

Abstract
A 0.9-kb open reading frame encoding a unique 32-kDa protein was identified downstream of the recA gene of Porphyromonas gingivalis. Reverse transcription-PCR and Northern blot analysis showed that both the recA gene and this open reading frame are part of the same transcriptional unit. This cloned fragment was insertionally inactivated using the ermF-ermAM antibiotic resistance cassette to create a defective mutant by allelic exchange. When plated on Brucella blood agar, the mutant strain, designated P. gingivalis FLL92, was non-black pigmented and showed significant reduction in beta-hemolysis compared with the parent strain, P. gingivalis W83. Arginine- and lysine-specific cysteine protease activities, which were mostly soluble, were approximately 90% lower than that of the parent strain. Expression of the rgpA, rgpB, and kgp protease genes was the same in P. gingivalis FLL92 as in the wild-type strain. In contrast to the parent strain, P. gingivalis FLL92 showed increased autoaggregration in addition to a significant reduction in hemagglutinating and hemolysin activities. In in vivo experiments using a mouse model, P. gingivalis FLL92 was dramatically less virulent than the parent strain. A molecular survey of this mutant and the parent strain using all known P. gingivalis insertion sequence elements as probes suggested that no intragenomic changes due to the movement of these elements have occurred in P. gingivalis FLL92. Taken together, these results suggest that the recA downstream gene, designated vimA (virulence-modulating gene), plays an important role in virulence modulation in P. gingivalis W83, possibly representing a novel posttranscriptional or translational regulation of virulence factors in P. gingivalis.
Porphyromonas gingivalis, a black-pigmented, gram-negative anaerobic bacterium, is an important etiological agent of chronic adult peridontitis, a chronic inflammatory condition affecting tooth-supporting tissues (reviewed in references 29, 36, and 60). This organism possesses several putative virulence factors, including proteases, adhesins, endotoxins, and cytotoxins, that can directly affect the periodontium or elicit host functions that result in destruction typical of advanced periodontitis (16, 51). The high proteolytic abilities of this organism have been the focus of much attention and appear to play an important role in virulence. The major proteases, called gingipains, are both extracellular and cell associated. They consist of arginine-specific protease (Arg-gingipain; RGP) and lysine-specific protease (Lys-gingipain; KGP) (45). RGP is encoded by rgpA and rgpB, and KGP is encoded by kgp (38, 41). Several reports (reviewed in reference 26) have documented the multiple effects of proteases, which, in addition to being essential for growth, play a role in complement and immunoglobulin degradation, inactivation of cytokines and their receptors, platelet aggregation, attenuation of neutrophil antibacterial activities, increase in vascular permeability, and prevention of blood clotting. Recently, the proteases which are not coordinately regulated (63) have been shown to act as major processing enzymes for various cell surface proteins, including individual proteases (2, 4, 21).
An ability to overcome oxidative stress is vital for colonization and survival of P. gingivalis in an inflammatory environment such as the periodontal pocket. Protease activity in P. gingivalis may play a role in oxidative stress. Some reports (25, 39) have suggested a role for P. gingivalis proteases in hemoglobin binding, adsorption, and heme accumulation. Iron accumulated on the cell surface can bind oxygen and its toxic derivatives (58). This could limit or reduce cell damage by facilitating maintenance of a local anaerobic environment and forming a protective barrier against oxygen and oxygen radicals generated by neutrophils and occasional exposure to air. P. gingivalis cells carrying a surface layer of bound heme are less sensitive to killing by toxic oxygen derivatives (57).
Previously, we reported the nucleotide sequence of the P. gingivalis W83 recA homolog and construction of a recA-defective mutant by allelic exchange mutagenesis (14). While the recA mutation resulted in increased sensitivity to UV irradiation, we found that it did not affect the virulence of P. gingivalis FLL33, a recA-defective mutant, in a murine model (14). Inactivation of the recA gene in P. gingivalis also produced a mutant (FLL32) that was nonpigmented and lacked beta-hemolytic activity on blood agar, in contrast to the wild-type strain and P. gingivalis FLL33 (1). Analysis of the nucleotide sequence of the recA gene together with its downstream flanking region (14) did not indicate the presence of a transcriptional terminator sequence. Thus, we could not rule out that a downstream gene is somehow responsible for the phenotype observed in FLL32. While P. gingivalis FLL32 had reduced Arg-X- and Lys-X-specific proteolytic activities, there was no change in the transcription of those genes compared to P. gingivalis W83 and FLL33 (1). There was also little or no membrane-bound Arg-X- and Lys-X-specific cysteine protease activity in P. gingivalis FLL32. This mutant was significantly less virulent than the wild-type strain in a murine model and partially protected the animals against a subsequent lethal challenge by the wild-type strain (1). P. gingivalis FLL32 is similar in proteolytic and virulence properties to spontaneous mutants of P. gingivalis described by others (9, 11, 32). In contrast to P. gingivalis FLL32, however, molecular characterizations of some of those mutants show alterations in the kgp protease gene which in those reports are explained by either a deletion (9) or a transposition event (32). To further examine the possibility that the phenotype in P. gingivalis FLL32 may be the result of polar mutation and not a spontaneous mutation, we undertook this investigation to determine the role of the recA locus in the proteolytic and virulence properties of this organism. Here we report the genetic analysis of a gene downstream of recA that encodes a unique 32-kDa protein. Using this cloned downstream gene, a defective mutant was constructed by allelic exchange. This strain demonstrated reduced Arg-X- and Lys-X-specific proteolytic activities, was nonpigmented, and lacked beta-hemolytic and hemagglutinating activities. Further, we found that this mutant showed increased autoaggregation and was nonvirulent when tested in the mouse model of virulence. There was no intragenomic movement of any of the known insertion sequence (IS) elements in this mutant compare to the wild-type strain. Our results suggest that the gene downstream of recA plays an important role in modulating the virulence potential of P. gingivalis; thus, we propose that this gene be designated vimA (virulence modulating).
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains and plasmids used in this study are listed in Table 1. P. gingivalis strains were grown in brain heart infusion (BHI) broth (Difco Laboratories, Detroit, Mich.) supplemented with hemin (5 μg/ml), vitamin K (0.5 μg/ml), and cysteine (1%). Escherichia coli strains were grown in Luria-Bertani broth (49). Unless otherwise stated, all cultures were incubated at 37°C. P. gingivalis strains were maintained in an anaerobic chamber (Coy Manufacturing, Ann Arbor, Mich.) in 10% H2–10% CO2–80% N2. Growth rates for P. gingivalis and E. coli strains were determined spectrophotometrically (optical density at 600 nm [OD600]). Antibiotics were used at the following concentrations: clindamycin, 0.5 μg/ml; erythromycin, 300 μg/ml; and carbenicillin, 100 μg/ml.
TABLE 1.
E. coli and P. gingivalis strains and plasmids used in this study
| Strain or plasmid | Genotype and phenotype | Reference |
|---|---|---|
| Strains | ||
| E. coli, NM522 | supE thi Δ(lac-proAB) Δ(hsdMS-mcrB)5 (F′ proAB+lacIqlacZΔM15) | 17 |
| P. gingivalis | ||
| W83 | Wild type (black pigmented, proteolytic) | |
| FLL32 | recA::ermF-ermAM (non-black pigmented, nonproteolytic) | 1 |
| FLL33 | recA::ermF-ermAM (black pigmented, proteolytic) | 14 |
| FLL92 | vimA::ermF-ermAM (non-black pigmented, nonproteolytic) | This work |
| FLL92.1 | vimA::ermF-ermAM (non-black pigmented, nonproteolytic) | This work |
| Plasmids | ||
| pT7-5 | Apr T7 φ10 | 61 |
| pFLL97 | pT7-5 carrying the vimA gene | This work |
| pUC19 | AprlacIPOZ′ | 66 |
| pVA2198 | Spr Emr | 15 |
| pFLL90 | Apr, pUC19 carrying 1.6-kb DNA fragment including the intact vimA gene | This work |
| pFLL91 | Apr Emr, pFLL90 with vimA gene interrupted with ermF-ermAM cassette | This work |
DNA isolation and analysis.
P. gingivalis chromosomal DNA was prepared by the method of Marmur (35). For plasmid DNA analysis, DNA extraction was performed by the alkaline lysis procedure of Birnboim and Doly (6). For large-scale preparation, plasmids were purified using a Qiagen (Santa Clarita, Calif.) plasmid maxi kit. DNA was digested with restriction enzymes as specified by the manufacturer (Boehringer Mannheim Corp., Indianapolis, Ind.). For DNA subcloning, the desired fragments were isolated from 0.7% agarose gels run in Tris-acetate-EDTA buffer (49) then purified using a Gene Clean kit as recommended by the manufacturer (Bio 101, Inc., La Jolla, Calif.). Southern blot transfer was done by the method of Smith and Summers (59). DNA hybridization, DNA labeling, and autoradiography were done as previously described (15). For the detection of IS elements, the IS DNA probes were amplified by PCR using specific primers (Table 2). These probes were 32P labeled using the Prime-a-Gene labeling system (Promega Corporation, Madison, Wis.). Hybridization was carried out in ExpressHyb hybridization solution (Clontech, Palo Alto, Calif.) according to the manufacturer's recommendations.
