

Abstract
Major progress has been made in defining the basis of the mitochondrial permeability transition, a Ca2+‐dependent permeability increase of the inner membrane that has puzzled mitochondrial research for almost 70 years. Initially considered an artefact of limited biological interest by most, over the years the permeability transition has raised to the status of regulator of mitochondrial ion homeostasis and of druggable effector mechanism of cell death. The permeability transition is mediated by opening of channel(s) modulated by matrix cyclophilin D, the permeability transition pore(s) (PTP). The field has received new impulse (a) from the hypothesis that the PTP may originate from a Ca2+‐dependent conformational change of F‐ATP synthase and (b) from the reevaluation of the long‐standing hypothesis that it originates from the adenine nucleotide translocator (ANT). Here, we provide a synthetic account of the structure of ANT and F‐ATP synthase to discuss potential and controversial mechanisms through which they may form high‐conductance channels; and review some intriguing findings from the wealth of early studies of PTP modulation that still await an explanation. We hope that this review will stimulate new experiments addressing the many outstanding problems, and thus contribute to the eventual solution of the puzzle of the permeability transition.
Keywords: adenine nucleotide translocator, ATP synthase, calcium transport, channels, cyclophilin, cyclosporine, mitochondria, permeability transition
The mechanistic basis for the mitochondrial permeability transition (an inner membrane permeability increase that is a causative event in cell death) has puzzled mitochondrial research for 70 years. Here, we review the field and discuss recent evidence on how a Ca2+‐dependent conformational change of F‐ATP synthase and of adenine nucleotide translocator may transform these energy‐conserving devices into energy‐dissipating multiconductance channels causing the permeability transition.
Abbreviations
- ANT
adenine nucleotide translocator
- ATR
atractylate
- BKA
bongkrekate
- Bz
benzodiazepine
- CsA
cyclosporine A
- CyP
cyclophilin
- DDM
dodecyl maltoside
- EM
electron microscopy
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- LMNG
lauryl maltose neopentyl glycol
- OMM
outer mitochondrial membrane
- OSCP
oligomycin sensitivity conferral protein
- PT
permeability transition
- PTP
permeability transition pore
- TSPO
translocator protein
- Ub
ubiquinone
- UCP1
uncoupling protein 1
- VDAC
voltage‐dependent anion channel
A bird’s eye view on the permeability transition
Although the term ‘permeability transition’ (PT) was introduced in 1976 [1] and further defined in 1979 [2, 3, 4], occurrence of an unselective permeability increase of mitochondria and its detrimental consequences on energy conservation had been known since the very first studies on isolated organelles [5, 6]. The permeability increase leading to swelling was widely considered to be a form of membrane damage [7] possibly caused by long‐chain fatty acid(s) [8] and by lysophospholipids generated by the Ca2+‐dependent activation of phospholipase A2, as suggested by the remarkable protective effects of nupercaine [9] and of N‐ethylmaleimide [10]. The far‐reaching hypothesis that the PT could rather be caused by opening of a regulated inner mitochondrial membrane (IMM) channel playing a role in physiology (the PT pore, PTP) [2, 3, 4] was generally met with scepticism also because of the acceptance of the chemiosmotic hypothesis of energy conservation [11]. Indeed, permeability defects with an estimated pore radius of 14 Å [7] seemed hard to reconcile with the low permeability of the coupling membrane to solutes and ions, as discussed in detail in previous reviews [12, 13, 14]. The channel hypothesis gained traction (a) with the electrophysiological identification of an IMM high‐conductance channel named mitochondrial multiconductance channel [15] or megachannel [16], which shares the key features of the PTP [17, 18], see [19] for a review and (b) with the discovery that the PT could be inhibited by nanomolar concentrations of cyclosporine A (CsA) [20, 21, 22, 23], a finding that was instrumental in establishing a pathogenic role for the PTP in cell and organ injury [24, 25, 26, 27, 28]. CsA had no effects on phospholipase A2 [29] while it inhibited a matrix peptidyl prolyl cis‐trans isomerase [23] that was later shown to be mitochondrial cyclophilin (CyP) D [30, 31]. There is no question that CyPD favours onset of the PT [30] because, like treatment with CsA, deletion of the Ppif gene (which encodes CyPD) desensitises the PTP to Ca2+ [32, 33, 34, 35]; and yet both CsA‐sensitive and CsA‐insensitive PTs with distinct features could be identified, with a synergistic effect of free fatty acids on PTP opening [36, 37].
The molecular nature of the PTP is still an open issue, but significant progress has been made in the recent years. Based on the effects of the selective inhibitors of the adenine nucleotide translocator (ANT) atractylate (ATR, which favours PTP opening) and bongkrekate (BKA, which favours PTP closure) the first proposed candidate for channel formation was the ANT itself [2]. Consistent with this hypothesis, ANT from bovine heart and Neurospora crassa formed Ca2+‐activated channels [38, 39] with many of the features displayed by the PTP at the patch‐clamp [17, 40, 41], including CsA‐inhibitable stimulation by yeast cyclophilin [39]. At about the same time, the ANT was shown to copurify with the outer mitochondrial membrane (OMM) proteins voltage‐dependent anion channel (VDAC) and translocator protein (TSPO, formerly called peripheral benzodiazepine receptor) [42]; and it was found that nanomolar concentrations of TSPO‐binding ligands (like benzodiazepines) promoted channel opening [43]. Although puzzling, given that the PT takes place at the IMM [44], these findings shifted the attention of the field to sites of contact between the OMM and IMM, with the former also providing accommodation for hexokinase II [45, 46] and for proteins of the Bcl‐2 family, linking the PTP to apoptosis and to regulated necrosis [47, 48].
Modulation by CyPD and inhibition by CsA considerably strengthened the idea that the PT was mediated by a single molecular entity. Given that genetic ablation of each of the putative components of the PTP (i.e., ANTs, VDACs and TSPO) did not prevent occurrence of the PT [49, 50, 51, 52, 53], we searched for novel PTP candidates using mitochondrial CyPD as the bait. A most interesting interactor turned out to be the mitochondrial F‐ATP synthase [54]. This observation was the starting point for a set of studies addressing the question of whether F‐ATP synthase can form a Ca2+‐dependent channel with the features of the PTP. Results were apparently conflicting.
On one hand, it was shown that partially purified F‐ATP synthase can generate Ca2+‐activated channels in a variety of eukaryotic mitochondria [55, 56, 57, 58, 59, 60]; that knockdown of subunit c [61] or point mutations that do not affect enzyme complex assembly or ATP synthesis caused specific changes in the properties of the PTP [56, 59, 62, 63, 64, 65, 66, 67, 68]; and that highly purified F‐ATP synthase preparations display features expected of the PTP in electrophysiological experiments [69, 70]. Remarkably, it was also discovered that benzodiazepines are actually not selective ligands of TSPO, as they also inhibit F‐ATP synthase [71, 72] while promoting the PT [55, 69], effects that mimic those of CyPD through a shared binding site on subunit oligomycin sensitivity conferral protein (OSCP) [55, 73].
On the other hand, ablation of individual subunits of F‐ATP synthase (which prevented its assembly) in HAP1 cells was not followed by disappearance of the PT, which persisted and maintained its sensitivity to CsA [74, 75, 76]. Yet, in the same cells ablation of subunit c led to the appearance of a channel sensitive to both CsA and BKA, while the channel of wild‐type cells was sensitive to CsA only [77]. These results can be explained by the existence of two CsA‐sensitive channels modulated by CyPD. One channel is formed from the F‐ATP synthase, the other from the ANT. Our recent work fully confirmed the dual nature of the PT and suggested that in wild‐type cells the F‐ATP synthase predominates, yet the two PTPs are interconnected [67] possibly by physical association at the ‘ATP synthasomes’, which may also involve the Pi carrier [78, 79, 80].
Despite these advances, many open questions remain including the molecular mechanisms through which the ANT and F‐ATP synthase can form high‐conductance channels, the role of the OMM, and the assignment of a variety of agents and factors that critically modulate the PT. The following discussion is meant to address these issues, with the hope that it can stimulate new experiments to solve the longest‐standing mystery of mitochondrial biology. Space limitations preclude discussion of the physiology and pharmacology of the PTP, and of its role in degenerative diseases, cancer and aging, important topics for which we refer the reader to published reviews [14, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92].
Adenine nucleotide translocator
The ANT is a 30 kDa protein of the IMM that was first identified in the early 1960s [93, 94]. It catalyses the equimolar exchange of Mg2+‐free ADP and ATP across the IMM. Transport of adenine nucleotides is determined by their concentration gradient and by the membrane potential existing across the IMM, since ADP/ATP exchange is electrogenic. In respiring mitochondria this results in uptake of ADP from the cytosol and release of newly synthesised ATP. Humans have four ANT isoforms, AAC1‐AAC4, with AAC4 being specific to germline and to pluripotent stem cells [95, 96]. ANTs belong to the SLC25 mitochondrial carrier family, which is the largest solute transporter family comprising 53 carriers in humans [97]. These carriers transport metabolites, inorganic ions and cofactors across the IMM and are thought to operate through a common mechanism, defined as alternating‐access mechanism [98]. It is based on three main functional elements, that is, one central substrate‐binding site and two gates with salt bridge networks on either side of the carrier [99].
Structure and catalytic mechanism
Most structural properties of the SLC25 family have been defined from studies of the ANT, which were favoured by its abundance and by the availability of two specific inhibitors, ATR [100] and BKA [101], which lock the carrier in two different conformational states. ATR traps the ANT in the cytoplasmic‐open state (c‐state) with the adenine‐binding site facing the cytosol, while BKA locks the ANT in the matrix‐open state (m‐state) with the adenine‐binding site facing the matrix [102]. It should be kept in mind that no structures of substrate‐bound states have yet been defined, so that the events taking place during adenine nucleotide translocation are not known. Information at atomic resolution was first provided by the crystallographic structure of the carboxyATR‐inhibited bovine ANT [103]. The protein contains three homologous domains of about 100 amino acids, each comprising an odd‐numbered transmembrane α‐helix (H1, H3 and H5), a loop with a short matrix α‐helix (h12, h34 and h56) lying in the plane of the IMM, and an even‐numbered transmembrane helix (H2, H4 and H6). This basic structural fold was confirmed for the yeast isoforms Aac2p and Aac3p [104].
In the c‐state (which favours the PT) the odd‐numbered transmembrane α‐helices, which contain the conserved Px[DE]xx[KR] motif, are close together towards the matrix side of the membrane, enabling the charged residues to establish inter‐domain salt‐bridges (the matrix salt‐bridge network) and closing the central substrate‐binding site from the matrix side [104]. In the m‐state (which inhibits the PT) the even‐numbered α‐helices make inter‐domain salt‐bridges (the cytoplasmic salt‐bridge network) through highly conserved [YF][DE]xx[KR] motifs, closing the central cavity from the cytoplasm side, while the matrix helices on the membrane surface are rotated outward opening the central cavity to the matrix side [105].
The proposed transport mechanism involves six mobile elements, two per domain. During the switch from the c‐ to the m‐state triggered by substrate binding, the core elements composed by the cytoplasmic ends of the H1, H3 and H5 helices would move outward as rigid bodies, opening up the substrate‐binding site to matrix, while the cytoplasmic gates formed by the H2, H4 and H6 helices rotate inwards, closing the cytoplasmic side. The reverse movements would occur in the m‐ to c‐state transition [99, 102]. According to this structural model, the carrier is monomeric because the large conformational changes preclude the formation of a stable dimerisation interface during the transport cycle. An outline of the ANT and of the overall ATP/ADP exchange process is reported in the left panel of Fig. 1.
Fig. 1.
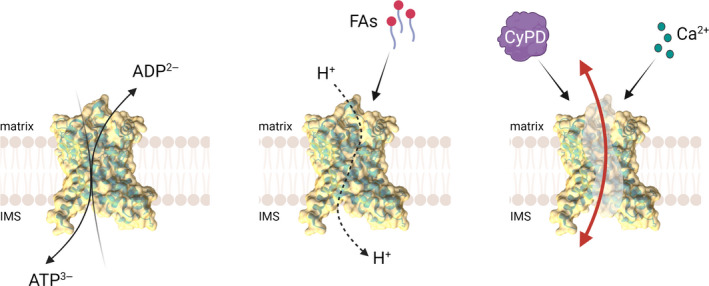
Schematic representation of the possible functions of ANT. (Left) The established function of ANT is to exchange adenine nucleotides, transporting matrix ATP to the cytosol and cytosolic ADP to the matrix in energised mitochondria. (Center) In response to fatty acids ANT mediates H+ currents, suggesting that nucleotide exchange and H+ transport are not mutually exclusive functions. (Right) In the presence of Ca2+ and CyPD, ANT may undergo a still undefined conformational change which allows the formation of a high‐conductance channel.
