

Abstract
Introduction
The apolipoprotein E (APOE) genotype is the strongest genetic risk factor for late‐onset Alzheimer's disease. However, its effect on lipid metabolic pathways, and their mediating effect on disease risk, is poorly understood.
Methods
We performed lipidomic analysis on three independent cohorts (the Australian Imaging, Biomarkers and Lifestyle [AIBL] flagship study, n = 1087; the Alzheimer's Disease Neuroimaging Initiative [ADNI] 1 study, n = 819; and the Busselton Health Study [BHS], n = 4384), and we defined associations between APOE ε2 and ε4 and 569 plasma/serum lipid species. Mediation analysis defined the proportion of the treatment effect of the APOE genotype mediated by plasma/serum lipid species.
Results
A total of 237 and 104 lipid species were associated with APOE ε2 and ε4, respectively. Of these 68 (ε2) and 24 (ε4) were associated with prevalent Alzheimer's disease. Individual lipid species or lipidomic models of APOE genotypes mediated up to 30% and 10% of APOE ε2 and ε4 treatment effect, respectively.
Discussion
Plasma lipid species mediate the treatment effect of APOE genotypes on Alzheimer's disease and as such represent a potential therapeutic target.
Keywords: APOE ε2, APOE ε4, Alzheimer's disease, lipidomics, lipid species, mass spectrometry
1. PART 1–NARRATIVE
The apolipoprotein E (APOE) gene is by far the largest genetic risk factor for sporadic Alzheimer's disease (AD). 1 , 2 Despite its identification and characterization nearly three decades ago, the mechanism by which the gene it influences sporadic AD onset and progression remains to be fully determined. There are two alleles of interest: the ε4 allele dramatically increases risk for sporadic AD, whereas the ε2 allele provides protection or resilience. The encoded protein (apolipoprotein E [apoE]) is involved in lipoprotein transport and metabolism. In peripheral circulation, apoE associates with triglyceride‐rich lipoprotein particles (chylomicrons and very low‐density lipoprotein). Despite its annotation as a causal genetic variant of sporadic AD, defining the underlying mechanism and the therapeutic potential remain elusive and are topics of considerable interest.
1.1. Current state of knowledge
Because amyloid beta (Aβ) is central to many hypotheses in both familial and sporadic AD pathogenesis, the relationship between APOE and sporadic AD has been largely investigated in the context of Aβ accumulation and clearance. Multiple studies have demonstrated a high proportion of brain Aβ in healthy ε4‐positive individuals relative to the other alleles, 3 , 4 with supporting evidence in human stem‐cell–derived neuronal studies highlighting increased Aβ production with the ε4 allele. 5 , 6 In vitro studies have indicated indirect roles for APOE in Aβ clearance via interaction with microglia 7 and other neuronal cells. 8 Despite evidence of the involvement of APOE with Aβ, no clear mechanism has been identified. Because therapeutics targeted toward Aβ have largely been unsuccessful, it is likely that several underlying pathways are involved in sporadic AD development.
As a key constituent of lipoproteins and lipid transport, a logical role for APOE variants in sporadic AD development would be through perturbations to lipid metabolism. The direct effect of APOE variants on human peripheral lipoprotein metabolism has been examined intensively. 9 , 10 , 11 , 12 Comprehensive Nuclear Magnetic Resonance (NMR) lipoprotein profiling shows that APOE ε4 leads to minor increases in nearly all lipoprotein subclasses, whereas APOE ε2 results in stronger changes to the lipoprotein profile. 13 In the central nervous system, apoE is the most abundant lipoprotein, playing an important role in lipid transport and cholesterol homeostasis. 14 Although lipoproteins are the main carriers of lipids and are studied more extensively in the context of AD, it has been proposed that lipid metabolism— represented by the complex lipid metabolic pathways responsible for the synthesis, interconversion, and catabolism of the small amphiphilic molecules that make up lipoprotein particles in addition to cellular membranes—plays a more critical role in AD pathogenesis. 15 , 16 The effects of APOE variants appear to mildly alter the relationship between peripheral lipid metabolites and the strength of association with AD. 17 , 18 These findings collectively support a potential relationship between lipid metabolism, APOE genotypes, and AD risk.
1.2. Knowledge gap, the study approach, and other alternatives
Sporadic AD is a complex disease unique to the human population, evident only through our relatively long life‐span and higher cognitive function. Thus studies in human populations are important and necessary to understand the complex relationships that exist. Lipidomics is a specialized field examining lipid metabolites in biological systems, and it has typically been limited to small sample sizes. Recent advances have paved the way for population‐level approaches that provide the power to examine associations within the variability of human diversity. Owing to the lack of large human studies conducted in the field, the associations between APOE genotypes, circulating lipid metabolites, and the relationship with sporadic AD risk have not been examined in detail.
To address this gap in knowledge, we examined the associations between plasma lipid species and APOE genotypes in three large cohorts comprising the Busselton Health Study (BHS), the Australian Imaging, Biomarkers and Lifestyle (AIBL) flagship study, and the Alzheimer's Disease Neuroimaging Initiative (ADNI) 1 study. The associations were determined independent of disease using the BHS, a largely healthy population cohort from Australia, to avoid reverse causation. Associations with APOE were then contrasted to lipid associations observed with prevalent AD. Finally, mediation analyses using individual lipid species or combined APOE lipid summary scores were performed to assess the role of lipid metabolism in mediating risk of APOE ε2 and ε4 alleles on AD (Figure 1).
FIGURE 1.
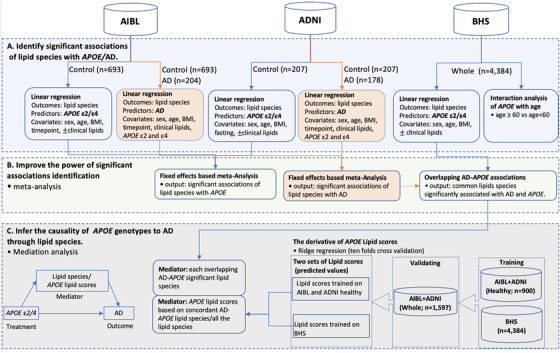
Study design. In this study, the analyses include three main sections: the identification of the significant associations of lipid species with the apolipoprotein E(APOE) gene and prevalent Alzheimer's disease (AD) (A), the improvement of the power of the associations by meta‐analysis combining Australian Imaging, Biomarkers and Lifestyle (AIBL) and Alzheimer's Disease Neuroimaging Initiative (ADNI) (B), and the causality inference of APOE genotypes to prevalent AD through lipid species by mediation analysis (C). (A) For each participant, we utilized available samples at their last acquired time point (n = 1087) to maximize the number of participants in the association studies. Lipid association studies with APOE were performed using only the control (CN) subset, whereas associations with AD prevalence was examined between the control and AD subsets. Covariates fitted into the models included age, sex, and BMI. Models for ADNI included fasting status, whereas models for AIBL included sample time point. To identify associations that were independent of lipoprotein metabolism, a second set of analyses was performed with further adjustment for clinical lipids (total cholesterol, HDL‐C, and triglycerides). Associations were corrected for multiple comparisons using the method of Benjamini Hochberg (BH). 60 In the Busselton Health Study (BHS), interaction of APOE genotypes and age was examined using a binary cut‐off of 60 years (Table S6). The “clinical lipids” means the linear regression was performed separately with/without clinical lipids adjustment. (B) Associations between APOE genotypes and lipid species and the associations between AD prevalence and lipid species were analyzed using a fixed‐effect inverse‐variance weighted meta‐analysis. Heterogeneity between AIBL and ADNI was assessed using Cochran Q. (C) The mediation analysis was performed on the combined AIBL and ADNI data sets, treating AD as the outcome and APOE genotypes as the treatment. There were two types of mediators: (1) individual lipid species (that showed concordant associations with APOE and AD from the previous analyze) and (2) APOE lipid scores. The lipid scores were created by ridge regression using either the lipid species concordant in association with AD/APOE or all the lipid species to predict APOE ε2/ε4. The models were trained on either the BHS cohort (n = 4384) or the combined AIBL and ADNI cohorts (Control; n = 900), followed with an external validation on the whole population of combined AIBL and ADNI cohorts (n = 1597). The resulting predicted values on the validate set were the APOE lipid scores that were treated as mediators for the mediation analysis
1.3. Findings
The mechanism by which APOE genotypes modulate risk has yet to be fully elucidated, and thus identifying this could pave way for additional modulatory therapeutic targets. We summarize the main findings into three major categories. (1) We identified multiple associations with specific lipid classes and species that were independent of clinical lipoprotein measurements, (2) we identified age‐specific interactions between the associations of lipid species and APOE genotypes, and (3) we demonstrated that lipid species partially mediate the AD risk resulting from inherited APOE genotypes.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using PubMed and Google to identify recent reports of the risk factors for Alzheimer's disease (AD) and the risk associated with the apolipoprotein E (APOE) genotypes. Although the relationship between APOE genotypes and lipoprotein levels has been reported previously, no reports on the relationship with the underlying lipid metabolism were found.
