

Abstract
Scope
Gut microbiota alterations are associated with obesity and type 2 diabetes. Yeast β‐glucans are potential modulators of the innate immune‐metabolic response, by impacting glucose, lipid, and cholesterol homeostasis. The study examines whether yeast β‐glucan interacts differentially with either an obese healthy or obese diabetic gut microbiome, to impact metabolic health through hepatic effects under high‐fat dietary challenge.
Methods and results
Male C57BL/6J mice are pre‐inoculated with gut microbiota from obese healthy (OBH) or obese type 2 diabetic (OBD) subjects, in conjunction with a high‐fat diet (HFD) with/without yeast β‐glucan. OBD microbiome colonization adversely impacts metabolic health compared to OBH microbiome engraftment. OBD mice are more insulin resistant and display hepatic lipotoxicity compared to weight matched OBH mice. Yeast β‐glucan supplementation resolves this adverse metabolic phenotype, coincident with increasing the abundance of health‐related bacterial taxa. Hepatic proteomics demonstrates that OBD microbiome transplantation increases HFD‐induced hepatic mitochondrial dysfunction, disrupts oxidative phosphorylation, and reduces protein synthesis, which are partly reverted by yeast β‐glucan supplementation.
Conclusions
Hepatic metabolism is adversely affected by OBD microbiome colonization with high‐fat feeding, but partially resolved by yeast β‐glucan. More targeted dietary interventions that encompass the interactions between diet, gut microbiota, and host metabolism may have greater treatment efficacy.
Keywords: gut microbiota, hepatic triacylglycerol (TAG), high‐fat diet, type 2 diabetes, yeast β‐glucan
Gut microbiota dysbiosis is associated with obesity and type 2 diabetes. Human obese diabetic microbiome transplantation specifically augments high‐fat diet induced hepatic steatosis and insulin resistance beyond obesity alone, which was resolved by yeast β‐glucan supplementation. Reverting adverse metabolic phenotypes requires precision nutrition ‐ understanding the interactions between diet, gut microbiota, and host metabolism to improve dietary management efficacy.
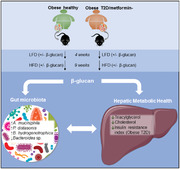
1. Introduction
The prevalence of diabetes continues to rise worldwide.[ 1 ] Type 2 diabetes accounts for 90–95% of diagnosed diabetes cases which affected more than 500 million people worldwide in 2018, which is an underestimation since many cases are undiagnosed.[ 2 , 3 ] Metformin is an effective therapeutic; however, there are opportunities for alternative diet‐mediated therapeutic regimes which may interact with the gut microbiome. Effective dietary intervention is an increasingly important alternative to drug therapy and there is growing interest in how this may interact with the gut microbiome to improve metabolic health.[ 4 ] Modification of dietary fatty acid composition improves several components of metabolic‐inflammation that typify insulin resistance and type 2 diabetes. Saturated fatty acids (SFA) drive insulin resistance, metabolic dysfunction, and inflammation.[ 5 , 6 ] Reducing dietary SFA attenuates insulin resistance in humans, improving both hepatic steatosis and adipose inflammation in animal models.[ 7 , 8 , 9 , 10 ] Dietary fiber also reduces insulin resistance and type 2 diabetes incidence, depending on fiber type.[ 11 ] The gut microbiome is a potential modifier of diabetes risk,[ 12 ] which in turn is modified by dietary composition.[ 13 ] Numerous studies suggest that metformin alleviates type 2 diabetes partly by reconfiguring gut microbiota composition.[ 14 ] It is reported that the human gut microbiota interacts with host metabolism to augment obesity, insulin resistance, and type 2 diabetes through several effectors, including lipopolysaccharide, short‐chain fatty acids, bile acids, and branched chain amino acids.[ 12 , 14 , 15 ]
β‐glucans, particularly β‐(1→3)‐glucan, are naturally occurring fibers with diverse functionality, dependent upon their source and structural features,[ 16 ] wherein some may improve insulin, glucose, lipid, and cholesterol metabolism.[ 17 , 18 , 19 ] Additionally, yeast β‐glucan has quite unique purported effects with respect to innate immune training. A supra‐physiological intervention demonstrated that such β‐glucan derived from Akkermansia spp. improved glycemic control, hepatic steatosis, and adipose inflammation in high‐fat diet (HFD) induced obese/type 2 diabetes mouse model.[ 20 ] However, that dose also attenuated weight gain, which may have influenced metabolic changes. Feeding an alternative barley β‐glucan lowered plasma cholesterol and elevated abundance of Bifidobacterium spp. and Akkermansia municiphila in patients with the metabolic syndrome, suggesting that the gut microbiota may modulate the diet‐induced metabolic response.[ 21 ] However, the potential interactions between yeast β‐glucan supplementation and the obese and/or diabetic subject's gut microbiome, in a weight‐neutral context, are not fully understood.
To address this question, we colonized antibiotic‐treated C57BL/6J mice with human microbiota from either obese healthy (OBH) or obese type 2 diabetic (OBD) subjects not receiving metformin. The murine recipients of the OBD human microbiome displayed insulin resistance, hepatic triacylglycerol (TAG), and cholesterol accumulation, with significant re‐configuration of the hepatic proteome, particularly with respect to protein synthesis and fatty acid metabolism pathway signatures. Gut microbiota composition and diversity responded positively to the consumption of β‐glucans accompanied by partial recovery of the OBH microbiome, amelioration of HFD‐induced insulin resistance, and normalization of hepatic TAG and cholesterol levels in this humanized murine model.
2. Experimental Section
2.1. Participant Recruitment and Fecal Sample Collection
Obese (body mass index (BMI) > 30 kg m–2) subjects were recruited from the Croí Heart and Stroke Centre, Galway, Ireland and written informed consent was obtained. Ethical approval was granted from the Clinical Research Ethics Committee at Galway University Hospitals (C.A. 1782) and adhered to the Declaration of Helsinki and Research Ethics Committee, Ireland guidelines. Fasting blood samples were obtained for metabolic biomarkers. Two groups, obese healthy and obese diabetic, were recruited based on their glucose tolerance. Glucose tolerance status was determined using HbA1c and fasting glucose; with OBH indicated by fasting glucose levels of <6 mmol L−1 and HbA1c of <42 mmol mol−1 and OBDdefined by a fasting glucose >7 mmol L−1 and HbA1c of ≥48 mmol mol−1. A freshly voided (same day) fecal sample was collected and maintained in an anaerobic environment from time of collection until same‐day processing and preparation for mice humanization.
2.2. Fecal Microbiota Transplantation
Fecal microbiota transplantation from humans to mice was performed as described previously by the lab.[ 22 ] In brief, fecal samples from the selected donors were collected immediately upon voiding and transferred to a plastic container from which oxygen was expunged by commercial anaerobic culture envelopes. The samples were moved immediately to the lab, opened in an anaerobic hood, diluted 1:10 mass/volume in pre‐reduced PBS containing 20% glycerol, and mixed gently till an even suspension was formed. Aliquots of 1 mL were flash‐frozen on dry ice, then stored at −80 °C till required.
2.3. Mice and Study Design
Male C57BL/6J mice were obtained from Envigo, United Kingdom. A schematic overview of the experimental study design is presented in Figure 1A. At 5 weeks of age, mice received an antibiotic cocktail of ampicillin (1 g L−1), metronidazole (1 g L−1), vancomycin (500 mg L−1), imipenem (250 mg L−1), and ciprofloxacin HCl (200 mg L−1) for 6 weeks in their drinking water to deplete the murine gut microbiota.[ 22 , 23 ] An anti‐fungal (Amphotericin B, 0.1 mg mL−1) was given by oral gavage (1 daily on the first and last 3 days of antibiotic treatment). Following antibiotic treatment, after 1 day of wash out, mice were inoculated with human microbiota derived from either an OBH or an OBD subject. Fecal material (300 µL of a prepared slurry) was orally administered in two 50 µL doses per day for 3 consecutive days. From 9 weeks old, mice were fed a low‐fat diet (LFD; 10% kcal from fat). After 2 weeks, half of the cohort was changed to a LFD with β‐glucan for 4 weeks, before being fed the high‐fat diet (HFD; 45% kcal from fat) with and without β‐glucan for 9 weeks. The primary objective was to determine the interaction between different microbiota and yeast β‐glucan supplementation on metabolic health. To this end, fecal microbiome transfer (FMT) was completed and initially mice were fed a LFD with/without yeast β‐glucan to acclimatize the animals. However, within any human OBH or OBD phenotype, it was most probable that a habitual HFD was consumed, therefore all animals were challenged with a HFD with/without β‐glucan. The β‐glucan dose was equivalent to 50 mg kg−1 day−1. While the recommended daily dose of Wellmune in human was 2.5 mg kg−1, the study fed a higher dose (50 mg kg−1) to investigate the microbiome interaction concept, over a short‐time frame as compared to long‐term human habitual consumption. Additionally, this dose was in line with previous animal studies yeast β‐glucan ingestion on microbiome and hepatic health.[ 20 ] All diets were purchased from Research Diets (New Brunswick, NJ, USA) with β‐glucan (Wellmune) supplied by Kerry. Mice were housed in metabolic cages in a 12:12‐h light‐dark cycle and fed ad libitum. Body weight and food intake were measured weekly. Fecal samples were collected at several time points. Mice were anesthetised (5% isoflurane gas inhalation), euthanized by cervical dislocation and tissues were then isolated for analysis. All experiments were approved by the Animal Ethics Experimentation Committee of University College Cork (AE19130/P072).
