

Abstract
Aims
Cardiac contractile dysfunction is prevalent in rheumatoid arthritis (RA), with an increased risk for heart failure. A hallmark of RA has increased levels of peptidyl arginine deaminases (PAD) that convert arginine to citrulline leading to ubiquitous citrullination, including in the heart. We aimed to investigate whether PAD‐dependent citrullination in the heart was linked to contractile function in a mouse model of RA during the acute inflammatory phase.
Methods
We used hearts from the collagen‐induced arthritis (CIA) mice, with overt arthritis, and control mice to analyze cardiomyocyte Ca2+ handling and fractional shortening, the force‐Ca2+ relationship in isolated myofibrils, the levels of PAD, protein post‐translational modifications, and Ca2+ handling protein. Then, we used an in vitro model to investigate the role of TNF‐α in the PAD‐mediated citrullination of proteins in cardiomyocytes.
Results
Cardiomyocytes from CIA mice displayed larger Ca2+ transients than controls, whereas cell shortening was similar in the two groups. Myofibrils from CIA hearts required higher [Ca2+] to reach 50% of maximum shortening, ie Ca2+ sensitivity was lower. This was associated with increased PAD2 expression and α‐actin citrullination. TNF‐α increased PAD‐mediated citrullination which was blocked by pre‐treatment with the PAD inhibitor 2‐chloroacetamide.
Conclusion
Using a mouse RA model we found evidence of impaired cardiac contractile function linked to reduced Ca2+ sensitivity, increased expression of PAD2, and citrullination of α‐actin, which was triggered by TNF‐α. This provides molecular and physiological evidence for acquired cardiomyopathy and a potential mechanism for RA‐associated heart failure.
Keywords: Ca2+ sensitivity, citrullination, heart failure, PAD, rheumatoid arthritis
1. INTRODUCTION
Patients with rheumatoid arthritis (RA) have a significantly higher risk to develop heart disease, a major contributor to the disease burden and mortality in RA. 1 , 2 , 3 Myocardial contractile function may be particularly vulnerable, as patients with RA have a greater risk of heart failure, 4 , 5 , 6 independent of traditional cardiovascular risk factors, medication and, prevalent and incident ischemic heart disease. 6 , 7 , 8 , 9 The mechanisms that link RA to heart failure are incompletely known. Impaired vascular smooth muscle contractility is described in animal models of RA. 10 , 11 We recently demonstrated myocardial hypertrophy, fibrosis, and impaired contractility due to defective Ca2+ handling in a mouse model of RA at a late stage after remission of the synovial inflammation. 12 The risk of heart failure increases early after the onset of RA, 6 indicating that systemic factors in active RA can promptly impair contractile function. How contractility and the contractile mechanisms are affected in the early acute phase, with overt arthritis, is yet unknown.
The myofibrillar contractile power is essentially graded by the concentration of free cytoplasmic Ca2+([Ca2+]i). Electrical stimulation of cardiomyocyte's plasma membrane leads to transient elevation of [Ca2+]i via the opening of the L‐type Ca2+ channel (also known as dihydropyridine receptor, DHPR), which triggers Ca2+ release from the sarcoplasmic reticulum (SR) via the ryanodine receptor 2 (RyR2) channel protein complex. 13 Binding of Ca2+ to troponin C triggers contraction by allowing the myosin cross‐bridges to bind to actin. All these steps are susceptible to diverse regulatory factors including post‐translational modifications of myofibrillar and Ca2+ handling proteins.
Citrullination is a post‐translational modification of proteins caused by peptidyl arginine deaminases (PADs), which convert the amino acid arginine to the non‐classical amino acid citrulline by replacing the primary positively charged guanidine group with a neutrally charged ureido group. While protein citrullination is a regulatory process under normal conditions, increased protein citrullination is a hallmark of RA and other inflammation‐linked diseases, such as multiple sclerosis and cancer. 14 , 15 , 16 Cellular Ca2+ and inflammation signals are important regulators of PAD expression and activity. 17 Interestingly, PAD2‐dependent citrullination of sarcomeric proteins has been observed in heart tissue of patients with cardiomyopathies, and the myofibrillar Ca2+ sensitivity was decreased in PAD2‐exposed mouse cardiomyocytes. 18
Contractile mechanisms and cardiac Ca2+ handling in an acute phase of RA have, to our knowledge, never been studied at a cellular level. We, therefore, aimed to investigate heart function in collagen‐induced arthritis (CIA) mouse, a model of human RA, which develops acute arthritis and reflects the early inflammatory phase. To study contractile regulation and PAD‐dependent modifications in the CIA heart we measured cardiomyocyte Ca2+ handling and shortening, Ca2+ sensitivity in isolated myofibrillar preparations, PAD2 expression, and protein citrullination (Figure 1).
FIGURE 1.

Development of the collagen‐induced arthritis (CIA) mouse model of Rheumatoid Arthritis. DBA mice were subcutaneously (s.c.) injected with 100 μg of type II collagen (CII) and 300 μg of M tuberculosis in 0.1 ml of the emulsion into the base of the tail. On day 5, the animals were boosted with a subcutaneous (s.c.) injection of 100 μg of CII in Freund's incomplete adjuvant. Control mice were injected with 0.1 ml saline on both occasions. The CIA mice were sacrificed when they had developed overt arthritis in at least 2 paws (day 10–15) and the hearts were collected for: cardiomyocytes isolation ([Ca2+]i transients), myofibrils (force‐Ca2+ relationship) and biochemistry (western blot).
2. RESULTS
2.1. Cardiomyocyte Ca2+ handling and contractility in CIA mice
Cardiomyocytes isolated from CIA mice displayed larger [Ca2+]i transient amplitudes than controls (Figure 2A,B). Moreover, the [Ca2+]i transient decay was faster in the CIA, as indicated by a shorter time constant (Tau; Figure 2C). Increased stimulation‐induced [Ca2+]i transient amplitudes might be due to increased SR Ca2+ stores. To test this, CIA and control cardiomyocytes were superfused with caffeine (10 mM), which fully opens the RyR2 and empties Ca2+ from the SR. 19 No difference in the caffeine‐induced increase in fluo‐3 fluorescence was seen between CIA and control cardiomyocytes (F/F0, control, 8.7 ± 0.6; CIA 9.0 ± 0.5, mean ± SEM, p = 0.7, Figure 2D). This suggests that the SR Ca2+ load was unaltered in CIA cardiomyocytes and hence could not explain the larger action potential‐induced [Ca2+]i transients seen in the CIA heart.
FIGURE 2.
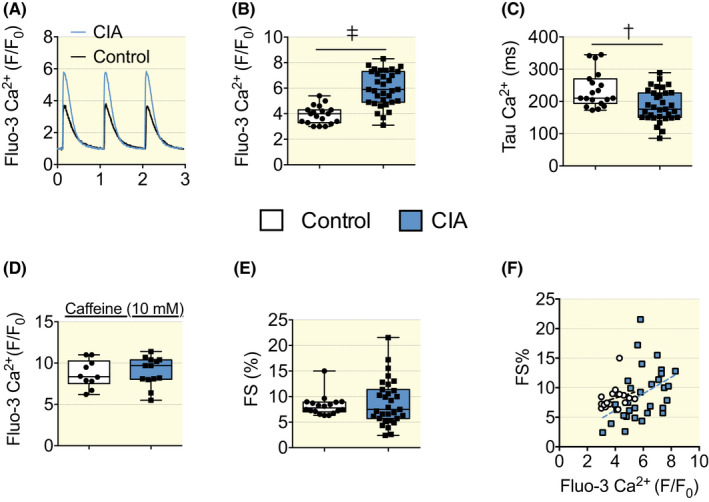
[Ca2+]i and fractional shortening measured in isolated cardiomyocytes, Fluo‐3 AM was used to measure [Ca2+]i transients in cardiomyocytes that were electrically paced (1 Hz) to elicit repeated contractions. Representative [Ca2+]i transient records (A). Mean data of [Ca2+]i transient amplitudes (B), Tau, [Ca2+]i decay time‐constant (C). Caffeine (10 mM) was used to trigger the opening of the RyR2 and cause the maximum release of SR Ca2+ (D). Fractional shortening (E) of cardiomyocytes stimulated at 1 Hz under control conditions (n = 19–33). (F) XY graph of Ca2+ fluorescence on the X axis and contractility (FS%) on the Y axis for each cardiomyocyte. Cardiomyocytes of the CIA group showed higher Ca2+ transients as they distributed on the right part of the graph and right shifting of the linear regression curve of the CIA group. Control r 2 0.069, p = 0.29, slope 0.731 ± 0.7, CIA r 2 0.183, p < 0.05, slope 0.139 ± 0.5. Data are presented as box and whiskers min and max including all points. *p < 0.05, †p < 0.01, ‡p < 0.001 with unpaired t test.