Possible mechanism of high‐conductance channel formation
Some clues on the requirements for channel formation by ANT may come from recent work on H+ transport by the structurally related uncoupling protein 1 (UCP1) and by ANT itself. In thermogenic brown fat H+ permeation is a highly regulated process under hormonal control requiring fatty acids and mediated by UCP1 [106, 107], which like ANTs is a member of the SLC25 mitochondrial carrier family [97]. H+ ions are transported by the fatty acyl anion, which shuttles within UCP1 itself [108]. Of note, deletion of amino acids 261–269 converts UCP1 into a pore allowing permeation of species with molecular mass up to 1 kDa [109]. The IMM has a measurable permeability to H+ (the ‘H+ leak’) also in tissues that do not express UCP1. The H+ leak is widely considered to result from passive H+ permeation through the lipid bilayer, which is compensated by basal respiration (oxygen consumption not coupled to ATP synthesis). Basal respiration is increased by fatty acids, an effect that can be prevented by ADP or by inhibitors of the ANT [110]. This interesting finding led to the suggestion that the H+ leak may be due, in part at least, to H+ transport through the ANT either through an allosteric effect of the fatty acid, or through transport of the fatty acyl anion coupled to passive diffusion of the protonated fatty acid through the lipid bilayer [110]. A recent electrophysiological study has demonstrated that a relevant fraction of the H+ leak indeed takes place through the ANT acting as a channel in which fatty acids play an essential role as cofactors in H+ transport without being actually translocated [111]. These studies demonstrate that the ANT can act as a (H+‐selective) channel (middle panel of Fig. 1).
At present, there is no obvious mechanism to explain channel formation by ANT. Inspection of the protein does not reveal the presence of Ca2+‐binding sites, suggesting the possible requirement for additional factors, such as cardiolipin [112] or for post‐translational modifications, for example, Ca2+‐dependent limited proteolysis. It has been suggested that the PT is favoured by oxidation of ANT residues C57 [113, 114] and C160 [114], which would cause formation of disulphide bridges followed by enhanced binding of CyPD [114]; yet, the PTP could be induced by thiol oxidants without dimerisation of the ANT [115] and Ca2+‐induced ANT channel opening has been documented in the absence of oxidants [38, 39]. It is hopeful that the contribution of ANT Cys residues will be studied by site‐directed mutagenesis and electrophysiology, and possibly by structure determination through high‐resolution cryo‐EM in the presence of Ca2+. It appears conceivable, however, that CyPD and Ca2+ could favour the channel transition of ANT, as seen in reconstitution experiments with the bovine [38] and yeast species [39], possibly in cooperation with fatty acids, which are well‐known activators of the PTP [116] (right panel of Fig. 1).
F‐ATP synthase
F‐ATP synthase is an abundant complex located in the IMM that functionally cooperates with ANT and the Pi carrier to produce ATP from cytosolic ADP in aerobic conditions. F‐ATP synthase makes ATP from ADP and Pi by rotary catalysis using the proton motive force generated by H+ pumping by the respiratory chain complexes. It consists of the water‐soluble, F1 head exposed to the mitochondrial matrix, which is responsible for the synthesis of ATP; and the membrane‐embedded F O sector involved in proton traslocation. The two sectors are connected by a central stalk rotating within F 1 and a stationary peripheral stalk essential in maintaining complex stability [117].
Structure and catalytic mechanism
Crystallography has revealed the structure of many F 1 and F O sub‐complexes from bovine heart [118, 119] and yeast [120]. Cryo‐EM studies have reported structures of the entire complex from Yarrowia lipolytica [121], Saccharomyces cerevisiae [122], Sus scrofa [123], Bos taurus [124] and Ovis aries [125], providing high‐resolution maps of all the F O subunits (upper panel of Fig. 2). The F 1 sector comprises three αβ pairs surrounding the γ subunit, which forms the central stalk by associating at the foot with subunits δ and ε. The three αβ pairs contain six nucleotide‐binding sites, only three of which are catalytically active and mostly contributed by the β subunits. A rotor ring with a variable number of copies of subunit c (8 in metazoans, 10 in yeast and a record 17 in Burkholderia pseudomallei) is located in the F O sector. The cavity of the c‐ring is occupied by phospholipids [125], in keeping with previous proposals [126]. The lipids at the intermembrane space (IMS) and matrix end of the c‐ring are functionally and chemically distinct. Only the matrix‐side lipids rotate with the c‐ring, and thus providing a ‘lubricated’ plug facilitating rotation [125]. The outer surface of the c‐ring is attached to subunit a, which is arranged in a concave four‐helix horizontal bundle around the c‐ring forming two aqueous half‐channels separated by a conserved arginine [117]. In mammals, subunit a is also tightly bound to subunits j (6.8PL), k (DAPIT) and 8 (A6L) forming a structure that has been denoted as ‘proton translocation cluster’ [124, 125]. The two half‐channels allow H+ to reach an essential ionised glutamate residue of subunit c that, once protonated, moves to a more hydrophobic environment generating rotation of the c‐ring and consequently of the central stalk. Rotation of γ subunit forces each of the three catalytic sites into three major conformations with different affinities for nucleotides, denoted as βE (empty), βDP (occupied by Mg2+‐ADP and Pi) and βTP (occupied by Mg2+‐ATP), thereby catalysing the synthesis of one ATP molecule during each 120° rotation of the γ subunit [127]. The enzyme can work in reverse pushing the central stalk and the c‐ring backward with the energy deriving from ATP hydrolysis, generating a H+ gradient. Catalysis requires the nucleotide in complex with Mg2+ (or with other metals with decreased efficiency) [128]. Of specific interest to the topic of this review, Ca2+ has the unique property to catalyse ATP hydrolysis not coupled to formation of a H+ gradient [129, 130] despite its ability to generate rotation of the γ subunit [131], a finding that suggests onset of permeabilisation.
Fig. 2.
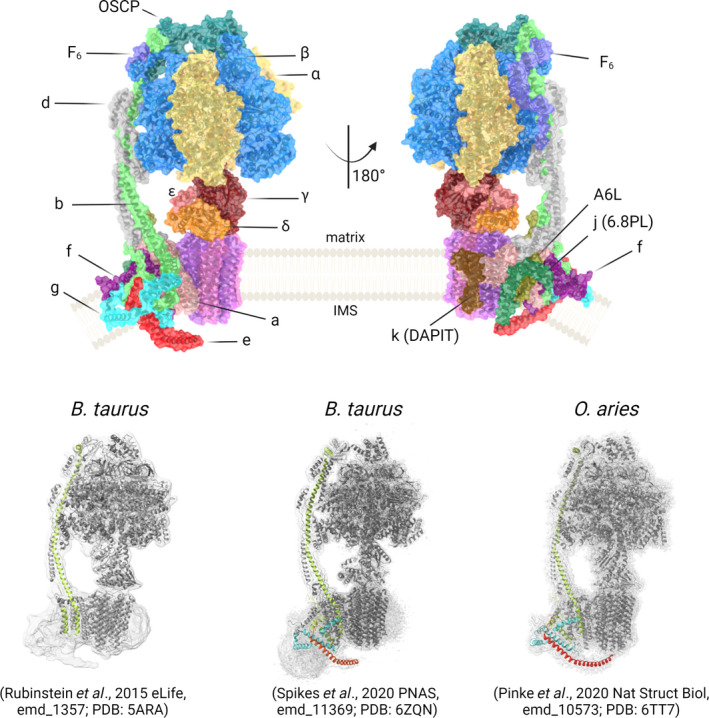
Structure of the F‐ATP synthase monomer. (Upper panel) F‐ATP synthase structure is based on the atomic model of Spikes et al. [124]. (Lower panel) Density maps of F‐ATP synthase derived by cryo‐EM in the indicated studies, with fitted atomic models. Subunits b, e and g are green, cyan and red, respectively.
The mitochondrial F O contains an additional subset of four conserved subunits (b, e, f, g) with subunit b reaching out of the membrane to form the core of the peripheral stalk. Through its helices H2 and H3 within the IMM, subunit b makes a ‘U‐turn’ forming a hairpin able to interact with the horizontal bundle of subunit a on one side, and on the other side to form a compact triple transmembrane helix bundle with subunits e and g [125]. This bundle is packed through GXXXG motifs located at N‐ and C‐termini of subunits e and g, respectively, with the C‐terminus of subunit e reaching out and attaching to the c‐ring lipids from the IMS to form a ‘hook’ [124, 125]. A structure protruding into the IMS, thought to arise from subunits e and g in contact with subunit b, had originally been shown in the cryo‐EM analysis of the bovine heart F‐ATP synthase (lower left panel of Fig. 2); this structure first revealed flexibility of the intact enzyme due to bending and twisting of the central and peripheral stalks [132]. Subunit b also contributes to form the membrane domain of the peripheral stalk together with subunits e, f, g, 8 and j [124]. In its extrinsic part subunit b forms a long, continuous α‐helix stiffened by interactions with subunits 8, F6 and d, finally reaching out to the C‐terminus of OSCP subunit on top of the F1 sector. OSCP forms many tight interactions with F 1 at three α, two β and with F6 and b subunits. These interactions result in a strong link between F O and F 1, which prevents co‐rotation of the αβ pairs with subunit γ. Anchoring is further strengthened by the already mentioned connection of subunit e with the lipid plug of the c‐ring at the opposite side [124, 125] (lower middle and right panels of Fig. 2). This complex structure makes F‐ATP synthase a ‘perfect chemomechanical coupling device’ between H+ translocation, rotor rotation and ATP synthesis/hydrolysis [133]. Based on the cryo‐EM structures of different rotational states [132, 134], cooperation between F O and F 1 appears ensured by elastic power transmission mediated by the flexible peripheral stalk, primarily at the inter‐domain hinge of the OSCP subunit, a well‐established target of drugs and matrix proteins including CyPD [135].
Oligomeric states of F‐ATP synthase
As mentioned earlier, a notable feature of the F‐ATP synthase complexes is their association into dimers that self‐assemble into long rows of oligomers to develop the typical cristae [136, 137], which have a direct impact on mitochondrial bioenergetics [138]. However, the structural details and functional roles of such supramolecular structures are still debated. Pioneering experiments in yeast established the existence of (a) a dimerisation interface stabilised through interactions of subunits a, b, e, g, F6 and i/j, which contribute in an additive way to the association of monomers; and (b) an oligomerisation interface mainly stabilised through e/e and g/g interactions [139]. Later, in situ cryo‐EM structures demonstrated that F‐ATP synthase forms V‐shaped (type I) dimers at an angle of 80–90° between the two central stalks, with the peripheral stalks turned away from one another [117]. The recent cryo‐EM structure of dimeric complexes from bovine heart shows that the interaction is mainly mediated by the two j subunits and is highly dynamic, that is, it changes over time to accommodate motions both dependent on and independent of catalysis [140]. This study also proposed that dimers associate into oligomers mainly through homo‐interactions between the matrix‐exposed part of subunit g together with subunit k, forming a ‘fluid interface’ able to follow the cristae curvature. Conversely, cryo‐EM analyses of the ovine and porcine tetrameric complexes showed that the ‘neighbours in a row’ monomers are linked by two IF1 molecules, and that the resulting tetrameric structures have a higher stability compared to the dimers [123, 125]. Such results suggest that different F‐ATP synthase oligomeric structures can coexist within a mitochondrion at the cristae edges. In addition, the presence of a sub‐population of monomeric F‐ATP synthase complexes probably prevailing in the inner boundary membrane has been proposed [141]. These monomeric complexes are characterised by higher mobility compared to the dimeric complexes localised at the cristae edges and may be working as ATPases. F‐ATP synthase thus seems able to undergo not only functional but also spatial and structural reorganisation to respond to different metabolic conditions [141], which also involve interactions with the cristae‐shaping protein OPA‐1 [142]. Finally, it has been proposed that F‐ATP synthase, ANT and the Pi carrier associate in a supramolecular structure, the ‘ATP synthasome’ [78, 79], the formation of which would depend on CyPD association and on the bioenergetic state of the mitochondria [80].
Possible mechanisms of channel formation
As mentioned in the Introduction, the first clue that the F‐ATP synthase could be involved in the PT was the demonstration that CyPD interacts with the peripheral stalk of F‐ATP synthase [54]. CyPD binding required relatively high concentrations (10 mm) of Pi and resulted in partial inhibition (about 30%) of the rate of oligomycin‐sensitive ATP synthesis and hydrolysis; CsA dissociated CyPD from the F‐ATP synthase removing this inhibitory effect [54]. A few years later clear evidence was obtained that Ca2+‐dependent channel formation from F‐ATP synthase can actually take place [55, 56] as now supported by a variety of studies based on reconstitution of highly purified preparations from bovine [69] and porcine hearts [70].
Based on extensive mutagenesis [56, 61, 62, 63, 64, 65, 66, 67, 68], reconstitution [55, 56, 57, 58, 59, 69, 70] and structural work [123, 124, 125] a plausible mechanism through which F‐ATP synthase could generate a channel is beginning to emerge. First proposed by Gerle as the ‘death finger’ hypothesis [143, 144], this model combines elements of two previous hypotheses, that is, that the PTP may form at the dimer interface of monomers through a conformational change originating at OSCP [55] or at the c‐ring after dissociation of F 1 [56, 61]. The following putative sequence of events takes into account most of the available information obtained in several laboratories (Fig. 3A).