Figure 1.
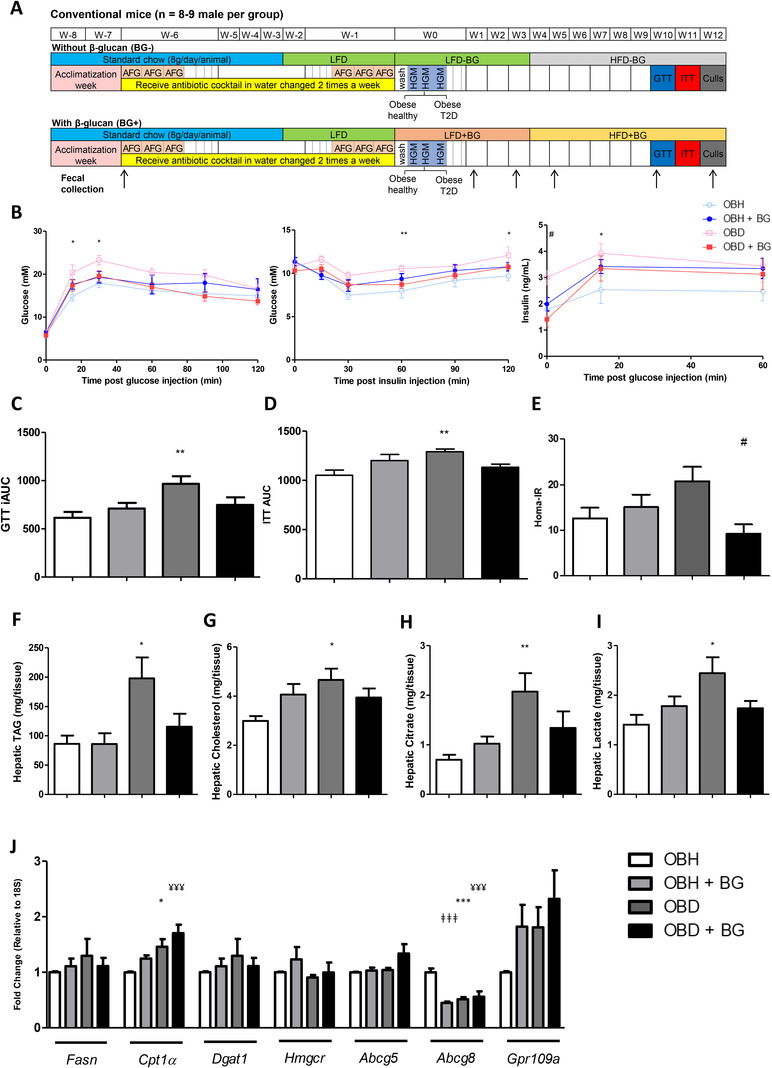
Mice that received obese diabetic microbiome displayed altered metabolic phenotype and increased hepatic lipid accumulation. A) Experimental design for murine intervention study. Conventional mice (N = 8–9) were inoculated with human gut microbiota (HGM) from either an obese healthy (OBH) or an obese type 2 diabetic (OBD) subject at week 0 for 3 days. Arrows indicate the week on which the fecal samples were collected. AFG, antifungal gavage; GTT, glucose tolerance test; HFD, high‐fat diet; ITT, insulin tolerance test; LFD, low‐fat diet. B) Response to glucose, insulin, and glucose stimulated insulin secretion, C) GTT incremental area under the curve (iAUC), D) ITT area under the curve (AUC), and E) HOMA‐IR following HFD. F) Hepatic TAG and G) cholesterol were measured. Citrate and lactate levels within the liver are shown in H) and I) respectively. J) Markers of hepatic lipid metabolism were assessed by RT‐PCR with 18S as the appropriate housekeeping gene. Data are represented as ±SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 with respect to (WRT) OBD versus OBH, #p ≤ 0.05 WRT OBD versus OBD+BG, ǂǂǂ p ≤ 0.001 WRT OBH versus OBH+BG, ¥¥¥ p ≤ 0.001 WRT OBH versus OBD+BG.
2.4. Microbiota Profiling and Metagenomics
Total genomic DNA extracted from human fecal and murine fecal samples was used as a template in library preparation for 16S rRNA gene amplicon sequencing of the V3–V4 variable region.[ 24 ] Purified amplicons were pooled in equal volumes. Sequencing (2 × 250 bp) of the pooled library was performed using Illumina MiSeq (Illumina, Inc., San Diego, CA, USA) in the Eurofins GATC Biotech GmbH (Constance, Germany). The study applied a similar bioinformatic analysis of 16S amplicon sequencing data as previously published.[ 22 ] Details of methods were presented in Supplementary Text.
Metagenomic libraries of human fecal samples were prepared using the Nextera XT DNA Library Prep Kit (Illumina) and sequenced using Illumina NextSeq 500/550 High Output v2 (300 cycles) at the National Irish Sequencing Centre (Teagasc Food Research Centre, Ireland) to generate 150 bp paired‐end read libraries.
2.5. Glucose and Insulin Tolerance Tests
Mice were fasted overnight for 12 h then injected intraperitoneally with 25% (wt/vol) glucose (1.5 g kg−1; B. Braun Medical, Dublin, Ireland) for glucose tolerance test (GTT). Mice were fasted for 6 h then injected intraperitoneally with insulin (0.5 U kg−1: Actrapid, Novo Nordisk, Denmark) for insulin tolerance test (ITT). Glucose levels were measured at baseline, 15, 60, 90, and 120 min post glucose/insulin challenge by an accu‐check glucometer (Roche, Dublin, Ireland). Tail‐vein bleeds were sampled at baseline as well as 15 and 60 min post glucose challenge to measure the insulin secretory response. ELISA (Crystal Chem, Inc., IL, USA) was used to quantify insulin levels post glucose challenge.
2.6. Hepatic TAG, Cholesterol, Citrate, and Lactate Quantification
Flash frozen liver tissue (50 mg) was homogenized in a TissueLyser II (Qiagen Cat. No./ID: 85300). For TAG and cholesterol assays, liver homogenate was prepared as previously described.[ 25 ] The dried sample was resuspended in water before TAG and cholesterol analysis. Hepatic TAG (Wako LabAssay Triglyceride kit, Fuggerstraße, Neuss, Germany) and hepatic cholesterol (Wako LabAssay Cholesterol kit) was measured as per manufacturer's instructions. Hepatic citrate (Sigma‐Aldrich Citrate Assay Kit) and lactate (Sigma‐Aldrich Lactate Assay Kit) were measured as per manufacturer's instructions.