Surprisingly, despite the increased [Ca2+]i transient amplitudes in the CIA cardiomyocytes, fractional shortening was similar to control (Figure 2E). We plotted the Ca2+ fluorescence on the X axis and the contractility (FS%) on the Y axis for each cardiomyocyte. Cardiomyocytes of the CIA group showed higher Ca2+ transients as their distribution is displayed on the right part of the graph (Figure 2F). This indicates impaired myofibrillar cross‐bridge function in CIA cardiomyocytes manifested as decreased Ca2+ sensitivity, although an additional general reduction in cross‐bridge contractility may also be present. To distinguish between these two possibilities, cardiomyocytes were exposed to the inotropic β‐adrenergic agonist isoproterenol (ISO, 100 nM). In the presence of ISO, [Ca2+]i transients became larger and faster with no difference in amplitude (Figure S1A) and little difference in decay rate (Figure S1B) between CIA and control cardiomyocytes. The fractional shortening in the presence of ISO was about twice as large as under control conditions and there was no significant difference between CIA and control cardiomyocytes (Figure S1C). The ISO‐exposure experiments indicate that the response to adrenergic stimulation was not affected in cardiomyocytes isolated from CIA animals. Indeed, upon maximal stimulation, there were no changes in Ca2+ transients amplitude with a slight but not significant decrease in fractional shortening. This supports the notion that decreased Ca2+ sensitivity rather than a generally reduced cross‐bridge contractility is the cause of the impaired shortening response to the [Ca2+]i as seen at baseline.
Inflammatory signals may alter mitochondrial function, including altered Ca2+ handling and increased production of oxygen radicals. 20 , 21 We measured mitochondrial Ca2+ using the fluorescent indicator Rhod‐2. 22 , 23 In the CIA cardiomyocytes, the increase in Rhod‐2 signals was larger than in control cardiomyocytes (Figure 3A,B). These results imply increased mitochondrial Ca2+ uptake in CIA cardiomyocytes, which would act to decrease, rather than increase, the amplitude of [Ca2+]i transients. However, mitochondria might play a minor role in the cytosolic Ca2+ buffering of ventricular cardiomyocytes. 24 We also measured reactive oxygen species (ROS) generation in the mitochondria with the mitochondrial ROS indicator MitoSOX Red, 22 , 25 and the results show higher MitoSOX Red fluorescence in CIA than in control cardiomyocytes (Figure 3C,D). Thus, mitochondria from CIA mice were more prone to take up Ca2+ and produce ROS during pacing.
FIGURE 3.
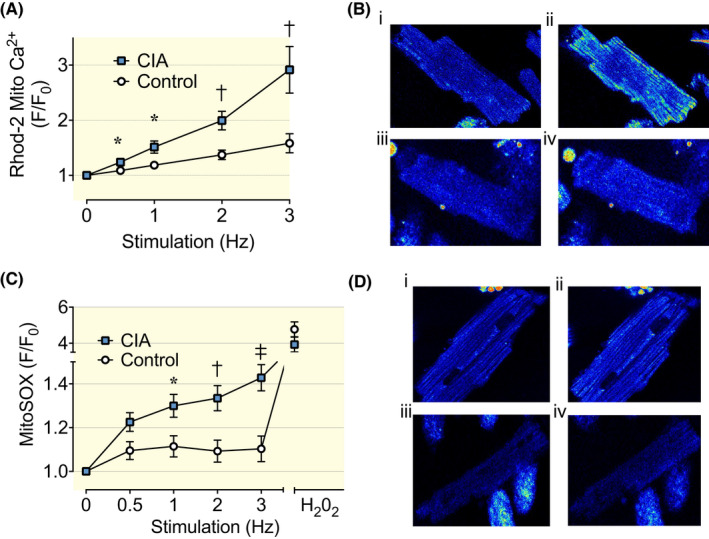
Mitochondrial Ca2+ uptake and ROS production in cardiomyocytes from CIA and control mice. (A) Mean (±SEM) of mitochondrial Ca2+ in cardiomyocytes electrically paced at 0.5–3 Hz by brief current pulses, n = 21–30. Rhod‐2 fluorescence is normalized to the resting value in each cardiomyocyte. (B) Representative images of Rhod‐2 fluorescence in CIA (i and ii) and control (iii and iv) cardiomyocytes at the end of 0.5 Hz (i and iii) and 3 Hz (ii and iv) pacing. (C) Mitochondrial ROS in cardiomyocytes was measured as MitoSOX Red fluorescence (mean data ± SEM, n = 12–22) at 0.5–3 Hz electrical stimulation. Hydrogen peroxide (H2O2; 1 μM) was used as a positive control at the end of the experiment. (D) Representative images of MitoSOX Red fluorescence in CIA (i and ii) and control (iii and iv) cardiomyocytes at the end of 0.5 Hz (i and iii) and 3 Hz (ii and iv) pacing. Data are presented as mean ± SEM. *p < 0.05, †p < 0.01, ‡p < 0.001 with Mann–Whitney non‐parametric test.
To address the mechanism causing the larger [Ca2+]i transients in CIA than in control cardiomyocytes, we measured the expression of proteins critical for cardiomyocyte Ca2+ handling. No difference in protein expression between CIA and control hearts was found for the Ca2+ channels RyR2 or DHPR, the SR Ca2+ buffer CSQ2, or the SERCA regulatory protein phospholamban (Figure 4A,B). On the contrary, the expression of the SR Ca2+ ATPase SERCA 2a was markedly higher in CIA than in control hearts (Figure 4A,B), which is consistent with the larger and faster [Ca2+]i transients in CIA cardiomyocytes. 26
FIGURE 4.
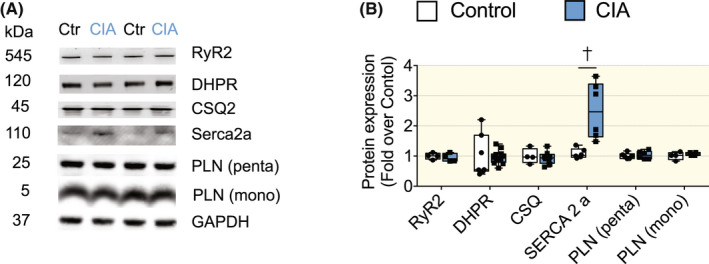
Ca2+ handling protein expression in hearts of CIA and control mice. (A) Representative immunoblots of, ryanodine receptor 2 (RyR2), dihydropyridin receptor (DHPR), calsequestrin 2 (CSQ2), SERCA2a, phospholamban (PLN, pentameric and monomeric form) and housekeeping protein glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). (B) Mean band densities normalized to GAPDH and the mean value in control hearts set to 1.0 (n = 6–8). Data are presented as box and whiskers min and max including all points. *p < 0.05, †p < 0.01, ‡p < 0.001 with unpaired t test. Data were analyzed with Mann–Whitney non‐parametric test.