Under basal conditions (enzyme turnover with Mg2+ at the catalytic site) rotation of the γ subunit is smooth, and elastic force is dissipated through OSCP and the peripheral stalk with little effects on the ‘hook apparatus’ connecting with the outer side of the lipid plug in the c‐ring (Fig. 3A, left).
Binding of Ca2+ occurs at the catalytic metal site contributed by both α and β subunits (Fig. 3A, centre), which is usually occupied by Mg2+; the larger van der Waals radius of Ca2+ causes a spatial rearrangement of the F 1 sector with increased overall rigidity [62].
The rearrangement is transmitted to the crown region, that is, the β‐barrel‐shaped ‘ring’ at the F 1/OSCP interface [119] through the long connecting loop made up by residues 82‐131 of subunit β [62].
The decreased compliance causes more mechanical stress on OSCP, overcoming its ‘shock absorber’ function during rotation of subunit γ [134] in a process that is favoured by binding of CyPD (Fig. 3A, centre) or of its chemical mimic benzodiazepine (Bz)‐423 [55], which decreases OSCP flexibility; CsA desensitises the PTP by displacing CyPD from OSCP rather than by a direct effect on F‐ATP synthase [54, 55].
The mechanical energy is transmitted from OSCP to the peripheral stalk relaying it at the point of entrance into the membrane [121, 132], where subunit b forms a hairpin that makes a tight association with the C‐ and N‐termini of subunits g and e, respectively, forming a strong ‘wedge’ [124] or ‘bundle’ [125].
The C‐terminus of subunit e, which extends outside the bundle region to make contact with the c‐ring in the ‘hook apparatus’ [125], exerts a pulling effect on the lipids allowing formation of a channel within the c‐ring by displacing the outer lipid plug [125].
At physiological, low levels of matrix Ca2+ the PTP oscillates between the closed and open states (Fig. 3A, centre); as matrix Ca2+ increases, or as the result of additional inducing agents, openings become more stable and lead to displacement of the inner lipids and of the central stalk (Fig. 3A, right). Of note, also the fully open state is reversible when Ca2+ is removed [145].
Fig. 3.
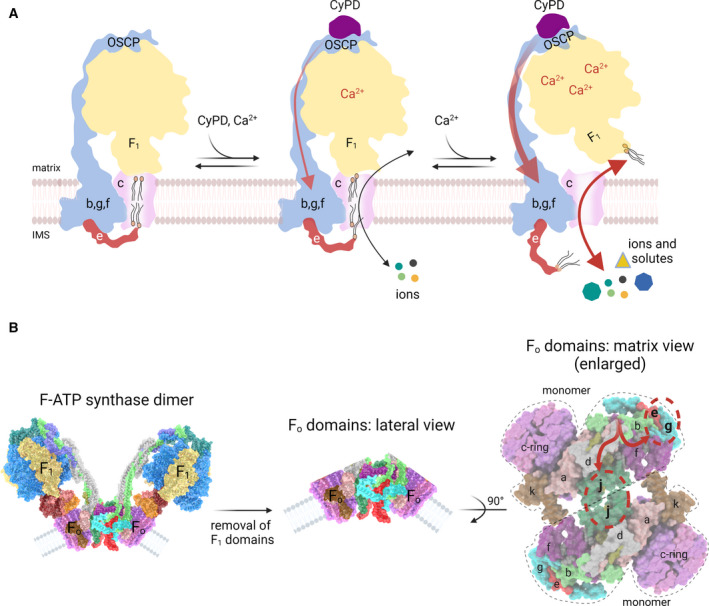
Hypothetical models for PTP formation by F‐ATP synthase. (A) The ‘death finger’ model proposes the transition of the c‐ring into the PTP channel on a conformational change of the peripheral stalk that eventually perturbs subunit e, the final transducer of pore opening. The C‐terminus of subunit e makes contacts with lipids of the plug within the c‐ring from the IMS (left). In the presence of physiological, low Ca2+ concentrations with CyPD bound to OSCP subunit (middle), the peripheral stalk transmits mechanical force generated by Ca2+ binding to subunit e (red arrow), which may exert a pulling action dragging some lipids out of the plug. This condition, together with a secondary relaxation of the central stalk/c‐ring connections would accommodate a low‐conductance mode of channel opening mediating the passage of ions but not of larger solutes, representing the ‘flickering’ mode of the PTP (middle). As the matrix Ca2+ levels rise, the mechanical force exerted on subunit e becomes stronger allowing removal of the lipid plug from the c‐ring and displacement of F 1 with formation of the high‐conductance PTP, which remains fully reversible if Ca2+ is removed [145]. (B) Entire F‐ATP synthase dimer (left), after removal of the F 1 domains (centre) and following a 90° rotation to show the two F O domains as viewed from the matrix side (right). The conformational change transmitted through the peripheral stalk (red arrows in the right panel) may affect both subunits g/e and the monomer–monomer interface, with possible channel formation at the point of contact of subunits j, which undergo a pivoting motion at their interface during catalysis [124].
Cryo‐EM structures of F‐ATP synthase prepared in the presence of 5 mm Ca2+ yielded 3D density classes specific to the Ca2+ dataset, that is, never observed with Mg2+. These could be arranged in a plausible sequence that may provide snapshots of the different stages in the process of PTP opening [125]. The mechanism through which the channel would reversibly form within the c‐ring [56], which in the fully open state also implies some rearrangement or even the displacement of F 1, remains shrouded in mystery. Importantly, it is not clear whether all conductance substates of the PTP can be explained by the above hypothesis.
In patch‐clamp experiments with native membranes the PTP exhibits a large variety of substates ranging from as little as 45 pS to as high as > 1000 pS [15, 16]. All these substates are also observed in highly purified dimeric F‐ATP synthase preparations reconstituted in lipid bilayers [69]. As just mentioned, it is possible that the low‐conductance flickering reflects reversible oscillations of the lipid plug within the c‐ring. An interesting (and not mutually exclusive) alternative, however, is suggested by the reconstruction of the dimer interface from the high‐resolution structure of monomers (Fig. 3B), where adjacent subunits j form a cavity that appears not to be filled by lipid [124] and that may provide an interface for channel formation in dimers (Fig. 3B). This could contribute to the reversible, transient flickering of the PTP observed both in isolated mitochondria and intact cells [146, 147, 148, 149].
It has been proposed that PTP formation requires dissociation of the dimers, in a study where isolated mitochondria were subjected to Ca2+‐dependent PTP opening leading to a small decrease of the dimer/monomer ratio, as measured by activity staining in native gels where the presence of oligomers and of partially assembled forms of the complex was not assessed [150]. The prevalent state of F‐ATP synthase was still the dimeric form, so that assigning occurrence of the PT to the monomers appears arbitrary. The key issue, however, is that these experiments cannot tell whether the partial disassembly of the dimers is the cause or rather the consequence of swelling, a problem that also applies to the interpretation of the in situ studies [150]. While PTP formation in monomers is possible, whether this is a requisite for pore opening remains very difficult to assess even in reconstituted systems, as will be discussed later.
Species‐specific features
The F‐ATP synthase is a highly conserved enzyme with a substantial degree of sequence identity from bacteria to mammals, which is particularly evident for the catalytic domain, where more than 60% residues of subunit β have been conserved [151]. In eukaryotes, the enzyme contains unique so‐called ‘supernumerary’ subunits anchored to the F O region, which in yeast include subunits e and g that are strictly associated with dimers [152, 153] and appear to mediate dimerisation of monomers in mammals as well [139]. Cryo‐EM images of dimers from different species confirmed a wide conservation of the central structure, while revealing extensive variations in the composition and structure of the peripheral stalk, especially in the dimeric interface [121, 154, 155].
The molecular connections between monomers or dimers vary among species, and most data were gathered by structural analysis of the yeast and bovine enzymes. In Yarrowia lipolytica, the dimeric interface was reported to be primarily occupied by the C‐terminus of subunit f, to include subunits a and 8 while the resulting wedge‐shaped space in between appears to be filled by lipids [121]. In the structure of the Saccharomyces cerevisiae F‐ATP synthase dimer, the density of subunit f was reassigned to subunit i/j (j in mammals), while occupancy of the dimer interface by subunit a was confirmed [156]. In both structures subunits e and g, which are connected with the N‐terminus of subunit b in a well‐defined bundle, locate at a more distal position. They confer a peculiar curvature to the membrane and do not participate in dimer formation but rather in dimer–dimer contacts. Subunit e protrudes straight out of the F o domain with its C‐terminus towards the IMS without any obvious implications in the organisation of higher‐order structures [121]. In Saccharomyces cerevisiae, it connects with subunit k in an arrangement that may be specific for yeast, given that subunit e has a completely different orientation in the mammalian enzyme [124, 125, 132]. However, it is important to note that the density for yeast subunit e (and g) was reconstructed using poly‐alanine models, which may camouflage the real structure and position of the protein. In the bovine and ovine F‐ATP synthases, the dimer interface includes subunit j, which spans from the matrix to the IMS and together with subunit f generates a cavity where the membrane has reduced thickness. Interestingly, subunit a appears to be completely covered by subunits j, 8 and partially by cardiolipin. As in yeast, subunit e locates in a proximal domain and coordinates dimer–dimer contacts, but it bends toward the lipid plug of the c‐ring.
Subunit e shows species‐specific sequence features (Fig. 4). As expected, the well‐characterised GXXXG motif (position 23‐27 in Homo sapiens), which allows tight packing with the α helices of subunits g and b, is highly conserved both in terms of sequence and of position within the protein. Sequence similarity is also visible in the transmembrane region. Prominent variations are instead present in the C‐terminus, with terminal sequences unique to various strains of Drosophila and yeast. These overall differences may provide an explanation for the different orientation of subunit e relative to the mammalian protein, and perhaps also for the channel‐forming ability of F‐ATP synthases. Indeed, it is of interest that the PTP displays distinct properties in different species [157] including the mean conductance, which ranges from 500 pS (bovine) [55] to 300 pS (yeast) [57], to a mere 53 pS in Drosophila [58], where the PTP operates as a highly selective Ca2+ release channel [158]. Whether these properties can be ascribed to the unique C‐termini of subunit e and/or to the specific features of the monomer/monomer interfaces is being actively investigated in our laboratories. These studies should provide interesting insights into the mechanism(s) of PTP formation and a potential test of the ‘death finger’ hypothesis [144].
Fig. 4.

Sequence alignment of subunit e from various species. Accession number of sequences used for the alignment: H. sapiens (P56385), B. taurus (Q00361), S. scrofa (Q9MYT8), R. norvegicus (P29419), M. musculus (Q06185), D. rerio (A7YY99), D. willistoni (B4N947), D. melanogaster (O77134), D. sechellia (B4HEN9), S. cerevisiae (P81449) and Y. lipolytica (B5FVG3). The multiple sequence alignment was performed with the CLUSTALW program and analysed by jalview software.
Effect of detergents
As already discussed, in the native IMM the F‐ATP synthase is organised in complex multimeric structures [159]. After assembly from preformed F1/c‐ring and peripheral stalk subcomplexes [160] monomers associate into dimers [152], the actual building blocks that by lateral association form long rows of oligomers, which in turn help to shape the IMM foldings at the cristae [161]. Purification of F‐ATP synthase disrupts its native conformation affecting the properties of the resulting enzyme complex in a manner that depends on the detergent(s) used, an issue that should always be kept in mind in the design and interpretation of reconstitution experiments.
Extraction with digitonin [162] or lauryl maltose neopentyl glycol (LMNG) [69] generated monomeric, dimeric and oligomeric states when F‐ATP synthase was analysed by non‐denaturing blue‐native [163] or clear‐native PAGE [164]. After elution, F‐ATP synthase dimers but not monomers displayed Ca2+‐induced channel activity in lipid bilayers [55, 69]. Protein elution from the gel slices was carried out with n‐heptyl β‐d‐thioglucopyranoside [55, 69], however; and we suspect that this step may have removed components of the hook apparatus (like subunit e) from monomers but not dimers, and thus preventing channel formation in the former.
In contrast with these results, it was convincingly shown that reconstituted, monomeric F‐ATP synthase generates currents similar to those of the PTP [70]. In these protocols, mitochondria were extracted with dodecyl maltoside (DDM), which removes the labile subunits j and k [165] together with the ‘dimerisation’ subunits e and g [70], thus generating the monomeric form only [166]. At variance from the case of the bona fide PTP, currents did not require added Ca2+ and were inhibited by oligomycin [70]. As mentioned earlier, the subunits removed by DDM are critically located in the hook apparatus [70, 165]; and yet, channel opening was seen [70]. A possible explanation is the delipidating effect of DDM [163], which may perturb the lipid plug thus bypassing the Ca2+ requirement for PTP activation. Preservation of the lipids by the milder detergents digitonin and LMNG could explain lack of channel formation in the reconstituted system with our monomer preparations [55, 69].