2.7. Gene Expression Analyses
RNA was extracted from 50 mg flash frozen hepatic tissue with Trizol. Chloroform was added, mixed by inversion and the colorless aqueous phase was transferred to a fresh tube. Isopropanol was added to precipitate the RNA overnight. RNA was quantified using a NanoDrop 8000 UV–vis spectrophotometer. Equal amounts of cDNA were synthesized using the Applied Biosystems High Capacity cDNA kit (Applied Biosystems by Thermo Fisher Scientific). Fasn, Cpt1α, Dgat1, Hmgcr, Abcg5, Abcg8, Gpr109a primers, and TaqMan Universal Mastermix were obtained from Applied Biosystems by Thermofisher Scientific. mRNA expression was measured by real‐time PCR on Applied Biosystems QuantStudio 7 Flex Real‐Time PCR System. Housekeeping gene 18S was used for hepatic gene expression. Comparison of 2‐(ΔΔCt) determined fold change as previously described.[ 26 ]
2.8. Fatty Acid Analysis
Fatty acid methyl esters (FAMEs) were prepared using the MARS 6 express 40 position microwave reaction system and quantified by gas chromatography (GC) as described previously.[ 27 ] Briefly, fatty acids were extracted from 50 mg flash frozen hepatic tissue using a microwave‐assisted preparation of FAMEs for GC analysis. Potassium hydroxide (10 mL, 2.5% w/v) in methanol and internal standard (ISTD) (tricasonaoic acid, 100 µL, 10 mg mL−1 in chloroform) were added for saponification, microwaved and heated to 130 °C, and held for 4 min. Methanolic acetyl chloride (15 mL, 5% v/v) was added for esterification, microwaved, heated to 120 °C in 4 min and held for 2 min. Pentane (10 mL) was added for fatty acid extraction and saturated sodium chloride (20 mL) was added to induce phase separation. FAMEs were measured using an Agilent 7890B gas chromatograph fitted with a flame ionization detector (FID). Analytes were separated using a CP‐Sil 88 capillary with a 100 m × 0.25 mm internal diameter × 0.2 µm film thickness column. Compounds were identified by comparing their retention times with FAME Supelco standards. Peak area analysis was conducted using Agilent OpenLAB CDS 2.1 Workstation. The content of each fatty acid (mg g−1 of tissue) was calculated according to the following equation:
| (1) |
2.9. Hepatic Proteomic Analysis
Hepatic proteomic samples were prepared as described in Supplementary Text. Briefly, liver protein was isolated from 50 mg tissue with trichloroacetic acid (20%) and protein pellets were twice washed in ice‐cold acetone before resuspension in 8 M urea in triethylammonium bicarbonate. Protein samples were in‐solution digested in trypsin overnight at 37 °C. After drying in vacuum centrifuge, peptides were acidified by acetic acid (AA), desalted with c18 STAGE tips,[ 28 ] and resuspended in 2.5% acetonitrile (ACN), 0.5% AA. Peptide fractions were analyzed on a quadrupole Orbitrap (Q‐Exactive, Thermo Scientific) mass spectrometer equipped with a reversed‐phase NanoLC UltiMate 3000 HPLC system (Thermo Scientific). Raw data were processed using MaxQuant version 1.6.3.4,[ 29 ] incorporating the Andromeda search engine.[ 30 ] To identify peptides and proteins, MS/MS spectra were matched to the Uniprot mouse database. For the generation of label free quantitative (LFQ) ion intensities for protein profiles, signals of corresponding peptides in different nano‐HPLC MS/MS runs were matched by MaxQuant in a maximum time window of 1 min.[ 31 ] The Perseus computational platform (version 1.6.2.3) was used to process MaxQuant results.[ 32 ] Data were log transformed. T‐test comparisons were carried out between liver proteomes. For visualization of data using heat maps, missing values were imputed with values from a normal distribution and the dataset was normalized by z‐score.
2.10. Bioinformatic Pathway Analysis
Bioinformatic analysis was performed to analyze differentiable expressed hepatic proteins. Briefly, t‐test differences from proteomic analysis were uploaded into Qiagen's Ingenuity Pathway Analysis (IPA) system for core analysis and overlaid with the Ingenuity pathway knowledge base. IPA was performed to identify canonical pathways, and putative upstream regulators that were the most significant in global molecular networks.[ 33 ] These results were ranked based on their p value (p ≤ 0.05) or activation score (z‐score) of pathway activation/inhibition.
2.11. Statistical Analysis
Statistical analysis was carried out using R v.3.5.5 software packages.[ 34 ] Significant differences in microbiome β‐diversity were detected using permutational multivariate analysis of variance (PERMANOVA) (vegan R package[ 35 ]). Differences of α‐diversity and different taxonomic levels were identified by Mann–Whitney U test for the comparison of two diet groups stratified by β‐glucan consumption (unpaired data) and Wilcoxon signed rank test for the comparison between time points (paired data). P‐values were adjusted for multiple comparisons using Benjamini–Hochberg correction.
Metabolic phenotype data were expressed as mean ± standard error of the mean (S.E.M.). For GTT, ITT, and insulin secretion a two‐way repeated measures Analysis of Variance (ANOVA) was completed followed by a post hoc Bonferroni test (if the ANOVA reached statistical significance). For between‐group comparisons, a one‐way ANOVA followed by post‐hoc Bonferroni test was performed. Analyses were completed using GraphPad Prism 5.0 software (GraphPad Prism Software, Inc, La Jolla, CA, USA).
3. Results
3.1. Microbiome Differences in Human Subjects with Obesity, with or without Type 2 Diabetes
The clinical characteristics and metformin treatment status of the OBD versus OBH subjects demonstrated no significant difference with respect to age, BMI, or sex, but displayed elevated fasting glucose and HbA1c, as expected (Tables S1, S2, Supporting Information). There was no significant separation by β‐diversity analysis of overall gut microbiota composition between OBH individuals and OBD subjects, with or without metformin treatment (unweighted and weighted UniFrac measures: p > 0.05; Figure S1A, Supporting Information). Although 16S rRNA amplicon sequencing was apparently biased towards higher proportions of Lachnospiraceae, compared with shotgun metagenomic sequencing, the proportional taxonomic composition between subjects was largely consistent by both amplicon sequencing and metagenomics (Figure S1B, Supporting Information). OBD subjects treated with metformin displayed significant loss of the Clostridiales order (unclassified Lachnospiraceae genus and Eubacterium eligens) and higher abundance of Streptococcus spp. and Lactobacillus spp. compared to OBH subjects and altered microbial metabolic pathway abundances (Figure S2A, Supporting Information). The increase in the relative abundance of Lactobacillus with metformin is established,[ 36 ] as well as other re‐configurations of the human gut microbiota. Therefore, we selected a non‐metformin treated OBD subject and a representative OBH subject as donors for the human‐to‐mouse microbiome transplantation. The fecal microbial community from the OBH donor was close to other OBH subjects but was distant from the OBD donor (Figure S1A, Supporting Information). Ruminococcaceae was present in higher abundance in OBH donor compared to OBD donor. The two donors exhibited differences in relative abundance at bacterial species level (Figure S2B, Supporting Information).
3.2. OBD Microbiome Transplantation Induced Insulin Resistance and Disrupted Hepatic TAG and Cholesterol Metabolism, which Was Resolved by β‐Glucan Supplementation
We humanized mice with the gut microbiome from the selected OBH or OBD human subjects (Figure 1A). OBD microbiome transplanted mice were more insulin resistant, glucose intolerant and hyperinsulinemic following HFD, compared to OBH inoculated mice. Interestingly, yeast β‐glucan supplementation partly corrected the adverse OBD/HFD‐induced metabolic phenotype, wherein fasting insulin concentrations and insulin resistance index (HOMA‐IR), were significantly lower in the OBD+β‐glucan group, compared to the OBD/HFD microbiome‐induced group (Figure 1B–E). Total weight gain, adipose depot weights, and energy intake were not different between groups (data not shown).
Hepatic TAG and cholesterol levels were significantly higher in OBD mice compared to OBH mice (Figure 1F,G). Metabolite measurement revealed that hepatic citrate (Figure 1H) and lactate (Figure 1I) levels increased in OBD mice, but not when supplemented with β‐glucan. Markers of lipid and cholesterol homeostasis were determined. Hepatic fatty acid synthesis (Fasn) was not significantly different. Hepatic Cpt1α mRNA, an indicator of mitochondrial fatty acid oxidation, was increased in OBD mice which was further enhanced by β‐glucan supplementation, compared to OBH. In terms of cholesterol regulation, expression of the cholesterol transporter Abcg8 was lower in OBD mice (Figure 1J). Hepatic fatty acid composition was not significantly affected by microbiome source (Table S3, Supporting Information).
3.3. The OBD Microbiota Composition Moves towards a Health‐Associated Composition upon Yeast β‐glucan Supplementation
Mouse fecal microbiome profiling by 16S rRNA gene amplicon sequencing was completed at key timepoints post‐transplantation (Figure 1A). Sequences assigned to 10 bacterial genera that were detected in the microbiome of at least 50% of the mice in the antibiotic pretreatment group were not found in the humanized mice, indicating their successful eradication. Meanwhile, of 17 genera of the human microbiome present in less than 50% in the pretreatment mouse group, only four genera including Faecalibacterium, Bilophila, Dialister, and Megamonas were not engrafted in the antibiotic‐treated mice. Twenty‐eight genera were detected in both the human donor and pretreatment mouse stools, only five of which (Desulfovibrio, Clostridium XlVb, Parasutterella, Bifidobacterium, and Gemmiger) were absent in the humanized mice (Figure S3, Supporting Information). Overall, the engraftment of human microbiota into antibiotic‐treated mice appeared successful with a significant change in the murine gut microbiota before and after antibiotic treatment and transfer.