TABLE 2.
Oligonucleotide primers used in this study
| Primer | Oligonucleotide sequence | Characteristic | Reference |
|---|---|---|---|
| P1 | 5′ TTCCTCAGGATAAGGAGGGC 3′ | Upstream recA right | 14 |
| P2 | 5′ GAGAAGAAACTGCAAGCCCTG 3′ | Intragenic recA right | 14 |
| P3 | 5′ AGCTGAACCACGATCCGCT 3′ | Intragenic recA left | 14 |
| P4 | 5′ TGAATGATGATGAAGCAACGC 3′ | Downstream 1 recA right | This work |
| P5 | 5′ CCATCCGTACGACATCGA 3′ | Downstream 1 recA left | This work |
| P6 | 5′ CGGTATCATGAAAGGCTCG 3′ | Downstream 2 recA left | This work |
| P7 | 5′ GCGAATGATGTAGTGGATTA 3′ | Downstream 3 recA left | This work |
| P8 | 5′ ATGACAAAAAAGAAATTGCC 3′ | ermF right | 48 |
| P9 | 5′ ATGAAAAACTTGAACAAGTT 3′ | rgpA right | 3 |
| P10 | 5′ CGTTGTAGTGGTCATTACCT 3′ | rgpA left | 3 |
| P11 | 5′ ATGAAAAAGAATTTTAGCAG 3′ | rgpB right | 54 |
| P12 | 5′ CGACCGATGAAGACTTCG 3′ | rgpB left | 56 |
| P13 | 5′ ATGAGGAAATTATTATTGCT 3′ | kgp right | 5 |
| P14 | 5′ CTGTAATACAAGTCGGTA 3′ | kgp left | 5 |
| P15 | 5′ ATGTATCATTCTCTATTTGAA 3′ | PGIS2 right | 65 |
| P16 | 5′ ATTAAAATTGTATTGTGATTA 3′ | PGIS2 left | 65 |
| P17 | 5′ ATGAGTACAAATATAAGCCTA 3′ | ISPg4 right | F. F. Dewhirst |
| P18 | 5′ CCTATGTGCCTAAAAGAGCTGA 3′ | ISPg left | F. F. Dewhirsta |
| P19 | 5′ ATGCAACCAGTATCCATTATGGGCA 3′ | ISPg5 right | 8 |
| P20 | 5′ TCTAATTTCGGTTGACACGAGCAA 3′ | ISPg5 left | 8 |
| P21 | 5′ ATGGATGTACAAGTTTATTTTTCT 3′ | ISPg6 right | F. F. Dewhirst |
| P22 | 5′ TTCAGAGCAGCTTTGAACCAA 3′ | ISPg6 left | F. F. Dewhirst |
| P23 | 5′ AGCTTTCATGGCTCTTTTGAA 3′ | ISPg7 right | F. F. Dewhirst |
| P24 | 5′ TATTGAGCAAATTAGTTTGCGAAA 3′ | ISPg7 left | F. F. Dewhirst |
| P25 | 5′ ATGAACACAAATATAGTTGA 3′ | IS195 right | 32 |
| P26 | 5′ ATCTAAGCGGAAATAGGAAA 3′ | IS195 left | 32 |
| P27 | 5′ ATGACGTGATTCTCTTATTCA 3′ | IS1126 right | 34 |
| P28 | 5′ TCAAGAATGTTTTGAGTA 3′ | IS1126 left | 34 |
Unpublished data.
DNA sequencing.
Nucleotide sequences were determined by the dideoxy-chain termination method (50) with a Perkin-Elmer (Foster City, Calif.) DNA sequencing kit and analyzed on an Applied Biosystems (Foster City, Calif.) model 373A DNA sequencing system at the DNA core facility of Loma Linda University (Loma Linda, Calif.); overlapping oligonucleotide primers were synthesized by the same facility. Nucleotide sequences were analyzed using the Genetics Computer Group sequence analysis software package (13).
In vitro transcription and translation of the cloned vimA gene.
Following PCR amplification, the vimA gene including its putative ribosome binding (Shine-Dalgarno) site was inserted into pT7-5 digested with SacI-SmaI (61). To inactivate the amp gene, whose product (β-lactamase; 31.5 kDa) could mask the vimA product after sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the recombinant plasmid designated pFLL97 was digested with ScaI. The linear pFLL97 plasmid was purified as described above. For in vitro transcription and translation we used the Promega E. coli S30 extract system for linear templates according to the manufacturer's instructions. The reaction mixture (50 μl) consisted of the purified linear DNA fragment, 5 μl of amino acid mixture without methionine, 20 μl of S30 premix, 1 μl of [35S]methionine (1,200 Ci/mmol at 15 mCi/ml), and 15 μl of the E. coli S30 extract. Nuclease-free water was added to a final volume of 50 μl. The reaction mixture was incubated at 37°C for at least 2 h and then placed in an ice bath for 5 min. The samples were then suspended in 2% SDS buffer and separated by SDS:PAGE on a 15% polyacrylamide gel by the method of Laemmli (27). To ensure good band resolution on SDS-PAGE, polyethylene glycol contained in the E. coli S30 extract was removed by acetone precipitation.
PCR analysis of RNase-treated chromosomal DNA from P. gingivalis.
PCR amplification was performed with a Perkin-Elmer Cetus (Norwalk, Conn.) DNA thermal cycler. The primers used (Table 2) were specific for a 0.8-kb intragenic region of recA (14), a 2.1-kb recA and downstream region (this study), and the 5′ region of the ermF gene (48). The reaction mixture (50 μl), containing 1 μl of template DNA (0.5 μg), 1 μM each primer, and 0.2 mM deoxynucleoside triphosphates in 1× Expand High Fidelity system buffer (Boehringer Mannheim), was denatured for 2 min at 94°C, and then 1.73 U of Expand High Fidelity system enzyme was added. The PCR program consisted of 40 cycles with a temperature profile of 30 s at 94°C, 1 min at 55°C, and 2 min at 72°C, with a final extension at 72°C for 7 min. The PCR-amplified DNA was then identified by 0.7% agarose gel electrophoresis.
RT-PCR and Northern blot analysis of DNase-treated RNA extracted from P. gingivalis strains.
Total RNA was extracted from P. gingivalis strains grown to mid-log phase (OD600 of 0.3 to 0.4) using a Qiagen RNeasy midi kit. The primers used for reverse transcription-PCR (RT-PCR) analysis (Table 2) were specific for a 0.8-kb intragenic region of recA (14), a 1-kb intragenic region of prpRI (rgpA) (3), prpRII (rgpB) (56), and prtP (kgp) (5). The reaction mixture (50 μl) contained 1 μg of template RNA, 1 μM each primer, 0.2 mM deoxynucleoside triphosphates, 1 mM MgSO4, 0.1 U of avian myeloblastosis virus reverse transcriptase for first-strand DNA synthesis, and 0.1 U of thermostable Tfl DNA polymerase (from Thermus flavus) for second-strand cDNA synthesis and DNA amplification (Access RT-PCR system; Promega). The reverse transcription reaction was performed at 48°C for 45 min. and then stopped by raising the temperature 94°C for 2 min. PCR amplification was performed with a Perkin-Elmer Cetus DNA thermal cycler, using 40 cycles of 30 s at 94°C, 1 min at 60°C, and 2 min at 68°C. The final products were analyzed by 0.7% agarose gel electrophoresis.
For Northern blot analysis, RNA samples (5 μg) were separated by 1% agarose gel electrophoresis and then transferred to a nylon membrane (Hybond-N; Amersham Pharmacia Biotech) according to the method of Sambrook et al. (49). The probes were 32P labeled using the Promega Prime-a-Gene labeling system. Hybridization was carried out in ExpressHyb hybridization solution (Clontech) as recommended by the manufacturer. Autoradiography was done as previously described (15).
Mutagenesis of the cloned vimA gene in P. gingivalis.
A 2.1-kb fragment carrying the intact recA and downstream gene was amplified by PCR using the P1 and P7 oligonucleotide primers (Table 2; Fig. 1). This fragment was treated with Klenow enzyme and then digested with SalI. The resulting 1.6-kb fragment was subcloned into pUC19 which was digested with SmaI and SalI in order to remove the BamHI site contained in the multiple cloning site. The ermF-ermAM cassette (which confers erythromycin/clindamycin resistance in E. coli and P. gingivalis) was isolated from pVA2198 digested with PstI and SacI (14) and treated with Klenow enzyme to fill in the single-stranded ends. The resulting plasmid, pFLL90, which carried a part of the recA gene and the intact vimA gene, was linearized at a unique BamHI site located within codon 55 of the vimA open reading frame and then ligated with the ermF-ermAM cassette. The resultant recombinant plasmid, pFLL91, was used as a donor in electroporation of P. gingivalis W83.
FIG. 1.