Cyclophilin D
CyPD is the unique mitochondrial isoform of the cyclophilins, a family with more than 15 mammalian members that exhibit peptidyl‐prolyl cis‐trans isomerase activity and share the ability to bind the inhibitory drug CsA. In mammals, CyPD is encoded by the Ppif gene [167, 168]. CyPD has emerged as central in the mitochondrial proteome. Early work documented an interaction between CyPD and the PTP putative components ANTs, VDACs and TSPO [23], the Pi carrier [169] and the F‐ATP synthase [54, 55]. Additional partners have been discovered including components of the electron transport chain [170], proteins involved in cellular signalling pathways including ERK [171, 172], GSK3β [171, 173] and SIRT3 [174, 175], and proteins involved in stress response pathways including HSP90 and TRAP1 [176]. Interestingly, only some of these interactions are sensitive to CsA, suggesting the existence of multiple binding modes that are yet to be explored [167]. The best‐characterised function of CyPD is sensitisation of the PTP through binding to OSCP [54, 55]. This association requires Pi, which probably acts by charge neutralisation favouring the electrostatic interactions between the two proteins [63]. Through this effect Pi is a PTP inducer [177] despite its lowering effect on the concentration of matrix free Ca2+ [178]. It is remarkable that in yeast and Drosophila mitochondria, where the PTP is insensitive to CsA, Pi is a PTP inhibitor [57, 158, 179]; and that in the absence of CyPD (or in the presence of CsA) Pi becomes an inhibitor also for the PTP of mammalian mitochondria [180].
Getting a crystal of CyPD suitable for X‐ray analysis has been challenging, due to the high water solubility and pI of the protein. A high‐resolution structure was obtained of a K133I mutant of human CyPD lacking the first 13 amino acids. The structure confirmed the high homology of CyPD with other CyPs, which all consist of eight antiparallel β‐sheets, two α‐helices and one 310 helix enclosing the sheets [181]. CyPD does not undergo conformational changes on binding of CsA, an interaction that mainly involves hydrophobic and hydrogen bonds [181]. It is important to note that the unique N‐terminus missing in the structure may impart specific features to the protein, also because this region is the target of many post‐translational modifications.
Post‐translational modifications in pore modulation
CyPD can undergo several post‐translational modifications that alter PTP regulation [167, 168]. CyPD is the target of phosphorylation, with functional effects that depend on the phosphorylated site and may result in opposite outcomes. In mice lacking the mitochondrial Ca2+ uniporter, phosphorylation at S42 increased association of CyPD with F‐ATP synthase resulting in PTP sensitisation [182]. Conversely, PI3K‐ and Akt2‐mediated phosphorylation at S31 preserved mitochondrial function; while expression of the CyPD‐S31A phosphomimetic mutant caused mitochondrial dysfunction consistent with PTP activation [183], as also observed after phosphorylation by GSK3β [171]. By combining in silico analysis of potential CyPD phosphorylation sites for GSK3β with genetic manipulations, a crucial role of CyPD residue S191 has been recently demonstrated in mouse hearts, with phosphorylation increasing CyPD binding to OSCP, increased PTP opening and myocardial damage [184].
CyPD can undergo (de)acetylation reactions, and these modifications significantly affect the PTP [174, 185]. Indeed, in Sirt3−/− mice CyPD is hyperacetylated at K166 (a residue conserved in the human protein) leading to PTP activation and cell death in a model of aortic constriction [174]. Consistently (a) hypoxia increased CyPD acetylation while SIRT3 overexpression was protective in rat heart‐derived H9C2 cells; (b) the acetylation mimic K166Q increased PTP sensitisation and cell death, while the K166R variant had the opposite effect; and (c) cardiac ischaemic postconditioning did not reduce infarct size and CyPD acetylation in mice lacking SIRT3, suggesting that attenuation of CyPD acetylation by SIRT3 could prevent lethal injury at reperfusion [186]. Finally, acetylation of OSCP residue K70 caused by NAD+ redox imbalance promotes interaction of F‐ATP synthase with CyPD and sensitises the PTP to opening in mouse hearts [187].
CyPD contains several Cys residues, some of which (C82 and C104 in the human protein) strongly influence the enzyme’s isomerase activity [188]. Site‐directed mutagenesis identified human C203 as an important redox‐sensitive residue, possibly through formation of a unique disulphide bridge with an yet‐undefined partner [188]. Consistently, mutation of the corresponding mouse C202 to a Ser residue desensitised the PTP in cells [189] and in Langendorff perfused mouse hearts subjected to ischaemia/reperfusion injury, with a matching decreased association of CyPD‐C202S with F‐ATP synthase [190]. CyPD C202 is also the target of S‐nitrosylation [191] and of S‐palmitoylation [190], and it has been proposed that oxidation of C202/3 favours PTP opening leading to ischaemia‐reperfusion damage, while its S‐nitrosylation/acylation confers cardioprotection [190].
Open questions
The progress discussed earlier has been substantial, and yet key open questions remain. These include the role of the OMM, the basis for modulation by quinones and activation by free fatty acids, the assignment of regulatory sulfhydryl residues mediating the effect of oxidants, and the existence of additional permeability pathways.
Outer membrane
The PT is an IMM event as it can take place in mitoplasts, which lack an intact OMM [44]; and yet there is evidence that the OMM (a) is involved in mediating the effect of maleimides because their inducing activity on the PTP is no longer seen in mitoplasts [192] and (b) is required for the inducing effects of photosensitisers like porphyrins [193]. Porphyrins can be excited by visible light to produce singlet oxygen, 1O2, which primarily affects amino acids (Trp, Tyr, His, Cys, Met) causing a variety of alterations in target proteins [194]. Porphyrins (like haematoporphyrin) tend to concentrate in mitochondria [195]. Remarkably, it was known that F‐ATP synthase and ANT are the most vulnerable targets of 1O2 [195, 196]. Photoirradiation of haematoporphyrin‐loaded mitochondria has been used to study the effects of 1O2 on the PTP [197, 198, 199]. At variance from the inducing effect generally observed with oxidants, short (≤ 100 s) irradiation times caused desensitisation of the PTP to Ca2+, the first example of pore inactivation by an oxidant [197]. If the irradiation time was extended to more than 100 s, a process of PTP reactivation ensued that was mediated by the OMM [199], as confirmed by the finding that mitoplasts were completely refractory to this reactivation of the PTP by high light doses [44]. These results are interesting also because they bear on the question of whether the PTP may form at the sites of contact between the OMM and IMM, as was suggested relatively early in the history of the PT [46]. Indeed, in the photoirradiation paradigm it is very unlikely that PTP activation is mediated by a diffusible species because 1O2 is very short‐lived particularly in biological systems, where it has an estimated lifetime of 100‐250 ns and a diffusion distance of the order of 10–20 nm [200, 201].
The major proteins of the OMM are VDACs [202], which have long been suggested to take part in PTP formation [45, 46, 203, 204, 205, 206, 207, 208, 209, 210], an issue that generated considerable discussion [211, 212, 213]. Ro 68‐3400, the first high‐affinity inhibitor of the PTP to be identified by functional screening of a chemical library, labelled a 32 kDa protein originally identified as VDAC1 by mass spectroscopy after hydroxyapatite chromatography [207]. Subsequent studies failed to confirm the identity of the labelled protein as VDAC1, 2 or 3 [50]; and a thorough characterisation of the PTP in mitochondria from VDAC1‐null mice revealed no differences in the effects of a variety of inducers and inhibitors, including CsA and ubiquinone (Ub) 0 [50]. The possible compensatory role of VDAC2 and 3 was ruled out by experiments in cells where all three VDAC isoforms had been genetically ablated [51]. As a result of these studies we think that VDACs cannot be considered essential components of the PTP. However, together with the earlier work mentioned earlier [203, 204, 205, 206, 207, 208, 209, 210], some recent studies are consistent with a modulatory role of VDACs, which may be the targets of signalling pathways and of drugs [214, 215, 216].
A role in the PT has also been described for other proteins that can reside in the OMM such as pro‐ and anti‐apoptotic members of the Bcl‐2 family [47, 48] and hexokinases [212, 217, 218, 219]. In the case of Bcl‐2 family members, the effect may be exerted more on PTP‐dependent swelling than on PTP modulation as such. Indeed, it has been shown that these proteins exert a mechanical effect on OMM resistance to stretching, which is decreased by proapoptotic Bax and Bak and increased by antiapoptotic Bcl‐2 [48]. In the case of hexokinase, the mechanism may instead be linked to Ca2+ diffusion through contact sites between the endoplasmic reticulum and the OMM, because displacement of the protein leads to PTP opening through a large yet localised process of Ca2+ influx [220].
Quinones and lipids
Quinones are among the most interesting modulators of the PT. After the discovery that Ub0 is a potent inhibitor [221], studies were performed to define the structure‐activity relationship of quinones with various side chains [222, 223], see [224] for a review. From these studies, three classes could be identified, that is, PTP inhibitors, PTP inducers and PTP‐inactive quinones that are able to compete with both inhibitors and inducers [224]. The structure‐activity relationship is extremely complex given that seemingly minor structural modifications profoundly affect the effects on the PTP. The most striking examples are perhaps those of decylUb (inhibitor) and OH‐decylUb (inducer); and of the isomers 3‐EtO‐decylUb (inhibitor) and 2‐EtO‐decylUb (inactive). It appears that the 1,4‐benzoquinone ring as such is not sufficient for pore regulation, and that specific substituents at carbons 2, 3, 5 and 6 may be essential for PTP modulation. It is also noteworthy that quinones with very different chemical structures may have similar effects on PTP regulation, suggesting that the spatial conformation may be more important than the nature of the substituents per se. The correlation between quinone structure and effects on the PTP, as well as their target(s), remain elusive although it is clear that neither the redox potential nor the hydrophobicity of the side chain are crucial factors [221, 222, 223, 224]. Overall, data are consistent with the existence of a common binding site, which however remains undefined [224].
A recent study with photoreactive, PTP‐inhibiting derivatives of Ub1 in Saccharomyces cerevisiae mitochondria has shown labelling between residues Phe221 and Lys234 (a segment that connects the 15th and 16th β‐strand sheets) in the C‐terminal region of VDAC1 [215]. No labelled proteins were detected in VDAC1‐null yeast, indicating that the interaction is selective for VDAC1 [215]. Whether quinone‐sensitive PTP opening occurred in yeast mitochondria lacking VDAC1 was not assessed, however, so it cannot be excluded that quinones bind to both VDAC1 and to a component of the IMM involved in the PT.
Another intriguing observation is that the crystallised, 14 subunit c‐ring of spinach chloroplasts resolved at 2.3 Å presents internal electron densities forming circles parallel to the membrane plane, which based on a variety of criteria were attributed to plastoquinones [225]. Similar structures have been observed in the c‐ring of other organisms [226, 227, 228, 229, 230], which could be formed by coenzyme Q in mitochondria and by menaquinone in bacteria [225]. Given the prominent effects of quinones on the PTP and the likely role of the c‐ring in PTP formation [56, 60, 61], it is tempting to speculate that short‐chain quinones may compete for binding with endogenous ubiquinone, affecting in turn the probability of channel formation by the PTP. The lipid plug of the c‐ring might also be the site of action of fatty acids, which have long been known to uncouple oxidative phosphorylation [8, 9, 10] with an effect largely mediated by the PTP [116, 231, 232].
Assigning a site of action to pore modulators
The number and variety of PT modulators remains puzzling. In their still very useful review of 1990, Gunter and Pfeiffer listed 43 classes of compounds and conditions affecting the PT [177], a list that has continued to grow and still represents a challenge to any mechanistic hypothesis to explain the PT. Significant progress was made with the discoveries that the PTP is voltage‐dependent [233] and inhibited by mildly acidic matrix pH [18, 234], and that the voltage threshold for opening is modulated by many agents [235] with a prominent effect of oxidative events affecting the status of critical thiols [236] and of matrix pyridine nucleotides [237]. These findings suggested that many inducers and inhibitors could converge on a smaller number of effector sites determining the stochastic probability of PTP opening [12]. The existence of more than one mechanism for the PT [67, 77] has complicated the picture, but some specific sites of regulation have been convincingly assigned to F‐ATP synthase (a) by genetic ablation or downregulation of subunits OSCP [55, 67], c [61], e and b [67], f [238] and of the bundle region made up by subunits b, e and g [59]; and (b) by site‐directed mutagenesis which led to identification of the Ca2+‐binding site in subunit β [62], of the H+‐sensing His residue in OSCP [63], of the conserved Arg residue modified by glyoxals in subunit g, which may play a role in voltage sensing [239], of the regulatory interaction between one Arg of subunit e and one Glu of subunit g [65], of the OSCP Cys residue responsible for the effects of the dithiol oxidant diamide [66] and of Gly residues involved in channel formation in subunit c [56, 68]. The location of at least two additional classes of Cys residues [115, 237] and of a Me2+‐binding site located on the cytosolic side, the occupancy of which results in PTP inhibition [240], remains to be identified.
