

Abstract
CDKL5 deficiency disorder (CDD) was first identified as a cause of human disease in 2004. Although initially considered a variant of Rett syndrome, CDD is now recognised as an independent disorder and classified as a developmental epileptic encephalopathy. It is characterised by early-onset (generally within the first 2 months of life) seizures that are usually refractory to polypharmacy. Development is severely impaired in patients with CDD, with only a quarter of girls and a smaller proportion of boys achieving independent walking; however, there is clinical variability, which is probably genetically determined. Gastrointestinal, sleep, and musculoskeletal problems are common in CDD, as in other developmental epileptic encephalopathies, but the prevalence of cerebral visual impairment appears higher in CDD. Clinicians diagnosing infants with CDD need to be familiar with the complexities of this disorder to provide appropriate counselling to the patients’ families. Despite some benefit from ketogenic diets and vagal nerve stimulation, there has been little evidence that conventional antiseizure medications or their combinations are helpful in CDD, but further treatment trials are finally underway.
Introduction
Although the X-linked CDKL5 gene (previously STK9)1 was first identified as a cause of human disease in 2004,1,2 the term CDKL5 deficiency disorder (CDD) appeared in the medical literature only in 2018.3 Its original association with Rett syndrome, and use of the nomenclature early-onset seizure variant (Hanefeld variant) of Rett syndrome,4,5 might have delayed recognition of the disorder in its own right.6 Thus, it is timely to reacquaint the neurological community with this condition, given what is now known about its diagnosis and clinical manifestations and the specific treatments that could be on the horizon. Nowadays, CDD is being recognised as a severe developmental epileptic encephalopathy with onset in early infancy,7 rather than by its relationship with Rett syndrome.4
The first aim of this Review is to synthesise current knowledge about epilepsy patterns, developmental course, functional abilities, and comorbidities associated with CDD. Second, we focus on clinical variability within the disorder and new knowledge accruing about genotype–phenotype relationships. Third, we comment on the impact CDD has on the quality of life of affected children and their families. Fourth, we discuss differential diagnoses and genetic testing, comparing CDD with other developmental epileptic encephalopathies at the time of diagnosis and subsequently. Finally, we summarise evidence regarding clinical management and describe the current landscape of clinical trials. Because CDD is now more commonly diagnosed in infancy rather than later, it is crucial that clinicians, especially paediatric neurologists who often have early or first contact, have access to the best available information to provide appropriate prognostic counselling to patients and their families.
Clinical features
CDD is an ultra-rare disorder with an estimated incidence of 2·36 per 100 000 livebirths (95% CI 0·805–5·59), based on four cases identified during 3 years of epilepsy gene panel testing of 333 Scottish children presenting with epilepsy before 36 months of age.8 It was on account of such rarity that in 2012 the International CDKL5 Disorder Database9 was established to accrue adequate case numbers in a way not feasible in an individual country. The data from the database were first published in 2015.10 One major shortcoming of this caregiver-reported database is the paucity of medical detail available in case reports and series.11–13 However, the strength of this database is its international scope, with close to 300 published cases of CDD,14 a number that could not be gleaned from a single centre or country. Different types of data are needed to build the body of knowledge on CDD. An emerging data source is a multicentre clinic-based infrastructure from the US Centers of Excellence, which has enabled the publication of data on more than 90 patients,15 that now includes approximately 200 patients from eight US centres and is recognised as being capable of bridging an important gap in the path to clinical trial readiness for patients with CDD.16 The International CDKL5 Clinical Research Network was established in 2019 to formalise a collaboration between the International CDKL5 Disorder Database and the US Centers of Excellence, linking these complementary data sources and thus combining caregiver perspectives with comprehensive clinical and diagnostic information in a potential dataset of more than 400 patients with CDD.
Epilepsy
As a developmental epileptic encephalopathy, the most prominent and presenting feature of CDD is the early onset of seizures (median age at onset 6 weeks [IQR 3–8],17 with onset by 12 months of age in 150 [90%] of 167 patients).18 In fewer than half of patients,15,17 early-onset seizures are followed by a characteristic honeymoon period (appendix pp 4–6) during which seizures appear to remit; although variable by age at onset, this phase lasted longer than 6 months in 23 (25%) of 92 patients in the US Centers of Excellence dataset.15 In a small case series of 12 patients,11 epilepsy was first described to occur in three stages: the first stage is early-onset convulsive seizures, despite the presence of a normal interictal EEG; the second stage is epileptic spasms (documented in 75 [82%] of 92 patients in the US Centers of Excellence dataset)15 and hypsarrhythmia (present in only half of patients with epileptic spasms);15 the third stage was ongoing treatment-refractory seizures, with 109 (71%) of 153 patients in the International CDKL5 Disorder Database having one or more seizure per day and nearly half of patients on three or more antiseizure medications.17 For patients in the US Centers of Excellence dataset (n=92), the most common initial seizure type at onset was tonic seizures, followed by epileptic spasms and generalised tonic-clonic seizures and focal seizures (figure 1; appendix pp 4–6).15 Over the disease course, however, the most common seizure types were epileptic spasms, followed by tonic, myoclonic, and then generalised tonic-clonic seizures.15 Findings from a 2021 Japanese series19 (n=29) and aggregated data from individual case reports of male patients (n=50) are mostly consistent with findings in the US Centers of Excellence dataset (appendix pp 4–6).24 A specific hypermotor-tonic-spasms sequence was first reported in four patients (aged between 6 months and 4 years),25 and this pattern, or similar patterns, of motor seizures with multiple phases were identified in additional case series26,27 and in 52 (57%) patients in a cohort of 92 patients from the US Centers of Excellence dataset.15 In those patients that did not have the classic hypermotor-tonic-spasms sequence, tonic seizures followed by spasms was the most common pattern and other patterns included tonic-vibratory contraction followed by spasms fading to distal myoclonic jerks and tonic-clonic-spasms.15 Only a quarter of patients in the US Centers of Excellence cohort had seizures that exactly fit the initially described hypermotor-tonic-spasms sequence.15 Consistent seizure classification is somewhat challenging in CDD, especially because of the mixed motor and sometimes non-motor features, and might account for differences between case series. Although most seizure types can be seen in patients with CDD, epileptic spasms (with or without hypsarrhythmia) and tonic seizures, at times with asymmetrical or focal features, are the most common.15,28
Figure 1: Distribution of CDKL5 deficiency disorder characteristics.