The α‐diversity (diversity within samples) measured by Phylogenetic Diversity and Observed Species was consistently lower at week 1, and remained stable or increased slightly from week 3 to week 12 in both OBH and OBD (Figure S4A, Supporting Information). The net increase in diversity during subsequent weeks was expected because the murine gut microbiota diversity had been lowered sharply by the administration of antibiotics and the engraftment of the human gut microbiota into antibiotic‐treated mice allowed some sub‐dominant taxa increase to more measurable proportions. In line with differences in the donor's gut microbiota, the α‐diversity in the recipient mice in the OBD group was lower than that of OBH, but both exhibited lower microbial diversity than their respective donors (Figure S4B, Supporting Information). Yeast β‐glucan supplementation increased α‐diversity of the gut microbiota in OBD mice at week 3 (phylogenetic diversity: p = 0.006, observed species: p = 0.01), but no statistically significant differences in diversity were associated with β‐glucan consumption from week 5 to week 12, when mice were receiving the HFD. However, the OBH+β‐glucan group showed lower phylogenetic diversity, compared to those without β‐glucan supplementation at week 3 (p = 0.001, Figure 2A). This result indicates that β‐glucan supplementation while on a LFD, prior to the HFD, temporarily lowered microbiota diversity in a normal microbiota, but increased bacterial diversity in low‐diversity communities.
Figure 2.
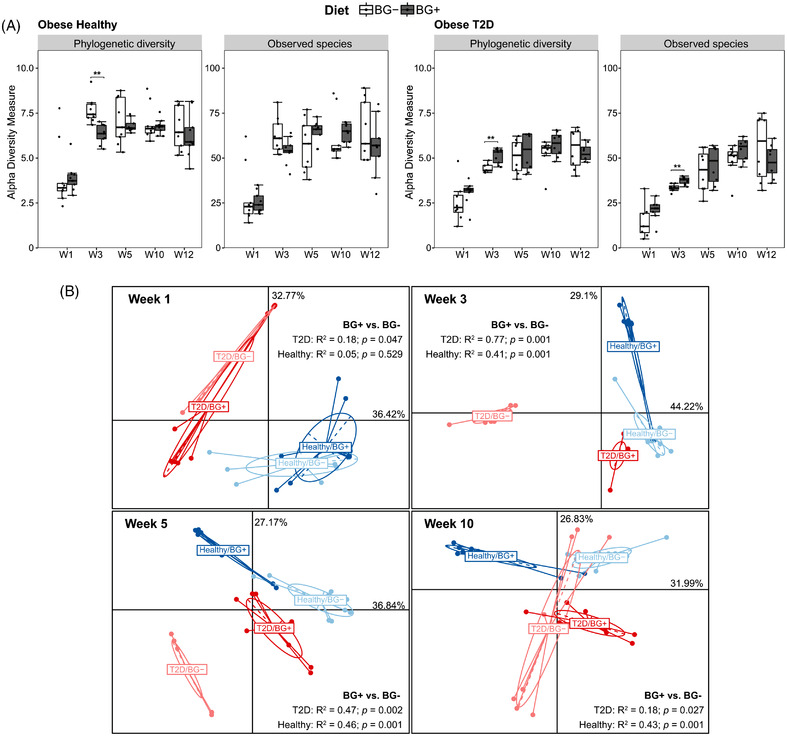
Dietary supplementation with β‐glucan drives gut microbiota diversity changes in obese healthy and obese type 2 diabetic inoculated mice. A) Difference in α‐diversity between BG− and BG+ were determined by Mann–Whitney U test adjusted using Benjamini–Hochberg correction, ** adjusted p ≤ 0.01. B) Principle coordinates analysis plots based on Bray–Curtis dissimilarity. The significant differences between groups were calculated by permutational multivariate analysis of variance (PERMANOVA) tests.
We next investigated the relatedness of the microbiota community structures (β‐diversity) between OBH and OBD, with or without β‐glucan supplementation (Figure 2B). Principal coordinates analysis (PCoA) based on a Bray–Curtis dissimilarity (which measures microbiota dissimilarity) at week 1 showed the main separation was between the human donor metabolic phenotype (PERMANOVA: R 2 = 0.31, p = 0.001). Supplementation with β‐glucan at this early time‐point had a marginal effect in OBD (PERMANOVA: R 2 = 0.18, p = 0.047), but no impact on OBH (PERMANOVA: R 2 = 0.05, p = 0.529). By week 3, the β‐glucan effect on the microbiota was significant for both donor types, but more strikingly, β‐glucan supplementation moved the OBD microbiota closer to that of the OBH group (Figure 2B). This effect was retained at week 5, but the β‐glucan supplementation effect on the microbiota was weaker within metabolic phenotypes (OBH vs OBD). By week 10 when mice had received a longer period of HFD, there was general convergence, although β‐glucan supplementation still had a significant effect within the OBD microbiota (PERMANOVA: R 2 = 0.18, p = 0.027). Yeast β‐glucan supplementation had a greater effect on OBD microbiota during the LFD phase than HFD phase. When all time points were mapped, the murine microbiota datasets were more similar to the respective human donor microbiota than the pretreatment mouse microbiota at week 6 (Figure S5A, Supporting Information). β‐glucan supplementation significantly separated mice in the PCoA, but distances were reduced upon HFD over time especially in the OBD group. Furthermore, changes in gut microbiota (measured with Bray–Curtis dissimilarity) throughout HFD versus LFD periods accompanied by β‐glucan consumption particularly in week 10 were significantly lower than in mice not supplemented with β‐glucan. This suggests that β‐glucan might reduce the effect of HFD on gut microbiota in both OBD and OBH (Figure S5B, Supporting Information).
3.4. β‐Glucan Responsive Taxa Differ Between OBD and OBH Inoculated Mice
Analysis of differentially abundant taxa showed increased relative abundance of Verrucomicrobiaceae, Ruminococcaceae, Desulfovibrionaceae, Porphyromonadaceae and a decrease in Streptococcaceae, Bacteroidaceae, Rhodospirillaceae, Enterobacteriaceae with β‐glucan supplementation. Flavonifractor, Akkermansia, Roseburia, Parabacteroides in the OBH group and Clostridium XlVa, Odoribacter, Anaerostipes in the OBD group were significantly higher after β‐glucan supplementation. Escherichia/Shigella abundance was consistently lower at all time points in the OBH+β‐glucan group (Supplementary Text, Figure S6, Supporting Information).
At the species level, 69 known species accounted for 73.2 ± 19% (mean ± SD) of the microbial composition abundance across all samples. Most samples were dominated by Akkermansia muciniphila, and Bacteroides vulgatus, although the OBH group was also dominated by Parabacteroides distasonis and Bacteroides xylanisolvens. Bacteroides uniformis was dominant in the OBD group, particularly when not supplemented with β‐glucan (Figure 3A). A. muciniphila was initially more abundant with β‐glucan supplementation, but this was not maintained in OBD mice on HFD. B. vulgatus, Alistipes massiliensis, Blautia hydrogenotrophica, Odoribacter splanchnicus were significantly more abundant in at least two consecutive time points in OBD+β‐glucan mice. The opposite applied for Clostridium paraputrificum, Clostridium ramosum, B. uniformis. In contrast, higher abundance of P. distasonis, A. muciniphila, and lower abundance of B. vulgatus, Bacteroides thetaiotaomicron, Bacteroides acidifaciens were associated with β‐glucan supplementation in the OBH group prior to and throughout the HFD (Figure 3B,C). This suggests the effect of β‐glucan on certain specific taxa was more stable in OBH than in OBD, even after HFD.
Figure 3.