Rotenone is a very effective inhibitor of the PTP, as reproducibly observed in succinate‐energised mitochondria [241, 242]. One possible explanation is that it inhibits reverse electron transfer through respiratory complex I, a major source of reactive oxygen species that may overwhelm antioxidant defences particularly in the phase of reperfusion after ischaemia [243]. However, the effects of rotenone and CsA are additive and complementary, in the sense that inhibition by rotenone increases as inhibition by CsA decreases and vice versa, with maximum inhibition being constant [242]. Genetic ablation of CyPD restores PTP inhibition by rotenone in tissues that are otherwise resistant to its effects, suggesting that inhibition by rotenone and CsA occurs through a common, still unidentified site masked by CyPD [242].
Conclusions and perspectives
We think that progress in defining the molecular bases of the PT has been substantial, particularly over the last 10 years; but it is also clear that several key questions still await an answer. The number of pores appears to be very limited compared with the abundance of F‐ATP synthase and ANT [244]. Can all F‐ATP synthase and ANT molecules form a PTP or rather a priming event, possibly a posttranslational modification, is required? Can the pores form anywhere, or are there privileged locations, for example, the interface with the OMM? What is the role of the interactions of F‐ATP synthase and ANT at the ATP synthasomes, given that lack of F‐ATP synthase leads to activation of the ANT channel [67, 77]? Are there additional mechanisms for the PT based on other proteins or molecular species such as polyphosphate [245, 246]? We would also like to mention a very important paper demonstrating that the salt‐ and anoxia‐tolerant brine shrimp Artemia franciscana does not undergo the permeability transition in spite of the presence of both ANT and F‐ATP synthase [247]. We expect that sequence analysis of the corresponding genes will provide an invaluable tool to address the basis for this resistance and to further test the molecular mechanisms of the PTP. Finally, we would also like to report a very elegant paper that appeared at the time of our submission, and that bears on the importance of the PTP in the lifespan of Caenorhabditis elegans. The study demonstrates that genetic ablation of OSCP in adult worms triggers the PTP and shortens lifespan, which can be restored by pharmacological or genetic pore inhibition [248]. We hope that this review will help address the numerous open questions and eventually help solve the 70‐year‐old mystery of the PT.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
All authors contributed to writing of the paper. PB edited the paper and reviewed its final version.
Acknowledgements
Work in our laboratories is supported by grants from Associazione Italiana Ricerca sul Cancro (IG23129), Fondation Leducq (16CVD04), Telethon (GGP17092) and the Italian Ministry for University and Research (2017LHFW42). Open Access Funding provided by Universita degli Studi di Padova within the CRUI‐CARE Agreement. [Correction added on 23 May 2022, after first online publication: CRUI funding statement has been added.]
References
- 1. Hunter DR, Haworth RA & Southard JH (1976) Relationship between configuration, function, and permeability in calcium‐treated mitochondria. J Biol Chem 251, 5069–5077. [PubMed] [Google Scholar]
- 2. Hunter DR & Haworth RA (1979) The Ca2+‐induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195, 453–459. [DOI] [PubMed] [Google Scholar]
- 3. Haworth RA & Hunter DR (1979) The Ca2+‐induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195, 460–467. [DOI] [PubMed] [Google Scholar]
- 4. Hunter DR & Haworth RA (1979) The Ca2+‐induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys 195, 468–477. [DOI] [PubMed] [Google Scholar]
- 5. Raaflaub J (1953) Die schwellung isolierter leberzell mitochondrien und ihre physikalisch beeinflußarkeit. Helv Physiol Pharmacol Acta 11, 142–156. [PubMed] [Google Scholar]
- 6. Raaflaub J (1953) Über den wirkungsmechanismus von adenosintriphosphat (ATP) als cofaktor isolierter mitochondrien. Helv Physiol Pharmacol Acta 11, 157–165. [PubMed] [Google Scholar]
- 7. Massari S & Azzone GF (1972) The equivalent pore radius of intact and damaged mitochondria and the mechanism of active shrinkage. Biochim Biophys Acta 283, 23–29. [DOI] [PubMed] [Google Scholar]
- 8. Lehninger AL & Remmert LF (1959) An endogenous uncoupling and swelling agent in liver mitochondria and its enzymic function. J Biol Chem 234, 2459–2464. [PubMed] [Google Scholar]
- 9. Scarpa A & Lindsay JG (1972) Maintenance of energy‐linked functions in rat liver mitochondria aged in the presence of nupercaine. Eur J Biochem 27, 401–407. [DOI] [PubMed] [Google Scholar]
- 10. Pfeiffer DR, Schmid PC, Beatrice MC & Schmid HH (1979) Intramitochondrial phospholipase activity and the effects of Ca2+ plus N‐ethylmaleimide on mitochondrial function. J Biol Chem 254, 11485–11494. [PubMed] [Google Scholar]
- 11. Mitchell P (1979) Keilin's respiratory chain concept and its chemiosmotic consequences. Science 206, 1148–1159. [DOI] [PubMed] [Google Scholar]
- 12. Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79, 1127–1155. [DOI] [PubMed] [Google Scholar]
- 13. Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly‐Dyson E, Di Lisa F & Forte MA (2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 273, 2077–2099. [DOI] [PubMed] [Google Scholar]
- 14. Bernardi P, Rasola A, Forte M & Lippe G (2015) The mitochondrial permeability transition pore: channel formation by F‐ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 95, 1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kinnally KW, Campo ML & Tedeschi H (1989) Mitochondrial channel activity studied by patch‐clamping mitoplasts. J Bioenerg Biomembr 21, 497–506. [DOI] [PubMed] [Google Scholar]
- 16. Petronilli V, Szabó I & Zoratti M (1989) The inner mitochondrial membrane contains ion‐conducting channels similar to those found in bacteria. FEBS Lett 259, 137–143. [DOI] [PubMed] [Google Scholar]
- 17. Szabó I, Bernardi P & Zoratti M (1992) Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem 267, 2940–2946. [PubMed] [Google Scholar]
- 18. Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabó I & Zoratti M (1992) Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267, 2934–2939. [PubMed] [Google Scholar]
- 19. Szabó I & Zoratti M (2014) Mitochondrial channels: ion fluxes and more. Physiol Rev 94, 519–608. [DOI] [PubMed] [Google Scholar]
- 20. Fournier N, Ducet G & Crevat A (1987) Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr 19, 297–303. [DOI] [PubMed] [Google Scholar]
- 21. Crompton M, Ellinger H & Costi A (1988) Inhibition by cyclosporin A of a Ca2+‐dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255, 357–360. [PMC free article] [PubMed] [Google Scholar]
- 22. Broekemeier KM, Dempsey ME & Pfeiffer DR (1989) Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem 264, 7826–7830. [PubMed] [Google Scholar]
- 23. Halestrap AP & Davidson AM (1990) Inhibition of Ca2+‐induced large‐amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial‐matrix peptidyl‐prolyl cis‐trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J 268, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Broekemeier KM, Carpenter Deyo L, Reed DJ & Pfeiffer DR (1992) Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett 304, 192–194. [DOI] [PubMed] [Google Scholar]
- 25. Imberti R, Nieminen AL, Herman B & Lemasters JJ (1992) Synergism of cyclosporin A and phospholipase inhibitors in protection against lethal injury to rat hepatocytes from oxidant chemicals. Res Commun Chem Pathol Pharmacol 78, 27–38. [PubMed] [Google Scholar]
- 26. Imberti R, Nieminen AL, Herman B & Lemasters JJ (1993) Mitochondrial and glycolytic dysfunction in lethal injury to hepatocytes by t‐butylhydroperoxide: protection by fructose, cyclosporin A and trifluoperazine. J Pharmacol Exp Ther 265, 392–400. [PubMed] [Google Scholar]
- 27. Duchen MR, McGuinness O, Brown LA & Crompton M (1993) On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res 27, 1790–1794. [DOI] [PubMed] [Google Scholar]
- 28. Griffiths EJ & Halestrap AP (1993) Protection by Cyclosporin A of ischemia/reperfusion‐induced damage in isolated rat hearts. J Mol Cell Cardiol 25, 1461–1469. [DOI] [PubMed] [Google Scholar]
- 29. Broekemeier KM, Schmid PC, Dempsey ME & Pfeiffer DR (1991) Generation of the mitochondrial permeability transition does not involve inhibition of lysophospholipid acylation. Acyl‐coenzyme A: 1‐acyllysophospholipid acyltransferase activity is not found in rat liver mitochondria. J Biol Chem 266, 20700–20708. [PubMed] [Google Scholar]
- 30. Nicolli A, Basso E, Petronilli V, Wenger RM & Bernardi P (1996) Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A‐sensitive channel. J Biol Chem 271, 2185–2192. [DOI] [PubMed] [Google Scholar]
- 31. Woodfield KY, Price NT & Halestrap AP (1997) cDNA cloning of rat mitochondrial cyclophilin. Biochim Biophys Acta 1351, 27–30. [DOI] [PubMed] [Google Scholar]
- 32. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW et al. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. [DOI] [PubMed] [Google Scholar]
- 33. Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA & Bernardi P (2005) Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem 280, 18558–18561. [DOI] [PubMed] [Google Scholar]
- 34. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T & Tsujimoto Y (2005) Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658. [DOI] [PubMed] [Google Scholar]
- 35. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA & Korsmeyer SJ (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102, 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Broekemeier KM & Pfeiffer DR (1989) Cyclosporin A‐sensitive and insensitive mechanisms produce the permeability transition in mitochondria. Biochem Biophys Res Commun 163, 561–566. [DOI] [PubMed] [Google Scholar]
- 37. Broekemeier KM & Pfeiffer DR (1995) Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry 34, 16440–16449. [DOI] [PubMed] [Google Scholar]
- 38. Brustovetsky N & Klingenberg M (1996) Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ . Biochemistry 35, 8483–8488. [DOI] [PubMed] [Google Scholar]
- 39. Brustovetsky N, Tropschug M, Heimpel S, Heidkamper D & Klingenberg M (2002) A large Ca2+‐dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry 41, 11804–11811. [DOI] [PubMed] [Google Scholar]
- 40. Szabó I & Zoratti M (1991) The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem 266, 3376–3379. [PubMed] [Google Scholar]
- 41. Szabó I & Zoratti M (1992) The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr 24, 111–117. [DOI] [PubMed] [Google Scholar]
- 42. McEnery MW, Snowman AM, Trifiletti RR & Snyder SH (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage‐dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA 89, 3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW & Tedeschi H (1993) Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci USA 90, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Šileikyte J, Petronilli V, Zulian A, Dabbeni‐Sala F, Tognon G, Nikolov P, Bernardi P & Ricchelli F (2011) Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J Biol Chem 286, 1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beutner G, Rück A, Riede B, Welte W & Brdiczka D (1996) Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett 396, 189–195. [DOI] [PubMed] [Google Scholar]
- 46. Beutner G, Rück A, Riede B & Brdiczka D (1998) Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim Biophys Acta 1368, 7–18. [DOI] [PubMed] [Google Scholar]
- 47. Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC & Kroemer G (1998) The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl‐2‐related proteins. J Exp Med 187, 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez‐Caballero S, Osinska H, Cheng EH, Robbins J et al. (2013) Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife Sciences 2, e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR & Wallace DC (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krauskopf A, Eriksson O, Craigen WJ, Forte MA & Bernardi P (2006) Properties of the permeability transition in VDAC1 ‐/‐ mitochondria. Biochim Biophys Acta 1757, 590–595. [DOI] [PubMed] [Google Scholar]
- 51. Baines CP, Kaiser RA, Sheiko T, Craigen WJ & Molkentin JD (2007) Voltage‐dependent anion channels are dispensable for mitochondrial‐dependent cell death. Nat Cell Biol 9, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Šileikyte J, Blachly‐Dyson E, Sewell R, Carpi A, Menabò R, Di Lisa F, Ricchelli F, Bernardi P & Forte M (2014) Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J Biol Chem 289, 13769–13781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM & Molkentin JD (2019) Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv 5, eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Giorgio V, Bisetto E, Soriano ME, Dabbeni‐Sala F, Basso E, Petronilli V, Forte MA, Bernardi P & Lippe G (2009) Cyclophilin D modulates mitochondrial F0F1‐ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284, 33982–33988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I et al. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110, 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M et al. (2014) An uncoupling channel within the c‐subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 111, 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carraro M, Giorgio V, Šileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabó I & Bernardi P (2014) Channel formation by yeast F‐ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 289, 15980–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. von Stockum S, Giorgio V, Trevisan E, Lippe G, Glick GD, Forte MA, Da‐Rè C, Checchetto V, Mazzotta G, Costa R et al. (2015) F‐ATPase of D. melanogaster Forms 53 Picosiemen (53‐pS) Channels Responsible for Mitochondrial Ca2+‐induced Ca2+ Release. J Biol Chem 290, 4537–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carraro M, Checchetto V, Sartori G, Kucharczyk R, di Rago J‐P, Minervini G, Franchin C, Arrigoni G, Giorgio V, Petronilli V et al. (2018) High‐conductance channel formation in yeast mitochondria is mediated by F‐ATP synthase e and g subunits. Cell Physiol Biochem 50, 1840–1855. [DOI] [PubMed] [Google Scholar]
