

Abstract
Nucleic acid therapeutics can be used to control virtually every aspect of cell behavior and therefore have significant potential to treat genetic disorders, infectious diseases, and cancer. However, while clinically approved to treat a small number of diseases, the full potential of nucleic acid therapeutics is hampered by inefficient delivery. Nucleic acids are large, highly charged biomolecules that are sensitive to degradation and so the approaches to deliver these molecules differ significantly from traditional small molecule drugs. Current studies suggest less than 1% of the injected nucleic acid dose is delivered to the target cell in an active form. This inefficient delivery increases costs and limits their use to applications where a small amount of nucleic acid is sufficient. In this review, we focus on two of the major barriers to efficient nucleic acid delivery: (1) delivery to the target cell and (2) transport to the subcellular compartment where the nucleic acids are therapeutically active. We explore how nanoparticles can be modified with targeting ligands to increase accumulation in specific cells, and how the composition of the nanoparticle can be engineered to manipulate or disrupt cellular membranes and facilitate delivery to the optimal subcellular compartments. Finally, we highlight how with intelligent material design, nanoparticle delivery systems have been developed to deliver nucleic acids that silence aberrant genes, correct genetic mutations, and act as both therapeutic and prophylactic vaccines.
This article is categorized under:
Nanotechnology Approaches to Biology > Cells at the Nanoscale
Therapeutic Approaches and Drug Discovery > Nanomedicine for Infectious Disease
Biology‐Inspired Nanomaterials > Lipid‐Based Structures
Keywords: endosomal escape, nanoparticles, nucleic acids, targeted delivery
There is significant potential to improve the delivery of nucleic acid theraputics by engineering smart, responsive and targeted nanoparticle delivery systems.
1. INTRODUCTION
Nucleic acids have broad potential to treat disease, with established clinical use in vaccination (Baden et al., 2021; Polack et al., 2020) and growing potential as gene therapies to treat infectious diseases (Richner et al., 2017), cancer (NCT03739931, 2018; NCT03897881, 2019), and hereditary conditions (Adams et al., 2018). As therapeutics, nucleic acids can directly or indirectly manipulate all cellular functions and thus can be engineered to address virtually any disease state. Therapeutic nucleic acids can use existing cellular processes (e.g., RNA interference) or can encode instructions for novel protein machinery (e.g., messenger RNA [mRNA] encoding recombinant proteins) to correct aberrant cellular processes. Furthermore, nucleic acids can be co‐delivered with other compounds (e.g., Cas9 protein and guide RNA, vaccine adjuvants, and mRNA) to work in a synergistic manner.
A major challenge common to all these therapeutic targets is the intracellular delivery of exogenous nucleic acids. Nucleic acids are only active if they are delivered the specific cellular compartments (cytoplasm, nucleus, and mitochondria) where they are required. However, nucleic acids are highly negatively charged and range in size from short oligomers to kilobases‐long protein‐encoding polymers. Thus, they are not able to permeate through the lipid bilayers that separate the different subcellular compartments. Furthermore, nucleic acids are relatively labile biomolecules that are susceptible to degradation by numerous cellular and environmental processes. A successful delivery system must therefore combine the ability to provide protection from premature degradation as well as transport nucleic acids to their target cells and across cellular membranes to their subcellular site of action.
In this review, we discuss nonviral, nanomaterials‐based technologies to deliver nucleic acids, and the cellular anatomy that must be overcome for them to succeed (Figure 1). A significant focus of nanomaterial research has been to increase circulation times and reduce immune clearance (Gustafson et al., 2015). However, as the application of nucleic acid therapies grows there is a need to look beyond organ and tissue‐based biodistribution studies. For the next generation of therapeutics there needs to be a focus on improved delivery to the specific cells where the therapeutics are required, and to ensure they are trafficked to the subcellular compartments where they are active. Here, we discuss how different classes of nucleic acids (Section 2) can be delivered using different nanoparticle delivery systems (Section 3). This can be achieved by engineering the surface of the nanoparticles to present targeting ligands that bind to receptors on the target cells (Section 4). We address the effect of these components on cellular trafficking and how formulating the core of the nanoparticle with lipids and polymers that manipulate or disrupt cellular membranes and facilitate transmembrane transport (Section 5). We discuss how nanoparticles can be engineered to control disassembly and cargo release (Section 6). Finally, we discuss the applications of these engineered nanoparticle systems for nucleic acid delivery (Section 7).
FIGURE 1.
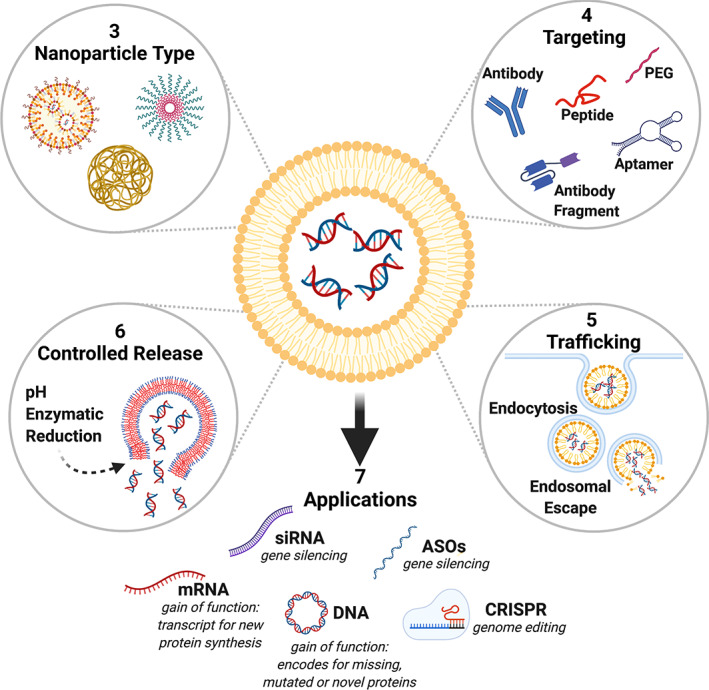
Nucleic acid therapeutics have the potential to encode for novel function (mRNA, pDNA), knockout aberrant function (siRNA, ASOs) or correct mutations in the genome (CRISPR). However, despite success in SARS‐CoV‐2 vaccination and treating hepatic conditions, the low efficiency of nucleic acid delivery remains an obstacle to widespread therapeutic use. In this review we focus on how nanoparticles can be modified to target the specific cells where they are required, and engineered to traffic the nucleic acids to the subcellular compartment where they are active.
2. SITES OF ACTION IN THE CELL
One of the main challenges of nucleic acid delivery systems is to transport nucleic acids to their site of action in the cell (cytoplasm, nucleus, or mitochondria). Each of these locations has biological machinery that can be manipulated by the delivered nucleic acids to modify the cellular responses (Figure 2, Table 1). These subcompartments are separated by phospholipid bilayer membranes, which act as a major physical barrier to the successful delivery of the nucleic acid cargo. For example, access into the nucleus requires traversal of the plasma membrane as well as the nuclear membrane, both of which have different properties and composition.
FIGURE 2.
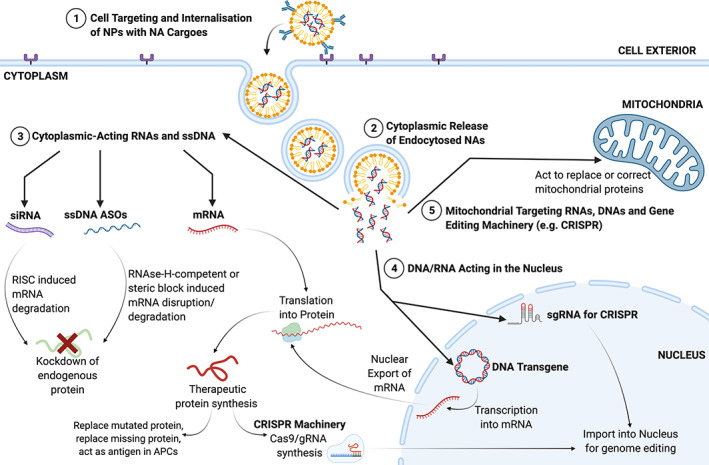
Nucleic acids must be delivered to specific subcellular compartments to be therapeutically active. 1) Nanoparticles carrying nucleic acid cargoes can be targeted to the correct cell using antibodies, promoting internalization by the cell. 2) Endocytosed nanoparticles need to undergo endosomal escape, to release the nucleic acid cargo into the cytoplasm. From here, nucleic acids can act in different areas depending on the nucleic acid type. 3) Cytoplasmic‐acting nucleic acids (siRNA, ASOs, and mRNA) act via different mechanisms. siRNA promotes RISC‐induced degradation of the target mRNA to knockdown expression of a target protein. ASOs can trigger RNAse‐H‐induced degradation of the mRNA or work to block mRNA interactions with the translation machinery; both resulting in knockdown of the endogenous protein. mRNA is translated into the therapeutic protein which can replace the function of an aberrant protein or act as a protein antigen. 4) DNA transgenes require transport to the nucleus where they are transcribed into mRNA. The mRNA is exported and translated into a therapeutic protein in the cytoplasm. 5) RNAs and DNAs can also be targeted to the mitochondria, where they can be transported into the mitochondrial matrix. Here, RNAs can be translated into therapeutic mitochondrial proteins by the local mitochondrial machinery, and DNAs can be used to replace mutated mitochondrial DNA.
TABLE 1.
Applications and examples of therapeutic nucleic acids delivered to different cellular compartments
| Location | Therapeutic | Application | Example |
|---|---|---|---|
| Cytoplasm | siRNA | Gene silencing
|
Onpattro™ (Adams et al., 2018) |
| ASOs | Gene silencing
|
Vitravene™ (Vitravene Study Group, 2002) | |
| mRNA | Gain of function
|
COVID‐19 mRNA Vaccines (Baden et al., 2021; Polack et al., 2020) | |
| Nucleus | DNA | Gain of function
|
pDNA endoded Cas9/sgRNA synthesis for in vivo gene editing (Luo et al., 2018) |
| ASOs | Gene silencing
|
Spinraza™ (Abbas et al., 2022) | |
| RNA | Template RNA
|
sgRNA CRISPR/Cas9 templates (Luo et al., 2018) | |
| Mitochondria | mtDNA | Gain of function
|
mtGFP DNA delivery (Bae et al., 2017) |
| RNA | Gain of function
|
tRNA therapy for mitochondrial mutations (Kawamura et al., 2020) |
2.1. The cytoplasm
The cytoplasm is the major intracellular target for nucleic acid delivery. As the primary site for protein translation, the cytoplasm is rich in mRNA and associated regulatory RNAs. Therapeutic nucleic acids can act in the cytoplasm to induce protein expression (mRNAs [Kole et al., 2012]), or to suppress the expression of aberrant proteins (RNAi and antisense oligonucleotides [ASOs] [Crooke et al., 2021]). In the cytoplasm RNA therapies are generally transient as most RNAs have relatively short half‐lives, although the advent of self‐amplifying RNA sequences can prolong activity to months (Steinle et al., 2017).
2.1.1. mRNA: Gain of function
Messenger RNA is the fundamental code for protein synthesis. Native human proteins (or protein variants) synthesized from mRNA can have a multitude of functions, including: (a) directly interacting with cellular signaling pathways, (b) acting as transcription factors to manipulate cell behaviors such as growth and survival, or (c) temporarily supplementing the activity of defective or missing proteins (Sahin et al., 2014). mRNA also allows delivery of “designer” fusion proteins combining domains from different proteins that could enable novel engineering of cell signaling or metabolism (e.g., targeted protein degradation using ubiquitinases [Ely et al., 2021]), and may simplify delivery of large custom nucleic acid modifying proteins used in gene editing (e.g. zinc fingers, Cas9 [Doudna & Charpentier, 2014]). Delivery of recombinant antigens as mRNA has come to the fore during the COVID‐19 pandemic (Lopez‐Cantu et al., 2022) and will quickly lead to the adoption of mRNA therapies for other infectious diseases and immunotherapy. In addition to acting as a transcript for protein synthesis, mRNA can also include untranslated regions that can confer other features for example, a protein‐encoding mRNA with an untranslated stem loop containing an siRNA sequence, or viral genome‐inspired secondary structures providing self‐replication abilities (Wolff & Herweijer, 1997).
2.1.2. RNA interference (RNAi): Gene silencing
Small interfering RNAs (siRNA) and microRNAs (miRNA) are shorter noncoding RNAs that function by inhibiting the expression of mRNA (known as RNA interference or RNAi). The process is initiated in the cytoplasm, where double‐stranded regions of the delivered RNA are cleaved into discrete siRNAs of 21–23 bp (Elbashir et al., 2001) by a cytoplasmic endonuclease “dicer” (Meister & Tuschl, 2004). The multiprotein RNA‐induced silencing complex (RISC) is then loaded with a “guide‐strand” from the siRNA duplex (Chernikov et al., 2019; Liu et al., 2004). These RISC complexes can recognize, bind and/or cleave mRNAs in a sequence‐specific manner, regulating subsequent translation of proteins by degrading the targeted mRNA or inhibiting its translation. Using this mechanism, synthetic siRNAs and miRNAs can be designed to target host proteins involved in disease.
2.1.3. Antisense oligonucleotides (ASOs): Gene silencing
Single‐stranded DNA sequences known as antisense oligonucleotides achieve gene silencing using different mechanisms. These can be divided into steric block or RNase‐competent ASOs, both of which inhibit target mRNA translation (Roberts et al., 2020). Steric block ASOs act on mature mRNAs in the cytoplasm or pre‐RNAs in the nucleus (Liang et al., 2016; Liang et al., 2019) and interfere with translation of the target transcript by blocking RNA–RNA or RNA–protein interactions. This disrupts ribosome binding, translation, transcript stability, or splicing in the nucleus (to either promote exon skipping or exon inclusion) (Liang et al., 2019). RNAse‐competent ASOs activate the enzyme RNase‐H (RNASEH1) (present in both the cytoplasm and nucleus [Liang et al., 2019]) which recognizes and degrades RNA/DNA heteroduplexes.
2.1.4. Other noncoding RNAs: Gene regulation
There is also a plethora of small, structured and noncoding RNA species present in cells that are poorly understood but may have future therapeutic importance. In the cytoplasm, Piwi‐interacting RNA and circular RNA are thought to provide addition transcriptional regulation (Ozata et al., 2019). Ribozymes, structured RNA which possesses catalytic activity (Tanner, 1999), are also poorly characterized but potentially powerful future nucleic acid therapeutics.
2.2. The nucleus
Genomic material and associated support machinery are enclosed within the nuclear membrane with import and export of materials regulated by nucleopores (Medina‐Kauwe et al., 2005). The nucleus is an obvious target as changes in DNA sequences are responsible for most heritable diseases and cancers. Genomic DNA is transcribed into mRNA and other noncoding RNAs involved in cellular processes, but the DNA sequence itself, chromatin structure, and other epigenetic features can also be manipulated for therapeutic effect.
2.2.1. DNA gene delivery: Gain of function
In its simplest form a gene encoded by DNA can be delivered to the cell, and once delivered to the nucleus is transcribed into mRNA for protein expression. Similar to mRNA, DNA can encode the wild‐type version of a mutated protein; a “helper” protein to help combat a disease state; or novel proteins with other functions. An advantage of delivering a gene coded by DNA over an mRNA sequence is that a single DNA template produces multiple copies of mRNA, leading to higher gene expression. Furthermore, DNA is less susceptible to hydrolysis than mRNA and thus persists in the cell longer (Walther et al., 2013). Plasmid DNA exhibits much greater stability than RNA and can easily accommodate multiple protein‐encoding genes, as well as regulatory elements such as promoters. While plasmid DNA (and most other exogenous forms of DNA) has increased stability, it lacks the ability to replicate and partition correctly during anaphase and is eventually lost (Walther et al., 2013). Rarely, DNA delivered to the nucleus can be incorporated into genomic DNA by recombination; however, at present this process is largely uncontrolled and may not be desirable (e.g. causing mutagenic insertion [Stapley et al., 2017]).
2.2.2. Therapeutic RNA in the nucleus: Sequence‐guided genome editing
At present the most prominent application of RNA delivery to the nucleus is for CRISPR‐mediated gene editing. CRISPR can be used to delete, mutate, or insert new sequences into genomic DNA to produce heritable changes in cell behavior (Doudna & Charpentier, 2014; Finn et al., 2018). This is achieved by targeted cleavage of DNA (e.g., to excise sequence) followed by repair of the breakage (mutagenesis by imperfect repair, or repair templated by novel sequence). DNA cleavage by Cas9 (and related enzymes, e.g., Cpf1) is guided by sequence‐specific binding of a short CRISPR RNA (crRNA) to target DNA sequences. To perform this function, crRNA complexes with a second RNA strand, the trans‐activating CRISPR RNA (tracrRNA), and Cas9 enzyme to form a ribonucleoprotein complex. Each component of this complex can be delivered to cells in different forms; the crRNA and tracrRNA are frequently combined into a single short guide RNA (sgRNA) that can be encoded in a plasmid (typically encoded along with Cas9) or can be delivered as an in vitro transcribed RNA precomplexed with recombinant Cas9 protein. Where template‐directed repair is being used to insert gene sequences using CRISPR editing, a synthetic template DNA also needs to be delivered into the nucleus with this complex.
Our understanding of other RNA species in the nucleus and their therapeutic application is less advanced. RNAi mechanisms in the nucleus can also silence gene expression and alter chromatin structure, which may result in heritable epigenetic effects (Castel & Martienssen, 2013). In addition to mRNA transcription and splicing activity, small noncoding RNAs have been implicated in maintaining the structure of chromatin microdomains and overall genome architecture, which influences gene expression (Zhang et al., 2021). Small nucleolar RNAs have further been implicated in guiding ribose modifications (e.g. pseudouridylation) which results in downstream effects on splicing and (in the cytoplasm) ribosomal activity (Bratkovic et al., 2020). Compared to gene editing, modulating gene expression through these RNAs may in future provide similarly durable but more easily reversible therapeutic effects.
2.3. Mitochondria
A third target location for nucleic acid therapeutics is the mitochondria. Many biochemical processes are governed by the mitochondria, and they can play an important role in different human diseases. While most mitochondrial proteins are synthesized in the cytoplasm, mitochondria also possess their own genome and limited protein synthesis machinery for the synthesis of unique mitochondrial proteins (Dard et al., 2020). Nucleic acid gene therapies can be used to replace or correct abnormal proteins caused by mutations arising in mitochondrial DNA (mtDNA), which cause several heritable diseases (Dard et al., 2020). These targeted therapies can be delivered in the form of RNAs and DNAs (especially mitochondrial transgenes and gene editing tools) for synthesis of new proteins, replacement of mtDNA or editing of mtDNA (Fox, 2012).
3. NANOPARTICLE DELIVERY SYSTEMS USED WITH NUCLEIC ACIDS
A range of nanoparticle delivery systems have been developed to deliver nucleic acid therapeutics, with a major focus on protecting the nucleic acids from degradation. However important consideration must also be given to engineering properties such as delivery to target cells and promoting cellular trafficking to reach their site of action. A diverse array of materials have been described for use as nucleic acid delivery systems, including lipid (Huang et al., 2021), polymer (Deirram et al., 2019), and inorganic/metallic nanoparticles (Gamrad et al., 2016). Most clinical applications have focused on the use of soft (lipid/polymer) nanoparticle delivery systems; however, inorganic/metallic delivery systems have been reviewed extensively elsewhere (Bobo et al., 2016; Ding et al., 2014). In this review, we will focus on four of the most prominent delivery systems: lipid nanoparticles, polyion complex (PIC) micelles, pH responsive polymer nanoparticles and biodegradable polymer nanoparticles (Figure 3). These versatile delivery systems can be adapted for the delivery of a range of nucleic acid therapeutics from short siRNA to large plasmid DNA.
FIGURE 3.
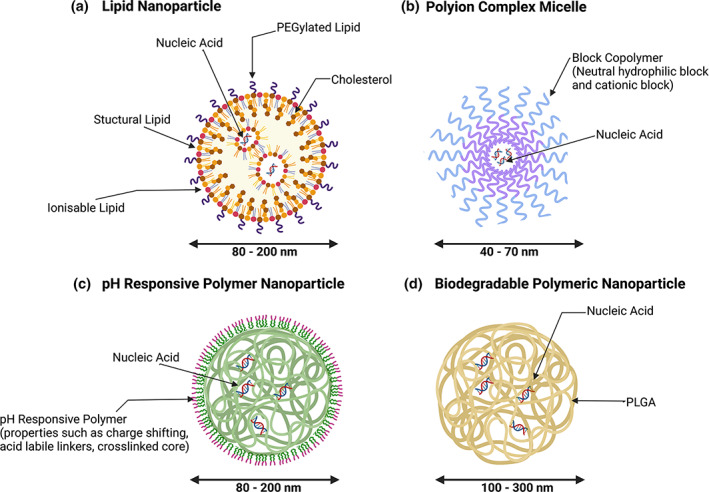
Nanoparticle delivery systems commonly used for the delivery of nucleic acids. When nucleic acids are encapsulated inside nanoparticles, they are protected from nucleases, which rapidly degrade free nucleic acids when they are delivered to the body. (a) Lipid nanoparticles – Formed from ionizable lipids, structural lipids, PEGylated lipids, and cholesterol, with the nucleic acids being complexed in the nanoparticle core. (b) Polyion complex micelles – Composed of block copolymers with a neutral hydrophilic block and a cationic block for encapsulating the nucleic acid cargo. (c) pH‐responsive polymer nanoparticles – Assembled from different polymers engineered with charge shifting ability, acid labile linkers or crosslinked cores. (d) Biodegradable polymeric nanoparticles – Typically synthesized from PLGA with nucleic acids physically entrapped within the particle.
3.1. Lipid nanoparticles
Lipid nanoparticle (LNP) delivery systems have recently come to the forefront of nucleic acid delivery systems, particularly through their use in the formulation of SARS‐CoV‐2 vaccines (Baden et al., 2021; Polack et al., 2020). They are generated by mixing ionizable cationic lipids, zwitterionic structural lipids, PEGylated lipids and cholesterol with the nucleic acids, which forms nanoparticles 80–200 nm in diameter (Huang et al., 2021). Ionizable lipids protect the nucleic acids from degradation by nucleases, oxidation, or hydrolysis by complexing the nucleic acids into the nanoparticle core. These lipids become ionized at a certain pH, dependent on their pKa, promoting interactions with the nucleic acids and subsequent complexation. Ionizable lipids are thought to facilitate cellular entry and endosomal escape, assisting release of the nucleic acids into the cytoplasm. Structural lipids (e.g., phospholipid) resembling endogenous cell membrane lipids are used to form lipid bilayers within the LNPs, and cholesterol stabilizes the lipid bilayer by facilitating lipid chain “packing,” decreasing permeability, and increasing fluidity of the membrane (Pardi et al., 2018). PEGylated lipids are also essential for LNP delivery systems, as they provide a hydrophilic “stealth” layer to the particle acting to improve colloidal stability and shielding the LNP from opsonization (Li & Huang, 2010). LNPs have very effective nucleic acid complexation and have thus been implemented for many gene therapies, specifically RNAi (siRNA patisiran [Adams et al., 2018]) and for mRNA vaccines (SARS‐CoV‐2 vaccines generated by Pfizer [Polack et al., 2020] and Moderna [Baden et al., 2021]). However, despite their success as vaccines, it is estimated that only 2% of the LNPs that reach a cell can deliver their cargo to the cytosol (Gilleron et al., 2013). This combined with possible immunotoxicity from the cationic lipids means there is significant opportunities to improve on these formulations.