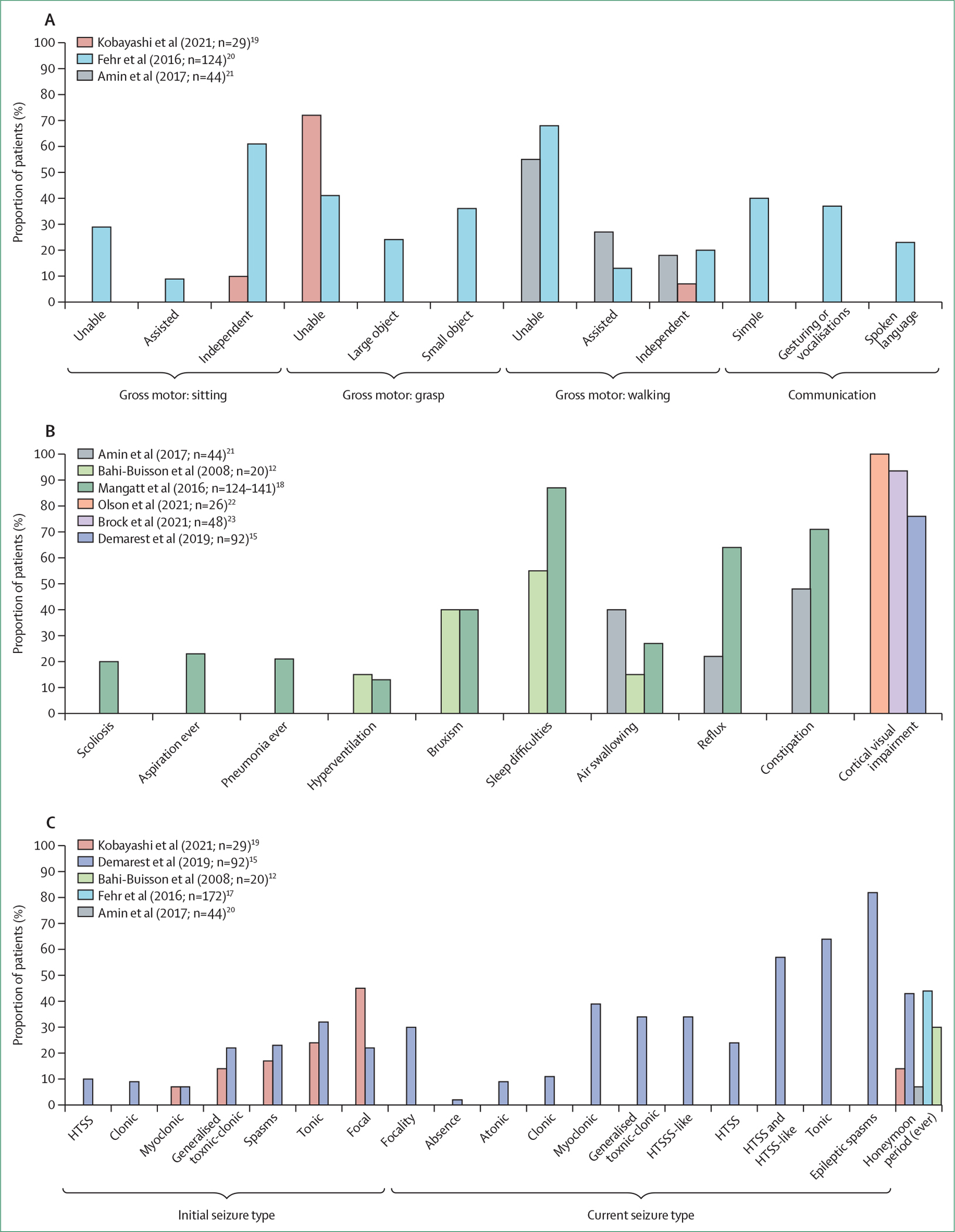
(A) Functional abilities. (B) Other comorbidities. (C) Seizures. HTSS=hypermotor-tonic-spasms sequence.
Published data about the electroclinical patterns in CDD are sparse, although case reports show that they range from mild EEG abnormalities to hypsarrhythmia at the initial presentation with seizures, with burst suppression being rare and atypical.29 In 2021, encephalopathy patterns (hypsarrhythmia, generalised slowing, discontinuity, slow spike and wave, continuous spike, and wave in sleep) were seen on EEG in all study patients older than 18 months (n=21).22 Unpublished US Centers of Excellence data showed abnormal background EEG patterns, most frequently hypsarrhythmia or generalised slowing with multifocal or generalised epileptiform activity, in 77 (87%) of 89 patients of all ages with CDD (Benke TA, Demarest S, Olson H, unpublished; Pestana Knight E M, Cleveland Clinic, OH, USA, personal communication). Longitudinal data from a large series of patients evaluated in association with other clinical features are urgently needed to better characterise the evolution of electroclinical features and to develop biomarkers in the future.
Although pathogenic CDKL5 variants are most often associated with refractory epilepsy, data from the International CDKL5 Disorder Database show that, consistent with murine models,30 seizures can also occasionally be absent in individuals harbouring such variants.17 For instance, a missense variant resulting in Ser215Arg in the CDKL5 catalytic domain, which was associated with reduced kinase activity in vitro (but less strongly associated with reduced kinase activity than a previously examined pathogenic CDKL5 variant resulting in Gln219Pro), was identified in a seizure-free woman with mild intellectual disability who was diagnosed with CDD.31,32
Development and functioning
Developmental milestones are severely delayed in affected children, with less than three-quarters of 109 girls and a third of 18 boys able to sit independently by the age of 5 years.10 Gross motor, fine motor, and communication skills are also extremely impaired (figure 1), with cross-sectional data showing that only two-thirds (n=66) of 97 girls could sit without support and just fewer than a quarter (n=22) could take steps independently.20 Although little has been published on developmental trajectories in CDD, data from the International CDKL5 Disorder Database have shown that there is considerable variation in the stability or otherwise (improvement or deterioration) of developmental status with time. Whereas about a quarter of patients had gained skills between initial contact with the database and follow-up, a third had lost skills in relation to gross motor function, hand function, or communication.33
A differentiating feature of CDD that was recognised early in the characterisation of the disorder is poor eye fixation and associated avoidance of eye gaze12 compared with the intensive eye communication seen in Rett syndrome.6 This symptom is classified as cerebral visual impairment and defined as visual dysfunction in the absence of ocular or anterior visual pathway abnormalities.34 Cerebral visual impairment, identified in 70 (76%) of 92 patients in the US Centers of Excellence cohort in Demarest and colleagues’ study (figure 1),15 was associated with poorer development even after adjusting for age and history of hypsarrhythmia.15 In a further study involving neurological and neuro-ophthalmological assessments, all 21 children with CDD who were older than 2 years were diagnosed with cerebral visual impairment, and visual acuity was found to correlate positively with gross motor ability.22 Another study showed a similar relationship between two different developmental scales; a novel cerebral visual impairment score, which was completed by paediatric neurologists, inversely correlated with developmental attainment.22,23 These studies suggest that cerebral visual impairment might be identifiable as an early and prominent feature of CDD and that measures of visual acuity or cerebral visual impairment might be useful outcome measures in future clinical trials, although better longitudinal data in larger cohorts are still needed.
Other comorbidities
Data on comorbidities other than epilepsy and cerebral visual impairment remain relatively sparse. However, sleep disturbances have been reported in 122 (87%) of 141 patients in a cohort of the International CDKL5 Disorder Database (figure 1).18 Abnormal sleep maintenance and duration have also been identified via the US Centers of Excellence database in a cohort of patients in Colorado, USA,29 and we have successfully used standard paediatric sleep treatments35 to support better sleep regulation. Given the probable burden of sleep disturbances clinically and the identification of a potential sleep breathing disorder in CDKL5 knockout mice,36 further research in this area is clearly needed.
Behavioural problems often co-occur with sleep problems in children with neurodevelopmental disorders37 but, to date, behavioural problems have not been investigated in patients with CDD. As is common with other severe forms of intellectual disability,38 122 (87%) of 141 patients in a cohort of the International CDKL5 Disorder Database were reported to have had gastrointestinal problems (figure 1; appendix pp 4–6), including constipation (100 [71%] of 141), reflux (82 [64%] of 128), and air swallowing (36 [27%] of 133).18 Feeding difficulties are a concern, with more than a quarter of patients in the International CDKL5 Disorder Database18 and fewer than a third of patients in a UK study21 requiring gastrostomy feeding.
