

Abstract
Introduction
Several investigations have argued for a strong relationship between neuroinflammation and amyloid metabolism but it is still unclear whether inflammation exerts a pro‐amyloidogenic effect, amplifies the neurotoxic effect of amyloid, or is protective.
Methods
Forty‐two patients with acute encephalitis (ENC) and 18 controls underwent an extended cerebrospinal fluid (CSF) panel of inflammatory, amyloid (Aβ40, 42, and 38, sAPP‐α, sAPP‐β), glial, and neuronal biomarkers. Linear and non‐linear correlations between CSF biomarkers were evaluated studying conditional independence relationships.
Results
CSF levels of inflammatory cytokines and neuronal/glial markers were higher in ENC compared to controls, whereas the levels of amyloid‐related markers did not differ. Inflammatory markers were not associated with amyloid markers but exhibited a correlation with glial and neuronal markers in conditional independence analysis.
Discussion
By an extensive CSF biomarkers analysis, this study showed that an acute neuroinflammation state, which is associated with glial activation and neuronal damage, does not influence amyloid homeostasis.
Keywords: acute inflammation, amyloid, glial cells, neuroinflammation, tau
1. BACKGROUND
In recent years, several lines of evidence have supported an important role of amyloid in neuroinflammation and in the pathogenesis of Alzheimer's disease (AD). 1 , 2 Indeed, in vitro and animal studies have demonstrated the activation of different inflammatory pathways is associated with a progressive increase of amyloid burden and tau‐related neurodegeneration. 3 , 4 Moreover, neuropathological studies have supported the claim that amyloid beta (Aβ) and tau‐phosphorylation are associated with activation of microglia and astrocytes, which are important mediators of neurotoxicity. 5 Further, it has been claimed that cytokines’ increase might potentially influence disease progression and severity by inducing either amyloidogenesis or neurodegeneration by activation of microglia. 6 , 7 Aβ has been involved in the immune response, and several works have hypothesized that Aβ may act as a defense mechanism against inflammatory triggers, thus suggesting that infection may be implicated in promoting amyloid production and poligomerization. 8 In fact, there is evidence that the exposure of human neuronal and extraneuronal cells to inflammatory cytokines may modulate proteins that are responsible for accumulation of Aβ in the brain. 9 , 10 , 11 Several studies have also argued for a potential effect of inflammatory cytokines on amyloid precursor protein (APP) metabolism by acting on β‐ and γ‐secretase. 12 , 13
In vivo, however, most studies addressing the relationships between neuroinflammation and amyloid pathology have been contradictory. Recent studies demonstrated a correlation between cerebrospinal fluid (CSF) neuroinflammation and tau biomarkers, but not with the amyloid markers CSF Aβ42, β‐secretase activity or sAβPPβ, 14 , 15 , 16 supporting the view that increased inflammatory activity rather might represent a consequence of accumulating amyloid pathology instead of being a causative factor for the development of brain amyloidosis. 15 In human low‐grade chronic neuroinflammatory or in neuroinfectious diseases, including multiple sclerosis, human immunodeficiency virus (HIV)‐associated neurocognitive dysfunction, Lyme disease, and bacterial meningitis, an acute reduction of CSF concentrations of both amyloidogenic and non‐amyloidogenic Aβ and AβPP fragments has been reported, 17 , 18 , 19 , 20 suggesting a general downregulation of APP expression and/or processing in these diseases. Additionally, in CSF from patients with bacterial meningitis, ex vivo 18O‐labelling mass spectrometry has demonstrated active Aβ degradation. 21 In contrast, however, several studies have found that, while glial inhibition or depletion may prevent neuronal loss, it may have no substantial effects on cerebral amyloid homeostasis. 3 , 22 This further suggests that amyloid production might much more depend on mechanisms other than neuroinflammation. Thus, neuroinflammation still represents a key controversial factor in the pathogenetic cascade of amyloid deposition, and more studies are needed to elucidate if there are causal relationships between different neuroinflammation states and Aβ homeostasis.
RESEARCH IN CONTEXT
Systematic review: Neuroinflammation has been associated with alterations within amyloid metabolism in several preclinical studies. In vivo, the impact of an acute inflammatory process on amyloid fragments and the interaction with neuronal and glial changes is still a theme of debate.
Interpretation: Our findings, based on extensive cerebrospinal fluid (CSF) analyses, showed that neuroinflammatory changes are not associated with amyloid markers but strongly related to neuronal and glial markers.
Future directions: We demonstrate that acute brain inflammation does not impact amyloid metabolism even in the presence of neuronal damage and glial activation. These findings questioned the role of amyloid in innate immunity and emphasize a glial mediated pathway linking neuroinflammation to neuronal degeneration. This result should stimulate larger studies on plasma and CSF biomarkers to understand the complex interaction among neuroinflammation, amyloid, and neuronal and glial activation in vivo.
HIGHLIGHTS
Encephalitis exhibits increased levels of inflammatory, neuronal, and glial cerebrospinal fluid (CSF) markers.
Amyloid markers are not altered during an acute inflammation due to encephalitis.
Neuroinflammation correlates with neuronal and glial but not amyloid markers in CSF.