3.2. PIC micelles
An alternative but analogous delivery system is PIC micelles. Instead of using charged lipids, PIC micelles are formed using block copolymers that possess a neutral hydrophilic block and a cationic block (Lee & Kataoka, 2009). The driving force for the assembly of nanoparticles is the electrostatic attraction between the ionic block and counter‐charged nucleic acids. The hydrophilic block is essential for the assembly of distinct nanoparticles as it prevents mass aggregation and precipitation of the cationic polymer/nucleic acid complexes. The ability to incorporate different functional groups into the copolymers allows PIC micelles to be engineered to respond to changes in biological environment such as pH, redox conditions, temperature, and salt concentration (Lee & Kataoka, 2009). Release of materials can be triggered in response to pH, either by the protonation‐deprotonation (based on the pKa) of the ionic block which disrupts the stabilizing charge and dissociating the PIC micelle, or through a pH‐sensitive bond between the block copolymers. PIC micelles have also been engineered with fusogenic peptides and polymers for improved membrane interaction for internalization and endosomal escape (Lee et al., 2001). While PIC micelles can be readily engineered with different properties, using these nanoparticles for nucleic acid delivery still requires improvement with clearance and degradation rates significantly higher for nucleic acid loaded PIC micelles than unloaded PIC micelles (Cabral et al., 2018).
3.3. pH‐responsive polymer nanoparticles
pH‐responsive nanoparticles are of specific interest to control the delivery of nucleic acids due to the change in pH that occurs when nanoparticles are endocytosed into a cell. When nanoparticles are endocytosed into the cell, the pH drops from 7.4 in the blood stream to ~pH 6.5 in early endosomal compartments, and down to <pH 5 in lysosomes (Casey et al., 2010). This drop in pH can be used to induce conformational changes in charge shifting polymers that results in the disassembly of the nanoparticles or release of a therapeutic cargo from the nanoparticle. Different pH‐responsive polymers and the mechanisms by which they self‐assemble and deliver cargo have been recently reviewed in detail by Deirram et al. (2019). One important class of charge shifting polymers is tertiary amines, such as poly(2‐(diisopropylamino)ethyl methacrylate (PDPA) and poly(2‐(diethylamino)ethyl methacrylate (PDEA). When these charge shifting polymers are mixed with a block co‐polymer (with a charge shifting block and a hydrophilic block) at pH 7.4, the amine is deprotonated, the polymers become hydrophobic and self‐assemble into 80–200 nm nanoparticles. However, when the pH drops below the pKa (~6.4 and ~ 7 for PDPA and PDEA, respectively), the polymers become charged, and the nanoparticles disassemble. Critically, the ratio of PDPA:PDEA plays an important role in the ability to disrupt the endosomal membrane and deliver cargo to the cytosol. Nucleic acids encapsulated in pH responsive nanoparticles and have been developed for oligonucleotide vaccine delivery (Wilson et al., 2013) and DNA delivery (Miyata et al., 2008).
Polymeric nanoparticles can also be synthesized with acid labile linkers, such as ortho esters, maleic acid amides and β‐thiopropionate linkers, to enable cleavage of surface‐conjugated nucleic acid therapeutics (Deirram et al., 2019). This has been investigated for delivery of DNA that can be slowly released at a pH of 4.0 at which the ortho ester is hydrolyzed (Xu et al., 2014). Significant work is still required to understand how to induce efficient cytosolic delivery and how these cationic materials interact with negatively charged nucleic acids and efficiently release their cargo once inside the cell (Deirram et al., 2019).
3.4. Biodegradable polymeric nanoparticles
Unlike LNPs, PIC micelles and pH‐responsive nanoparticles, which typically rely on electrostatic interactions to encapsulate the nucleic acids, biodegradable polymer nanoparticles can be used to physically trap the nucleic acid cargo. Poly(d, l‐lactide‐co‐glycolic acid) (PLGA) is a biodegradable polyester extensively investigated for delivery of nucleic acids through encapsulation of the nucleic acids during the particle synthesis. PLGA undergoes hydrolysis in the body, breaking down the polymer into lactic acid and glycolic acid monomers (which are readily metabolized by the body) releasing the nucleic acid cargo (Dinarvand et al., 2011). PLGA nanoparticles have been successfully loaded with a range of nucleic acid therapies, including RNAi and plasmid DNA (Panyam & Labhasetwar, 2003). PLGA nanoparticles can readily protect nucleic acids from degradation, allowing controlled release of the nucleic acid in the cytoplasm resulting in gene expression that can be sustained for over a month. This has been shown by different groups both in vitro and in vivo (Cohen et al., 2000; Hedley et al., 1998). PLGA nanoparticles have the ability to encapsulate large (plasmid DNA) (Jo et al., 2020) and small (siRNA) nucleic acid cargo; however, short oligos often exhibited burst‐release kinetics, which may result in off target in vivo delivery (Essa et al., 2020). While PLGA polymers are readily available, chemically modifying PLGA to tune its properties can be synthetically challenging, and the degradation products of PLGA can cause inflammation and adverse immune responses.
4. CONTROLLING DELIVERY TO TARGET CELLS
Before delivery of nucleic acids to their site of action within the cell, a delivery system must access the target cell of interest. Target cells in different tissues will present different challenges to delivery. To maximize therapeutic effect, minimize dose, and reduce off‐target side effects, an ideal delivery system must target nucleic acids only to cells requiring the therapeutic. For rare or sparsely distributed cells or metastases, this typically requires intravenous injection with nanoparticles engineered with a long circulation time to allow the delivery system to perfuse through tissues, in addition to active targeting of a cell‐specific surface receptor to enhance cellular internalization during a brief contact window. In contrast, delivery to hepatic tissue requires minimal targeting and blood circulation time due to the innate deposition of nanoparticles in this blood‐filtering organ. For vaccines, intramuscular injection is desirable (Carrasco et al., 2021). Following intramuscular administration, the bolus of therapeutic can interact with cells at the immediate injection site but will also rapidly begin to diffuse away and will travel interstitially toward draining lymph nodes and their associated immune cells. With both intramuscular and intravenous methods, nanoparticle contact with circulating humoral or lymph proteins results in deposition of a surface “corona” of host proteins, which begins a clearance process mediated primarily by innate immune mechanisms (Mahon et al., 2012). Minimizing these interactions can increase systemic distribution and promote nanoparticle accumulation at target destinations, but conversely must be balanced as to not prevent intentional interactions completely.
4.1. Pegylation and stealth
A major hurdle in nanoparticle mediated therapeutic delivery is nonspecific interaction with the mononuclear phagocytic system (MPS), which recognizes and rapidly clears foreign macromolecules through opsonization (Gustafson et al., 2015). Upon contact with blood, nanoparticles accumulate opsonin proteins on their surface, which are rapidly recognized and cleared via phagocytosing macrophages. Opsonization occurs in a time‐dependent manner and is specific to the surface chemistry and topography of nanoparticles (Gustafson et al., 2015). The reduction of nanoparticle association with blood opsonins can extend circulation time and reduce nonspecific uptake and is commonly referred to as particle “stealth” property. Most commonly this is achieved by the addition of an outer layer of hydrophilic poly(ethylene glycol)‐lipid PEG, which provides a hydrophilic “stealth” layer to improve colloidal stability, employing steric hindrance to prevent nonspecific interactions, including opsonization (Li & Huang, 2010). PEG‐lipid is used in LNP formulations, as well as other nanoparticle formulations, and was implemented in the first clinical nanoformulation Doxil™ to improve systemic circulation (Papahadjopoulos et al., 1991).
While beneficial to limit undesired nonspecific interactions with the immune system, and enable the nanoparticles to diffuse away from the injection site, the presence of PEG also inhibits therapeutic delivery to the target cells. PEGylation reduces cellular uptake by limiting interactions with the cellular membranes (Abstiens et al., 2019; Shi et al., 2021; Song et al., 2002). The structure and amount of PEGylation can be tuned to control these interactions, with a study by Pozzi et al. showing that dense PEGylation (5 kDa) on the surface of cationic liposomes reduced cellular uptake in PC3 cells by a factor of 2 compared to moderate PEGylation (1 and 2 kDa) (Pozzi et al., 2014).
The “PEG dilemma” has prompted the generation of more sophisticated nanoparticles that can trigger “PEG shedding” to overcome the steric interference of PEG at the point of cellular entry. In LNP formulations, the dissociation of the outer PEG layer renders particles vulnerable to opsonization; however, likely increases particle cellular association. Chen et al. showed that substitution of more‐rapidly dissociating DMG‐PEG; which is included in Moderna's SARS‐CoV‐2 vaccine, with longer alkyl tailed DSG‐PEG lead to a marked increase in particle circulation times, reduced hepatic uptake and decreased dissociation of other lipid components (Chen et al., 2016). It is possible that shedding of the PEG layer from LNP formulations soon after injection can be beneficial, as PEG enables the LNPs to diffuse away from the injection site, but once shed, enables the LNPs to bind to and be internalized by a range of different cells.
PEG‐lipids are commonly attached via amide or ether bonds, which are highly stable and thus difficult to cleave (Fang et al., 2017). Cleavable PEG‐lipids, which “shed” from the nanoparticle surface when triggered by conditions of the cellular microenvironment (e.g. reducing conditions, pH, or enzymatic activity), to allow for membrane interactions required for internalization and endosomal escape. Cleavable PEG moieties have been implemented in a range of LNP formulations. Hatakeyama et al. used a PEG‐peptide‐DOPE (PPD) conjugated LNP loaded with siRNA, possessing a matrix metalloproteinase (MMP‐2) sensitive peptide for PEG cleavage (Hatakeyama et al., 2011). The PPD‐LNP showed 60% cellular association with HT1080‐luc cells while PEG‐LNP demonstrated only 20% cellular association. Furthermore, PEG‐LNP did not demonstrate any siRNA‐mediated gene silencing, while PPD‐LNP showed a 40% reduction in luciferase expression. Inducing PEG shedding to maximize cellular association and internalization of targeting ligands and nanoparticles is essential to maximize therapeutic efficiency (Juang et al., 2019; Li & Huang, 2010).
4.2. Methods to target‐specific cells
The improved circulation half‐life of nanoparticles formulated with stealth surfaces can result in passive targeting/distribution to the tissues of interest. For tumor targeting, the size of nanoparticle formulations can be optimized to exploit the enhanced permeability and retention (EPR) effect (Fang et al., 2011). The EPR effect exploits the abnormal tumor vasculature which develops large gaps in rapidly growing endothelium. A combination of tumor microenvironment factors including intratumoral pressure, angiogenesis and vascular “leakiness” (Attia et al., 2019) can lead to enhanced accumulation in tumors, however tumors need to be a significant size before they exhibit the EPR effect. For noncancer applications, distribution to target tissue is more reliant on route of administration. A study by Pardi et al. of examined the distribution of firefly luciferase mRNA‐LNPs administered by various common routes in mice. As expected, in systemically administered formulations (IV, intraperitoneal, deep IM) high luciferase activity was observed in liver and spleen tissues, while sustained luciferase expression (8 days) was localized to the administration site for subcutaneous and intramuscularly administered LNPs. Additionally short‐term (<2 days) luciferase expression was observed in IM administered LNPs and is consistent with the partial clearance and systemic circulation of LNPs from IM injection site (Pardi et al., 2015). The accumulation in liver and spleen tissues following systemic administration is consistent with findings from studies of ionizable LNPs, which have shown hepatocytes uptake through ApoE‐dependent, receptor‐mediated endocytosis (Akinc et al., 2010; Gilleron et al., 2013; Pardi et al., 2015), as uptake by MPS (Kimura et al., 2021; Moghimi & Szebeni, 2003). The exploitation of ApoE‐dependent receptor‐mediated endocytosis of LNPs led to the development and approval of the first siRNA therapeutic; patisiran (Onpattro™), for the treatment of transthyretin‐mediated amyloidosis(Adams et al., 2018), and while this delivery system represents an important advancement in nanomedicine, approved nanoparticle‐based nucleic acid therapeutics are confined to passive targeting for mode of action, limiting the potential of nonliver and spleen associated targets.
Ideally, nucleic acid loaded nanoparticles should be specific to pathological tissues to minimize off target effects and maximize the therapeutic efficacy of nucleic acid cargo. The development of targeting ligands which are specific to receptors exclusive to or upregulated on the target cells has the potential to limit off target effects and improve therapeutic efficacy. There are several considerations including ligand choice, biocompatibility, cell specificity, conjugation method, and stability that are critical for optimizing delivery. The targeting ligands can be classified into several broad categories (Figure 4): small molecules, peptides, aptamers, and antibodies (Abs) and associated derivatives, each with distinct advantages and disadvantages (Friedman et al., 2013).
FIGURE 4.
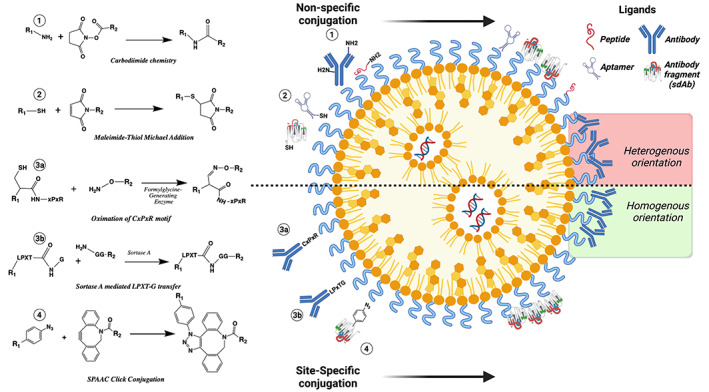
Overview of the conjugation chemistry used to attach targeting ligands to nanoparticles, which enables delivery to specific cells. 1) Amine‐carboxylic acid: Carboxylic acids activated with EDC/NHS (NHS‐ester) on the nanoparticle react with primary amines on lysine residues and N‐terminus of proteins, resulting in heterogeneous ligand orientation. 2) Maleimide‐thiol: Michael addition of a maleimide (typically attached to the particle) and a thiol (either engineered into the peptide/aptamer of interest or from a reduced cysteine residue). 3a) Formylglycine‐generating enzyme (FGE) mediated conjugation: The cysteine thiol group in the peptide sequence CxPxR is posttranslationally modified by (FGE) into aldehyde which can react with aminooxy groups on the nanoparticle. 3b) Sortase A‐mediated conjugation: Proteins with C‐terminal LPxTG motif undergo transpeptidation with oligoglycine residues on the nanoparticle for site specific conjugation, enabling homogenous ligand orientation via the C‐terminus. 4) Strain‐promoted azide–alkyne cycloaddition (SPAAC) conjugation: Dibenzocyclooctyne (DBCO) functionalized particles undergo copper‐free click SPAAC conjugation with unnatural amino acid AzPhe (which can be site selectively introduced during protein synthesis) for site‐specific conjugation of proteins.
4.2.1. Small molecule ligands
Small molecular ligands offer a simple way increasing accumulation of nanoparticles with their target cells. These ligands are typically low cost, stable, and are selected for simple conjugation to nanoparticles. Folic acid (Vitamin B9) has been widely investigated for the targeting tumor cells due to the upregulation of folate receptor alpha (FRα) in many forms of cancer (Cheung et al., 2016). Folate‐PEG5000 targeted cyclodextrin‐siRNA nanoparticles showed increased uptake in PSMA+ cells in vitro via a folate dependent internalization mechanism (Evans et al., 2016). Compared to untargeted particles, which resulted in no significant gene silencing, folate‐targeted cyclodetxrin‐siRNA particles reduced target gene expression by as much as 44% in PSMA+ cells.
Carbohydrate interactions with certain cellular glycoprotein receptors have also been explored for targeting. Notably, asialoglycoprotein receptors (ASPGRs) expressed on hepatocytes can be targeted with trilvalent N‐acetylgalactoseamine‐ (GalNAc) conjugated siRNA and siRNA‐loaded LNPs (Akinc et al., 2010). GalNAc‐directed LNPs and siRNA both showed improved activity of siRNA in hepatocytes, and subsequently several GalNAc‐siRNA conjugates have entered clinical trials for the treatment of liver associated conditions (Holm et al., 2021). GalNAc‐targeting relies on the specific high surface expression of ASPGR on hepatocytes (~500,000 receptors/cell) which allows for rapid receptor mediated endocytosis of GalNAc conjugates.
4.2.2. Peptide and nucleic acid ligands
Short peptides, while larger than small molecule ligands, still offer similar advantages of low cost and simple conjugation procedures, while benefiting from a greater array of potential targets. The use of cell targeting peptides has been explored extensively in the past decade, most prevalently in the context of cancer and more specifically tumor‐associated antigens (TAAs) (Dissanayake et al., 2017).
Targeting peptides can be derived from native receptor ligands or identified from synthetic library screens. One of the most widely used naturally occurring peptides is arginylglycylaspartic acid (RGD), which is derived from a sequence motif in fibronectin which binds cell membrane integrins (Dehaini et al., 2016). These RGD peptides have garnered interest due to the role of a v β 3 integrin in tumor angiogenesis, with RGD targeted nucleic acid therapies showing improved therapeutic efficacy (Yonenaga et al., 2012). However there have been additional complications with increased uptake by RES and off‐target effects due to interaction of RGD peptides with integrins in healthy organs (Zylberberg et al., 2017). Other notable natural peptides used for targeting have included EGF (Xin et al., 2011) and Angiopep‐2 (Xin et al., 2011) in the delivery of chemotherapeutics and may be readily adapted for nucleic acid therapeutics.
Synthetic peptide ligands can be identified through bacteriophage peptide display screening. Phage display uses a library of randomized peptide sequences with diverse binding properties to identify sequences that bind to the target epitope. Phage display is a widely available technique and has been used to develop CTPs including: anti‐MCF‐7 breast cancer peptide for siRNA‐liposome delivery (Bedi et al., 2011) and anti‐neovascular peptide APRPG (Oku et al., 2002), which has been investigated in targeting both siRNA (Ando et al., 2014) and miRNA (Ando et al., 2014) nanoparticles to tumor microenvironments.
Like polypeptides, both DNA and RNA chains can fold into complex three‐dimensional (3D) structures and can mediate several intracellular functions (known as aptamers) (Dunn et al., 2017). Aptamers are synthetic oligonucleotides 15–100 bases long that can fold to form complex tertiary structures, allowing for the binding of many biological targets with high affinity (Khan et al., 2022). Aptamers are typically functionally selected for using systematic evolution of ligands by exponential amplification (SELEX) (Wilson & Szostak, 1999). The key benefit of this selection method is library construction and screening both occur completely in vitro, allowing a greater number (>1015) of molecules to be screened than transformation‐reliant techniques. Additionally, aptamers can be easily functionalized with conjugation moieties such as aldehyde, amino, and carboxylate at one end of the nucleic acid, or alternatively hybridized with a surface‐bound oligonucleotide anchor, allowing for facile homogenous conjugation to nanoparticle systems for targeting, as well as typically possessing low immunogenetic profiles (Friedman et al., 2013).
Like peptide and protein‐based targeting strategies, most aptamer targeted nanoparticle therapeutics are focused towards cancer cells and tumor markers including nucleolin (L. Li, Hou, et al., 2014), PSMA (Farokhzad et al., 2004) and protein tyrosine kinase 7 (PTK7) (Fang et al., 2019) to improve the therapeutic efficiency of existing chemotherapeutic agents (Urmann & Walter, 2020). Recently an aptamer targeting the cancer membrane protein PTK7 was coupled to glutathione (GSH)‐responsive polymeric micelles for the delivery of a chemotherapeutic (cytarabine) to treat acute lymphoblastic leukemia (Fang et al., 2019). Compared to free cytarabine, aptamer‐targeted micelles exhibited significantly increased cellular uptake and cell apoptosis without any apparent toxicity to normal organs.
4.2.3. Antibodies as ligands
Antibodies (Abs) are one of the most useful biopharmaceutical tools to target specific cells and show excellent capability as targeting ligands in antibody‐drug conjugates due to their hyper‐specificity, availability, and in vivo stability, and therefore have routinely been investigated for use as nanoparticle targeting moieties (Richards et al., 2017). There are already several examples of nucleic acid delivery systems to otherwise intractable tissues in vivo using antibody targeting including intracellular delivery of plasmid DNA to ischemic cardiac myosin with mAb‐2G4‐targeted lipoplexes (Ko et al., 2009), and plasmid DNA delivery across the blood brain barrier in a rat glioma model using dual targeted liposomes coated with anti‐transferrin monoclonal Ab OX26 and anti‐glioma peptide chlorotoxin (Yue et al., 2014).
4.2.4. Antibody fragments
While the complex structure of Abs imparts target specificity, they can be hindered by their large complex structure (150 kDa), potential immunogenicity, production cost, and potential disruption of antibody‐target binding when conjugated to nanoparticles. Additionally, nonspecific uptake of Ab‐targeted formulations by immune cells via Fcg‐receptor‐mediated endocytosis (Gessner et al., 1998) potentially undermines the therapeutic benefits of targeting.
To mitigate these complications, smaller fragments of Abs, lacking Fc‐domains and/or several constant domains have been developed as targeting moieties including single chain variable fragments (scFv), and single‐domain antibodies (sdAb, also referred to as nanobodies) (Bates & Power, 2019). The relative simplicity of these smaller fragments allows for their synthesis by microbial systems, reducing production costs and potential immunogenicity (Pardon et al., 2014). scFvs have shown excellent potential in directing nanoparticle formulations for gene delivery, with two separate anti‐transferrin receptor scFv conjugated liposomes in clinical trials for delivery of gene therapies to tumors (Camp et al., 2013; Pirollo et al., 2008). The lack of Fc‐region in scFv prevents innate immune cell effector functions such as antibody‐dependent cellular cytotoxicity and complement‐dependent cytotoxicity which are normally facilitated through FcgR engagement and uptake, which generally for nucleic acid delivery is disadvantageous. Additionally, with a lack of supporting framework domains, scFv can exhibit poor thermostability and are more aggregation prone than full length counterparts, with the potential for misfolding between VH‐VL domains (Worn & Pluckthun, 1999).
The development of single domain antibody fragments has further advantages over full length Abs. As with scFvs, sdAbs have the Fc‐domains removed, but as they are derived from Camelidae heavy chain Abs, they also lack the variable light chain. This limits potential misfolding between domains as seen in scFv and larger antibody scaffolds (Worn & Pluckthun, 1999), and results in robust antigen binding in a range of conditions, including wide pH range and high temperatures (Ewert et al., 2002). While early in development, these small molecules have demonstrated efficacy in the delivery of gene‐based therapeutics. Gujrati et al. developed sdAb‐coated membrane vesicles for delivery of siRNA to HER2+ HCC‐1954 mouse xenografts, which showed significant inhibition of tumor growth compared to passively targeted siRNA vesicles (Gujrati et al., 2014).