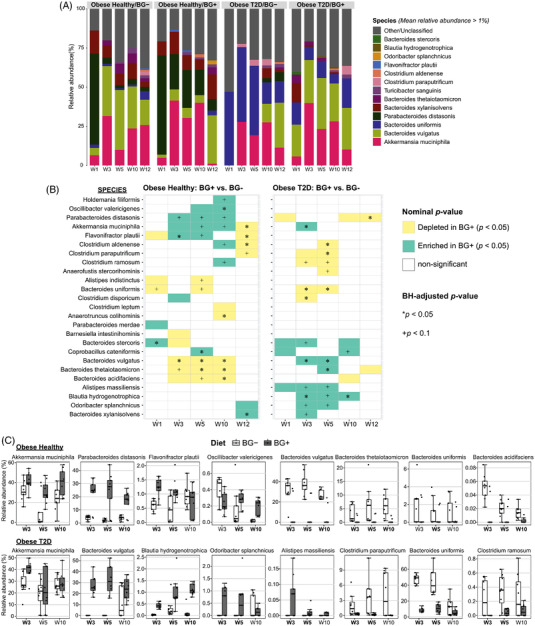
The β‐glucan responsive taxa at species level differed between obese type 2 diabetic inoculated mice and obese healthy inoculated mice. A) Mean relative abundance of the represented microbial taxa at species level. All known bacterial taxa with mean relative abundance less than 1% and unclassified taxa were merged to form the Other/Unclassified group. B) Significantly differentially abundant taxa between two diet groups were determined by Mann–Whitney U test adjusted using Benjamini–Hochberg correction. C) Box plot of the relative abundance distribution of selected species associated with β‐glucan consumption from week 3 to week 5. Time points in bold indicates nominal p‐value ≤ 0.05.
We found moderate correlations between the abundance of several gut microbial OTUs and the host expression of Cpt1α, Abcg8 genes, and HOMA‐IR (Spearman's correlation analysis: |rho| < 0.62, nominal p‐values < 0.05) but these associations lost significance after adjustment for multiple testing except the association of OTU_1183 (unclassified Clostridium XlVa) and Cpt1α (Figure S7, Supporting Information). In particular, OTU_1290 (A. massiliensis) and OTU_501 (B. vulgatus) belonging to the species that differed significantly in abundance upon yeast β‐glucan supplementation were negatively correlated with HOMA‐IR. Similarly, negative correlations were observed between Abcg8 expression gene and the abundances of OTU_456 (Flavonifractor plautii), OTU_75 (Oscillibacter valericigenes), OTU_612 (P. distasonis). The hepatic Cpt1α mRNA level was positively correlated with OTU_265 (B. uniformis) but inversely with OTU_239 (B. thetaiotaomicron). Collectively, these findings suggest the impact of gut microbiota upon hepatic gene expression as well as the insulin resistance score HOMA‐IR, by currently unknown mechanisms.
3.5. Hepatic Proteomic Expression Is Altered by both Microbiome Type and β‐Glucan Supplementation
The liver is a central organ that integrates signals from the gut, pivotal for regulating metabolism. To identify mechanisms underpinning diet/microbiome induced changes in metabolism, hepatic mass spectrometry (MS)‐based proteomics was performed, comparing the OBD mice, the most adverse metabolic phenotype, with OBH as the control or relative comparator. A total of 102 proteins were differentially expressed between OBD and OBH livers, 25 increased in abundance and 77 proteins were decreased (Figure 4A). In conjunction with the improved phenotype, 66 differentially expressed proteins (20 increased and 46 reduced) involved in fatty acid metabolism, mitochondrial dysfunction, and inflammation between OBD and OBD+β‐glucan livers (Figure 4B). Hepatic proteomic comparison between OBH+β‐glucan versus OBH displayed 132 increased and 161 decreased proteins totaling 293 differentially expressed proteins (data not shown). However, the hepatic metabolic phenotype was not significantly different in OBH mice fed β‐glucan compared to those who were not.
Figure 4.
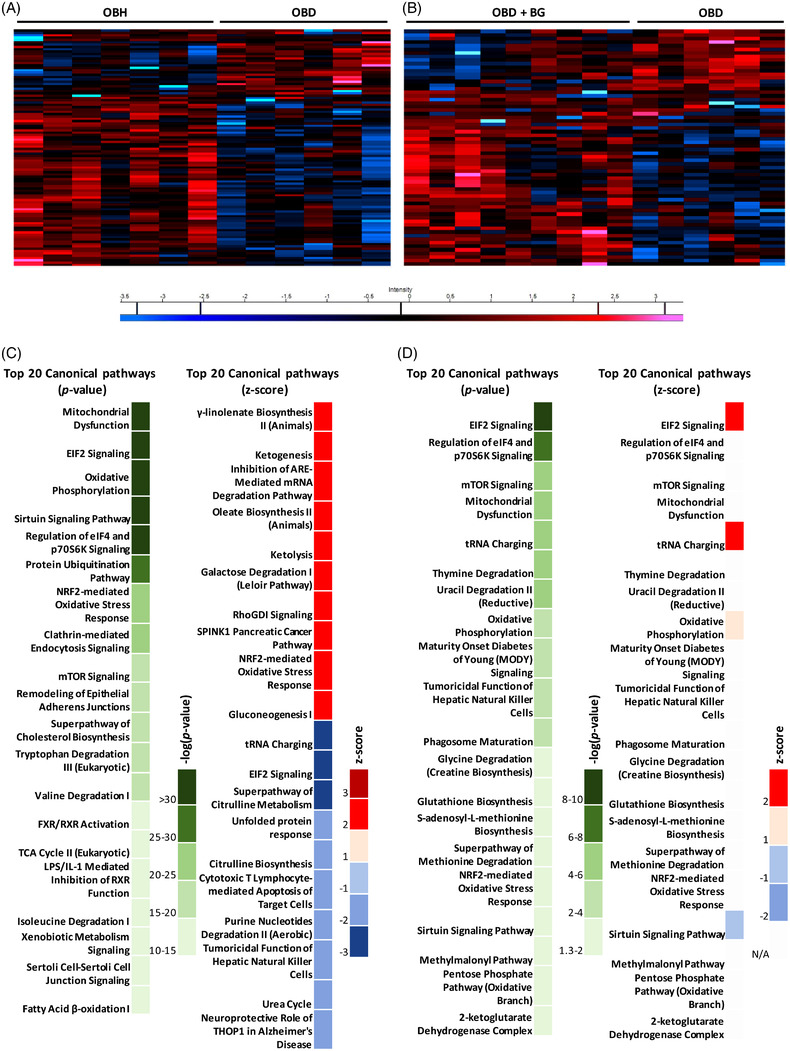
Microbiome and β‐glucan alter hepatic protein signatures. A) OBH and OBD hepatic proteomic signature heat map. B) OBD and OBD+β‐glucan (BG) hepatic proteomic signature heat map. Red and blue bars indicate proteins significantly up or down regulated respectively (p ≤ 0.05). Canonical pathway analysis of differentially expressed hepatic protein using IPA. C) Top 20 canonical pathways as per −log(p‐value) and as per z‐score in OBD with respect to OBH. D) Top 20 canonical pathways as per −log(p‐value) and as per z‐score in OBD+β‐glucan with respect to OBD.
IPA identified key pathways that were differentially expressed between OBD and OBH (Figure 4C), and between OBD mice fed β‐glucan and OBD hepatic proteomes (Figure 4D). Interestingly, the pathways most modulated by OBD versus OBH microbiome colonization differed by magnitude of alteration, compared to those associated with β‐glucan supplementation in OBD mice, wherein hepatic steatosis and insulin resistance was partly resolved. Specifically, mitochondrial dysfunction, EIF2 signaling, oxidative phosphorylation, the sirtuin signaling pathway, and regulation of EIF4 and p7056K signaling were significantly affected in the OBD group (Table S4, Supporting Information). The most significant pathways in OBD+β‐glucan included EIF2 signaling, regulation of EIF4 and p7056K signaling, mTOR signaling, mitochondrial dysfunction, and tRNA charging (Table S6, Supporting Information).
The top canonical pathways that were differentially up‐ or down‐regulated in OBD versus OBH livers are listed in Table S4, Supporting Information. Key pathways inhibited in OBD livers include tRNA charging, EIF2 signaling, citrulline metabolism, citrulline biosynthesis, and unfolded protein response. In contrast, galactose degradation, RhoGDI signaling, SPINK1 Pancreatic Cancer Pathway, NRF2‐mediated oxidative stress response, and gluconeogenesis were up‐regulated in OBD livers (Table S4, Supporting Information). IPA also identified putative upstream regulators of changes in the proteomic dataset. MYCN, MYC, Glucagon, ACOX1, and CLPP were potential upstream regulators that were down‐regulated in OBD mice (Table S5, Supporting Information). Upstream regulators upregulated in OBD were PPARA PNPLA2, ACSS2, PCGEM1, FOXA2, and PPARG (Table S5, Supporting Information). Importantly, several proteins, predominantly transcription factors, CEBPA, CEBPB, SREBF1, FOX01, RXRA, and SIRT1, were shared between key pathways. This indicates important coincident dysregulated regulation of hepatic mitochondrial, fatty acid, insulin, and glucose metabolism following diabetic microbiome inoculation and high‐fat feeding in OBD liver.