More than a third of individuals in the International CDKL5 Disorder Database were reported to have had a lower respiratory tract infection in their first 5 years of life; lower respiratory tract infections became less frequent with age, possibly related to subsequent initiation of gastrostomy feeding, which reduces the risk of infection caused by inhaled food.18 In one cohort of the International CDKL5 Disorder Database, 27 (21%) of 126 patients were reported to have had pneumonia and 28 (23%) of 124 were reported to have had aspiration (figure 1). Episodes of apnoea and hyperventilation also occur in CDD, but less frequently than in Rett syndrome.18 Stereotypical hand movements have also been reported in more than three-quarters of patients in three different studies, but are less common in male patients than in female patients.6,12,39 In a 2021 Japanese case series of 29 patients with pathogenic CDKL5 variants, chorea affected seven (24%) patients and dystonia affected three (10%) patients.19 Musculoskeletal problems like scoliosis can also occur in CDD, with a 31·5% (95% CI 20·3–46·7) likelihood of being affected by 10 years of age.18 There is an urgent need to pool or harmonise data from multiple sources to allow the systematic investigation of comorbidities not yet adequately characterised. Given that clinical trials exploring the amelioration of CDD’s effects are expected in the future, it is imperative that the symptoms that cause the most concern to patients and their families are identified.40
Neuroimaging
Reports of neuroimaging in patients with CDD are sparse and often include small patient numbers; results too can be variable and non-specific, sometimes involving brain atrophy and white matter hyperintensities, as can be seen in other developmental epileptic encephalopathies (eg, Ohtahara syndrome).9,19,29,39,41 In CDD, abnormal brain imaging appears to be more evident in male than in female patients.9,19,29,39 A 2021 study that used quantitative morphological analyses to compare patients with CDD with age-matched and sex-matched healthy controls found significant reductions in total and subcortical grey matter and posterior cortical thickness in three individuals with CDD.42
Determinants of clinical variability
Most of the variation in CDD examined to date relates to the acquisition of developmental milestones,10 levels of gross and fine motor functioning and communication,20 and epilepsy burden.17 In the first investigation of functional abilities in patients with CDD, boys, who also have a higher prevalence of MRI abnormalities,39 had poorer gross motor function than girls.20 Similar disparities between sexes were seen for fine motor skills and communication.20 No differences were detected in parent-reported skill level when younger patients (aged from >18 months to ≤7 years) were compared with older patients (aged from >7 years to ≤13 years or >13 years). 20
Additional to sex and age group, genotype is another probable determinant of clinical variability in CDD.43 The CDKL5 gene, located at chromosome Xp22.13, comprises 22 exons and encodes CDKL5, a serine/threonine kinase with a role in regulating axon outgrowth, dendritic morphogenesis, and synapse formation in early postnatal life, and in maintaining synaptic function in the adult brain.44 CDKL5 is subject to X chromosome inactivation, and the protein has multiple transcripts due to alternative splicing, with the primary brain-expressed transcript not including exons 17 or 20–22. The N-terminus of the protein contains a highly conserved catalytic domain where pathogenic missense variants are exclusively clustered, but pathogenic truncating variants occur throughout the coding region of the gene.45 In the International CDKL5 Disorder Database, 285 affected individuals harboured more than 200 different CDKL5 variants,14 making genotype–phenotype relationships particularly challenging to study. Therefore, initial studies categorised variants into four groups according to their position on the gene and functional consequences46 (figure 2).10,17,18,20 Using this framework, patients with truncating variants after amino acid 781 were found to reach gross motor,10 fine motor, and communication milestones earlier than patients in other variant groups, results that were recapitulated in a cross-sectional analysis of functional abilities.20 When comparing seizure rates, the lowest rates were found in patients with truncations between amino acid 172 and amino acid 781.17 Another study found no relationships between variant group and seizure type, hypsarrhythmia, periods of seizure freedom, or the presence of cerebral visual impairment among 92 patients from the US Centers of Excellence dataset.15 Few other genotype–phenotype relationships have been identified between the aforementioned genetic classification and comorbidities in patients with CDD.18
Figure 2: Location of recurrent* variants on the CDKL5 protein.
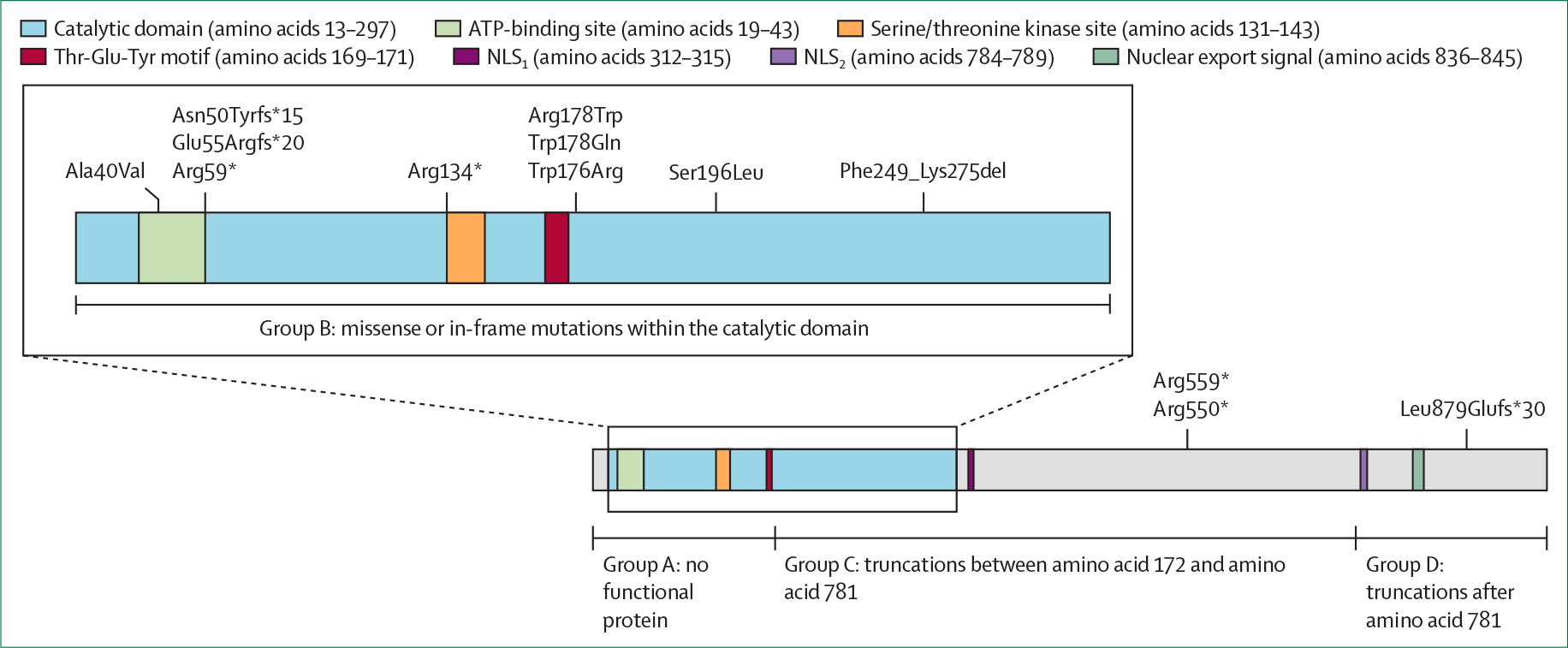
The inset shows an expanded diagram of the catalytic domain. NLS1=nuclear localisation signal 1. NLS2=nuclear localisation signal 2. *Variants affecting three or more individuals.
Adjusting for age and sex, a 2021 study14 explored the relationships between 13 recurrent (affected three or more patients) CDKL5 variants and phenotype (developmental outcomes and clinical severity) by use of the CDKL5 Developmental Scale15 and a modified version of the CDKL5 Clinical Severity Assessment47 among patients in the International CDKL5 Disorder Database. Surprisingly, given the low number of recurrent variants, small numbers of patients were identified at the high or low ends of the spectrum of severity. Individuals with the missense variant resulting in Arg178Trp had a high mean severity (panel 1) and a low mean developmental score, as did individuals with variants resulting in Arg559* or Arg178Gln. Variants associated with milder phenotypes (both in clinical severity and developmental score) included Arg134* (panel 2), Arg550* and Glu55Argfs*20.