4.2.5. Conjugation strategies
To ensure efficient and stable targeted delivery systems, the targeting ligands must be covalently coupled to the nanoparticle. One commonly employed strategy is EDC or NHS chemistry to couple primary amines to activated carboxylic acids (Sivaram et al., 2018). While this chemistry is widely performed due to the prevalence of amines and carboxylic acids in both the nanoparticles and targeting ligands, the prevalence of the reactive groups is also a significant limitation. When multiple amine and carboxylic acid groups are present, unwanted side reactions can result in cross‐linking of the system, or conjugation of the ligands in suboptimal conformations. Furthermore, the NHS esters are highly susceptible to hydrolysis, making the conjugation reaction very inefficient. Thiol‐maleimide coupling offers a more controlled alternative to attach compounds with a reactive maleimide group to free thiols via a Michael addition (Cal et al., 2014). The Michael addition is more efficient than EDC/NHC reactions, however, thiol groups are susceptible to oxidation and the reducing the disulfides to thiols can change the tertiary structure of proteins, affecting their activity.
To overcome these limitations bio‐orthogonal chemistry can be used. Bio‐orthogonal reactions can occur in biological systems without undergoing side reactions (Sivaram et al., 2018). Strain‐promoted azide–alkyne cycloaddition (SPAAC) and trans‐cyclooctene/tetrazine ligation are two such bio‐orthogonal chemistries that have recently gained popularity due to their rapid reaction rates and readily available linkers to enable this chemistry (Koo et al., 2012). A further advantage of bio‐orthogonal chemistry is that enables site‐specific conjugation of biomolecules to the nanoparticles. This is especially important for the conjugation of proteins, which have a defined 3D structure which is critical for target binding. Conjugation methods that rely on reactions with amines, carboxylic acids, and cysteine residues result in conjugation of proteins to nanoparticles with random and often suboptimal orientations that lower the targeting efficiency. The incorporation of reactive groups in specific positions in the protein has the potential to significantly improve targeting efficiency (Montenegro et al., 2013).
One way to achieve this is to incorporate peptide motifs that induce site‐specific posttranslational enzyme modification that incorporates bio‐orthogonal groups for attachment to the nanoparticles (Wu et al., 2009). Alternatively, peptide motifs that are recognized by transferase enzymes (such as sortase) can be used to modify the C‐terminus of the protein (Hagemeyer et al., 2015). However, the incorporation of these additional peptide motifs can interfere with the protein secondary and tertiary structure, thus limiting the sites where these sequences can be inserted.
This limitation can be overcome by the incorporation of synthetic amino acid residues through genetic code expansion in prokaryotic expression systems, further emphasizing the adaptability of Ab fragments over their full‐length counterparts. By “reprogramming” the amber stop codon, UAG, to incorporate a synthetic amino acid (Mukai et al., 2015), unique functional groups can be incorporated at any desired location within the antibody fragment structure. The site‐specific incorporation of synthetic amino acid p‐azido‐l‐phenylalanine (azPhe) into an anti‐EGFR sdAb resulted in a 6‐fold improvement in binding of conjugated QDots to A549 cells versus randomly orientated sdAb‐conjugated QDots (Yong et al., 2019), demonstrating the importance of considering both conjugation chemistry and protein architecture for targeting purposes.
5. INTRACELLULAR TRAFFICKING
Typically, nanoparticles enter the cell via endocytosis. This cellular process results in the nanoparticle being enclosed in membrane and engulfed into an intracellular vesicle which then matures through the endo/lysosomal pathway (Rennick et al., 2021). There are five main mechanisms by which nanoparticles can be taken up by cells: (1) Clathrin‐mediated endocytosis (CME), (2) fast endophilin‐mediated endocytosis (FEME), (3) clathrin‐independent carrier (CLIC)/glycosylphosphatidylinositol‐anchored protein enriched early endocytic compartment (GEEC) endocytosis, (4) macropinocytosis, or (5) phagocytosis. A sixth mechanism of uptake, caveolin‐mediated endocytosis, has been widely reported in the literature; however, there is growing evidence to suggest this is not a major pathway for cellular uptake (Kiss & Botos, 2009). For targeted systems, endocytosis in nonphagocytic cells is normally linked to CME, FEME, or CLIC/GEEC, whereas if particles nonspecifically bind to cells internalization occurs generally through micropinocytosis and may be associated with membrane turnover. Each of these pathways is distinct, however, it is unclear if the subsequent transport of the cargo is influenced by the internalization mechanism, as there is convergence of these pathways into endosomes and multivesicular bodies.
Less commonly, delivery systems are capable of direct interaction with plasma membrane lipids and can directly deliver their cargo to the cytoplasm (Lee et al., 2020). For nanoparticles enclosed in endosomes, a similar membrane interaction must occur to release the endosomal contents into the cytoplasm, however, this must occur before the lysosomal maturation and cargo destruction. Several physiological changes to the endosome occur during this maturation process, and these can be used to trigger delivery mechanisms built into nanoparticles to achieve stimuli‐responsive release. Nanoparticle entry can be characterized in cultured cells, however, delivery in vivo may be more complex as further complexities in cell physiology are likely to exist in a live organism. Importantly, detection of association or even internalization of nanoparticles into cells (e.g. by flow cytometry or fluorescence microscopy [Smith et al., 2019]) does not imply successful cytoplasmic (or subsequent nuclear or mitochondrial) delivery of a nucleic acid cargo. Cytosolic delivery can be detected by measuring subsequent therapeutic effects; however this gives limited information on how the process occurs and where potential inefficiencies in the process may lie. The successful development of a nucleic acid delivery system should leverage an understanding of these cellular internalization pathways and use feedback generated by these assays to iterate and improve designs.
5.1. Endosomal escape
Materials internalized into the cell must escape the endo−/lysosomal pathway to be therapeutically active. Although escaping the endosome is among the most important barriers to improved therapeutic efficiency, the mechanisms that govern this process remain poorly understood (Selby et al., 2017). Most delivery systems are very inefficient and delivering their cargo to the cytosol, with only approximately 1% of cargo delivered to the cytosol (Gilleron et al., 2013). There are three proposed strategies for endosomal escape (Figure 5): endosomal membrane fusion and endosomal membrane destabilization (by polymers or peptides), and the proton sponge effect (although there is growing consensus that this is unlikely to be a major mechanism for endosomal escape) (Selby et al., 2017).
FIGURE 5.
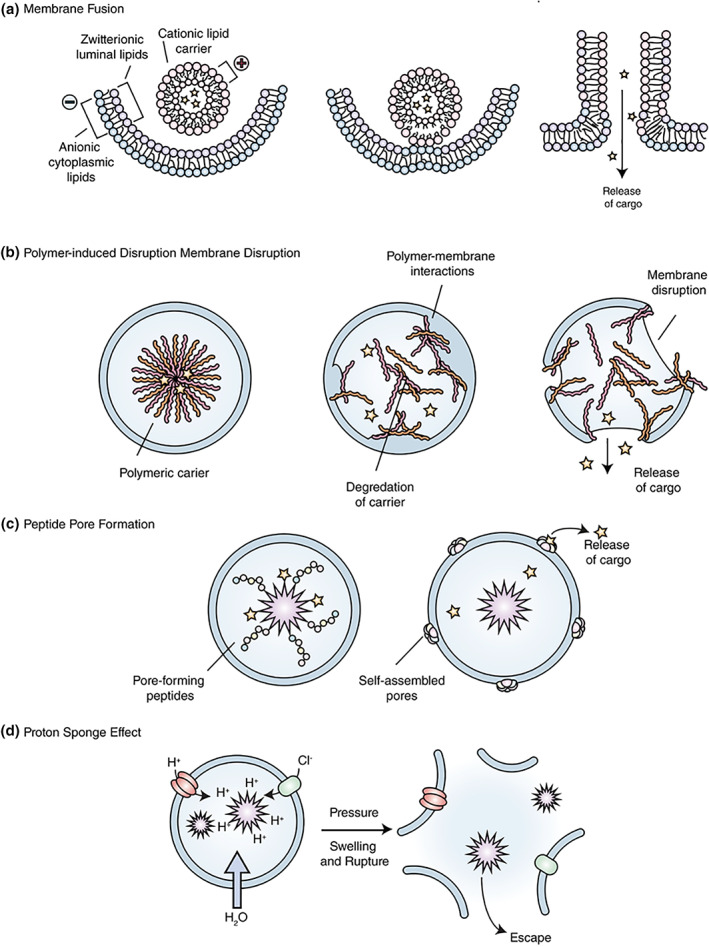
Endosomal escape mechanisms. (a) Membrane fusion – Lipids or amphiphilic materials that can fuse with the endosomal membrane, prompting transfer of the nucleic acid cargo to the cytoplasm. (b) Polymer‐induced membrane disruption – Polymers can interact with the endosomal membrane prompting disruption and cargo escape. (c) Peptide pore formation – Certain peptides interact with the membrane to form pores for the escape of low molecular weight cargoes. (d) Proton sponge effect – Polymers with buffering capacity proposed to induce osmotic rupture of the endosome for cytosolic delivery of the nucleic acid cargo. Figure adapted from Selby et al. (2017).
Membrane fusion is proposed to occur through certain lipids or amphiphilic materials that can fuse with the endosomal membrane, causing inversion of the lipid‐based nanoparticle and delivery of the cargo into the cytosol. Materials can also be engineered to destabilize the endosomal membrane, leading to a local change in membrane stability and permeability. Different pH‐responsive polymers and cell penetrating peptides have been proposed to exploit this pathway. Polymers such as poly(ethylenimine) (Behr, 1997) or poly(amidoamine) (Patil et al., 2009) have been proposed to induce osmotic rupturing of the endosome (proton sponge effect) leading to cytosolic release of the materials. However, evidence for this pathway conflicts with the most recent studies, which suggest membrane fusion or destabilization are the major mechanisms to induce endosomal escape (Selby et al., 2017; Won et al., 2009).
5.2. Measuring endosomal escape
The lack of consensus on the best way to engineer efficient delivery to the cytosol is partly due to limitations with how cytosolic delivery is measured. The focus of most studies has been end‐point assays, such as gene expression or gene knockdown. While these assays provide important information regarding therapeutic response, they neither measure delivery efficiency, nor can they identify where the inefficiencies in the delivery process lie. There is a fundamental need to directly measure endosomal escape in order to determine the most efficient method for improving cytosolic delivery.
Basic ex vivo assays can be employed to rapidly screen materials for the ability to disrupt membrane. Dye‐loaded liposomes can offer a simple way to mimic endosomal membranes and membrane disruption (Smith et al., 2019). Similarly, red blood cell lysis assays can confirm a material's ability to cause membrane disruption. Compared to simpler liposomal models, red blood cell membranes are more complex composed of proteins and polysaccharides in addition to lipids, potentially acting as a better biological model (Wang, 2021), although the lipid and protein composition of the endo/lysosomal membrane differs significantly from the plasma membrane of red blood cells.
In‐vitro fluorescence localization assays attempt to either directly visualize the endosomal escape process or measure changes in colocalization with in the endosomal (e.g. EEA1) or lysosomal (e.g. LAMP1) markers (Johnston, 2017). Visualizing endosomal escape is challenging for several reasons. First, fixation of cells can easily disrupt cell membranes leading to false identification of endosomal escape. Therefore, live cell imaging must be employed. Second, as the efficiency of endosomal escape is so low, detecting very low signal against a high background signal in the endo/lysosomal pathway is challenging. Finally, the lack of colocalization with endosomal markers such as EEA1 or LAMP1 is not evidence of endosomal escape, as the cargo may be compartmentalized in a different vesicle (Johnston, 2017). Incubation with calcein, a membrane impermeable fluorescent dye is also commonly used to screen for endosomal escape (Jones et al., 2003). Calcein appears as punctuate fluorescence when it is internalized into cells, but if endosomal escape occurs, it exhibits bright diffuse signal. Identifying the diffuse signal is highly subjective, and as with all the other techniques, it does not quantify the amount or efficiency of endosomal escape.
Recently, more sophisticated methods have been developed to directly measure endosomal escape. Techniques such as fluorescence correlation spectroscopy (Deprey et al., 2019), split‐GFP complementation (Cabantous et al., 2005; Cabantous & Waldo, 2006), and NanoClick (Peier et al., 2021) have been developed to measure cytosolic delivery, and while they are a significant improvement on indirect methods, they tend to be hampered by low sensitivity, potentially subjective measurements, and can only measure the amount delivered to the cytosol, not the efficiency of delivery. More recently our group developed a Split Luciferase Endosomal Escape Quantification (SLEEQ) assay which can quantify both the amount of material delivered to the cytosol and the endosomal escape efficiency. This technique uses a split luciferase (cytoplasmic LgBit and material tagged HiBit) (Teo et al., 2021) to measure picomolar concentrations of materials delivered to the cytosol and is four orders of magnitude more sensitive than fluorescence assays. Importantly this technique is also able to measure the efficiency of endosomal escape and not just the amount delivered to the cytosol.
5.3. Methods employed to induce endosomal escape
5.3.1. Membrane interaction/fusion
Many enveloped viruses are thought to access the cytosol by lipids in the viral envelope fusing with the phospholipid membrane of the cell, allowing the viral capsid to enter the cytoplasm (White & Whittaker, 2016). In the context of nucleic acid delivery, exploiting these viral mechanisms show significant promise. Lipids like DOPE (1,2‐Dioleoyl‐sn‐glycero‐3‐phosphoethanolamine) have been shown to induce membrane fusion by lipid inversion. This phenomenon has been demonstrated through synchrotron small angle X‐ray scattering (Koltover et al., 1998) and NMR spectroscopy (Hafez et al., 2001). Simulated environments have also been used to observe the fusion of unilamellar vesicles (Mora et al., 2020). It has been proposed that using charged lipids to form nanoparticles (like LNPs or liposomes) stimulates membrane inversion of the lipid‐based nanoparticle structure and release of the therapeutic directly into the cytoplasm (Zelphati & Szoka Jr., 1996).
The use of LNPs for SARS‐CoV‐2 vaccines has highlighted the potential of this technique. Both Pfizer/BioNTech (BNT162b2) and Moderna (mRNA1273) use proprietary lipid formulations and key to their success is incorporation of lipids that can induce cytosolic delivery (Carrasco et al., 2021). There are multiple mechanisms proposed for how this may occur. Cationic lipids could complex to the anionic lipids in the membrane bilayer, causing membrane inversion (Hafez et al., 2001). Ion pairs generated by the cationic lipid interaction with the anionic membrane have been shown to adopt a “cone‐shaped” conformation, prompting inversion of the lipid bilayer, forming a hexagonal (HII) phase (Hafez et al., 2001), as seen in inverted liposomes. Ionizable lipids commonly used for LNP assembly have also been found to induce membrane fusion by becoming ionized at endosomal pH and inducing the same mechanism as cationic lipids (Schlich et al., 2021). While LNPs can efficiently protect nucleic acids from degradation, their endosomal escape ability remains poor. Studies conducted on LNP delivery systems found LNP‐siRNA cytosolic delivery to be <2% and LNP‐mRNA cytosolic delivery to be <1%, both attributing this to poor escape and rapid recycling kinetics (Gilleron et al., 2013; Maugeri et al., 2019; Sahay et al., 2013). Methods to improve this have been considered, with studies focusing on altering lipid properties, such as hydrophilic head group or hydrophobic lipid chain or linker to influence the endosomal escape of the LNPs (Semple et al., 2010), promoting membrane destabilization by ionic interactions of lipids or cationic lipid‐induced membrane fusion (Wittrup et al., 2015). Libraries of lipids have been generated to test the effect of changing lipid characteristics on endosomal escape. Ramishetti et al. conducted a recent study using a combinatorial lipid library in which they designed lipids with novel structural components. They used this library to investigate changes in the ionizable lipid head group and the subsequent effect on endosomal escape (Ramishetti et al., 2020). Ultimately, they found that transfection efficiency was improved when using LNPs generated from ionizable lipids with two ionizable amine groups (piperazine groups) compared to those with only one tertiary amine.
5.3.2. pH‐responsive polymer membrane disruption
Polymers engineered to promote membrane interactions have significant potential to improve endosomal escape. One of the earliest examples of pH‐responsive endosomal escape polymers are poly(propylacrylic acid) (PPAA) and poly(butyl acrylate) (Pack et al., 2005). PPAA contains pendent hydrophobic propyl (‐C3 alkyl) groups hypothesized to coat the membrane through nondestructive intercalation. This combined with the carboxylate moieties of PPAA which become protonated at acidic pH, trigger a change in solubility toward a more hydrophobic, membrane disruptive state (Evans et al., 2019). The membrane destabilizing properties of PPAA was shown by Murthy et al. using red blood cells to assess the hemolytic activity of PPAA (Murthy et al., 1999). From these experiments PPAA was found to induce hematolysis at acidic pH, which was thought to occur by the formation of membrane pores.
Evans et al. have investigated the use of PPAA for induced membrane destabilization and cytoplasmic delivery (Evans et al., 2019; Evans, Hocking, Kilchrist, et al., 2015; Evans, Hocking, Osgood, et al., 2015). Most recently, they looked at PPAA poly‐complexes formed with peptides, nucleic acids, gene‐editing proteins and nanoparticles (Evans et al., 2019). Using a split‐GFP assay, they demonstrated that PPAA rapidly triggers endosomal disruption, driven by the pH change in the endosome, driving PPAA to adopt a hydrophobic membrane‐active confirmation. GFP peptide co‐delivered with PPAA showed a 5.6‐fold increase in cytoplasmic delivery compared to free GFP peptide. However, cells pretreated with PPAA before GFP peptide showed a 13.6‐fold increase in cytoplasmic accumulation compared to the free GFP peptide. Because this peptide is amphipathic, they hypothesized that the more hydrophobic nature of the peptide may compete with the PPAA interaction with the endosomal membrane, reducing the potentiation of endosomal escape, explaining why pretreatment sees superior cytoplasmic delivery.
PDEA and PDPA are part of a family of pH‐responsive polymers that contain tertiary amines that are being increasingly employed for use in nanoparticle delivery systems. PDEA‐PEG block‐copolymers were initially developed for self‐assembly into polymeric micelles, which showed high stability at pH 7.4 but rapid disassembly at endosomal pH (pH 5.8) (Convertine et al., 2010). Membrane disruption was confirmed using a red blood cell lysis assay, where nanoparticles only induced lysis at low pH. Continuing on from this work, by tuning the ratio of PDPA to PDEA, Such et al. constructed an library of pH‐responsive polymer nanoparticles with controlled disassembly between pH 4.9 and 7.2 (Kongkatigumjorn et al., 2018). All the particles induced red blood cell lysis at ~0.5 pH unit above their disassembly pH. However interestingly, particles that disassembled at pH 7.2 or pH 4.9 induced more endosomal escape of calcein than those that disassembled between pH 5.8 and 6.6. To further investigate this phenomenon we applied the SLEEQ technique to quantify the endosomal escape efficiency of nanoparticles (Beach et al., 2022). This work demonstrated that by optimizing the PDEA/PDPA ratio, the endosomal escape efficiency could be improved by more than 5‐fold. Interestingly, even the optimized nanoparticles only showed approximately 10% endosomal escape efficiency, indicating that there is significant scope to further improve the endosomal escape efficiency of polymer nanoparticles (Figure 6).
FIGURE 6.
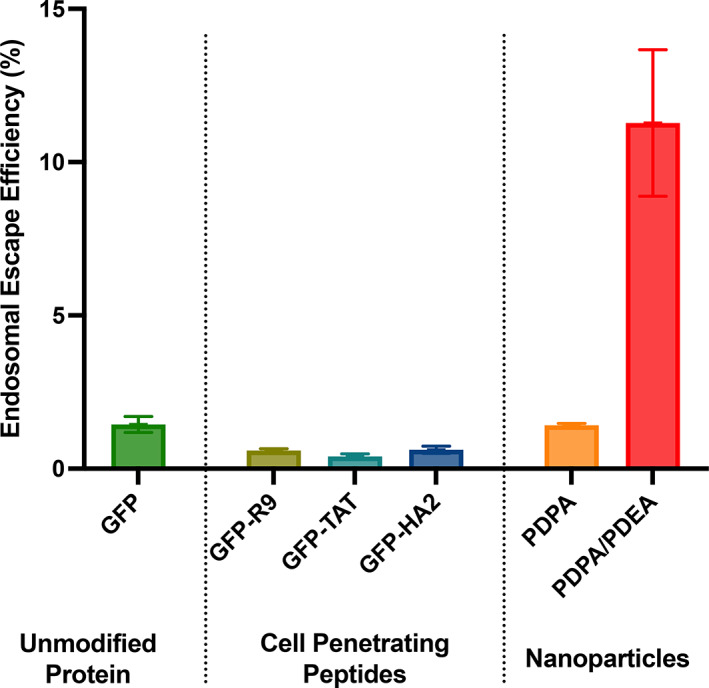
Endosomal escape is an inefficient process. Only a small amount of protein (e.g., GFP) endocytosed by a cell reaches the cytosol. Fusing cell penetrating peptides to GFP can increase the amount of protein delivered to the cytosol, but this is largely due to greater nonspecific association rather than improving the endosomal escape efficiency. Optimizing the composition of nanoparticles can increase endosomal escape efficiency, but the process remains inefficient. Data adapted from Teo et al. (2021) and Beach et al. (2022).
5.3.3. Peptide‐induced escape
An alternative approach is to incorporate cell‐penetrating peptides (CPPs) into the delivery system to facilitate cytosolic delivery. CPPs are typically derived from viruses and mimic the native pathways these infective agents use to enter cells. The term CPPs broadly refers to peptides that increase delivery to cells, however in many cases the increased accumulation results mostly in accumulation in lysosomes, rather than delivering the cargo to the cytosol.
Arginine‐rich CPPs, derived from viral or bacterial proteins are commonly employed to facilitate endosomal escape (Appelbaum et al., 2012). The guanidinium functional group from arginine is thought to complex to the membrane phospholipids' phosphate groups, through a combination of hydrogen bonding and electrostatic interactions. Numerous studies have demonstrated the ability of functional arginine groups on CPPs and other arginine‐functionalized materials to interact with phospholipid bilayers (Herce et al., 2009; Pantos et al., 2008). The most common example of this class of CPP is TAT, which is derived from the Trans‐Activator of Transcription protein of human immunodeficiency virus (Romani et al., 2010). It is thought that upon internalization, TAT is trafficked to Rab5+ early endosomes, transitioning into Rab7+ late endosomes before it can induce escape (Appelbaum et al., 2012; Brock et al., 2018; Erazo‐Oliveras et al., 2016). Brock et al. demonstrated that TAT‐induced endosomal disruption is dependent on the number of TAT peptides, with 2 or 3 TATs significantly more likely to induce endosomal escape than a single TAT (Brock et al., 2018). While TAT alone appears to provoke reasonable endosomal escape, TAT‐functionalized materials have not seen the same degree of success (Erazo‐Oliveras et al., 2014; Teo et al., 2021). This inefficiency could suggest the need for TAT to be untethered to induce escape or indicates that TAT may remain bound in the endosomal membrane inhibiting the escape of the attached material.