Interpretation: We report detailed associations between APOE genotypes and plasma lipid species, indicating an effect on lipid metabolism. The APOE ε2 allele showed stronger associations with ether lipid species than the APOE ε4 allele, with these lipid species mediating up to 30% of APOE ε2 treatment effect, leading to increased resilience.
Future Directions: Understanding the relationship between ether lipid metabolism, AD risk and resilience presents new therapeutic options to delay or prevent the onset of disease.
1.4. Lipid metabolites are strongly associated with APOE genotype
We noted that APOE genotypes were associated with circulating lipoprotein levels in all three cohorts only in healthy individuals, in particular, higher high‐density lipoprotein cholesterol (HDL‐C) and lower total cholesterol were evident in individuals with the APOE ε2 allele (Table 1). These associations have been observed previously 9 , 10 , 11 , 12 , 19 , 20 and highlight the importance of considering clinical lipids when examining lipidomic associations. Without adjusting for clinical lipids, associations with lipid species were influenced by the relative levels of lipoproteins in circulation. However, after adjustment for clinical lipids, the resulting associations highlight altered lipid species composition as a potential effect of APOE genotype. Of note, APOE ε2 exhibited stronger associations with the plasma lipidome than the APOE ε4 .
TABLE 1.
Basic characteristics of participants from the Australian Imaging, Biomarkers and Lifestyle (AIBL) flagship study, Alzheimer's Disease Neuroimaging Initiative (ADNI), and the Busselton Health Study (BHS)
| AIBL | ||||||||
|---|---|---|---|---|---|---|---|---|
| Number of APOE ε2 alleles | Number of APOE ε4 alleles | |||||||
| 0 | 1 | 2 | P value | 0 | 1 | 2 | P‐value | |
| N | 564 | 124 | 5 | 520 | 163 | 10 | ||
| Age (years) | 75.2 (6.5) | 75.5 (6.9) | 75.4 (5.2) | .927 | 75.7 (6.7) | 74.2 (5.8) | 71.6 (6.2) | .01 |
| Gender (% female) | 327 (58.0) | 77 (62.1) | 4 (80.0) | .44 | 303 (58.3) | 100 (61.3) | 5 (50.0) | .665 |
| BMI (kg/m2) | 26.30(4.4) | 26.3 (4.4) | 25.6 (5.1) | .927 | 26.3 (4.2) | 26.3 (4.8) | 26.6 (4.2) | .966 |
| HDL‐C (mmol/L) | 1.56 (0.41) | 1.67 (0.47) | 1.58 (0.54) | .029 | 1.58 (0.43) | 1.56 (0.42) | 1.68 (0.46) | .656 |
| Total Cholesterol (mmol/L) | 5.28 (1.12) | 5.15 (1.05) | 5.52 (2.41) | .444 | 5.23 (1.11) | 5.35 (1.16) | 5.51 (0.94) | .4 |
| Triglycerides (mmol/L) | 1.27 (0.61) | 1.19 (0.54) | 2.30 (2.42) | .001 | 1.25 (0.63) | 1.32 (0.65) | 1.15 (0.46) | .4 |
| 1 | 37 (6.6) | 8 (6.5) | 0 (0.0) | 38 (7.3) | 7 (4.3) | 0 (0.0) | ||
| 2 | 57 (10.1) | 15 (12.1) | 1 (20.0) | 55 (10.6) | 17 (10.4) | 1 (10.0) | ||
| 3 | 28 (5.0) | 5 (4.0) | 0 (0.0) | 23 (4.4) | 10 (6.1) | 0 (0.0) | ||
| 4 | 63 (11.2) | 9 (7.3) | 0 (0.0) | 53 (10.2) | 17 (10.4) | 2 (20.0) | ||
| 5 | 379 (67.2) | 87 (70.2) | 4 (80.0) | 351 (67.5) | 112 (68.7) | 7 (70.0) | ||
| ADNI | ||||||||
|---|---|---|---|---|---|---|---|---|
| N | 174 | 31 | 2 | 152 | 50 | 5 | ||
| Age (years) | 75.8 (4.8) | 75.2 (5.6) | 73.5 (4.7) | .661 | 75.7 (5.0) | 76.0 (5.0) | 74.6 (3.8) | .812 |
| Gender (%female) | 82 (47.1) | 19 (61.3) | 0 (0.0) | .133 | 76 (50.0) | 23 (46.0) | 2 (40.0) | .819 |
| BMI (kg/m2) | 26.8 (4.4) | 26.5 (4.0) | 28.5 (1.2) | .782 | 27.2 (4.5) | 25.8 (3.9) | 24.8 (2.3) | .087 |
| HDL‐C (mmol/L) | 1.36 (0.43) | 1.43 (0.58) | 1.32 (0.44) | .73 | 1.39 (0.44) | 1.35 (0.49) | 1.24 (0.44) | .693 |
| Cholesterol (mmol/L) | 4.74 (0.93) | 4.76 (0.95) | 3.37 (1.23) | .119 | 4.72 (0.94) | 4.81 (0.96) | 4.37 (0.66) | .566 |
| Triglycerides (mmol/L) | 1.42 (0.76) | 1.54 (1.06) | 0.93 (0.54) | .516 | 1.37 (0.68) | 1.61 (1.11) | 1.36 (0.64) | .19 |
| Fasting status (% fasting) | 16 (9.2) | 3 (9.7) | 0 (0.0) | .9 | 17 (11.2) | 0 (0.0) | 2 (40.0) | .003 |
| BHS | ||||||||
|---|---|---|---|---|---|---|---|---|
| N | 3625 | 733 | 26 | 3221 | 1061 | 102 | ||
| Age (years) | 50.4 (17.3) | 50.7 (17.6) | 50.7 (16.7) | .9 | 50.67 (17.5) | 50.2 (17.0) | 47.9 (17.0) | .228 |
| Gender (% female) | 2032 (56.1) | 413 (56.3) | 16 (61.5) | .848 | 1836 (57.0) | 572 (53.9) | 53 (52.0) | .147 |
| BMI (kg/m2) | 26.0 (4.2) | 26.1 (4.2) | 25.6 (4.3) | .844 | 26.0 (4.2) | 26.0 (4.2) | 25.4 (4.0) | .315 |
| HDL‐C (mmol/L) | 1.38 (0.39) | 1.44 (0.41) | 1.45 (0.40) | .001 | 1.40 (0.39) | 1.37 (0.40) | 1.32 (0.37) | .024 |
| Cholesterol (mmol/L) | 5.65 (1.10) | 5.26 (1.10) | 4.37 (1.14) | <.001 | 5.52 (1.12) | 5.74 (1.08) | 5.65 (1.09) | <.001 |
| Triglycerides(mmol/L) | 1.29 (0.91) | 1.34 (0.90) | 1.38 (1.01) | .43 | 1.28 (0.89) | 1.38 (0.91) | 1.33 (1.25) | .009 |
In this study, we only used the records at the last time point in AIBL cohort.
In the BHS study, we observed 29 lipid classes and 347 lipid species significantly associated with APOE ε2 after correction for multiple comparisons (Figure 2; Tables S1 and S2). Adjustment for clinical lipids, to identify associations independent of lipoprotein metabolism, resulted in 28 lipids classes and 237 lipid species significantly associated. There were 20 classes and 133 species associated with APOE ε2 in the meta‐analysis of the fully adjusted AIBL and ADNI models (including healthy individuals only). Comparison of the two analyses identified 120 concordant lipid species, nominally significant in both analyses, including species of 18 lipid classes including ceramide, hexosylceramide, sphingomyelin, plasmalogens, alkyldiacylglycerol, and cholesteryl esters.