- 60. Amodeo GF, Lee BY, Krilyuk N, Filice CT, Valyuk D, Otzen DE, Noskov S, Leonenko Z & Pavlov EV (2021) C subunit of the ATP synthase is an amyloidogenic calcium dependent channel‐forming peptide with possible implications in mitochondrial permeability transition. Sci Rep 11, 8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A et al. (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, Petronilli V, Argenton F, Forte M, Tosatto S, Lippe G et al. (2017) Ca2+ binding to F‐ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep 18, 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Antoniel M, Jones K, Antonucci S, Spolaore B, Fogolari F, Petronilli V, Giorgio V, Carraro M, Di Lisa F, Forte M et al. (2018) The unique histidine in OSCP subunit of F‐ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep 19, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guo L, Carraro M, Sartori G, Minervini G, Eriksson O, Petronilli V & Bernardi P (2018) Arginine 107 of yeast ATP synthase subunit g mediates sensitivity of the mitochondrial permeability transition to phenylglyoxal. J Biol Chem 293, 14632–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guo L, Carraro M, Carrer A, Minervini G, Urbani A, Masgras I, Tosatto SCE, Szabó I, Bernardi P & Lippe G (2019) Arg‐8 of yeast subunit e contributes to the stability of F‐ATP synthase dimers and to the generation of the full‐conductance mitochondrial megachannel. J Biol Chem 294, 10987–10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Carraro M, Jones K, Sartori G, Schiavone M, Antonucci S, Kucharczyk R, di Rago J‐P, Franchin C, Arrigoni G, Forte M et al. (2020) The unique cysteine of F‐ATP Synthase OSCP subunit participates in modulation of the permeability transition pore. Cell Rep 32, 108095. [DOI] [PubMed] [Google Scholar]
- 67. Carrer A, Tommasin L, Šileikyte J, Ciscato F, Filadi R, Urbani A, Forte E, Rasola A, Szabò I, Carraro M et al. (2021) Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F‐ATP synthase. Nat Commun 12, 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morciano G, Pedriali G, Bonora M, Pavasini R, Mikus E, Calvi S, Bovolenta M, Lebiedzinska‐Arciszewska M, Pinotti M, Albertini A et al. (2021) A naturally occurring mutation in ATP synthase subunit c is associated with increased damage following hypoxia/reoxygenation in STEMI patients. Cell Rep 35, 108983. [DOI] [PubMed] [Google Scholar]
- 69. Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa‐Itoh K, Bogers JFM et al. (2019) Purified F‐ATP synthase forms a Ca2+‐dependent high‐conductance channel matching the mitochondrial permeability transition pore. Nat Commun 10, 4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ & Jonas EA (2019) A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat Commun 10, 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Johnson KM, Chen X, Boitano A, Swenson L, Opipari AW Jr & Glick GD (2005) Identification and validation of the mitochondrial F1F0‐ATPase as the molecular target of the immunomodulatory benzodiazepine Bz‐423. Chem Biol 12, 485–496. [DOI] [PubMed] [Google Scholar]
- 72. Cleary J, Johnson KM, Opipari AW Jr & Glick GD (2007) Inhibition of the mitochondrial F1F0‐ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg Med Chem Lett 17, 1667–1670. [DOI] [PubMed] [Google Scholar]
- 73. Stelzer AC, Frazee RW, Van Huis C, Cleary J, Opipari AW Jr, Glick GD & Al‐Hashimi HM (2010) NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F1F0‐ATPase. Biopolymers 93, 85–92. [DOI] [PubMed] [Google Scholar]
- 74. He J, Ford HC, Carroll J, Ding S, Fearnley IM & Walker JE (2017) Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci USA 114, 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. He J, Carroll J, Ding S, Fearnley IM & Walker JE (2017) Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci USA 114, 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Carroll J, He J, Ding S, Fearnley IM & Walker JE (2019) Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc Natl Acad Sci USA 116, 12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA & Pavlov EV (2019) ATP Synthase C‐Subunit‐deficient mitochondria have a small cyclosporine a‐sensitive channel, but lack the permeability transition pore. Cell Rep 26, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ko YH, Delannoy M, Hullihen J, Chiu W & Pedersen PL (2003) Mitochondrial ATP Synthasome. Cristae‐enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J Biol Chem 278, 12305. [DOI] [PubMed] [Google Scholar]
- 79. Nusková H, Mrácek T, Mikulová T, Vrbacký M, Kovárová N, Kovalcíková J, Pecina P & Houštek J (2015) Mitochondrial ATP synthasome: Expression and structural interaction of its components. Biochem Biophys Res Commun 464, 787–793. [DOI] [PubMed] [Google Scholar]
- 80. Beutner G, Alanzalon RE & Porter GA Jr (2017) Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci Rep 7, 14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rasola A & Bernardi P (2014) The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 56, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Halestrap AP & Richardson AP (2015) The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78, 129–141. [DOI] [PubMed] [Google Scholar]
- 83. Šileikyte J & Forte M (2016) Shutting down the pore: The search for small molecule inhibitors of the mitochondrial permeability transition. Biochim Biophys Acta 1857, 1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zulian A, Schiavone M, Giorgio V & Bernardi P (2016) Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol Res 113, 563–573. [DOI] [PubMed] [Google Scholar]
- 85. Quintanilla RA, Tapia C & Pérez MJ (2017) Possible role of mitochondrial permeability transition pore in the pathogenesis of Huntington disease. Biochem Biophys Res Commun 483, 1078–1083. [DOI] [PubMed] [Google Scholar]
- 86. Britti E, Delaspre F, Tamarit J & Ros J (2018) Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal 2, 20180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Briston T, Selwood DL, Szabadkai G & Duchen MR (2019) Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends Pharmacol Sci 40, 50–70. [DOI] [PubMed] [Google Scholar]
- 88. Belosludtsev KN, Belosludtseva NV & Dubinin MV (2020) Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+‐Dependent Permeability Transition Pore. Int J Mol Sci 21, 6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Esteras N & Abramov AY (2020) Mitochondrial calcium deregulation in the mechanism of beta‐amyloid and tau pathology. Cells 9, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jia K & Du H (2021) Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer's Disease. Cells 10, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rottenberg H & Hoek JB (2021) The mitochondrial permeability transition: nexus of aging. Disease and Longevity. Cells 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhao L, Tang M, Bode AM, Liao W & Cao Y (2021) ANTs and cancer: Emerging pathogenesis, mechanisms, and perspectives. Biochim Biophys Acta Rev Cancer 1875, 188485. [DOI] [PubMed] [Google Scholar]
- 93. Duee ED & Vignais PV (1965) Éxchange entre adenine‐nucleotides extra‐ et intramitochondriaux. Biochim Biophys Acta 107, 184–188. [DOI] [PubMed] [Google Scholar]
- 94. Heldt HW & Klingenberg M (1965) Endogenous nucleotides of mitochondria participating in phosphate transfer reactions as studied with 32P labelled orthophosphate and ultramicro scale ion exchange chromatography. Biochem Z 343, 433–451. [PubMed] [Google Scholar]
- 95. Stepien G, Torroni A, Chung AB, Hodge JA & Wallace DC (1992) Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem 267, 14592–14597. [PubMed] [Google Scholar]
- 96. Rodic N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, Maatouk DM, Resnick JL, Li E & Terada N (2005) DNA methylation is required for silencing of Ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 23, 1314–1323. [DOI] [PubMed] [Google Scholar]
- 97. Monné M & Palmieri F (2014) Antiporters of the mitochondrial carrier family. Curr Top Membr 73, 289–320. [DOI] [PubMed] [Google Scholar]
- 98. Klingenberg M (1979) The ADP, ATP shuttle of the mitochondrion. Trends Biochem Sci 4, 249–252. [Google Scholar]
- 99. Ruprecht JJ & Kunji ERS (2021) Structural Mechanism of Transport of Mitochondrial Carriers. Annu Rev Biochem 90, 535–558. [DOI] [PubMed] [Google Scholar]
- 100. Bruni A & Luciani S (1962) Effects of atractyloside and oligomycin on magnesium‐stimulated adenosine triphosphatase and on adenosine triphosphate‐induced contraction of swollen mitochondria. Nature 196, 578–580. [DOI] [PubMed] [Google Scholar]
- 101. Henderson PJ & Lardy HA (1970) Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem 245, 1319–1326. [PubMed] [Google Scholar]
- 102. Ruprecht JJ & Kunji ER (2019) Structural changes in the transport cycle of the mitochondrial ADP/ATP carrier. Curr Opin Struct Biol 57, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pebay‐Peyroula E, Dahout‐Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ & Brandolin G (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426, 39–44. [DOI] [PubMed] [Google Scholar]
- 104. Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ & Kunji ER (2014) Structures of yeast mitochondrial ADP/ATP carriers support a domain‐based alternating‐access transport mechanism. Proc Natl Acad Sci USA 111, E426–E434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ruprecht JJ, King MS, Zogg T, Aleksandrova AA, Pardon E, Crichton PG, Steyaert J & Kunji ERS (2019) The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell 176, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nicholls DG (1976) Hamster brown‐adipose‐tissue mitochondria. Purine nucleotide control of the ion conductance of the inner membrane, the nature of the nucleotide binding site. Eur J Biochem 62, 223–228. [DOI] [PubMed] [Google Scholar]
- 107. Rafael J & Heldt HW (1976) Binding of guanine nucleotides to the outer surface of the inner membrane of guinea pig brown fat mitochondria in correlation with the thermogenic activity of the tissue. FEBS Lett 63, 304–308. [DOI] [PubMed] [Google Scholar]
- 108. Fedorenko A, Lishko PV & Kirichok Y (2012) Mechanism of fatty‐acid‐dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gonzalez‐Barroso MM, Fleury C, Levi‐Meyrueis C, Zaragoza P, Bouillaud F & Rial E (1997) Deletion of amino acids 261–269 in the brown fat uncoupling protein converts the carrier into a pore. Biochemistry 36, 10930–10935. [DOI] [PubMed] [Google Scholar]
- 110. Andreyev AY, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP & Volkov NI (1988) Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett 226, 265–269. [DOI] [PubMed] [Google Scholar]
- 111. Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, Vidoni S, Garrity R, Cho J, Terada N et al. (2019) H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 571, 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Beyer K & Klingenberg M (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24, 3821–3826. [DOI] [PubMed] [Google Scholar]
- 113. Costantini P, Belzacq AS, La VH, Larochette N, de Pablo M , Zamzami N, Susin SA, Brenner C & Kroemer G (2000) Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl‐2‐independent permeability transition pore opening and apoptosis. Oncogene 19, 307–314. [DOI] [PubMed] [Google Scholar]
- 114. McStay GP, Clarke SJ & Halestrap AP (2002) Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J 367, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Costantini P, Colonna R & Bernardi P (1998) Induction of the mitochondrial permeability transition by N‐ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper‐ortho‐phenanthroline without dimerization of the adenine nucleotide translocase. Biochim Biophys Acta 1365, 385–392. [DOI] [PubMed] [Google Scholar]
- 116. Penzo D, Tagliapietra C, Colonna R, Petronilli V & Bernardi P (2002) Effects of fatty acids on mitochondria: implications for cell death. Biochim Biophys Acta 1555, 160–165. [DOI] [PubMed] [Google Scholar]
- 117. Kühlbrandt W (2019) Structure and Mechanisms of F‐Type ATP Synthases. Annu Rev Biochem 88, 515–549. [DOI] [PubMed] [Google Scholar]
- 118. Abrahams JP, Leslie AG, Lutter R & Walker JE (1994) Structure at 2.8 Å resolution of F1‐ATPase from bovine heart mitochondria. Nature 370, 621–628. [DOI] [PubMed] [Google Scholar]
- 119. Rees DM, Leslie AG & Walker JE (2009) The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci USA 106, 21597–21601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Stock D, Leslie AG & Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700–1705. [DOI] [PubMed] [Google Scholar]