Another class of CPP is those that do not possess arginine residues and are instead thought to enable endosomal disruption by inserting themselves into the phospholipid membrane. Many viruses possess a unique ability to facilitate infection through fusogenic peptides. The influenza virus can do so through its surface presented hemagglutinin (HA) protein which induces receptor‐mediated endocytosis (Erazo‐Oliveras et al., 2012). HA2, the second subunit of the HA protein, possesses the highly conserved N‐terminal region (residues 1–23) responsible for mediating membrane fusion. Endosomal escape peptides derived from the HA2 subunit have shown success in inducing escape when in the free form. However, the same efficiency of endosomal escape is not seen for HA2 functionalized protein cargoes (Lee et al., 2011). Lee et al. found that while endosomal destabilization was induced by the HA2 peptide, the HA2‐protein fusion remained localized in the endosome, likely a result of the HA2 remaining anchored in the membrane (Lee et al., 2011). Synthetic escape peptides are also used to promote endosomal escape, such as GALA, a synthetic 30‐residue amphiphilic peptide. It is designed to have a repeat sequence Glu‐Ala‐Leu‐Ala which becomes protonated at low pH causing it to adopt an alpha‐helical conformation at low pH allowing it to provoke membrane destabilization (Li et al., 2004).
Efficacy of endosomal escape induced by CPPs when fused to proteins has been investigated by our group using the SLEEQ assay (Teo et al., 2021). While free CPPs may show efficient endosomal escape, when fused to GFP, none of the CPPs tested improved on the endosomal escape efficiency of unmodified GFP (which showed ~2% endosomal escape efficiency, Figure 6). Cationic CPPs increased the total amount material delivered to the cytosol, but they appeared to do this by increasing total association to the cell, rather than improving the efficiency of cytosolic delivery. Nonspecific association of CPPs to the cell membrane may improve cytosolic delivery in vitro, however in vivo these materials are likely to become trapped at the injection site and have poor biodistribution.
5.4. Delivery to the nucleus
While there is an array of nucleic acid therapeutics that act in the cytoplasm, some ASOs (ssDNA) and double‐stranded DNAs (plasmid DNA) act in the nucleus. Engineering materials to cross the nuclear envelope, especially in nondividing cells remains challenging. Nuclear entry in dividing cells is largely facilitated by the disassembly of the nuclear envelope during mitosis which enables nuclear entry (Dean et al., 2005). However, many cells targeted for gene therapy do not undergo division during gene transfer, which means active transport mechanisms are required.
Active transport into the nucleus may be gained through a nuclear pore complex; a group of proteins that tightly regulate what passes through into the nucleus (Bastos et al., 1995; Knockenhauer & Schwartz, 2016). Traversing the nuclear pore remains a significant challenge for nucleic acid delivery. Microinjection studies using “naked” nonviral vectors have been used to demonstrate the difficulty of nuclear entry. Graessman et al. showed that following cytoplasmic microinjection of 1000–3000 vector copies, gene expression was <3% of the levels found following nuclear microinjection (Graessmann et al., 1989). Findings such as this have been confirmed by other studies in different mammalian cell types (Capecchi, 1980; Mirzayans et al., 1992; Thorburn & Alberts, 1993). Poor gene expression seen following cytoplasmic injection is in part attributed to the inability to cross through the nuclear pore complex but is also the result of the rapid degradation of DNA trapped in the cytoplasm by cytoplasmic nucleases. A study conducted by Lechardeur et al. found that the half‐life of cytoplasmic DNA in HeLa cells to be approximately 90 min (Lechardeur et al., 1999). This rapid degradation of DNA ultimately reduces the DNA available for nuclear entry.
Initiating transport through the nuclear pore can be achieved by a nuclear localization signal. These are short peptides that can be composed of 8–40 amino acids depending on the species from which the signal was derived (Lu et al., 2021). Most NLS appear to execute nuclear import in a similar fashion, being recognized by multiple cytoplasmic importins which allow passing through the nuclear pore and release in the nucleus by nucleoporins (Lu et al., 2021).
5.5. Delivery to the mitochondria
Mitochondria play an essential role in regulating an array of cellular functions including energy (ATP) transduction, REDOX signaling, hormone biosynthesis, inflammation, or cell death (Herst et al., 2017). Mutations in mtDNA can result in abnormal proteins or deficiency in a protein, causing changes in electron‐transfer chain enzymes leading to production of reactive oxygen species (Dard et al., 2020). Many diseases such as neurodegenerative diseases, diabetes, cardiovascular diseases and cancers, as well as mitochondrial disease, have been linked to mtDNA mutations (Fulda et al., 2010; Slone & Huang, 2020). As such, effectively targeting gene therapies to the mitochondria can enable long‐term treatment of these diseases.
Abnormal mitochondrial genes exported from the nucleus and signaled mitochondrial localization can be silenced or replaced in the cytoplasm by cytoplasmic acting gene therapies (see Section 2.1). However, targeting mitochondrial DNA or RNA needed for proteins that are synthesized in the mitochondrial matrix requires entry into the mitochondria. This can be achieved using a leader sequence derived from that of nuclear‐encoded mitochondrial DNAs, which are recognized by mitochondrial membrane translocators, made up of inner mitochondrial membrane and outer mitochondrial membrane translocases (Jaremko et al., 2014; Lee et al., 2008). Early studies conducted on this process by Seibel et al. showed that dsDNA (up to 322 bp) conjugated to an ornithine carbamoyl transferase leader sequence was able to enter the mitochondrial matrix through the translocator (Seibel et al., 1995). In the mitochondrial matrix, DNA (either imported from outside the matrix or mtDNA) is transcribed into RNAs by mitochondrial machinery and in subsequently synthesized into the encoded protein (Fox, 2012). Correcting the effects of mutated mtDNA can be achieved by importing DNAs and RNAs for synthesis of functional proteins.
5.6. Intercellular transport
Intercellular transport, the process of transferring material from one cell to another, also plays an important role in the function of nucleic acid trafficking. There is evidence to suggest that nucleic acid therapeutics delivered to a target cell can be repackaged and transported to other different cell (Ridder et al., 2015). Packaging of the nucleic acid into extracellular vesicles is dependent on the sequence motifs and/or secondary configuration of the nucleic acids (O'Brien et al., 2020). These sequences are recognized by nucleic acid‐sensing molecules, which initialize transport of the nucleic acid to the cell surface where they are packaged into exosomes. Multiple mechanisms have been proposed to explain recipient cell uptake of extracellular vesicles; however, how a particular cell will respond to an incoming vesicle is not clear (Mathieu et al., 2019). This process is likely linked to the presence of specific proteins and lipids on the vesicular surface, triggering different cellular internalization mechanisms into the recipient cell (O'Brien et al., 2020). Nucleic acid therapeutics can be repackaged and transported intercellularly by the same pathways (Ridder et al., 2015). It is however worth noting that extracellular vesicles act to recycle intracellular material that already contain a number of proteins and nucleic acids, thus it is unclear how much additional cargo therapeutic cargo can be loaded inside them (O'Brien et al., 2020).
6. ENGINEERING PARTICLE DISASSEMBLY AND CARGO RELEASE
To elicit a therapeutic effect, the carrier vehicle must release the nucleic acid cargo at the site of action. This is challenging as many of these material nanoparticle systems rely on electrostatic interactions to encapsulate the nucleic acid cargoes, which are not easily switched off. Different methods have been used to attempt to promote dissociation of the material from the nucleic acid using degradation of the nanoparticle. There is an array of interesting material science studies conducted on releasing the nucleic acid cargo from the “smart” nanoparticle carrier system (Yu et al., 2021). Different mechanisms to do this include external application of light (H.‐J. Li, Wang, et al., 2014; Liu et al., 2009), ultrasound (Kumar et al., 2021), different temperatures (Yang et al., 2015), or magnetic fields (Yang et al., 2016). While there are specific applications for using these external stimuli to trigger release, applying these triggers for most vaccines and gene therapies remains unfeasible. As such, release mechanisms induced by changes in the cellular microenvironment, such as changes in pH, redox environment, or the presence of enzymes have potential to maximize release at the site of action within the cell.
6.1. pH‐induced release
As outlined in Section 5.3.2, pH‐responsive nanoparticles can induce endosomal escape, a process which is often coupled with the release of cargo from the nanoparticle. Other pH‐sensitive components can also be engineered onto the nanoparticle to facilitate particle degradation and cargo release. One approach is to incorporate pH‐sensitive linkers to attach targeting moieties or PEG. Xu et al. developed block copolymer PEG‐Dlinkm‐PLGA nanoparticles which contains an acid‐sensitive Dlinkm linkage between the two blocks (Xu et al., 2016). These siRNA‐loaded nanoparticles accumulate at the tumor site via the EPR effect. The low pH of the tumor environment induces cleavage of the PEG layer which facilitates cellular uptake of the particles. Degradation of the PLGA once the particles are internalized into the cell then induces release of siRNA. PEG was effectively released at pH 6.5 (~60%), mimicking the tumor microenvironment, whereas less PEG was released at pH 7.4 (~18%). In vitro studies using different polymers was also shown to affect the intracellular release of siRNA, whereby nanoparticles with a PLGA layer showed faster release (~30%) of siRNA than control nanoparticles (~11%) with a PLA layer.
6.2. Reduction‐triggered release
Nanoparticle systems can be engineered with redox sensitive linkages such as disulfides. Disulfides are readily reduced in the cytoplasm, where the reduction potential is ~100‐fold higher than the extracellular environment due to the high concentration of reducing agents like GSH (Du et al., 2010). Xu et al. developed a redox‐responsive poly(disulfide amide) (PDSA) nanoparticle with PEG groups and encapsulated siRNA against tumor proteins KIF11 (Xu et al., 2018). Redox responsive nanoparticles decreased expression of model target (luciferase) by ~95%, while nonredox responsive nanoparticles loaded with siRNA only showed ~60% knockdown. Similar nanoparticles were loaded with siRNA against KIF11 and tested in prostate cancer (PC3) cells and showed suppression of KIF11 expression (red stain) with a low (5 nM) siRNA dose (Figure 7a, b). In vivo results showed similar knockdown efficiency, with PEG‐PDSA‐siKIF11 nanoparticles achieving ~85% knockdown of KIF11 expression following consecutive 3‐day IV injection dosing period.
FIGURE 7.
Silencing of an oncogene (KIF11) using redox‐responsive PEG‐PDSA nanoparticles. Redox‐sensitive polymeric PDSA nanoparticles were loaded with siRNA (siKIF11) engineered to degrade and release siKIF11 upon exposure to cytoplasmic GSH. KIF11 expression levels were analyzed in PC3 cells treated with siKIF11‐PDSA nanoparticles by (a) Western blot and (b) immunofluorescence analysis with (i) control nanoparticles and (ii) siKIF11‐loaded PDSA nanoparticles at a 5 × 10−9 m siRNA dose. Scale is 30 μm. Reprinted with permission from Small (Xu et al., 2018).
6.3. Release triggered by enzymatic cleavage
Nanoparticle delivery systems can also be engineered to include enzyme cleavage sites that induce nanoparticle degradation upon exposure to the relevant enzyme. A range of enzymatic cleavage sites can be engineered into nanoparticles, including peptidase, protease, lipase, or glycosidase sites (de la Rica et al., 2012). A class of enzymes commonly used for enzyme responsive nanoparticles are matrix metalloproteinases (MMPs); a class of endopeptidase that cleaves peptide bonds between nonterminal amino acids (Mathew et al., 2017). These enzymes are over‐expressed in inflammatory diseases and cancers. A number of enzyme‐responsive nanoparticle delivery systems exploit upregulation of specific enzymes at the target site to induce PEG‐shedding from the nanoparticle and promote subsequent cell association and internalization. This has been achieved using MMP‐cleavable peptide‐linked block copolymers to form micelles for vector delivery (Li et al., 2013), MMP‐cleavable PEGylated pDNA‐nanoparticles (Yingyuad et al., 2013), and MMP‐cleavable PEGylated liposomes for siRNA delivery (Hatakeyama et al., 2011), all of which saw improved tumor penetration and therapeutic efficacy of the nucleic acids.
Combination approaches can also be implemented, whereby different mechanisms are triggered at different stages. This is especially relevant in the tumor microenvironment, where the differences from the normal cell microenvironment can be exploited (J. Li, Yu, et al., 2014; Wang et al., 2016). Wang et al. developed a reduction‐ and pH‐sensitive polymeric nanoparticle for tumor‐targeted delivery of siRNA (Wang et al., 2016). In this study, triblock folate‐PEG‐PAsp(MEA)‐PAsp(DIP) polymer nanoparticles were formed, encapsulating siRNA in the core. Folate was employed to target over expression of the folate receptor on the cells of interest, and the PAsp(DIP) block was engineered to promote endosomal escape. The central PAsp(MEA) block was engineered to degrade in the presence of cytoplasmic GSH, which enables siRNA release. In this system the nanoparticles are stable at pH 7.4, which allows siRNA encapsulation and delivery to target cells. Once internalized into the cell, the pH of the endosome drops to 5.0, where PAsp(DIP) induces endosomal rupture, and nanoparticle release into the cytoplasm, where GSH triggers siRNA release. In vitro studies showed that delivery of the anti‐luciferase siRNA using these nanoparticles to Bel‐7402 cells showed ~70% luciferase knockdown (Wang et al., 2016).
7. APPLICATIONS OF NUCLEIC ACID THERAPIES
7.1. Cytoplasmic therapies
7.1.1. RNA and ssDNA gene therapies
siRNAs and ssDNA ASOs act in the cytoplasm to degrade or block the activity of a target mRNA sequence linked to a cancer or genetic disease. With siRNAs inducing more efficient knockdown than ASOs (Roberts et al., 2020), they have become the prevalent choice for silencing nucleic acid therapies. siRNAs can be implemented for a wide range of disease states and have been extensively tested in the treatment of cancer (Gao et al., 2021). The versatility of siRNA is broad, having been investigated in a range of cancer models spanning from liver cancer (Springer & Dowdy, 2018) to Triple‐Negative breast cancer tumors in mice (Vaidya et al., 2019). siRNA is also approved for treatment of genetic conditions such as hereditary amyloidogenic transthyretin (hATTR) and acute hepatic porphyria, with the potential for use in many other conditions (Hu et al., 2020).
Due to the propensity of nanoparticles to accumulate in the liver, siRNA has been used for treatment of hepatocellular carcinoma in mouse models. Khan et al. used PEG and GalNAc functionalized PLGA nanoparticles to load anticancer siRNA, specifically with the ability to silence survivin, an overexpressed protein in liver cancer cells (Khan et al., 2019). siRNA delivered using the nanoparticles induced a ~50% reduction in survivin mRNA (Figure 8a), extending the survival of the mice by 12 weeks compared to those treated only with free siRNA. This is aided by the stealth properties of PEG and the highly efficient targeting of liver cells using GalNAc functionalization as demonstrated in vitro (Figure 8b).
FIGURE 8.
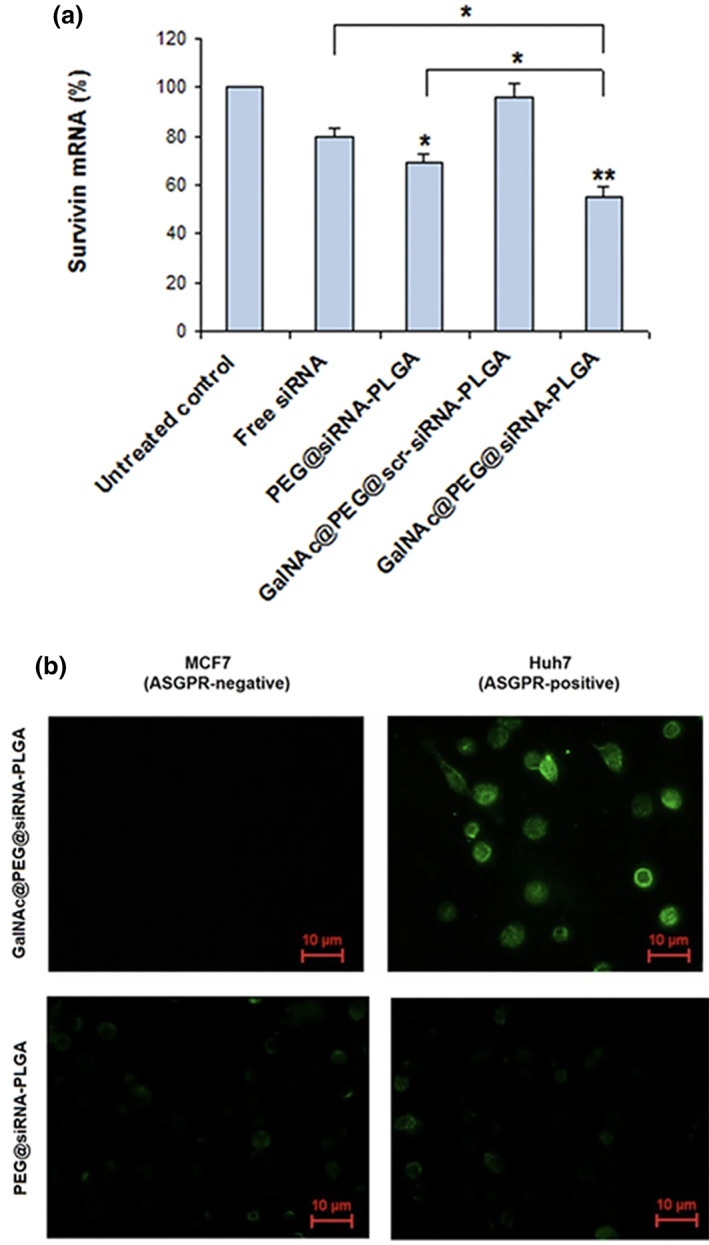
The combination of encapsulation in PLGA nanoparticles and GalNAc targeting improves the therapeutic response from siRNA in vivo. (a) in vivo knockdown of survivin expression in the liver of mice treated with GalNAc and PEG functionalized PLGA nanoparticles with encapsulated siRNA (GalNAc@PEG@siRNA‐PLGA) (b) specific targeting of GalNAc@PEG@siRNA‐PLGA to Huh7 and MCF7 cells incubated with fluorescently labeled (green) PEG@siRNA‐PLGA or fluorescently labeled GalNAc@PEG@siRNA‐PLGA. Scale bar is 10 μm. Reprinted with permission from Khan et al. (2019).
Nonliver targets remain difficult due to the biodistribution of nanoparticle formulations and the low dose of siRNA received by nonhepatic cells. However, the conjugation of targeting ligands to siRNA delivery systems can improve delivery beyond the liver. To bolster activity and improve delivery of chemotherapeutic DOX for anaplastic large‐cell lymphoma therapy, Zhao et al. developed a multifunctional self‐assembling CD30 DNA aptamer targeted siRNA/DOX nanomedicine (Apt‐NMed). In a mouse xenograft model, Apt‐NMed specifically bound and internalized into tumor cells via CD30 aptamer targeting, triggering concomitant doxorubicin release and anaplastic lymphoma kinase oncogene silencing, resulting in an average tumor volume 6‐fold lower than in mice treated with free doxorubicin following the treatment regime, as well as no noticeable changes to body weight (Zhao et al., 2018). These Apt‐NMed particles are an excellent example of cutting‐edge multifunctional particles, which can exploit gene modulation by nucleic acid therapeutics for the enhancement of existing formulations (Sangtani et al., 2017). Such formulations have been investigated more extensively in the context of cancer, specifically for the resensitization of tumors to chemotherapeutic agents such as doxorubicin (Cheng et al., 2012).
7.1.2. RNA vaccines
The most prominent application of nucleic acids as therapeutics has been the development of mRNA vaccines in response to the COVID‐19 pandemic. mRNA vaccines can be rapidly tailored to virtually any disease state, as highlighted by the development of two mRNA vaccines against SARS‐CoV‐2 by Pfizer/BioNTech (BNT162b2; Polack et al., 2020) and Moderna (mRNA1273; Baden et al., 2021) in less than a year. Unlike traditional protein antigen‐based vaccines, nucleic acids are a consistent and biochemically defined payload, regardless of the proteins encoded in their sequence. This enables the antigens that the mRNA encodes for to be tweaked in response to mutations, without the need to completely reformulate the delivery system. There is significant potential to adapt the mRNA vaccines developed in response to SARS‐CoV‐2 and apply them to other infectious diseases like influenza and to develop therapeutic vaccines for cancer (Pardi et al., 2018).
The initial recognition and subsequent signaling by the innate immune systems is prerequisite in establishing an adaptive immune response. Central to this innate signaling are dendritic cells (DCs), which specialize in the capture and presentation of foreign antigens to T lymphocytes (Steinman & Hemmi, 2006). Antigen presentation occurs via two pathways mediated either by MHC‐I or MHC‐II. The MHC‐II response aids the humoral (antibody) response by activating CD4+ T‐cells and is triggered when antigen is loaded onto MHC‐II molecules in the lysosome (Roche & Furuta, 2015). In contrast, MHC‐I triggers a cytotoxic T‐cell response by activating CD8+ T‐cells and antigen is loaded into MHC‐I in the cytosol (Reuter et al., 2015). Most protein antigen‐based vaccines rely primarily on the MHC‐II mediated humoral response; however, there is significant potential to improve the effectiveness of vaccines if a cytotoxic T‐cell response can be induced as well. This is particularly important for the development of therapeutic anti‐cancer vaccines.
The LNP‐mRNA vaccines for COVID‐19 are not targeted to specific cells and are thought to exploit both the sustained expression of antigen at the site of administration as well as delivery to antigen presenting cells. A proportion of the LNP‐mRNA vaccines are delivered to myocytes at the site of administration (intramuscular), which can act as factories that produce SARS‐CoV‐2 spike protein (S protein). Antigenic protein expressed in this manner works in a similar fashion to conventional protein‐based vaccines, where exogenous antigens are internalized, processed and presented by APCs on MHC‐II complexes. Recognition causes subsequent activation of CD4+ T cells, which help modulate B cell antibody production (humoral response) (Painter et al., 2021). The CD4+ response generated by the LNP‐mRNA vaccines is likely a result of the translated protein antigens being released from myocytes, either through excretion or cell lysis, and subsequently reinternalized by professional APCs causing MHC class II antigen presentation (Leung, 2015; Rijkers et al., 2021; Wadhwa et al., 2020).
Antigen‐encoding mRNA delivered to the cytosol of APCs also can induce an MHC‐I response (Wadhwa et al., 2020), and subsequent CD8+ T cell activation. LNPs that are not taken up by myocytes at the injection site can encounter APCs either by drainage to lymph nodes or peripheral APCs within muscle. Interestingly, LNP‐mRNA vaccine generated in response to SARS‐CoV‐2 show variable, steadily increasing CD8+ antigen specific responses following prime boost doses (Painter et al., 2021), and appear to confer long‐term infectious immunity through a more robust antigen‐specific CD4+ responses (Anderson et al., 2020; Oberhardt et al., 2021; Sahin et al., 2020).
In addition to antigen sampling by MHC‐I in the cytosol, exogenous antigen can be loaded onto MHC‐I by specialized cross‐presenting dendritic cells, however the mechanism by how this occurs is not fully understood (Cruz et al., 2017). Myocyte‐derived antigens have previously been implicated in a DC‐dependent CD8+ cross‐priming mechanism and may involve release of antigen‐associated apoptopic bodies from transfected myocytes, contributing to antigen‐specific CD8+ priming (Lazzaro et al., 2015). Robust antigen‐specific CTL responses require effective delivery of LNP‐mRNA to DCs either in the periphery tissues or lymph node resident DCs. It is not yet clear which route of delivery contributes more to eliciting SARS‐CoV‐2 CD8+ responses, although it is likely that both play a role in MHC‐I cross‐presentation and must be considered in both future prophylactic and therapeutic vaccine designs.
mRNA has also been investigated for cancer immunotherapy, whereby TAA‐encoding mRNA is delivered to DCs to initiate antigen‐specific immune responses (Pardi et al., 2018). mRNA has shown durable anticancer immunity when injected intratumorally (Hewitt et al., 2019). Hewitt et al. used LNP‐encapsulated mRNA encoding for three different TAAs to act on tumor localized professional antigen‐presenting cells (Hewitt et al., 2019). In vivo intratumor delivery of the triplet mRNA showed early activation of T lymphocytes in the tumor draining lymph nodes, with increased CD4+ and CD8+ T cells detected 7 days following injection compared to negative controls (Figure 9).