FIGURE 2.
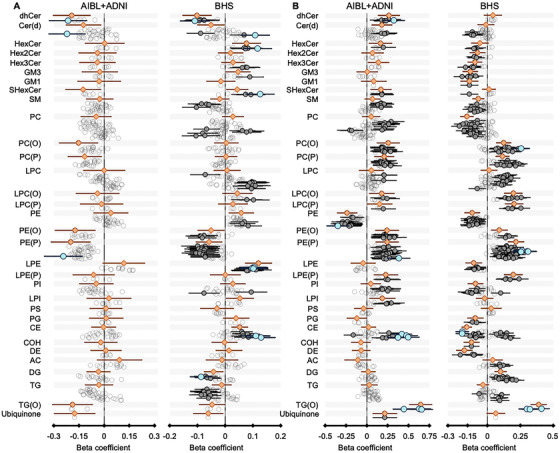
Association of APOE ε4 (A) and APOE ε2 (B) with lipid species in the Australian imaging, Biomarkers and Lifestyle (AIBL); Alzheimer's Disease Neuroimaging Initiative (ADNI); and Busselton Health Study (BHS) cohorts. ***Linear regression analyses of APOE ε4/ε2 against lipid species were performed adjusting for age, sex, BMI, total cholesterol, HDL‐C, triglycerides, timepoint (specific for AIBL), and fasting status (specific for ADNI). Meta‐analyses were performed by combining AIBL and ADNI data. Gray open circles, corrected P > .05; gray closed circles, corrected P < .05; blue circles, top 10 species ranked by P‐value; orange diamonds, lipid classes
Fewer significant associations with APOE ε4 were observed in both the BHS and meta‐analysis of the AIBL and ADNI cohorts. There were 23 lipid classes and 223 lipid species significantly associated with APOE ε4 in BHS, after correction for multiple comparisons (Figure S1; Tables S3 and S4). When we further adjusted for clinical lipids, we observed 18 lipid classes and 104 lipid species significantly associated with APOE ε4, after correction for multiple comparisons (Figure 2, Tables S3 and S4). Meta‐analysis of the fully adjusted models in the AIBL and ADNI cohorts identified three lipid species associated with APOE ε4, after correction, and 91 lipid species that were nominally significant, of which 43 species were also nominally significant in the BHS cohort.
1.5. APOE lipid associations are weaker with increasing age
We observed stronger associations within the BHS cohort than the AIBL and ADNI combined meta‐analysis, beyond the expected differences from the larger sample size. Owing to the larger age range of the BHS compared to the AIBL and ADNI cohorts, we hypothesized that age might influence how APOE genotype associates with plasma lipids. Interaction analysis with a binary cut‐off at age 60 using the BHS cohort (age <60, n = 2884; age ≥ 60, n = 1368; Tables S5) identified a nominal significant interaction of age with the association of APOE ε2 with 48 lipid species from 12 classes (Figure 3, Table S6). A greater number of the associations of APOE ε4 with lipid species were observed to have interaction effects of age (88 lipid species from 18 classes; Figure 4, Table S7). These included species of phosphatidylethanolamine, alkylphosphatidylethanolamine, alkenylphosphatidylethanolamine, and lysophosphatidylethanolamine
FIGURE 3.
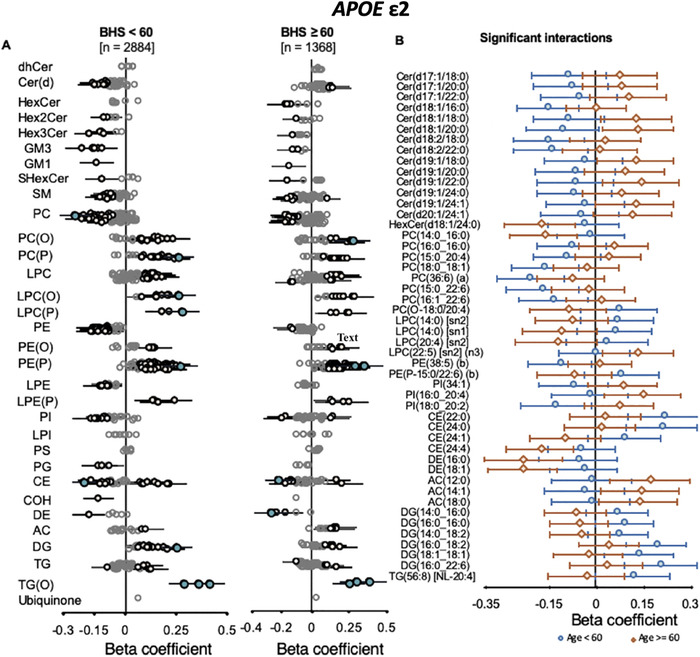
Interaction of age on the associations of APOE ε2 with peripheral lipid species. Linear regression analyses of APOE ε2 against lipid species, adjusted for APOE ε4, age, sex, BMI, total cholesterol, HDL‐C, and triglycerides with the interaction terms of age (binary cut‐off at 60‐years‐old, Panel A) was performed in the Busselton Health Study (BHS) cohort. Beta coefficients and 95% CI for each group were plotted (left panels). Beta coefficients and 95% CI for lipid species showing significant interaction (P value < .05) are plotted together (right panel)
FIGURE 4.
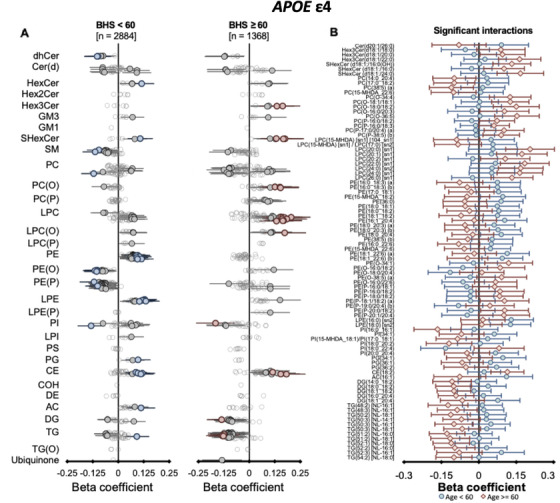
Interaction of age on the associations of APOE ε4 with peripheral lipid species. Linear regression analyses of APOE ε4 against lipid species, adjusted for APOE ε2, age, sex, BMI, total cholesterol, HDL‐C, and triglycerides with the interaction terms of age (binary cut‐off at 60‐years‐old, Panel A) was performed in the Busselton Health Study (BHS) cohort. Beta coefficients and 95% CI for each group were plotted (left panels). Beta coefficients and 95% CI for lipid species showing significant interaction (P value < .05) are plotted together (right panel)
Of interest, these results highlight the amelioration of the genotype effect in the older group (≥60), particularly for the phosphatidylethanolamine classes that show a strong association with AD. 16 , 17 This coincides with observations in the healthy AIBL and ADNI population where lipid associations with the APOE genotype, particularly the ε4 allele, were weaker. The large meta‐analysis conducted by Farrer et al., described a similar age–APOE relationship, where the risk of AD from ε4 allele was considerably reduced at ages > 65 to 70. 1 Although the exact mechanism behind the reduced association with increasing age remains to be determined, one possibility is that individuals who ultimately maintain the lower level of ether lipids progress to develop MCI, AD, or possibly other metabolic diseases including cardiovascular disease, where plasmalogens have shown a negative association, 21 , 22 leading to a survivorship bias. Because APOE ε4 associations were determined with healthy controls only, a survivorship bias may result a reduction in the strength of the associations between APOE ε4 genotype and plasma lipids with increasing age.
1.6. APOE ε2 resilience to AD is mediated via the peripheral lipidome
Ether lipids showed concordant risk profiles for APOE genotype and AD. Ether lipid associations with AD have been reported previously, 23 , 24 , 25 also within the AIBL and ADNI studies. 16 , 26 Some studies have suggested that these lipids confer protection and may become depleted in disease progression (see reviews 27 , 28 , 29 ). Species from the ether lipid group are structurally different from other lipids species by having the characteristic fatty alcohol instead of a carboxylic acid in the sn1 position of the glycerol backbone. Plasmalogens are a subclass of ether lipids, with the characteristic vinyl‐ether bond linking the alcohol to the glycerol backbone. These lipids are peroxisome‐dependent species that have been highly implicated in AD.