The aim of the study was to determine the involvement of amyloid processing in the acute inflammatory reaction and to assess its relationship with glial activation and neuronal damage. To do this, we examined a panel of CSF inflammatory and amyloid‐related, glial, and neuronal biomarkers in a series of patients with acute encephalitis and a group of cognitively unimpaired individuals.
2. METHODS
2.1. Patients
The study included patients who underwent CSF analyses for the diagnostic work‐up of encephalitis (ENC) at the Neurology and Infectious Disease Department of ASST Spedali Civili of Brescia and Neurology Department of ASST Cremona. Inclusion criteria were (1) diagnosis of probable ENC according to current criteria, 23 , 24 (2) no neurological disease prior the onset of acute encephalitis, and (3) absence of concomitant immunomodulator treatment (such as steroid or immunoglobulin) or antibiotics at the time of CSF sampling. Each patient with suspected encephalitis underwent brain magnetic resonance imaging (MRI), standard electroencephalography (EEG), thyroid function and antibodies (anti‐thyroglobulin, anti‐thyroid peroxidase), immunoglobulin (Ig)M and IgG for Borrelia burgdorferi, and CSF analyses. CSF viral screening included herpes simplex virus (HSV‐1, HSV‐2, HSV‐6, HSV‐8, cytomegalovirus, Epstein‐Barr virus, varicella zoster virus), adenovirus, and enterovirus. For biomarker comparison, neurologically healthy controls (HCs; n = 18) with available CSF were retrospectively selected from the Neurology Department of ASST Spedali Civili of Brescia. Healthy subjects were selected from individuals hospitalized because of acute onset headache to exclude brain hemorrhages, intracranial hypertension, or meningo‐encephalitis. The group consisted of those with normal neurological examination, MRI, and CSF biochemical analyses. The Institutional Ethical Standards Committee on human experimentation at Brescia University Hospital provided approval for the study (NP 4067).
2.2. CSF immunological and biochemical analyses
At enrollment, 3 mL of CSF from each participant was collected, centrifuged, and processed for standard biochemical analyses. Two milliliters of CSF were stored in cryotubes at −80°C before testing. All CSF samples were analyzed for inflammatory and neuronal/glial markers at the Clinical Neurochemistry Laboratory at Sahlgrenska University Hospital (Mölndal, Sweden). CSF cytokine concentrations (including interleukin [IL]‐6, IL‐8, tumor necrosis factor alpha [TNF‐α]) were measured using a Mesoscale Discovery (MSD) multiplexed immunoassay (MSD, Rockville, MD). CSF β2‐microglobulin (β2M) concentration was measured using an immunoassay on an Atellica instrument (Siemens Healthcare GmbH). CSF total and phosphorylated tau (t‐tau and p‐tau) concentrations were measured by Lumipulse (Fujirebio, Ghent, Belgium). CSF neurofilament light (NfL) and glial fibrillary acidic protein (GFAP) concentrations were measured using in‐house enzyme‐linked immunosorbent assays (ELISAs). 25 , 26 CSF sTREM‐2 concentration was measured using an in‐house immunoassay with electrochemiluminescent detection. 27 CSF chitinase‐3‐like protein 1 (YKL‐40) concentration was measured using the Human Chitinase 3‐like 1 Quantikine kit (R&D Systems,Minneapolis, MN). CSF Aβ1–38 (Aβ38), Aβ1–40 (Aβ40), and Aβ1–42 (Aβ42) concentrations were measured using the MSD Triplex Assay (MSD, Rockville, MD). 27 CSF sAPP‐α and sAPP‐β concentrations were measured using commercial ELISAs (IBL). Board‐certified laboratory technicians who were blinded to clinical data performed all analyses.
2.3. Statistical analyses
Data are presented as median, interquartile ranges for continuous variables, and number (%) for categorical variables. For the comparison of demographic characteristics and CSF biomarker levels between groups (HC vs. ENC and within different subtypes of encephalitis) we used the Fisher test and nonparametric Kruskal‐Wallis test adjusted for the effects of age and sex. Post hoc comparisons were performed using Bonferroni correction. In the encephalitis subgroup, the correlations among specific inflammatory markers, amyloid, and glial and neuronal markers were evaluated using Pearson partial correlations adjusted for the effect of age and sex; the strength of correlation was indicated by r and P values.
To investigate the possible dependence, including non‐linear relations, or lack thereof between the different biomarkers and subjects’ covariates we studied the conditional independence relationships following a graphical model approach. 28 In graphical models each variable is associated with a node in a graph and edges connecting such nodes represent conditional dependency. Specifically, we used a Bayesian Gaussian copula graphical model 29 , 30 (which can handle non‐Gaussian data including continuous, discrete, and qualitative variables). Further benefits of this are the good performance for small samples and the ability to handle missing data. The analysis was carried out with the R package “BDgraph” 30 via Markov chain Monte Carlo simulation setting the prior expectation for the probability of connection to 0.25 and running the algorithm for 10 random initializations to improve robustness. As output, we reported the probability that two variables were connected, estimated as the number of times in the Markov chain Monte Carlo simulation that a non‐null partial correlation was drawn in the copula model.