FIGURE 9.
Tumor‐associated antigen LNP‐mRNA vaccine. Lymphocyte numbers (CD4+ and CD8+ T‐cells) increased in response to intratumoral administration of LNP‐mRNA (either 5 μg OX40L, 2.5 μg of combination IL‐23/IL‐36γ mRNAs or 1.67 μg of triplet mRNAs [IL‐23/IL‐36γ/OX40L]). The high immune cell recruitment into tumors triggered by triplet mRNA vaccine enabled effective tumor destruction, suggesting that all three mRNA components are required to elicit maximal and durable T cell infiltration into tumors. EM, effector memory; CM, central memory; N, naïve. Reprinted with permission from Science (Hewitt et al., 2019).
The targeting of DC endocytic receptors has been investigated in attempts improve ex vivo DC cancer therapies and in vivo cytosolic delivery of cancer vaccines in attempts to bolster tumor‐specific CTL responses (Fossum et al., 2020). Notably, peptide‐antigens internalized by C‐type lectin receptors on DCs, namely DEC205 (Bonifaz et al., 2004; Dhodapkar et al., 2014) and CLEC9A (Caminschi et al., 2008), have shown the ability to induce robust antigen‐specific CD8+ T lymphocyte cross‐priming. These targets have since been investigated as means for delivery of peptide‐based DC‐targeted nanoparticle vaccines for several cancers (Nagy et al., 2021; Saluja et al., 2014).
The in vivo targeting of DC receptors with nanoparticle formulations has the potential to improve existing mRNA vaccine platforms. Active targeting through ligand‐conjugation may increase the uptake of nucleic‐acid therapeutics by DCs, and importantly modulate intracellular trafficking pathways following receptor binding for a more desirable immune outcome. DC‐targeted nucleic acid therapies may have applications outside to context of cancers and infectious diseases, such as the induction of tolerance in auto‐immune conditions (Petzold et al., 2012), or immunomodulation by siRNA for treatment of pro‐inflammatory diseases (Katakowski et al., 2016).
7.2. Nuclear therapies
7.2.1. DNA‐based gene therapies
Delivery of a DNA transgene for expression requires trafficking through the cytoplasm and then delivery into the nucleus. Due to the rapid degradation kinetics of unprotected DNA, delivery systems are often included features to improve nuclear localization and transgene expression. Kataoka et al. have conducted several studies using PIC micelles for DNA delivery (Han et al., 2009; Harada‐Shiba et al., 2002). In one study, they used PIC micelles with a luciferase transgene for in vivo delivery in mice (Harada‐Shiba et al., 2002). They showed that when injected into mice tail veins, most of injected ‘naked’ DNA was degraded within 5 min. However, when delivered using PIC micelles, DNA remained present in the blood for up to 3 h and gene expression was observed in the liver for 3 days postinjection.
DNA in the form of ASOs can also require delivery to the nucleus. Unlike cytoplasmic‐acting ASOs which target mature mRNAs directly, ASOs in the nucleus target pre‐RNA for degradation. As a result, RNA levels are not replenished, which ultimately reduces the levels of cytoplasmic mature mRNA through natural RNA decay pathways (Clark et al., 2012). Splice‐altering ASOs, such as those already approved by the FDA require direct injection to the site of action (e.g. Spinraza™ [Abbas et al., 2022] for SMA requiring intrathecal injection), due to the extent of degradation that occurs if intravenously administered (Roberts et al., 2020). As such, other material‐based delivery systems are being investigated to improve delivery and efficacy of ASOs. Guan et al. used iRGD (a tumor penetrating peptide) functionalized liposomes for splice altering ASO delivery to cancerous tumors in vivo (Guan et al., 2021). ASO encapsulation into the iRGD‐liposomes significantly enhanced ASO tumor suppression, with 4× more tumor growth observed when using free ASO.
7.2.2. CRISPR genome editing
In addition to targeting therapeutic DNA to the nucleus for transgene expression, genomic DNA located in the nucleus can act as the target for gene editing therapies. CRISPR gene editing systems require at least two components: CRISPR endonuclease Cas9 and an RNA guide strand (gRNA) to target the Cas9 component to the correct position in the DNA (O'Driscoll & Jeggo, 2006). A repair template is also sometimes required (Aird et al., 2018). Alterations to targeted DNA occur by endonuclease‐introduced double stranded breaks which can be repaired by one of two mechanisms: homology‐directed repair or nonhomologous end‐joining (O'Driscoll & Jeggo, 2006), introducing small insertions or deletions. Different methods can be used to deliver the CRISPR/Cas9 system. A plasmid‐based strategy can be employed whereby the Cas9 gene, and the gRNA are encoded and synthesized in the cell (Doudna & Charpentier, 2014). Cas9 can also be delivered as mRNA for translation or as the protein alongside the gRNA strand (Li et al., 2020). In all instances, transport of the Cas9 protein and gRNA require transport into the nucleus. In vivo gene editing by Cas9/gRNA pDNA delivery has been achieved in mouse models for liver protein using LNPs (Finn et al., 2018) and in mouse macrophages using polymeric nanoparticles (Luo et al., 2018).
7.2.3. DNA vaccines
DNA‐based vaccines have mostly been developed with viral vectors. While viruses do permit facile entry into the nucleus, cytotoxicity issues associated with viral vector delivery systems have prompted the investigation into alternative delivery systems for DNA‐based vaccines (Patil et al., 2005). The immune response generated by DNA‐vaccines is similar to the response generated by mRNA vaccines, however the antigen response is typically slightly delayed (as they require nuclear translocation and transcription into mRNA) and last longer (as DNA is less susceptible to degradation than mRNA).
DNA vaccines have been widely explored as therapeutic cancer treatments, specifically for delivery of anti‐cancer TAAs. Moku et al. used DC‐targeting LNPs to deliver plasmid DNA encoding for a melanoma TAA (Human MelanA antigen) (Moku et al., 2021). Mice vaccinated with the three doses of LNP‐DNA vaccine exhibited no tumor growth when challenged with the B16F10 tumor cells, an effect lasting up to 120 days in vaccinated mice, while all mice in the control groups (PBS and LNPs with scrambled DNA) saw tumor growth. Cellular immune responses also showed approximately four times higher IFN‐y and IL‐4 were measured in mice vaccinated with LNP‐MelanA compared to the LNP‐DNA control. With IFN‐y being secreted by Th1 cells, high levels of this cytokine were attributed to the LNP‐DNA vaccine ability to activate DC MHC Class I and II presentation, leading to downstream CD4 T helper cell (Th1 and Th2) activation.
7.3. Mitochondrial therapies
An often‐neglected target for nucleic acid therapies are the mitochondria. Liposome‐like vesicles modified with dequalinium (termed DQAsomes) have been investigated for mitochondrial delivery of nucleic acids. DQA possesses a positively charged quaternary ammonium and has been shown to prompt targeting and fusion with the negatively charged mitochondrial membrane (Bae et al., 2017; Lyrawati et al., 2011; Weissig et al., 2021). Bae et al. developed DQA80somes for the transport of plasmid DNA encoding a GFP into mitochondria of HeLa cells (Figure 10) (Bae et al., 2017). DQA80somes (dequalinium‐DOPE‐DOTAP nanosome) showed strong mitochondrial localization in HeLa cells, confirmed by TEM and confocal microscopy (Figure 10a,c). Delivery of a mtGFP (mtZsGreen) gene using the DQA80somes was used to confirm the ability to induce mitochondrial gene expression by using rtPCR (Figure 10b). Confocal microscopy confirmed that DQA80s‐pmtZsGreen exhibited three times the fluorescence intensity of the DQAsomes (dequalinium‐only nanosome) control (Figure 10d), with the ZsGreen signal observed in the same region of the cell as the mitochondria. This study confirmed the ability to both target and enter the mitochondria to promote gene replacement and expression of mtDNA.
FIGURE 10.
DQAsomes used for targeted delivery of pDNA to the mitochondria. (a) TEM image of distributions DQA80somes/pDNA complexes localized to the mitochondria (scale bar: 500 nm). (b) Expression of hmtZsGreen by DQA80s in HeLa cells using RT‐PCR. (c) Confocal images of HeLa cells 24 h posttransfection with CF‐labeled DQA80somes loaded with empty pDNA vector. (d) Confocal images of HeLa cells expressing hmtZsGreen 24 h after transfection with DQA80somes. Nuclei were stained with NucBlue live ready probes. Mitochondria were stained with Mito tracker red. Scale bar: 50 μm. Reprinted with permission from Bae et al. (2017).
Gene editing can also target and degrade mutated mtDNA to increase the amount of functional mtDNA. Unlike the nucleus, mitochondria cannot repair double‐stranded breaks in mtDNA, meaning linearized mtDNAs will eventually degrade (Peeva et al., 2018). CRISPR/Cas9 can be used to selectively edit mtDNA in vitro (Jo et al., 2015); however, the mechanism by which the components would be imported into the mitochondrial matrix were not thoroughly investigated. Although extensively used for nuclear DNA repair, using a two‐component system for editing of mtDNA is challenging, as both components, Cas9 and gRNA, in addition to a repair template if required, need to be imported into the mitochondrial matrix (Slone & Huang, 2020). Specifically, gRNA is very poorly transported into the mitochondrial matrix (Gammage, Viscomi, et al., 2018; Slone & Huang, 2020), with a recent study conducted by Hussain et al. further confirming the inefficient localization of unmodified gRNAs into the mitochondria (Hussain et al., 2021). Less complex endonuclease systems have thus been investigated which use restriction endonucleases that selectively cut mutated mtDNA while leaving healthy mtDNA untouched. Transcription Activator‐Like Effector Nucleases (Bacman et al., 2018; Lee et al., 2021) and Zinc‐Finger Nucleases (Gammage, Moraes, & Minczuk, 2018) have shown promise in mouse models, both of which use AAV vectors to promote delivery to the mitochondria.
8. CONCLUSION AND OUTLOOK
For the past 20 years, nucleic acid therapeutics have held great therapeutic potential, but until the COVID‐19 pandemic their application was limited to a small number of disease states. However, the great success of mRNA vaccines to rapidly and effectively generate strong protection against SARS‐CoV‐2 has paved the way to apply nucleic acid therapeutics to a wider array of diseases. While the success of mRNA vaccines has been remarkable, there is still significant scope to further improve nucleic acid delivery. Specific applications where large amounts of protein are required, or where off target effects could have undesirable side effects, the current delivery systems are not efficient or specific enough.
The success of future nucleic acid delivery systems will depend on understanding the biological barriers that limit the effectiveness of current nucleic acid‐based therapies. As outlined in here, targeted delivery to the specific cells where the nucleic acids are required and engineering the nanoparticles to facilitate transmembrane trafficking to the subcellular compartment where they are active has significant potential to improve the effectiveness of nucleic acid delivery. Beyond this, targeting permissive entry pathways (such as highly endocytic surface receptors) or manipulating cellular signaling following membrane disruption (e.g. galectins) may in practice be as crucial as penetrating the biophysical barrier of lipid membranes. The cellular context (target cell biology, disease, or application) also plays a critical role in the design of the delivery system, which means an RNA vaccine requires a different approach to gene editing in hemopoietic stem cells. Improvements in nanomaterials as well as our understanding of cellular events surrounding nucleic acid‐delivery will be required to advance these therapeutics.
AUTHOR CONTRIBUTIONS
Lara M. Mollé: Conceptualization (supporting); writing – original draft (lead); writing – review and editing (equal). Cameron H. Smyth: Conceptualization (supporting); writing – original draft (supporting); writing – review and editing (equal). Daniel Yuen: Conceptualization (supporting); writing – original draft (supporting); writing – review and editing (supporting). Angus Johnston: Conceptualization (lead); writing – review and editing (supporting).
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
RELATED WIREs ARTICLES
Nanoescapology: progress towardunderstanding the endosomalescape of polymeric nanoparticles
Mollé, L. M. , Smyth, C. H. , Yuen, D. , & Johnston, A. P. R. (2022). Nanoparticles for vaccine and gene therapy: Overcoming the barriers to nucleic acid delivery. WIREs Nanomedicine and Nanobiotechnology, 14(6), e1809. 10.1002/wnan.1809
Edited by: Nancy Monteiro‐Riviere, Associate Editor and Nils G. Walter, Co‐Editor‐in‐Chief
Funding information Australian Research Council, Grant/Award Numbers: DP200100475, DP210103174; National Health and Medical Research Council, Grant/Award Number: GNT2011963
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Abbas, K. S. , Eltaras, M. M. , El‐Shahat, N. A. , Abdelazeem, B. , Shaqfeh, M. , & Brasic, J. R. (2022). The safety and efficacy of Nusinersen in the treatment of spinal muscular atrophy: A systematic review and meta‐analysis of randomized controlled trials. Medicina (Kaunas, Lithuania), 58(2), 213. 10.3390/medicina58020213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abstiens, K. , Gregoritza, M. , & Goepferich, A. M. (2019). Ligand density and linker length are critical factors for multivalent nanoparticle‐receptor interactions. ACS Applied Materials & Interfaces, 11(1), 1311–1320. 10.1021/acsami.8b18843 [DOI] [PubMed] [Google Scholar]
- Adams, D. , Gonzalez‐Duarte, A. , O'Riordan, W. D. , Yang, C. C. , Ueda, M. , Kristen, A. V. , Tournev, I. , Schmidt, H. H. , Coelho, T. , Berk, J. L. , Lin, K. P. , Vita, G. , Attarian, S. , Plante‐Bordeneuve, V. , Mezei, M. M. , Campistol, J. M. , Buades, J. , Brannagan, T. H., 3rd , Kim, B. J. , … Suhr, O. B. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. The New England Journal of Medicine, 379(1), 11–21. 10.1056/NEJMoa1716153 [DOI] [PubMed] [Google Scholar]
- Aird, E. J. , Lovendahl, K. N. , St Martin, A. , Harris, R. S. , & Gordon, W. R. (2018). Increasing Cas9‐mediated homology‐directed repair efficiency through covalent tethering of DNA repair template. Communications Biology, 1(1), 54. 10.1038/s42003-018-0054-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc, A. , Querbes, W. , De, S. , Qin, J. , Frank‐Kamenetsky, M. , Jayaprakash, K. N. , Jayaraman, M. , Rajeev, K. G. , Cantley, W. L. , Dorkin, J. R. , Butler, J. S. , Qin, L. , Racie, T. , Sprague, A. , Fava, E. , Zeigerer, A. , Hope, M. J. , Zerial, M. , Sah, D. W. , … Maier, M. A. (2010). Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand‐based mechanisms. Molecular Therapy, 18(7), 1357–1364. 10.1038/mt.2010.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E. J. , Rouphael, N. G. , Widge, A. T. , Jackson, L. A. , Roberts, P. C. , Makhene, M. , Chappell, J. D. , Denison, M. R. , Stevens, L. J. , Pruijssers, A. J. , McDermott, A. B. , Flach, B. , Lin, B. C. , Doria‐Rose, N. A. , O'Dell, S. , Schmidt, S. D. , Corbett, K. S. , Swanson, P. A., 2nd , Padilla, M. , … Beigel, J. H. (2020). Safety and immunogenicity of SARS‐CoV‐2 mRNA‐1273 vaccine in older adults. The New England Journal of Medicine, 383(25), 2427–2438. 10.1056/NEJMoa2028436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando, H. , Asai, T. , Koide, H. , Okamoto, A. , Maeda, N. , Tomita, K. , Dewa, T. , Minamino, T. , & Oku, N. (2014). Advanced cancer therapy by integrative antitumor actions via systemic administration of miR‐499. Journal of Controlled Release, 181, 32–39. 10.1016/j.jconrel.2014.02.019 [DOI] [PubMed] [Google Scholar]
- Appelbaum, J. S. , LaRochelle, J. R. , Smith, B. A. , Balkin, D. M. , Holub, J. M. , & Schepartz, A. (2012). Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chemistry & Biology, 19(7), 819–830. 10.1016/j.chembiol.2012.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia, M. F. , Anton, N. , Wallyn, J. , Omran, Z. , & Vandamme, T. F. (2019). An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. The Journal of Pharmacy and Pharmacology, 71(8), 1185–1198. 10.1111/jphp.13098 [DOI] [PubMed] [Google Scholar]
- Bacman, S. R. , Kauppila, J. H. K. , Pereira, C. V. , Nissanka, N. , Miranda, M. , Pinto, M. , Williams, S. L. , Larsson, N. G. , Stewart, J. B. , & Moraes, C. T. (2018). MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nature Medicine, 24(11), 1696–1700. 10.1038/s41591-018-0166-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden, L. R. , El Sahly, H. M. , Essink, B. , Kotloff, K. , Frey, S. , Novak, R. , Diemert, D. , Spector, S. A. , Rouphael, N. , Creech, C. B. , McGettigan, J. , Khetan, S. , Segall, N. , Solis, J. , Brosz, A. , Fierro, C. , Schwartz, H. , Neuzil, K. , Corey, L. , … Group, C. S . (2021). Efficacy and safety of the mRNA‐1273 SARS‐CoV‐2 vaccine. The New England Journal of Medicine, 384(5), 403–416. 10.1056/NEJMoa2035389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, Y. , Jung, M. K. , Song, S. J. , Green, E. S. , Lee, S. , Park, H. S. , Jeong, S. H. , Han, J. , Mun, J. Y. , Ko, K. S. , & Choi, J. S. (2017). Functional nanosome for enhanced mitochondria‐targeted gene delivery and expression. Mitochondrion, 37, 27–40. 10.1016/j.mito.2017.06.005 [DOI] [PubMed] [Google Scholar]
- Bastos, R. , Pante, N. , & Burke, B. (1995). Nuclear pore complex proteins. International Review of Cytology, 162B, 257–302. 10.1016/s0074-7696(08)62619-4 [DOI] [PubMed] [Google Scholar]
- Bates, A. , & Power, C. A. (2019). David vs. Goliath: The structure, function, and clinical prospects of antibody fragments. Antibodies (Basel), 8(2), 28. 10.3390/antib8020028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach, M. A. , Teo, S. L. Y. , Chen, M. Z. , Smith, S. A. , Pouton, C. W. , Johnston, A. P. R. , & Such, G. K. (2022). Quantifying the endosomal escape of pH‐responsive nanoparticles using the split luciferase endosomal escape quantification assay. ACS Applied Materials & Interfaces, 14(3), 3653–3661. 10.1021/acsami.1c18359 [DOI] [PubMed] [Google Scholar]
- Bedi, D. , Musacchio, T. , Fagbohun, O. A. , Gillespie, J. W. , Deinnocentes, P. , Bird, R. C. , Bookbinder, L. , Torchilin, V. P. , & Petrenko, V. A. (2011). Delivery of siRNA into breast cancer cells via phage fusion protein‐targeted liposomes. Nanomedicine, 7(3), 315–323. 10.1016/j.nano.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr, J. P. (1997). The proton sponge: A trick to enter cells the viruses did not exploit [conference paper]. Chimia, 51(1–2), 34–36. [Google Scholar]
- Bobo, D. , Robinson, K. J. , Islam, J. , Thurecht, K. J. , & Corrie, S. R. (2016). Nanoparticle‐based medicines: A review of FDA‐approved materials and clinical trials to date. Pharmaceutical Research, 33(10), 2373–2387. 10.1007/s11095-016-1958-5 [DOI] [PubMed] [Google Scholar]
- Bonifaz, L. C. , Bonnyay, D. P. , Charalambous, A. , Darguste, D. I. , Fujii, S. , Soares, H. , Brimnes, M. K. , Moltedo, B. , Moran, T. M. , & Steinman, R. M. (2004). In vivo targeting of antigens to maturing dendritic cells via the DEC‐205 receptor improves T cell vaccination. The Journal of Experimental Medicine, 199(6), 815–824. 10.1084/jem.20032220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratkovic, T. , Bozic, J. , & Rogelj, B. (2020). Functional diversity of small nucleolar RNAs. Nucleic Acids Research, 48(4), 1627–1651. 10.1093/nar/gkz1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock, D. J. , Kustigian, L. , Jiang, M. , Graham, K. , Wang, T. Y. , Erazo‐Oliveras, A. , Najjar, K. , Zhang, J. , Rye, H. , & Pellois, J. P. (2018). Efficient cell delivery mediated by lipid‐specific endosomal escape of supercharged branched peptides [article]. Traffic, 19(6), 421–435. 10.1111/tra.12566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous, S. , Terwilliger, T. C. , & Waldo, G. S. (2005). Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nature Biotechnology, 23(1), 102–107. 10.1038/nbt1044 [DOI] [PubMed] [Google Scholar]
- Cabantous, S. , & Waldo, G. S. (2006). In vivo and in vitro protein solubility assays using split GFP. Nature Methods, 3(10), 845–854. 10.1038/nmeth932 [DOI] [PubMed] [Google Scholar]
- Cabral, H. , Miyata, K. , Osada, K. , & Kataoka, K. (2018). Block copolymer micelles in Nanomedicine applications. Chemical Reviews, 118(14), 6844–6892. 10.1021/acs.chemrev.8b00199 [DOI] [PubMed] [Google Scholar]