2.2. Myofibrillar force‐Ca2 + relationship
The [Ca2+]i‐shortening data described above, where a higher [Ca2+]i did not increase shortening in CIA cardiomyocytes, suggests that myofibrillar Ca2+ sensitivity was reduced. To study this further, we used atomic force cantilevers to directly measure the force generated by myofibrils activated at different concentrations of Ca2+. The results showed markedly lower submaximal forces in CIA than in control myofibrils, whereas there was no difference in maximal force (Figure 5A). The [Ca2+] required to achieve 50% of the maximum force (Ca50) was significantly higher in CIA than in control myofibrils, i.e. the Ca2+ sensitivity was decreased in CIA myofibrils (Figure 5B). The rates of force development at the onset of maximum contractions (KAct) and redevelopment after the shortening step (KTr) were both lower in CIA than in control myofibrils (Figure 5C,D). The rate of relaxation following deactivation did not differ between CIA and control myofibrils (Figure 5E).
FIGURE 5.
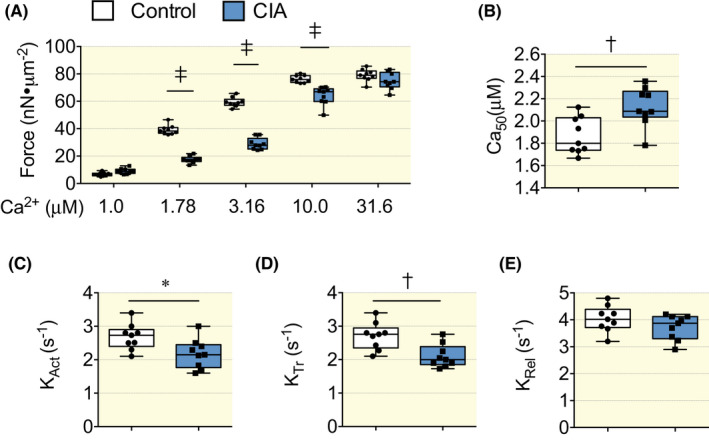
Reduced Ca2+ sensitivity in myofibrils isolated from CIA hearts. (A) The submaximal Ca2+ activated force was lower in isolated myofibrils from CIA hearts than in controls, whereas maximal Ca2+ activated force was not different between the groups. (B) The [Ca2+] needed to achieve 50% of maximal myofibrillar force (Ca50) was significantly increased in the CIA group, showing that myofibrillar Ca2+ sensitivity was lower in the CIA hearts. (C,D) The rates of force development at the onset of maximum contractions (KAct) and redevelopment after a shortening step (KTr) were both reduced in CIA than in control myofibrils. (E) The rate of relaxation following deactivation (KRel) did not differ between CIA and control myofibrils. Average data in (A–E) is displayed as box and whiskers min and max including all points., n = 9. *p < 0.05, †p < 0.01, ‡p < 0.001, repeated measurements anova (A), t test (B–E).
2.3. PAD2‐dependent citrullination of the sarcomeric protein α‐actin and SR Ca2+ channel RyR2
PAD enzymes, which catalyze the conversion of arginine to citrulline in proteins, can increase under inflammatory conditions. Accordingly, PAD2 expression was significantly higher in the CIA than in control hearts (Figure 6A). Interestingly, impaired myofilament Ca2+ sensitivity in heart failure has been linked to increased PAD‐dependent citrullination of sarcomeric proteins. 18 We assessed citrullination by measuring the binding of an anti‐citrulline specific antibody to immunoprecipitated preparations of α‐actin from heart tissue of CIA and control mice and observed a significantly higher level of citrullination in the CIA than in the control group (p < 0.001) (Figure 6B).
FIGURE 6.
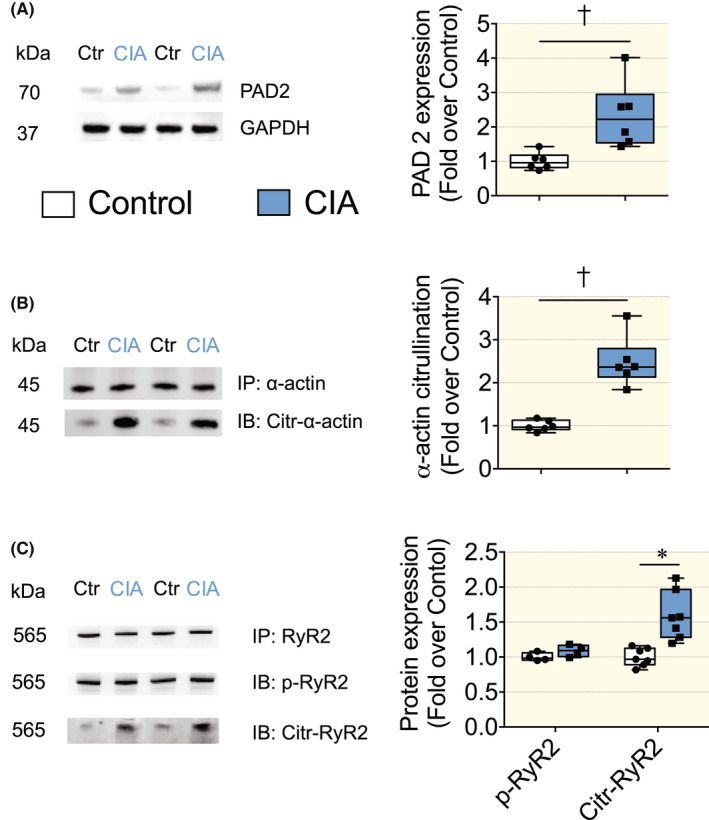
Expression of PAD2 and post‐translational modification of α‐Actin and RyR2 in hearts of CIA and control mice. (A) (left) Expression of PAD2 in hearts of CIA and control mice; (right) mean band densities normalized to GAPDH and the mean value in control hearts set to 1.0 (n = 6). (B) (left) Representative immunoblots from immunoprecipitated α‐Actin; (right) mean citrulline band densities normalized to total α‐Actin and the mean value in control hearts set to 1.0 (n = 6). (C) (left) Representative immunoblots from immunoprecipitated RyR2; (right) mean phosphorylated (p‐RyR2; PKA phosphorylation site RyR2‐S2808) and citrullinated RyR2 bands densities normalized to total RyR2 and the mean value in control hearts set to 1.0 (n = 7). Data are presented as box and whiskers min and max including all points. *p < 0.05, †p < 0.01, ‡p < 0.001 with Mann–Whitney non‐parametric test.
Functional alterations in excitation‐contraction coupling are often due to post‐translational protein modifications (i.e. oxidation, phosphorylation). We used heart lysates from WT and CIA mice hearts and blotted for oxidation marker such as Malondialdehyde (MDA). Our results showed no difference in MDA levels between the control and CIA groups (Figure S2). This suggests that oxidative stress mediated by peroxidation cannot explain the difference in Ca2+ sensitivity.
The SR Ca2+ channel RyR2 can regulate the amplitude of Ca2+ transients, where protein kinases A (PKA)‐dependent phosphorylation of the RyR2, is an important post‐translational modification that can increase SR Ca2+ release under conditions of adrenergic stress. 27 Therefore, we measured in heart biopsies of mice, the phosphorylation levels of RyR2 using a specific antibody (RyR2‐S2808) targeting the PKA phosphorylation site. 27 However, our results showed no significant difference between the control and CIA groups suggesting that the increased cytoplasmic Ca2+ transient amplitude in the CIA group was not a consequence of PKA‐mediated adrenergic stress.
Furthermore, we checked the citrullination levels of RyR2 in cardiac tissues during the inflammatory phase of RA. Our results showed that the CIA group has a higher grade of citrullination in RyR2 compared to the control group (Figure 6C), which could potentially affect the permeability of Ca2+ through this channel.
2.4. TNF‐α‐induced PAD‐dependent citrullination of proteins in cardiomyocytes
To investigate the role of proinflammatory cytokines in PAD‐mediated citrullination and provide more mechanistic insights, an in vitro model to mimic RA was employed (Figure 7A).
FIGURE 7.
