKI interaction with the primary miRNA transcript. (e) In the cytoplasm QKI interacts with PAPD4 (TENT2) and coordinates cytoplasmic polyadenylation of specific transcripts. (f) QKI can also repress mRNA stability through unknown mechanisms. (g) QKI can enhance or repress translation of mRNAs, in some cases in specific cellular locations. QKI, RNA‐binding protein Quaking
3.5.1. The balance of the QKI isoforms determine subcellular localization of target mRNAs
Early work on QKI‐6 and QKI‐7 in oligodendrocytes found that both isoforms associate with microtubules, while QKI‐6 alone interacts with actin filaments (Wu et al., 2001). It was suggested that both isoforms could direct traffic of mRNAs along microtubules away from the cell body while QKI‐6 might also direct mRNAs to the cell periphery via the actin cytoskeleton (Wu et al., 2001). More recently, studies of the QKI isoforms in astrocytes found that many mRNAs are locally translated at the peripheral processes and that these mRNAs had higher occurrence of the QKI motif in their 3′ UTRs, with QKI‐6 binding many of these mRNAs (Sakers et al., 2017; Sakers et al., 2021). It is possible that the ability of QKI‐6 and QKI‐7 to associate with the cytoskeleton allows them to shuttle mRNAs to differentially localize their translation at specific locations in the cell.
The balance of QKI‐5 to QKI‐6 and QKI‐7 has been shown to control the nuclear export of target mRNAs. In oligodendrocytes, the balance in the QKI isoforms is important for MBP mRNA to be exported from the nucleus where it can be translated into protein (Larocque et al., 2002). This phenomenon is discussed in greater detail in Section 4.1.2. It is currently unknown how widespread the QKI‐controlled nuclear export of mRNA is and what the consequences are for different physiological and disease states.
3.5.2. The QKI isoforms have complex roles in regulating mRNA stability and translation
QKI‐5, QKI‐6, and QKI‐7 have been shown to stabilize or destabilize many different mRNAs (Azam et al., 2019; Cochrane et al., 2017; Doukhanine et al., 2010; Guo et al., 2014; Larocque et al., 2005; S. Li, Lin, et al., 2018; Thangaraj et al., 2017; Zhang et al., 2016). Similarly, the QKI isoforms have been implicated in both translational repression and activation (De Bruin, Van Der Veer, et al., 2016; Saccomanno et al., 1999; Sakers et al., 2017; Yamagishi et al., 2016; Zhao, Ku, et al., 2006). Recently, QKI‐7, but not QKI‐5 or QKI‐6, was found to directly promote translation of bound mRNAs by recruiting PAP‐associated domain containing 4 (PAPD4, also known as Terminal nucleotidyltransferase 2 or TENT2) to increase poly(A) tail length (Yamagishi et al., 2016). The complex and sometimes contradictory effects of QKI isoforms likely arise from recruitment of different partner proteins. However, due to the complication of heterodimerization, nuclear‐cytoplasmic shuttling, and autoregulation between the isoforms, it is difficult to determine the precise isoform contribution to mRNA regulation in contexts where all isoforms are expressed. This highlights the importance of finding methods to study the QKI isoforms in isolation, to identify mechanisms of RNA regulation that are specific to each isoform.
3.5.3. Functions for QKI in regulation of microRNAs
The QKI isoforms have also been implicated in the regulation of microRNAs. QKI‐5 and QKI‐6 have been shown to bind a QKI motif in the primary transcript (pri‐miR) encoding miR‐7‐1 in glial cells where it repressed the formation of mature miR‐7‐1 (Wang et al., 2013). The pri‐miR‐7‐1 was retained in the nucleus and tightly bound to Drosha in the presence of QKI‐5 and QKI‐6, but not QKI‐7 (Wang et al., 2013). Conversely, QKI‐5 has been shown to promote the maturation of pri‐miR‐124‐1 during erythropoiesis by facilitating its interaction with the microprocessor component, DGCR8, through directly binding the QKI‐5 KH domain (F. Wang, Song, et al., 2017). More recently, QKI‐7 was shown to stabilize the mature miR‐122 by promoting adenylation at its 3′ end. This occurs through the formation of a ternary structure involving Ago2, which recognizes the QUA2 domain, and PAPD4, which recognizes the C‐terminal region of QKI‐7 (Hojo et al., 2020). Lastly, the co‐localization of QKI‐6 with Ago2 within stress granules has been previously reported, providing further evidence of a role for the QKI isoforms in regulating miRNAs (Wang et al., 2010).
3.5.4. QKI‐regulated alternative splicing
One of the most established functions of QKI is the regulation of pre‐mRNA alternative splicing (Wu et al., 2002). QKI has been found to regulate many alternative splicing events in different contexts including during EMT (Conn et al., 2015; Pillman et al., 2018; Yang et al., 2016), monocyte differentiation (De Bruin, Shiue, et al., 2016), myelination (Darbelli et al., 2016; Darbelli et al., 2017; Mandler et al., 2014), microglia function (Lee et al., 2020; J. Ren, Dai, et al., 2021), muscle differentiation (Hall et al., 2013), and various aspects of cancer progression (Brosseau et al., 2014; de Miguel et al., 2016; Zong et al., 2014). The nuclear isoform, QKI‐5, has been found to bind primarily within introns, suggesting that its major function is regulation of splicing (Pillman et al., 2018). Interestingly, QKI‐6 has been found to bind the first intron of HDAC7 and regulate its splicing (Caines et al., 2019). This suggests that although QKI‐5 is the predominant regulator of alternative splicing, the other isoforms might have a minor role in regulating splicing, possibly though the formation of heterodimers with QKI‐5.
Whether QKI‐5 promotes inclusion or exclusion of a cassette exon appears dependent on its location of binding, promoting skipping when it binds within or 100 base pairs upstream from a cassette exon or inclusion when it binds in the downstream intron (Pillman et al., 2018). This has been shown in muscle cell differentiation, where QKI‐5 promotes the retention of Exon 9 of Capzb by binding its motif in the downstream intron (Hall et al., 2013). Another RBP, PTBP1 was found to repress this exon by binding in the same intron as QKI‐5, which suggests that the two proteins could compete to regulate the exon in opposite directions (Hall et al., 2013). Further studies are required to define clear rules for how QKI promotes exon inclusion.
In the opposite case, an explanation for how QKI‐5 might promote skipping of a target exon is through direct competition with the U2 snRNP or SF1 for the branchpoint (Zong et al., 2014). The QKI motif bares striking resemblance to the branchpoint sequence and has been shown to compete with SF1 for binding to the branchpoint of the target gene NUMB (Zong et al., 2014). It is currently not known how often QKI‐5 interacts with branchpoint sites and the extent of this mechanism of regulation.
3.6. The QKI isoforms engage in complex autoregulation
A feature of the QKI isoforms that complicates their study is autoregulation. Expression of QKI‐5 is required to allow the formation of QKI‐6 and QKI‐7, which is suggested to be controlled though alternative splicing (Darbelli et al., 2017; Fagg et al., 2017). This is consistent with the presence of QKI motifs within introns of the QKI pre‐mRNA; however, the mechanism behind this phenomenon has not been determined. The 3′ end of the QKI locus is organized in an unusual manner, where last exons that are specific for each isoform are alternatively included (see Figure 1). This phenomenon of ALE is thought to have emerged late in evolution (Dutertre et al., 2014) and occurs frequently in neuronal cells, with alternative isoforms bearing distal exons often localizing to neurites (Taliaferro et al., 2016). Some evidence for RBP‐mediated regulation of ALE exists, such as ELAV1/HUR promoting widespread inclusion of ALEs through direct exon binding (Dutertre et al., 2014). Furthermore, regulation of alternative polyadenylation through direct binding of polyadenylation sites by RBPs is an established phenomenon (Goering et al., 2021; Tian & Manley, 2017) and is a possible mechanism of regulating ALEs. Further study is required to determine the specific mechanism by which QKI‐5 controls splicing of the cytoplasmic isoforms.
Recent work has uncovered more complex interactions between the QKI isoforms. QKI‐5 was found to inhibit its own expression by binding a QKI motif in its 3′ UTR (Fagg et al., 2017). Interestingly, QKI‐6 was found to promote its own translation though binding its 3′ UTR and to repress QKI‐5 protein production, potentially through binding its coding region (Fagg et al., 2017). The full extent of autoregulation by the QKI isoforms remains to be determined.
3.7. QKI as a transcriptional regulator and chromatin‐binding protein
More recently, the realm of QKI functions was expanded to include transcriptional co‐activation and single‐stranded DNA‐binding. QKI was found to regulate the structural lipid components of myelin, through interaction with the peroxisome proliferator‐activated receptor β–retinoid X receptor α complex, leading to the differential regulation of genes associated with lipid metabolism (Zhou et al., 2020). The same group discovered that QKI also regulates cholesterol metabolism in myelinogenesis, again through transcription, in this instance through co‐activation with the transcriptional activator, Sterol regulatory element‐binding protein 2 (Srebp2) (Zhou et al., 2021). This work was expanded to show that QKI co‐activates Srebp2‐mediated transcription of cholesterol‐metabolism genes in eye lens cells, where mice deficient in QKI have loss of lens transparency (Shin et al., 2021). Strikingly, in this context QKI was shown to function by directly binding single‐stranded DNA (ssDNA), which has been suggested for KH domain‐containing proteins (Galarneau & Richard, 2009), but is a novel finding for QKI (Shin et al., 2021). Similar to the RNA binding motif, ACUAAC, the central trinucleotide TAA was uncovered as being selective for ssDNA‐binding by QKI (Shin et al., 2021). More recently, a large‐scale analysis of RBPs with functions in chromatin‐binding uncovered QKI‐5 as directly binding to promotors of genes associated with hematopoiesis (Y. Ren, Huo, et al., 2021). QKI‐5 was able to facilitate differentiation of monocytes by transcriptionally activating CXCL2, a cytokine required for monocyte differentiation (Y. Ren, Huo, et al., 2021). However, an alternative possibility is that QKI may also bind nascent transcripts that anchor it to chromatin and facilitate its transcriptional activity (Lee et al., 2015; Pompon & Garcia‐Blanco, 2015), The combined functions of RNA and DNA‐binding increases the repertoire of QKI functions in gene expression and raises the possibility of QKI regulating a target gene from transcription to translation, and as such it would be worthwhile exploring whether such targets exist.