3. RESULTS
The study recruited 42 cases of encephalitis (ENC, median age 62, 17 females) and 18 HCs. The group with ENC included infectious viral encephalitis (INF‐ENC n = 10); autoimmune encephalitis (AI‐ENC, n = 11); encephalitis during SARS‐CoV‐2 infection (COV‐ENC, n = 11); and encephalitis of unknown origin, defined as patients with described infectious and autoimmune negative screening (UO‐ENC, n = 10). Blood biochemical testing at the time of CSF sampling showed similar white blood cell count but mild increase in granulocytes and decrease in lymphocytes in ENC compared to controls, whereas C‐reactive protein levels did not differ between groups (Table 1). In ENC, the CSF samples were collected during the diagnostic assessment after a mean of 7.5 ± 4 days from symptom onset.
TABLE 1.
Demographic, CSF, and blood standard biomarkers according to the clinical diagnosis
| HC (n = 18) | ENC (n = 42) | P | |
|---|---|---|---|
| Demographics | |||
| Age, years | 40 (28‐52) | 62 (49‐73) | <.001 |
| Sex female, n (%) | 12 (66.7%) | 17 (40.4%) | .06 |
| Blood analyses | |||
| White‐cell count/ mm3 | 7.4 (6.2‐8.9) | 8.6 (6.3‐8.6) | .443 |
| Granulocyte count/ mm3 | 4.3 (3.3‐5.9) | 5.5 (3.6‐5.5) | .160 |
| Lymphocyte count/ mm3 | 2.1 (1.7‐2.6) | 1.6 (1.2‐2.2) | .024 |
| C‐reactive protein, mg/L | 2.9 (2.9‐3.0) | 2.9 (2.1‐7.2) | .639 |
| CSF biochemical analyses | |||
| Protein, mg/L | 38.5 (32.2‐46.0) | 54.8 (36.4‐78.7) | .001 |
| Albumin, mg/L | 183 (147‐258) | 365 (222‐459) | .003 |
| Cells, total count/uL | 1 (0‐2) | 9 (4‐26) | .003 |
| Glucose, mg/dL | 63 (55‐68) | 65 (54‐72) | .45 |
Abbreviations: CSF, cerebrospinal fluid; ENC, encephalitis; HC, healthy controls.
Notes: Data are presented as median (interquartile ranges).
P‐values were calculated by Kruskal‐Wallis test or Fisher's exact test, as appropriate.
Compared to HC, ENC showed significantly higher CSF levels of specific inflammatory markers, namely IL‐6, IL‐8, TNF‐α, and β2M (P < .002, Table 1). In contrast, CSF levels of amyloid‐related markers (Aβ38, Aβ40, Aβ42, sAPP‐α, sAPP‐β) did not differ between ENC and HC (Table 2). Glial‐related markers (GFAP, sTREM‐2, and YKL‐40) and neuronal markers (NfL and t‐tau) were significantly increased ENC compared to HC (P from < .004 to .001 adjusting for age and sex).
TABLE 2.
CSF biomarkers according to the clinical diagnosis
| HC (n = 18) | ENC (n = 42) | P | |
|---|---|---|---|
| Inflammatory markers | |||
| IL‐6, pg/mL | 1.05 (0.56–1.64) | 2.36 (0.99–8.74) | .002 |
| IL‐8, pg/mL | 33 (28–47) | 121 (59–516) | <.001 |
| TNF‐α, pg/mL | 0.17 (0.17–0.17) | 0.37 (0.25–1.88) | <.001 |
| β‐2 microglobulin, mg/L | 0.90 (0.71–1.01) | 1.82 (1.37–2.94) | <.001 |
| Amyloid markers | |||
| Aβ38, pg/mL | 1045 (858–1568) | 1338 (815–1652) | .67 |
| Aβ40, pg/mL | 2961 (2806–4455) | 3672 (2627–4626) | .97 |
| Aβ42, pg/mL | 243 (165–358) | 262 (136–373) | .92 |
| Aβ42/40 ratio | 0.073 (0.058–0.083) | 0.068 (0.048–0.086) | .28 |
| sAPP‐α, pg/mL | 131 (88–241) | 154 (103–227) | .64 |
| sAPP‐β, pg/mL | 432 (291–786) | 409 (296‐580) | .44 |
| Glial markers | |||
| GFAP, pg/mL | 109 (76–151) | 323 (214–629) | <.001 |
| sTREM‐2, pg/mL | 589 (478–1005) | 2573 (1465–4104) | <.001 |
| YKL‐40, ng/mL | 64 (45–97) | 199 (145–422) | .004 |
| Neuronal markers | |||
| NfL, pg/mL | 297 (229–437) | 1388 (496–3935) | <.001 |
| T‐tau, pg/mL | 165 (127–183) | 327 (238–559) | <.001 |
| P‐tau, pg/mL | 18.2 (14.2–24.1) | 25.5 (19.7‐45.7) | .02 |
Abbreviations: ENC, encephalitis; GFAP, glial fibrillary acidic protein; HC, healthy controls; IL‐6, interleukin 6; IL‐8, interleukin 8; NfL, neurofilament light chain; p‐tau, phosphorylated tau; sAPP‐α, soluble amyloid precursor protein alpha, sAPP‐β, soluble amyloid precursor protein beta; sTREM‐2, triggering receptor expressed on myeloid cells 2; TNF‐α, tumor necrosis factor alpha; t‐tau, total tau; YKL‐40, chitinase‐3‐like protein 1.
Notes: Data are presented as median (interquartile ranges).
P‐values were calculated by Mann‐Whitney test or Fisher's exact test, as appropriate.