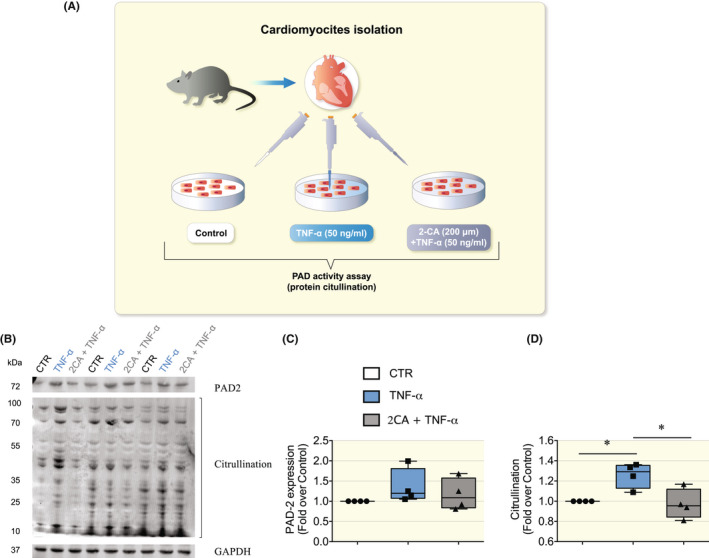
TNF‐α triggered PAD‐dependent citrullination in cardiomyocytes. (A) Cartoon depicting the in vitro model to mimic the inflammatory phase of RA. We used cardiomyocytes isolated from DBA mice (N = 4 mice) treated for 20 h with TNF‐α (50 ng/ml), pretreated with or without 2‐chloroacetamidine (2‐CA, 200 μM) a small molecule that inhibits PADs active‐sites. Then, PAD‐dependent citrullination was measured and normalized to baseline levels (e.g. cells untreated, CTR). (B) Representative immunoblots from cardiomyocytes lysates blotted for PAD2, citrullination, and GAPDH. (C) Mean band density of PAD2 normalized to GAPDH. (D) The mean band density of protein citrullination normalized to GAPDH, which appeared to be significantly increased in cardiomyocytes treated with TNF‐α. Data are presented as box and whiskers min and max including all points. *p < 0.05, †p < 0.01, ‡p < 0.001 with one‐way anova test, multiple comparisons.
We used cardiomyocytes isolated from DBA mice treated for 20 h with TNF‐α, with or without 2‐chloroacetamidine (2‐CA), a small molecule that inhibits all PADs active‐site. 15 , 28 , 29 Our results showed that cardiomyocytes treated with TNF‐α presented significantly higher levels of PAD‐mediated citrullination, while pretreatment with 2‐chloroacetamide maintained PAD activity at basal levels (Figure 7B–D). These findings suggest that a major pro‐inflammatory cytokine that is elevated in RA such as TNF‐α plays a role in modulating PAD activity in cardiomyocytes, which could impact the function of myofibrillar proteins during RA.
3. DISCUSSION
Inflammation is a key pathogenic mediator of cardiovascular disease and it is increasingly recognized particularly in heart failure. In this context, it is clear that myocardial disease is prevalent in RA, which is seen by a high incidence of clinical heart failure as well as sub‐clinically impaired cardiac function. 6 Yet, mechanisms of cardiac dysfunction in RA have not been thoroughly addressed. Intriguingly, heart failure with preserved ejection fraction is a diagnosis overrepresented in RA patients. 30 Moreover, patients with rheumatoid arthritis are susceptible to more rapid subclinical changes in diastolic function than the general population. 31 Here, we found that mouse hearts from a model of acute CIA‐induced RA, display PAD‐dependent post‐translational modifications in core components of contractile regulation actin myofilaments, which were linked to reduced myofilament Ca2+ sensitivity.
Enhanced cardiomyocyte Ca2+ release could be due to augmented opening of the RyR2, and/or increased driving force for Ca2+ across the SR caused by an increase in SR luminal [Ca2+]. Our results do not support the second alternative, as the [Ca2+]i amplitude of a maximal SR Ca2+ release induced by caffeine stimulation was similar CIA and control cardiomyocytes. Furthermore, we observed no difference in the expression of the major SR Ca2+ buffering protein, calsequestrin‐2, between CIA and control hearts, which indicates that the SR Ca2+ buffering capacity was unchanged in CIA cardiomyocytes. However, molecular forms of calsequestrin‐2 and its abundance in junctional SR via altered trafficking may affect the SR Ca2+ buffering capacity, although mechanisms governing these phenomena remain unresolved. 32
The enhanced [Ca2+]i transients in CIA cardiomyocytes cannot be explained by altered expressions of RyR2, DHPR, CSQ2, or phospholamban since these did not differ between CIA and control cardiomyocytes. In contrast, the markedly increased expression of SERCA2a in CIA hearts would result in enhanced Ca2+ cycling and could explain the larger and faster Ca2+ transients. 26
An increased [Ca2+]i transient amplitude may also be due to decreased cytosolic Ca2+ buffering. 33 Accordingly, the decreased myofibrillar Ca2+ sensitivity in CIA cardiomyocytes can reflect reduced Ca2+ binding to troponin C, which would act toward increased [Ca2+]i transient amplitudes. In contrast, we observed an increased mitochondrial Ca2+ uptake during pacing in CIA cardiomyocytes, which would tend to decrease the amplitude of [Ca2+]i transients, although the potential of mitochondria to serve as an effective cytosolic Ca2+ buffer has not yet reached unanimous scientific consensus. 24 To sum up, augmented RyR2 opening appears to be the most likely explanation for the increased [Ca2+]i transient amplitude in CIA cardiomyocytes but further experiments are required to reveal the exact mechanism.