Fewer canonical pathways were enriched or unenriched, when comparing OBD to OBD+β‐glucan (Table S6, Supporting Information). The pathways include EIF2 signaling, tRNA charging, and sirtuin signaling pathway, suggesting oxidative phosphorylation and fatty acid metabolism were altered by β‐glucan. Putative upstream regulators inhibited within these pathways were RICTOR, INS1, BDNF and activated were MYC, MYN, SYVN1, XBP1, and NFE2l2 (Table S7, Supporting Information). Of note, we found that the MYC family was inhibited in OBD livers but activated in OBD+β‐glucan. This could be a mechanism by which yeast β‐glucan increased insulin sensitivity and reduced hepatic steatosis.
4. Discussion
The adverse metabolic impact of high‐fat feeding was greatly accentuated after obese diabetic microbiome transplantation, compared to obese but metabolically healthy microbiome transplantation, in a humanized mouse model. Interestingly this adverse phenotype was partly rescued by yeast β‐glucan supplementation. The adverse OBD microbiome induced metabolic phenotype was characterized by hyperinsulinemia, glucose intolerance, and increased hepatic TAG and cholesterol levels, despite being weight matched. Interestingly, β‐glucan supplementation reverted the OBD‐induced hyperinsulinemia and HOMA‐IR, despite high‐fat feeding. Yeast derived β‐glucan is a relatively novel functional food which is purported to enhance metabolism and innate immune training, as previously reviewed.[ 16 ] Dietary β‐glucan functionality is highly dependent upon structure and source. The metabolic effects of a yeast β‐glucan are less well defined compared to the well characterized impact of oat derived β‐glucan which improves plasma lipid metabolism and circulating insulin levels.[ 37 , 38 ] In humans, daily consumption of yeast β‐(1,3/1,6)‐d‐glucan (15 g day−1 for 8 weeks) lowered total cholesterol concentrations.[ 39 ] Another group has demonstrated that feeding very high doses of yeast β‐glucan improved metabolic health, but that dose also resulted in increased fecal fat excretion that significantly impeded weight gain, which presumably confounded metabolic readouts.[ 40 ] Our findings are noteworthy because metabolic reconfigurations were in the obese state and dependent upon human microbiome source, without any weight change.
The liver is a first pass organ after the gut. Our data suggest synergy between the OBD microbiome and HFD, to induce greater hepatic TAG and cholesterol levels, despite equal weight gain as OBH microbiome recipients. Interestingly hepatic citrate and lactate levels increased in OBD–HFD mice. This suggests that the TCA cycle was disrupted, wherein citrate could be shuttled off into Acyl‐CoA promoting de novo lipogenesis (DNL), which in conjunction with hyperinsulinemia might explain elevated hepatic TAG levels. In addition, HFD increased hepatic cholesterol levels in OBD microbiome recipients. Interestingly, cholesterol transporter Abcg8 expression, which promotes hepatic cholesterol excretion, was reduced which may account for increased cholesterol levels.[ 41 ]
This study was conducted using a limited number of individuals as fecal donors, because of the ethical, logistical, and financial limits that necessarily pertain when transferring human microbiomes into animal models. Despite this restriction, our data confirm the evidence for a link between the gut microbiome and both type 2 diabetes and metformin treatment, suggesting that gut microbiota modulation could be a key target in type 2 diabetes management. FMT from humans to mice showed that several bacterial species were significantly differentially abundant between OBD or OBH microbiome inoculated mice. A. muciniphila a well characterized mucin‐degrading bacterium associated with metabolic health[ 42 ] rather than type 2 diabetes,[ 43 , 44 ] was significantly increased following β‐glucan supplementation.[ 20 , 21 ] In humans, direct A. muciniphila supplementation for 3 months improved insulin sensitivity and reduced total cholesterol levels in overweight/obese subjects.[ 45 ] OBH mice displayed a proportional reduction in total Bacteroides species. Specifically, B. vulgatus abundance has been associated with branched‐chain amino acids biosynthesis and insulin resistance.[ 12 ] Here we observed that OBD β‐glucan supplemented mice had increased abundance of B. vulgatus, A. massiliensis, B. hydrogenotrophica, O. splanchnicus and decreased abundance of C. paraputrificum, C. ramosum, B. uniformis. In terms of speculating whether these taxa explain the improved metabolic phenotype of OBD+β‐glucan mice, C. ramosum is associated with diet‐induced obesity and type 2 diabetes.[ 46 ] O. splanchnicus is a well‐characterized butyrate‐producing bacterium[ 47 ] and B. hydrogenotrophica, which can produce acetate,[ 48 ] may also contribute to modulating metabolism via short‐chain fatty acid metabolites. In terms of relating the microbiome to key phenotypes in our study, some gut microbial OTUs from Lachnospiraceae sp., A. massiliensis, B. vulgatus associated with yeast β‐glucan supplementation, were negatively correlated with HOMA‐IR, although these associations did not pass multiple testing correction. These bacteria can produce beneficial short‐chain fatty acids which may regulate hepatic gluconeogenesis via the TCA cycle, to subsequent lower levels of HOMA‐IR.[ 49 ] While the abundance of different bacterial OTUs were also correlated with hepatic Cpt1α, Abcg8 gene expression, a potential functional relationship is, as yet, unknown. Indeed, these hepatic genes are affected by hepatic lipid, cholesterol, and insulin concentrations,[ 50 , 51 ] therefore direct modulation via the microbiome is not necessarily the case. Future work is required to further validate if/how OBD microbiota and related metabolites, have functional effects in response to β‐glucan supplementation.
Hepatic proteomic signatures were determined to elucidate mechanisms underpinning the impact of different microbiota and β‐glucan supplementation on metabolic health. Several hepatic pathways were differentially regulated between OBD and OBH recipients in response to high‐fat feeding. Those inhibited include the tRNA charging pathway which is responsible for attaching amino acids to tRNA to during protein synthesis. The EIF2 signaling pathway regulates mRNA translation, both specifically and globally, within the cell. The unfolded protein response (UPR) pathway is activated in response to endoplasmic reticulum (ER) stress resulting from unfolded or misfolding proteins. This suggests protein synthesis was inhibited in OBD livers. While we did not investigate plasma amino acid profiles, it is well established that dysregulated amino acid metabolism plays a key role in type 2 diabetes.[ 52 , 53 ] The citrulline metabolism and biosynthesis pathways were also inhibited in OBD livers. Citrulline is downstream of Acyl‐CoA and glutamate in the urea cycle in hepatic mitochondria. This indicates inhibition of the urea cycle in OBD livers. Pathways which were activated in the OBD versus OBH groups include the galactose degradation pathway which is responsible for the degradation of d‐galactose to enter the glycolysis pathway. Gluconeogenesis was also activated, which generates glucose from non‐carbohydrate precursors. Signaling of RhoGDI, a chaperone which prevents Rho protein degradation and alters cellular growth and regeneration patterns was activated. NRF2‐mediates oxidative stress response which is a regulator of cellular resistance to oxidants within the liver. This indicates higher glucose formation and utilization in conjunction with altered cellular growth patterns and increased oxidative stress within the OBD liver.
Pathway analysis also indicated putative upstream regulators that changed in OBD recipients in comparison to OBH recipients. MYC downregulation indicates inhibition of protein synthesis. Additional downregulated targets suggest increased glycolysis (glucagon), fatty acid accumulation (ACOX1), and alterations in mitochondria protein biogenesis, trafficking, and degradation (CLPP). Activated upstream regulators in OBD hepatic tissue display altered gene transcriptions patterns specific to both fatty acid β‐oxidation (PPARA, PNPLA2) and fatty acid storage (ACSS2). Thus, fatty acid metabolism/storage was greatly altered in OBD livers, as evidenced by higher TAG and cholesterol levels.
Even though β‐glucan supplementation had important phenotypic affects with lower HOMA‐IR and hepatic TAG accumulation, relatively few canonical pathways were predicted to be enriched or depleted, compared to the OBD group, without β‐glucan supplementation. Of note is the predicted upstream regulation activation of MYC with β‐glucan supplementation which was inhibited in OBD without β‐glucan supplementation. The MYC proto‐oncogene induces genes involved in glycolysis and stimulates lipid synthesis genes.[ 54 ] Overexpression of MYC can lower lipid accumulation in murine livers.[ 55 ] Additionally, MYC inhibition is associated with increased lipid accumulation due to mitochondrial dysfunction.[ 56 ] This could be a mechanism through which β‐glucan may increase insulin sensitivity and decrease hepatic steatosis.