Construction of site-specific mutanttions by allelic exchange. pFLL91 contains the vimA gene interrupted by an ermF-ermAM cassette. The vimA gene and flanking sequences were amplified by PCR; the ermF-ermAM cassette was purified from pVA2298 (15). The circular recombinant plasmid pFLL91 was integrally introduced into P. gingivalis W83 by electroporation. A reciprocal recombination event between areas of homology on the chromosome and regions flanking the ermAM-ermF cassette on pFLL91 replaced the intact vimA with an inactivated copy.
Electroporation.
Electroporation of cells was performed as previously reported (14). Briefly, 1 ml of an actively growing culture of P. gingivalis was used to inoculate 10 ml of BHI broth supplemented with hemin and vitamin K, which was then incubated overnight at 37°C. Seventy milliliters of the same prewarmed medium (37°C) was then inoculated with 3 ml of the overnight culture and incubated for an additional 4 h. The cells were harvested by centrifugation at 2,600 × g for 7 min at 4°C and washed in 70 ml of electroporation buffer (10% glycerol, 1 mM MgCl2; filter sterilized; stored at 4°C), and the pellet was resuspended in 0.5 ml of electroporation buffer A 100-μl sample of cells to which 1 μg of DNA was added was placed in a sterile electrode cuvette (0.2-cm gap). The cells were pulsed with a Bio-Rad gene pulser at 2,500 V for 9.5 ms and then incubated on ice for 3 min. The cell suspension was then added to 0.5 ml of BHI broth supplemented with hemin and vitamin K and incubated for approximately 16 h. A 100-μl sample was plated on solid medium containing clindamycin and incubated anaerobically at 37°C for 7 to 10 days.
UV sensitivity measurements.
Aliquots (0.1 ml) of exponentially growing cells at a 10−5 dilution were plated on BHI agar, placed in the dark, and irradiated with increasing doses (from 500 to 2,000 μJ) of UV using a Stratalinker-2400 (Stratagene, La Jolla, Calif.). To avoid photoactivation, the plates were immediately wrapped in foil and incubated for 7 to 10 days at 37°C in 10% H2–10% CO2–80% N2.
Fraction preparation and protease assay.
Whole-cell culture, cell-free medium, cell suspension, vesicles, and particle-free medium were prepared as previously reported (46). The presence of Arg-X- and Lys-X-specific cysteine protease activities was determined using a microplate reader (Bio-Rad) by the method of Potempa et al. (44).
Hemolytic activity assay.
Hemolytic activity was determined as previously reported (10). Briefly, bacterial cells from overnight cultures were harvested by centrifugation (10,000 × g for 30 min) using a Sorvall RC5C centrifuge, washed three times with NCN buffer (3 mM sodium citrate, 0.9% NaCl [pH 6.9]), and then resuspended to a final concentration of 2 × 1010 bacteria per ml. Sheep erythrocytes (Hemostat Laboratories, Dixon, Calif.) were harvested by centrifugation (4,400 × g for 25 min) and washed with NCN buffer until the supernatant was visually free of hemoglobin pigment. The washed erythrocytes were suspended in NCN buffer to a concentration of 2 × 109/ml. Hemolytic activity was determined by mixing 2 × 1010 bacterial cells with 2 × 109 erythrocytes in NCN buffer. This mixture was then slowly mixed in a water bath at 37°C. Samples (100 μl) were withdrawn every 2 h, immediately diluted with 900 μl of NCN buffer, and then centrifuged (1,300 × g for 5 min) in an Eppendorf 5403 centrifuge. The resulting supernatant was diluted another 10-fold with NCN buffer, and the OD was determined by spectrophotometry at 405 nm. As a negative control, erythrocytes were used alone.
Hemagglutination assays.
Hemagglutination activity was assayed by the method of Chu et al. (10). Bacterial cells from overnight cultures were harvested by centrifugation (10,000 × g for 30 min) in a Sorvall RC5C centrifuge, washed three times with NCN buffer, and then resuspended to a final concentration of 2 × 1010 bacteria per ml. The cells (100-μl volumes) were twofold serially diluted in 96-well microtiter plates. To each dilution, 100 μl of 1% sheep erythrocyte suspension was added. Sheep erythrocytes (Hemostat Laboratories) were harvested by centrifugation (3,800 × g for 25 min) in an Eppendorf 5403 centrifuge and then washed with NCN buffer until the supernatant was visually free of hemoglobin pigment. The washed erythrocytes were suspended in NCN buffer to a 1% suspension. The plates were incubated at 5°C for 3 h. Hemagglutination was assessed visually, and a positive reaction was taken as the reciprocal of the last dilution showing complete hemagglutination.
Virulence testing.
P. gingivalis wild-type strain W83 and mutant strain FLL92 were tested for invasiveness in a murine model as previously described (15). These experiments were performed under authorization of an institutionally approved animal use protocol (33).
Nucleotide sequence accession number.
The nucleotide sequence reported here has been assigned GenBank accession number AF064682.
RESULTS
Nucleotide sequencing of the gene downstream of the recA homolog gene in P. gingivalis.
Inactivation of the recA gene in P. gingivalis FLL32 not only affected the capacity for DNA repair but also resulted in increased autoaggregation, decreased protease activity, and reduced virulence in a mouse model (1). Analysis of the recA and flanking downstream sequences did not show any putative transcription terminator (14), which suggests that the recA gene could be part of an operon resulting in a polar mutation affecting a downstream gene(s). Using a forward oligonucleotide primer from the recA gene (P1 [Table 2; Fig. 1]) in combination with a reverse primer from the downstream flanking region (P5 [Table 2; Fig. 1]), a 1.4-kb fragment was amplified by RT-PCR using total DNase-treated RNA from the wild-type strain W83 (Fig. 2, lane C). As expected, the intragenic primers of the recA gene (P2 and P3 [Table 2; Fig. 1]) led to the amplification of a 0.86-kb fragment (Fig. 2, lane B). No amplified fragments were observed when reverse transcriptase was omitted from the reaction (data not shown). The putative recA gene is 1 kb and thus may be associated with some other gene(s) on the same transcriptional unit.
FIG. 2.

RT-PCR analysis of DNase-treated RNA extracted from P. gingivalis. Total RNA was extracted from W83 grown to mid-log phase (OD600 of 0.3 to 0.4) using a Qiagen RNeasy midi kit. Samples (1 μg) were subjected to RT-PCR (Access RT PCR system; Promega). Lanes: A, Promega kanamycin positive control RNA; B, intragenic primers for recA (P2 and P3 Table 2; Fig. 1); C, forward primer from recA and reverse primer from the downstream gene (P2 and P5 (Table 2; Fig. 1); D, 1-kb ladder. Expected sizes of the transcripts are 0.323, 0.86, and 1.4 kb for the positive control, recA, and recA/downstream genes, respectively. Sizes of the PCR products are given at the left.
Phage L10, which is part of a P. gingivalis W83 genomic lambda library, carries the recA gene together with its downstream region (14). Using overlapping oligonucleotide primers, both strands of a 1.2-kb region downstream of the recA gene were sequenced. A putative 0.9-kb open reading frame (called vimA) corresponding to a 32-kDa protein was detected 113 bases downstream of the stop codon of the recA gene. This sequence was confirmed by comparison with the P. gingivalis genome sequence in the databank (Forsyth Dental Center/The Institute of Genome Research). A purine-rich sequence found in E. coli ribosome binding sites was also seen one base upstream from the translation initiation site. Further, partial direct repeat sequences (bp 2820 to 2832 and 2848 to 2860) starting 6 bp downstream of the translation stop were observed. The calculated G+C content for this gene was 52%, which is close to the value of 46 to 48% previously reported for P. gingivalis genomic DNA (53). Hydrophobicity analysis using the Kyte-Doolittle scale (PROTSCALE) predicted the putative vimA gene product to be hydrophilic. NNPREDICT and DAS queries indicted no transmembrane and DNA binding domains, respectively. Comparison of the nucleotide and amino acid sequences of this gene with entries in the National Center for Biotechnology Information data bank did not reveal any significant similarity with any known genes.
In vitro expression of the vimA gene.
To confirm the predicted size of the VimA protein, chromosomal DNA from the wild type was subjected to PCR analysis using two primers (P4 and P7; Table 2; Fig. 1) that would amplify the open reading frame plus the putative ribosome binding site. This purified DNA fragment was inserted into pT7-5 under control of the φ10 promoter. The recombinant plasmid pFLL97 was linearized at the ScaI site contained in the amp gene and then expressed in an in vitro transcription-translation system. As shown in Fig. 3 (lane B), the predicted 32-kDa protein was observed. In contrast, no protein was seen with the nonrecombinant plasmid pT7-5 (lane A). Taken together, these results suggest that the vimA gene that is present downstream of the recA gene encodes a 32-kDa protein.
FIG. 3.