Modifications of RyR2 leading to increased channel open probability can mediate increased Ca2+ transients, 27 , 34 but also lead to SR Ca2+ leak that over time reduces SR Ca2+ stores and diminishes Ca2+ release. 35 , 36 If RyR2 modifications lead to increased or decreased SR Ca2+ release seems to be reliant on, for instance, the type of modification, local redox balance, duration, and changes in the interaction between RyR2 and associated proteins. 37 , 38 In this study, we investigated post‐translational modifications of RyR2 that could explain the increased Ca2+ transient amplitude at unchanged SR Ca2+ load observed in the acute inflammatory phase of RA. Our results showed that in the hearts of CIA mice, the levels of PKA‐mediated phosphorylation were unchanged. On the contrary, we found increased PAD‐dependent post‐translational modifications of RyR2, which might be linked to the enhanced Ca2+ permeability. Moreover, the RyR2 protein sequence is particularly abundant in arginine, which is a positively charged amino acid that has high aqueous pKa's (13.8 pKa) 39 indicating a strong propensity to carry charge at physiological pH affecting protein structure and function that involve electrostatic interaction and protein solvation. Therefore, the conversion of arginine to citrulline by removing the positively charged amine group (NH2, weak base) might have consequences on the permeation properties of the channel, facilitating Ca2+ flux when the gate opens or affects the core and cytoplasmic shell portion of the channel, which controls the gating activity. 40
Moreover, the larger [Ca2+]i transients in CIA cardiomyocytes were counteracted by the reduced myofibrillar Ca2+ sensitivity; hence, the fractional shortening of cardiomyocytes was similar between the two groups. In our preclinical model of acute inflammatory RA, where heart remodeling might not have occurred yet, the contractility observed in vitro was not different between groups, then we did not expect to see major differences in systolic function between CIA and control in vivo. Nevertheless, we previously showed that a preclinical model of chronic RA presented cardiac remodeling and systolic function impairment both in vitro and in vivo. 12
3.1. Reduced Ca2+ sensitivity in myofibrils
Analyses at the myofibrillar level using atomic force cantilevers (AFC) revealed lower submaximal forces in CIA than in control myofibrils, whereas the maximum force was similar in the two groups. This fits with a reduced myofibrillar Ca2+ sensitivity in CIA myofibrils and can explain the inability of CIA cardiomyocytes to respond to the larger [Ca2+]i transients with greater shortening. Moreover, the unaltered maximum force in CIA myofibrils fits with the results obtained when isolated cardiomyocytes were exposed to the β‐adrenergic agonist isoproterenol, 41 which showed similar fractional shortenings in CIA and control cells at presumably saturating [Ca2+]i transient amplitudes. The lower rates of force development in myofibrils from CIA than from control hearts further illustrates an impaired myofibrillar function in CIA hearts. The rate of force development represents the sum of the rates of myosin cross‐bridge attachment (f app) and detachment (g app) to the actin filament, whereas the rate of relaxation only reflects the detachment rate. 42 The rate of relaxation was similar in CIA and control myofibrils, which means that the reduced rate of force development in CIA myofibrils reflects a decreased f app combined with an unaltered g app. Interestingly, a decreased f app/g app ratio can be linked to reduced forces at submaximal [Ca2+] and hence decreased myofibrillar Ca2+ sensitivity. 42 The fact that K Act, which involves Ca2+ activation of the actin filament, and K Tr, where the actin filament is already active, were equally decreased in CIA myofibrils indicates that the major mechanism behind the reduced myofibrillar Ca2+ sensitivity in CIA cardiomyocytes was altered cross‐bridge kinetics rather than decreased activation due to impaired Ca2+ binding to troponin C. 42
3.2. Citrullination during rheumatoid arthritis
Circulating anti‐citrulline protein autoantibodies (ACPA) and anti‐PAD2 antibodies is a common finding used to diagnose seropositive RA. 16 The suggested background to the formation of ACPA in RA is an immune reaction to the increased PAD activity and presence of citrullinated proteins. 16 Moreover, the levels of ACPA in the blood of naïve RA patients correlate with the worsening of global longitudinal systolic strain and LV systolic function. 43 , 44 Although the formation of circulating ACPA may possibly be triggered by the increased presence of citrullinated proteins with RA, it should be noted that circulating ACPA is not a clear‐cut biomarker for effects mediated by protein citrullination. Interestingly, a cardiac postmortem immunohistology study showed higher protein citrullination in heart tissue of RA patients. 45 In our RA model, we found higher expression levels of PAD2 in the heart together with significantly increased levels of citrullination in α‐actin. Other post‐translational modifications in myofibrillar proteins including PKA‐mediated phosphorylation and oxidation‐mediated hypo‐phosphorylation of cTnI may affect the Ca2+ sensitivity of cardiomyocytes. 46 , 47 However, citrullination of sarcomeric proteins has been reported to reduce Ca2+ sensitivity of myofibrils during heart failure, 18 and our study provided supporting evidence that citrullination of cardiac α‐actin and RyR2 also occurred in a preclinical model of the acute inflammatory phase of RA. Furthermore, our in vitro experiments showed that the proinflammatory cytokine TNF‐α plays a key role in the PAD‐mediated citrullination of proteins in cardiomyocytes. These novel findings suggest that TNF‐α is not only a valuable predictive biomarker of disease activity in RA patients, 48 , 49 , 50 but it is also involved in modulating excitation‐contraction coupling in cardiomyocytes through PAD‐dependent signaling.
3.3. Myocardial function in RA patients
The reduced myofilament Ca2+ sensitivity in the CIA cardiomyocyte would result in a reduced contractile reserve in vivo. This mechanism fits with the evidence that myocardial function in RA patients, also without clinical signs of heart failure, is slightly impaired when measured with sensitive methods such as myocardial wall strain in speckle tracking echocardiography. 51 , 52 Moreover, this myocardial functional detriment fits with the increased incidence of developing clinical heart failure in RA, independent of ischemic heart disease. 6 It remains unknown whether RA predisposes more to heart failure with reduced or with preserved ejection fraction, but in both syndromes, systolic strain is reduced, thus RA can conceivably be a risk factor for both.
3.4. Conclusion
In conclusion, hearts from mice with acute experimental RA display increased PAD‐dependent citrullination of the sarcomeric protein α‐actin and reduced myofilament Ca2+ sensitivity (Figure 8). In this acute RA model, the decreased Ca2+ sensitivity is counterbalanced by increased [Ca2+]i transients. In chronic RA, on the contrary, citrullination of contractile proteins together with impaired SR Ca2+ release might lead to declined contractile function and pumping capacity, which could explain the higher incidence of heart failure and mortality amongst patients with RA, 6 , 9 thus highlighting the importance of monitoring RA patients for the development of myocardial dysfunction. Remarkably, a late‐stage RA model presented reduced Ca2+ transient amplitudes and reduced contractility in isolated cardiomyocytes as a consequence of diminished SR Ca2+ stores linked to oxidative post‐translation modifications of Ca2+ handling proteins. 12 Our present results, on the other hand, showed that in the acute inflammatory phase of RA, citrullination rather than oxidative modification of proteins plays a crucial role in the excitation‐contraction coupling of cardiomyocytes. Therefore, we propose that the grade of inflammation and the stage of RA represent key factors in the modulation of cardiac contractile function.
FIGURE 8.
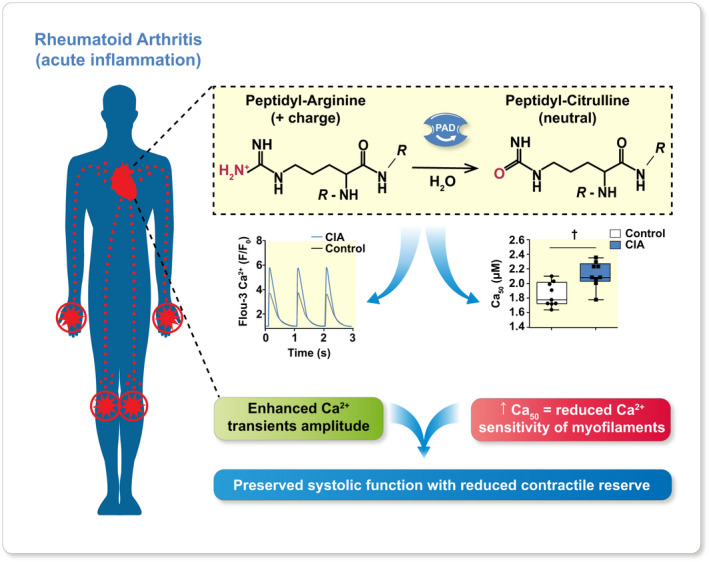
Graphical abstract. In the acute inflammation phase of rheumatoid arthritis, the expression and activity of peptidyl arginine deaminase 2 (PAD2) in the heart increases. This links to aggravated PAD‐dependent citrullination of proteins involved in contractile regulation, which causes reduced myofilament Ca2+ sensitivity (e.g. increased amount of Ca2+ needed to elicit 50% of maximal myofibrillar force, Ca50). This reduction is counteracted by increased [Ca2+]i transient amplitudes, yet it decreases the contractile reserve. A declined contractile reserve that sensitizes the heart to insults is a factor that could explain the high prevalence of heart failure amongst patients with RA and shows the importance of monitoring RA patients for the development of HF.
3.5. Study limitation
From a translational point of view, there is supportive evidence that citrullination occurs in heart of patients with heart failure and that PAD‐dependent citrullination affects the hearts of RA patients. Whether there is citrullination of alpha‐actin in heart of RA patients is not known yet. A limitation of our study is the absence of human cardiac biopsies to confirm our preclinical findings. However, our preclinical results reveal mechanisms that are likely to apply also in human patients.
Citrullination of sarcomeric proteins in cardiac biopsies of heart failure patients was associated with decreased Ca2+ sensitivity of myocytes. 18 Preventing citrullination by knocking down PAD2 in RA is potentially an experiment that might provide mechanistic insights. However, based on the current literature, we believe that it will not provide conclusive results because preclinical models of chronic inflammatory diseases show that protein citrullination can occur independently of the presence of PAD‐2, 53 , 54 suggesting that ablation of citrullination requires knockout of all PAD activity (e.g. blocking the PAD active site with 2‐Chloroacetamide).