Panel 1: Case study of a patient with a severe phenotype.
A now 16-year-old girl with CDKL5 deficiency disorder (CDKL5 variant 532C→T, resulting in Arg178Trp) was born by vaginal delivery at 40 weeks’ gestation and suspected seizure onset occurred on her second day of life. She was non-dysmorphic and had normal growth parameters. She had an onset of tonic seizures at 3 months, infantile spasms with hypsarrhythmia were diagnosed at 4 months, and she subsequently developed generalised myoclonic and tonic-clonic seizures in the first year of life. Multiple generalised seizure types contributed to the later diagnosis of Lennox–Gastaut syndrome. Brain MRI at 10 months showed delayed myelination and global atrophy. Over time, seizure control has been variable. She experienced periods of seizure control during her first 10 years of life, but then the seizures became severe and refractory, with daily clusters of generalised tonic seizures lasting up to 10 min each. At age 11 years, she underwent complete corpus callosotomy for treatment of refractory seizures. After corpus callosotomy, seizure frequency reduced by approximately 50% and seizure severity improved. Trials of multiple antiseizure medications before and after corpus callosotomy were ineffective; a ketogenic diet introduced at 13 months of life reduced seizure frequency temporarily. At the most recent follow-up at 15 years of age, she continued to have daily brief myoclonic seizures or brief tonic seizures. EEGs showed generalised slowing with multifocal and generalised epileptiform activity.
In addition to intractable seizures, she presented with developmental delay and hypotonia. Cortical visual impairment was diagnosed at 3 months and, on examination at 5 years of age, she did not have any visual or auditory tracking. Diffuse choreoathetoid movements in her arms and legs were first described at 11 months. She was able to roll for 2–3 months as an infant but lost the skill at 6 months of age. At the most recent follow-up at 15 years of age, she had diffuse hypotonia, poor head and trunk control, and was dependent on a wheelchair for all mobility. She has been non-verbal without vocalisations throughout life. She had a Nissen and gastrostomy tube placed when she was 2 years old because she was having difficulties with oral feeding. She had spinal fusion for scoliosis 1 month after the corpus callosotomy at 11 years of age.
Panel 2: Case study of a patient with a mild phenotype.
This girl with CDKL5 deficiency disorder (CDKL5 variant 400C→T, resulting in Arg134*) was born after a normal pregnancy by caesarean section due to fetal decelerations at 41 weeks’ gestation. She first presented at 2 months of age with infantile spasms. Her seizures later became predominantly bilateral tonic and refractory to treatment and occurred multiple times per day. At the most recent follow-up at 9 years of age, she had between four and seven bilateral tonic seizures per day. Non-medication options for management had been offered but not used thus far, and she had received eight antiseizure drugs, including levetiracetam, prescription cannabidiol, and lorazepam. Her most recent EEG showed multifocal and generalised epileptiform activity, generalised slowing with superimposed fast activity, an absence of normal sleep features (no well formed vertex waves or sleep spindles), and electrographic generalised seizures. Brain MRI was normal.
She had global developmental delay and cortical visual impairment and strabismus but did visually track at times. She sat independently at 12 months of age, crawled on her belly at 3 years, and walked with assistance at 3 years and 8 months. With continued physical and occupational therapy, she learnt to sit up and used a stander, a gait trainer, and an adaptive bike. She said her first words at age 4 years and has a limited vocabulary of up to five single words, including “mama”, “dada”, and “hi.” She could pick up large objects with a palmar grasp, but did not have a pincer grasp. At the most recent follow-up at 9 years of age, she used a sippy cup and also helped to dress herself and brush her teeth. She showed increased engagement in self-feeding and took all food by mouth. Precocious puberty first presented at 5 years of age in addition to a diagnosis of central hypothyroidism. Scoliosis was first diagnosed at 7 years of age (53° left lumbar curve and 42° right thoracic curve), for which bracing (with thoracic lumbar sacral orthosis) was prescribed, and was described as moderate thoracolumbar scoliosis at 9 years of age. At examination at 9 years of age, hypertonia, athetoid movements, and dyskinesias were reported to be present. The child was breathing with regular rhythm and no other autonomic disturbances, like cold hands or feet, or gastrointestinal concerns, were reported.
Two of the severe variants were located in the Arg178 codon where, in vitro, the Arg178Trp change markedly reduces CDKL5 activity, which then fails to phosphorylate various target proteins.32,48,49 The ability to investigate individual genotype–phenotype relationships clinically could have implications not only for the provision of prognostic information but also for the understanding of CDKL5 functioning.
When the originally used gene groupings10 were re-examined in a larger dataset,14,50 patients with truncations after amino acid 781 still had the highest CDKL5 Developmental Scale scores, indicating less severely impaired development, although statistical significance was not reached.14 Moreover, such individuals also had a higher clinical severity score than did patients in other variant groups, possibly related to worse epilepsy.17 The influence of mosaicism could not be assessed. Consistent with data from epilepsy gene panels,51 mosaicism has now been detected in five of 47 male patients and in one of 238 female patients in the International CDKL5 Disorder Database,14 and in eight of 50 male individuals in a 2021 review of the literature on CDD in male patients.24 Both male patients (who are more likely to walk) and female patients with mosaicism are likely to function better than those without (Leonard H, Olson H, unpublished; panel 3). Future research needs to evaluate the effect of sex and somatic mosaicism on disease severity. Given genetic heterogeneity, it is crucial that researchers combine or harmonise data rather than depend on individual inadequately powered datasets.
Panel 3: Case study of a patient with mosaicism and a mild phenotype.
This girl with CDKL5 deficiency disorder (CDKL5 variant 541G→A, resulting in Glu181Lys; mosaicism present in 15·25% of blood reads) was born by vaginal delivery at 39 weeks’ gestation after an uneventful pregnancy. She was first evaluated in genetic and neurology clinics at 15 months of age due to concerns of developmental delay. Her first seizures occurred at 18 months and included tonic-clonic seizures followed by clusters of probable myoclonic seizures. At the most recent follow-up at 2·5 years of age, she had bilateral tonic stiffening seizures between four and five nights per week, after having had a period of seizure freedom for 3 months. EEG background activity was normal and brain MRI was normal.
Regarding her development, she sat at 10 months of age, crawled at 14 months, and walked at 17 months. Mild hypotonia was reported at 15 months. At 19 months, she walked fast and was able to run. She had good fine motor adaptive skills that were normal for her age, such as manipulating small objects with her hands and fingers. She also spoke several single words at 19 months, including “hi” and “momma”. At 24 months of age, bruxism was reported. At 2·5 years, she could walk independently and run slowly. Her vision was age appropriate. She spoke in three-word sentences and also used hand signing and gesturing for communication. She was able to throw a ball and had good fine motor manipulation of small objects with both hands. She liked using her hands, picking things up, and playing with toys. These skills were continually strengthened during regular speech and occupational therapy sessions. She showed social engagement with shared attention and excitement, especially with her mother, but could be aggressive with others, had a poor sense of danger, and had sensory seeking behaviour. No feeding difficulties, autonomic disturbances, or gastrointestinal concerns were reported.