Here we demonstrate that ether lipids, in particular, are lower in individuals with the APOE ε4 allele, and higher in individuals with the APOE ε2 allele, independent of changes in circulating lipoprotein levels. Furthermore, the same lipid species were associated with AD (Figure 5; Tables S8‐S10). In mediation analyses, we observed that up to 36% of the AD risk from APOE ε2 was mediated through peripheral lipid species, notably the alkyldiacylglycerol and plasmalogens species, with TG (O‐52:2) [NL‐16:0] showing the strongest mediation effect (Figure S2, Table S11), whereas lipid scores created against AD mediated up to 24% the AD risk from APOE ε2 (Table 2). This mediation of AD risk by peripheral lipid species or lipid scores was much lower for the APOE ε4 allele (Table 2; Figure S2; Table S12). These mediation analyses strengthen the evidence for the involvement of ether lipids in AD etiology. Because lipids constitute modifiable risk factors, and dietary supplements are available to increase circulating ether lipids in humans, 27 this raises the possibility of risk reduction through ether lipid modulation.
FIGURE 5.
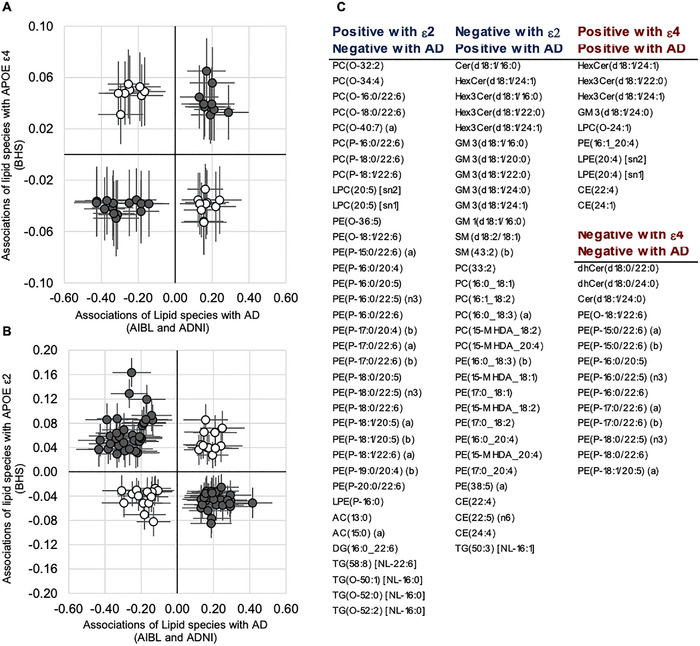
Lipid species profiles significantly associated with apolipoprotein E (APOE) gene and prevalent Alzheimer's disease (AD). Meta‐analysis of the Australian Imaging, Biomarkers and Lifestyle (AIBL) and Alzheimer's disease Neuroimaging Initiative (ADNI) cohorts was performed to identify lipid species associated with prevalent AD (linear regression of AD against lipid species, adjusted for APOE ε2, APOE ε4, age, sex, BMI, total cholesterol, HDL‐C, and triglycerides). Linear regression of APOE ε4 (A) or APOE ε2 (B) against lipid species, adjusted for age, sex, BMI, total cholesterol, HDL‐C, and triglycerides was performed in the in Busselton Health Study (BHS) cohort. The beta‐coefficients for lipid species significant in both analyses were plotted against each other. Dark closed circles highlight species that are in a concordant direction with ε4 risk increase (A) or ε2 risk reduction (B)
TABLE 2.
Mediation analysis of lipid species–based lipid scores for prevalent Alzheimer's disease (AD) cases and APOE ε4 (A)/ε2 (B)
| Concordant lipid species–based lipid scores | All lipid species–based lipid scores | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipid score trained on combined AIBL and ADNI (Healthy) | Lipid score trained on BHS | Lipid score trained on combined AIBL and ADNI (Healthy) | Lipid score trained on BHS | |||||||||
| Est. | 95% CI | P‐value | Est. | 95% CI | P‐value | Est. | 95% CI | P‐value | Est. | 95% CI | P‐value | |
| A. APOE ε2 | ||||||||||||
| Total effect | −0.13 | (−0.18 ‐ −0.07) | <2e‐16 | −0.13 | (−0.18 ‐ −0.07) | <2e‐16 | −0.12 | (−0.18 ‐ −0.07) | <2e‐16 | −0.13 | (−0.18 ‐ −0.07) | <2e‐16 |
| Prop. mediated (Cont) | 0.28 | (0.12 ‐ 0.55) | 8.0e‐04 | 0.24 | (0.13 ‐ 0.43) | <2e‐16 | −0.09 | (−0.4 ‐ 0.15) | 4.3e‐01 | 0.27 | (0.15 ‐ 0.51) | <2e‐16 |
| Prop. mediated (AD) | 0.19 | (0.06 ‐ 0.48) | 8.0e‐04 | 0.16 | (0.07 ‐ 0.37) | <2e‐16 | −0.06 | (−0.25 ‐ 0.1) | 4.3e‐01 | 0.19 | (0.08 ‐ 0.45) | <2e‐16 |
| ACME (Avg) | −0.03 | (−0.05 ‐ −0.01) | 8.0e‐04 | −0.03 | (−0.04 ‐ −0.01) | <2e‐16 | 0.01 | (−0.02 ‐ 0.03) | 4.3e‐01 | −0.03 | (−0.04 ‐ −0.02) | <2e‐16 |
| ADE (Avg) | −0.10 | (−0.15 ‐ −0.04) | 1.6e‐03 | −0.10 | (−0.16 ‐ −0.05) | 1.2e‐03 | −0.13 | (−0.19 ‐ −0.07) | <2e‐16 | −0.10 | (−0.15 ‐ −0.04) | 2.8e‐03 |
| Prop. mediated (Avg) | 0.24 | (0.09 ‐ 0.52) | 8.0e‐04 | 0.20 | (0.1 ‐ 0.4) | <2e‐16 | −0.08 | (−0.32 ‐ 0.13) | 4.3e‐01 | 0.23 | (0.12 ‐ 0.48) | <2e‐16 |
| B. APOE ε4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total effect | 0.19 | (0.16 ‐ 0.22) | <2e‐16 | 0.19 | (0.16 ‐ 0.22) | <2e‐16 | 0.18 | (0.16 ‐ 0.22) | <2e‐16 | 0.19 | (0.16 ‐ 0.22) | <2e‐16 |
| Prop. mediated (Cont) | 0.05 | (0.02 ‐ 0.08) | 1.4e‐03 | 0.07 | (0.04 ‐ 0.11) | <2e‐16 | −0.01 | (−0.05 ‐ 0.03) | 8.1e‐01 | 0.05 | (0.03 ‐ 0.09) | <2e‐16 |
| Prop. mediated (AD) | 0.08 | (0.03 ‐ 0.13) | 1.4e‐03 | 0.11 | (0.07 ‐ 0.17) | <2e‐16 | −0.01 | (−0.09 ‐ 0.07) | 8.1e‐01 | 0.09 | (0.04 ‐ 0.13) | <2e‐16 |
| ACME (Avg) | 0.01 | (0 ‐ 0.02) | 1.4e‐03 | 0.02 | (0.01 ‐ 0.03) | <2e‐16 | 0.00 | (−0.01 ‐ 0.01) | 8.1e‐01 | 0.01 | (0.01 ‐ 0.02) | <2e‐16 |
| ADE (Avg) | 0.18 | (0.15 ‐ 0.21) | <2e‐16 | 0.17 | (0.14 ‐ 0.2) | <2e‐16 | 0.19 | (0.16 ‐ 0.23) | <2e‐16 | 0.18 | (0.14 ‐ 0.21) | <2e‐16 |
| Prop. mediated (Avg) | 0.06 | (0.02 ‐ 0.11) | 1.4e‐03 | 0.09 | (0.06 ‐ 0.14) | <2e‐16 | −0.01 | (−0.07, 0.06) | 8.1e‐01 | 0.07 | (0.03 ‐ 0.11) | <2e‐16 |
Alkyl diacylglycerols is notable owing to its ether‐linkage in the sn1 position (with fatty acyl linkages in the sn2 and sn3 positions), placing them in the same family as ether lipids and plasmalogens, a lipid class reported to be negatively associated with AD. 16 , 23 , 30 , 31 Although the biosynthetic origin of the alkyldiacylglycerols in the periphery remains largely uncharacterized, it is clear that they are derived from the peroxisomal pathway that synthesizes 1‐O‐alkyl‐dihydroxyacetone phosphate, which is then converted into 1‐O‐alkyl‐2‐acylglycerol in the endoplasmic reticulum, at which point it can be converted into either plasmalogens or alkyl diacylglycerol. APOE ε2 dramatically elevates the levels of both of these lipid classes, above those of typical phospholipid or triglyceride species. The APOE ε2 polymorphism may have selective preference for alkyldiacylglycerols and other ether lipids in incorporation into lipoproteins. Alternatively, the turnover rate of lipoproteins with APOE variants has been highlighted previously, where changes to the metabolic flux rate of lipoproteins due to their interactions with the low‐density lipoprotein (LDL) receptor may influence its composition. 32 Although ether lipids are a diverse group of lipids, it is well known that plasmalogens represent the more biologically active lipid class, having been linked to anti‐oxidative and anti‐inflammatory properties. 27 , 33 , 34 Potentially the alkyldiacylglycerol species rather represent sensitive markers of ether lipid synthesis or turnover perturbation. In contrast to APOE ε2, the APOE ε4 allele showed a weaker association with lipid species, and those same species and the lipid scores mediated only a small proportion of the APOE ε4 risk on AD (7% and 9%, respectively), although here also the plasmalogen species were the strongest mediators (Table 2; Figure S2; Table S12). This raises the possibility that the two common polymorphisms of APOE mediate risk through alternate mechanisms, with the resilient effect of APOE ε2 being more strongly influenced by its effect on ether lipid metabolism in the periphery.