4. MATERIALS AND METHODS
All the material submitted is conformed with good publishing practice in physiology. 55
4.1. Ethics approval
All experimental procedures were approved by the Stockholm North Ethical Committee on Animal Experiments. Animal experiments complied with the Swedish Animal Welfare Act, the Swedish Welfare ordinance, and recommendations and applicable regulations from Swedish authorities.
4.2. Animals
We used female DBA mice 12–16 weeks old for CIA‐induction (N = 17) and control mice (N = 15). The generation of the CIA mice has been previously described. 56 In short, 100 μg of type II collagen (CII) and 300 μg of M tuberculosis in 0.1 ml of emulsion were subcutaneously injected into the base of the tail. On day 5, the animals were boosted with a subcutaneous injection of 100 μg of CII in Freund's incomplete adjuvant (0.1 ml). Control mice were injected with 0.1 ml saline (same administration route) on both occasions (cartoon Figure 1). The development of arthritis was followed daily by assessing erythema and swelling of the metatarsophalangeal and ankle joints. When a CIA mouse displayed overt arthritis, in at least 2 paws, it was sacrificed by rapid neck disarticulation.
4.3. Cardiomyocyte isolation
Single viable cardiomyocytes were isolated freshly from cardiac ventricles of 3 CIA mice and 3 control mice for each experiment (including Ca2+ transient, contractility experiments mitochondrial Ca2+ and MitoSox experiments) following the protocol developed by the Alliance for Cellular Signaling (AfCS Procedure Protocol ID PP00000125). 22 , 57
4.4. Confocal imaging
Cardiomyocytes were plated on laminin‐coated glass bottom dishes (Mattek). The dishes were placed in a custom‐built perfusion/stimulation chamber and continuously perfused with O2/CO2 (95/5%) bubbled Tyrode solution (room temperature) with the following composition (in mM): NaCl 121, KCl 5.0, CaCl2 1.8, MgCl2 0.5, NaH2PO4 0.4, NaHCO3 24, EDTA 0.1, glucose 5.5. Throughout the experiments, cardiomyocytes were generally stimulated to contract at 1 Hz with supra‐threshold electrical current pulses delivered via two platinum electrodes attached to the perfusion/stimulation chamber. Measurements were only performed in cardiomyocytes that contracted upon electrical stimulation and displayed normal morphology (e.g. striated, “brick shaped”). Cells that displayed spontaneous contractions were discarded. Fluorescence was measured using a confocal microscope (Biorad 1024; 40× oil immersion lenses). Fiji‐ImageJ software was used to quantify changes in fluorescence.
4.5. Measurement of [Ca2+]i with Fluo‐3 AM
Cardiomyocytes were loaded with the fluorescent indicator Fluo‐3 AM (~5 μM; Invitrogen) for ~20 min. [Ca2+]i transients was measured using confocal microscopy in line scan mode. A wavelength of 491 nm was used for excitation and emitted light was collected at >515 nm. The line scan was oriented along the long axis of the cardiomyocyte. The fluorescent signal (F/Fo) was calculated as the ratio of the maximum fluorescence (F) and resting fluorescence before the start (F0) of the [Ca2+]i transient. 57 The fractional shortening (FS) of cardiomyocytes was measured as the length in diastole (Max length) and systole (Min length). FS% was calculated as: (Max length−Min length)/Max length × 100. In subset experiments, isolated myocytes were acutely exposed to the β‐receptor agonist isoproterenol (ISO; 100 nM in Tyrode solution) as a positive control for an inotropic response. In other experiments, isolated cardiomyocytes were exposed to caffeine (10 mM in Tyrode solution) that triggers the opening of RyR2 allowing the total SR Ca2+ load to be assessed. 12 All the experiments were conducted blinded regarding the treatment of the mice.
4.6. Measurement of mitochondrial Ca2+ using Rhod‐2 AM
Isolated cardiomyocytes were incubated with the fluorescent indicator Rhod‐2 AM (5 μM; Invitrogen) for ~1 h at room temperature, followed by washout. The cardiac cells were perfused with Tyrode solution and electrically paced to contract with incremental pacing frequencies (0.5, 1, 2, and 3 Hz). The Rhod‐2 fluorescence was measured using excitation at 568 nm and the emitted signal was collected through a bandpass filter (585 nm). Confocal images were taken at rest and then at regular intervals after a series of contractions of cardiomyocytes, while pacing was temporarily stopped. The fluorescent signal in each cardiomyocyte normalized to that at the start of the experiment.
4.7. Confocal imaging of ROS production using MitoSOX Red
The fluorescent indicator MitoSOX Red was used to measure mitochondrial ROS production. Cardiomyocytes, isolated as above, were loaded with MitoSOX Red (~5 μM; Invitrogen) in Tyrode for ~20 min at room temperature, followed by washout. Confocal images were obtained by excitation at 488 nm and measuring the emitted light at >515 nm. Cardiomyocytes were electrically paced to contract with incremental stimulation frequencies (0.5, 1, 2, and 3 Hz). Confocal images were taken at rest and then at regular intervals after a series of contractions of cardiomyocytes, while pacing was temporarily stopped. The fluorescent signal in each cardiomyocyte normalized to that at the start of the experiment.
4.8. Measurement of the myofibrillar force–Ca2+ relationship
Small pieces of the heart samples from CIA and control mice, were homogenized following standard procedures, 58 yielding a solution containing isolated myofibrils. The myofibrils were transferred into an experimental chamber and placed on top of an inverted microscope equipped with a system of atomic force cantilevers (AFC). 59 In this system, a laser shone upon and reflected from a pre‐calibrated AFC, which acts as a force transducer. The cantilever deflection caused by myofibril activation was detected and recorded using a photo‐quadrant detector. Since the stiffness of the AFC (k) was known and we measured the amount of cantilever displacement (Δd), the force (F) can be calculated as F = k × Δd.
Myofibrils chosen for mechanical testing displayed an evenly distributed striation pattern and between 10 and 30 sarcomeres in series. Under high magnification, the contrast between the dark bands of myosin (A‐bands) and the light bands of actin (I‐bands) provided a dark–light intensity pattern, representing the striation pattern produced by the sarcomeres, which allowed measurements of sarcomere length during the experiments. Using micromanipulators, the myofibrils were attached between the AFC and a rigid glass needle. The rigid glass needle was connected to a piezoelectric motor and length changes during the experiments were induced by moving the needle. A computer‐controlled, multichannel fluidic system connected to a double‐barreled pipette was used for activation of the myofibril in solutions (15°C) with different Ca2+ concentrations: 1.0, 1.78, 3.16, 10.0, and 31.6 μM.
Once the myofibrils were fully activated and maximal force for a given Ca2+ concentration was obtained, they underwent a fast shortening (amplitude 30% of sarcomere length; speed 10 μm/s) during which the force declined and then rapidly redeveloped to reach a new force plateau. The force produced by the myofibrils after force re‐development and stabilization were averaged for a period of 2 s. All forces were normalized to the myofibril cross‐sectional area and expressed relative to the force produced at saturating [Ca2+]. The [Ca2+] required to achieve 50% of the maximum force (Ca50) was, for each myofibril, obtained by interpolation between the force immediately below and above 50% force. Rates of force development at the onset of contraction (KAct) and during the force redevelopment after the shortening (KTr) at saturating [Ca2+] were analyzed with a two‐exponential equation (a × (1−exp(−K × t)−exp(−l × t)) + b), and the rate of relaxation (KRel) was analyzed with a single‐exponential equation (a × exp(−K × [t−c]) + b). For both equations, t is time, K is the rate constant, a is the amplitude of the force change, and b and c are constants. All the experiments were conducted blinded regarding the treatment of the mice.