UO‐ENC exhibited higher cell count compared to INF‐ENC, AI‐ENC, and COV‐ENC. No differences in distribution of inflammatory, amyloid, and neuronal and glial biomarkers were observed according to the subdiagnosis of encephalitis (Table S1 in supporting information and Figure 1).
FIGURE 1.
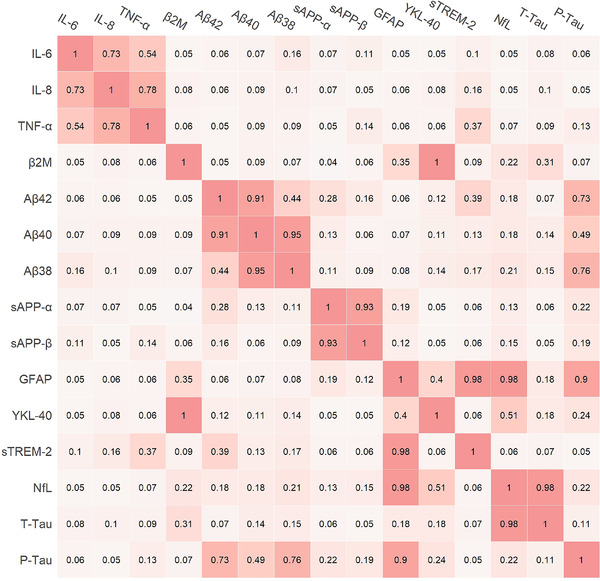
Estimated probability of connection between biomarkers according to the non‐linear model. β2M, β2‐microglobulin; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; IL‐8, interleukin 8; NfL, neurofilament light chain; p‐tau, phosphorylated tau; sAPP‐α, soluble amyloid precursor protein alpha, sAPP‐β, soluble amyloid precursor protein beta; sTREM‐2, triggering receptor expressed on myeloid cells 2; TNF‐α, tumor necrosis factor alpha; t‐tau, total Tau protein; YKL‐40, chitinase‐3‐like protein
3.1. Partial correlation analyses
In ENC, partial correlation analyses adjusted for age and sex failed to show a significant correlation between inflammatory markers (TNF‐α, IL‐6, IL‐8, and β2M) and amyloid markers (Aβ42, Aβ38, Aβ40, sAPP‐α, sAPP‐β).
Conversely, TNF‐α showed a positive correlation with GFAP (r = 0.86, P = .001), YKL‐40 (r = 0.91, P = .001), and t‐tau (r = 0.68, P = .001). IL‐6 exhibited a positive correlation with GFAP (r = 0.54, P = .001), YKL‐40 (r = 0.51, P = .003), and t‐tau (r = 0.47, P = .006), whereas IL‐8 did not show significant correlations with neuronal or glial markers. β2M showed a positive correlation with GFAP (r = 0.46, P = .001), sTREM‐2 (r = 0.73, P = .001), YKL‐40 (r = 0.41, P = .019), and t‐tau (r = 0.43, P = .001; Table S2 in supporting information).
3.2. Graphical model analysis
Figures 1 and 2 show the results of the graphical model analysis, which is able to consider also non‐linear relations between biomarkers. The model showed a strong intra‐correlation between IL‐8, IL‐6, and TNF‐α, whereas β2M appeared to be an independent marker of cellular immune response. The model confirms the lack of association between cytokines or β2M and amyloid markers (i.e., linking the probability lower than the prior expected value of 0.25). Amyloid‐related markers specifically showed a strong intra‐correlation but exhibited low probability of correlation with any other markers, including glial and neuronal biomarkers. Among neuroinflammatory markers, TNF‐α exhibited a positive correlation with sTREM‐2, whereas β2M showed a positive correlation with GFAP, YKL‐40, and t‐tau. Separate analyses using the Aβ40/42 ratio confirmed the lack of association between neuroinflammatory mediators and amyloid‐related markers (Figures S2 and S3 in supporting information).
FIGURE 2.