4. FUNCTIONS OF QKI IN CELL DIFFERENTIATION
4.1. QKI in NSC fate and the maturation of cells of glial lineage
4.1.1. Role of QKI in neural cells and differentiation of oligodendrocytes
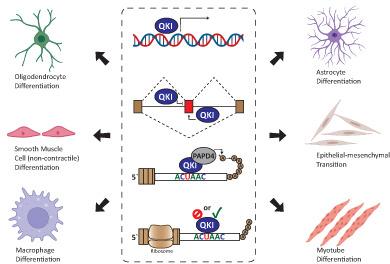
During embryogenesis, NSCs differentiate into neural progenitor cells, which either progress down a neuron‐restricted pathway, or a glial restricted pathway. QKI‐5 and QKI‐6 expression is detected in NSCs and is retained in cells that differentiate down the glial lineage, but is lost in cells that differentiate into neurons (Hardy, 1998; Hayakawa‐Yano et al., 2017) (Figure 3). In primary NSCs, QKI was found to contribute to the alternative splicing of a large number of genes, and to maintain a gene expression profile consistent with apical radial glia, which suggests that QKI maintains the NSC state and prevents differentiation toward the neuron lineage (Hayakawa‐Yano et al., 2017). Furthermore, Sox10, an important transcription factor for NSC differentiation to glial cells (Pozniak et al., 2010), contains a QKI motif within its 3′ UTR and is reduced in expression along with QKI in the brains of schizophrenic patients (Åberg et al., 2006). Expression of QKI remerges in glial cells and is one of the earliest markers of glial cell differentiation, leading to the proposition that QKI contributes to neuron‐glial cell fate determination (Hardy, 1998; Takeuchi et al., 2020).
FIGURE 3.
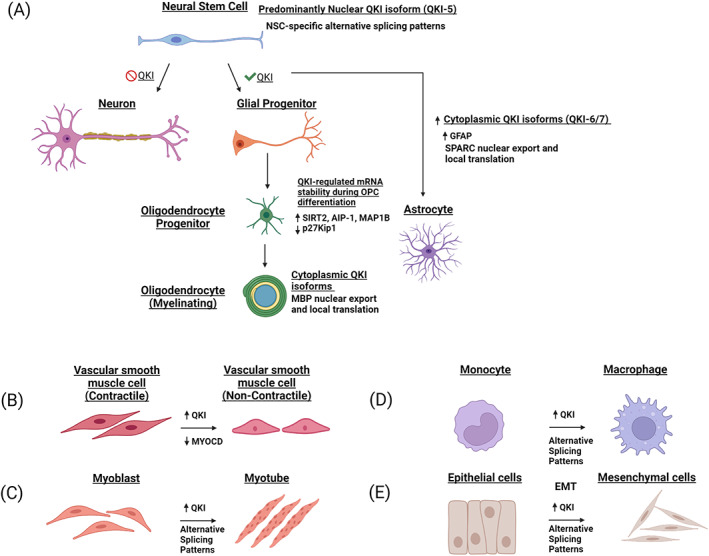
Overview of QKI regulation of cellular differentiation. (a) QKI mediates alternative splicing in neural stem cells and is reduced during differentiation to mature neurons. QKI exhibits a range of metabolic functions in glial to oligodendrocyte differentiation and in astrocyte maturation. (b–e) Upregulation of QKI influences alternative splicing patterns that facilitate differentiation of smooth muscle, non‐smooth muscle, monocyte to macrophage differentiation and EMT. Created with BioRender.com. EMT, epithelial to mesenchymal transition; QKI, RNA‐binding protein Quaking
Expression of QKI is also vital for oligodendrocyte progenitor cell (OPC) differentiation into mature oligodendrocytes (Chen et al., 2007). The QKI isoforms are known to affect the stability of several mRNAs during OPC differentiation (Figure 3). Interestingly, while loss of QKI prevents OPC differentiation, QKI‐5 and QKI‐6 can only partially restore it, suggesting that multiple isoforms are likely required for OPC differentiation (Chen et al., 2007). The QKI target, p27Kip1 is a regulator of the cell cycle and promotes cell cycle exit to decrease proliferation, which is a key step for the differentiation of OPCs into mature oligodendrocytes (Casaccia‐Bonnefil et al., 1999; Durand et al., 1998; Tikoo et al., 1998). In primary rat OPC cells, QKI‐6 and QKI‐7 stabilize p27Kip1 mRNA, promoting cell cycle arrest, a key step in OPC differentiation (Larocque et al., 2005). Overexpression of QKI‐6 and QKI‐7 in mouse embryos is sufficient to induce OPC differentiation (Larocque et al., 2005), however, ectopic expression of p27Kip1 alone cannot induce OPC differentiation (Tang et al., 2000), suggesting that QKI‐6‐ and QKI‐7‐induced differentiation of OPCs is controlled by multiple QKI targets.
Two other genes with known roles in OPC differentiation, MAP1B (Zhao, Ku, et al., 2006) and Sirtuin 2 (SIRT2; Thangaraj et al., 2017) also produce mRNAs that are stabilized by QKI. The NAD+‐dependent tubulin deacetylase (North et al., 2003), SIRT2, is expressed in OPCs prior to myelination (Zhu et al., 2012), and it is known to promote OPC differentiation and to facilitate the growth of extensions (Ji et al., 2011). MAP1B mRNA is also stabilized by QKI‐6 in OPCs, and, like SIRT2, its expression precedes differentiation into mature oligodendrocytes (Vouyioukiis & Brophy, 1993). In neurons, MAP1B has functions in axon regeneration and neurite branching (Barnat et al., 2016). It is feasible that MAP1B could participate in OPC differentiation through a similar mechanism involving regulation of microtubules to affect morphology.
Unlike SIRT2 and MAP1B, AIP‐1 mRNA stability is decreased by QKI‐6 during OPC differentiation into mature oligodendrocytes (Doukhanine et al., 2010). AIP‐1 expression is required earlier in OPC cells where it modulates actin disassembly and its premature loss is associated with impaired process outgrowth (Doukhanine et al., 2010). These findings highlight the complex and dynamic regulation of mRNAs by QKI in the context of late OPC differentiation, although further work is required to determine its full repertoire of targets and to elucidate the rules for QKI‐mediated alteration to target mRNA stability.
4.1.2. The Qkv mouse
Most of what we know about the function of QKI in oligodendrocyte differentiation and myelination is through the study of the Qkv . Comparative examination of QKI isoforms between wild type and Qkv mouse brains uncovered cell type‐specific alterations in their expression (Hardy et al., 1996). Oligodendrocytes express all three QKI isoforms during myelination, but expression of QKI‐6 and QKI‐7 is lost in Qkv oligodendrocytes (Hardy et al., 1996). QKI‐5 expression is retained in most cells; however, it is entirely absent in oligodendrocytes derived from areas of the brain that had the most severe dysmyelination phenotype (Hardy et al., 1996). The cell type specific loss of QKI‐6 and QKI‐7 suggests that the deletion causing the Qkv phenotype affects an enhancer region that is active in oligodendrocytes alone (Hardy et al., 1996), however, the function of the region deleted in the Qkv mouse has not yet been determined.
Loss of QkI is now known to cause the Qkv mouse dysmyelination phenotype by altering posttranscriptional regulation of mRNAs that are required for oligodendrocyte function. The most studied and important of these mRNAs is the transcript for MBP. All three major QKI isoforms bind MBP mRNA via QKI motifs in its 3′ UTR, leading to different consequences for the fate of the transcript (Larocque et al., 2002;Li et al., 2000; Zhang & Feng, 2001). Accumulation of MBP mRNA is prevented in the Qkv mouse brain through destabilization (Li et al., 2000; Zhang & Feng, 2001), and ectopic QKI‐6 expression in HEK293T cells increases stability of mRNA from an MBP reporter (Zhang et al., 2003), suggesting that QKI‐6 stabilizes MBP mRNA. In oligodendrocytes and Schwann cells, MBP mRNA is locally translated at the myelin sheath, where the MBP protein can incorporate itself in the sheath immediately posttranslation (Ainger et al., 1993; Colman et al., 1982; Trapp et al., 1987). Interestingly, in Qkv oligodendrocytes, which have lost expression of QKI‐6 and QKI‐7, the MBP transcript is predominantly localized in the nucleus, preventing translation to protein (Larocque et al., 2002). Similarly, ectopic expression of QKI‐5 in wild‐type oligodendrocytes forces nuclear retention of MBP mRNAs, preventing its translation (Larocque et al., 2002), suggesting that the disrupted balance between QKI‐5 and the cytoplasmic isoforms prevents nuclear export of MBP mRNA in Qkv oligodendrocytes. This was supported when the effect was replicated by transfecting ectopic MBP and QKI‐5 in COS cells, which do not express QKI, while co‐transfection of QKI‐5 with QKI‐6, QKI‐7, or both, rescued export to the cytoplasm and promoted their translation (Larocque et al., 2002). In fact, expression of QKI‐6 alone is able to restore expression of MBP and partially rescue the Qkv phenotype in oligodendrocytes (Zhao, Tian, et al., 2006). As QKI‐6 and QKI‐7 localize to peripheral processes of oligodendrocytes (Hardy et al., 1996; Wu et al., 2001), it is possible that controlled shuttling of MBP mRNA by these isoforms is necessary for their correct local translation at the myelin sheath. It is not currently known if other targets are regulated by QKI in the same manner.
Alternative splicing is also affected in the Qkv mouse brain, suggesting that QKI might regulate alternative splicing in to facilitate myelination. In the brains harvested from Qkv mice, aberrant splicing of MBP, myelin proteolipid protein (PLP) and myelin‐associated glycoprotein (MAG) is detected (Wu et al., 2002). QKI‐5 expression was found to cause dose‐dependent exclusion of MAG Exon 12, dependent on the presence of a 53 nucleotide region in the downstream intron, which QKI‐5 interacted with (Wu et al., 2002). However QKI‐6 was found to repress translation of another RBP, hnRNPA1, which promotes inclusion of MAG Exon 12 (Zhao et al., 2010), suggesting that QKI regulates MAG splicing both directly and indirectly. Lastly, in an oligodendrocyte‐specific conditional QKI knockout mouse, alternative splicing of several genes were detected including MBP, PLP, SIRT2 and neurofascin (Darbelli et al., 2016).