4.9. In vitro model of inflammatory phase during RA
Cardiomyocytes were isolated from DBA mice (12 weeks old, N = 4) and aliquoted in tubes (~100 000 cells/tube). The cells were washed with Tyrode's buffer and incubated for 20 h at RT with TNF‐α (50 ng/ml, R&D 410‐MT‐025/CF), pre‐treated with or without the PAD inhibitor 2‐chloroacetamide (200 μM, 2‐CA, Sigma Aldrich C0267‐500G). PAD activity was measured through quantification of protein citrullination normalized to the baseline levels of cardiomyocytes incubated with Tyrode's solution (Control, CTR).
4.10. Protein immunoblotting
Crude tissue homogenate from cardiac ventricles was separated by electrophoresis and transferred onto PVDF membranes. Membranes were incubated with primary antibodies: anti‐PAD2 (1:1000, Abcam, ab16478), anti‐RyR2 (1:5000, Abcam, ab2728), anti‐phosphorylated‐S2808‐RyR (1:5000, donated by Andrew R Marks, Columbia University), anti‐dihydropyridine receptor (1:1000, Abcam, ab2864), anti‐calsequestrin 2 (1:1000, Abcam, ab3516), anti‐SR Ca2+‐ATPase 2a (SERCA 2a; 1:1000, Abcam ab2861), anti‐phospholamban (1:1000, AbCam, ab2865), α‐actin (1:1000, Abcam ab28052), anti‐citrulline antibody (1:1000, Millipore, 07–377), Malondialdehyde (MDA) antibody (1: 1000, Abcam, ab27642), GAPDH (1:1000, Abcam, ab9485), and infrared‐labeled secondary antibodies (1:5000, Licor IRDye 680 and IRDye 800). Immunoreactive bands were analyzed using the Odyssey Infrared Imaging System. Band densities were quantified with Image J and normalized to GAPDH. All the experiments were conducted blinded regarding the treatment of the mice.
4.11. Protein immunoprecipitation
Dynabeads Protein G (Invitrogen) were incubated with anti‐α‐actin (Abcam, ab28052) or RyR2 antibody (Abcam, ab2728) for 40 min at RT to allow the cross‐binding between beads and antibody, then the beads were washed three times with phosphate buffer solution (PBS). Cardiac ventricle lysate (500 μg) was incubated at 4°C overnight with the beads conjugated to the anti‐α‐actin antibody. The immunocomplex was washed once with lysis buffer and then three times with PBS. Finally, the immunoprecipitate was collected in 50 μl of Laemmli buffer 2× (Bio‐Rad #161–0737, with β‐Mercaptoethanol 10% and DTT 0.05 M). Proteins were separated by electrophoresis and immunoblots were performed as described above and quantified relative to the RyR2 or α‐actin expression.
4.12. Statistics
Data in graphs are presented as box and whiskers min and max including all individual points. The sample size was calculated using a significance level of 5% and 80% power in order to detect any difference between the control and CIA groups. The normality distribution of the data was tested with D'Agostino and Pearson test. Statistical difference between the two groups was calculated using the Mann‐Withney test for non‐normally distributed data or unpaired Student's t test repeated measurements or anova as indicated. p < 0.05 was regarded as statistically significant. In the figures p values were indicated as follows: *p < 0.05, † p < 0.01, †† p < 0.001.
AUTHOR CONTRIBUTIONS
G.P. and D.C.A conceived the study. G.P., S.G., D. E. R., J.T.L, L.H.L, H. W. T.Y., and D. C. A. contributed to the acquisition, analysis, or interpretation of data. G.P., H. W., and D. C. A. drafted the manuscript together with S.G., M.C., D. E. R., L. H. L., and T. Y. that revised it and contributed to its intellectual content. All the authors approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
CONFLICT OF INTEREST
There is no conflict of interest.
PHYSIOLOGIC RELEVANCE
These results provide a potential mechanism as to why RA patients are prone to develop heart failure, providing a potential molecular target to impact the Ca2+ sensitivity of cardiomyocytes. Our findings emphasize the contribution of systemic inflammation to the development of heart failure.
Supporting information
Figure S1
Figure S2
ACKNOWLEDGMENTS
This study was supported by grants from the Swedish Research Council (HW and LHL), the Swedish Heart Lung Foundation (DCA and LHL), the Swedish Society for Medical Research (DCA), the Jeansson Family Foundation (DCA), the Swedish Society of Medicine (DCA), and the Lars Hierta Memorial Foundation (GP).
Pironti G, Gastaldello S, Rassier DE, et al. Citrullination is linked to reduced Ca2+ sensitivity in hearts of a murine model of rheumatoid arthritis. Acta Physiol. 2022;236:e13869. doi: 10.1111/apha.13869
Contributor Information
Gianluigi Pironti, Email: gianluigi.pironti@ki.se.
Daniel C. Andersson, Email: daniel.c.andersson@ki.se.
REFERENCES
- 1. Giles JT, Post W, Blumenthal RS, Bathon JM. Therapy insight: managing cardiovascular risk in patients with rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:320‐329. [DOI] [PubMed] [Google Scholar]
- 2. John H, Kitas G. Inflammatory arthritis as a novel risk factor for cardiovascular disease. Eur J Intern Med. 2012;23:575‐579. [DOI] [PubMed] [Google Scholar]
- 3. Giles JT, Fernandes V, Lima JA, Bathon JM. Myocardial dysfunction in rheumatoid arthritis: epidemiology and pathogenesis. Arthritis Res Ther. 2005;7:195‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicola PJ, Crowson CS, Maradit‐Kremers H, et al. Contribution of congestive heart failure and ischemic heart disease to excess mortality in rheumatoid arthritis. Arthritis Rheum. 2006;54:60‐67. [DOI] [PubMed] [Google Scholar]
- 5. Nicola PJ, Maradit‐Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: a population‐based study over 46 years. Arthritis Rheum. 2005;52:412‐420. [DOI] [PubMed] [Google Scholar]
- 6. Mantel A, Holmqvist M, Andersson DC, Lund LH, Askling J. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J Am Coll Cardiol. 2017;69:1275‐1285. [DOI] [PubMed] [Google Scholar]
- 7. Crowson CS, Nicola PJ, Kremers HM, et al. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52:3039‐3044. [DOI] [PubMed] [Google Scholar]
- 8. del Rincon ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737‐2745. [DOI] [PubMed] [Google Scholar]
- 9. Davis JM 3rd, Roger VL, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58:2603‐2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanghera C, Wong LM, Panahi M, Sintou A, Hasham M, Sattler S. Cardiac phenotype in mouse models of systemic autoimmunity. Dis Model Mech. 2019;12:dmm036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reynolds S, Williams AS, Williams H, et al. Contractile, but not endothelial, dysfunction in early inflammatory arthritis: a possible role for matrix metalloproteinase‐9. Br J Pharmacol. 2012;167:505‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pironti G, Bersellini‐Farinotti A, Agalave NM, et al. Cardiomyopathy, oxidative stress and impaired contractility in a rheumatoid arthritis mouse model. Heart. 2018;104:2026‐2034. [DOI] [PubMed] [Google Scholar]
- 13. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23‐49. [DOI] [PubMed] [Google Scholar]
- 14. Gyorgy B, Toth E, Tarcsa E, Falus A, Buzas EI. Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol. 2006;38:1662‐1677. [DOI] [PubMed] [Google Scholar]
- 15. Falcao AM, Meijer M, Scaglione A, et al. PAD2‐mediated citrullination contributes to efficient oligodendrocyte differentiation and myelination. Cell Rep. 2019;27:1090‐1102.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Curran AM, Naik P, Giles JT, Darrah E. PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol. 2020;16:301‐315. [DOI] [PubMed] [Google Scholar]
- 17. Bicker KL, Thompson PR. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers. 2013;99:155‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fert‐Bober J, Giles JT, Holewinski RJ, et al. Citrullination of myofilament proteins in heart failure. Cardiovasc Res. 2015;108:232‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tavi P, Hansson A, Zhang SJ, Larsson NG, Westerblad H. Abnormal ca(2+) release and catecholamine‐induced arrhythmias in mitochondrial cardiomyopathy. Hum Mol Genet. 2005;14:1069‐1076. [DOI] [PubMed] [Google Scholar]