- Cal, P. M. , Bernardes, G. J. , & Gois, P. M. (2014). Cysteine‐selective reactions for antibody conjugation. Angewandte Chemie (International Ed. in English), 53(40), 10585–10587. 10.1002/anie.201405702 [DOI] [PubMed] [Google Scholar]
- Caminschi, I. , Proietto, A. I. , Ahmet, F. , Kitsoulis, S. , Shin Teh, J. , Lo, J. C. , Rizzitelli, A. , Wu, L. , Vremec, D. , van Dommelen, S. L. , Campbell, I. K. , Maraskovsky, E. , Braley, H. , Davey, G. M. , Mottram, P. , van de Velde, N. , Jensen, K. , Lew, A. M. , Wright, M. D. , … Lahoud, M. H. (2008). The dendritic cell subtype‐restricted C‐type lectin Clec9A is a target for vaccine enhancement. Blood, 112(8), 3264–3273. 10.1182/blood-2008-05-155176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp, E. R. , Wang, C. , Little, E. C. , Watson, P. M. , Pirollo, K. F. , Rait, A. , Cole, D. J. , Chang, E. H. , & Watson, D. K. (2013). Transferrin receptor targeting nanomedicine delivering wild‐type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Therapy, 20(4), 222–228. 10.1038/cgt.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi, M. R. (1980). High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell, 22(2 Pt 2), 479–488. 10.1016/0092-8674(80)90358-x [DOI] [PubMed] [Google Scholar]
- Carrasco, M. J. , Alishetty, S. , Alameh, M. G. , Said, H. , Wright, L. , Paige, M. , Soliman, O. , Weissman, D. , Cleveland, T. E.t. , Grishaev, A. , & Buschmann, M. D. (2021). Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Communications Biology, 4(1), 956. 10.1038/s42003-021-02441-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey, J. R. , Grinstein, S. , & Orlowski, J. (2010). Sensors and regulators of intracellular pH. Nature Reviews. Molecular Cell Biology, 11(1), 50–61. 10.1038/nrm2820 [DOI] [PubMed] [Google Scholar]
- Castel, S. E. , & Martienssen, R. A. (2013). RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nature Reviews. Genetics, 14(2), 100–112. 10.1038/nrg3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Tam, Y. Y. C. , Lin, P. J. C. , Sung, M. M. H. , Tam, Y. K. , & Cullis, P. R. (2016). Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. Journal of Controlled Release, 235, 236–244. 10.1016/j.jconrel.2016.05.059 [DOI] [PubMed] [Google Scholar]
- Cheng, D. , Cao, N. , Chen, J. , Yu, X. , & Shuai, X. (2012). Multifunctional nanocarrier mediated co‐delivery of doxorubicin and siRNA for synergistic enhancement of glioma apoptosis in rat. Biomaterials, 33(4), 1170–1179. 10.1016/j.biomaterials.2011.10.057 [DOI] [PubMed] [Google Scholar]
- Chernikov, I. V. , Vlassov, V. V. , & Chernolovskaya, E. L. (2019). Current development of siRNA bioconjugates: From research to the clinic [review]. Frontiers in Pharmacology, 10, 444. 10.3389/fphar.2019.00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, A. , Bax, H. J. , Josephs, D. H. , Ilieva, K. M. , Pellizzari, G. , Opzoomer, J. , Bloomfield, J. , Fittall, M. , Grigoriadis, A. , Figini, M. , Canevari, S. , Spicer, J. F. , Tutt, A. N. , & Karagiannis, S. N. (2016). Targeting folate receptor alpha for cancer treatment. Oncotarget, 7(32), 52553–52574. 10.18632/oncotarget.9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. B. , Johnston, R. L. , Inostroza‐Ponta, M. , Fox, A. H. , Fortini, E. , Moscato, P. , Dinger, M. E. , & Mattick, J. S. (2012). Genome‐wide analysis of long noncoding RNA stability. Genome Research, 22(5), 885–898. 10.1101/gr.131037.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, H. , Levy, R. J. , Gao, J. , Fishbein, I. , Kousaev, V. , Sosnowski, S. , Slomkowski, S. , & Golomb, G. (2000). Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Therapy, 7(22), 1896–1905. 10.1038/sj.gt.3301318 [DOI] [PubMed] [Google Scholar]
- Convertine, A. J. , Diab, C. , Prieve, M. , Paschal, A. , Hoffman, A. S. , Johnson, P. H. , & Stayton, P. S. (2010). pH‐responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules, 11(11), 2904–2911. 10.1021/bm100652w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke, S. T. , Baker, B. F. , Crooke, R. M. , & Liang, X. H. (2021). Antisense technology: An overview and prospectus. Nature Reviews. Drug Discovery, 20(6), 427–453. 10.1038/s41573-021-00162-z [DOI] [PubMed] [Google Scholar]
- Cruz, F. M. , Colbert, J. D. , Merino, E. , Kriegsman, B. A. , & Rock, K. L. (2017). The biology and underlying mechanisms of Cross‐presentation of exogenous antigens on MHC‐I molecules. Annual Review of Immunology, 35(1), 149–176. 10.1146/annurev-immunol-041015-055254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dard, L. , Blanchard, W. , Hubert, C. , Lacombe, D. , & Rossignol, R. (2020). Mitochondrial functions and rare diseases. Molecular Aspects of Medicine, 71, 100842. 10.1016/j.mam.2019.100842 [DOI] [PubMed] [Google Scholar]
- de la Rica, R. , Aili, D. , & Stevens, M. M. (2012). Enzyme‐responsive nanoparticles for drug release and diagnostics. Advanced Drug Delivery Reviews, 64(11), 967–978. 10.1016/j.addr.2012.01.002 [DOI] [PubMed] [Google Scholar]
- Dean, D. A. , Strong, D. D. , & Zimmer, W. E. (2005). Nuclear entry of nonviral vectors. Gene Therapy, 12(11), 881–890. 10.1038/sj.gt.3302534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehaini, D. , Fang, R. H. , & Zhang, L. (2016). Biomimetic strategies for targeted nanoparticle delivery. Bioengineering & Translational Medicine, 1(1), 30–46. 10.1002/btm2.10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deirram, N. , Zhang, C. , Kermaniyan, S. S. , Johnston, A. P. R. , & Such, G. K. (2019). pH‐responsive polymer nanoparticles for drug delivery. Macromolecular Rapid Communications, 40(10), e1800917. 10.1002/marc.201800917 [DOI] [PubMed] [Google Scholar]
- Deprey, K. , Becker, L. , Kritzer, J. , & Pluckthun, A. (2019). Trapped! A critical evaluation of methods for measuring total cellular uptake versus cytosolic localization. Bioconjugate Chemistry, 30(4), 1006–1027. 10.1021/acs.bioconjchem.9b00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar, M. V. , Sznol, M. , Zhao, B. , Wang, D. , Carvajal, R. D. , Keohan, M. L. , Chuang, E. , Sanborn, R. E. , Lutzky, J. , Powderly, J. , Kluger, H. , Tejwani, S. , Green, J. , Ramakrishna, V. , Crocker, A. , Vitale, L. , Yellin, M. , Davis, T. , & Keler, T. (2014). Induction of antigen‐specific immunity with a vaccine targeting NY‐ESO‐1 to the dendritic cell receptor DEC‐205. Science Translational Medicine, 6(232), 232ra251. 10.1126/scitranslmed.3008068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarvand, R. , Sepehri, N. , Manoochehri, S. , Rouhani, H. , & Atyabi, F. (2011). Polylactide‐co‐glycolide nanoparticles for controlled delivery of anticancer agents. International Journal of Nanomedicine, 6, 877–895. 10.2147/IJN.S18905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. , Jiang, Z. , Saha, K. , Kim, C. S. , Kim, S. T. , Landis, R. F. , & Rotello, V. M. (2014). Gold nanoparticles for nucleic acid delivery. Molecular Therapy, 22(6), 1075–1083. 10.1038/mt.2014.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake, S. , Denny, W. A. , Gamage, S. , & Sarojini, V. (2017). Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. Journal of Controlled Release, 250, 62–76. 10.1016/j.jconrel.2017.02.006 [DOI] [PubMed] [Google Scholar]
- Doudna, J. A. , & Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR‐Cas9. Science, 346(6213), 1258096. 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- Du, F.‐S. , Wang, Y. , Zhang, R. , & Li, Z.‐C. (2010). Intelligent nucleic acid delivery systems based on stimuli‐responsive polymers [10.1039/B915020J]. Soft Matter, 6(5), 835–848. 10.1039/b915020j [DOI] [Google Scholar]
- Dunn, M. R. , Jimenez, R. M. , & Chaput, J. C. (2017). Analysis of aptamer discovery and technology. Nature Reviews Chemistry, 1(10), 0076. 10.1038/s41570-017-0076 [DOI] [Google Scholar]
- Elbashir, S. M. , Harborth, J. , Lendeckel, W. , Yalcin, A. , Weber, K. , & Tuschl, T. (2001). Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411(6836), 494–498. 10.1038/35078107 [DOI] [PubMed] [Google Scholar]
- Ely, A. , Singh, P. , Smith, T. S. , & Arbuthnot, P. (2021). In vitro transcribed mRNA for expression of designer nucleases: Advantages as a novel therapeutic for the management of chronic HBV infection. Advanced Drug Delivery Reviews, 168, 134–146. 10.1016/j.addr.2020.05.010 [DOI] [PubMed] [Google Scholar]
- Erazo‐Oliveras, A. , Muthukrishnan, N. , Baker, R. , Wang, T. Y. , & Pellois, J. P. (2012). Improving the endosomal escape of cell‐penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals (Basel), 5(11), 1177–1209. 10.3390/ph5111177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo‐Oliveras, A. , Najjar, K. , Dayani, L. , Wang, T. Y. , Johnson, G. A. , & Pellois, J. P. (2014). Protein delivery into live cells by incubation with an endosomolytic agent [article]. Nature Methods, 11(8), 861–867. 10.1038/nmeth.2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo‐Oliveras, A. , Najjar, K. , Truong, D. , Wang, T. Y. , Brock, D. J. , Prater, A. R. , & Pellois, J. P. (2016). The late endosome and its lipid BMP act as gateways for efficient cytosolic access of the delivery agent dfTAT and its macromolecular cargos. Cell Chemical Biology, 23(5), 598–607. 10.1016/j.chembiol.2016.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essa, D. , Kondiah, P. P. D. , Choonara, Y. E. , & Pillay, V. (2020). The design of poly(lactide‐co‐glycolide) Nanocarriers for medical applications [review]. Frontiers in Bioengineering and Biotechnology, 8, 48. 10.3389/fbioe.2020.00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, B. C. , Fletcher, R. B. , Kilchrist, K. V. , Dailing, E. A. , Mukalel, A. J. , Colazo, J. M. , Oliver, M. , Cheung‐Flynn, J. , Brophy, C. M. , Tierney, J. W. , Isenberg, J. S. , Hankenson, K. D. , Ghimire, K. , Lander, C. , Gersbach, C. A. , & Duvall, C. L. (2019). An anionic, endosome‐escaping polymer to potentiate intracellular delivery of cationic peptides, biomacromolecules, and nanoparticles. Nature Communications, 10(1), 5012. 10.1038/s41467-019-12906-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, B. C. , Hocking, K. M. , Kilchrist, K. V. , Wise, E. S. , Brophy, C. M. , & Duvall, C. L. (2015). Endosomolytic Nano‐Polyplex platform technology for cytosolic peptide delivery to inhibit pathological vasoconstriction. ACS Nano, 9(6), 5893–5907. 10.1021/acsnano.5b00491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, B. C. , Hocking, K. M. , Osgood, M. J. , Voskresensky, I. , Dmowska, J. , Kilchrist, K. V. , Brophy, C. M. , & Duvall, C. L. (2015). MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Science Translational Medicine, 7(291), 291ra295. 10.1126/scitranslmed.aaa4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, J. C. , Malhotra, M. , Guo, J. , O'Shea, J. P. , Hanrahan, K. , O'Neill, A. , Landry, W. D. , Griffin, B. T. , Darcy, R. , Watson, R. W. , & O'Driscoll, C. M. (2016). Folate‐targeted amphiphilic cyclodextrin.siRNA nanoparticles for prostate cancer therapy exhibit PSMA mediated uptake, therapeutic gene silencing in vitro and prolonged circulation in vivo. Nanomedicine, 12(8), 2341–2351. 10.1016/j.nano.2016.06.014 [DOI] [PubMed] [Google Scholar]
- Ewert, S. , Cambillau, C. , Conrath, K. , & Plückthun, A. (2002). Biophysical properties of camelid VHH domains compared to those of human VH3 domains. Biochemistry, 41(11), 3628–3636. [DOI] [PubMed] [Google Scholar]
- Fang, J. , Nakamura, H. , & Maeda, H. (2011). The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced Drug Delivery Reviews, 63(3), 136–151. 10.1016/j.addr.2010.04.009 [DOI] [PubMed] [Google Scholar]
- Fang, Y. , Xue, J. , Gao, S. , Lu, A. , Yang, D. , Jiang, H. , He, Y. , & Shi, K. (2017). Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Delivery, 24(sup1), 22–32. 10.1080/10717544.2017.1388451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, Z. , Wang, X. , Sun, Y. , Fan, R. , Liu, Z. , Guo, R. , & Xie, D. (2019). Sgc8 aptamer targeted glutathione‐responsive nanoassemblies containing Ara‐C prodrug for the treatment of acute lymphoblastic leukemia. Nanoscale, 11(47), 23000–23012. 10.1039/c9nr07391d [DOI] [PubMed] [Google Scholar]
- Farokhzad, O. C. , Jon, S. , Khademhosseini, A. , Tran, T. N. , Lavan, D. A. , & Langer, R. (2004). Nanoparticle‐aptamer bioconjugates: A new approach for targeting prostate cancer cells. Cancer Research, 64(21), 7668–7672. 10.1158/0008-5472.CAN-04-2550 [DOI] [PubMed] [Google Scholar]
- Finn, J. D. , Smith, A. R. , Patel, M. C. , Shaw, L. , Youniss, M. R. , van Heteren, J. , Dirstine, T. , Ciullo, C. , Lescarbeau, R. , Seitzer, J. , Shah, R. R. , Shah, A. , Ling, D. , Growe, J. , Pink, M. , Rohde, E. , Wood, K. M. , Salomon, W. E. , Harrington, W. F. , … Morrissey, D. V. (2018). A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Reports, 22(9), 2227–2235. 10.1016/j.celrep.2018.02.014 [DOI] [PubMed] [Google Scholar]
- Fossum, E. , Tesfaye, D. Y. , Bobic, S. , Gudjonsson, A. , Braathen, R. , Lahoud, M. H. , Caminschi, I. , & Bogen, B. (2020). Targeting antigens to different receptors on conventional type 1 dendritic cells impacts the immune response. Journal of Immunology, 205(3), 661–673. 10.4049/jimmunol.1901119 [DOI] [PubMed] [Google Scholar]
- Fox, T. D. (2012). Mitochondrial protein synthesis, import, and assembly. Genetics, 192(4), 1203–1234. 10.1534/genetics.112.141267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, A. D. , Claypool, S. E. , & Liu, R. (2013). The smart targeting of nanoparticles. Current Pharmaceutical Design, 19(35), 6315–6329. 10.2174/13816128113199990375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda, S. , Galluzzi, L. , & Kroemer, G. (2010). Targeting mitochondria for cancer therapy. Nature Reviews. Drug Discovery, 9(6), 447–464. 10.1038/nrd3137 [DOI] [PubMed] [Google Scholar]
- Gammage, P. A. , Moraes, C. T. , & Minczuk, M. (2018). Mitochondrial genome engineering: The revolution may not be CRISPR‐Ized. Trends in Genetics, 34(2), 101–110. 10.1016/j.tig.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage, P. A. , Viscomi, C. , Simard, M. L. , Costa, A. S. H. , Gaude, E. , Powell, C. A. , Van Haute, L. , McCann, B. J. , Rebelo‐Guiomar, P. , Cerutti, R. , Zhang, L. , Rebar, E. J. , Zeviani, M. , Frezza, C. , Stewart, J. B. , & Minczuk, M. (2018). Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nature Medicine, 24(11), 1691–1695. 10.1038/s41591-018-0165-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamrad, L. , Rehbock, C. , Westendorf, A. M. , Buer, J. , Barcikowski, S. , & Hansen, W. (2016). Efficient nucleic acid delivery to murine regulatory T cells by gold nanoparticle conjugates. Scientific Reports, 6(1), 28709. 10.1038/srep28709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, H. , Cheng, R. , & Santos, H. A. (2021). Nanoparticle‐mediated siRNA delivery systems for cancer therapy. Viewpoints, 2(3), 20200111. 10.1002/viw.20200111 [DOI] [Google Scholar]
- Gessner, J. E. , Heiken, H. , Tamm, A. , & Schmidt, R. E. (1998). The IgG fc receptor family. Annals of Hematology, 76(6), 231–248. 10.1007/s002770050396 [DOI] [PubMed] [Google Scholar]
- Gilleron, J. , Querbes, W. , Zeigerer, A. , Borodovsky, A. , Marsico, G. , Schubert, U. , Manygoats, K. , Seifert, S. , Andree, C. , Stoter, M. , Epstein‐Barash, H. , Zhang, L. , Koteliansky, V. , Fitzgerald, K. , Fava, E. , Bickle, M. , Kalaidzidis, Y. , Akinc, A. , Maier, M. , & Zerial, M. (2013). Image‐based analysis of lipid nanoparticle‐mediated siRNA delivery, intracellular trafficking and endosomal escape. Nature Biotechnology, 31(7), 638–646. 10.1038/nbt.2612 [DOI] [PubMed] [Google Scholar]
- Graessmann, M. , Menne, J. , Liebler, M. , Graeber, I. , & Graessmann, A. (1989). Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Research, 17(16), 6603–6612. 10.1093/nar/17.16.6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, J. , Guo, H. , Tang, T. , Wang, Y. , Wei, Y. , Seth, P. , Li, Y. , Dehm, S. M. , Ruoslahti, E. , & Pang, H. B. (2021). iRGD‐liposomes enhance tumor delivery and therapeutic efficacy of antisense oligonucleotide drugs against primary prostate cancer and bone metastasis. Advanced Functional Materials, 31(24), 2100478. 10.1002/adfm.202100478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujrati, V. , Kim, S. , Kim, S. H. , Min, J. J. , Choy, H. E. , Kim, S. C. , & Jon, S. (2014). Bioengineered bacterial outer membrane vesicles as cell‐specific drug‐delivery vehicles for cancer therapy. ACS Nano, 8(2), 1525–1537. 10.1021/nn405724x [DOI] [PubMed] [Google Scholar]
- Gustafson, H. H. , Holt‐Casper, D. , Grainger, D. W. , & Ghandehari, H. (2015). Nanoparticle uptake: The phagocyte problem. Nano Today, 10(4), 487–510. 10.1016/j.nantod.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafez, I. M. , Maurer, N. , & Cullis, P. R. (2001). On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Therapy, 8(15), 1188–1196. 10.1038/sj.gt.3301506 [DOI] [PubMed] [Google Scholar]
- Hagemeyer, C. E. , Alt, K. , Johnston, A. P. , Such, G. K. , Ta, H. T. , Leung, M. K. , Prabhu, S. , Wang, X. , Caruso, F. , & Peter, K. (2015). Particle generation, functionalization and sortase A‐mediated modification with targeting of single‐chain antibodies for diagnostic and therapeutic use. Nature Protocols, 10(1), 90–105. 10.1038/nprot.2014.177 [DOI] [PubMed] [Google Scholar]
- Han, M. , Oba, M. , Nishiyama, N. , Kano, M. R. , Kizaka‐Kondoh, S. , & Kataoka, K. (2009). Enhanced percolation and gene expression in tumor hypoxia by PEGylated polyplex micelles. Molecular Therapy, 17(8), 1404–1410. 10.1038/mt.2009.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada‐Shiba, M. , Yamauchi, K. , Harada, A. , Takamisawa, I. , Shimokado, K. , & Kataoka, K. (2002). Polyion complex micelles as vectors in gene therapy‐‐pharmacokinetics and in vivo gene transfer. Gene Therapy, 9(6), 407–414. 10.1038/sj.gt.3301665 [DOI] [PubMed] [Google Scholar]
- Hatakeyama, H. , Akita, H. , Ito, E. , Hayashi, Y. , Oishi, M. , Nagasaki, Y. , Danev, R. , Nagayama, K. , Kaji, N. , Kikuchi, H. , Baba, Y. , & Harashima, H. (2011). Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor‐specific cleavable PEG‐lipid. Biomaterials, 32(18), 4306–4316. 10.1016/j.biomaterials.2011.02.045 [DOI] [PubMed] [Google Scholar]
- Hedley, M. L. , Curley, J. , & Urban, R. (1998). Microspheres containing plasmid‐encoded antigens elicit cytotoxic T‐cell responses. Nature Medicine, 4(3), 365–368. 10.1038/nm0398-365 [DOI] [PubMed] [Google Scholar]
- Herce, H. D. , Garcia, A. E. , Litt, J. , Kane, R. S. , Martin, P. , Enrique, N. , Rebolledo, A. , & Milesi, V. (2009). Arginine‐rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell‐penetrating peptides. Biophysical Journal, 97(7), 1917–1925. 10.1016/j.bpj.2009.05.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herst, P. M. , Rowe, M. R. , Carson, G. M. , & Berridge, M. V. (2017). Functional mitochondria in health and disease [review]. Frontiers in Endocrinology (Lausanne), 8, 296. 10.3389/fendo.2017.00296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt, S. L. , Bai, A. , Bailey, D. , Ichikawa, K. , Zielinski, J. , Karp, R. , Apte, A. , Arnold, K. , Zacharek, S. J. , Iliou, M. S. , Bhatt, K. , Garnaas, M. , Musenge, F. , Davis, A. , Khatwani, N. , Su, S. V. , MacLean, G. , Farlow, S. J. , Burke, K. , & Frederick, J. P. (2019). Durable anticancer immunity from intratumoral administration of IL‐23, IL‐36gamma, and OX40L mRNAs. Science Translational Medicine, 11(477), eaat9143. 10.1126/scitranslmed.aat9143 [DOI] [PubMed] [Google Scholar]
- Holm, A. , Løvendorf, M. B. , & Kauppinen, S. (2021). Development of siRNA therapeutics for the treatment of liver diseases. In Ditzel H. J., Tuttolomondo M., & Kauppinen S. (Eds.), Design and delivery of SiRNA therapeutics (pp. 57–75). Springer US. 10.1007/978-1-0716-1298-9_5 [DOI] [PubMed] [Google Scholar]
- Hu, B. , Zhong, L. , Weng, Y. , Peng, L. , Huang, Y. , Zhao, Y. , & Liang, X. J. (2020). Therapeutic siRNA: State of the art. Signal Transduction and Targeted Therapy, 5(1), 101. 10.1038/s41392-020-0207-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Yuen, D. , Mintern, J. D. , & Johnston, A. P. R. (2021). Opportunities for innovation: Building on the success of lipid nanoparticle vaccines. Current Opinion in Colloid & Interface Science, 55, 101468. 10.1016/j.cocis.2021.101468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain, S. A. , Yalvac, M. E. , Khoo, B. , Eckardt, S. , & McLaughlin, K. J. (2021). Adapting CRISPR/Cas9 system for targeting mitochondrial genome [brief research report]. Frontiers in Genetics, 12(402), 627050. 10.3389/fgene.2021.627050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaremko, L. , Jaremko, M. , Giller, K. , Becker, S. , & Zweckstetter, M. (2014). Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science, 343(6177), 1363–1366. 10.1126/science.1248725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, A. , Ham, S. , Lee, G. H. , Lee, Y. I. , Kim, S. , Lee, Y. S. , Shin, J. H. , & Lee, Y. (2015). Efficient mitochondrial genome editing by CRISPR/Cas9. BioMed Research International, 2015, 305716. 10.1155/2015/305716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, A. , Ringel‐Scaia, V. M. , McDaniel, D. K. , Thomas, C. A. , Zhang, R. , Riffle, J. S. , Allen, I. C. , & Davis, R. M. (2020). Fabrication and characterization of PLGA nanoparticles encapsulating large CRISPR‐Cas9 plasmid. Journal of Nanobiotechnology, 18(1), 16. 10.1186/s12951-019-0564-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, A. P. R. (2017). Life under the microscope: Quantifying live cell interactions to improve nanoscale drug delivery. ACS Sensors, 2(1), 4–9. 10.1021/acssensors.6b00725 [DOI] [PubMed] [Google Scholar]