In vitro transcription-translation of the linear pFLL97 plasmid carrying the vimA gene. Five microliters of each 50-μl reaction mix was subjected to acetone precipitation and analyzed by SDS-PAGE. The plasmid vector (pT7-5 [61]) and the recombinant plasmid pFLL97 were linearized by ScaI to prevent expression of the amp gene. Molecular masses are shown on the left. Lanes: A, pT7-5; B, pFLL97 carrying the vimA gene. Proteins were labeled with [35S]methionine and visualized by autoradiography.
Inactivation of the vimA gene in P. gingivalis W83 by allelic exchange mutagenesis.
Isogenic vimA-defective derivatives of P. gingivalis W83 were constructed by allelic exchange mutagenesis. The circular recombinant plasmid pFLL91, which carries the ermF-ermAM cassette in the unique BamHI restriction site (bp 165 of the open reading frame) of the vimA gene, was used as a donor in electroporation of P. gingivalis W83 (Fig. 1). Because the plasmid was unable to replicate in P. gingivalis, we predicted that two double-crossover events between the regions flanking the erm marker and the wild-type gene on the chromosome would result in replacement of a segment of the wild-type gene with a fragment conferring clindamycin resistance.
Following electroporation and plating on selective medium, we detected 200 clindamycin-resistant colonies after a 7-day incubation period. To compare their phenotypic properties with those of wild-type strain W83, all mutants were plated on Brucella blood agar plates (Anaerobic Systems Inc., San Jose, Calif.). In contrast to the wild-type strain, all mutants displayed a non-black-pigmented, non-beta-hemolytic phenotype. These mutant strains grown in BHI broth showed increased autoaggregation, in contrast to the wild-type strain.
The non-black-pigmented, non-beta-hemolytic phenotype of the vimA mutants shows some similarity to the phenotype of spontaneous mutants of P. gingivalis (9, 11, 32) and thus could have arisen by a similar mechanism. To determine the likelihood for selection of this phenotype independent of inactivation of the vimA gene or to rule out the possibility of the effect of electroporation on selection of this phenotype, the recombinant plasmid pVA2295 (15), which carries the ermF-ermAM cassette with the rgpA gene flanking region, was used as a donor in electroporation of P. gingivalis W83. Inactivation in the rgpA gene does not give rise to mutants with a non-black-pigmented and non-beta-hemolytic phenotype (15), in contrast to kgp-defective mutants, which are non-black pigmented and non-beta-hemolytic (9, 32). We detected 80 clindamycin-resistant colonies following a 7-day incubation. When plated on Brucella blood agar plates, all of these mutants displayed a black-pigmented, beta-hemolytic phenotype similar to that of the wild type. When the recombinant plasmid pFLL91, which carries the ermF-ermAM cassette in the vimA gene, was used as a donor in electroporation of P. gingivalis W83 in an independent experiment, all of the clindamycin-resistant mutants again displayed a non-black-pigmented and non-beta-hemolytic phenotype. Growth of these mutant strains in BHI broth also resulted in increased autoaggregation compared to the wild-type strain. Five mutant strains were randomly chosen from the two independent experiments and used for further studies.
Characterization of vimA-defective P. gingivalis.
To confirm the changes in the vimA gene and to verify the sequence of the intact recA gene, chromosomal DNA from five strains (from two independent experiments), P. gingivalis FLL32 (recA::ermF-ermAM), P. gingivalis FLL33 (recA::ermF-ermAM), and the wild type were probed with 32P-labeled recA or ermF-ermAM fragments. A predicted 2.1-kb fragment should be seen in the wild-type and vimA-defective mutants when digested with EcoRI and PstI and probed with the recA-specific fragment. Since the ermF-ermAM cassette is missing EcoRI and PstI sites, a 4.2-kb fragment should be seen in the recA-defective mutants as previously reported (1). As shown in Fig. 4A, the wild type (lane 1) and the vimA-defective mutants (lanes 4 to 8) showed a 2.1-kb DNA fragment. In contrast, a 4.2-kb fragment was present only in the recA mutants that contained the 2.1-kb ermF-ermAM cassette in the recA gene (P. gingivalis FLL32 [lane 2] and P. gingivalis FLL33 [lane 3]) as reported previously (1). A similar blot probed with the ermF-ermAM cassette (Fig. 4B) revealed an identical 4.2-kb hybridizing band in the recA mutants (P. gingivalis FLL32) [lane 2] and P. gingivalis FLL33 [lane 3]); however, a 7.2-kb hybridizing fragment was present in the vimA-defective mutants (lanes 4 to 8). No hybridizing sequences were seen in the wild-type strain (lane 1). The ermF-ermAM cassette and its orientation in the vimA gene were further verified by PCR analysis (data not shown). These data indicated that the predicted recombination had occurred and vimA was interrupted by the ermF-ermAM cassette. One strain from the five vimA-defective mutants, designated P. gingivalis FLL92, was randomly chosen for further studies.
FIG. 4.

Southern blot analysis of allelic exchange mutants of P. gingivalis. Total chromosomal DNA from P. gingivalis was cleaved with EcoRI and PstI, electrophoresed through 0.7% agarose, and bidirectionally transferred to a nitrocellulose membrane. Lanes: 1, W83; 2, FLL32; 3, FLL33; 4 to 8, vimA-defective mutants. The probes used were 32P-labeled fragments representing an intragenic region of the recA gene (A) and erythromycin cassette purified from plasmid pVA2198 (12) (B). A 2.1-kb fragment was detected in strains W83 and FLL92, which have the intact recA gene. A 4.2-kb fragment was detected in FLL32 and FLL33, which contain the recA gene inactivated with the ermF-ermAM cassette, in contrast to a 7.2-kb fragment in the vimA mutants.
The recA gene in P. gingivalis plays a role in DNA repair (14). To confirm the integrity of the RecA protein function in P. gingivalis FLL92, cells from P. gingivalis FLL92 and wild-type strain W83 were exposed to UV irradiation. Both P. gingivalis W83 and P. gingivalis FLL92 showed 77% survival after 1,000 μJ and 43% survival after 2,000 μJ of UV irradiation (data not shown). However, in the previous study, less than 1% of the recA-defective mutants survived after a 2,000-μJ dose of UV irradiation (1).
To confirm the presence of the recA transcript, oligonucleotide primers (P2 and P3) specific for an intragenic region of the recA gene (Table 2; Fig. 1) were used in RT-PCR to amplify that gene. In contrast to the recA-defective mutants (P. gingivalis FLL32 and P. gingivalis FLL33) that did not show any amplified fragments, the recA intragenic primers amplified a 0.86-kb region in both P. gingivalis FLL92 and the wild-type strains (data not shown). These data confirm that the function of RecA protein is intact in P. gingivalis FLL92 compared to the recA-defective mutants (P. gingivalis FLL32 and P. gingivalis FLL33).
Northern blot analysis of the P. gingivalis recA locus.
To confirm that the recA and vimA genes are part of the same transcriptional unit, total RNA was isolated from the wild-type strain W83, the isogenic recA-defective mutants, and the vimA-defective mutant grown to mid-log phase. Intragenic probes from the recA and vimA genes were amplified by PCR from the wild-type W83 chromosomal DNA, using the primers listed in Table 2. Since the sizes of the recA and vimA genes are 1.0 and 0.9 kb, respectively, a 1.9-kb transcript is expected. As shown in Fig. 5 (lane A), hybridizing bands of approximately of 1, 1.9, and 2.8 kb were observed in the wild-type strain when the recA gene was used as a probe. For the recA-defective mutants (lanes B and C), a hybridizing band of approximately 1 kb was observed. In contrast, a 1.9-kb band was observed for a mutant that carried a defect in the vimA gene. A similar hybridization profile was observed when the vimA gene was used as a probe (data not shown). Taken together, these results suggest that the recA and vimA genes are a part of the same transcriptional unit. Sequence analysis of the DNA region upstream of the recA gene identified a putative 0.5-kb open reading frame with the stop codon located 20 bp from the recA start codon. No typical transcriptional stop was observed. Preliminary in silico analysis of this open reading frame, using Blast and Alignment (http://www.ncbi.nlm.nih.gov), suggests that this gene may encode a 18-kDa protein with homology to the bacterioferritin comigratory protein (BCP) (20).
FIG. 5.

Autoradiograph of Northern hybridization filter of total RNA from mid-log-phase P. gingivalis isogenic strains. Lanes: A, P. gingivalis W83; B, recA-defective mutant P. gingivalis FLL32; C, recA-defective mutant P. gingivalis FLL33; D, vimA-defective mutant P. gingivalis FLL92. The probe used was the 32P-labeled intragenic region of the recA gene. Sizes of the transcripts are given at the left.
Hemolytic activity of P. gingivalis FLL92.
Cell- and vesicle-associated hemolysins which will liberate hemoglobin from erythrocytes are produced in P. gingivalis apparently by two distinct genes (10, 21). The effects of P. gingivalis W83, P. gingivalis FLL32, and P. gingivalis FLL92 on erythrocyte lysis are shown in Fig. 6. P. gingivalis W83 showed a significant increase in hemolytic activity. However, similar to results for the negative control, hemolytic activity was missing in P. gingivalis FLL32 and P. gingivalis FLL92.