4.1.3. QKI functions in astrocytes
QKI has also been implicated in astrocyte function through several mechanisms. The astrocyte marker, GFAP, appears to have its mRNA stabilized by QKI‐7 in primary human astrocytes (Radomska et al., 2013) (Figure 3). Although knockdown of QKI‐7 represses GFAP mRNA, loss of total QKI does not, which suggests that affecting the balance of QKI isoforms is important in this context (Radomska et al., 2013). Next‐generation sequencing of RNAs captured from the peripheral processes of astrocytes of mice brains has uncovered a significant enrichment of QKI motifs (Sakers et al., 2017). One of these mRNAs, Sparc, which regulates the morphology of astrocytes (Au et al., 2007), was detected after immunoprecipitation of QKI‐5 and QKI‐6 suggesting that QKI promotes nuclear export of Sparc mRNA to mediate astrocyte functions.
Using QKI‐6 QKI CLIP‐Seq performed on developing mouse forebrains brains, it was found that many astrocyte‐associated mRNAs are bound by QKI‐6 through their 3′ UTRs, and a large subset of these transcripts are locally translated in the peripheral processes of astrocytes (Sakers et al., 2021). Conditional KO of QKI in mice leads to decreased association of QKI‐6 target mRNAs on the ribosomes of GFAP+ astrocytes, affecting mRNAs important for astrocyte maturation, suggesting that QKI affects the stability or translation of these mRNAs. These observations suggest a significant and complex role for QKI in the regulation of RNA in astrocytes.
4.2. QKI in vascular smooth muscle and endothelial cell differentiation
Unlike the QkV mutation, several N‐ethyl‐N‐nitrosourea‐induced mutations were generated that affected the coding sequence of the QKI gene, and these are embryonically lethal for homozygous mice before the dysmyelination phenotype has an opportunity to emerge (Justice & Bode, 1988; Shedlovsky et al., 1988). Although the mutations vary in consequence for QKI function, they either impact the ability of QKI to dimerize (Chen & Richard, 1998; Ebersole et al., 1996), bind RNA, ablate all QKI function, or prevent the specific production of QKI‐5 (Cox et al., 1999), suggesting that functional QKI‐5 is vital for embryogenesis. A study of vascular development in the qkk2 mouse embryos uncovered defective blood vessel formation, which is the proposed mechanism for embryonic lethality (Noveroske et al., 2002). Similarly, mice homozygous for the qk I−1 mutation, which causes loss of QKI‐5, also died during midgestation from vascular defects (Bohnsack et al., 2006). The generation of a QKI null mouse reproduced the lethal defective vascular formation, and the cause of death was determined to be disturbed vascular remodeling in angiogenesis, presumably from loss of QKI expression in both vascular smooth muscle cells (VSMCs) and endothelial cells (Li et al., 2003).
QKI also has a role in the regulation of mature VSMCs where it promotes de‐differentiation to a non‐contractile phenotype (van der Veer et al., 2013). In injured blood vessels, where VSMC cells lack the ability to contract and blood vessels are narrowed, QKI expression is increased and promotes skipping of myocardin (Myocd) Exon 2a, removing the MEF‐interacting domain important for its function (van der Veer et al., 2013) (Figure 3). Myocd is known to regulate transcription of genes associated with VSMC differentiation, and its expression is sufficient to induce a contractile phenotype (Chen et al., 2002; Long et al., 2008). Furthermore, another RBP strongly associated with VSMC differentiation, was found to compete against QKI to promote Myocd Exon 2a inclusion, highlighting the significance of Myocd splicing in VSMC function (Nakagaki‐Silva et al., 2019). Treatment of mouse inducible pluripotent stem cells (iPSCs) with platelet‐derived growth factor BB to promote differentiation into VSMCs increases QKI‐6, but not QKI‐5, expression (Caines et al., 2019). Interestingly, QKI‐6 was localized in the nucleus in this context and found to regulate splicing of HDAC7 from a cytoplasmic (HDAC7u) to a nuclear (HDAC7s) isoform, which interacts with serum response factor and Myocd to promote VSMC differentiation (Margariti et al., 2009).
In endothelial cells, QKI has been shown to regulate both endothelial barrier function and angiogenesis. Mechanistically, QKI‐5 binds the 3′ UTRs of beta‐catenin and VE‐cadherin (CD144) mRNA to promote their translation and to STAT3 mRNA to promote its stability (Cochrane et al., 2017; De Bruin, Van Der Veer, et al., 2016), which impacts these cellular processes. QKI‐7 expression was found to be high in diabetic human iPSC‐derived endothelial cells, as well as high‐glucose treated mouse iPSC‐derived endothelial cells, with its overexpression mimicking features of endothelial dysfunction that are seen in diabetic patients (Yang et al., 2020). Expression of QKI‐7 was found to be controlled by a balance between the RBPs CUGBP Elav‐like family member 1 and heterogeneous nuclear ribonucleoprotein M (Yang et al., 2020). Interestingly, in contrast to QKI‐5, QKI‐7 expression caused destabilization of VE‐cadherin mRNA (as well as NLGN1 and TSG6), suggesting that these isoforms compete for binding of the VE‐cadherin 3′ UTR, leading to opposing outcomes (Yang et al., 2020). Recently, QKI was found to act as a promoter of angiogenesis in the tumor micro‐environment by stabilizing CCND1 mRNA and facilitating endothelial cell cycle progression (Azam et al., 2019). In this case, QKI levels in endothelial cells were modulated by the miR‐200 family (Azam et al., 2019; Gregory, 2019), which also regulates QKI activity and alternative splicing in the context of EMT (Pillman et al., 2018).
4.3. QKI in nonsmooth muscle cell differentiation
QKI also has an emerging role in cardiac and skeletal muscle differentiation. During myoblast differentiation into myotubes, QKI‐5 and QKI‐6 increase in expression (Fagg et al., 2017; Hall et al., 2013) and influence alternative splicing patterns, including Exon 9 of the Capzb, which regulates growth of actin filaments in muscle cells (Hall et al., 2013) (Figure 3). In competition with QKI, PTB promotes skipping of Exon 9, and an estimated 39% of alternative exons are controlled by either of these proteins during myogenesis (Hall et al., 2013). RNA‐seq and CLIP‐seq performed on C2C12 mouse myoblast cells uncovered multiple putative QKI splicing targets, as well as mRNAs that are either upregulated or downregulated in an isoform‐dependent manner by QKI‐6 and QKI‐7 (Fagg et al., 2017).
In addition, QKI has also been implicated in cardiomyocyte development. Human embryonic stem cells with QKI knocked out can still differentiate into early cardiogenic progenitor cells, but fail to differentiate into functional cardiomyocytes (Chen et al., 2021). In this context, QKI‐5 was found to be strongly upregulated in differentiated cardiomyocytes, and necessary for alternative splicing of genes associated with contractile physiology (Chen et al., 2021). QKI also controls the formation of muscle myofibrils by binding and stabilizing the muscle‐specific tropomyosin 3 transcript, promoting its accumulation (Bonnet et al., 2017). Collectively, these reports highlight important roles for QKI in the splicing and stabilization of mRNAs in nonsmooth muscle cells.
4.4. QKI and differentiation of monocytes
Several studies have implicated functions for QKI in the differentiation and maturation of monocytes into macrophages. In committed macrophage progenitors, QKI represses colony‐stimulating factor 1 receptor tyrosine kinase (CSF1R) mRNA (Fu et al., 2012), a key regulator of macrophage differentiation (Bourette & Rohrschneider, 2000; Pixley & Stanley, 2004). Intriguingly, transcriptional activation of QKI and CSF1R during macrophage progenitor maturation is both governed by CCAAT/enhancer‐binding protein α in macrophage progenitor cells (Fu et al., 2012). This keeps CSF1R transcriptionally active while posttranscriptionally repressed by QKI, suggesting that a disruption of QKI‐mediated repression of CSF1R is likely required before macrophage maturation can continue (Fu et al., 2012).
During monocyte differentiation into pro‐atherosclerotic macrophages, QKI causes changes expression of many transcripts enriched in monocyte–macrophage differentiation pathways, as well as cause changes in splicing of mRNAs including Adducin 3 (ADD3), Fc fragment of IgG receptor IIb (FCGR2B), very low density lipoprotein receptor, Protein bicaudal D homolog 2, and Kinesin family member 13A (De Bruin, Shiue, et al., 2016) (Figure 3). Two transcription factors associated with macrophage development, PU.1 (Zhang et al., 1994) and Kruppel‐like factor 4 (Shankman et al., 2015), were found to bind the QKI promotor region, suggesting a possible mechanism for how QKI is up‐regulated during macrophage differentiation (De Bruin, Shiue, et al., 2016; Feinberg et al., 2007; Gertz et al., 2013). Furthermore, QKI was shown to have functions in the macrophage lipid metabolism and the formation of foam cells (De Bruin, Shiue, et al., 2016). An important foam cell marker, Scavenger receptor A, was found to be directly targeted and repressed by QKI during monocyte differentiation (Wang et al., 2015). In addition, functions for QKI have also been implicated in mature macrophages (L. Wang, Zhai, et al., 2017; W. Wang, Zhai, et al., 2021), re‐enforcing the notion that QKI participates in the differentiation and maintenance of macrophage phenotypes.
4.5. QKI as a regulator of EMT
EMT is a dynamic cell differentiation process that facilitates embryonic development and occurs during cancer progression, aiding cancers cells to acquire invasive properties (Thiery et al., 2009). QKI was recently shown to be induced during EMT through loss of the epithelial miRNA family, miR‐200 (Pillman et al., 2018). The miR‐200 family is critical regulators of EMT, operating within a reciprocal feedback loop with the mesenchymal transcription factors Zinc finger E‐box‐binding homeobox (ZEB) 1 and ZEB2 to control cell plasticity (Burk et al., 2008; Gregory et al., 2008; Gregory et al., 2011). Numerous miR‐200 target mRNAs have been uncovered that are relevant to EMT, particularly mRNAs coding for proteins involved in the actin cytoskeleton (Bracken et al., 2014). Using large cancer datasets, QKI‐5 was found to be amongst the strongest targets negatively correlated with the miR‐200 family and miR‐375, both of which are repressed during EMT and allow for increased expression of QKI‐5 (Kim et al., 2019; Pillman et al., 2018).