- 121. Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W & Meier T (2016) Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol Cell 63, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Srivastava AP, Luo M, Zhou W, Symersky J, Bai D, Chambers MG, Faraldo‐Gomez JD, Liao M & Mueller DM (2018) High‐resolution cryo‐EM analysis of the yeast ATP synthase in a lipid membrane. Science 360, eaas9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gu J, Zhang L, Zong S, Guo R, Liu T, Yi J, Wang P, Zhuo W & Yang M (2019) Cryo‐EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science 364, 1068–1075. [DOI] [PubMed] [Google Scholar]
- 124. Spikes TE, Montgomery MG & Walker JE (2020) Structure of the dimeric ATP synthase from bovine mitochondria. Proc Natl Acad Sci USA 117, 23519–23526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pinke G, Zhou L & Sazanov LA (2020) Cryo‐EM structure of the entire mammalian F‐type ATP synthase. Nat Struct Mol Biol 27, 1077–1085. [DOI] [PubMed] [Google Scholar]
- 126. Watt IN, Montgomery MG, Runswick MJ, Leslie AG & Walker JE (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA 107, 16823–16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Noji H, Ueno H & McMillan DGG (2017) Catalytic robustness and torque generation of the F1‐ATPase. Biophys Rev 9, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nesci S, Trombetti F, Ventrella V, Pirini M & Pagliarani A (2017) Kinetic properties of the mitochondrial F1FO‐ATPase activity elicited by Ca2+ in replacement of Mg2+ . Biochimie 140, 73–81. [DOI] [PubMed] [Google Scholar]
- 129. Papageorgiou S, Melandri AB & Solaini G (1998) Relevance of divalent cations to ATP‐driven proton pumping in beef heart mitochondrial F0F1‐ATPase. J Bioenerg Biomembr 30, 533–541. [DOI] [PubMed] [Google Scholar]
- 130. De Col V, Petrussa E, Casolo V, Braidot E, Lippe G, Filippi A, Peresson C, Patui S, Bertolini A, Giorgio V et al. (2018) Properties of the permeability transition of pea stem mitochondria. Front Physiol 9, 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tucker WC, Schwarz A, Levine T, Du Z, Gromet‐Elhanan Z, Richter ML & Haran G (2004) Observation of calcium‐dependent unidirectional rotational motion in recombinant photosynthetic F1‐ATPase molecules. J Biol Chem 279, 47415–47418. [DOI] [PubMed] [Google Scholar]
- 132. Zhou A, Rohou A, Schep DG, Bason JV, Montgomery MG, Walker JE, Grigorieff N & Rubinstein JL (2015) Structure and conformational states of the bovine mitochondrial ATP synthase by cryo‐EM. eLife Sciences 4, e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Soga N, Kimura K, Kinosita K Jr, Yoshida M & Suzuki T (2017) Perfect chemomechanical coupling of FoF1‐ATP synthase. Proc Natl Acad Sci USA 114, 4960–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Murphy BJ, Klusch N, Langer J, Mills DJ, Yildiz Ö & Kühlbrandt W (2019) Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1‐Fo coupling. Science 364, eaaw9128. [DOI] [PubMed] [Google Scholar]
- 135. Giorgio V, Fogolari F, Lippe G & Bernardi P (2019) OSCP subunit of mitochondrial ATP synthase: Role in regulation of enzyme function and of its transition to a pore. Br J Pharmacol 176, 4247–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Allen RD (1995) Membrane tubulation and proton pumps. Protoplasma 189, 1–8. [Google Scholar]
- 137. Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brèthes D, di Rago JP & Velours J (2002) The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cogliati S, Enriquez JA & Scorrano L (2016) Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41, 261–273. [DOI] [PubMed] [Google Scholar]
- 139. Habersetzer J, Ziani W, Larrieu I, Stines‐Chaumeil C, Giraud MF, Brèthes D, Dautant A & Paumard P (2013) ATP synthase oligomerization: from the enzyme models to the mitochondrial morphology. Int J Biochem Cell Biol 45, 99–105. [DOI] [PubMed] [Google Scholar]
- 140. Spikes TE, Montgomery MG & Walker JE (2021) Interface mobility between monomers in dimeric bovine ATP synthase participates in the ultrastructure of inner mitochondrial membranes. Proc Natl Acad Sci USA 118, e2021012118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Salewskij K, Rieger B, Hager F, Arroum T, Duwe P, Villalta J, Cogliati S, Richter CP, Psathaki OE, Enriquez JA et al. (2020) The spatio‐temporal organization of mitochondrial F1FO ATP synthase in cristae depends on its activity mode. Biochim Biophys Acta 1861, 148091. [DOI] [PubMed] [Google Scholar]
- 142. Quintana‐Cabrera R, Quirin C, Glytsou C, Corrado M, Urbani A, Pellattiero A, Calvo E, Vazquez J, Enriquez JA, Gerle C et al. (2018) The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat Commun 9, 3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gerle C (2016) On the structural possibility of pore‐forming mitochondrial FoF1 ATP synthase. Biochim Biophys Acta 1857, 1191–1196. [DOI] [PubMed] [Google Scholar]
- 144. Gerle C (2020) Mitochondrial F‐ATP synthase as the permeability transition pore. Pharmacol Res 160, 105081. [DOI] [PubMed] [Google Scholar]
- 145. Petronilli V, Nicolli A, Costantini P, Colonna R & Bernardi P (1994) Regulation of the permeability transition pore, a voltage‐dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta 1187, 255–259. [DOI] [PubMed] [Google Scholar]
- 146. Hüser J, Rechenmacher CE & Blatter LA (1998) Imaging the permeability pore transition in single mitochondria. Biophys J 74, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hüser J & Blatter LA (1999) Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J 343, 311–317. [PMC free article] [PubMed] [Google Scholar]
- 148. Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P & Di Lisa F (1999) Transient and long‐lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sambri I, Massa F, Gullo F, Meneghini S, Cassina L, Patanella L, Carissimo A, Iuliano A, Santorelli F, Codazzi F et al. (2020) Impaired flickering of the permeability transition pore causes SPG7 spastic paraplegia. EBioMed 61, 103050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bonora M, Morganti C, Morciano G, Pedriali G, Lebiedzinska‐Arciszewska M, Aquila G, Giorgi C, Rizzo P, Campo G, Ferrari R et al. (2017) Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C‐ring conformation. EMBO Rep 18, 1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Yoshida M, Muneyuki E & Hisabori T (2001) ATP synthase–a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2, 669–677. [DOI] [PubMed] [Google Scholar]
- 152. Arnold I, Pfeiffer K, Neupert W, Stuart RA & Schägger H (1998) Yeast mitochondrial F1F0‐ATP synthase exists as a dimer: identification of three dimer‐specific subunits. EMBO J 17, 7170–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Arselin G, Vaillier J, Salin B, Schaeffer J, Giraud MF, Dautant A, Brèthes D & Velours J (2004) The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J Biol Chem 279, 40392–40399. [DOI] [PubMed] [Google Scholar]
- 154. Vinothkumar KR, Montgomery MG, Liu S & Walker JE (2016) Structure of the mitochondrial ATP synthase from Pichia angusta determined by electron cryo‐microscopy. Proc Natl Acad Sci USA 113, 12709–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bohnert M, Zerbes RM, Davies KM, Muhleip AW, Rampelt H, Horvath SE, Boenke T, Kram A, Perschil I, Veenhuis M et al. (2015) Central role of Mic10 in the mitochondrial contact site and cristae organizing system. Cell Metab 21, 747–755. [DOI] [PubMed] [Google Scholar]
- 156. Guo H, Bueler SA & Rubinstein JL (2017) Atomic model for the dimeric FO region of mitochondrial ATP synthase. Science 358, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA & Bernardi P (2010) The mitochondrial permeability transition from yeast to mammals. FEBS Lett 584, 2504–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. von Stockum S, Basso E, Petronilli V, Sabatelli P, Forte MA & Bernardi P (2011) Properties of Ca2+ Transport in Mitochondria of Drosophila melanogaster . J Biol Chem 286, 41163–41170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Allen RD, Schroeder CC & Fok AK (1989) An investigation of mitochondrial inner membranes by rapid‐freeze deep‐etch techniques. J Cell Biol 108, 2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. He J, Carroll J, Ding S, Fearnley IM, Montgomery MG & Walker JE (2020) Assembly of the peripheral stalk of ATP synthase in human mitochondria. Proc Natl Acad Sci USA 117, 29602–29608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Dudkina NV, Sunderhaus S, Braun HP & Boekema EJ (2006) Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett 580, 3427–3432. [DOI] [PubMed] [Google Scholar]
- 162. Wittig I & Schägger H (2008) Structural organization of mitochondrial ATP synthase. Biochim Biophys Acta 1777, 592–598. [DOI] [PubMed] [Google Scholar]
- 163. Wittig I, Braun HP & Schägger H (2006) Blue native PAGE. Nat Protoc 1, 418–428. [DOI] [PubMed] [Google Scholar]
- 164. Wittig I, Karas M & Schägger H (2007) High resolution clear native electrophoresis for in‐gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics 6, 1215–1225. [DOI] [PubMed] [Google Scholar]
- 165. Meyer B, Wittig I, Trifilieff E, Karas M & Schägger H (2007) Identification of two proteins associated with mammalian ATP synthase. Mol Cell Proteomics 6, 1690–1699. [DOI] [PubMed] [Google Scholar]
- 166. Lutter R, Saraste M, Van Walraven HS, Runswick MJ, Finel M, Deatherage JF & Walker JE (1993) F1F0‐ATP synthase from bovine heart mitochondria: development of the purification of a monodisperse oligomycin‐sensitive ATPase. Biochem J 295, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Porter GA Jr & Beutner G (2018) Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 8, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Amanakis G & Murphy E (2020) Cyclophilin D: an integrator of mitochondrial function. Front Physiol 11, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Leung AW, Varanyuwatana P & Halestrap AP (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 283, 26312–26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Etzler JC, Bollo M, Holstein D, Deng JJ, Perez V, Lin DT, Richardson A, Bai Y & Lechleiter JD (2017) Cyclophilin D over‐expression increases mitochondrial complex III activity and accelerates supercomplex formation. Arch Biochem Biophys 613, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS & Bernardi P (2010) Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci USA 107, 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bigi A, Beltrami E, Trinei M, Stendardo M, Pelicci PG & Giorgio M (2016) Cyclophilin D counteracts P53‐mediated growth arrest and promotes Ras tumorigenesis. Oncogene 35, 5132–5143. [DOI] [PubMed] [Google Scholar]
- 173. Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL et al. (2004) Glycogen synthase kinase‐3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113, 1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A & Sinclair DA (2010) Regulation of the mPTP by SIRT3‐mediated deacetylation of CypD at lysine 166 suppresses age‐related cardiac hypertrophy. Aging (Albany NY) 2, 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Li P, Meng X, Bian H, Burns N, Ks Z & Song R (2015) Activation of sirtuin 1/3 improves vascular hyporeactivity in severe hemorrhagic shock by alleviation of mitochondrial damage. Oncotarget 6, 36998–37011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ & Altieri DC (2007) Regulation of tumor cell mitochondrial homeostasis by an organelle‐specific Hsp90 chaperone network. Cell 131, 257–270. [DOI] [PubMed] [Google Scholar]
- 177. Gunter TE & Pfeiffer DR (1990) Mechanisms by which mitochondria transport calcium. Am J Physiol 258, C755–C786. [DOI] [PubMed] [Google Scholar]
- 178. Chalmers S & Nicholls DG (2003) The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 278, 19062–19070. [DOI] [PubMed] [Google Scholar]
- 179. Jung DW, Bradshaw PC & Pfeiffer DR (1997) Properties of a cyclosporin‐insensitive permeability transition pore in yeast mitochondria. J Biol Chem 272, 21104–21112. [DOI] [PubMed] [Google Scholar]
- 180. Basso E, Petronilli V, Forte MA & Bernardi P (2008) Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem 283, 26307–26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kajitani K, Fujihashi M, Kobayashi Y, Shimizu S, Tsujimoto Y & Miki K (2008) Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96‐Å resolution. Proteins 70, 1635–1639. [DOI] [PubMed] [Google Scholar]
- 182. Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T & Murphy E (2019) Cyclophilin D‐mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res 115, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC et al. (2015) Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 107, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hurst S, Gonnot F, Dia M, Crola Da Silva C, Gomez L & Sheu SS (2020) Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia‐reperfusion. Cell Death Dis 11, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Nguyen TT, Wong R, Menazza S, Sun J, Chen Y, Wang G, Gucek M, Steenbergen C, Sack MN & Murphy E (2013) Cyclophilin D modulates mitochondrial acetylome. Circ Res 113, 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Bochaton T, Crola‐Da‐Silva C, Pillot B, Villedieu C, Ferreras L, Alam MR, Thibault H, Strina M, Gharib A, Ovize M et al. (2015) Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3‐mediated deacetylation of cyclophilin D. J Mol Cell Cardiol 84, 61–69. [DOI] [PubMed] [Google Scholar]