Child and caregiver quality of life
Caregiver wellbeing, family quality of life,52,53 and, more recently, parent-reported child quality of life50 have been examined. A qualitative study54 found that domains previously shown to be relevant to Rett syndrome, and subsequently incorporated into the Quality of Life Inventory-Disability measure (appendix p 4),55 were also applicable to individuals with CDD. This instrument was then administered to families in the International CDKL5 Disorder Database to explore the determinants of quality of life among children with CDD.50,55 Overall, low levels of child functioning, polypharmacy (a factor impairing quality of life in adults with epilepsy56), and increased sleep difficulties had the greatest adverse impact on child quality of life.50 An earlier report52 had investigated parental wellbeing by use of the 12-Item Short Form Survey and family quality of life by use of the Beach Family Quality of Life Scale. The mean mental, but not physical, component score on the 12-Item Short Form Survey was lower among parents of children with CDD compared with the general population. Caregivers whose child had gastrostomy feeding had poorer physical but better mental wellbeing scores than caregivers whose child was only feeding orally, suggesting that, although additional physical tasks are required to manage this intervention, it could alleviate mental stress. As with child quality of life, increasing child sleep disturbances also had a negative impact on caregiver emotional wellbeing.
Furthermore, in a subsequent study, parental emotional wellbeing for mothers of children with CDD was found to be generally poorer than that of the general population, and parental emotional wellbeing for all caregivers of children with CDD was found to be significantly worse than for caregivers of children with Rett syndrome or Down syndrome.53 Although a direct comparison of quality of life among children with CDD versus children with Rett syndrome or Down syndrome has not been reported, total quality of life scores50 were also poorer among children with CDD compared with children with Rett syndrome or Down syndrome57 (appendix p 9).
Clinicians and researchers should attend closely to such findings, which represent the caregiver voice and reveal much about the burden of CDD for the child and their family. For instance, it is possible that the deleterious effects of polypharmacy could sometimes outweigh any benefit in seizure control, as also reported by adults with epilepsy.56 Clearly, child sleep disturbances are also adversely affecting both child and caregiver wellbeing, reinforcing the urgent need for further research into sleep assessments and treatments.
Differential diagnosis
Genetic testing in epilepsy is now largely done through epilepsy gene panels and exome sequencing, in which CDKL5 is one of the high-yield genes,58–60 and there is a growing need to develop formal criteria for gene and variant curation.61 Differential diagnosis of CDD varies according to age at presentation (appendix p 10) and, when confirmed, there are considerable implications for medical providers who have to counsel and prognosticate on their infant patient’s probable clinical course. When presentation is neonatal, differential diagnoses include KCNQ2, SCN2A, and STXBP1 encephalopathies, all of which are often accompanied by a more severely abnormal EEG than is CDD, which includes, in some cases, burst suppression.62 When spasms present after the neonatal period (1 month of life; eg, in West syndrome), differential diagnoses include SCN8A or STXBP1 encephalopathies and other genetic causes of spasms.63,64 Although epileptic spasms, hypsarrhythmia, and developmental regression are more commonly seen in CDD than in other early life genetic epilepsies, the prevalence of these three signs and symptoms occurring together as West syndrome appears the same in CDD as in other early life genetic epilepsies (7%).65
Before 2015, CDD was often diagnosed in girls under the name of early-onset seizure variant of Rett syndrome.4 These individuals would have severe global delay and severely impaired gross motor function with abnormal muscle tone.6 As with typical Rett syndrome (which has a median age at seizure onset of 48 months [IQR 24–90]),66 sleep disturbances and gastrointestinal issues were common;18 hand stereotypies, breathing disturbances, and laughing and screaming spells were sometimes present in patients with CDD, but were less frequent than in patients with Rett syndrome.6 Reported in more than a third of patients with CDD,67 and often associated with seizures, the severity and timing of regression in CDD is more variable than in Rett syndrome, in which regression characteristically occurs between 12 months of age and 24 months of age. Overall clinical severity has been shown to be greater in CDD and FOXG1 syndrome than in Rett syndrome, whereas microcephaly is more likely to occur in Rett syndrome than in CDD.67 Although these core features were singled out as discriminatory across the four Rett syndrome-related disorders studied by Cutri-French and colleagues67 (appendix p 10), there are other differentiating characteristics and morbidities that have not yet been fully investigated,18 including cerebral visual impairment, sleep dysregulation, dysautonomia, gastrointestinal dysfunction and gastrointestinal growth, musculoskeletal abnormalities, and respiratory infections.
The genetic differential diagnosis of early-onset epileptic encephalopathy is broad but early features suggestive of CDD include seizures with multiple motor phases, prominent hypotonia, cerebral visual impairment, and progressively worse encephalopathy on EEG. In childhood and beyond, symptoms of CDD might overlap with those of Rett syndrome18 or one of the related developmental epileptic encephalopathies.67
Clinical management
Evidence-based information on the clinical management of patients with CDD is relatively sparse and limited to treatments for epilepsy. In the International CDKL5 Disorder Database, median seizure rate was calculated to be two per day (range 0–20), with 86 (56%) of 153 individuals having between one and five seizures per day on average and 23 (15%) having more than five per day.17 Only 12 (8%) had been seizure free during the previous 6 months,17 consistent with the six (7%) of 92 patients who were seizure free at their last visit to a US Center of Excellence.15 Findings from a more recent but smaller (n=22) Japanese study were similar.19
Patients with CDD do not show consistent responses to specific combinations of antiseizure medications, with any improvement generally being only short-term. Polypharmacy, reflecting the refractory nature of the epilepsy, is common and associated with poorer quality of life.50 Nearly half (57 [47%]) of 122 patients in a cohort of the International CDKL5 Disorder Database were on three or more antiseizure medications,50 and the median lifetime number of antiseizure medications was found to be six (range 0–18).28 In the International CDKL5 Disorder Database, the most frequently used antiseizure medications are broad-spectrum, including clobazam, valproate, topiramate, levetiracetam, and vigabatrin, but no benefits from particular antiseizure medications have been identified.17 One mostly European multicentre study (39 patients) showed that a subset of commonly prescribed medications (namely felbamate, vigabatrin, clobazam, and valproate) reduced seizure frequency by 50% or more, but response after 3 months was not sustained. With some medications, including carbamazepine, there was also some worsening of seizure frequency.68 Another study reported positive responses in seizure number to antiseizure medications that block sodium channels in a subset of patients with CDD and more focal epilepsy.69 In a Japanese study, seizure frequency was reduced by 50% or more in four of 29 patients with CDD (one received lamotrigine, two received valproate, and one received vigabatrin), although seizure frequency was reduced by at least 25% but less than 50% in a higher proportion of patients (it is unclear whether these drugs were being used as monotherapy or in combination with other antiseizure medications).19 A Georgian case series (n=8) provided encouraging results, with five patients with CDD responding with the best reduced seizure frequency to a combination of vigabatrin and zonisamide, but these findings require replication.27
Use of purified cannabidiol has also been examined in an open-label study (17 patients with CDD), showing a 41% reduction in convulsive seizures at 12 weeks and a 60% reduction by 48 weeks.3 Cannabidiol seems to be a treatment option that families consider beneficial.70 A 2020 study of the National Infantile Spasms Consortium database investigated the management of epileptic spasms and found that responses (defined as electroclinical remission) to conventional treatment with adrenocorticotropic hormone, prednisone, and vigabatrin were worse in patients with CDD than in a control group of patients with infantile spasms of other aetiologies, but that the best response was with vigabatrin (27% 14-day response; 11% 3-month response).71 The initial efficacy of ketogenic diets for refractory spasms in patients with CDD was 20% at 1 month after diet initiation and 17% at 3 months after diet initiation.71 In the International CDKL5 Disorder Database (n=204), more than half of patients (n=104) had used the ketogenic diet, with a median duration of 17 months (95% CI 9–24).72 Of the 69 caregivers who reported associated changes in seizure activity, 61 (88%) reported beneficial changes; side-effect profiles (mostly gastrointestinal) were as expected. Only a third were still following the diet at ascertainment to the database, with half of the caregivers who discontinued the diet citing lack of efficacy as their reason for withdrawal. The ketogenic diet appears to provide some short-term benefit for seizure control that might not be sustained in the long term.