- Jones, R. A. , Cheung, C. Y. , Black, F. E. , Zia, J. K. , Stayton, P. S. , Hoffman, A. S. , & Wilson, M. R. (2003). Poly(2‐alkylacrylic acid) polymers deliver molecules to the cytosol by pH‐sensitive disruption of endosomal vesicles. The Biochemical Journal, 372(Pt 1), 65–75. 10.1042/BJ20021945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang, V. , Chang, C. H. , Wang, C. S. , Wang, H. E. , & Lo, Y. L. (2019). pH‐responsive PEG‐shedding and targeting peptide‐modified nanoparticles for dual‐delivery of Irinotecan and microRNA to enhance tumor‐specific therapy. Small, 15(49), e1903296. 10.1002/smll.201903296 [DOI] [PubMed] [Google Scholar]
- Katakowski, J. A. , Mukherjee, G. , Wilner, S. E. , Maier, K. E. , Harrison, M. T. , DiLorenzo, T. P. , Levy, M. , & Palliser, D. (2016). Delivery of siRNAs to dendritic cells using DEC205‐targeted lipid nanoparticles to inhibit immune responses. Molecular Therapy, 24(1), 146–155. 10.1038/mt.2015.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura, E. , Maruyama, M. , Abe, J. , Sudo, A. , Takeda, A. , Takada, S. , Yokota, T. , Kinugawa, S. , Harashima, H. , & Yamada, Y. (2020). Validation of gene therapy for mutant mitochondria by delivering mitochondrial RNA using a MITO‐Porter. Molecular Therapy ‐ Nucleic Acids, 20, 687–698. 10.1016/j.omtn.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. A. , Alanazi, A. M. , Jabeen, M. , Chauhan, A. , & Ansari, M. A. (2019). Therapeutic potential of functionalized siRNA nanoparticles on regression of liver cancer in experimental mice. Scientific Reports, 9(1), 15825. 10.1038/s41598-019-52142-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. , Hussain, A. , Fahimi, H. , Aliakbari, F. , Haj Bloukh, S. , Edis, Z. , Mahdi Nejadi Babadaei, M. , Izadi, Z. , Shiri Varnamkhasti, B. , Jahanshahi, F. , Lin, Y. , Hao, X. , Hasan Khan, R. , Rasti, B. , Vaghar‐Lahijani, G. , Hua, L. , Derakhshankhah, H. , Sharifi, M. , & Falahati, M. (2022). A review on the therapeutic applications of aptamers and aptamer‐conjugated nanoparticles in cancer, inflammatory and viral diseases. Arabian Journal of Chemistry, 15(2), 103626. 10.1016/j.arabjc.2021.103626 [DOI] [Google Scholar]
- Kimura, S. , Khalil, I. A. , Elewa, Y. H. A. , & Harashima, H. (2021). Novel lipid combination for delivery of plasmid DNA to immune cells in the spleen. Journal of Controlled Release, 330, 753–764. 10.1016/j.jconrel.2021.01.005 [DOI] [PubMed] [Google Scholar]
- Kiss, A. L. , & Botos, E. (2009). Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? Journal of Cellular and Molecular Medicine, 13(7), 1228–1237. 10.1111/j.1582-4934.2009.00754.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer, K. E. , & Schwartz, T. U. (2016). The nuclear pore complex as a flexible and dynamic gate. Cell, 164(6), 1162–1171. 10.1016/j.cell.2016.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, Y. T. , Hartner, W. C. , Kale, A. , & Torchilin, V. P. (2009). Gene delivery into ischemic myocardium by double‐targeted lipoplexes with anti‐myosin antibody and TAT peptide. Gene Therapy, 16(1), 52–59. 10.1038/gt.2008.135 [DOI] [PubMed] [Google Scholar]
- Kole, R. , Krainer, A. R. , & Altman, S. (2012). RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nature Reviews. Drug Discovery, 11(2), 125–140. 10.1038/nrd3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltover, I. , Salditt, T. , Radler, J. O. , & Safinya, C. R. (1998). An inverted hexagonal phase of cationic liposome‐DNA complexes related to DNA release and delivery. Science, 281(5373), 78–81. 10.1126/science.281.5373.78 [DOI] [PubMed] [Google Scholar]
- Kongkatigumjorn, N. , Smith, S. A. , Chen, M. , Fang, K. , Yang, S. , Gillies, E. R. , Johnston, A. P. R. , & Such, G. K. (2018). Controlling endosomal escape using pH‐responsive nanoparticles with tunable disassembly. ACS Applied Nano Materials, 1(7), 3164–3173. 10.1021/acsanm.8b00338 [DOI] [Google Scholar]
- Koo, H. , Lee, S. , Na, J. H. , Kim, S. H. , Hahn, S. K. , Choi, K. , Kwon, I. C. , Jeong, S. Y. , & Kim, K. (2012). Bioorthogonal copper‐free click chemistry in vivo for tumor‐targeted delivery of nanoparticles. Angewandte Chemie (International Ed. in English), 51(47), 11836–11840. 10.1002/anie.201206703 [DOI] [PubMed] [Google Scholar]
- Kumar, S. U. , Wang, H. , Telichko, A. V. , Natarajan, A. , Bettinger, T. , Cherkaoui, S. , Massoud, T. F. , Dahl, J. J. , & Paulmurugan, R. (2021). Ultrasound triggered co‐delivery of therapeutic MicroRNAs and a triple suicide gene therapy vector by using biocompatible polymer nanoparticles for improved cancer therapy in mouse models. Advanced Therapeutics, 4(5), 2000197. 10.1002/adtp.202000197 [DOI] [Google Scholar]
- Lazzaro, S. , Giovani, C. , Mangiavacchi, S. , Magini, D. , Maione, D. , Baudner, B. , Geall, A. J. , De Gregorio, E. , D'Oro, U. , & Buonsanti, C. (2015). CD8 T‐cell priming upon mRNA vaccination is restricted to bone‐marrow‐derived antigen‐presenting cells and may involve antigen transfer from myocytes. Immunology, 146(2), 312–326. 10.1111/imm.12505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechardeur, D. , Sohn, K. J. , Haardt, M. , Joshi, P. B. , Monck, M. , Graham, R. W. , Beatty, B. , Squire, J. , O'Brodovich, H. , & Lukacs, G. L. (1999). Metabolic instability of plasmid DNA in the cytosol: A potential barrier to gene transfer. Gene Therapy, 6(4), 482–497. 10.1038/sj.gt.3300867 [DOI] [PubMed] [Google Scholar]
- Lee, H. , Jeong, J. H. , & Park, T. G. (2001). A new gene delivery formulation of polyethylenimine/DNA complexes coated with PEG conjugated fusogenic peptide. Journal of Controlled Release, 76((1–2)), 183–192. 10.1016/s0168-3659(01)00426-6 [DOI] [PubMed] [Google Scholar]
- Lee, H. , Lee, S. , Baek, G. , Kim, A. , Kang, B. C. , Seo, H. , & Kim, J. S. (2021). Mitochondrial DNA editing in mice with DddA‐TALE fusion deaminases. Nature Communications, 12(1), 1190. 10.1038/s41467-021-21464-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. , Choi, J. S. , & Ko, K. S. (2008). Mitochondria targeting delivery of nucleic acids. Expert Opinion on Drug Delivery, 5(8), 879–887. 10.1517/17425247.5.8.879 [DOI] [PubMed] [Google Scholar]
- Lee, Y. , & Kataoka, K. (2009). Biosignal‐sensitive polyion complex micelles for the delivery of biopharmaceuticals. Soft Matter, 5(20), 3810–3817. 10.1039/b909934d [DOI] [Google Scholar]
- Lee, Y. J. , Johnson, G. , Peltier, G. C. , & Pellois, J. P. (2011). A HA2‐fusion tag limits the endosomal release of its protein cargo despite causing endosomal lysis. Biochimica et Biophysica Acta, 1810(8), 752–758. 10.1016/j.bbagen.2011.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. W. , Luther, D. C. , Goswami, R. , Jeon, T. , Clark, V. , Elia, J. , Gopalakrishnan, S. , & Rotello, V. M. (2020). Direct cytosolic delivery of proteins through coengineering of proteins and polymeric delivery vehicles. Journal of the American Chemical Society, 142(9), 4349–4355. 10.1021/jacs.9b12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, C. S. (2015). Endogenous antigen presentation of MHC class II epitopes through non‐autophagic pathways [mini review]. Frontiers in Immunology, 6, 464. 10.3389/fimmu.2015.00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Yang, Y. , Hong, W. , Huang, M. , Wu, M. , & Zhao, X. (2020). Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduction and Targeted Therapy, 5(1), 1. 10.1038/s41392-019-0089-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H.‐J. , Wang, H.‐X. , Sun, C.‐Y. , Du, J.‐Z. , & Wang, J. (2014). Shell‐detachable nanoparticles based on a light‐responsive amphiphile for enhanced siRNA delivery. RSC Advances, 4(4), 1961–1964. 10.1039/c3ra44866e [DOI] [Google Scholar]
- Li, J. , Ge, Z. , & Liu, S. (2013). PEG‐sheddable polyplex micelles as smart gene carriers based on MMP‐cleavable peptide‐linked block copolymers. Chemical Communications (Cambridge), 49(62), 6974–6976. 10.1039/c3cc43576h [DOI] [PubMed] [Google Scholar]
- Li, J. , Yu, X. , Wang, Y. , Yuan, Y. , Xiao, H. , Cheng, D. , & Shuai, X. (2014). A reduction and pH dual‐sensitive polymeric vector for long‐circulating and tumor‐targeted siRNA delivery. Advanced Materials, 26(48), 8217–8224. 10.1002/adma.201403877 [DOI] [PubMed] [Google Scholar]
- Li, L. , Hou, J. , Liu, X. , Guo, Y. , Wu, Y. , Zhang, L. , & Yang, Z. (2014). Nucleolin‐targeting liposomes guided by aptamer AS1411 for the delivery of siRNA for the treatment of malignant melanomas. Biomaterials, 35(12), 3840–3850. 10.1016/j.biomaterials.2014.01.019 [DOI] [PubMed] [Google Scholar]
- Li, S. D. , & Huang, L. (2010). Stealth nanoparticles: High density but sheddable PEG is a key for tumor targeting. Journal of Controlled Release, 145(3), 178–181. 10.1016/j.jconrel.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Nicol, F. , & Szoka, F. C., Jr. (2004). GALA: A designed synthetic pH‐responsive amphipathic peptide with applications in drug and gene delivery. Advanced Drug Delivery Reviews, 56(7), 967–985. 10.1016/j.addr.2003.10.041 [DOI] [PubMed] [Google Scholar]
- Liang, X. H. , Nichols, J. G. , Hsu, C. W. , Vickers, T. A. , & Crooke, S. T. (2019). mRNA levels can be reduced by antisense oligonucleotides via no‐go decay pathway. Nucleic Acids Research, 47(13), 6900–6916. 10.1093/nar/gkz500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. H. , Shen, W. , Sun, H. , Migawa, M. T. , Vickers, T. A. , & Crooke, S. T. (2016). Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nature Biotechnology, 34(8), 875–880. 10.1038/nbt.3589 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Carmell, M. A. , Rivas, F. V. , Marsden, C. G. , Thomson, J. M. , Song, J. J. , Hammond, S. M. , Joshua‐Tor, L. , & Hannon, G. J. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science, 305(5689), 1437–1441. 10.1126/science.1102513 [DOI] [PubMed] [Google Scholar]
- Liu, Y. C. , Le Ny, A. L. , Schmidt, J. , Talmon, Y. , Chmelka, B. F. , & Lee, C. T., Jr. (2009). Photo‐assisted gene delivery using light‐responsive catanionic vesicles. Langmuir, 25(10), 5713–5724. 10.1021/la803588d [DOI] [PubMed] [Google Scholar]
- Lopez‐Cantu, D. O. , Wang, X. , Carrasco‐Magallanes, H. , Afewerki, S. , Zhang, X. , Bonventre, J. V. , & Ruiz‐Esparza, G. U. (2022). From bench to the clinic: The path to translation of nanotechnology‐enabled mRNA SARS‐CoV‐2 vaccines. Nano‐Micro Letters, 14(1), 41. 10.1007/s40820-021-00771-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Wu, T. , Zhang, B. , Liu, S. , Song, W. , Qiao, J. , & Ruan, H. (2021). Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Communication and Signaling: CCS, 19(1), 60. 10.1186/s12964-021-00741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. L. , Xu, C. F. , Li, H. J. , Cao, Z. T. , Liu, J. , Wang, J. L. , Du, X. J. , Yang, X. Z. , Gu, Z. , & Wang, J. (2018). Macrophage‐specific in vivo gene editing using cationic lipid‐assisted polymeric nanoparticles. ACS Nano, 12(2), 994–1005. 10.1021/acsnano.7b07874 [DOI] [PubMed] [Google Scholar]
- Lyrawati, D. , Trounson, A. , & Cram, D. (2011). Expression of GFP in the mitochondrial compartment using DQAsome‐mediated delivery of an artificial mini‐mitochondrial genome. Pharmaceutical Research, 28(11), 2848–2862. 10.1007/s11095-011-0544-0 [DOI] [PubMed] [Google Scholar]
- Mahon, E. , Salvati, A. , Baldelli Bombelli, F. , Lynch, I. , & Dawson, K. A. (2012). Designing the nanoparticle‐biomolecule interface for “targeting and therapeutic delivery”. Journal of Controlled Release, 161(2), 164–174. 10.1016/j.jconrel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- Mathew, A. P. , Cho, K. H. , Uthaman, S. , Cho, C. S. , & Park, I. K. (2017). Stimuli‐regulated smart polymeric systems for gene therapy. Polymers (Basel), 9(4), 152. 10.3390/polym9040152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Thery, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology, 21(1), 9–17. 10.1038/s41556-018-0250-9 [DOI] [PubMed] [Google Scholar]
- Maugeri, M. , Nawaz, M. , Papadimitriou, A. , Angerfors, A. , Camponeschi, A. , Na, M. , Holtta, M. , Skantze, P. , Johansson, S. , Sundqvist, M. , Lindquist, J. , Kjellman, T. , Martensson, I. L. , Jin, T. , Sunnerhagen, P. , Ostman, S. , Lindfors, L. , & Valadi, H. (2019). Linkage between endosomal escape of LNP‐mRNA and loading into EVs for transport to other cells. Nature Communications, 10(1), 4333. 10.1038/s41467-019-12275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina‐Kauwe, L. K. , Xie, J. , & Hamm‐Alvarez, S. (2005). Intracellular trafficking of nonviral vectors. Gene Therapy, 12(24), 1734–1751. 10.1038/sj.gt.3302592 [DOI] [PubMed] [Google Scholar]
- Meister, G. , & Tuschl, T. (2004). Mechanisms of gene silencing by double‐stranded RNA. Nature, 431(7006), 343–349. 10.1038/nature02873 [DOI] [PubMed] [Google Scholar]
- Mirzayans, R. , Aubin, R. A. , & Paterson, M. C. (1992). Differential expression and stability of foreign genes introduced into human fibroblasts by nuclear versus cytoplasmic microinjection. Mutation Research, 281(2), 115–122. 10.1016/0165-7992(92)90045-j [DOI] [PubMed] [Google Scholar]
- Miyata, K. , Oba, M. , Nakanishi, M. , Fukushima, S. , Yamasaki, Y. , Koyama, H. , Nishiyama, N. , & Kataoka, K. (2008). Polyplexes from poly(aspartamide) bearing 1,2‐diaminoethane side chains induce pH‐selective, endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. Journal of the American Chemical Society, 130(48), 16287–16294. 10.1021/ja804561g [DOI] [PubMed] [Google Scholar]
- Moghimi, S. M. , & Szebeni, J. (2003). Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein‐binding properties. Progress in Lipid Research, 42(6), 463–478. 10.1016/s0163-7827(03)00033-x [DOI] [PubMed] [Google Scholar]
- Moku, G. , Vangala, S. , Gulla, S. K. , & Yakati, V. (2021). In vivo targeting of DNA vaccines to dendritic cells via the mannose receptor induces Long‐lasting immunity against melanoma. Chembiochem, 22(3), 523–531. 10.1002/cbic.202000364 [DOI] [PubMed] [Google Scholar]
- Montenegro, J. M. , Grazu, V. , Sukhanova, A. , Agarwal, S. , de la Fuente, J. M. , Nabiev, I. , Greiner, A. , & Parak, W. J. (2013). Controlled antibody/(bio‐) conjugation of inorganic nanoparticles for targeted delivery. Advanced Drug Delivery Reviews, 65(5), 677–688. 10.1016/j.addr.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Mora, N. L. , Boyle, A. L. , Kolck, B. J. V. , Rossen, A. , Pokorna, S. , Koukalova, A. , Sachl, R. , Hof, M. , & Kros, A. (2020). Controlled peptide‐mediated vesicle fusion assessed by simultaneous dual‐colour time‐lapsed fluorescence microscopy. Scientific Reports, 10(1), 3087. 10.1038/s41598-020-59926-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T. , Yamaguchi, A. , Ohtake, K. , Takahashi, M. , Hayashi, A. , Iraha, F. , Kira, S. , Yanagisawa, T. , Yokoyama, S. , Hoshi, H. , Kobayashi, T. , & Sakamoto, K. (2015). Reassignment of a rare sense codon to a non‐canonical amino acid in Escherichia coli. Nucleic Acids Research, 43(16), 8111–8122. 10.1093/nar/gkv787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy, N. , Robichaud, J. R. , Tirrell, D. A. , Stayton, P. S. , & Hoffman, A. S. (1999). The design and synthesis of polymers for eukaryotic membrane disruption. Journal of Controlled Release, 61(1–2), 137–143. 10.1016/s0168-3659(99)00114-5 [DOI] [PubMed] [Google Scholar]
- Nagy, N. A. , de Haas, A. M. , Geijtenbeek, T. B. H. , van Ree, R. , Tas, S. W. , van Kooyk, Y. , & de Jong, E. C. (2021). Therapeutic liposomal vaccines for dendritic cell activation or tolerance [review]. Frontiers in Immunology, 12(1711), 674048. 10.3389/fimmu.2021.674048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT03739931, I . (2018. ‐, 21/12/21). Dose escalation study of mRNA‐2752 for intratumoral injection to participants in advanced malignancies . https://clinicaltrials.gov/ct2/show/NCT03739931
- NCT03897881, I . (2019. ‐, 21/12/21). An efficacy study of adjuvant treatment with the personalized cancer vaccine mRNA‐4157 and pembrolizumab in participants with high‐risk melanoma . https://clinicaltrials.gov/ct2/show/record/NCT03897881
- Oberhardt, V. , Luxenburger, H. , Kemming, J. , Schulien, I. , Ciminski, K. , Giese, S. , Csernalabics, B. , Lang‐Meli, J. , Janowska, I. , Staniek, J. , Wild, K. , Basho, K. , Marinescu, M. S. , Fuchs, J. , Topfstedt, F. , Janda, A. , Sogukpinar, O. , Hilger, H. , Stete, K. , … Hofmann, M. (2021). Rapid and stable mobilization of CD8(+) T cells by SARS‐CoV‐2 mRNA vaccine. Nature, 597(7875), 268–273. 10.1038/s41586-021-03841-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, K. , Breyne, K. , Ughetto, S. , Laurent, L. C. , & Breakefield, X. O. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews. Molecular Cell Biology, 21(10), 585–606. 10.1038/s41580-020-0251-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll, M. , & Jeggo, P. A. (2006). The role of double‐strand break repair ‐ insights from human genetics. Nature Reviews. Genetics, 7(1), 45–54. 10.1038/nrg1746 [DOI] [PubMed] [Google Scholar]
- Oku, N. , Asai, T. , Watanabe, K. , Kuromi, K. , Nagatsuka, M. , Kurohane, K. , Kikkawa, H. , Ogino, K. , Tanaka, M. , Ishikawa, D. , Tsukada, H. , Momose, M. , Nakayama, J. , & Taki, T. (2002). Anti‐neovascular therapy using novel peptides homing to angiogenic vessels. Oncogene, 21(17), 2662–2669. 10.1038/sj.onc.1205347 [DOI] [PubMed] [Google Scholar]
- Ozata, D. M. , Gainetdinov, I. , Zoch, A. , O'Carroll, D. , & Zamore, P. D. (2019). PIWI‐interacting RNAs: Small RNAs with big functions. Nature Reviews. Genetics, 20(2), 89–108. 10.1038/s41576-018-0073-3 [DOI] [PubMed] [Google Scholar]
- Pack, D. W. , Hoffman, A. S. , Pun, S. , & Stayton, P. S. (2005). Design and development of polymers for gene delivery. Nature Reviews. Drug Discovery, 4(7), 581–593. 10.1038/nrd1775 [DOI] [PubMed] [Google Scholar]
- Painter, M. M. , Mathew, D. , Goel, R. R. , Apostolidis, S. A. , Pattekar, A. , Kuthuru, O. , Baxter, A. E. , Herati, R. S. , Oldridge, D. A. , Gouma, S. , Hicks, P. , Dysinger, S. , Lundgreen, K. A. , Kuri‐Cervantes, L. , Adamski, S. , Hicks, A. , Korte, S. , Giles, J. R. , Weirick, M. E. , … Wherry, E. J. (2021). Rapid induction of antigen‐specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS‐CoV‐2 mRNA vaccination. Immunity, 54(9), 2133–2142 e2133. 10.1016/j.immuni.2021.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantos, A. , Tsogas, I. , & Paleos, C. M. (2008). Guanidinium group: A versatile moiety inducing transport and multicompartmentalization in complementary membranes. Biochimica et Biophysica Acta, 1778(4), 811–823. 10.1016/j.bbamem.2007.12.003 [DOI] [PubMed] [Google Scholar]
- Panyam, J. , & Labhasetwar, V. (2003). Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Advanced Drug Delivery Reviews, 55(3), 329–347. 10.1016/s0169-409x(02)00228-4 [DOI] [PubMed] [Google Scholar]