FIG. 6.

Kinetic analysis of the hemolytic activity of whole cells of P. gingivalis W83 (◊) and derivative mutants strains FLL32 (▵) and FLL92 (■). Erythrocytes were prepared by the method of Chu et al. (9). Bacterial cells were grown, pelleted, and assayed for hemolytic activity as described in the text. As a negative control (○), erythrocytes were used alone. Each value indicates average ± standard deviation (assays were performed in triplicate). Using Student's t test (Statistical Package for the Social Sciences, version 9), there is a significance difference (P > 0.05) between the wild-type strain and isogenic mutants.
Proteolytic activity of P. gingivalis FLL92.
The proteolytic activity of P. gingivalis FLL32, a recA-defective mutant that was non-black pigmented, was more than 90% lower than that of the wild-type strain (1). Thus, it is possible that the vimA locus affects proteolytic activity in P. gingivalis. P. gingivalis strains W83, FLL32, and FLL92 were assayed for proteolytic activity using N-α-benzoyl-dl-arginine p-nitroanilide (BAPNA) and Z-lysine-dl-p-nitroanilide. In late-exponential-growth-phase cultures, proteolytic activity in P. gingivalis FLL92, as in P. gingivalis FLL32, was almost 90% lower than the wild-type level (Fig.7). In addition, most of the Arg-X (Fig. 8A) and Lys-X (Fig. 8B) specific activity in P. gingivalis FLL92 as in P. gingivalis FLL32 (Fig. 8C and D) was found to be soluble. In contrast, most of the Arg-X (Fig. 8E) and lysine-X (Fig. 8F)-specific activity in P. gingivalis W83 was membrane bound. Taken together, these data suggest that under the same physiological conditions, the proteolytic profiles for P. gingivalis FLL32 and P. gingivalis FLL92 were similar, and this proteolytic activity could be severely altered by mutations affecting the vimA gene expression in P. gingivalis.
FIG. 7.

Proteolytic activity of P. gingivalis mutants. P. gingivalis was grown to late log phase (OD600 of 1.1) in 500 ml of BHI broth supplemented with hemin and vitamin K. Activity against BAPNA (A) and Z-lysine-dl-p-nitroanilide (B) was tested in whole-cell culture by the method of Potempa et al. (38). The results shown are representative of four independent experiments.
FIG. 8.

Distribution of cysteine protease activity in P. gingivalis. Activities against BAPNA (A, FLL92; C, FLL32; E, W83) and Z-lysine-dl-p-nitroanilide (B, FLL92; D, FLL32; F, W83) were tested in whole-cell culture (WC), cell-free medium (CF), cell suspension (CS), vesicles (V), and particle-free medium (VF). The fractions were prepared by the method of Potempa et al. (38). The activity of each strain in the whole-cell fraction was assumed to be 100%; this activity measured in P. gingivalis FLL32 and FLL92 represents 10% of the activity measured in the wild-type strain W83. The results are representative of three independent experiments performed on late-log-phase (OD600 = 1.1) growth cultures.
Transcription of the gingipain genes in strain FLL92 is similar to that in the wild-type strain.
The reduced proteolytic activity in P. gingivalis FLL92 could have been the result of alteration in transcription of the gingipain genes. To determine the presence of mRNA transcripts for the major protease genes, total RNA was isolated using a Qiagen RNeasy kit from the wild-type strain W83 and P. gingivalis FLL92 grown to mid-log phase. Unique oligonucleotide primers for prtP (kgp) (5) prpRI (rgpA) (3), and prRII (rgpB) (56) (Table 2) were used in RT-PCR to amplify a 1-kb region of the transcripts. When reverse transcriptase was present in the reaction, amplified products of the predicted size (1.0 kb) were observed for all three protease gene transcripts in both strains (data not shown). Since non-black pigmentation in P. gingivalis can arise from a defect in the kgp gene (9, 32), the level of expression of that gene was further examined in P. gingivalis FLL92. In Northern blot analysis as shown in Fig. 9, there was no observed difference between the wild type and P. gingivalis FLL92 when probed with the kgp gene. These data are consistent with previous results for P. gingivalis FLL32, a recA-defective mutant which was non-black pigmented and had reduced proteolytic activity compared to the wild-type strain (1).
FIG. 9.

Northern blot analysis of the kgp protease gene from isogenic mutants of P. gingivalis. Lanes: A, P. gingivalis W83; B, P. gingivalis FLL32; C, P. gingivalis FLL92. The probe used was the 32P-labeled intragenic region of the kgp gene. The size of the corresponding kgp transcript is given at the left.
Hemagglutination studies.
We assessed the hemagglutination potential of P. gingivalis W83, the recA-defective mutants FLL32 and FLL33, and the vimA-defective mutant FLL92. While the activity of P. gingivalis FLL33 (a proteolytic recA mutant) was similar to the wild-type level, the hemagglutination capability of P. gingivalis FLL32 (a nonproteolytic recA mutant) and P. gingivalis FLL92 (vimA::ermF-ermAM) was reduced (Fig. 10).
FIG. 10.

Hemagglutinating activity of P. gingivalis. Titers were determined by twofold serial dilution of 100-μl volumes of suspension in microtiter plates as previously described (9). To each dilution, 100 μl of 1% sheep erythrocyte suspension was added. The plates were incubated at 5°C for 3 h. Reciprocals of the dilutions are shown at the left; the strains tested are indicated above the columns of wells. The experiments were carried out at least three times; the results reported are representative of four independent experiments.
Analysis of isogenic mutants for the presence all known P. gingivalis IS elements.
Some of the phenotypic properties of the vimA-defective mutant are similar to the IS element-induced properties of P. gingivalis spontaneous mutants previously described (9, 32, 55, 65). To determine the movement of any IS element in P. gingivalis FLL92, chromosomal DNA isolated from two vimA mutants (P. gingivalis FLL92 and P. gingivalis FLL92.1 [from two independent experiments]), P. gingivalis FLL32 (recA::ermF-ermAM), P. gingivalis FLL33 (recA::ermF-ermAM), and P. gingivalisW83 was digested with BamHI and probed with 32P-labeled intragenic DNA from PGIS2 (65), ISPg4 (F. F. Dewhirst, unpublished data), ISPg5 (8), ISPg6 (Dewhirst, unpublished), ISPg7 (Dewhirst, unpublished), IS195 (32) and IS1126 (34). As shown in Fig. 11, P. gingivalis FLL92 and P. gingivalis FLL92.1 exhibited similar profiles, for all IS elements compared to the wild-type strain W83 or the recA-defective mutants P. gingivalis FLL32 and P. gingivalis FLL33. Taken together, these data suggest that no detectable intragenomic changes due to the movement of the known P. gingivalis IS elements have occurred in the isogenic mutants.
FIG. 11.

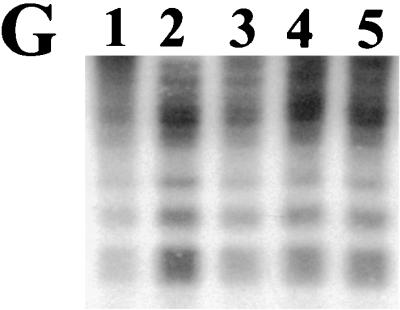
Southern blot analysis of isogenic mutants of P. gingivalis to determine intragenomic movement of IS elements. Total chromosomal DNA from P. gingivalis was cleaved with BamHI, electrophoresed through 0.7% agarose, and bidirectionally transferred to nitrocellulose. Lanes: 1, P. gingivalis W83; 2, P. gingivalis FLL32 (ermF-ermAM∷recA); 3, P. gingivalis FLL33 (ermF-ermAM∷recA); 4, P. gingivalis FLL92 (ermF-ermAM∷vimA); 5, P. gingivalis FLL92.1 (ermF-ermAM∷vimA). The probes used were the 32P-labeled intragenic region of the IS elements. (A) PGIS2; (B) ISPg4; (C) ISPg5; (D) ISPg6; (E) ISPg7; (F) IS195; (G) IS1126.
Virulence testing of P. gingivalis FLL92.
The virulence of P. gingivalis FLL92 was assessed, as reduced proteolytic activity in P. gingivalis can give rise to attenuation in virulence in a mouse model (15). At 36 h, two of five mice challenged with P. gingivalis W83 (wild type) at a dose of 1010 bacteria/animal died. Surviving animals appeared cachectic and hunched, with ruffled hair. Although the mice did not display lesions at the dorsal surface site of injection, all had developed spreading, uncerative abdominal lesions. By 48 h all animals had died. In contrast, all of the five mice challenged with P. gingivalis FLL92 at a dose of 1010 bacteria/animal survived the 14-day observation period. No lesions of any sort appeared, and none of the animals in this group appeared cachectic.