During EMT, QKI‐5 causes changes in alternative splicing of many genes by directly binding within introns adjacent to the alternative exon or within the alternative exon itself (Pillman et al., 2018; Yang et al., 2016) (Figure 3). Similar to miR‐200 target genes, QKI‐5 was predominantly found to regulate genes that control the dynamics of the actin cytoskeleton (Pillman et al., 2018). This allows miR‐200 to directly manipulate the actin cytoskeleton by altering gene expression while indirectly affecting splicing through QKI‐5. Furthermore, QKI regulation of alternative splicing of the actin‐associated protein Filamin B (FLNB) was found to promote an intermediate mesenchymal state and enhance tumorigenicity in breast cancer models (J. Li, Choi, et al., 2018).
Functional studies in mesenchymal breast cancer cell lines show QKI enhances features of the mesenchymal phenotype including promoting an elongated cell morphology with increased cell migration and invasion, and mammosphere forming potential (J. Li, Choi, et al., 2018; Pillman et al., 2018). In other contexts, such as lung and oral squamous cell carcinoma, QKI appears to repress EMT‐associated properties, and in some cases these involve splicing of transcripts, such as NUMB and ADD3, that are promoted during EMT (Kim et al., 2019; J. Z. Wang, Fu, et al., 2021; S. Wang, Tong, et al., 2021; Zong et al., 2014). These apparent discrepancies likely reflect contextual differences, including relative levels of QKI isoform, target genes, and associated splicing factors, that are important for QKIs function in cancer cells.
5. CONCLUSION
Upon reviewing the literature, a consistent theme emerges where QKI isoforms promote or affect cell differentiation. Early studies in the mutant QKI mouse models demonstrated improper development of the vasculature and nervous system, with subsequent investigation revealing underlying changes in a range of cell differentiation processes may mediate these effects. The balance of QKI isoforms is an important determinant of QKI function, for example, during myelination cytoplasmic QKI isoforms influence nuclear export and local translation of MBP mRNA (Larocque et al., 2002; Zhao, Tian, et al., 2006). It is possible that similar mechanisms occur in other physiological contexts, where the stages of gene expression during differentiation and maturation are coordinated by QKI isoforms. Transcription factors associated with cell type‐specific differentiation, including QKI‐5 itself, could increase transcription of genes while QKI‐5 affects their alternative splicing and regulates their nuclear export. Following this, cytoplasmic expression of QKI‐6 and QKI‐7 could control cytoplasmic localization, stability, and translation of these transcripts. This could allow QKI to temporally control posttranscriptional regulation of genes driven by different transcription factors.
QKI is not the only RBP that regulates cell differentiation and altered expression of RBPs is often observed during differentiation. For instance, Muscleblind‐like splicing regulators 1/2 (Han et al., 2013), PTBP1/2 (Linares et al., 2015), NOVA (Leggere et al., 2016), fragile X mental retardation protein (Liu et al., 2018), and Pumilio (PUM) 1/2 (Zhang et al., 2017) have all been reported to have roles in differentiation including alternative splicing and mRNA localization. The reported overlap in function between RBPs during cell differentiation suggests they engage in a complex regulatory network. The full extent of how QKI cooperates and competes with other RBPs to affect cell differentiation is yet to be determined and warrants further exploration.
One important observation is the dynamic regulation of QKI expression through different stages of cell differentiation. During glial (Hardy, 1998; Takeuchi et al., 2020) and monocyte (De Bruin, Shiue, et al., 2016; Fu et al., 2012) differentiation, QKI is expressed highly in progenitor cell populations and is reduced after differentiation but is re‐expressed upon terminal differentiation when the cell gains its functional identity. These studies indicate QKI has distinct effects on gene expression in different stages of development and reinforces the role that QKI and other RBPs have in coordinating the complexity involved in cellular differentiation.
More research is likely to identify additional examples of alternate localization and local translation of mRNAs by the QKI isoforms. The complicated nature of autoregulation between the isoforms and the limited number of specialized techniques are available to detect these modes of regulation likely pose challenges. However, as cancer EMT‐associated features like migration and cell morphology are controlled through localization and local translation of target mRNAs (Liao et al., 2015; Mardakheh et al., 2015), QKI is a promising candidate regulator of this phenomenon, warranting further exploration.
It is also important to characterize the QKI‐specific posttranscriptional regulation across multiple cell types to find a core QKI‐specific signature. Although QKI is expressed in many different cell lineages, there is likely a consistent overlap in target transcripts between these contexts. For instance, the splicing event of ADD3 Exon 14 exclusion is regulated by QKI in monocyte to macrophage differentiation (De Bruin, Shiue, et al., 2016), breast and prostate cancer EMT (Pillman et al., 2018) and lung cancer (Wang, Fu, et al., 2021; Zong et al., 2014). It might be that ADD3 Exon 14 splicing is part of a core QKI‐directed transcriptomic signal that exists across multiple contexts of tumorigenesis and cellular differentiation alongside cell type‐specific regulation of genes like MBP or GFAP. Identifying such a signature and characterizing the functions of its constituent genes could lead to a better understanding of how posttranscriptional regulation and splicing broadly facilitates differentiation.
Uncovering the binding partners and co‐regulators that associate with QKI during the regulation of alternative splicing will also be a fruitful area of exploration. Recent work characterizing alternative splicing events during development uncovered distinct patterns for putative QKI targets during development of the brain and heart (Mazin et al., 2021). This highlights the important and complex role that QKI plays in regulation of tissue development and differentiation through alternative splicing. Elucidating the mechanisms governing this process, including identifying RBPs that co‐regulate target genes with QKI (e.g., RNA‐binding fox‐1 homolog 1/2; J. Li, Choi, et al., 2018), or antagonize (e.g., PTB; Hall et al., 2013) will help to generate a mechanistic model of how QKI directly regulates splicing.
QKI is consistently identified as regulating cell fate decisions, playing a role in cellular differentiation in development or in cancer. The balance of the QKI isoforms during oligodendrocyte appears to be tightly and temporally regulated for healthy maturation of myelin producing cells (Larocque et al., 2002; Larocque et al., 2005; Takeuchi et al., 2020; Thangaraj et al., 2017). Studies on cellular differentiation using single‐cell transcriptomics have uncovered the stochastic nature of cell fate decisions (Zhang et al., 2019). As most transcriptional analyses rely on bulk methods, which provide a gene expression average that ignores intratissue heterogeneity, it is possible that the majority of QKI's function in differentiation and cancer has not yet been identified. Future studies using single‐cell and spatial transcriptomics to uncover cell type and tissue niche‐specific expression will help further unravel functions of QKI isoforms.
AUTHOR CONTRIBUTIONS
Daniel Neumann: Conceptualization (equal); writing – original draft (lead); writing – review and editing (equal). Gregory Goodall: Writing – review and editing (equal). Philip Gregory: Conceptualization (equal); funding acquisition (lead); writing – original draft (supporting); writing – review and editing (equal).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
RELATED WIREs ARTICLES
Emerging functions of the Quaking RNA‐binding proteins and link to human diseases
Alternative splicing programming of axon formation
Classification and function of RNA‐protein interactions
Making sense of mRNA landscapes: Translation control in neurodevelopment
ACKNOWLEDGMENTS
This work was supported by grants from the National Health and Medical Research Council of Australia to Philip A. Gregory and Gregory J. Goodall (1128479, 1164669) and a grant from the Hospital Research Foundation to Philip A. Gregory. Philip A. Gregory is supported by a Principal Cancer Research Fellowship (PRF2518) awarded by Cancer Council of South Australia's Beat Cancer project on behalf of its donors, the state Government through the Department of Health, and the Australian Government through the Medical Research Future Fund.
Open access publishing facilitated by University of South Australia, as part of the Wiley – University of South Australia agreement via the Council of Australian University Librarians. [Correction added on 17 May 2022, after first online publication: CAUL funding statement has been added.]