A further mode of treatment used less commonly is vagal nerve stimulation. Only 38 (17%) of 224 individuals in the International CDKL5 Disorder Database have had vagal nerve stimulation at a median age of 4·9 years (IQR 3·1–8·0), with early termination secondary to side-effects reported in three patients.73 About two-thirds of caregivers reported improvements in the frequency, duration, or intensity of seizures. Despite these benefits, there was no reduction in the use of antiseizure medications. However, improved behaviours, increased alertness, or both were reported for a small number of patients (n=9). A few patients have undergone corpus callosotomy29 or, generally before genetic diagnosis, resective epilepsy surgery. Overall, the highly refractory nature of seizures in CDD and the negative effects of daily seizures and polypharmacy on quality of life make CDD a key disorder for which to strive for disease-modifying therapies.
CDD requires a multidisciplinary approach to care. Most patients have debilitating developmental challenges necessitating life-long therapy to maintain developmental achievements and adaptive care needs. Unpublished data from the International CDKL5 Disorder Database suggest that caregivers generally feel that their children are showing benefit from physical and occupational therapies. Because of its high prevalence, cerebral visual impairment generally needs to be accommodated for in psychoeducational activities and therapies.15,22,23 Furthermore, challenges related to sleep,18 gastrointestinal symptoms,18,21 and growth status are important to address given their potential effects on quality of life.50,52
Future treatments
Several drugs are in development for CDD (table). Further to a phase 2, proof-of-concept trial,74 the double-blind phase of a phase 3, randomised, placebo-controlled trial of the neurosteroid GABAA agonist ganaxolone79 (NCT03572933) was recently completed and met its primary endpoint, finding a 30 7% reduction in 28-day major motor seizure frequency in the ganaxolone group versus a 6 9% reduction in the placebo group (p=0·0036).28,75 The US Food and Drug Administration approved ganaxolone in March, 2022, for the treatment of seizures associated with CDD in patients aged 2 years or older. Soticlestat (OV935/TAK-935) is a selective inhibitor of the enzyme CH24H, which converts cholesterol to 24S-hydroxycholesterol, a positive allosteric modulator of the NMDA receptor. The recently completed phase 2 trial of soticlestat in patients with CDD or 15q duplication syndrome showed a 24% reduction in major motor seizure frequency with soticlestat (NCT03694275) and a 50% reduction in the long-term extension of the trial for patients with CDD; treatment-emergent side-effects were reported in the majority of patients (n=19) but were only severe in three (NCT03635073).28,77 Ataluren, a readthrough compound, did not show efficacy in the treatment of CDD (NCT02758626),76 consistent with cell-based data.76,80 Given its demonstrated efficacy in Dravet syndrome81 and despite mixed results in developmental epileptic encephalopathies more broadly, the recently published results of a small (n=6) open-label trial of fenfluramine in patients with CDD are encouraging, especially in relation to the management of tonic-clonic seizures.78
Table:
Therapeutic interventions in clinical trials for patients with CDD
| Study design | Participants | Intervention | Comparator | Outcomes |
Results | ||
|---|---|---|---|---|---|---|---|
| Primary | Secondary | ||||||
|
| |||||||
| Highly purified cannabidiol | |||||||
| Devinsky et al (2018)3 | Open-label, non-randomised, uncontrolled trial with a single group assignment | 55 participants aged 1–30 years with various epileptic encephalopathies, including 20 patients with CDD | Highly purified cannabidiol (oral sesame oil-based solution) at an initial dose of 5 mg/kg per day, which was titrated every 2 weeks by increments of 2–10 mg/kg per day until intolerance or a maximum dose of 25 mg/kg per day* | No placebo group; participants were compared with their own 4-week baseline | Change in monthly frequency of convulsive seizures | Change in monthly frequency of all seizure types and subtypes | Median monthly convulsive seizure frequency in the CDD group decreased from 66 (IQR 26–212) at baseline to 36 (9–142) at week 12 (p=0·032) and remained stable (26, 7–75) at week 48; 4 participants withdrew due to adverse effects |
| Ganaxolone | |||||||
| Specchio et al (2021; NCT02358538)74 | Phase 2, openlabel, nonrandomised, uncontrolled, proof-of-concept trial with single group assignment | 30 participants aged 2–18 years with various epileptic encephalopathies, including seven patients with CDD | Ganaxolone (oral suspension or capsules) at a maximum dose of 1800 mg/day (patients weighing >28 kg) or 63 mg/kg per day (patients weighing ≤28 kg) | No placebo group; participants were compared with their own baseline | Change in 28-day seizure frequency | Clinician-assessed and caregiver-assessed Global Impression of Change scores; safety and tolerability of ganaxolone | Only preliminary results are available;74 patients with CDD on ganaxolone had a 44·4% median reduction in 28-day seizure frequency versus baseline; 4 patients progressed to the open-label extension phase and continued to have reduced seizure frequency until 18 months (study end) |
| Pestana Knight et al (2022; NCT03572933)75 | Phase 3, doubleblind, randomised, placebo-controlled trial | 101 participants with CDD aged 2–21 years | Enteral ganaxolone (maximum dose 63 mg/kg per day for patients weighing ≤28 kg or 1800 mg/day for patients weighing >28 kg) for 17 weeks | Matching enteral placebo for 17 weeks | Change in median 28-day major motor seizure frequency | Proportion with seizure frequency reduction of at least 50%; Clinical Global Impression of Improvement score; change in percentage of seizure-free days; Caregiver Global Impression of Change in Seizure Intensity/ Duration score; Caregiver Global Impression of Change in Attention score; Caregiver Global Impression of Change score in a target behaviour | 28-day major motor seizure frequency reduced by 30·7% in the ganaxolone group compared with 6·9% in the placebo group (p=0·0036); the discontinuation rate was 4% in the ganaxolone group and 8% in the placebo group |
| Ataluren | |||||||
| Devinsky et al (2021; NCT02758626)76 | Phase 2, doubleblind, randomised, placebocontrolled, crossover trial | 16 participants aged 2–12 years with CDD or Dravet syndrome, with the number of participants with CDD unclear | Participants were randomly assigned to oral ataluren (10 mg/kg in the morning and midday and 20 mg/kg in the evening; treatment period 1; 12 weeks) followed by a 4-week washout and then placebo (treatment period 2; 12 weeks) or placebo (treatment period 1; 12 weeks) followed by a 4-week washout and then oral ataluren (same dose and schedule; treatment period 2; 12 weeks) | Participants were randomly assigned to oral ataluren (10 mg/kg in the morning and midday and 20 mg/kg in the evening; treatment period 1; 12 weeks) followed by a 4-week washout and then placebo (treatment period 2; 12 weeks) or placebo (treatment period 1; 12 weeks) followed by a 4-week washout and then oral ataluren (same dose and schedule; treatment period 2; 12 weeks) | Number of adverse events and serious adverse events | Change in seizure frequency; quality of life (as assessed by the Quality of Life of Childhood Epilepsy scale); and change in cognitive behaviour (as assessed by the Behavior Assessment System for Children) | During the blinded phase, seven of eight patients with CDD had adverse events while on ataluren (two patients had adverse events leading to study withdrawal [one due to gastrointestinal symptoms of moderate severity possibly related to the study drug and one due to increased seizures of mild severity not likely to be related to the study drug]); no efficacy was shown |
| Soticlestat | |||||||
| Demarest et al (2021; NCT03694275)77 | Phase 2, openlabel, nonrandomised, uncontrolled, pilot trial with single group assignment | 20 participants aged 2–55 years with CDD (n=12) or 15q duplication syndrome | Soticlestat tablets or mini-tablets; 8-week dose optimisation and then a 12-week maintenance period | No placebo group; participants were compared with their own baseline | Change in motor seizure frequency during the maintenance period (weeks 9–20) | Percentage of participants classified as treatment responders; change in motor seizure frequency during the treatment period (weeks 0–20); change from baseline in Clinician’s Global Impression of Severity Responses of Investigator; proportion of participants with Clinical Global Impression of Change responses as per the investigator-reported impression; correlation between plasma 24S-hydroxycholesterol concentrations and motor seizure frequency; and proportion of participants with Care Clinical Global Impression of Change responses assessed by parent or family | Participants with CDD showed a mean reduction in motor seizure frequency of 24% after the 12-week maintenance period |
| Fenfluramine | |||||||
| Devinsky et al (2018; NCT03861871)78 | Phase 2, openlabel, nonrandomised, uncontrolled trial with single group assignment | 6 participants aged 2–26 years with CDD | Oral fenfluramine titrated to 0·8 mg/kg per day or a maximum dose of 30 mg/day for 14 days | No placebo group; participants were compared with their own baseline | Median monthly convulsive seizure frequency | Caregiver Global Impression of Change; Quality of Life of Childhood Epilepsy scale; and Pediatric Quality of Life Inventory | 90% reduction in seizure frequency in 5 participants with tonic-clonic seizures and 50–60% reduction in seizure frequency in 2 participants with tonic seizures (preliminary results) |
CDD=CDKL5 deficiency disorder. FDA=Food and Drug Administration.