DISCUSSION
In this study we used a genetic approach to further examine the recA locus in P. gingivalis. Several studies have reported that the recA gene in some bacteria can be associated with other genes in an operon structure (40, 42, 62). Northern blot analysis of the P. gingivalis recA region using the recA gene as a probe revealed a 2.8-kb transcript for the wild-type strain W83. Since recA and the putative downstream gene are 1 and 0.9 kb, respectively, this result suggested that recA is part of a multigene operon. Furthermore, the recA-defective mutants P. gingivalis FLL32 and P. gingivalis FLL33 and the vimA-defective mutant P. gingivalis FLL92 exhibited transcripts of 1 and 1.9 kb, respectively, suggesting that recA and vimA genes are associated with a third gene that should be localized upstream of recA, A 0.5-kb open reading frame was detected upstream of the recA gene. In silico analysis of this open reading frame has shown that the putative gene may encode a 18-kDa protein with homology to BCP, which is known to have hydroperoxide peroxidase activity (20). Further confirmation of this gene as part of the recA operon and examination of its functional role in P. gingivalis are in progress.
The recA gene in bacteria can be associated with genes of different functions, such as resistance to low pH in Helicobacter pylori (62), competence induction in Streptococcus pneumoniae (42), or regulation of RecA function in mycobacteria (40). In P. gingivalis, however, the recA gene may be associated with a bcp-like gene (upstream) and the vimA gene (downstream), which shows no significant similarity with any known genes. The vimA gene is not involved in DNA repair, instead P. gingivalis FLL92, the isogenic mutant defective in that gene, showed reduced proteolytic activity and was non-black pigmented. This phenotype of P. gingivalis FLL92, which is similar to that of P. gingivalis FLL32, a recA-defective mutant (1), could be related to its reduced proteolytic activity and, in particular, reduced membrane-bound activity. This would be consistent with other reports of the involvement of gingipains with hemoglobin binding, absorption, and heme accumulation (21, 37, 39). More recently, KGP, the Lys-X-specific protease in P. gingivalis has been shown to be a hemoglobinase which plays a role in iron uptake by effecting the accumulation of iron protoporphyrin IX on bacterial cell surface (31).
Consistent with a previous report on P. gingivalis FLL32, a recA-defective mutant with reduced proteolytic activity (1), there was no detectable alteration of the gingipain genes in P. gingivalis FLL92. It is unclear whether the low activity is a result of an inefficient processing of the gingipains or other proteases (12) that could lead to inefficient substrate cleavage. However, as has been suggested for P. gingivalis FLL32 (1), the decreased proteolytic activity observed in P. gingivalis FLL92 may result from posttranscriptional regulation of those genes. Furthermore, in contrast to extracellular protein extracts from the wild-type strain, preliminary experiments in the laboratory have demonstrated P. gingivalis FLL92 high-molecular-weight proteins immunoreacting with antibodies against the gingipains but lacking proteolytic activity (data not shown). This could suggest a role for the vimA gene in posttranslational regulation of the gingipains. Posttranslation modification of proteases has been documented in P. gingivalis W50/BE1, an avirulent spontaneous mutant that is non-black pigmented with reduced Arg-X-specific protease activity (11). The source of the mutation in P. gingivalis W50/BE1 is unknown.
The vimA gene appears to also modulate hemolysin activity in P. gingivalis. P. gingivalis FLL92, the vimA-defective mutant used in this study, showed no hemolytic activity when grown on Brucella blood plates or incubated with sheep erythrocytes. These results are not surprising due to the reduced proteolytic activity in P. gingivalis FLL92. Gingipains, especially the Lys-X-specific gingipain, play a role in erythrocyte degradation (52). Mutations abolishing Lys-X-specific activity have resulted in loss of black pigmentation and a reduction in the hemolytic potential of the mutant strains (31). In addition to the effects of the proteolytic activity in P. gingivalis on its hemolysin potential, the presence of two genetically distinct hemolysins have been documented (22). Thus, the reduced proteolytic and hemolysin activities in P. gingivalis FLL92 may implicate the regulation of those genes or gene products by the vimA gene.
The ability for hemagglutination is a significant characteristic of P. gingivalis (reviewed in reference 28). The vimA gene analyzed in this study appears to also affect hemagglutination in P. gingivalis. Hemagglutination of sheep erythrocytes was reduced in P. gingivalis FLL92, the vimA-defective mutant used in this study. Again, these results are not surprising due to the reduced proteolytic activity in P. gingivalis FLL92. The RGP-adhesin and KGP-adhesin complexes have hemagglutinating activity (43). Han et al. (19) reported that the adhesin domain of the gingipains was also encoded by hagA, a hemagglutinin gene in P. gingivalis. Further, a monoclonal antibody (61BG1.3) that inhibited hemagglutination and selectively prevented the recolonization of P. gingivalis in periodontal patients was found to recognize a peptide within the adhesin domain encoded by rgpA, kgp, and hagA (7, 23). In addition to the association of the gingipains with hemagglutination in P. gingivalis, the presence of several genetically distinct genes including hemagglutinin genes hagB, hagC, and hagD has been reported (18, 30, 47). Shi et al. (54) reported construction of an a rgpA kgp hagA triple mutant and confirmed that the rgpA-, kgp-, and hagA-encoded adhesin domains play a role in the hemagglutination of P. gingivalis. The effects of the vimA gene product on hemagglutination in P. gingivalis may implicate the regulation of those genes or gene products by this gene.
In contrast to P. gingivalis W50/BE1, a spontaneous mutant which did not show autoaggregation (M. A. Curtis, personal communications), P. gingivalis FLL92 grown in BHI broth showed increase autoaggregation compare to the parent strain W83. This phenotype is similar to that of isogenic recA-defective mutants previously reported (1). Inactivation of the vimA gene suggests that vimA plays a role in autoaggregation. It is noteworthy that inactivation of the vimA gene also resulted in reduced membrane-bound proteolytic activity. While the reduced activity could give rise to incomplete processing of surface membrane proteins resulting in aggregation, our results are in contrast to a previous report of an rgpA-defective mutant (64). This mutant displayed approximately 40% of the Arg-X-specific cysteine protease activity of the wild-type strain and was severely retarded in its ability to hemagglutinate and autoaggregate. Our results, however, do not rule out the possibility that the vimA gene is a regulatory gene whose product could negatively affect the expression of other genes that facilitate autoaggregation.
The correlation of intragenomic changes due to the movement of IS elements and their effect on the phenotypic properties of P. gingivalis is well documented (9, 32, 55, 65). While most of the P. gingivalis spontaneous mutants that were non-black pigmented and had reduced proteolytic activities and virulence were IS element induced (9, 32), no movement of the these elements was observed in the vimA-defective mutant compared to the wild-type strain. Although we cannot rule out any other genetic rearrangement in the vimA-defective isogenic mutants, these results suggest that the phenotypic changes observed in P. gingivalis FLL92 could be due to the lack of expression of the vimA gene. While in vitro translation of the vimA gene in E. coli may verify the predicted size in that system, it is still unclear whether a similar product is made in P. gingivalis. Also, we cannot rule out the possibility that the vimA-induced effects observed in P. gingivalis FLL92 could be due to a regulatory RNA molecule transcribed from that gene. Complementation is needed to confirm the role of vimA in the observed phenotype of P. gingivalis FLL92.
Reduced proteolytic activity is correlated with reduced virulence in P. gingivalis (15). Consistent with this report, P. gingivalis FLL92 has shown lower virulence in a murine model than the wild-type W83 strain. These results have also confirmed that the vimA gene is involved in virulence modulation in P. gingivalis. Based on the properties of P. gingivalis FLL92 in this study, the phenotype of P. gingivalis FLL32, a non-black-pigmented non-virulent recA-defective mutant, might be the result of a polar mutation (1). This possibility raises questions as to the source of the mutation of P. gingivalis FLL33, another isogenic recA-defective mutant that was black pigmented and similar in virulence to the wild-type strain (15). While both isogenic recA-defective mutants appear to have the ermF-ermAM cassette in similar locations (1), it is possible that differences during the recombination event could give rise to a gene product of the recA locus that has partial activity in P. gingivalis FLL33. Further, we cannot rule out the occurrence in P. gingivalis FLL33 of a suppressor mutation that has restored a wild-type phenotype. Complete molecular characterization of the recA locus is under way in our laboratory.
The modulation of virulence in P. gingivalis may be coordinated via an ability to modulate proteolytic activity, although we cannot rule out any other direct or indirect effect of vimA on the expression of other virulence factors. It is possible, therefore, that the recA locus is important for the survival of P. gingivalis in the inflammatory microenvironment of the periodontal pocket. It is interesting that recA, which plays a role in DNA repair, is upstream of a gene that is involved in modulating proteolytic activity. Such an association may be significant, since a response to oxidative stress involves binding of oxygen and its toxic derivatives to iron accumulated on the cell surface (58). The bound heme can be involved in the catalytic destruction of the toxic oxygen derivative species (57). Since proteolytic activity in P. gingivalis is associated with heme accumulation, it may be an important strategy for the organism to coordinate its oxidative stress and proteolytic activities.