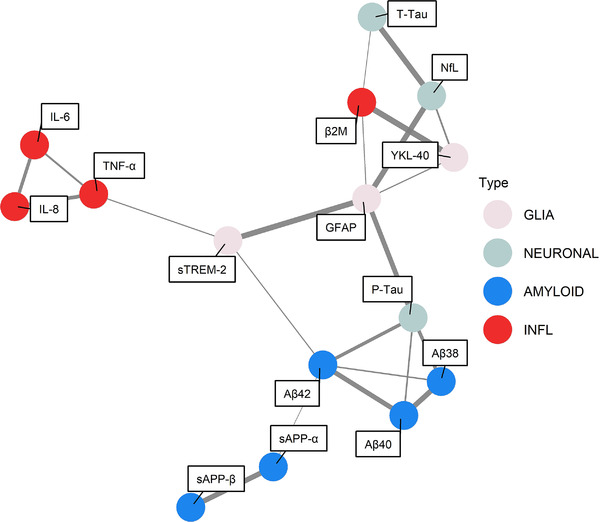
Graphical representation of estimated probability of connection in encephalitis obtained by fixing the position of the nodes using a Fruchterman–Reingold force‐direct algorithm. The edges’ thicknesses are proportional to the estimated probabilities of connection. Values below the prior expected value of 0.25 are not reported for graphical purposes. β2M, β2‐microglobulin; GFAP, glial fibrillary acidic protein; IL‐1β, interleukin‐1β; IL‐6, interleukin 6; IL‐8, interleukin 8; NfL, neurofilament light chain; p‐tau, phosphorylated tau; sAPP‐α, soluble amyloid precursor protein alpha, sAPP‐β, soluble amyloid precursor protein beta; sTREM‐2, triggering receptor expressed on myeloid cells 2; TNF‐α, tumor necrosis factor alpha; t‐tau, total tau protein; YKL‐40, chitinase‐3‐like protein 1
4. DISCUSSION
In a series of patients with acute encephalitis, this study investigated whether acute neuroinflammation influences amyloid metabolism by addressing the complex interplay among inflammatory, amyloid, and glial and neuronal markers. We found that CSF levels of inflammatory markers (IL‐6, IL‐8, TNF‐α, and β2M) and measures of glial activation (GFAP, YKL‐40, sTREM‐2) and neuronal damage (NfL and t‐tau) were all increased in patients with acute neuroinflammation due to encephalitis. However, CSF levels of amyloid‐related biomarkers (Aβ38, Aβ40, Aβ42, sAPP‐α, sAPP‐β) did not vary and were not associated with CSF levels of inflammatory cytokines or β2M (as markers of central nervous system [CNS] immune response and blood–brain barrier integrity). In fact, CSF acute inflammatory markers did correlate to each other and were significantly associated with CSF levels of glial biomarkers and markers of neuronal damage but not any amyloid‐related biomarkers.
The study included subjects without explicit neurological diseases prior to the onset of encephalitis, thus providing a unique perspective for evaluating the complex interactions among neuroinflammatory mediators, cellular responses, and amyloid pathways in normal conditions in vivo.
Evidence from in vitro and animal models, as well as human CSF studies, claimed that inflammatory mediators influence amyloid production and clearance. 17 , 18 , 19 , 31 However, it is still debated whether an acute neuroinflammation might impact Aβ pathology or whether inflammation influences APP expression and/or processing upstream of Aβ deposition. 3 , 4 , 32 According to our results, an acute inflammatory response was associated neither with Aβ peptides nor with the cleavage products of APP, namely sAPP‐α and sAPP‐β. Conversely, inflammatory cytokines and β2M correlated with microglia/astrocytes and—to a lesser extent—with the neuronal damage marker t‐tau.
These results strongly support the concept that acute neuroinflammation, at least in the acute stage, does not primarily influence Aβ processing whereas they argue for its involvement in glial activation and neuronal damage independently from amyloid processing. 33 Such findings seem to support the claim that microglial activation might protect patients with amyloidosis from deterioration but have an opposite role on tau pathology. 34 , 35 , 36 , 37 In fact, there is evidence that chronic inflammation and cytokine upregulation induce tau hyperphosphorylation in a prepathological mice model. 38 Accordingly, the overall Aβ pathological features did not change in a mice experimental model of either acute or chronic inflammation whereas there was a marked exacerbation of the tau pathological characteristics, which was associated in an age‐related aberrant activation of GSK‐3β. 39 In fact, the cellular crosstalk between microglia and astrocytes might form a positive feedback loop under the inflammatory milieu in the brain, which might result in a dysregulated and self‐amplifying inflammatory response increasing the vulnerability of neurons to neurodegeneration, independently of Aβ or APP involvement. 40 , 41 Accordingly, a very recent study on non‐demented older people identified a cluster of subjects characterized by increased levels of inflammatory markers in CSF and by higher levels of t‐tau and p‐tau levels in CSF but no difference in CSF Aβ42 levels. 42
Further, the lack of CSF variations of levels of different amyloid‐related biomarkers in this series of patients affected by acute encephalitis of different etiologies, including viral infection, might shed some light on the role of amyloid in innate inflammation. 8 , 43 Accordingly, this study showed that there was a clear increase of CSF levels of GFAP, TREM2, and YKL‐40 indicating an innate reaction, which was highly associated with both acute neuroinflammatory markers and neuronal markers but amyloid. These data do not substantiate the growing body of literature supporting the view that Aβ may be regarded as an early responder cytokine and might be mobilized to act as an antimicrobial peptide (AMP), which is considered the first line of defense against pathogens and a potent broad‐spectrum antibiotic and immunomodulator. 43 , 44 Indeed, most of these data were generated in experimental animal models, 45 while human studies linking infections and amyloid accumulation have been mostly correlational in nature. However, the lack of clustering with amyloid markers of this study may have different explanations including a delay in amyloid processing (i.e., the change in amyloid‐related biomarkers levels not necessarily occur during the acute stage of encephalitis requiring time to manifest). Indeed, it is also conceivable that the involvement of amyloid processing builds up with aging due to immunosenescence and breakdown of the blood–brain barrier. 46 , 47 , 48 Again, it is still possible that chronic asymptomatic infection may accelerate amyloid burden, and that aging itself might lead to a self‐perpetuating immune response starting from a transient infection. 8 , 47 Finally, the previously reported association of acute and chronic neuroinflammation with reduced CSF levels of amyloid‐related markers in diseases like multiple sclerosis, HIV‐associated neurocognitive dysfunction, Lyme disease, and bacterial meningitis 17 , 18 , 19 , 20 , 21 might well support the view that there are different innate inflammatory pathways and that amyloid species might be specifically involved in some of these, eventually depending on white matter involvement or some other chronic‐related pathogenic mechanisms yet to be defined.