1.7. Next steps
Our analyses demonstrate that up to 36% of the AD resilience associated with APOE ε2 is mediated by lipid species (primarily ether lipids, alkyl diacylglycerol, and plasmalogen) and, to a lesser extent, the increased AD risk associated with APOE ε4 is also mediated by some of the same lipid species. These lipid species then represent a potential therapeutic target to reduce the risk of AD. However, it will be important to understand the mechanism(s) by which such ether lipids may attenuate disease risk.
Alkyl diacylglycerol is a naturally occurring class of lipids, particularly enriched in the livers of several species of sharks 35 and relatively abundant in human breast milk, 36 which, upon ingestion, can be metabolized into plasmalogens and other ether lipid species, to increase the levels of these lipids in circulation and within immune cells. 37 The use of alkyldiacylglycerol as a nutraceutical has been examined in the context of immune modulation 38 and a potential treatment for specific cancers, 39 and the immune‐modulating properties of ether lipids has been reviewed. 27 , 40
There is growing evidence of an immune component in AD pathogenesis 41 and genetic evidence linking both immunity and lipid metabolism involvement with AD risk. 42 The involvement of APOE in the immune response 43 , 44 suggests that its potential role in risk reduction may be via modulation of immune cell function and behavior. More recently, ether lipids have been linked directly to ferroptosis, 45 , 46 a novel cell death mechanism that links together immunity, 47 iron metabolism, and AD. 48 , 49 Plasmalogens, an end product of the ether lipid biosynthetic pathway resulting from the formation of the vinyl ether bond by the desaturase PEDS1/TMEM189, appear to play complex and possibly contradicting roles in ferroptosis. 45 Although our data do not directly link ferroptosis, APOE polymorphisms and AD together, the critical role of ether lipids in mediating these biological processes necessitates further examination. In vitro and in vivo studies exploring the role of ether lipids, ferroptosis, and immune cell function in the context of AD will likely provide some of the answers to the mechanisms by which ether lipids may be mediating the risk reduction afforded by APOE ε2.
With this insight, a next logical step would be to modulate ether lipid species with a view toward preventing or attenuating AD onset and progression, or to influence surrogate AD risk markers (Aβ, phosphorylated tau, or cognition) in the early stages of disease. Modulation of ether lipid species in humans has been demonstrated in several studies, 31 , 37 where the biologically active precursor, alkylglycerols, that can be synthesized 50 or derived from natural sources in various marine animals 27 , 51 has been used to bypass the rate‐limiting peroxisomal step to upregulate plasmalogen synthesis. Because the vinyl‐ether bond of plasmalogen species is highly susceptible to acid hydrolysis, ingestion of these species may not be the optimal approach to raise plasmalogen levels. Thus the non‐plasmalogen precursors may serve as better and more‐stable dietary interventions for raising plasmalogens.
The expected development of sporadic AD likely spans decades prior to the onset of symptoms 52 ; therefore, early intervention will be required. Nutraceuticals comprising alkyldiacylglycerols are potentially low‐cost, low‐risk dietary supplements that may provide tangible risk reductions and so represent prime targets as a proactive preventative measure for AD. To demonstrate efficacy for such an intervention will require substantial investment in clinical trials of sufficient size and duration to reach statistical significance. However, an additional application of our findings is the development of these ether lipids as biomarkers to not only identify those at increased risk (for inclusion in clinical studies) but also those who will most benefit from ether lipid‐modulation therapy.
1.8. Limitations and remaining questions
Our study examines the relationship between APOE polymorphisms and plasma lipid species in the context of AD using three large independent cohorts. The classification of AD and non‐AD dementia is clinically difficult and is confirmed only through post‐mortem examination. Such misclassification could lead to confounding and underestimation of effect sizes in our analyses. Further to this, in neurological diseases, the importance of peripheral biomarkers remains contentious, as they may not accurately reflect the neurological pathophysiology. However, there are many biological processes that can ultimately influence the pathogenesis of neurological diseases, including the innate and adaptive immune systems. Additional research into lipid metabolic changes within both the immune system and the brain, in relation to AD and APOE variants, will shed light on the mechanisms by which dysregulated lipid metabolism may influence AD risk.
Finally, although we were able to validate many of our analyses across cohorts, the interaction of age with the association between plasma lipid species and APOE variants requires external validation on an independent cohort. The sample size and age range required meant that this effect could be explored only in the BHS cohort in this study.
1.9. Conclusion
Here, we combine the power of two large clinical studies of AD with an Australian population study to elucidate the relationship between APOE variants and lipid metabolism. We demonstrate a strong relationship of APOE ε2 and ε4 alleles with ether lipid species. We further demonstrate that these same lipid species strongly mediate the resilient effects of APOE ε2 on AD risk, thereby presenting a therapeutic opportunity.
2. PART 2–CONSOLIDATED RESULTS AND STUDY DESIGN
2.1. Study design
This study includes three main sections of analyze (Figure 1). First, lipid association studies with APOE were performed using only the cognitively healthy individuals to avoid any associations driven by reverse causation, whereas associations with AD prevalence were examined between the CN and AD subsets. In BHS, interaction of APOE alleles and age was examined using a binary cut‐off at 60 years (Table S5).
Next, associations between APOE genotypes and lipid species and the associations between AD prevalence and lipid species were analyzed using a fixed‐effect inverse‐variance weighted meta‐analysis. Heterogeneity between AIBL and ADNI was assessed using Cochran Q.
Then, the mediation analysis was performed on the aligned AIBL and ADNI data sets to assess whether lipid species mediate the effects of APOE on AD. We investigated two types of mediators: (1) individual lipid species (which showed concordant associations with APOE and AD from the previous analyses) and (2) APOE lipid scores. The lipid scores were created by ridge regression using either the lipid species concordant in association with AD/APOE or all the lipid species to predict APOE ε2/ε4. Causal mediation analysis was then performed to estimate the proportion of risk in the outcome model explained by a direct effect of APOE genotype on prevalent AD and the proportion that was mediated by individual lipid species or lipid scores.
2.2. Results
2.2.1. The association of APOE genotypes with plasma lipid species
In the BHS study, we observed 29 lipid classes and 347 lipid species significantly associated with APOE ε2 after correction for multiple comparisons (Figure 2; Tables S1 and S2). Adjustment for clinical lipids, to identify associations that are independent of lipoprotein metabolism, resulted in 28 lipids classes and 237 lipid species significantly associated. There were 20 classes and 133 species associated with APOE ε2 in the meta‐analysis of the fully adjusted AIBL and ADNI models. A total of 120 concordant lipid species from 18 lipid classes including ceramide, hexosylceramide, sphingomyelin, plasmalogen, alkyl diacylglycerol, and cholesteryl ester, were identified to be nominally significant in both analyses.