We have constructed an isogenic mutant of P. gingivalis that is defective in vimA, a gene, downstream of recA. While this mutant had reduced proteolytic activity, there was no detectable modification of the gingipain genes. Further, this mutant, in contrast to the wild-type strain, showed increased autoaggregration, reduced hemolytic and hemagglutinating activities, and no virulence when tested in a mouse model. Identification of the vimA gene represents a potentially new avenue for regulating virulence in P. gingivalis.
ACKNOWLEDGMENTS
We thank Zubaida Jhuma for technical assistance.
This work was supported by the Loma Linda University School of Medicine and National Institute of Dental Research grant DE11864-01A2 (to H.M.F.).
REFERENCES
- 1.Abaibou H, Ma Q, Olango G J, Potempa J, Travis J, Fletcher H M. Unaltered expression of the major protease genes in a non-virulent recA-defective mutant of Porphyromonas gingivalis W83. Oral Microbiol Immunol. 2000;15:40–47. doi: 10.1034/j.1399-302x.2000.150107.x. [DOI] [PubMed] [Google Scholar]
- 2.Abe N, Kadowaki T, Okamoto K, Nakayama K, Ohishi M, Yamamoto K. Biochemical and functional properties of lysine-specific cysteine proteinase (Lys-gingipain) as a virulence factor of Porphyromonas gingivalis in periodontal disease. J Biochem (Tokyo) 1998;123:305–312. doi: 10.1093/oxfordjournals.jbchem.a021937. [DOI] [PubMed] [Google Scholar]
- 3.Aduse-Opoku J, Muir J, Slaney J M, Rangarajan M, Curtis M A. Characterization, genetic analysis, and expression of a protease antigen (PrpRI) of Porphyromonas gingivalis W50. Infect Immun. 1995;63:4744–4754. doi: 10.1128/iai.63.12.4744-4754.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aduse-Opoku J, Rangarajan M, Young K A, Curtis M A. Maturation of the arginine-specific proteases of Porphyromonas gingivalis W50 is dependent on a functional prR2 protease gene. Infect Immun. 1998;66:1594–1600. doi: 10.1128/iai.66.4.1594-1600.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barkocy-Gallagher G A, Han N M, Patti J M, Whitlock J, Progulske-Fox A, Lantz M S. Analysis of the prtP gene encoding porphypain, a cysteine proteinase of Porphyromonas gingivalis. J Bacteriol. 1996;178:2734–2741. doi: 10.1128/jb.178.10.2734-2741.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birnboim H C, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booth V, Lehner T. Characterization of the Porphyromonas gingivalis antigen recognized by a monoclonal antibody which prevents colonization by the organism. J Periodontal Res. 1997;32:54–60. doi: 10.1111/j.1600-0765.1997.tb01382.x. [DOI] [PubMed] [Google Scholar]
- 8.Califano J V, Kitten T, Lewis J P, Macrina F L, Fleischmann R D, Fraser C M, Duncan M J, Dewhirst F E. Characterization of Porphyromonas gingivalis insertion sequence-like element ISPg5. Infect Immun. 2000;68:5247–5253. doi: 10.1128/iai.68.9.5247-5253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen W, Kuramitsu H K. Molecular mechanism for the spontaneous generation of pigmentless Porphyromonas gingivalis mutants. Infect Immun. 1999;67:4926–4930. doi: 10.1128/iai.67.9.4926-4930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu L, Bramanti T E, Ebersole J L, Holt S C. Hemolytic activity in the periodontopathogen Porphyromonas gingivalis: kinetics of enzyme release and localization. Infect Immun. 1991;59:1932–1940. doi: 10.1128/iai.59.6.1932-1940.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collinson L M, Rangarajan M, Curtis M A. Altered expression and modification of proteases from an avirulent mutant of Porphyromonas gingivalis W50 (W50/BE1) Microbiology. 1998;144:2487–2496. doi: 10.1099/00221287-144-9-2487. [DOI] [PubMed] [Google Scholar]
- 12.Curtis M A, Kuramitsu H K, Lantz M, Macrina F L, Nakayama K, Potempa J, Reynolds E C, Aduse-Opoku J. Molecular genetics and nomenclature of proteases of Porphyromonas gingivalis. J Periodont Res. 1999;34:464–472. doi: 10.1111/j.1600-0765.1999.tb02282.x. [DOI] [PubMed] [Google Scholar]
- 13.Devereux J, Haeberli P, Smithies O. A complete set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher H M, Morgan R M, Macrina F L. Nucleotide sequence of Porphyromonas gingivalis W83 recA homolog and construction of a recA-deficient mutant. Infect Immun. 1997;65:4592–4597. doi: 10.1128/iai.65.11.4592-4597.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher H M, Schenkein H A, Morgan R M, Bailey K A, Berry C R, Macrina F L. Virulence of a mutant of Porphyromonas gingivalis W83 that is defective in the prtH gene. Infect Immun. 1995;63:1521–1528. doi: 10.1128/iai.63.4.1521-1528.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genco R J. Host responses in periodontal diseases: current concepts. J Periodontol. 1992;63:338–355. doi: 10.1902/jop.1992.63.4s.338. [DOI] [PubMed] [Google Scholar]
- 17.Gough J A, Murray N E. Sequence diversity among related genes for recognition of specific targets in DNA molecules. J Mol Biol. 1983;166:1–19. doi: 10.1016/s0022-2836(83)80047-3. [DOI] [PubMed] [Google Scholar]
- 18.Han N, Lepine G, Whitlock J, Wojciechowski L, Progulske-Fox A. The Porphyromonas gingivalis prtP/kgp homologue exists as two open reading frames in strain 381. Oral Dis. 1998;4:170–179. doi: 10.1111/j.1601-0825.1998.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 19.Han N M, Whitlock J, Progulske-Fox A. The hemagglutinin gene A (hagA) of Porphyromonas gingivalis 381 contains four large, contiguous, direct repeats. Infect Immun. 1996;64:4000–4007. doi: 10.1128/iai.64.10.4000-4007.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong W, Cha M K, Kim I H. Thioredoxin-dependent hydroperoxide peroxidase activity of bacterioferritin comigratory protein (BCP) as a new member of the thiol-specific antioxidant protein (TSA)/alkyl hydroperoxide peroxidase C (AhpC) family. J Biol Chem. 2000;275:2924–2930. doi: 10.1074/jbc.275.4.2924. [DOI] [PubMed] [Google Scholar]
- 21.Kadowaki T, Nakayama K, Yoshimura F, Okamoto K, Abe N, Yamamoto K. Arg-gingipain acts as a major processing enzyme for various cell surface proteins in Porphyromonas gingivalis. J Biol Chem. 1998;273:29072–29076. doi: 10.1074/jbc.273.44.29072. [DOI] [PubMed] [Google Scholar]
- 22.Karunakaran T, Holt S C. Cloning of two distinct hemolysin genes from Porphyromonas (Bacteroides) gingivalis in Escherichia coli. Microb Pathog. 1993;15:37–49. doi: 10.1006/mpat.1993.1055. [DOI] [PubMed] [Google Scholar]
- 23.Kelly C G, Booth V, Kendal H, Slaney J M, Curtis M A, Lehner T. The relationship between colonization and haemagglutination inhibiting and B cell epitopes of Porphyromonas gingivalis. Clin Exp Immunol. 1997;110:285–291. doi: 10.1111/j.1365-2249.1997.tb08329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kittle J D, Simons R W, Lee J, Kleckner N. Insertion sequence IS10 anti-sense pairing initiates by an interaction between the 5′ end of the target RNA and a loop in the anti-sense RNA. J Mol Biol. 1989;210:561–572. doi: 10.1016/0022-2836(89)90132-0. [DOI] [PubMed] [Google Scholar]
- 25.Kuboniwa M, Amano A, Shizukuishi S. Hemoglobin-binding protein purified from Porphyromonas gingivalis is identical to lysine-specific cysteine proteinase (Lys-Gingipain) Biochem Biophys Res Commun. 1998;249:38–43. doi: 10.1006/bbrc.1998.8958. [DOI] [PubMed] [Google Scholar]
- 26.Kuramitsu H K. Protease of Porphyromonas gingivalis: what don't they do? Oral Microbiol Immunol. 1998;13:263–270. doi: 10.1111/j.1399-302x.1998.tb00706.x. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Lamont R J, Jenkinson H F. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lantz M S. New insights into mechanisms of bacterial pathogenesis in periodontitis. Curr Opin Periodontol. 1996;3:10–18. [PubMed] [Google Scholar]
- 30.Lepine G, Progulske-Fox A. Duplication and differential expression of hemagglutinin genes in Porphyromonas gingivalis. Oral Microbiol Immunol. 1996;11:65–78. doi: 10.1111/j.1399-302x.1996.tb00339.x. [DOI] [PubMed] [Google Scholar]