- 20. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221‐225. [DOI] [PubMed] [Google Scholar]
- 21. Tschopp J. Mitochondria: sovereign of inflammation? Eur J Immunol. 2011;41:1196‐1202. [DOI] [PubMed] [Google Scholar]
- 22. Andersson DC, Fauconnier J, Yamada T, et al. Mitochondrial production of reactive oxygen species contributes to the beta‐adrenergic stimulation of mouse cardiomycytes. J Physiol. 2011;589:1791‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aydin J, Andersson DC, Hanninen SL, et al. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet. 2009;18:278‐288. [DOI] [PubMed] [Google Scholar]
- 24. Hamilton S, Terentyeva R, Clements RT, Belevych AE, Terentyev D. Sarcoplasmic reticulum‐mitochondria communication; implications for cardiac arrhythmia. J Mol Cell Cardiol. 2021;156:105‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fauconnier J, Andersson DC, Zhang SJ, et al. Effects of palmitate on ca(2+) handling in adult control and Ob/Ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes. 2007;56:1136‐1142. [DOI] [PubMed] [Google Scholar]
- 26. Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation‐contraction coupling in the heart. Circ Res. 2017;121:181‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shan J, Kushnir A, Betzenhauser MJ, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388‐4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moscarello MA, Lei H, Mastronardi FG, et al. Inhibition of peptidyl‐arginine deiminases reverses protein‐hypercitrullination and disease in mouse models of multiple sclerosis. Dis Model Mech. 2013;6:467‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawaguchi H, Matsumoto I, Osada A, et al. Peptidyl arginine deiminase inhibition suppresses arthritis via decreased protein citrullination in joints and serum with the downregulation of interleukin‐6. Mod Rheumatol. 2019;29:964‐969. [DOI] [PubMed] [Google Scholar]
- 30. Liang KP, Myasoedova E, Crowson CS, et al. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1665‐1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis JM 3rd, Lin G, Oh JK, et al. Five‐year changes in cardiac structure and function in patients with rheumatoid arthritis compared with the general population. Int J Cardiol. 2017;240:379‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kiarash A, Kelly CE, Phinney BS, Valdivia HH, Abrams J, Cala SE. Defective glycosylation of calsequestrin in heart failure. Cardiovasc Res. 2004;63:264‐272. [DOI] [PubMed] [Google Scholar]
- 33. Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW. Balanced changes in ca buffering by SERCA and troponin contribute to ca handling during beta‐adrenergic stimulation in cardiac myocytes. Cardiovasc Res. 2014;104:347‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci USA. 2010;107:10274‐10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andersson DC, Marks AR. Fixing ryanodine receptor ca leak ‐ a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov Today Dis Mech. 2010;7:e151‐e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. 2014;76:107‐127. [DOI] [PubMed] [Google Scholar]
- 37. Wagner S, Rokita AG, Anderson ME, Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal. 2013;18:1063‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kohler AC, Sag CM, Maier LS. Reactive oxygen species and excitation‐contraction coupling in the context of cardiac pathology. J Mol Cell Cardiol. 2014;73:92‐102. [DOI] [PubMed] [Google Scholar]
- 39. Fitch CA, Platzer G, Okon M, Garcia‐Moreno BE, McIntosh LP. Arginine: its pKa value revisited. Protein Sci. 2015;24:752‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miotto MC, Weninger G, Dridi H, et al. Structural analyses of human ryanodine receptor type 2 channels reveal the mechanisms for sudden cardiac death and treatment. Sci Adv. 2022;8:eabo1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lazzerini PE, Acampa M, Capecchi PL, et al. Association between high sensitivity C‐reactive protein, heart rate variability and corrected QT interval in patients with chronic inflammatory arthritis. Eur J Intern Med. 2013;24:368‐374. [DOI] [PubMed] [Google Scholar]
- 42. Brenner B. Effect of Ca2+ on cross‐bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA. 1988;85:3265‐3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Logstrup BB, Deibjerg LK, Hedemann‐Andersen A, Ellingsen T. Left ventricular function in treatment‐naive early rheumatoid arthritis. Am J Cardiovasc Dis. 2014;4:79‐86. [PMC free article] [PubMed] [Google Scholar]
- 44. Logstrup BB, Masic D, Laurbjerg TB, et al. Left ventricular function at two‐year follow‐up in treatment‐naive rheumatoid arthritis patients is associated with anti‐cyclic citrullinated peptide antibody status: a cohort study. Scand J Rheumatol. 2017;46:432‐440. [DOI] [PubMed] [Google Scholar]
- 45. Giles JT, Fert‐Bober J, Park JK, et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. 2012;14:R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12‐21. [DOI] [PubMed] [Google Scholar]
- 47. Budde H, Hassoun R, Tangos M, et al. The interplay between S‐Glutathionylation and phosphorylation of cardiac troponin I and myosin binding protein C in end‐stage human failing hearts. Antioxidants (Basel). 2021;10:1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Inam Illahi M, Amjad S, Alam SM, Ahmed ST, Fatima M, Shahid MA. Serum tumor necrosis factor‐alpha as a competent biomarker for evaluation of disease activity in early rheumatoid arthritis. Cureus. 2021;13:e15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tetta C, Camussi G, Modena V, Di Vittorio C, Baglioni C. Tumour necrosis factor in serum and synovial fluid of patients with active and severe rheumatoid arthritis. Ann Rheum Dis. 1990;49:665‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shrivastava AK, Singh HV, Raizada A, et al. Inflammatory markers in patients with rheumatoid arthritis. Allergol Immunopathol (Madr). 2015;43:81‐87. [DOI] [PubMed] [Google Scholar]
- 51. Sitia S, Tomasoni L, Cicala S, et al. Detection of preclinical impairment of myocardial function in rheumatoid arthritis patients with short disease duration by speckle tracking echocardiography. Int J Cardiol. 2012;160:8‐14. [DOI] [PubMed] [Google Scholar]
- 52. Fine NM, Crowson CS, Lin G, Oh JK, Villarraga HR, Gabriel SE. Evaluation of myocardial function in patients with rheumatoid arthritis using strain imaging by speckle‐tracking echocardiography. Ann Rheum Dis. 2014;73:1833‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wood DD, Ackerley CA, Brand B, et al. Myelin localization of peptidylarginine deiminases 2 and 4: comparison of PAD2 and PAD4 activities. Lab Invest. 2008;88:354‐364. [DOI] [PubMed] [Google Scholar]
- 54. Coudane F, Mechin MC, Huchenq A, et al. Deimination and expression of peptidylarginine deiminases during cutaneous wound healing in mice. Eur J Dermatol. 2011;21:376‐384. [DOI] [PubMed] [Google Scholar]
- 55. Persson PB. Good publication practice in physiology 2019. Acta Physiol (Oxf). 2019;227:e13405. [DOI] [PubMed] [Google Scholar]
- 56. Yamada T, Place N, Kosterina N, et al. Impaired myofibrillar function in the soleus muscle of mice with collagen‐induced arthritis. Arthritis Rheum. 2009;60:3280‐3289. [DOI] [PubMed] [Google Scholar]
- 57. Pironti G, Ivarsson N, Yang J, et al. Dietary nitrate improves cardiac contractility via enhanced cellular ca(2)(+) signaling. Basic Res Cardiol. 2016;111:34. [DOI] [PubMed] [Google Scholar]
- 58. Rassier DE. Pre‐power stroke cross bridges contribute to force during stretch of skeletal muscle myofibrils. Proc Biol Sci. 2008;275:2577‐2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Labuda A, Brastaviceanu T, Pavlov I, Paul W, Rassier DE. Optical detection system for probing cantilever deflections parallel to a sample surface. Rev Sci Instrum. 2011;82:013701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2