Some sites included patients who received up to a maximum dose of 50 mg/kg per day.
Epilepsy is not routinely observed in mouse models of CDD, which presents a key challenge in treatment development.82,83 However, various efforts are underway to develop disease-modifying therapies that would go beyond seizure control to address the neurobiology44 and developmental challenges84–90 of CDD (appendix p 8). These efforts include gene replacement therapy in rodents via an adeno-associated virus.89 Additional cell culture-based and murine model-based strategies are being piloted that involve repurposed drugs,91 pregnenolone and derivatives,92 RNA-based approaches,93 and alteration of X chromosome inactivation.94 Many of these approaches, such as those based around X chromosome inactivation or RNA, remain as proof of concept and have not progressed to in vivo models. Notably, animal models often fail to fully recapitulate human disease, and models of CDD are no exception.95
A considerable challenge for clinical trials in CDD (as well as other developmental epileptic encephalopathies) is the choice of validated and psychometrically sound outcome measures. The domains affected in animal models suggest which domains will be essential to evaluate in clinical trials (appendix p 8). Existing measures, such as the Adaptive Vineland Behavior Scales, that do not take into account the unique features of CDD, will be hampered by their floor effects and unknown reliability, validity, and capacity to show responsiveness to change. Floor effects were found with the Adaptive Vineland Behavior Scales in a study of 64 children with SCN2A encephalopathy96 and this pattern could be replicated in CDD trials in the future. Although the CDKL5 Clinical Severity Assessment was developed specifically for CDD,47 the clinical portion of the assessment has recently been refined, providing evidence of content validity.97 The Quality of Life Inventory-Disability measure has been examined for content54 and known-groups50 validation but its reliability and responsiveness to change in CDD remain unknown. A clinical trial readiness grant in the USA (the National Institute of Neurological Disorders and Stroke of the National Institutes of Health [NIH]; grant number U01NS114312 [awarded to Tim A Benke]) aims to validate clinical outcome measures and biomarkers (EEG and evoked potentials) in patients with CDD for use in future clinical trials. Seizures, developmental delay (eg, in communication), sleep disturbances, and gastrointestinal problems have been identified by caregivers of individuals with CDD as being of major concern.40 The specific aspects of CDD that can be modulated by gene replacement therapies are unknown.89,98 Hence, it is a imperative to validate outcome measures for a range of domains in CDD to enable the evaluation of efficacy in clinical trials and meaningful monitoring in clinical practice.
Conclusion and future directions
Research is ongoing on the pathophysiology of CDD, including the role of CDKL5 in microtubular dynamics,48,49,99 and efforts to understand the clinical picture and factors influencing the variability of CDD continue. Progress is limited by the rarity8,45 and genetic heterogeneity (with >200 individual pathogenic CDKL5 variants) of CDD and is therefore dependent on the few existing large-scale case aggregations,14,15 underscoring the need for data harmonisation (panel 4). Thus, literature informing the management of this complex disorder remains scant, although clinical trials of new potential antiseizure treatments are now underway. However, many of the symptoms that caregivers have highlighted as mattering most are going to require disease-modifying, rather than symptomatic, treatments to target the cause of CDD. Although gene therapy is on the horizon, a successful programme of clinical trial readiness is still needed to make such therapy a reality.89,98
Panel 4: Research priorities for CDKL5 deficiency disorder (CDD).
Clinical features
Data on the evolution of electroclinical (EEG and evoked potentials) features, which could be used for the future development of biomarkers
Development of assessments that measure the impact of seizures on daily functions
Evaluation of the nature and magnitude of sleep disturbances given their probable burden on children with CDD and their families
Investigation of the prevalence and clinical features of movement disorders, musculoskeletal issues, and other comorbidities that have not yet been characterised (eg, behavioural profile and cardiac rhythm abnormalities)
Better understanding of the factors that predict variability by pooling or harmonising data from multiple sources
Data on natural history and mortality, including the rate of sudden unexpected death in epilepsy
Clinical management
Replication of small studies showing benefit for particular antiseizure medications initially through large-scale observational studies accounting for age, seizure type, and combinations of treatments used
CDD-specific clinical trials of new antiseizure medications that, like fenfluramine, have shown efficacy in other disorders
Evaluation of the impact of different therapeutic approaches on developmental progress
Animal-based and cell-based models to identify potential disease-modifying treatments that go beyond only seizure control
Clinical assessments and biomarkers that are translatable from animal models to a human population with CDD
Programme of clinical trial readiness to ensure that validated outcome measures are available for the domains (including quality of life) that are likely to be affected in CDD
Search strategy and selection criteria
We searched Web of Science Core Collection, MEDLINE, and Embase for potentially relevant articles related to CDKL5 deficiency disorder published in English between Jan 1, 2016, and Dec 12, 2021, using the search terms “CDKL5”, “cyclin-dependent kinase-like 5”, “STK9”, “Serine-Protein-Threonine kinase”, “Epileptic syndromes”, “Infantile spasms”, and “Rett syndrome”. There were no language restrictions. The final reference list was generated on the basis of relevance to the topics covered in this Review.
Supplementary Material
Acknowledgments
The primary funding source for this Review was the NHMRC Senior Research Fellowship (number 1117105) awarded to HL. HO was funded through the National Institute of Neurological Disorders and Stroke (K23 NS107646-04). TAB was funded through the NIH (U54HD061222 and U01NS114312) and the Children’s Hospital Foundation Ponzio Family Chair in Neurology Research. JD was funded through the NIH (U01NS114312) and the International Foundation for CDKL5 Research. LS was funded through the International Foundation for CDKL5 Research. Other funding sources did not have a role in this Review. We thank Conor Mackay for his painstaking efforts in the formatting of the tables and Mohammed Junaid for his assistance. We thank Elia Pestana-Knight and her team at the Cleveland Clinic CDKL5 Center of Excellence (Cleveland, OH, USA) for allowing the inclusion of their unpublished data.