Neumann, D. P. , Goodall, G. J. , & Gregory, P. A. (2022). The Quaking RNA‐binding proteins as regulators of cell differentiation. Wiley Interdisciplinary Reviews: RNA, 13(6), e1724. 10.1002/wrna.1724
Edited by: Jeff Wilusz, Editor‐in‐Chief
Funding information Cancer Council South Australia, Grant/Award Number: PRF2518; Hospital Research Foundation; National Health and Medical Research Council, Grant/Award Numbers: 1128479, 1164669
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study
REFERENCES
- Åberg, K. , Saetre, P. , Jareborg, N. , & Jazin, E. (2006). Human QKI, a potential regulator of mRNA expression of human oligodendrocyte‐related genes involved in schizophrenia. Proceedings of the National Academy of Sciences, 103(19), 7482–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainger, K. , Avossa, D. , Morgan, F. , Hill, S. J. , Barry, C. , Barbarese, E. , & Carson, J. H. (1993). Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. The Journal of Cell Biology, 123(2), 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artzt, K. , & Wu, J. I. (2010). Star Trek. In Post‐transcriptional regulation by STAR proteins (pp. 1–24). Springer. [Google Scholar]
- Au, E. , Richter, M. W. , Vincent, A. J. , Tetzlaff, W. , Aebersold, R. , Sage, E. H. , & Roskams, A. J. (2007). SPARC from olfactory ensheathing cells stimulates Schwann cells to promote neurite outgrowth and enhances spinal cord repair. Journal of Neuroscience, 27(27), 7208–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam, S. H. , Porrello, A. , Harrison, E. B. , Leslie, P. L. , Liu, X. , Waugh, T. A. , Belanger, A. , Mangala, L. S. , Lopez‐Berestein, G. , & Wilson, H. L. (2019). Quaking orchestrates a post‐transcriptional regulatory network of endothelial cell cycle progression critical to angiogenesis and metastasis. Oncogene, 38(26), 5191–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat, M. , Benassy, M.‐N. , Vincensini, L. , Soares, S. , Fassier, C. , Propst, F. , Andrieux, A. , von Boxberg, Y. , & Nothias, F. (2016). The GSK3–MAP1B pathway controls neurite branching and microtubule dynamics. Molecular and Cellular Neuroscience, 72, 9–21. [DOI] [PubMed] [Google Scholar]
- Bennett, W. I. , Gall, A. M. , Southard, J. L. , & Sidman, R. L. (1971). Abnormal spermiogenesis in quaking, a myelin‐deficient mutant mouse. Biology of Reproduction, 5(1), 30–58. [DOI] [PubMed] [Google Scholar]
- Berglund, J. A. , Abovich, N. , & Rosbash, M. (1998). A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes & Development, 12(6), 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack, B. L. , Lai, L. , Northrop, J. L. , Justice, M. J. , & Hirschi, K. K. (2006). Visceral endoderm function is regulated by quaking and required for vascular development. Genesis, 44(2), 93–104. [DOI] [PubMed] [Google Scholar]
- Bonnet, A. , Lambert, G. , Ernest, S. , Dutrieux, F. X. , Coulpier, F. , Lemoine, S. , Lobbardi, R. , & Rosa, F. M. (2017). Quaking RNA‐binding proteins control early myofibril formation by modulating tropomyosin. Developmental Cell, 42(5), 527–541.e524. [DOI] [PubMed] [Google Scholar]
- Bourette, R. P. , & Rohrschneider, L. R. (2000). Early events in M‐CSF receptor signaling. Growth Factors, 17(3), 155–166. [DOI] [PubMed] [Google Scholar]
- Bracken, C. P. , Li, X. , Wright, J. A. , Lawrence, D. M. , Pillman, K. A. , Salmanidis, M. , Anderson, M. A. , Dredge, B. K. , Gregory, P. A. , Tsykin, A. , Neilsen, C. , Thomson, D. W. , Bert, A. G. , Leerberg, J. M. , Yap, A. S. , Jensen, K. B. , Khew‐Goodall, Y. , & Goodall, G. J. (2014). Genome‐wide identification of miR‐200 targets reveals a regulatory network controlling cell invasion. The EMBO Journal, 33(18), 2040–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseau, J.‐P. , Lucier, J.‐F. , Nwilati, H. , Thibault, P. , Garneau, D. , Gendron, D. , Durand, M. , Couture, S. , Lapointe, E. , & Prinos, P. (2014). Tumor microenvironment–associated modifications of alternative splicing. RNA, 20(2), 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk, U. , Schubert, J. , Wellner, U. , Schmalhofer, O. , Vincan, E. , Spaderna, S. , & Brabletz, T. (2008). A reciprocal repression between ZEB1 and members of the miR‐200 family promotes EMT and invasion in cancer cells. EMBO Reports, 9(6), 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caines, R. , Cochrane, A. , Kelaini, S. , Vila‐Gonzalez, M. , Yang, C. , Eleftheriadou, M. , Moez, A. , Stitt, A. W. , Zeng, L. , & Grieve, D. J. (2019). The RNA‐binding protein QKI controls alternative splicing in vascular cells, producing an effective model for therapy. Journal of Cell Science, 132(16), jcs230276. [DOI] [PubMed] [Google Scholar]
- Casaccia‐Bonnefil, P. , Hardy, R. , Teng, K. , Levine, J. , Koff, A. , & Chao, M. (1999). Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development, 126(18), 4027–4037. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Kitchen, C. M. , Streb, J. W. , & Miano, J. M. (2002). Myocardin: A component of a molecular switch for smooth muscle differentiation. Journal of Molecular and Cellular Cardiology, 34(10), 1345–1356. [DOI] [PubMed] [Google Scholar]
- Chen, T. , Damaj, B. B. , Herrera, C. , Lasko, P. , & Richard, S. (1997). Self‐association of the single‐KH‐domain family members Sam68, GRP33, GLD‐1, and Qk1: Role of the KH domain. Molecular and Cellular Biology, 17(10), 5707–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. , & Richard, S. (1998). Structure‐function analysis of Qk1: A lethal point mutation in mouse quaking prevents homodimerization. Molecular and Cellular Biology, 18(8), 4863–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Liu, Y. , Xu, C. , Ba, L. , Liu, Z. , Li, X. , Huang, J. , Simpson, E. , Gao, H. , & Cao, D. (2021). QKI is a critical pre‐mRNA alternative splicing regulator of cardiac myofibrillogenesis and contractile function. Nature Communications, 12(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Tian, D. , Ku, L. , Osterhout, D. J. , & Feng, Y. (2007). The selective RNA‐binding protein quaking I (QKI) is necessary and sufficient for promoting oligodendroglia differentiation. Journal of Biological Chemistry, 282(32), 23553–23560. [DOI] [PubMed] [Google Scholar]
- Côté, J. , Boisvert, F. O.‐M. , Boulanger, M.‐C. , Bedford, M. T. , & Richard, S. (2003). Sam68 RNA binding protein is an in vivo substrate for protein arginine N‐methyltransferase 1. Molecular Biology of the Cell, 14(1), 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane, A. , Kelaini, S. , Tsifaki, M. , Bojdo, J. , Vilà‐González, M. , Drehmer, D. , Caines, R. , Magee, C. , Eleftheriadou, M. , & Hu, Y. (2017). Quaking is a key regulator of endothelial cell differentiation, neovascularization, and angiogenesis. Stem Cells, 35(4), 952–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman, D. R. , Kreibich, G. , Frey, A. B. , & Sabatini, D. D. (1982). Synthesis and incorporation of myelin polypeptides into CNS myelin. The Journal of Cell Biology, 95(2), 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn, S. J. , Pillman, K. A. , Toubia, J. , Conn, V. M. , Salmanidis, M. , Phillips, C. A. , Roslan, S. , Schreiber, A. W. , Gregory, P. A. , & Goodall, G. J. (2015). The RNA binding protein quaking regulates formation of circRNAs. Cell, 160(6), 1125–1134. [DOI] [PubMed] [Google Scholar]
- Cox, R. D. , Hugill, A. , Shedlovsky, A. , Noveroske, J. K. , Best, S. , Justice, M. J. , Lehrach, H. , & Dove, W. F. (1999). Contrasting effects of ENU induced embryonic lethal mutations of the quaking gene. Genomics, 57(3), 333–341. [DOI] [PubMed] [Google Scholar]
- Darbelli, L. , Choquet, K. , Richard, S. , & Kleinman, C. L. (2017). Transcriptome profiling of mouse brains with qkI‐deficient oligodendrocytes reveals major alternative splicing defects including self‐splicing. Scientific Reports, 7(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbelli, L. , Vogel, G. , Almazan, G. , & Richard, S. (2016). Quaking regulates neurofascin 155 expression for myelin and axoglial junction maintenance. Journal of Neuroscience, 36(14), 4106–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner, G. M. , Brümmer, A. , Tocchini, C. , Gerhardy, S. , Ciosk, R. , Zavolan, M. , & Allain, F. H.‐T. (2014). Structural and functional implications of the QUA2 domain on RNA recognition by GLD‐1. Nucleic Acids Research, 42(12), 8092–8105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruin, R. G. , Shiue, L. , Prins, J. , De Boer, H. C. , Singh, A. , Fagg, W. S. , Van Gils, J. M. , Duijs, J. M. , Katzman, S. , & Kraaijeveld, A. O. (2016). Quaking promotes monocyte differentiation into pro‐atherogenic macrophages by controlling pre‐mRNA splicing and gene expression. Nature Communications, 7(1), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruin, R. G. , Van Der Veer, E. P. , Prins, J. , Lee, D. H. , Dane, M. J. , Zhang, H. , Roeten, M. K. , Bijkerk, R. , De Boer, H. C. , & Rabelink, T. J. (2016). The RNA‐binding protein quaking maintains endothelial barrier function and affects VE‐cadherin and β‐catenin protein expression. Scientific Reports, 6(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miguel, F. J. , Pajares, M. J. , Martínez‐Terroba, E. , Ajona, D. , Morales, X. , Sharma, R. D. , Pardo, F. J. , Rouzaut, A. , Rubio, A. , & Montuenga, L. M. (2016). A large‐scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Molecular Oncology, 10(9), 1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doukhanine, E. , Gavino, C. , Haines, J. D. , Almazan, G. , & Richard, S. (2010). The QKI‐6 RNA binding protein regulates actin‐interacting protein‐1 mRNA stability during oligodendrocyte differentiation. Molecular Biology of the Cell, 21(17), 3029–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, B. , Fero, M. L. , Roberts, J. M. , & Raff, M. C. (1998). p27Kip1 alters the response of cells to mitogen and is part of a cell‐intrinsic timer that arrests the cell cycle and initiates differentiation. Current Biology, 8(8), 431–440. [DOI] [PubMed] [Google Scholar]
- Dutertre, M. , Chakrama, F. Z. , Combe, E. , Desmet, F.‐O. , Mortada, H. , Espinoza, M. P. , Gratadou, L. , & Auboeuf, D. (2014). A recently evolved class of alternative 3′‐terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nature Communications, 5(1), 1–12. [DOI] [PubMed] [Google Scholar]