The differential effect of the OBD microbiome on insulin resistance, hepatic lipid, and metabolite profiles, with associated hepatic proteome profiles compared to an OBH microbiome, is novel. We show that obesity alone is not sufficient to impair metabolic health and indeed, within equivalent obese states the metabolic impact of diabetes on the microbiota can be substantial. Additionally, functional food ingredients such as β‐glucan supplementation can partly reverse the adverse OBD induced metabolic phenotype. We acknowledge that the fecal microbiota transplantation experiment is limited to one human donor per metabolic health type due to ethical, practical, and financial limitations of pre‐clinical models. Additionally, fecal energy measurement following β‐glucan supplementation should be a focus in future studies. Also given the inherent link between metabolism and inflammation, more detailed work in relation to liver associated macrophage biology is warranted. Further studies are required to validate these finding and determine if/how therapeutic effect of β‐glucan supplementation were coincidental or directly ascribed to microbiome modulation.
Conflict of Interest
The authors declare no conflict of interests, except that up until 2017, F.M. Finucane received honoraria, travel grants, and have served on advisory boards for Novo Nordisk, Eli Lilly, Ethicon, Pfizer Inc., Sanofi‐Aventis, Astra Zeneca, Merck‐Serono, Boehringer Ingelheim, Janssen, and Novartis.
Author Contributions
K.A.J.M. and T.T.T.T. contributed equally to this work. H.M.R. and P.W. O'T. are Senior authors and contributed equally to this work. Conceptualization, H.M.R. and P.W.O'T.; Methodology, E.M.O'C, H.M.R., and P.W.O'T.; Formal analysis, K.A.J.M., T.T.T.T., E.T.D., K.V., and S.M.H.; Investigation, K.A.J.M., K.V., and A. N.; Resources, E.M.O'C., H.M.R., and P.W.O'T.; Data Curation, K.A.J.M., T.T.T.T, E.T.D., and S.M.H.; Writing – Original Draft, K.A.J.M., T.T.T.T., H.M.R., and P.W.O'T.; Writing – Review & Editing, all authors; Visualization, K.A.J.M., T.T.T.T; Supervision, E.M.O'C, H.M.R., and P.W.O'T.; Funding Acquisition, E.M.O'C, H.M.R., and P.W.O'T.
Supporting information
Supporting Information
Acknowledgements
The authors would like to thank APC colleagues, Edel M. Cormac, and Werner Frei for technical assistance in DNA isolation and sequencing of human fecal samples, and Cara M. Hueston, Céline Ribière, and Marta Perez for assistance with the mouse experiment. They gratefully acknowledge the study participants for their interest and contributing their samples. This work was principally funded by the Irish Department of Agriculture, Food and the Marine (14/F/828"ImmunoMet’’ Programme) to K.A.J.M., P.W.O'T., and H.M.R. K.A.J.M. was also funded by Precision Oncology Ireland (18/SPP/3522), a Science Foundation Ireland Strategic Partnership Programme. F.M.F. is supported by a Career Development Award from the Saolta University Healthcare Group. Work in H.M.R.’s laboratory is also funded by Science Foundation Ireland Frontiers Award Programme (SFI 19/FFP/6625).
Open access funding provided by IReL.
Correction added on 28 November 2022, after first online publication: IReL funding statement has been added.
Mitchelson K. A. J., Tran T. T. T., Dillon E. T., Vlckova K., Harrison S. M., Ntemiri A., Cunningham K., Gibson I., Finucane F. M., O'Connor E. M., Roche H. M., O'Toole P. W., Yeast β‐Glucan Improves Insulin Sensitivity and Hepatic Lipid Metabolism in Mice Humanized with Obese Type 2 Diabetic Gut Microbiota. Mol. Nutr. Food Res. 2022, 66, 2100819. 10.1002/mnfr.202100819
Data Availability Statement
Raw 16S rRNA gene sequencing data and associated metadata for this study are available at the National Center for Biotechnology Information Sequence Read Archive (SRA), under BioProject PRJNA669448. This study did not generate new unique codes.
References
- 1. Saeedi P., Petersohn I., Salpea P., Malanda B., Karuranga S., Unwin N., Colagiuri S., Guariguata L., Motala A. A., Ogurtsova K., Shaw J. E., Bright D., Williams R., Committee I. D. A., Diabetes Res. Clin. Pract. 2019, 157, 107843. [DOI] [PubMed] [Google Scholar]
- 2. Kaiser A. B., Zhang N., Pluijm W., Diabetes 2018, 67, 202‐LB. [Google Scholar]
- 3. Centers for Disease Control and Prevention, National diabetes statistics report: estimates of diabetes and its burden in the United States 2017, https://www.cdc.gov/diabetes/pdfs/data/statistics/national‐diabetes‐statistics‐report.pdf
- 4. Umirah F., Neoh C. F., Ramasamy K., Lim S. M., Diabetes Res. Clin. Pract. 2021, 173, 108689. [DOI] [PubMed] [Google Scholar]
- 5. Tierney A. C., McMonagle J., Shaw D. I., Gulseth H. L., Helal O., Saris W. H. M., Paniagua J. A., Gołąbek‐Leszczyñska I., Defoort C., Williams C. M., Karsltröm B., Vessby B., Dembinska‐Kiec A., López‐Miranda J., Blaak E. E., Drevon C. A., Gibney M. J., Lovegrove J. A., Roche H. M., Int. J. Obes. 2010, 35, 800. [DOI] [PubMed] [Google Scholar]
- 6. Ralston J. C., Lyons C. L., Kennedy E. B., Kirwan A. M., Roche H. M., Annu. Rev. Nutr. 2017, 37, 77. [DOI] [PubMed] [Google Scholar]
- 7. Finucane O. M., Lyons C. L., Murphy A. M., Reynolds C. M., Klinger R., Healy N. P., Cooke A. A., Coll R. C., McAllan L., Nilaweera K. N., O'Reilly M. E., Tierney A. C., Morine M. J., Alcala‐Diaz J. F., Lopez‐Miranda J., O'Connor D. P., O'Neill L. A., McGillicuddy F. C., Roche H. M., Diabetes 2015, 64, 2116. [DOI] [PubMed] [Google Scholar]
- 8. Vessby B., Uusitupa M., Hermansen K., Riccardi G., Rivellese A. A., Tapsell L. C., Nälsén C., Berglund L., Louheranta A., Rasmussen B. M., Calvert G. D., Maffetone A., Pedersen E., Gustafsson I. B., Storlien L. H., Study K., Diabetologia 2001, 44, 312. [DOI] [PubMed] [Google Scholar]
- 9. Rosqvist F., Iggman D., Kullberg J., Cedernaes J., Johansson H.‐E., Larsson A., Johansson L., Ahlström H., Arner P., Dahlman I., Risérus U., Diabetes 2014, 63, 2356. [DOI] [PubMed] [Google Scholar]
- 10. Yubero‐Serrano E. M., Delgado‐Lista J., Tierney A. C., Perez‐Martinez P., Garcia‐Rios A., Alcala‐Diaz J. F., Castaño J. P., Tinahones F. J., Drevon C. A., Defoort C., Blaak E. E., Dembinska‐Kieć A., Risérus U., Lovegrove J. A., Perez‐Jimenez F., Roche H. M., Lopez‐Miranda J., Am. J. Clin. Nutr. 2015, 102, 1509. [DOI] [PubMed] [Google Scholar]
- 11. Weickert M. O., Pfeiffer A. F. H., J. Nutr. 2018, 148, 7. [DOI] [PubMed] [Google Scholar]
- 12. Pedersen H. K., Gudmundsdottir V., Nielsen H. B., Hyotylainen T., Nielsen T., Jensen B. A., Forslund K., Hildebrand F., Prifti E., Falony G., Le Chatelier E., Levenez F., Dore J., Mattila I., Plichta D. R., Poho P., Hellgren L. I., Arumugam M., Sunagawa S., Vieira‐Silva S., Jorgensen T., Holm J. B., Trost K., Meta H. I. T. C., Kristiansen K., Brix S., Raes J., Wang J., Hansen T., Bork P., et al., Nature 2016, 535, 376.27409811 [Google Scholar]
- 13. Zmora N., Suez J., Elinav E., Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 35. [DOI] [PubMed] [Google Scholar]
- 14. Forslund K., Hildebrand F., Nielsen T., Falony G., Le Chatelier E., Sunagawa S., Prifti E., Vieira‐Silva S., Gudmundsdottir V., Pedersen H. K., Arumugam M., Kristiansen K., Voigt A. Y., Vestergaard H., Hercog R., Costea P. I., Kultima J. R., Li J., Jorgensen T., Levenez F., Dore J., Meta H. I. T. C., Nielsen H. B., Brunak S., Raes J., Hansen T., Wang J., Ehrlich S. D., Bork P., Pedersen O., Nature 2015, 528, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allin K. H., Nielsen T., Pedersen O., Eur. J. Endocrinol. 2015, 172, R167. [DOI] [PubMed] [Google Scholar]