A lower number of significant associations with APOE ε4 were observed in both the BHS and meta‐analysis of the AIBL and ADNI cohorts. There were 23 lipid classes and 223 lipid species significantly associated with APOE ε4 in BHS, after correction for multiple comparisons (Figure S1; Tables S3 and S4). When we further adjusted for clinical lipids, we observed 18 lipid classes and 104 lipid species significantly associated with APOE ε4, after correction for multiple comparisons (Figure 2, Tables S3 and S4). Meta‐analysis of the fully adjusted models in the AIBL and ADNI cohorts identified only three lipid species associated with APOE ε4, after correction. Ninety‐one lipid species were nominally significant, of which 43 species were also nominally significant in the BHS cohort.
2.2.2. Interaction of age with the associations between APOE and lipid species in the BHS cohort
We observed considerably stronger associations within the BHS cohort than the AIBL and ADNI meta‐analysis, beyond that expected from the larger sample size. Owing to the larger age range of the BHS compared to the AIBL and ADNI cohorts, we hypothesized that age might influence how APOE genotype associates with plasma lipids. Interaction analysis with a binary cut‐off at age 60 using the BHS cohort (age < 60, n = 2884; age ≥ 60, n = 1368) identified a nominal significant interaction of age with the association of APOE ε2 with 48 lipid species from 12 classes (Figure 3, Table S6). A greater number of the associations of APOE ε4 with lipid species was observed to have interaction effects of age (88 lipid species from 18 classes; Figure 4, Table S7). These included species of phosphatidylethanolamine, alkylphosphatidylethanolamine, alkenylphosphatidylethanolamine, and lysophosphatidylethanolamine. Of interest, these results highlight weaker associations between lipid species and APOE ε4 genotype with increasing age.
2.2.3. Concordance of the associations of APOE alleles and prevalent AD with lipid species
We rationalized that lipids increased by the ε2 allele, but negatively associated with AD may be involved in mediating risk reduction. Similarly, lipids that associate in the same direction with AD and APOE ε4 are likely important in disease pathology. After aligning the associations of lipids species with AD (Table S8) and APOE ε2, we observed 68 lipid species displaying an inverse association with AD diagnosis and APOE ε2 (putative protective lipid species, Figure 4; Table S9). These lipid species included 37 species of ether phospholipids and 29 lipid species of sphingolipids, phosphatidylethanolamine, and dehydrocholesterol species.
We observed fewer lipid species (24) showing concordant associations with AD and APOE ε4, which fall predominantly within the ceramide, trihexosylceramide, alkenylphosphatidylethanolamine, and lysophosphatidylethanolamine classes (Figure 4; Table S10).
2.2.4. Mediating role of the plasma lipidome on the effects of APOE on AD risk
We identified 11 lipid species mediating the effect of APOE ε2 on AD, after correcting for multiple comparisons (Figure S2; Table S11). These lipid species differed slightly from those mediating the effects of APOE ε4, largely comprising the alkyl phosphatidylcholine and phosphatidylethanolamine and alkenyl phosphatidylethanolamine classes. Of particular interest were three species of alkyldiacylglycerol TG (O‐50:1) [NL ‐16:0], TG (O‐52:0) [NL ‐16:0], and TG (O‐52:2) [NL ‐16:0], which showed strong mediating effects, accounting for up to 30% of the total effects of APOE ε2 on AD. There were 14 lipid species that showed a significant mediation effect between APOE ε4 and AD, after correcting for multiple comparisons (Figure S2; Table S12). These lipid species were of the dihydroceramide, alkylphosphatidylethanolamine, and alkenylphosphatidylethanolamine classes.
The APOE ε2 lipid scores derived from BHS and healthy AIBL/ADNI both showed strong mediating effects on AD risk of 20% and 24%, respectively (Table 2). In contrast, the mediating effects of the APOE ε4 lipid scores were smaller, but still significant, at 9% and 6% for the BHS and healthy AIBL/ADNI derived scores, respectively (Table 2). Mediation analysis with lipid scores derived from all lipid species showed similar performance when built using the BHS cohort, with a 23% and 7% mediation effect for APOE ε2 and APOE ε4, respectively.
3. PART 3–DETAILED METHODS AND RESULTS
More details of methodology including the data and statistical methods are described.
3.1. Study cohorts
3.1.1. The Busselton Health study (BHS)
BHS is a community‐based population study for which participants were recruited in Western Australia in the 1960s. The BHS holds extensive phenotype data (eg, cardiovascular disease traits), high‐density single‐nucleotide polymorphism (SNP) panels, and plasma lipidomic profiling data. 53 , 54 , 55 In this study, we used data from 4492 participants who provided plasma samples at the 1994 to 1995 recall.
3.1.2. The Australian Imaging, Biomarkers and Lifestyle (AIBL) flagship study
AIBL is a longitudinal study that initially recruited 1112 participants who are older than 60 years of age. This included 768 cognitive normal individuals (CN), 133 with mild cognitive impairment (MCI), and 211 with AD at the last time point. The participants were recalled at 18‐month intervals for up to 72 months. APOE genotype and other biochemical data were collected. Lipidomics analysis was performed on all available plasma samples, with 4106 fasted plasma samples examined from baseline up to the fifth time point. 56
3.1.3. Alzheimer's Disease Neuroimaging Initiative (ADNI)
ADNI is a multi‐site longitudinal study using a non‐randomized, natural history, non‐treatment design. The first phase (ADNI‐1), launched in 2004, aimed to track disease progression using biomarkers and to identify features of MCI that may predict cognitive decline. Clinical follow‐up is available for up to 2 to 3 years post screening in the ADNI‐1 study, with participants carried forward into subsequent ADNI2‐GO studies. The ADNI‐1 cohort includes 819 participants (229 CN, 398 MCI, and 192 AD). Lipidomics was performed on all participants with available samples at baseline.
3.1.4. Ethics approval and consent to participate
For all the above cohorts, written informed consent was obtained from all participants before protocol‐specific procedures were performed.
3.2. Lipid extraction and mass spectrometry analysis
Extensive details on the lipidomic profiling of the BHS, ADNI, and AIBL cohorts have been published previously. 16 , 26 , 55 Lipid extractions were performed on plasma (AIBL) and serum (ADNI, BHS) samples as described previously. 57 Lipidomic profiling (569 lipid species from 32 classes) was carried out using scheduled multiple reaction monitoring on an Agilent 6490 QqQ mass spectrometer. 57
3.3. Definition of AD state
In AIBL, clinical criteria used to determine disease status included a Mini Mental State Examination score of <28, failure on the Logical Memory test, other evidence of possible significant cognitive difficulty on neuropsychological testing, a Clinical Dementia Rating score of ≥0.5, a medical history suggestive of the presence of illnesses likely to impair cognitive function, and an informant or personal history suggestive of impaired cognitive function. 56 The definition of possible AD in ADNI followed the the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINDS‐ADRDA) criteria, 58 whereas the classification of MCI is defined according to the criteria proposed by Petersen et al. 59
3.4. Statistical analysis
3.4.1. Cohort stratification
We sought to examine the association of APOE genotypes with lipid species independent of disease to avoid any associations driven by reverse causation. Thus we utilized only the cognitively healthy individuals in the AIBL and ADNI cohorts. After removing samples with missing records, we had a total of 5284 participants: AIBL (n = 693), ADNI (n = 207), and BHS (n = 4384). The characteristics of each cohort are presented in Table 1. All the lipid species were log10 transformed, followed by normalization to zero mean and one‐unit standard deviation.
3.4.2. Association of APOE genotypes with lipid species
The associations between APOE genotype (ε2 and ε4) and lipid species were determined by linear regression in healthy individuals in each cohort separately. BHS is a population cohort with voluntary enrolment and a median age of 48; we treated the BHS population as non‐AD. Adjustment for covariates and meta‐analyses are described in Figure 1.
3.4.3. Identifying concordant associations with AD
Linear regression was used to determine the associations between lipid species and AD, relative to healthy control. To identify associations independent of APOE genotype, these analyses were adjusted for both APOE ε2 and ε4. Other covariates included age, sex, BMI, and clinical lipids. We then selected the lipid species that were associated with both AD and APOE genotype (ε2 or ε4).