The current findings might be further validated in a wider sample of acute and chronic infections by using longitudinal CSF assessment to characterize the temporal role of the immune response role of amyloid and to clarify the relationship among cytokines, glial activation, and neuronal dysfunction in neuroinflammation.
Strengths of this study are the large panel of inflammatory, neuronal, glial, and amyloid markers known to play a role in the response to damage in the CNS. The main limitation is the relatively small sample size of subjects with encephalitis and the differences in age with controls. To address this, we adjusted all the analyses for demographics and we implemented a parallel evaluation of partial correlation analyses and graph models enabled the confirmation of the findings considering different models of interactions—according to the complex interplay we observed between CSF markers.
Our findings may also have important consequences for a deeper understanding of the results of new pharmacological approaches targeting neuroinflammation, and amyloid or tau pathologies in neurodegeneration.
Specifically, we observed a lack of association between TNF‐α and amyloid pathways in vivo. This suggests that the effects of anti‐TNF antibodies on APP recently described is probably mediated by neuronal and non‐neuronal responses instead of changes in amyloid production or clearance. 49 According to this model, new pharmacological approaches targeting Aβ and tau pathologies might exert an additional benefit through the reduction of neuroinflammation in AD acting on the vulnerability of neurons and glial cells to neurodegeneration. The inefficacy of non‐steroidal anti‐inflammatory drugs in trials targeting neurodegenerative diseases 50 might also argue for a much more complex relationship between inflammatory mediators and amyloid pathways, requiring a deeper modulation of glial and neuronal response to be more effective. Targeting microglial activation, such as modulating sTREM‐2, has been indeed recently suggested in AD, 41 but further studies are needed to exclude that glial activation might be the first essential response for counteracting Aβ accumulation.
We need to acknowledge that some limitations may affect the interpretation of our data. First, the study has limited the observation to the acute phase of neuroinflammation, thus not allowing the assessment of the effects of increased levels of cytokines through persistent activation of microglia, astrocytes, and neuronal cells. This is a very important issue, as the impact of chronic low‐level inflammation could not be addressed in this study and might be different for amyloid pathways and biomarkers. Moreover, the study lack longitudinal data, as it relied on the assessment of CSF biomarkers limited to the acute stage of encephalitis. This leaves open the question of subsequent biomarkers’ changes, which would be helpful to better clarify the temporal pattern of amyloid involvement in the immune response. Finally, the study did not include elderly patients nor patients with dementia and apolipoprotein E genotyping was not performed. Thus, further studies including subjects with neurodegenerative changes during acute, subacute, and chronic neuroinflammation are warranted to extend these findings. In fact, in this study we did not include elderly and demented patients, as the immune responses has been reported to be dysfunctional during aging and in AD. 31 , 51 Therefore, it is possible that acute neuroinflammation may not be detrimental in young individuals when the immune responses can be properly regulated, but it affects neuronal homeostasis in older individuals.
Notwithstanding these limits, this study showed that the assessment of different CSF markers may allow us to investigate in vivo the correlates of acute neuroinflammation due to encephalitis and might contribute to assessing the complex interplay among inflammatory, amyloid, and glial and neuronal markers.
In conclusion, our main findings showed that acute neuroinflammation in ENC is characterized by a CSF increase of inflammatory biomarkers in association with glial and neuronal biomarkers, whereas amyloid‐related markers specifically showed a strong intra‐correlation but exhibited low probability of correlation with any other markers, including glial and neuronal biomarkers. Future studies investigating follow‐up/later time point assessment of both plasma and CSF samples, by studying specific cellular responses (such as macrophages or lymphocyte subtypes) are needed to further clarify the relationship between the activation of specific immunological responses and amyloid pathways.