- Ebersole, T. , Rho, O. , & Artzt, K. (1992). The proximal end of mouse chromosome 17: New molecular markers identify a deletion associated with quakingviable. Genetics, 131(1), 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole, T. A. , Chen, Q. , Justice, M. J. , & Artzt, K. (1996). The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nature Genetics, 12(3), 260–265. [DOI] [PubMed] [Google Scholar]
- Fagg, W. S. , Liu, N. , Fair, J. H. , Shiue, L. , Katzman, S. , Donohue, J. P. , & Ares, M. (2017). Autogenous cross‐regulation of Quaking mRNA processing and translation balances Quaking functions in splicing and translation. Genes & Development, 31(18), 1894–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg, M. W. , Wara, A. K. , Cao, Z. , Lebedeva, M. A. , Rosenbauer, F. , Iwasaki, H. , Hirai, H. , Katz, J. P. , Haspel, R. L. , & Gray, S. (2007). The Kruppel‐like factor KLF4 is a critical regulator of monocyte differentiation. The EMBO Journal, 26(18), 4138–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, H. , Yang, G. , Wei, M. , Liu, L. , Jin, L. , Lu, X. , Wang, L. , Shen, L. , Zhang, J. , & Lu, H. (2012). The RNA‐binding protein QKI5 is a direct target of C/EBPα and delays macrophage differentiation. Molecular Biology of the Cell, 23(9), 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau, A. , & Richard, S. (2005). Target RNA motif and target mRNAs of the Quaking STAR protein. Nature Structural & Molecular Biology, 12(8), 691–698. [DOI] [PubMed] [Google Scholar]
- Galarneau, A. , & Richard, S. (2009). The STAR RNA binding proteins GLD‐1, QKI, Sam68 and SLM‐2 bind bipartite RNA motifs. BMC Molecular Biology, 10(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz, J. , Savic, D. , Varley, K. E. , Partridge, E. C. , Safi, A. , Jain, P. , Cooper, G. M. , Reddy, T. E. , Crawford, G. E. , & Myers, R. M. (2013). Distinct properties of cell‐type‐specific and shared transcription factor binding sites. Molecular Cell, 52(1), 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goering, R. , Engel, K. L. , Gillen, A. E. , Fong, N. , Bentley, D. L. , & Taliaferro, J. M. (2021). LABRAT reveals association of alternative polyadenylation with transcript localization, RNA binding protein expression, transcription speed, and cancer survival. BMC Genomics, 22(1), 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, P. A. (2019). The miR‐200‐Quaking axis functions in tumour angiogenesis. Oncogene, 38(41), 6767–6769. [DOI] [PubMed] [Google Scholar]
- Gregory, P. A. , Bert, A. G. , Paterson, E. L. , Barry, S. C. , Tsykin, A. , Farshid, G. , Vadas, M. A. , Khew‐Goodall, Y. , & Goodall, G. J. (2008). The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biology, 10(5), 593–601. [DOI] [PubMed] [Google Scholar]
- Gregory, P. A. , Bracken, C. P. , Smith, E. , Bert, A. G. , Wright, J. A. , Roslan, S. , Morris, M. , Wyatt, L. , Farshid, G. , & Lim, Y.‐Y. (2011). An autocrine TGF‐β/ZEB/miR‐200 signaling network regulates establishment and maintenance of epithelial‐mesenchymal transition. Molecular Biology of the Cell, 22(10), 1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Jiang, T. , Lian, C. , Wang, H. , Zheng, Q. , & Ma, H. (2014). QKI deficiency promotes FoxO1 mediated nitrosative stress and endoplasmic reticulum stress contributing to increased vulnerability to ischemic injury in diabetic heart. Journal of Molecular and Cellular Cardiology, 75, 131–140. [DOI] [PubMed] [Google Scholar]
- Hafner, M. , Landthaler, M. , Burger, L. , Khorshid, M. , Hausser, J. , Berninger, P. , Rothballer, A. , Ascano, M., Jr. , Jungkamp, A.‐C. , & Munschauer, M. (2010). Transcriptome‐wide identification of RNA‐binding protein and microRNA target sites by PAR‐CLIP. Cell, 141(1), 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, M. P. , Nagel, R. J. , Fagg, W. S. , Shiue, L. , Cline, M. S. , Perriman, R. J. , Donohue, J. P. , & Ares, M. (2013). Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA, 19(5), 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, H. , Irimia, M. , Ross, P. J. , Sung, H. K. , Alipanahi, B. , David, L. , Golipour, A. , Gabut, M. , Michael, I. P. , Nachman, E. N. , Wang, E. , Trcka, D. , Thompson, T. , O'Hanlon, D. , Slobodeniuc, V. , Barbosa‐Morais, N. L. , Burge, C. B. , Moffat, J. , Frey, B. J. , … Blencowe, B. J. (2013). MBNL proteins repress ES‐cell‐specific alternative splicing and reprogramming. Nature, 498(7453), 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, R. J. (1998). QKI expression is regulated during neuron‐glial cell fate decisions. Journal of Neuroscience Research, 54(1), 46–57. [DOI] [PubMed] [Google Scholar]
- Hardy, R. J. , Loushin, C. L. , Friedrich, V. L., Jr. , Chen, Q. , Ebersole, T. A. , Lazzarini, R. A. , & Artzt, K. (1996). Neural cell type‐specific expression of QKI proteins is altered in quakingviable mutant mice. Journal of Neuroscience, 16(24), 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa‐Yano, Y. , Suyama, S. , Nogami, M. , Yugami, M. , Koya, I. , Furukawa, T. , Zhou, L. , Abe, M. , Sakimura, K. , & Takebayashi, H. (2017). An RNA‐binding protein, Qki5, regulates embryonic neural stem cells through pre‐mRNA processing in cell adhesion signaling. Genes & Development, 31(18), 1910–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo, H. , Yashiro, Y. , Noda, Y. , Ogami, K. , Yamagishi, R. , Okada, S. , Hoshino, S.‐I. , & Suzuki, T. (2020). The RNA‐binding protein QKI‐7 recruits the poly (A) polymerase GLD‐2 for 3′ adenylation and selective stabilization of microRNA‐122. Journal of Biological Chemistry, 295(2), 390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier, J.‐M. , Ibáñez, P. , Mena, M. A. , Abbas, N. , Cohen‐Salmon, C. , Bohme, G. A. , Laville, M. , Pratt, J. , Corti, O. , & Pradier, L. (2003). Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Human Molecular Genetics, 12(18), 2277–2291. [DOI] [PubMed] [Google Scholar]
- Ji, S. , Doucette, J. R. , & Nazarali, A. J. (2011). Sirt2 is a novel in vivo downstream target of Nkx2. 2 and enhances oligodendroglial cell differentiation. Journal of Molecular Cell Biology, 3(6), 351–359. [DOI] [PubMed] [Google Scholar]
- Justice, M. J. , & Bode, V. C. (1988). Three ENU‐induced alleles of the murine quaking locus are recessive embryonic lethal mutations. Genetics Research, 51(2), 95–102. [DOI] [PubMed] [Google Scholar]
- Justice, M. J. , & Hirschi, K. K. (2010). The role of quaking in mammalian embryonic development. In Post‐transcriptional regulation by STAR proteins (pp. 82–92). Springer. [DOI] [PubMed] [Google Scholar]
- Kim, E. J. , Kim, J. S. , Lee, S. , Lee, H. , Yoon, J. S. , Hong, J. H. , Chun, S. H. , Sun, D. S. , Won, H. S. , & Hong, S. A. (2019). QKI, a miR‐200 target gene, suppresses epithelial‐to‐mesenchymal transition and tumor growth. International Journal of Cancer, 145(6), 1585–1595. [DOI] [PubMed] [Google Scholar]
- Kondo, T. , Furuta, T. , Mitsunaga, K. , Ebersole, T. A. , Shichiri, M. , Wu, J. , Artzt, K. , Yamamura, K.‐I. , & Abe, K. (1999). Genomic organization and expression analysis of the mouse qkI locus. Mammalian Genome, 10(7), 662–669. [DOI] [PubMed] [Google Scholar]
- Larocque, D. , Galarneau, A. , Liu, H.‐N. , Scott, M. , Almazan, G. , & Richard, S. (2005). Protection of p27 Kip1 mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nature Neuroscience, 8(1), 27–33. [DOI] [PubMed] [Google Scholar]
- Larocque, D. , Pilotte, J. , Chen, T. , Cloutier, F. , Massie, B. , Pedraza, L. , Couture, R. , Lasko, P. , Almazan, G. , & Richard, S. (2002). Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron, 36(5), 815–829. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Villarreal, O. D. , Chen, X. , Zandee, S. , Young, Y. K. , Torok, C. , Lamarche‐Vane, N. , Prat, A. , Rivest, S. , & Gosselin, D. (2020). QUAKING regulates microexon alternative splicing of the rho GTPase pathway and controls microglia homeostasis. Cell Reports, 33(13), 108560. [DOI] [PubMed] [Google Scholar]
- Lee, N. , Moss, W. N. , Yario, T. A. , & Steitz, J. A. (2015). EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell, 160(4), 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggere, J. C. , Saito, Y. , Darnell, R. B. , Tessier‐Lavigne, M. , Junge, H. J. , & Chen, Z. (2016). NOVA regulates Dcc alternative splicing during neuronal migration and axon guidance in the spinal cord. eLife, 5, e14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Choi, P. S. , Chaffer, C. L. , Labella, K. , Hwang, J. H. , Giacomelli, A. O. , Kim, J. W. , Ilic, N. , Doench, J. G. , & Ly, S. H. (2018). An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. eLife, 7, e37184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Lin, C. , Zhang, J. , Tao, H. , Liu, H. , Yuan, G. , & Chen, Z. (2018). Quaking promotes the odontoblastic differentiation of human dental pulp stem cells. Journal of Cellular Physiology, 233(9), 7292–7304. [DOI] [PubMed] [Google Scholar]
- Li, Z. , Takakura, N. , Oike, Y. , Imanaka, T. , Araki, K. , Suda, T. , Kaname, T. , Kondo, T. , Abe, K. , & Yamamura, K. (2003). Defective smooth muscle development in qkI‐deficient mice. Development, Growth & Differentiation, 45(5–6), 449–462. [DOI] [PubMed] [Google Scholar]
- Li, Z. , Zhang, Y. , Li, D. , & Feng, Y. (2000). Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA‐binding proteins. Journal of Neuroscience, 20(13), 4944–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, G. , Mingle, L. , Van De Water, L. , & Liu, G. (2015). Control of cell migration through mRNA localization and local translation. Wiley Interdisciplinary Reviews: RNA, 6(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares, A. J. , Lin, C.‐H. , Damianov, A. , Adams, K. L. , Novitch, B. G. , & Black, D. L. (2015). The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. eLife, 4, e09268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Li, Y. , Stackpole, E. E. , Novak, A. , Gao, Y. , Zhao, Y. , Zhao, X. , & Richter, J. D. (2018). Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proceedings of the National Academy of Sciences, 115(48), E11397–E11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Luyten, I. , Bottomley, M. J. , Messias, A. C. , Houngninou‐Molango, S. , Sprangers, R. , Zanier, K. , Krämer, A. , & Sattler, M. (2001). Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science, 294(5544), 1098–1102. [DOI] [PubMed] [Google Scholar]