- Papahadjopoulos, D. , Allen, T. M. , Gabizon, A. , Mayhew, E. , Matthay, K. , Huang, S. K. , Lee, K. D. , Woodle, M. C. , Lasic, D. D. , & Redemann, C. (1991). Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proceedings of the National Academy of Sciences of the United States of America, 88(24), 11460–11464. 10.1073/pnas.88.24.11460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi, N. , Hogan, M. J. , Porter, F. W. , & Weissman, D. (2018). mRNA vaccines ‐ A new era in vaccinology. Nature Reviews. Drug Discovery, 17(4), 261–279. 10.1038/nrd.2017.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi, N. , Tuyishime, S. , Muramatsu, H. , Kariko, K. , Mui, B. L. , Tam, Y. K. , Madden, T. D. , Hope, M. J. , & Weissman, D. (2015). Expression kinetics of nucleoside‐modified mRNA delivered in lipid nanoparticles to mice by various routes. Journal of Controlled Release, 217, 345–351. 10.1016/j.jconrel.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon, E. , Laeremans, T. , Triest, S. , Rasmussen, S. G. , Wohlkonig, A. , Ruf, A. , Muyldermans, S. , Hol, W. G. , Kobilka, B. K. , & Steyaert, J. (2014). A general protocol for the generation of Nanobodies for structural biology. Nature Protocols, 9(3), 674–693. 10.1038/nprot.2014.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, M. L. , Zhang, M. , Taratula, O. , Garbuzenko, O. B. , He, H. , & Minko, T. (2009). Internally cationic polyamidoamine PAMAM‐OH dendrimers for siRNA delivery: Effect of the degree of quaternization and cancer targeting. Biomacromolecules, 10(2), 258–266. 10.1021/bm8009973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, S. D. , Rhodes, D. G. , & Burgess, D. J. (2005). DNA‐based therapeutics and DNA delivery systems: A comprehensive review. The AAPS Journal, 7(1), E61–E77. 10.1208/aapsj070109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeva, V. , Blei, D. , Trombly, G. , Corsi, S. , Szukszto, M. J. , Rebelo‐Guiomar, P. , Gammage, P. A. , Kudin, A. P. , Becker, C. , Altmuller, J. , Minczuk, M. , Zsurka, G. , & Kunz, W. S. (2018). Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nature Communications, 9(1), 1727. 10.1038/s41467-018-04131-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peier, A. , Ge, L. , Boyer, N. , Frost, J. , Duggal, R. , Biswas, K. , Edmondson, S. , Hermes, J. D. , Yan, L. , Zimprich, C. , Sadruddin, A. , Kristal Kaan, H. Y. , Chandramohan, A. , Brown, C. J. , Thean, D. , Lee, X. E. , Yuen, T. Y. , Ferrer‐Gago, F. J. , Johannes, C. W. , … Partridge, A. W. (2021). NanoClick: A high throughput, target‐agnostic peptide cell permeability assay. ACS Chemical Biology, 16(2), 293–309. 10.1021/acschembio.0c00804 [DOI] [PubMed] [Google Scholar]
- Petzold, C. , Schallenberg, S. , Stern, J. N. , & Kretschmer, K. (2012). Targeted antigen delivery to DEC‐205(+) dendritic cells for tolerogenic vaccination. The Review of Diabetic Studies, 9(4), 305–318. 10.1900/RDS.2012.9.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirollo, K. F. , Rait, A. , Zhou, Q. , Zhang, X. Q. , Zhou, J. , Kim, C. S. , Benedict, W. F. , & Chang, E. H. (2008). Tumor‐targeting nanocomplex delivery of novel tumor suppressor RB94 chemosensitizes bladder carcinoma cells in vitro and in vivo. Clinical Cancer Research, 14(7), 2190–2198. 10.1158/1078-0432.CCR-07-1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack, F. P. , Thomas, S. J. , Kitchin, N. , Absalon, J. , Gurtman, A. , Lockhart, S. , Perez, J. L. , Perez Marc, G. , Moreira, E. D. , Zerbini, C. , Bailey, R. , Swanson, K. A. , Roychoudhury, S. , Koury, K. , Li, P. , Kalina, W. V. , Cooper, D. , Frenck, R. W., Jr. , Hammitt, L. L. , … Group, C. C. T . (2020). Safety and efficacy of the BNT162b2 mRNA Covid‐19 vaccine. The New England Journal of Medicine, 383(27), 2603–2615. 10.1056/NEJMoa2034577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi, D. , Colapicchioni, V. , Caracciolo, G. , Piovesana, S. , Capriotti, A. L. , Palchetti, S. , De Grossi, S. , Riccioli, A. , Amenitsch, H. , & Lagana, A. (2014). Effect of polyethyleneglycol (PEG) chain length on the bio‐nano‐interactions between PEGylated lipid nanoparticles and biological fluids: From nanostructure to uptake in cancer cells. Nanoscale, 6(5), 2782–2792. 10.1039/c3nr05559k [DOI] [PubMed] [Google Scholar]
- Ramishetti, S. , Hazan‐Halevy, I. , Palakuri, R. , Chatterjee, S. , Naidu Gonna, S. , Dammes, N. , Freilich, I. , Kolik Shmuel, L. , Danino, D. , & Peer, D. (2020). A combinatorial library of lipid nanoparticles for RNA delivery to leukocytes. Advanced Materials, 32(12), e1906128. 10.1002/adma.201906128 [DOI] [PubMed] [Google Scholar]
- Rennick, J. J. , Johnston, A. P. R. , & Parton, R. G. (2021). Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nature Nanotechnology, 16(3), 266–276. 10.1038/s41565-021-00858-8 [DOI] [PubMed] [Google Scholar]
- Reuter, A. , Panozza, S. E. , Macri, C. , Dumont, C. , Li, J. , Liu, H. , Segura, E. , Vega‐Ramos, J. , Gupta, N. , Caminschi, I. , Villadangos, J. A. , Johnston, A. P. , & Mintern, J. D. (2015). Criteria for dendritic cell receptor selection for efficient antibody‐targeted vaccination. Journal of Immunology, 194(6), 2696–2705. 10.4049/jimmunol.1402535 [DOI] [PubMed] [Google Scholar]
- Richards, D. A. , Maruani, A. , & Chudasama, V. (2017). Antibody fragments as nanoparticle targeting ligands: A step in the right direction. Chemical Science, 8(1), 63–77. 10.1039/c6sc02403c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner, J. M. , Himansu, S. , Dowd, K. A. , Butler, S. L. , Salazar, V. , Fox, J. M. , Julander, J. G. , Tang, W. W. , Shresta, S. , Pierson, T. C. , Ciaramella, G. , & Diamond, M. S. (2017). Modified mRNA vaccines protect against Zika virus infection. Cell, 168(6), 1114–1125 e1110. 10.1016/j.cell.2017.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder, K. , Sevko, A. , Heide, J. , Dams, M. , Rupp, A. K. , Macas, J. , Starmann, J. , Tjwa, M. , Plate, K. H. , Sultmann, H. , Altevogt, P. , Umansky, V. , & Momma, S. (2015). Extracellular vesicle‐mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology, 4(6), e1008371. 10.1080/2162402X.2015.1008371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers, G. T. , Weterings, N. , Obregon‐Henao, A. , Lepolder, M. , Dutt, T. S. , van Overveld, F. J. , & Henao‐Tamayo, M. (2021). Antigen presentation of mRNA‐based and virus‐vectored SARS‐CoV‐2 vaccines. Vaccines (Basel), 9(8), 848. 10.3390/vaccines9080848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, T. C. , Langer, R. , & Wood, M. J. A. (2020). Advances in oligonucleotide drug delivery. Nature Reviews. Drug Discovery, 19(10), 673–694. 10.1038/s41573-020-0075-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, P. A. , & Furuta, K. (2015). The ins and outs of MHC class II‐mediated antigen processing and presentation. Nature Reviews. Immunology, 15(4), 203–216. 10.1038/nri3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani, B. , Engelbrecht, S. , & Glashoff, R. H. (2010). Functions of tat: The versatile protein of human immunodeficiency virus type 1. The Journal of General Virology, 91(Pt 1), 1–12. 10.1099/vir.0.016303-0 [DOI] [PubMed] [Google Scholar]
- Sahay, G. , Querbes, W. , Alabi, C. , Eltoukhy, A. , Sarkar, S. , Zurenko, C. , Karagiannis, E. , Love, K. , Chen, D. , Zoncu, R. , Buganim, Y. , Schroeder, A. , Langer, R. , & Anderson, D. G. (2013). Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nature Biotechnology, 31(7), 653–658. 10.1038/nbt.2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, U. , Kariko, K. , & Tureci, O. (2014). mRNA‐based therapeutics—Developing a new class of drugs. Nature Reviews. Drug Discovery, 13(10), 759–780. 10.1038/nrd4278 [DOI] [PubMed] [Google Scholar]
- Sahin, U. , Muik, A. , Derhovanessian, E. , Vogler, I. , Kranz, L. M. , Vormehr, M. , Baum, A. , Pascal, K. , Quandt, J. , Maurus, D. , Brachtendorf, S. , Lorks, V. , Sikorski, J. , Hilker, R. , Becker, D. , Eller, A. K. , Grutzner, J. , Boesler, C. , Rosenbaum, C. , … Tureci, O. (2020). COVID‐19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature, 586(7830), 594–599. 10.1038/s41586-020-2814-7 [DOI] [PubMed] [Google Scholar]
- Saluja, S. S. , Hanlon, D. J. , Sharp, F. A. , Hong, E. , Khalil, D. , Robinson, E. , Tigelaar, R. , Fahmy, T. M. , & Edelson, R. L. (2014). Targeting human dendritic cells via DEC‐205 using PLGA nanoparticles leads to enhanced cross‐presentation of a melanoma‐associated antigen. International Journal of Nanomedicine, 9, 5231–5246. 10.2147/IJN.S66639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangtani, A. , Nag, O. K. , Field, L. D. , Breger, J. C. , & Delehanty, J. B. (2017). Multifunctional nanoparticle composites: Progress in the use of soft and hard nanoparticles for drug delivery and imaging. WIREs. Nanomedicine and Nanobiotechnology, 9(6), e1466. 10.1002/wnan.1466 [DOI] [PubMed] [Google Scholar]
- Schlich, M. , Palomba, R. , Costabile, G. , Mizrahy, S. , Pannuzzo, M. , Peer, D. , & Decuzzi, P. (2021). Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles. Bioengineering & Translational Medicine, 6(2), e10213. 10.1002/btm2.10213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibel, P. , Trappe, J. , Villani, G. , Klopstock, T. , Papa, S. , & Reichmann, H. (1995). Transfection of mitochondria: Strategy towards a gene therapy of mitochondrial DNA diseases. Nucleic Acids Research, 23(1), 10–17. 10.1093/nar/23.1.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby, L. I. , Cortez‐Jugo, C. M. , Such, G. K. , & Johnston, A. P. R. (2017). Nanoescapology: Progress toward understanding the endosomal escape of polymeric nanoparticles. WIREs. Nanomedicine and Nanobiotechnology, 9(5), e1452. 10.1002/wnan.1452 [DOI] [PubMed] [Google Scholar]
- Semple, S. C. , Akinc, A. , Chen, J. , Sandhu, A. P. , Mui, B. L. , Cho, C. K. , Sah, D. W. , Stebbing, D. , Crosley, E. J. , Yaworski, E. , Hafez, I. M. , Dorkin, J. R. , Qin, J. , Lam, K. , Rajeev, K. G. , Wong, K. F. , Jeffs, L. B. , Nechev, L. , Eisenhardt, M. L. , … Hope, M. J. (2010). Rational design of cationic lipids for siRNA delivery. Nature Biotechnology, 28(2), 172–176. 10.1038/nbt.1602 [DOI] [PubMed] [Google Scholar]
- Shi, L. , Zhang, J. , Zhao, M. , Tang, S. , Cheng, X. , Zhang, W. , Li, W. , Liu, X. , Peng, H. , & Wang, Q. (2021). Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale, 13(24), 10748–10764. 10.1039/d1nr02065j [DOI] [PubMed] [Google Scholar]
- Sivaram, A. J. , Wardiana, A. , Howard, C. B. , Mahler, S. M. , & Thurecht, K. J. (2018). Recent advances in the generation of antibody‐nanomaterial conjugates. Advanced Healthcare Materials, 7(1), 1700607. 10.1002/adhm.201700607 [DOI] [PubMed] [Google Scholar]
- Slone, J. , & Huang, T. (2020). The special considerations of gene therapy for mitochondrial diseases. NPJ Genomic Medicine, 5(1), 7. 10.1038/s41525-020-0116-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. A. , Selby, L. I. , Johnston, A. P. R. , & Such, G. K. (2019). The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjugate Chemistry, 30(2), 263–272. 10.1021/acs.bioconjchem.8b00732 [DOI] [PubMed] [Google Scholar]
- Song, L. Y. , Ahkong, Q. F. , Rong, Q. , Wang, Z. , Ansell, S. , Hope, M. J. , & Mui, B. (2002). Characterization of the inhibitory effect of PEG‐lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochimica et Biophysica Acta, 1558(1), 1–13. 10.1016/s0005-2736(01)00399-6 [DOI] [PubMed] [Google Scholar]
- Springer, A. D. , & Dowdy, S. F. (2018). GalNAc‐siRNA conjugates: Leading the way for delivery of RNAi therapeutics. Nucleic Acid Therapeutics, 28(3), 109–118. 10.1089/nat.2018.0736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapley, J. , Feulner, P. G. D. , Johnston, S. E. , Santure, A. W. , & Smadja, C. M. (2017). Recombination: The good, the bad and the variable. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 372(1736), 20170279. 10.1098/rstb.2017.0279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle, H. , Behring, A. , Schlensak, C. , Wendel, H. P. , & Avci‐Adali, M. (2017). Concise review: Application of in vitro transcribed messenger RNA for cellular engineering and reprogramming: Progress and challenges. Stem Cells, 35(1), 68–79. 10.1002/stem.2402 [DOI] [PubMed] [Google Scholar]
- Steinman, R. M. , & Hemmi, H. (2006). Dendritic cells: Translating innate to adaptive immunity. Current Topics in Microbiology and Immunology, 311, 17–58. 10.1007/3-540-32636-7_2 [DOI] [PubMed] [Google Scholar]
- Tanner, N. K. (1999). Ribozymes: The characteristics and properties of catalytic RNAs. FEMS Microbiology Reviews, 23(3), 257–275. 10.1111/j.1574-6976.1999.tb00399.x [DOI] [PubMed] [Google Scholar]
- Teo, S. L. Y. , Rennick, J. J. , Yuen, D. , Al‐Wassiti, H. , Johnston, A. P. R. , & Pouton, C. W. (2021). Unravelling cytosolic delivery of cell penetrating peptides with a quantitative endosomal escape assay. Nature Communications, 12(1), 3721. 10.1038/s41467-021-23997-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn, A. M. , & Alberts, A. S. (1993). Efficient expression of miniprep plasmid DNA after needle micro‐injection into somatic cells. BioTechniques, 14(3), 356–358. [PubMed] [Google Scholar]
- Urmann, K. , & Walter, J.‐G. (2020). Aptamers in biotechnology (1st ed.). Springer International Publishing. [Google Scholar]
- Vaidya, A. M. , Sun, Z. , Ayat, N. , Schilb, A. , Liu, X. , Jiang, H. , Sun, D. , Scheidt, J. , Qian, V. , He, S. , Gilmore, H. , Schiemann, W. P. , & Lu, Z. R. (2019). Systemic delivery of tumor‐targeting siRNA nanoparticles against an oncogenic LncRNA facilitates effective triple‐negative breast cancer therapy. Bioconjugate Chemistry, 30(3), 907–919. 10.1021/acs.bioconjchem.9b00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitravene Study Group . (2002). A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. American Journal of Ophthalmology, 133(4), 467–474. 10.1016/s0002-9394(02)01327-2 [DOI] [PubMed] [Google Scholar]
- Wadhwa, A. , Aljabbari, A. , Lokras, A. , Foged, C. , & Thakur, A. (2020). Opportunities and challenges in the delivery of mRNA‐based vaccines. Pharmaceutics, 12(2), 102. 10.3390/pharmaceutics12020102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther, W. , Schmeer, M. , Kobelt, D. , Baier, R. , Harder, A. , Walhorn, V. , Anselmetti, D. , Aumann, J. , Fichtner, I. , & Schleef, M. (2013). A seven‐year storage report of good manufacturing practice‐grade naked plasmid DNA: Stability, topology, and in vitro/in vivo functional analysis. Human Gene Therapy. Clinical Development, 24(4), 147–153. 10.1089/humc.2013.067 [DOI] [PubMed] [Google Scholar]
- Wang, S. (2021). pH‐responsive amphiphilic carboxylate polymers: Design and potential for endosomal escape [mini review]. Frontiers in Chemistry, 9(145), 645297. 10.3389/fchem.2021.645297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Xiao, H. , Fang, J. , Yu, X. , Su, Z. , Cheng, D. , & Shuai, X. (2016). Construction of negatively charged and environment‐sensitive nanomedicine for tumor‐targeted efficient siRNA delivery. Chemical Communications (Cambridge), 52(6), 1194–1197. 10.1039/c5cc09181k [DOI] [PubMed] [Google Scholar]
- Weissig, V. , Lozoya, M. , Yu, N. , & D'Souza, G. G. M. (2021). DQAsomes as the prototype of mitochondria‐targeted pharmaceutical Nanocarriers: An update. Methods in Molecular Biology, 2275, 13–25. 10.1007/978-1-0716-1262-0_2 [DOI] [PubMed] [Google Scholar]
- White, J. M. , & Whittaker, G. R. (2016). Fusion of enveloped viruses in endosomes. Traffic, 17(6), 593–614. 10.1111/tra.12389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, D. S. , & Szostak, J. W. (1999). In vitro selection of functional nucleic acids. Annual Review of Biochemistry, 68(1), 611–647. 10.1146/annurev.biochem.68.1.611 [DOI] [PubMed] [Google Scholar]
- Wilson, J. T. , Keller, S. , Manganiello, M. J. , Cheng, C. , Lee, C. C. , Opara, C. , Convertine, A. , & Stayton, P. S. (2013). pH‐responsive nanoparticle vaccines for dual‐delivery of antigens and immunostimulatory oligonucleotides. ACS Nano, 7(5), 3912–3925. 10.1021/nn305466z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup, A. , Ai, A. , Liu, X. , Hamar, P. , Trifonova, R. , Charisse, K. , Manoharan, M. , Kirchhausen, T. , & Lieberman, J. (2015). Visualizing lipid‐formulated siRNA release from endosomes and target gene knockdown. Nature Biotechnology, 33(8), 870–876. 10.1038/nbt.3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff, J. A. , & Herweijer, H. (1997). Self‐amplifying vectors for gene delivery. Advanced Drug Delivery Reviews, 27(1), 5–16. 10.1016/s0169-409x(97)00018-5 [DOI] [PubMed] [Google Scholar]
- Won, Y. Y. , Sharma, R. , & Konieczny, S. F. (2009). Missing pieces in understanding the intracellular trafficking of polycation/DNA complexes. Journal of Controlled Release, 139(2), 88–93. 10.1016/j.jconrel.2009.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worn, A. , & Pluckthun, A. (1999). Different equilibrium stability behavior of ScFv fragments: Identification, classification, and improvement by protein engineering. Biochemistry, 38(27), 8739–8750. 10.1021/bi9902079 [DOI] [PubMed] [Google Scholar]
- Wu, P. , Shui, W. , Carlson, B. L. , Hu, N. , Rabuka, D. , Lee, J. , & Bertozzi, C. R. (2009). Site‐specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proceedings of the National Academy of Sciences of the United States of America, 106(9), 3000–3005. 10.1073/pnas.0807820106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, H. , Jiang, X. , Gu, J. , Sha, X. , Chen, L. , Law, K. , Chen, Y. , Wang, X. , Jiang, Y. , & Fang, X. (2011). Angiopep‐conjugated poly(ethylene glycol)‐co‐poly(epsilon‐caprolactone) nanoparticles as dual‐targeting drug delivery system for brain glioma. Biomaterials, 32(18), 4293–4305. 10.1016/j.biomaterials.2011.02.044 [DOI] [PubMed] [Google Scholar]
- Xu, C. F. , Zhang, H. B. , Sun, C. Y. , Liu, Y. , Shen, S. , Yang, X. Z. , Zhu, Y. H. , & Wang, J. (2016). Tumor acidity‐sensitive linkage‐bridged block copolymer for therapeutic siRNA delivery. Biomaterials, 88, 48–59. 10.1016/j.biomaterials.2016.02.031 [DOI] [PubMed] [Google Scholar]
- Xu, X. , Wu, J. , Liu, S. , Saw, P. E. , Tao, W. , Li, Y. , Krygsman, L. , Yegnasubramanian, S. , De Marzo, A. M. , Shi, J. , Bieberich, C. J. , & Farokhzad, O. C. (2018). Redox‐responsive nanoparticle‐mediated systemic RNAi for effective cancer therapy. Small, 14(41), e1802565. 10.1002/smll.201802565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Lai, J. , Tang, R. , Ji, W. , Wang, R. , Wang, J. , & Wang, C. (2014). Synthesis and characterization of homopolymers bearing acid‐cleavable cationic side‐chains for pH‐modulated release of DNA. Macromolecular Bioscience, 14(7), 1015–1024. 10.1002/mabi.201400004 [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Xie, X. , Xu, X. , Xia, X. , Wang, H. , Li, L. , Dong, W. , Ma, P. , Yang, Y. , Liu, Y. , & Mei, X. (2016). Thermal and magnetic dual‐responsive liposomes with a cell‐penetrating peptide‐siRNA conjugate for enhanced and targeted cancer therapy. Colloids and Surfaces. B, Biointerfaces, 146, 607–615. 10.1016/j.colsurfb.2016.07.002 [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Cheng, Q. , Jiang, Q. , Deng, L. , Liang, Z. , & Dong, A. (2015). Thermo‐sensitive nanoparticles for triggered release of siRNA. Journal of Biomaterials Science. Polymer Edition, 26(4), 264–276. 10.1080/09205063.2014.997559 [DOI] [PubMed] [Google Scholar]
- Yingyuad, P. , Mevel, M. , Prata, C. , Furegati, S. , Kontogiorgis, C. , Thanou, M. , & Miller, A. D. (2013). Enzyme‐triggered PEGylated pDNA‐nanoparticles for controlled release of pDNA in tumors. Bioconjugate Chemistry, 24(3), 343–362. 10.1021/bc300419g [DOI] [PubMed] [Google Scholar]
- Yonenaga, N. , Kenjo, E. , Asai, T. , Tsuruta, A. , Shimizu, K. , Dewa, T. , Nango, M. , & Oku, N. (2012). RGD‐based active targeting of novel polycation liposomes bearing siRNA for cancer treatment. Journal of Controlled Release, 160(2), 177–181. 10.1016/j.jconrel.2011.10.004 [DOI] [PubMed] [Google Scholar]
- Yong, K. W. , Yuen, D. , Chen, M. Z. , Porter, C. J. H. , & Johnston, A. P. R. (2019). Pointing in the right direction: Controlling the orientation of proteins on nanoparticles improves targeting efficiency. Nano Letters, 19(3), 1827–1831. 10.1021/acs.nanolett.8b04916 [DOI] [PubMed] [Google Scholar]
- Yu, C. , Li, L. , Hu, P. , Yang, Y. , Wei, W. , Deng, X. , Wang, L. , Tay, F. R. , & Ma, J. (2021). Recent advances in stimulus‐responsive Nanocarriers for gene therapy. Advanced Science (Weinheim), 8(14), 2100540. 10.1002/advs.202100540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, P. J. , He, L. , Qiu, S. W. , Li, Y. , Liao, Y. J. , Li, X. P. , Xie, D. , & Peng, Y. (2014). OX26/CTX‐conjugated PEGylated liposome as a dual‐targeting gene delivery system for brain glioma. Molecular Cancer, 13, 191. 10.1186/1476-4598-13-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelphati, O. , & Szoka, F. C., Jr. (1996). Mechanism of oligonucleotide release from cationic liposomes. Proceedings of the National Academy of Sciences of the United States of America, 93(21), 11493–11498. 10.1073/pnas.93.21.11493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Zhang, J. , Diao, L. , & Han, L. (2021). Small non‐coding RNAs in human cancer: Function, clinical utility, and characterization. Oncogene, 40(9), 1570–1577. 10.1038/s41388-020-01630-3 [DOI] [PubMed] [Google Scholar]
- Zhao, N. , Zeng, Z. , & Zu, Y. (2018). Self‐assembled aptamer‐nanomedicine for targeted chemotherapy and gene therapy. Small, 14(4), 1702103. 10.1002/smll.201702103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylberberg, C. , Gaskill, K. , Pasley, S. , & Matosevic, S. (2017). Engineering liposomal nanoparticles for targeted gene therapy. Gene Therapy, 24(8), 441–452. 10.1038/gt.2017.41 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.