- 31.Lewis J P, Dawson J A, Hannis J C, Muddiman D, Macrina F L. Hemoglobinase activity of the lysine gingipain protease (Kgp) of Porphyromonas gingivalis W83. J Bacteriol. 1999;181:4905–4913. doi: 10.1128/jb.181.16.4905-4913.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis J P, Macrina F L. IS195, an insertion sequence-like element associated with protease genes in Porphyromonas gingivalis. Infect Immun. 1998;66:3035–3042. doi: 10.1128/iai.66.7.3035-3042.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loma Linda University Animal Research Committee. Approved protocol OSR# 95558. Loma Linda, Calif: Loma Linda University; 1996. [Google Scholar]
- 34.Maley J, Roberts I S. Characterisation of IS1126 from Porphyromonas gingivalis W83: a new member of the IS4 family of insertion sequence elements. FEMS Microbiol Lett. 1994;123:219–224. doi: 10.1111/j.1574-6968.1994.tb07225.x. [DOI] [PubMed] [Google Scholar]
- 35.Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol. 1961;3:208–218. [Google Scholar]
- 36.Mayrand D, Holt S C. Biology of asaccharolytic black-pigmented Bacteroides species. Microbiol Rev. 1988;52:134–152. doi: 10.1128/mr.52.1.134-152.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakayama K, Ratnayake D B, Tsukuba T, Kadowaki T, Yamamoto K, Fujimura S. Haemoglobin receptor protein is intragenically encoded by the cysteine proteinase-encoding genes and the haemaglutinin-encoding gene of Porphyromonas gingivalis. Mol Microbiol. 1998;27:51–61. doi: 10.1046/j.1365-2958.1998.00656.x. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto K, Kadowaki T, Nakayama K, Yamamoto K. Cloning and sequencing of the gene encoding a novel lysine-specific cysteine proteinase (Lys-gingipain) in Porphyromonas gingivalis: structural relationship with the arginine-specific cysteine proteinase (Arg-gingipain) J Biochem (Tokyo) 1996;120:398–406. doi: 10.1093/oxfordjournals.jbchem.a021426. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto K, Nakayama K, Kadowaki T, Abe N, Ratnayake D B, Yamamoto K. Involvement of a lysine-specific cysteine proteinase in hemoglobin adsorption and heme accumulation by Porphyromonas gingivalis. J Biol Chem. 1998;273:21225–21231. doi: 10.1074/jbc.273.33.21225. [DOI] [PubMed] [Google Scholar]
- 40.Papavinasasundaram K G, Movahedzadeh F, Keer J T, Stroker N G, Colston M J, Davis E O. Mycobacterial recA is contranscribed with a potential regulatory gene called recX. Mol Microbiol. 1997;24:141–153. doi: 10.1046/j.1365-2958.1997.3441697.x. [DOI] [PubMed] [Google Scholar]
- 41.Pavloff N, Pemberton P A, Potempa J, Chen W C A, Pike R N, Prochazka V, Kiefer M C, Travis J, Barr P J. Molecular cloning and characterization of Porphyromonas gingivalis lysine-specific gingipain—a new member of an emerging family of pathogenic bacterial cysteine proteinases. J Biol Chem. 1997;272:1595–1600. doi: 10.1074/jbc.272.3.1595. [DOI] [PubMed] [Google Scholar]
- 42.Pearce B J, Naughton A M, Campbell E A, Masure H R. The rec locus, a competence-induced operon in Streptococcus pneumoniae. J Bacteriol. 1995;177:86–93. doi: 10.1128/jb.177.1.86-93.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pike R, McGraw W, Potempa J, Travis J. Lysine- and arginine-specific proteinases from Porphyromonas gingivalis. Isolation, characterization, and evidence for the existence of complexes with hemagglutinins. J Biol Chem. 1994;269:406–411. [PubMed] [Google Scholar]
- 44.Potempa J, Mikolajczyk-Pawlinska J, Brassell D, Nelson D, Thogersen I B, Enghild J J, Travis J. Comparative properties of two cysteine proteinases (gingipains R), the products of two related but individual genes of Porphyromonas gingivalis. J Biol Chem. 1998;273:21648–21657. doi: 10.1074/jbc.273.34.21648. [DOI] [PubMed] [Google Scholar]
- 45.Potempa J, Pavloff N, Travis J. Porphyromonas gingivalis: a proteinase/gene accounting audit. Trends Microbiol. 1995;3:430–434. doi: 10.1016/s0966-842x(00)88996-9. [DOI] [PubMed] [Google Scholar]
- 46.Potempa J, Pike R, Travis J. The multiple forms of trypsin-like activity present in various strains of Porphyromonas gingivalis are due to the presence of either Arg-gingipain or Lys-gingipain. Infect Immun. 1995;63:1176–1182. doi: 10.1128/iai.63.4.1176-1182.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Progulske-Fox A, Tumwasorn S, Lepine G, Whitlock J, Savett D, Ferretti J J, Banas J A. The cloning, expression and sequence analysis of a second Porphyromonas gingivalis gene that codes for a protein involved in hemagglutination. Oral Microbiol Immunol. 1995;10:311–318. doi: 10.1111/j.1399-302x.1995.tb00160.x. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen J L, Odelson D A, Macrina F L. Complete nucleotide sequence and transcription of ermF, a macrolide-lincosimide-streptogramin B resistance determinant from Bacteroides fragilis. J Bacteriol. 1986;168:523–533. doi: 10.1128/jb.168.2.523-533.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 50.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schenkein H A. The role of complement in periodontal diseases. Crit Rev Oral Biol Med. 1991;2:65–81. doi: 10.1177/10454411910020010501. [DOI] [PubMed] [Google Scholar]
- 52.Shah H N, Gharbia S E. Lysis of erythrocytes by the secreted cysteine protease of Porphyromonas gingivalis W83. FEMS Microbiol Lett. 1989;61:213–218. doi: 10.1016/0378-1097(89)90199-7. [DOI] [PubMed] [Google Scholar]
- 53.Shah H N, Collins M D. Proposal for reclassification of Bacteroides asaccharolyticus, Bacteroides gingivalis and Bacteroides endodontalis in a new genus, Porphyromonas. Int J Syst Bacteriol. 1988;38:128–131. [Google Scholar]
- 54.Shi Y, Ratnayake D B, Okamoto K, Abe N, Yamamoto K, Nakayama K. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem. 1999;274:17955–17960. doi: 10.1074/jbc.274.25.17955. [DOI] [PubMed] [Google Scholar]
- 55.Simpson W, Wang C Y, Mikolajczyk-Pawlinska J, Potempa J, Travis J, Bond V C, Genco C A. Transposition of the endogenous insertion sequence element IS1126 modulates gingipain expression in Porphyromonas gingivalis. Infect Immun. 1999;67:5012–5020. doi: 10.1128/iai.67.10.5012-5020.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slakeski N, Bhogal P S, O'Brien-Simpson N M, Reynolds E C. Characterization of a second cell-associated Arg-specific cysteine proteinase of Porphyromonas gingivalis and identification of an adhesin-binding motif involved in association of the prtR and prtK proteinases and adhesins into large complexes. Microbiology. 1998;144:1583–1592. doi: 10.1099/00221287-144-6-1583. [DOI] [PubMed] [Google Scholar]
- 57.Smalley J W, Birss A J, Silver J. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the μ-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol Lett. 2000;183:159–164. doi: 10.1111/j.1574-6968.2000.tb08951.x. [DOI] [PubMed] [Google Scholar]
- 58.Smalley J W, Silver J, Marsh P J, Birss A J. The periodontopathogen Porphyromonas gingivalis binds iron protoporphyrin IX in the μ-oxo dimeric form: an oxidative buffer and possible pathogenic mechanism. Biochem J. 1998;331:681–685. doi: 10.1042/bj3310681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith G E, Summers M D. The bidirectional transfer of DNA and RNA to nitrocellulose or diazobenzyloxymethyl-paper. Anal Biochem. 1980;109:123–129. doi: 10.1016/0003-2697(80)90019-6. [DOI] [PubMed] [Google Scholar]
- 60.Sundqvist G. Pathogenicity and virulence of black-pigmented Gram-negative anaerobes. FEMS Immunol Med Microbiol. 1993;6:125–138. doi: 10.1111/j.1574-695X.1993.tb00315.x. [DOI] [PubMed] [Google Scholar]
- 61.Tabor S, Richardson C C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson S A, Blaser M J. Isolation of the Helicobacter pylori recA gene and involvement of the recA region in resistance to low pH. Infect Immun. 1995;63:2185–2193. doi: 10.1128/iai.63.6.2185-2193.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tokuda M, Chen W, Karunakaran T, Kuramitsu H K. Regulation of protease expression in Porphyromonas gingivalis. Infect Immun. 1998;66:5232–5237. doi: 10.1128/iai.66.11.5232-5237.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tokuda M, Karunakaran T, Duncan M, Hamada N, Kuramitsu H. Role of Arg-gingipain A in virulence of Porphyromonas gingivalis. Infect Immun. 1998;66:1159–1166. doi: 10.1128/iai.66.3.1159-1166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang C Y, Bond V C, Genco C A. Identification of a second endogenous Porphyromonas gingivalis insertion element. J Bacteriol. 1997;179:3808–3812. doi: 10.1128/jb.179.11.3808-3812.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]