- Lock, P. , Fumagalli, S. , Polakis, P. , McCormick, F. , & Courtneidge, S. A. (1996). The human p62 cDNA encodes Sam68 and not the RasGAP‐associated p62 protein. Cell, 84(1), 23–24. [DOI] [PubMed] [Google Scholar]
- Long, X. , Bell, R. D. , Gerthoffer, W. T. , Zlokovic, B. V. , & Miano, J. M. (2008). Myocardin is sufficient for a smooth muscle‐like contractile phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(8), 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzetti, D. , Antalffy, B. , Vogel, H. , Noveroske, J. , Armstrong, D. , & Justice, M. (2004). The neurological mutant quaking viable is Parkin deficient. Mammalian Genome, 15(3), 210–217. [DOI] [PubMed] [Google Scholar]
- Lu, Z. , Ku, L. , Chen, Y. , & Feng, Y. (2005). Developmental abnormalities of myelin basic protein expression in fyn knock‐out brain reveal a role of Fyn in posttranscriptional regulation. Journal of Biological Chemistry, 280(1), 389–395. [DOI] [PubMed] [Google Scholar]
- Mandler, M. D. , Ku, L. , & Feng, Y. (2014). A cytoplasmic quaking I isoform regulates the hnRNP F/H‐dependent alternative splicing pathway in myelinating glia. Nucleic Acids Research, 42(11), 7319–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardakheh, F. K. , Paul, A. , Kümper, S. , Sadok, A. , Paterson, H. , Mccarthy, A. , Yuan, Y. , & Marshall, C. J. (2015). Global analysis of mRNA, translation, and protein localization: Local translation is a key regulator of cell protrusions. Developmental Cell, 35(3), 344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margariti, A. , Xiao, Q. , Zampetaki, A. , Zhang, Z. , Li, H. , Martin, D. , Hu, Y. , Zeng, L. , & Xu, Q. (2009). Splicing of HDAC7 modulates the SRF‐myocardin complex during stem‐cell differentiation towards smooth muscle cells. Journal of Cell Science, 122(4), 460–470. [DOI] [PubMed] [Google Scholar]
- Mazin, P. V. , Khaitovich, P. , Cardoso‐Moreira, M. , & Kaessmann, H. (2021). Alternative splicing during mammalian organ development. Nature Genetics, 53(6), 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov, A. , Denisov, S. , Gress, A. , Kalinina, O. V. , & Pervouchine, D. D. (2021). An extended catalogue of tandem alternative splice sites in human tissue transcriptomes. PLoS Computational Biology, 17(4), e1008329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagaki‐Silva, E. E. , Gooding, C. , Llorian, M. , Jacob, A. G. , Richards, F. , Buckroyd, A. , Sinha, S. , & Smith, C. W. (2019). Identification of RBPMS as a mammalian smooth muscle master splicing regulator via proximity of its gene with super‐enhancers. eLife, 8, e46327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, M. , & Berget, S. M. (1991). Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes & Development, 5(11), 2086–2095. [DOI] [PubMed] [Google Scholar]
- Niwa, M. , Rose, S. D. , & Berget, S. M. (1990). In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes & Development, 4(9), 1552–1559. [DOI] [PubMed] [Google Scholar]
- North, B. J. , Marshall, B. L. , Borra, M. T. , Denu, J. M. , & Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+‐dependent tubulin deacetylase. Molecular Cell, 11(2), 437–444. [DOI] [PubMed] [Google Scholar]
- Noveroske, J. K. , Lai, L. , Gaussin, V. , Northrop, J. L. , Nakamura, H. , Hirschi, K. K. , & Justice, M. J. (2002). Quaking is essential for blood vessel development. Genesis, 32(3), 218–230. [DOI] [PubMed] [Google Scholar]
- Pedersen, S. K. , Mitchell, S. M. , Graham, L. D. , McEvoy, A. , Thomas, M. L. , Baker, R. T. , Ross, J. P. , Xu, Z.‐Z. , Ho, T. , & LaPointe, L. C. (2014). CAHM, a long non‐coding RNA gene hypermethylated in colorectal neoplasia. Epigenetics, 9(8), 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillman, K. A. , Phillips, C. A. , Roslan, S. , Toubia, J. , Dredge, B. K. , Bert, A. G. , Lumb, R. , Neumann, D. P. , Li, X. , Conn, S. J. , Liu, D. , Bracken, C. P. , Lawrence, D. M. , Stylianou, N. , Schreiber, A. W. , Tilley, W. D. , Hollier, B. G. , Khew‐Goodall, Y. , Selth, L. A. , … Gregory, P. A. (2018). miR‐200/375 control epithelial plasticity‐associated alternative splicing by repressing the RNA‐binding protein Quaking. The EMBO Journal, 37(13), e99016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte, J. , Larocque, D. , & Richard, S. (2001). Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes & Development, 15(7), 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pixley, F. J. , & Stanley, E. R. (2004). CSF‐1 regulation of the wandering macrophage: Complexity in action. Trends in Cell Biology, 14(11), 628–638. [DOI] [PubMed] [Google Scholar]
- Pompon, J. , & Garcia‐Blanco, M. A. (2015). RNA: Jack of all trades and master of all. Cell, 160(4), 579–580. [DOI] [PubMed] [Google Scholar]
- Pozniak, C. D. , Langseth, A. J. , Dijkgraaf, G. J. , Choe, Y. , Werb, Z. , & Pleasure, S. J. (2010). Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proceedings of the National Academy of Sciences, 107(50), 21795–21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska, K. J. , Halvardson, J. , Reinius, B. , Lindholm Carlström, E. , Emilsson, L. , Feuk, L. , & Jazin, E. (2013). RNA‐binding protein QKI regulates Glial fibrillary acidic protein expression in human astrocytes. Human Molecular Genetics, 22(7), 1373–1382. [DOI] [PubMed] [Google Scholar]
- Ren, J. , Dai, C. , Zhou, X. , Barnes, J. A. , Chen, X. , Wang, Y. , Yuan, L. , Shingu, T. , Heimberger, A. B. , & Chen, Y. (2021). Qki is an essential regulator of microglial phagocytosis in demyelination. Journal of Experimental Medicine, 218(1), e20190348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Y. , Huo, Y. , Li, W. , He, M. , Liu, S. , Yang, J. , Zhao, H. , Xu, L. , Guo, Y. , & Si, Y. (2021). A global screening identifies chromatin‐enriched RNA‐binding proteins and the transcriptional regulatory activity of QKI5 during monocytic differentiation. Genome Biology, 22(1), 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, S. , Yu, D. , Blumer, K. J. , Hausladen, D. , Olszowy, M. W. , Connelly, P. A. , & Shaw, A. S. (1995). Association of p62, a multifunctional SH2‐and SH3‐domain‐binding protein, with src family tyrosine kinases, Grb2, and phospholipase C gamma‐1. Molecular and Cellular Biology, 15(1), 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo, F. , & Martinson, H. G. (2008). Functional coupling of last‐intron splicing and 3′‐end processing to transcription in vitro: The poly (A) signal couples to splicing before committing to cleavage. Molecular and Cellular Biology, 28(2), 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberson, B. L. , Cote, G. J. , & Berget, S. M. (1990). Exon definition may facilitate splice site selection in RNAs with multiple exons. Molecular and Cellular Biology, 10(1), 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccomanno, L. , Loushin, C. , Jan, E. , Punkay, E. , Artzt, K. , & Goodwin, E. B. (1999). The STAR protein QKI‐6 is a translational repressor. Proceedings of the National Academy of Sciences, 96(22), 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakers, K. , Lake, A. M. , Khazanchi, R. , Ouwenga, R. , Vasek, M. J. , Dani, A. , & Dougherty, J. D. (2017). Astrocytes locally translate transcripts in their peripheral processes. Proceedings of the National Academy of Sciences, 114(19), E3830–E3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakers, K. , Liu, Y. , Llaci, L. , Lee, S. M. , Vasek, M. J. , Rieger, M. A. , Brophy, S. , Tycksen, E. , Lewis, R. , & Maloney, S. E. (2021). Loss of Quaking RNA binding protein disrupts the expression of genes associated with astrocyte maturation in mouse brain. Nature Communications, 12(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman, L. S. , Gomez, D. , Cherepanova, O. A. , Salmon, M. , Alencar, G. F. , Haskins, R. M. , Swiatlowska, P. , Newman, A. A. , Greene, E. S. , & Straub, A. C. (2015). KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine, 21(6), 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlovsky, A. , King, T. R. , & Dove, W. F. (1988). Saturation germ line mutagenesis of the murine t region including a lethal allele at the quaking locus. Proceedings of the National Academy of Sciences, 85(1), 180–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, S. , Zhou, H. , He, C. , Wei, Y. , Wang, Y. , Shingu, T. , Zeng, A. , Wang, S. , Zhou, X. , & Li, H. (2021). Qki activates Srebp2‐mediated cholesterol biosynthesis for maintenance of eye lens transparency. Nature Communications, 12(1), 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman, R. L. , Dickie, M. M. , & Appel, S. H. (1964). Mutant mice (quaking and jimpy) with deficient myelination in the central nervous system. Science, 144(3616), 309–311. [DOI] [PubMed] [Google Scholar]
- Takeuchi, A. , Takahashi, Y. , Iida, K. , Hosokawa, M. , Irie, K. , Ito, M. , Brown, J. , Ohno, K. , Nakashima, K. , & Hagiwara, M. (2020). Identification of Qk as a glial precursor cell marker that governs the fate specification of neural stem cells to a glial cell lineage. Stem Cell Reports, 15(4), 883–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro, J. M. , Vidaki, M. , Oliveira, R. , Olson, S. , Zhan, L. , Saxena, T. , Wang, E. T. , Graveley, B. R. , Gertler, F. B. , & Swanson, M. S. (2016). Distal alternative last exons localize mRNAs to neural projections. Molecular Cell, 61(6), 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. M. , Beesley, J. , Grinspan, J. , Seth, P. , Kamholz, J. , & Cambi, F. (2000). Cell cycle arrest induced by ectopic expression of p27 is not sufficient to promote oligodendrocyte differentiation. Journal of Cellular Biochemistry, 76(2), 270–279. [DOI] [PubMed] [Google Scholar]
- Teplova, M. , Hafner, M. , Teplov, D. , Essig, K. , Tuschl, T. , & Patel, D. J. (2013). Structure–function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes & Development, 27(8), 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangaraj, M. P. , Furber, K. L. , Gan, J. K. , Ji, S. , Sobchishin, L. , Doucette, J. R. , & Nazarali, A. J. (2017). RNA‐binding protein quaking stabilizes Sirt2 mRNA during oligodendroglial differentiation. Journal of Biological Chemistry, 292(13), 5166–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery, J. P. , Acloque, H. , Huang, R. Y. , & Nieto, M. A. (2009). Epithelial‐mesenchymal transitions in development and disease. Cell, 139(5), 871–890. [DOI] [PubMed] [Google Scholar]