- 187. Lee CF, Chavez JD, Garcia‐Menendez L, Choi YS, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE et al. (2016) Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Linard D, Kandlbinder A, Degand H, Morsomme P, Dietz KJ & Knoops B (2009) Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Arch Biochem Biophys 491, 39–45. [DOI] [PubMed] [Google Scholar]
- 189. Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN & Murphy E (2011) Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 286, 40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Amanakis G, Sun J, Fergusson MM, McGinty S, Liu C, Molkentin JD & Murphy E (2021) Cysteine 202 of Cyclophilin D is a site of multiple post‐translational modifications and plays a role in cardioprotection. Cardiovasc Res 117, 212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M & Steenbergen C (2011) Characterization of potential S‐nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol 300, H1327–H1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Lê‐Quôc K & Lê‐Quôc D (1985) Crucial role of sulfhydryl groups in the mitochondrial inner membrane structure. J Biol Chem 260, 7422–7428. [PubMed] [Google Scholar]
- 193. Ricchelli F, Šileikyte J & Bernardi P (2011) Shedding light on the mitochondrial permeability transition. Biochim Biophys Acta 1807, 482–490. [DOI] [PubMed] [Google Scholar]
- 194. Davies MJ (2003) Singlet oxygen‐mediated damage to proteins and its consequences. Biochem Biophys Res Commun 305, 761–770. [DOI] [PubMed] [Google Scholar]
- 195. Hilf R (2007) Mitochondria are targets of photodynamic therapy. J Bioenerg Biomembr 39, 85–89. [DOI] [PubMed] [Google Scholar]
- 196. Atlante A, Passarella S, Quagliariello E, Moreno G & Salet C (1989) Haematoporphyrin derivative (Photofrin II) photosensitization of isolated mitochondria: inhibition of ADP/ATP translocator. J Photochem Photobiol B 4, 35–46. [DOI] [PubMed] [Google Scholar]
- 197. Salet C, Moreno G, Ricchelli F & Bernardi P (1997) Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J Biol Chem 272, 21938–21943. [DOI] [PubMed] [Google Scholar]
- 198. Moreno G, Poussin K, Ricchelli F & Salet C (2001) The effects of singlet oxygen produced by photodynamic action on the mitochondrial permeability transition differ in accordance with the localization of the sensitizer. Arch Biochem Biophys 386, 243–250. [DOI] [PubMed] [Google Scholar]
- 199. Petronilli V, Šileikyte J, Zulian A, Dabbeni‐Sala F, Jori G, Gobbo S, Tognon G, Nikolov P, Bernardi P & Ricchelli F (2009) Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin‐mediated photooxidative stress. Unmasking pore‐regulating external thiols. Biochim Biophys Acta 1787, 897–904. [DOI] [PubMed] [Google Scholar]
- 200. Moan J (1990) On the diffusion length of singlet oxygen in cells and tissues. J Photochem Photobiol B: Biol 6, 334–343. [Google Scholar]
- 201. Moan J & Berg K (1991) The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol 53, 549–553. [DOI] [PubMed] [Google Scholar]
- 202. De Pinto V (2021) Renaissance of VDAC: new insights on a protein family at the interface between mitochondria and cytosol. Biomolecules 11, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Szabó I & Zoratti M (1993) The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett 330, 201–205. [DOI] [PubMed] [Google Scholar]
- 204. Szabó I, De Pinto V & Zoratti M (1993) The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 330, 206–210. [DOI] [PubMed] [Google Scholar]
- 205. Priault M, Chaudhuri B, Clow A, Camougrand N & Manon S (1999) Investigation of bax‐induced release of cytochrome c from yeast mitochondria permeability of mitochondrial membranes, role of VDAC and ATP requirement. Eur J Biochem 260, 684–691. [DOI] [PubMed] [Google Scholar]
- 206. Shimizu S, Shinohara Y & Tsujimoto Y (2000) Bax and Bcl‐xL independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene 19, 4309–4318. [DOI] [PubMed] [Google Scholar]
- 207. Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P & Kemp JA (2003) The voltage‐dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem 278, 49812–49818. [DOI] [PubMed] [Google Scholar]
- 208. Gincel D & Shoshan‐Barmatz V (2004) Glutamate interacts with VDAC and modulates opening of the mitochondrial permeability transition pore. J Bioenerg Biomembr 36, 179–186. [DOI] [PubMed] [Google Scholar]
- 209. Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H & Chen Q (2004) Essential role of the voltage‐dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 23, 1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Gutierrez‐Aguilar M, Perez‐Vazquez V, Bunoust O, Manon S, Rigoulet M & Uribe S (2007) In yeast, Ca2+ and octylguanidine interact with porin (VDAC) preventing the mitochondrial permeability transition. Biochim Biophys Acta 1767, 1245–1251. [DOI] [PubMed] [Google Scholar]
- 211. Halestrap AP, McStay GP & Clarke SJ (2002) The permeability transition pore complex: another view. Biochimie 84, 153–166. [DOI] [PubMed] [Google Scholar]
- 212. Azoulay‐Zohar H, Israelson A, Abu‐Hamad S & Shoshan‐Barmatz V (2004) In self‐defence: hexokinase promotes voltage‐dependent anion channel closure and prevents mitochondria‐mediated apoptotic cell death. Biochem J 377, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Rostovtseva TK, Tan W & Colombini M (2005) On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr 37, 129–142. [DOI] [PubMed] [Google Scholar]
- 214. Tanno M, Kuno A, Ishikawa S, Miki T, Kouzu H, Yano T, Murase H, Tobisawa T, Ogasawara M, Horio Y et al. (2014) Translocation of GSK‐3β, a trigger of permeability transition, is kinase activity‐dependent and mediated by interaction with VDAC2. J Biol Chem 289, 29285–29296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Murai M, Okuda A, Yamamoto T, Shinohara Y & Miyoshi H (2017) Synthetic ubiquinones specifically bind to mitochondrial voltage‐dependent anion channel 1 (VDAC1) in Saccharomyces cerevisiae Mitochondria. Biochemistry 56, 570–581. [DOI] [PubMed] [Google Scholar]
- 216. Koushi M, Aoyama Y, Kamei Y & Asakai R (2020) Bisindolylpyrrole triggers transient mitochondrial permeability transitions to cause apoptosis in a VDAC1/2 and cyclophilin D‐dependent manner via the ANT‐associated pore. Sci Rep 10, 16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Pastorino JG & Hoek JB (2003) Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr Med Chem 10, 1535–1551. [DOI] [PubMed] [Google Scholar]
- 218. Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P & Rasola A (2008) Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage‐dependent anion channels. PLoS One 3, e1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Sun L, Shukair S, Naik TJ, Moazed F & Ardehali H (2008) Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 28, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Ciscato F, Filadi R, Masgras I, Pizzi M, Marin O, Damiano N, Pizzo P, Gori A, Frezzato F, Chiara F et al. (2020) Hexokinase 2 displacement from mitochondria‐associated membranes prompts Ca2+‐dependent death of cancer cells. EMBO Rep 21, e49117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Fontaine E, Eriksson O, Ichas F & Bernardi P (1998) Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex I. J Biol Chem 273, 12662–12668. [DOI] [PubMed] [Google Scholar]
- 222. Fontaine E, Ichas F & Bernardi P (1998) A ubiquinone‐binding site regulates the mitochondrial permeability transition pore. J Biol Chem 273, 25734–25740. [DOI] [PubMed] [Google Scholar]
- 223. Walter L, Nogueira V, Leverve X, Bernardi P & Fontaine E (2000) Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. J Biol Chem 275, 29521–29527. [DOI] [PubMed] [Google Scholar]
- 224. Walter L, Miyoshi H, Leverve X, Bernardi P & Fontaine E (2002) Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A Progress Report. Free Radic Res 36, 405–412. [DOI] [PubMed] [Google Scholar]
- 225. Vlasov AV, Kovalev KV, Marx SH, Round ES, Gushchin IY, Polovinkin VA, Tsoy NM, Okhrimenko IS, Borshchevskiy VI, Buldt GD et al. (2019) Unusual features of the c‐ring of F1FO ATP synthases. Sci Rep 9, 18547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Preiss L, Yildiz Ö, Hicks DB, Krulwich TA & Meier T (2010) A new type of proton coordination in an F1Fo‐ATP synthase rotor ring. PLoS Biol 8, e1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Symersky J, Osowski D, Walters DE & Mueller DM (2012) Oligomycin frames a common drug‐binding site in the ATP synthase. Proc Natl Acad Sci USA 109, 13961–13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Symersky J, Pagadala V, Osowski D, Krah A, Meier T, Faraldo‐Gomez JD & Mueller DM (2012) Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol 19, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Preiss L, Langer JD, Hicks DB, Liu J, Yildiz Ö, Krulwich TA & Meier T (2014) The c‐ring ion binding site of the ATP synthase from Bacillus pseudofirmus OF4 is adapted to alkaliphilic lifestyle. Mol Microbiol 92, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Preiss L, Langer JD, Yildiz Ö, Eckhardt‐Strelau L, Guillemont JE, Koul A & Meier T (2015) Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti‐TB drug bedaquiline. Sci Adv 1, e1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Scorrano L, Penzo D, Petronilli V, Pagano F & Bernardi P (2001) Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor‐α apoptotic signaling. J Biol Chem 276, 12035–12040. [DOI] [PubMed] [Google Scholar]
- 232. Penzo D, Petronilli V, Angelin A, Cusan C, Colonna R, Scorrano L, Pagano F, Prato M, Di Lisa F & Bernardi P (2004) Arachidonic acid released by phospholipase A2 activation triggers Ca2+‐dependent apoptosis through the mitochondrial pathway. J Biol Chem 279, 25219–25225. [DOI] [PubMed] [Google Scholar]
- 233. Bernardi P (1992) Modulation of the mitochondrial cyclosporin A‐sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem 267, 8834–8839. [PubMed] [Google Scholar]
- 234. Nicolli A, Petronilli V & Bernardi P (1993) Modulation of the mitochondrial cyclosporin A‐sensitive permeability transition pore by matrix pH. Evidence that the pore open‐closed probability is regulated by reversible histidine protonation. Biochemistry 32, 4461–4465. [DOI] [PubMed] [Google Scholar]
- 235. Petronilli V, Cola C, Massari S, Colonna R & Bernardi P (1993) Physiological effectors modify voltage sensing by the cyclosporin A‐sensitive permeability transition pore of mitochondria. J Biol Chem 268, 21939–21945. [PubMed] [Google Scholar]
- 236. Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S & Bernardi P (1994) The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation‐reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem 269, 16638–16642. [PubMed] [Google Scholar]
- 237. Costantini P, Chernyak BV, Petronilli V & Bernardi P (1996) Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem 271, 6746–6751. [DOI] [PubMed] [Google Scholar]
- 238. Galber C, Minervini G, Cannino G, Boldrin F, Petronilli V, Tosatto S, Lippe G & Giorgio V (2021) The f subunit of human ATP synthase is essential for normal mitochondrial morphology and permeability transition. Cell Rep 35, 109111. [DOI] [PubMed] [Google Scholar]
- 239. Johans M, Milanesi E, Franck M, Johans C, Liobikas J, Panagiotaki M, Greci L, Principato G, Kinnunen PKJ, Bernardi P et al. (2005) Modification of permeability transition pore arginine(s) by phenylglyoxal derivatives in isolated mitochondria and mammalian cells: Structure‐function relationship of arginine ligands. J Biol Chem 280, 12130–12136. [DOI] [PubMed] [Google Scholar]
- 240. Bernardi P, Veronese P & Petronilli V (1993) Modulation of the mitochondrial cyclosporin A‐sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem 268, 1005–1010. [PubMed] [Google Scholar]
- 241. Chauvin C, De Oliveira F, Ronot X, Mousseau M, Leverve X & Fontaine E (2001) Rotenone inhibits the mitochondrial permeability transition‐induced cell death in U937 and KB cells. J Biol Chem 276, 41394–41398. [DOI] [PubMed] [Google Scholar]
- 242. Li B, Chauvin C, De Paulis D, De Oliveira F, Gharib A, Vial G, Lablanche S, Leverve X, Bernardi P, Ovize M et al. (2012) Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate‐sensitive inhibitory site masked by cyclophilin D. Biochim Biophys Acta 1817, 1628–1634. [DOI] [PubMed] [Google Scholar]
- 243. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Neginskaya MA, Strubbe JO, Amodeo GF, West BA, Yakar S, Bazil JN & Pavlov EV (2020) The very low number of calcium‐induced permeability transition pores in the single mitochondrion. J Gen Physiol 152, e202012631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Solesio ME, Elustondo PA, Zakharian E & Pavlov EV (2016) Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem Soc Trans 44, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Baev AY, Negoda A & Abramov AY (2017) Modulation of mitochondrial ion transport by inorganic polyphosphate ‐ essential role in mitochondrial permeability transition pore. J Bioenerg Biomembr 49, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Menze MA, Hutchinson K, Laborde SM & Hand SC (2005) Mitochondrial permeability transition in the crustacean Artemia franciscana: absence of a calcium-regulated pore in the face of profound calcium storage. Am J Physiol 289, R68–R76. [DOI] [PubMed] [Google Scholar]
- 248. Angeli S, Foulger A, Chamoli M, Peiris TH, Gerencser A & Shahmirzadi AA (2021) The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. eLife Sciences 10, e63453 [DOI] [PMC free article] [PubMed] [Google Scholar]