- 16. De Marco Castro E., Calder P. C., Roche H. M., Mol. Nutr. Food Res. 2020, 65, e1901071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kusmiati, Dhewantara F. X., Sci. Pharm. 2016, 84, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Silva Vde O., Lobato R. V., Andrade E. F., de Macedo C. G., Napimoga J. T., Napimoga M. H., Messora M. R., Murata R. M., Pereira L. J., PLoS One 2015, 10, e0134742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aoki S., Iwai A., Kawata K., Muramatsu D., Uchiyama H., Okabe M., Ikesue M., Maeda N., Uede T., Sci. Rep. 2015, 5, 10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao Y., Zou S., Xu H., Li M., Tong Z., Xu M., Xu X., Mol. Nutr. Food Res. 2016, 60, 2678. [DOI] [PubMed] [Google Scholar]
- 21. Velikonja A., Lipoglavsek L., Zorec M., Orel R., Avgustin G., Anaerobe 2019, 55, 67. [DOI] [PubMed] [Google Scholar]
- 22. Tran T. T. T., Cousin F. J., Lynch D. B., Menon R., Brulc J., Brown J. R. M., O'Herlihy E., Butto L. F., Power K., Jeffery I. B., O'Connor E. M., O'Toole P. W., Microbiome 2019, 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heimesaat M. M., Bereswill S., Fischer A., Fuchs D., Struck D., Niebergall J., Jahn H. K., Dunay I. R., Moter A., Gescher D. M., Schumann R. R., Gobel U. B., Liesenfeld O., J. Immunol. 2006, 177, 8785. [DOI] [PubMed] [Google Scholar]
- 24. Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glockner F. O., Nucleic Acids Res. 2013, 41, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ralston J. C., Mitchelson K. A. J., Lynch G. M., Tran T. T. T., Wang H., Strain C. R., Lenighan Y. M., Kennedy E. B., Stanton C., McGillicuddy F. C., Su Q., O'Toole P. W., Roche H. M., Mol. Nutr. Food Res. 2021, 65, 2000202. [DOI] [PubMed] [Google Scholar]
- 26. McGillicuddy F. C., Harford K. A., Reynolds C. M., Oliver E., Claessens M., Mills K. H., Roche H. M., Diabetes 2011, 60, 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lenighan Y. M., Nugent A. P., Moloney A. P., Monahan F. J., Walton J., Flynn A., Roche H. M., McNulty B. A., Public Health Nutr. 2020, 23, 2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rappsilber J., Mann M., Ishihama Y., Nat. Protoc. 2007, 2, 1896. [DOI] [PubMed] [Google Scholar]
- 29. Cox J., Mann M., Nat. Biotechnol. 2008, 26, 1367. [DOI] [PubMed] [Google Scholar]
- 30. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M., J. Proteome Res. 2011, 10, 1794. [DOI] [PubMed] [Google Scholar]
- 31. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., Mann M., Mol. Cell. Proteomics 2014, 13, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., Cox J., Nat. Methods 2016, 13, 731. [DOI] [PubMed] [Google Scholar]
- 33. Krämer A., Green J., Pollard J., Tugendreich S., Bioinformatics 2014, 30, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. R. Core Team , R Foundation for Statistical Computing, Vienna, Austria: 2016. [Google Scholar]
- 35. Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O'Hara R. B., Simpson G. L., Solymos P., Stevens M. H. H., Wagner H., vegan: Community Ecology Package. R package version 2.5‐5. https://CRAN.R‐project.org/package=vegan 2019.
- 36. Bauer P. V., Duca F. A., Waise T. M. Z., Rasmussen B. A., Abraham M. A., Dranse H. J., Puri A., O'Brien C. A., Lam T. K. T., Cell Metab. 2018, 27, 101. [DOI] [PubMed] [Google Scholar]
- 37. El Khoury D., Cuda C., Luhovyy B. L., Anderson G. H., J. Nutr. Metab. 2012, 2012, 851362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ho H. V., Sievenpiper J. L., Zurbau A., Blanco Mejia S., Jovanovski E., Au‐Yeung F., Jenkins A. L., Vuksan V., Br. J. Nutr. 2016, 116, 1369. [DOI] [PubMed] [Google Scholar]
- 39. Nicolosi R., Bell S. J., Bistrian B. R., Greenberg I., Forse R. A., Blackburn G. L., Am. J. Clin. Nutr. 1999, 70, 208. [DOI] [PubMed] [Google Scholar]
- 40. Cao Y., Sun Y., Zou S., Li M., Xu X., J. Agric. Food Chem. 2017, 65, 9665. [DOI] [PubMed] [Google Scholar]
- 41. Yu L., Li‐Hawkins J., Hammer R. E., Berge K. E., Horton J. D., Cohen J. C., Hobbs H. H., J. Clin. Invest. 2002, 110, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dao M. C., Everard A., Aron‐Wisnewsky J., Sokolovska N., Prifti E., Verger E. O., Kayser B. D., Levenez F., Chilloux J., Hoyles L., Consortium M. I.‐O., Dumas M. E., Rizkalla S. W., Dore J., Cani P. D., Clement K., Gut 2016, 65, 426. [DOI] [PubMed] [Google Scholar]
- 43. Zhang X., Shen D., Fang Z., Jie Z., Qiu X., Zhang C., Chen Y., Ji L., PLoS One 2013, 8, e71108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghosh T. S., Das M., Jeffery I. B., O'Toole P. W., Elife 2020, 9, e50240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Depommier C., Everard A., Druart C., Plovier H., Van Hul M., Vieira‐Silva S., Falony G., Raes J., Maiter D., Delzenne N. M., de Barsy M., Loumaye A., Hermans M. P., Thissen J. P., de Vos W. M., Cani P. D., Nat. Med. 2019, 25, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Woting A., Pfeiffer N., Loh G., Klaus S., Blaut M., MBio 2014, 5, e01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vital M., Howe A. C., Tiedje J. M., mBio 2014, 5, e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rey F. E., Faith J. J., Bain J., Muehlbauer M. J., Stevens R. D., Newgard C. B., Gordon J. I., J. Biol. Chem. 2010, 285, 22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krisko T. I., Nicholls H. T., Bare C. J., Holman C. D., Putzel G. G., Jansen R. S., Sun N., Rhee K. Y., Banks A. S., Cohen D. E., Cell Metab. 2020, 31, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kajani S., Curley S., O'Reilly M. E., Yin X., Dillon E. T., Guo W., Nilaweera K. N., Brennan L., Roche H. M., McGillicuddy F. C., Mol. Metab. 2022, 56, 101425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun W., Nie T., Li K., Wu W., Long Q., Feng T., Mao L., Gao Y., Liu Q., Gao X., Ye D., Yan K., Gu P., Xu Y., Zhao X., Chen K., Loomes K. M., Lin S., Wu D., Hui X., Diabetes 2021, 71, 31. [Google Scholar]
- 52. White P. J., Lapworth A. L., An J., Wang L., McGarrah R. W., Stevens R. D., Ilkayeva O., George T., Muehlbauer M. J., Bain J. R., Trimmer J. K., Brosnan M. J., Rolph T. P., Newgard C. B., Mol. Metab. 2016, 5, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Newgard C. B., Cell Metab. 2017, 25, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dang C. V., Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xin B., Yamamoto M., Fujii K., Ooshio T., Chen X., Okada Y., Watanabe K., Miyokawa N., Furukawa H., Nishikawa Y., Oncogene 2017, 36, 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zirath H., Frenzel A., Oliynyk G., Segerström L., Westermark U. K., Larsson K., Munksgaard Persson M., Hultenby K., Lehtiö J., Einvik C., Påhlman S., Kogner P., Jakobsson P. J., Henriksson M. A., Proc. Natl. Acad. Sci. U S A 2013, 110, 10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Raw 16S rRNA gene sequencing data and associated metadata for this study are available at the National Center for Biotechnology Information Sequence Read Archive (SRA), under BioProject PRJNA669448. This study did not generate new unique codes.