To identify lipids of potential biological relevance, we highlighted lipid species that were concordant in their association with APOE and AD. Concordant species between APOE ε2 and AD were negatively associated with APOE ε2 but positively associated with AD, and vice versa. Conversely, concordant species between APOE ε4 and AD were positively or negatively associated with both APOE ε4 and AD.
3.4.4. Mediation of the APOE genotype effect on AD by lipid species
To assess whether lipid species mediate the effect of APOE on AD, we performed mediation analysis (using the R package “mediation”) on the combined AIBL and ADNI data sets (n = 1597). The analysis was conducted using either individual lipid species that showed concordant associations with APOE and AD, or lipid scores for APOE genotypes. Two lipid scores (Figure 1, lower panel) were created by ridge regression (R package “glmnet”) using either: (1) the lipid species concordant in association with AD/APOE; or (2) all lipid species. Penalty parameters were optimized using internal 10‐fold cross‐validation. As illustrated in Figure 1 (lower panel), models were created using the BHS cohort (n = 4384) or the combined AIBL and ADNI cohorts (healthy individuals; n = 900), adjusting for age, sex, BMI, fasting status, HDL‐C, total cholesterol, and triglycerides. The resulting predicted values on the whole population of combined AIBL and ADNI cohorts (n = 1597) were the APOE lipid scores that were treated as mediators in the mediation analysis.
Causal mediation analysis (Figure 1, lower panel) was performed by first estimating the total effect of APOE genotypes on prevalent AD using logistic regression, adjusted for age, sex, BMI, HDL‐C, total cholesterol, and triglycerides. The mediator model is constructed, looking at the association of APOE genotype with lipid species and lipid scores, adjusting for the same covariates. Causal mediation analysis was then used to estimate the proportion of risk in the outcome model explained by a direct effect of APOE genotype on prevalent AD—the average direct effect (ADE)—and the proportion that was mediated by lipid species or lipid scores—the average causal mediation effect (ACME). To test for moderation effects, an interaction term was introduced between lipid species and APOE genotype. Confidence intervals were estimated using resampling (10,000 empirical bootstraps).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest with the contents of this manuscript.
AUTHOR CONTRIBUTIONS
Meikle and Kaddurah‐Daouk led the study design team. Wang, Huynh, and Giles led the statistical analyses presented in this study. Mellett, Duong, Nguyen, Lim, Smith, Olshansky, Huynh, and Giles supported the acquisition and processing of the lipidomic data for the three cohorts. Cadby, Hung, Hui, Beilby, Watts, and Moses were key members of the Busselton Health Study team. Chatterjee, I Martins, Laws, Bush, Rowe, Villemagne, Ames, Masters, Taddei, Doré, Fripp, and Martins were key members of the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing team. Arnold, Kastenmüller, Nho, Saykin, Baillie, Han, and Kaddurah‐Daouk were key members of the Alzheimer's Disease Neuroimaging Initiative team and represent the Alzheimer's Disease Metabolomics consortium (ADMC): A complete listing of ADMC investigators can be found at https://sites.duke.edu/adnimetab/who‐we‐are/.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
Support was provided by the National Health and Medical Research Council (NHMRC) of Australia (no. 1101320 and 1157607). Kevin Huynh was supported by a Dementia Australia Research Foundation Scholarship and NHMRC investigator grant (1197190) . This work was also supported in part by the Victorian Government's Operational Infrastructure Support Program. The Busselton Health Study (BHS) acknowledges the generous support for the 1994/95 Busselton follow‐up studies from HealthWay, the Department of Health, PathWest Laboratory Medicine of Western Australia (WA), and the Busselton community volunteers, who assisted with data collection and the study participants from the Shire of Busselton. Funding for the AIBL study was provided in part by the study partners (Commonwealth. Scientific Industrial and Research Organization [CSIRO], Edith Cowan University [ECU], Mental Health Research institute [MHRI], National Ageing Research Institute [NARI], Austin Health, and CogState Ltd). The Australian Imaging, Biomarkers and Lifestyle (AIBL) flagship study has also received support from the NHMRC and the Dementia Collaborative Research Centre (DCRC2), as well as funding from the Science and Industry Endowment Fund (SIEF) and the Cooperative Research Centre (CRC) for Mental Health—funded through the CRC Program (Grant ID:20100104), an Australian Government Initiative. Support for the metabolomics sample processing, assays and analytics reported here was provided by grants from the National Institute on Aging (NIA); NIA supported the Alzheimer's Disease Metabolomics Consortium, which is a part of NIA's national initiatives AMP‐AD and M2OVE‐AD (R01 AG046171, RF1 AG051550, RF1 AG057452, and 3U01 AG024904‐09S4). Additional United States National Institutes of Health (NIH) support from the NIA, National Library of Medicine (NLM), and National Cancer Institute (NCI) for analysis includes P30 AG10133, R01 AG19771, R01 LM012535, R03 AG054936, R01 AG061788, K01 AG049050, and R01 CA129769. Matthias Arnold is supported by NIA grants RF1 AG057452, RF1 AG058942, RF1 AG059093, and U01 AG061359. Matthias Arnold is also supported by funding from Qatar National Research Fund NPRP8‐061‐3‐011. Kwangsik Nho is supported by NLM R01 LM012535 and NIA R03AG054936. Data collection and sharing for the Alzheimer's Disease Neuroimaging Initiative (ADNI) was supported by NIH grant U01 AG024904. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This study was only possible with the help of the AIBL research group. The authors who made direct contribution to this study have been listed as authors in this article. Members of the AIBL group who did not participate in the analysis or writing of this report are listed here: https://aibl.csiro.au/about/aibl‐research‐team/. Part of the data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The authors who made direct contribution to this study have been listed as authors in this article. As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Part of the data used in preparation of this article were generated by the Alzheimer's Disease Metabolomics Consortium (ADMC). The authors who made direct contribution to this study have been listed as authors in this article. Investigators within the ADMC provided data but did not participate in analysis or writing of this report can be found at https://sites.duke.edu/adnimetab/team/. Metabolomics data and results from the ADNI study have been made accessible through the AMP‐AD Knowledge Portal (https://ampadportal.org). The AMP‐AD Knowledge Portal is the distribution site for data, analysis results, analytical methodology, and research tools generated by the AMP‐AD Target Discovery and Preclinical Validation Consortium and multiple Consortia and research programs supported by the National Institute on Aging. Funding sources that contributed to the cohort studies or directly to the analyses presented in the study are described in the Acknowledgements section. The funding sources had no role in the collection, analysis, or interpretation of the data; in the writing of the report; or in the decision to submit this article for publication.
Wang T, Huynh K, Giles C, et al. APOE ε2 resilience for Alzheimer's disease is mediated by plasma lipid species: Analysis of three independent cohort studies. Alzheimer's Dement. 2022;18:2151–2166. 10.1002/alz.12538
Tingting Wang, Kevin Huynh, and Corey Giles joint first authors.
Rima Kaddurah‐Daouk and Peter J Meikle joint senior authors.