CONFLICTS OF INTEREST
L. S., S.M., A.I, B.R., G.B., V.D.G., N.J.A. have no conflict of interest.
AUTHOR CONTRIBUTIONS
Alessandro Padovani and Andrea Pilotto contributed to the conception and design of the study; Alessandro Padovani, Antonio Canale, Lorenzo Schiavon, Stefano Masciocchi, Alberto Imarisio, Barbara Risi, Giulio Bonzi, Valeria De Giuli, Monica Di Luca, Nicholas J. Ashton, Kaj Blennow, Henrik Zetterberg, and Alessandro Padovani contributed to the acquisition and analyses of data; Alessandro Padovani and Andrea Pilotto contributed to drafting the text; Antonio Canale, Lorenzo Schiavon, contributed to statistical analyses and preparing the figures.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors thank the patients who participated in the study and the health personnel involved in the clinical assistance and care of patients. Alessandro Padovani received grant support from Ministry of Health (MINSAL) and Ministry of Education, Research and University (MIUR), from CARIPLO Foundation; personal compensation as a consultant/scientific advisory board member for Biogen 2019‐2020‐2021, Roche 2019‐2020, Nutricia 2020‐2021, General Healthcare (GE) 2019; he received honoraria for lectures at meeting ADPD2020 from Roche, lecture at meeting of the Italian society of Neurology 2020 from Biogen and from Roche, lecture at meeting AIP 2020 and 2021 from Biogen and from Nutricia, Educational Consulting 2019‐2020‐2021 from Biogen. Antonio Canale is supported by STAR GRANT, Grantor University of Padova, Grant and 2018 Visiting program, grantor Department of statistical sciences, University of Padova, payed to the institution. Monica Di Luca is supported by Ministry for Research Italy, European Commission H2020; she served on the advisory board of Roche, Senate DZNE Scientific Advisory Board LIN–payment made to M.D.L. as an individual; and she served as president, European Brain Council. Kaj Blennow has served as a consultant, on advisory boards, or on data monitoring committees or served as a consultant or on advisory boards for Abcam (payment made to KB as an individual), Axon (payment made to KB as an individual), Biogen (payment made to KB as an individual), JOMDD/Shimadzu (payment made to KB as an individual), Lilly )payment made to KB as an individual, MagQu (payment made to KB as an individual), Prothena (payment made to KB as an individual), Roche Diagnostics (payment made to KB as an individual), Siemens Healthineers (payment made to KB as an individual); served on data monitoring committees for Julius Clinical (payment made to KB as an individual), Novartis (payment made to KB as an individual). He is co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. Hans Zetterberg is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), and the UK Dementia Research Institute at UCL. Payments made to Institution. Hans Zetterberg has served pn scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx (all payments made to HZ as an individual). He has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen (all payments made to HZ). He is chair of the Alzheimer's Association Global Biomarker Standardization Consortium and the Alzheimer's Association Biofluid‐Based Biomarker Professional Interest Area. He is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work; payments made to HZ). Andrea Pilotto is supported by IMI H2020 initiative (MI2‐2018‐15‐06) paid to the university of Brescia Italian Ministry of Health; he received lecture honoraria from Bial, Biomarin, Abbvie, CHiesi, and Zambon pharmaceuticals (payments made to AP as an individual); he received research support from Bial, Biomarin, Abbvie, CHiesi, and Zambon pharmaceuticals (payment made to the Institution University of Brescia).
Open Access Funding provided by Universita degli Studi di Brescia within the CRUI‐CARE Agreement.
Padovani A, Canale A, Schiavon L, et al. Is amyloid involved in acute neuroinflammation? A CSF analysis in encephalitis. Alzheimer's Dement. 2022;18:2167–2175. 10.1002/alz.12554
[Correction added on May 12, 2022, after first online publication: CRUI‐CARE funding statement has been added.]
REFERENCES
- 1. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer's disease: where do we go from here? Nat Rev Neurol. 2021;17:157‐172. [DOI] [PubMed] [Google Scholar]
- 3. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimer's Dement. 2016;12:719‐732. [DOI] [PubMed] [Google Scholar]
- 4. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer's disease. Nat Rev Neurosci. 2015;16:358‐372. [DOI] [PubMed] [Google Scholar]
- 5. Perez‐Nievas BG, Stein TD, Tai HC, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. 2013;136:2510‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer's disease. J Neuroinflammation. 2005:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimer's. Dement. 2018;14:1602‐1614. [DOI] [PubMed] [Google Scholar]
- 9. Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin‐1. Mol Brain Res. 1992;16:128‐134. [DOI] [PubMed] [Google Scholar]
- 10. Blasko I, Marx I, Steiner E, Hartmann T. Gruebeck‐Loebenstein B. TNFα plus IFNγ induce the production of Alzheimer's β‐amyloid peptides and decrease the secretion of APPs. FASEB J. 1999;13:63‐68. [DOI] [PubMed] [Google Scholar]
- 11. Sutinen EM, Pirttilä T, Anderson G, Salminen A, Ojala JO. Pro‐inflammatory interleukin‐18 increases Alzheimer's disease‐associated amyloid‐β production in human neuron‐like cells. J Neuroinflammation. 2012:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto M, Kiyota T, Horiba M, et al. Interferon‐γ and tumor necrosis factor‐α regulate amyloid‐β plaque deposition and β‐secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paouri E, Tzara O, Zenelak S, Georgopoulos S. Genetic deletion of tumor necrosis factor‐α attenuates amyloid‐β production and decreases amyloid plaque formation and glial response in the 5xfad model of Alzheimer's disease. J Alzheimer's Dis. 2017;60:165‐181. [DOI] [PubMed] [Google Scholar]
- 14. Popp J, Oikonomidi A, Dayon L, et al. Markers of neuroinflammation associated with Alzheimer ’ s disease pathology in older adults. Brain, Behavior, and Immunity. 2017;62:203‐211. [DOI] [PubMed] [Google Scholar]
- 15. Alcolea D, Carmona‐Iragui M, Suárez‐Calvet M, et al. Relationship between β‐Secretase, inflammation and core cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimer's Dis. 2014;42:157‐167. [DOI] [PubMed] [Google Scholar]