- Tian, B. , & Manley, J. L. (2017). Alternative polyadenylation of mRNA precursors. Nature Reviews Molecular Cell Biology, 18(1), 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikoo, R. , Osterhout, D. J. , Casaccia‐Bonnefil, P. , Seth, P. , Koff, A. , & Chao, M. V. (1998). Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell‐cycle growth arrest. Journal of Neurobiology, 36(3), 431–440. [PubMed] [Google Scholar]
- Timney, B. L. , Raveh, B. , Mironska, R. , Trivedi, J. M. , Kim, S. J. , Russel, D. , Wente, S. R. , Sali, A. , & Rout, M. P. (2016). Simple rules for passive diffusion through the nuclear pore complex. Journal of Cell Biology, 215(1), 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, B. , Moench, T. , Pulley, M. , Barbosa, E. , Tennekoon, G. , & Griffin, J. (1987). Spatial segregation of mRNA encoding myelin‐specific proteins. Proceedings of the National Academy of Sciences, 84(21), 7773–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde, R. , Edwards, L. , & Regan, L. (2008). Structure and function of KH domains. The FEBS Journal, 275(11), 2712–2726. [DOI] [PubMed] [Google Scholar]
- van der Veer, E. P. , de Bruin, R. G. , Kraaijeveld, A. O. , de Vries, M. R. , Bot, I. , Pera, T. , Segers, F. M. , Trompet, S. , van Gils, J. M. , & Roeten, M. K. (2013). Quaking, an RNA‐binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circulation Research, 113(9), 1065–1075. [DOI] [PubMed] [Google Scholar]
- Van Nostrand, E. L. , Pratt, G. A. , Shishkin, A. A. , Gelboin‐Burkhart, C. , Fang, M. Y. , Sundararaman, B. , Blue, S. M. , Nguyen, T. B. , Surka, C. , & Elkins, K. (2016). Robust transcriptome‐wide discovery of RNA‐binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods, 13(6), 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet, C. , & Artzt, K. (1997). STAR, a gene family involved in signal transduction and activation of RNA. Trends in Genetics, 13(12), 479–484. [DOI] [PubMed] [Google Scholar]
- Vouyioukiis, D. , & Brophy, P. (1993). Microtubule‐associated protein MAP1B expression precedes the morphological differentiation of oligodendrocytes. Journal of Neuroscience Research, 35(3), 257–267. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Song, W. , Zhao, H. , Ma, Y. , Li, Y. , Zhai, D. , Pi, J. , Si, Y. , Xu, J. , & Dong, L. (2017). The RNA‐binding protein QKI5 regulates primary miR‐124‐1 processing via a distal RNA motif during erythropoiesis. Cell Research, 27(3), 416–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. Z. , Fu, X. , Fang, Z. , Liu, H. , Zong, F. Y. , Zhu, H. , Yu, Y. F. , Zhang, X. Y. , Wang, S. F. , Huang, Y. , & Hui, J. (2021). QKI‐5 regulates the alternative splicing of cytoskeletal gene ADD3 in lung cancer. Journal of Molecular Cell Biology, 13(5), 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Zhai, D.‐S. , Ruan, B.‐J. , Xu, C.‐M. , Ye, Z.‐C. , Lu, H.‐Y. , Jiang, Y.‐H. , Wang, Z.‐Y. , Xiang, A. , & Yang, Y. (2017). Quaking deficiency amplifies inflammation in experimental endotoxemia via the aryl hydrocarbon receptor/signal transducer and activator of transcription 1–NF‐κB pathway. Frontiers in Immunology, 8, 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , & Brattain, M. G. (2007). The maximal size of protein to diffuse through the nuclear pore is larger than 60 kDa. Federation of European Biochemical Societies Letters, 581(17), 3164–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Tong, X. , Li, C. , Jin, E. , Su, Z. , Sun, Z. , Zhang, W. , Lei, Z. , & Zhang, H. T. (2021). Quaking 5 suppresses TGF‐beta‐induced EMT and cell invasion in lung adenocarcinoma. EMBO Reports, 22(6), e52079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Zan, J. , Wu, M. , Zhao, W. , Li, Z. , Pan, Y. , Sun, Z. , & Zhu, J. (2015). miR‐29a promotes scavenger receptor A expression by targeting QKI (quaking) during monocyte‐macrophage differentiation. Biochemical and Biophysical Research Communications, 464(1), 1–6. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Zhai, D. , Bai, Y. , Xue, K. , Deng, L. , Ma, L. , Du, T. , Ye, Z. , Qu, D. , & Xiang, A. (2021). Loss of QKI in macrophage aggravates inflammatory bowel disease through amplified ROS signaling and microbiota disproportion. Cell Death Discovery, 7(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Lacroix, G. , Haines, J. , Doukhanine, E. , Almazan, G. , & Richard, S. (2010). The QKI‐6 RNA binding protein localizes with the MBP mRNAs in stress granules of glial cells. PLoS One, 5(9), e12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Vogel, G. , Yu, Z. , & Richard, S. (2013). The QKI‐5 and QKI‐6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Molecular and Cellular Biology, 33(6), 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. Y. , Dawson, M. R. , Reynolds, R. , & Hardy, R. J. (2001). Expression of QKI proteins and MAP1B identifies actively myelinating oligodendrocytes in adult rat brain. Molecular and Cellular Neuroscience, 17(2), 292–302. [DOI] [PubMed] [Google Scholar]
- Wu, J. , Zhou, L. , Tonissen, K. , Tee, R. , & Artzt, K. (1999). The quaking I‐5 protein (QKI‐5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. Journal of Biological Chemistry, 274(41), 29202–29210. [DOI] [PubMed] [Google Scholar]
- Wu, J. I. , Reed, R. B. , Grabowski, P. J. , & Artzt, K. (2002). Function of quaking in myelination: Regulation of alternative splicing. Proceedings of the National Academy of Sciences, 99(7), 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi, R. , Tsusaka, T. , Mitsunaga, H. , Maehata, T. , & Hoshino, S.‐I. (2016). The STAR protein QKI‐7 recruits PAPD4 to regulate post‐transcriptional polyadenylation of target mRNAs. Nucleic Acids Research, 44(6), 2475–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Eleftheriadou, M. , Kelaini, S. , Morrison, T. , González, M. V. , Caines, R. , Edwards, N. , Yacoub, A. , Edgar, K. , & Moez, A. (2020). Targeting QKI‐7 in vivo restores endothelial cell function in diabetes. Nature Communications, 11(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Park, J. W. , Bebee, T. W. , Warzecha, C. C. , Guo, Y. , Shang, X. , Xing, Y. , & Carstens, R. P. (2016). Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial‐to‐mesenchymal transition. Molecular and Cellular Biology, 36(11), 1704–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.‐E. , Hetherington, C. J. , Chen, H.‐M. , & Tenen, D. G. (1994). The macrophage transcription factor PU.1 directs tissue‐specific expression of the macrophage colony‐stimulating factor receptor. Molecular and Cellular Biology, 14(1), 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Nie, Q. , & Zhou, T. (2019). Revealing dynamic mechanisms of cell fate decisions from single‐cell transcriptomic data. Frontiers in Genetics, 10, 1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Chen, D. , Xia, J. , Han, W. , Cui, X. , Neuenkirchen, N. , Hermes, G. , Sestan, N. , & Lin, H. (2017). Post‐transcriptional regulation of mouse neurogenesis by Pumilio proteins. Genes & Development, 31(13), 1354–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R.‐L. , Yang, J.‐P. , Peng, L.‐X. , Zheng, L.‐S. , Xie, P. , Wang, M.‐Y. , Cao, Y. , Zhang, Z.‐L. , Zhou, F.‐J. , & Qian, C.‐N. (2016). RNA‐binding protein QKI‐5 inhibits the proliferation of clear cell renal cell carcinoma via post‐transcriptional stabilization of RASA1 mRNA. Cell Cycle, 15(22), 3094–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , & Feng, Y. (2001). Distinct molecular mechanisms lead to diminished myelin basic protein and 2′, 3′‐cyclic nucleotide 3′‐phosphodiesterase in qkv dysmyelination. Journal of Neurochemistry, 77(1), 165–172. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Lu, Z. , Ku, L. , Chen, Y. , Wang, H. , & Feng, Y. (2003). Tyrosine phosphorylation of QKI mediates developmental signals to regulate mRNA metabolism. The EMBO Journal, 22(8), 1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Ku, L. , Chen, Y. , Xia, M. , LoPresti, P. , & Feng, Y. (2006). QKI binds MAP1B mRNA and enhances MAP1B expression during oligodendrocyte development. Molecular Biology of the Cell, 17(10), 4179–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Mandler, M. D. , Yi, H. , & Feng, Y. (2010). Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin‐associated glycoprotein. Proceedings of the National Academy of Sciences, 107(44), 19061–19066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Tian, D. , Xia, M. , Macklin, W. B. , & Feng, Y. (2006). Rescuing qkV dysmyelination by a single isoform of the selective RNA‐binding protein QKI. Journal of Neuroscience, 26(44), 11278–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , He, C. , Ren, J. , Dai, C. , Stevens, S. R. , Wang, Q. , Zamler, D. , Shingu, T. , Yuan, L. , & Chandregowda, C. R. (2020). Mature myelin maintenance requires Qki to coactivate PPARβ‐RXRα‐mediated lipid metabolism. The Journal of Clinical Investigation, 130(5), 2220–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Shin, S. , He, C. , Zhang, Q. , Rasband, M. N. , Ren, J. , Dai, C. , Zorrilla‐Veloz, R. I. , Shingu, T. , & Yuan, L. (2021). Qki regulates myelinogenesis through Srebp2‐dependent cholesterol biosynthesis. eLife, 10, e60467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , Zhao, L. , Wang, E. , Dimova, N. , Liu, G. , Feng, Y. , & Cambi, F. (2012). The QKI‐PLP pathway controls SIRT2 abundance in CNS myelin. Glia, 60(1), 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong, F.‐Y. , Fu, X. , Wei, W.‐J. , Luo, Y.‐G. , Heiner, M. , Cao, L.‐J. , Fang, Z. , Fang, R. , Lu, D. , & Ji, H. (2014). The RNA‐binding protein QKI suppresses cancer‐associated aberrant splicing. PLoS Genetics, 10(4), e1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn, A. M. , & Krieg, P. A. (1997). The KH domain protein encoded by quaking functions as a dimer and is essential for notochord development inXenopus embryos. Genes & Development, 11(17), 2176–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study