REFERENCES
- 1. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta‐analysis. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 2. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 3. Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid‐beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fleisher AS, Chen K, Liu X, et al. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging. 2013;34:1‐12. [DOI] [PubMed] [Google Scholar]
- 5. Lin Y‐T, Seo J, Gao F, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC‐derived brain cell types. Neuron. 2018;98:1141‐1154. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang Y‐WA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell. 2017;168:427‐441. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee CD, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes β‐amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J, Kanekiyo T, Shinohara M, et al. Differential regulation of amyloid‐β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms*♦. J Biol Chem. 2012;287:44593‐44601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. Association of higher levels of high‐density lipoprotein cholesterol in elderly individuals and lower risk of late‐onset Alzheimer disease. Arch Neurol. 2010;67:1491‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moroney JT, Tang MX, Berglund L, et al. Low‐density lipoprotein cholesterol and the risk of dementia with stroke. JAMA. 1999;282:254‐260. [DOI] [PubMed] [Google Scholar]
- 11. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anstey KJ, Ashby‐Mitchell K, Peters R. Updating the evidence on the association between serum cholesterol and risk of late‐life dementia: review and meta‐analysis. J Alzheimers dis. 2017;56:215‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karjalainen J‐P, Mononen N, Hutri‐Kähönen N, et al. The effect of apolipoprotein E polymorphism on serum metabolome—A population‐based 10‐year follow‐up study. Sci Rep. 2019;9:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahley RW. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chatterjee P, Lim WL, Shui G, et al. Plasma phospholipid and sphingolipid alterations in presenilin1 mutation carriers: a pilot study. J Alzheimers dis. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huynh K, Lim WLF, Giles C, et al. Concordant peripheral lipidome signatures in two large clinical studies of Alzheimer's disease. Nat Commun. 2020;11:5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lim WLF, Huynh K, Chatterjee P, et al. Relationships between plasma lipids species, gender, risk factors, and alzheimer's disease. J Alzheimers dis. 2020;76:303‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arnold M, Nho K, Kueider‐Paisley A, et al. Sex and APOE ε4 genotype modify the Alzheimer's disease serum metabolome. Nat Commun. 2020;11:1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anstey KJ, Lipnicki DM, Low LF. Cholesterol as a risk factor for dementia and cognitive decline: a systematic review of prospective studies with meta‐analysis. Am J Geriatr Psychiatry. 2008;16:343‐354. [DOI] [PubMed] [Google Scholar]
- 20. Koch M, Jensen MK. HDL‐cholesterol and apolipoproteins in relation to dementia. Curr Opin Lipidol. 2016;27:76‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sutter I, Velagapudi S, Othman A, et al. Plasmalogens of high‐density lipoproteins (HDL) are associated with coronary artery disease and anti‐apoptotic activity of HDL. Atherosclerosis. 2015;241:539‐546. [DOI] [PubMed] [Google Scholar]
- 22. Sutter I, Klingenberg R, Othman A, et al. Decreased phosphatidylcholine plasmalogens–A putative novel lipid signature in patients with stable coronary artery disease and acute myocardial infarction. Atherosclerosis. 2016;246:130‐140. [DOI] [PubMed] [Google Scholar]
- 23. Goodenowe DB, Cook LL, Liu J, et al. Peripheral ethanolamine plasmalogen deficiency: a logical causative factor in Alzheimer's disease and dementia. J Lipid Res. 2007;48:2485‐2598. [DOI] [PubMed] [Google Scholar]
- 24. Kling MA, Goodenowe DB, Senanayake V, et al. Circulating ethanolamine plasmalogen indices in Alzheimer's disease: relation to diagnosis, cognition, and CSF tau. Alzheimers Dement. 2020;16:1234‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barupal DK, Baillie R, Fan S, et al. Sets of coregulated serum lipids are associated with Alzheimer's disease pathophysiology. Alzheimers Dement. 2019;11:619‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lim WLF, Huynh K, Chatterjee P, et al. Relationships between plasma lipids species, gender, risk factors, and Alzheimer's disease. J Alzheimers Dis. 2020;76(1):303‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paul S, Lancaster GI, Meikle PJ. Plasmalogens: a potential therapeutic target for neurodegenerative and cardiometabolic disease. Prog Lipid Res. 2019;74:186‐195. [DOI] [PubMed] [Google Scholar]
- 28. Farooqui AA, Horrocks LA. Plasmalogens: workhorse lipids of membranes in normal and injured neurons and glia. Neuroscientist. 2001;7:232‐245. [DOI] [PubMed] [Google Scholar]
- 29. Su XQ, Wang J, Sinclair AJ. Plasmalogens and Alzheimer's disease: a review. Lipids Health Dis. 2019;18:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goodenowe DB, Senanayake V. Relation of serum plasmalogens and APOE genotype to cognition and dementia in older persons in a cross‐sectional study. Brain Sci. 2019;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fujino T, Yamada T, Asada T, et al. Efficacy and blood plasmalogen changes by oral administration of plasmalogen in patients with mild Alzheimer's disease and mild cognitive impairment: a multicenter, randomized, double‐blind, placebo‐controlled trial. EBioMedicine. 2017;17:199‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruiz J, Kouiavskaia D, Migliorini M, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46:1721‐1731. [DOI] [PubMed] [Google Scholar]
- 33. Ifuku M, Katafuchi T, Mawatari S, et al. Anti‐inflammatory/anti‐amyloidogenic effects of plasmalogens in lipopolysaccharide‐induced neuroinflammation in adult mice. J Neuroinflammation. 2012;9:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wallner S, Schmitz G. Plasmalogens the neglected regulatory and scavenging lipid species. Chem Phys Lipids. 2011;164:573‐589. [DOI] [PubMed] [Google Scholar]
- 35. Wetherbee BM, Nichols PD. Lipid composition of the liver oil of deep‐sea sharks from the Chatham Rise, New Zealand. Comp Biochem Physiol B Biochem Mol Biol. 2000;125:511‐521. [DOI] [PubMed] [Google Scholar]
- 36. Hallgren B, Larsson S. The glyceryl ethers in man and cow*. J Lipid Res. 1962;3:39‐43. [Google Scholar]
- 37. Paul S, Smith AAT, Culham K, et al. Shark liver oil supplementation enriches endogenous plasmalogens and reduces markers of dyslipidaemia and inflammation. J Lipid Res. 2021;62:100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berdel WE, Bausert WR, Weltzien HU, Modolell ML, Widmann KH, Munder PG. The influence of alkyl‐lysophospholipids and lysophospholipid‐activated macrophages on the development of metastasis of 3‐Lewis lung carcinoma. Eur J Cancer. 1980;16:1199‐1204. [DOI] [PubMed] [Google Scholar]
- 39. Pedrono F, Martin B, Leduc C, et al. Natural alkylglycerols restrain growth and metastasis of grafted tumors in mice. Nutr Cancer. 2004;48:64‐69. [DOI] [PubMed] [Google Scholar]
- 40. Iannitti T, Palmieri B. An update on the therapeutic role of alkylglycerols. Mar Drugs. 2010;8:2267‐2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc Natl Acad Sci. 2016;113:E1316‐E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vitek MP, Brown CM, Colton CA. APOE genotype‐specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jofre‐Monseny L, Minihane AM, Rimbach GJMn, research f. . Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 2008;52:131‐145. [DOI] [PubMed] [Google Scholar]
- 45. Cui W, Liu D, Gu W, Chu BJCD. Differentiation . Peroxisome‐driven ether‐linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021;28(8):2536‐2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zou Y, Henry WS, Ricq EL, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen X, Kang R, Kroemer G, Tang DJJoEM. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218:e20210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bao W‐D, Pang P, Zhou X‐T, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ. 2021;28:1548‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gleason A, Bush AIJN. Iron and ferroptosis as therapeutic targets in Alzheimer's disease. Neurotherapeutics. 2021;18:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Magnusson CD, Gudmundsdottir AV, Haraldsson GG. Chemoenzymatic synthesis of a focused library of enantiopure structured 1‐O‐alkyl‐2,3‐diacyl‐sn‐glycerol type ether lipids. Tetrahedron. 2011;67:1821‐1836. [Google Scholar]
- 51. Deniau AL, Mosset P, Pedrono F, Mitre R, Le Bot D, Legrand AB. Multiple beneficial health effects of natural alkylglycerols from shark liver oil. Mar Drugs. 2010;8:2175‐2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357‐367. [DOI] [PubMed] [Google Scholar]
- 53. James AL, Knuiman MW, Divitini ML, et al. Changes in the prevalence of asthma in adults since 1966: the Busselton Health study. Eur Respir J. 2010;35:273‐278. [DOI] [PubMed] [Google Scholar]
- 54. Beyene HB, Olshansky G, T Smith AA, et al. High‐coverage plasma lipidomics reveals novel sex‐specific lipidomic fingerprints of age and BMI: evidence from two large population cohort studies. PLoS Biol. 2020;18:e3000870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cadby G, Melton PE, McCarthy NS, et al. Heritability of 596 lipid species and genetic correlation with cardiovascular traits in the Busselton Family Heart Study. J Lipid Res. 2020;61:537‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr. 2009;21:672‐687. [DOI] [PubMed] [Google Scholar]
- 57. Huynh K, Barlow CK, Jayawardana KS, et al. High‐throughput plasma lipidomics: detailed mapping of the associations with cardiometabolic risk factors. Cell Chem Biol. 2018;26(1):71‐84. [DOI] [PubMed] [Google Scholar]
- 58. Weiner MW, Veitch DP, Aisen PS, et al. Impact of the Alzheimer's disease neuroimaging initiative, 2004 to 2014. Alzheimers Dement. 2015;11:865‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289‐300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information