- 16. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer's disease. Neurology. 2018;91:e867‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gisslén M, Krut J, Andreasson U, et al. Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol. 2009:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krut JJ, Zetterberg H, Blennow K, et al. Cerebrospinal fluid Alzheimer's biomarker profiles in CNS infections. J Neurol. 2013;260:620‐626. [DOI] [PubMed] [Google Scholar]
- 19. Augutis K, Axelsson M, Portelius E, et al. Cerebrospinal fluid biomarkers of β‐amyloid metabolism in multiple sclerosis. Mult Scler J. 2013;19:543‐552. [DOI] [PubMed] [Google Scholar]
- 20. Mattsson N, Bremell D, Anckarsäter R, et al. Neuroinflammation in Lyme neuroborreliosis affects amyloid metabolism. BMC Neurol. 2010:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Portelius E, Mattsson N, Pannee J, et al. Ex vivo 18O‐labeling mass spectrometry identifies a peripheral amyloid β clearance pathway. Mol Neurodegener. 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spangenberg EE, Lee RJ, Najafi AR, et al. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid‐β pathology. Brain. 2016;139:1265‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Venkatesan A, Tunkel AR, Bloch KC, et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis. 2013;57:1114‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pilotto A, Masciocchi S, Volonghi I, et al. Clinical presentation and outcomes of severe acute respiratory syndrome coronavirus 2‐Related encephalitis: the ENCOVID multicenter study. J Infect Dis. 2021;223:28‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaetani L, Höglund K, Parnetti L, et al. A new enzyme‐linked immunosorbent assay for neurofilament light in cerebrospinal fluid: analytical validation and clinical evaluation. Alzheimer's Res Ther. 2018:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pilotto A, Masciocchi S, Volonghi I, et al. SARS‐CoV‐2 encephalitis is a cytokine release syndrome: evidences from cerebrospinal fluid analyses. Clin Infect Dis. 2021. 10.1093/cid/ciaa1933. Clin Infect Dis . 2021 Jan 4;ciaa1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Banerjee G, Ambler G, Keshavan A, et al. Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy. J Alzheimer's Dis. 2020;74:1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lauritzen SL. Graphical Models ‐ 1996. Oxford University Press. [Google Scholar]
- 29. Dobra A, Lenkoski A, Rodriguez A. Bayesian inference for general gaussian graphical models with application to multivariate lattice data. J Am Stat Assoc. 2011;106:1418‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mohammadi R, Wit EC. Bayesian structure learning in sparse gaussian graphical models. Bayesian Anal. 2015;10:109‐138. [Google Scholar]
- 31. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015;16:229‐236. [DOI] [PubMed] [Google Scholar]
- 32. Alasmari F, Alshammari MA, Alasmari AF, Alanazi WA, Alhazzani K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed Res Int. 2018. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bacher M, Deuster O, Aljabari B, et al. The role of macrophage migration inhibitory factor in Alzheimer's disease. Mol Med. 2010;16:116‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hamelin L, Lagarde J, Dorothée G, et al. Early and protective microglial activation in Alzheimer's disease: a prospective study using 18F‐DPA‐714 PET imaging. Brain. 2016;139:1252‐1264. [DOI] [PubMed] [Google Scholar]
- 35. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer's disease trajectory. Brain. 2017;140:792‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parbo P, Ismail R, Sommerauer M, et al. Does inflammation precede tau aggregation in early Alzheimer's disease? A PET study Neurobiol Dis. 2018;117:211‐216. [DOI] [PubMed] [Google Scholar]
- 37. Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer's disease. Brain. 2018;141:2740‐2754. [DOI] [PubMed] [Google Scholar]
- 38. Lee DC, Rizer J, Selenica MLB, et al. LPS‐ induced inflammation exacerbates phospho‐tau pathology in rTg4510 mice. J Neuroinflammation. 2010:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sy M, Kitazawa M, Medeiros R, et al. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811‐2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Felsky D, Roostaei T, Nho K, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. 2019:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milà‐Alomà M, Salvadó G, Gispert JD, et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimer's. Dement. 2020;16:1358‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng Y, Chen B, Chi L, Zhou Q, Shi Z. Patterns of CSF inflammatory markers in non‐demented older people: a cluster analysis. Front Aging Neurosci. 2020:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar DKV, Choi HS, Washicosky KJ, et al. Amyloid‐β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gosztyla ML, Brothers HM, Robinson SR. Alzheimer's Amyloid‐β is an antimicrobial peptide: a review of the evidence. J Alzheimer's Dis. 2018;62:1495‐1506. [DOI] [PubMed] [Google Scholar]
- 45. Weaver DF. Amyloid beta is an early responder cytokine and immunopeptide of the innate immune system. Alzheimer's Dement. 2020;6:e12100. 10.1002/trc2.12100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sweeney MD, Sagare AP, Zlokovic BV. Blood‐brain barrier breakdown in Alzheimer's disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu F, Schillinger JA, Sternberg MR, et al. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988‐1994. J Infect Dis. 2002;185:1019‐1024. [DOI] [PubMed] [Google Scholar]
- 48. Itzhaki RF, Lathe R, Balin BJ, et al. Microbes and Alzheimer's disease. J Alzheimer's Dis. 2016;51:979‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steeland S, Gorlé N, Vandendriessche C, et al. Counteracting the effects of TNF receptor‐1 has therapeutic potential in Alzheimer's disease. EMBO Mol Med. 2018:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miguel‐Álvarez M, Santos‐Lozano A, Sanchis‐Gomar F, et al. Non‐steroidal anti‐inflammatory drugs as a treatment for Alzheimer's disease: a systematic review and meta‐analysis of treatment effect. Drugs Aging. 2015;32:139‐147. [DOI] [PubMed] [Google Scholar]
- 51. Fülöp T, Larbi A, Witkowski JM. Human inflammaging. Gerontology. 2019;65:495‐504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information