Declaration of interests
HL has received support from the Australian National Health and Medical Research Council (NHMRC; NHMRC Senior Research Fellowship [number 1117105]) in relation to this Review; has received funding from the NIH, the International Foundation for CDKL5 Research, the Orphan Disease Centre, the University of Pennsylvania, and Marinus in relation to this subject matter; and has consulted for Ovid Therapeutics on a related subject matter. JD has received funding from the NIH, the International Foundation for CDKL5 Research, the Orphan Disease Centre, the University of Pennsylvania, and Marinus in relation to this subject matter and has consulted for Ovid Therapeutics on a related subject matter. TAB has received funding from the NIH and the Children’s Hospital Foundation for a related subject matter and has consulted for Neuren/Acadia, Ovid/Takeda, AveXis, Marinus Taysha, Alcyone, and Marinus (all compensation was made to his department). SD has funding from the NIH and the International Foundation for CDKL5 Research related to this subject matter; has consulted for Marinus and Ovid Therapeutics on a related subject matter; and is a member of the Scientific Advisory Board for Families SCN2A and SLC6A1 Connect. HO has received funding from the National Institute of Neurological Disorders and Stroke and the International Foundation for CDKL5 Research related to this subject matter; has received funding from the Manton Center for Rare Disease Research and Lou Lou for unrelated research; has consulted for Takeda, Zogenix, and Ovid regarding clinical trials in CDD or information about CDD; and has consulted for the FOXG1 Research Foundation on an unrelated matter to CDD. LS declares no competing interests.
Footnotes
See Online for appendix
Contributor Information
Helen Leonard, Telethon Kids Institute, The University of Western Australia, Perth, WA, Australia.
Jenny Downs, Telethon Kids Institute, The University of Western Australia, Perth, WA, Australia; Curtin School of Allied Health, Curtin University, Perth, WA, Australia.
Tim A Benke, Department of Neurology, Children’s Hospital Colorado, Aurora, CO, USA; Department of Pediatrics, University of Colorado at Denver, Aurora, CO, USA; Department of Pharmacology, University of Colorado at Denver, Aurora, CO, USA; Department of Otolaryngology, University of Colorado at Denver, Aurora, CO, USA.
Lindsay Swanson, Department of Neurology, Boston Children’s Hospital, Boston, MA, USA.
Heather Olson, Department of Neurology, Boston Children’s Hospital, Boston, MA, USA.
Scott Demarest, Department of Neurology, Children’s Hospital Colorado, Aurora, CO, USA; Department of Pediatrics, University of Colorado at Denver, Aurora, CO, USA; Department of Neurology, University of Colorado at Denver, Aurora, CO, USA.
References
- 1.Tao J, Van Esch H, Hagedorn-Greiwe M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 2004; 75: 1149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004; 75: 1079–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devinsky O, Verducci C, Thiele EA, et al. Open-label use of highly purified CBD (Epidiolex) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav 2018; 86: 131–37. [DOI] [PubMed] [Google Scholar]
- 4.Hanefeld F The clinical pattern of the Rett syndrome. Brain Dev 1985; 7: 320–25. [DOI] [PubMed] [Google Scholar]
- 5.Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68: 944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehr S, Wilson M, Downs J, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet 2013; 21: 266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arican P, Gencpinar P, Olgac Dundar N. A new cause of developmental and epileptic encephalopathy with continuous spike-and-wave during sleep: CDKL5 disorder. Neurocase 2019; 25: 59–61. [DOI] [PubMed] [Google Scholar]
- 8.Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain 2019; 142: 2303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakimiec M, Paprocka J, Śmigiel R. CDKL5 deficiency disorder—a complex epileptic encephalopathy. Brain Sci 2020; 10: E107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fehr S, Leonard H, Ho G, et al. There is variability in the attainment of developmental milestones in the CDKL5 disorder. J Neurodev Disord 2015; 7: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahi-Buisson N, Kaminska A, Boddaert N, et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 2008; 49: 1027–37. [DOI] [PubMed] [Google Scholar]
- 12.Bahi-Buisson N, Nectoux J, Rosas-Vargas H, et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008; 131: 2647–61. [DOI] [PubMed] [Google Scholar]
- 13.Bernardo P, Ferretti A, Terrone G, et al. Clinical evolution and epilepsy outcome in three patients with CDKL5-related developmental encephalopathy. Epileptic Disord 2019; 21: 271–77. [DOI] [PubMed] [Google Scholar]
- 14.MacKay CI, Wong K, Demarest ST, Benke TA, Downs J, Leonard H. Exploring genotype-phenotype relationships in the CDKL5 deficiency disorder using an international dataset. Clin Genet 2021; 99: 157–65. [DOI] [PubMed] [Google Scholar]
- 15.Demarest ST, Olson HE, Moss A, et al. CDKL5 deficiency disorder: relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019; 60: 1733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mefford HC. Phenotype to genotype and back again. Epilepsy Curr 2020; 20: 88–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fehr S, Wong K, Chin R, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology 2016; 87: 2206–13. [DOI] [PubMed] [Google Scholar]
- 18.Mangatt M, Wong K, Anderson B, et al. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J Rare Dis 2016; 11: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi Y, Tohyama J, Takahashi Y, et al. Clinical manifestations and epilepsy treatment in Japanese patients with pathogenic CDKL5 variants. Brain Dev 2021; 43: 505–14. [DOI] [PubMed] [Google Scholar]
- 20.Fehr S, Downs J, Ho G, et al. Functional abilities in children and adults with the CDKL5 disorder. Am J Med Genet A 2016; 170: 2860–69. [DOI] [PubMed] [Google Scholar]
- 21.Amin S, Majumdar A, Mallick AA, et al. Caregiver’s perception of epilepsy treatment, quality of life and comorbidities in an international cohort of CDKL5 patients. Hippokratia 2017; 21: 130–35. [PMC free article] [PubMed] [Google Scholar]
- 22.Olson HE, Costantini JG, Swanson LC, et al. Cerebral visual impairment in CDKL5 deficiency disorder: vision as an outcome measure. Dev Med Child Neurol 2021; 63: 1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brock D, Fidell A, Thomas J, Juarez-Colunga E, Benke TA, Demarest S. Cerebral visual impairment in CDKL5 deficiency disorder correlates with developmental achievement. J Child Neurol 2021; 36: 974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siri B, Varesio C, Freri E, et al. CDKL5 deficiency disorder in males: five new variants and review of the literature. Eur J Paediatr Neurol 2021; 33: 9–20. [DOI] [PubMed] [Google Scholar]
- 25.Klein KM, Yendle SC, Harvey AS, et al. A distinctive seizure type in patients with CDKL5 mutations: hypermotor-tonic-spasms sequence. Neurology 2011; 76: 1436–38. [DOI] [PubMed] [Google Scholar]
- 26.Melani F, Mei D, Pisano T, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol 2011; 53: 354–60. [DOI] [PubMed] [Google Scholar]
- 27.Melikishvili G, Epitashvili N, Tabatadze N, et al. New insights in phenomenology and treatment of epilepsy in CDKL5 encephalopathy. Epilepsy Behav 2019; 94: 308–11. [DOI] [PubMed] [Google Scholar]
- 28.Olson HE, Daniels CI, Haviland I, et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J Neurodev Disord 2021; 13: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson HE, Demarest ST, Pestana-Knight EM, et al. Cyclin-dependent kinase-like 5 deficiency disorder: clinical review. Pediatr Neurol 2019; 97: 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulcahey PJ, Tang S, Takano H, et al. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp Neurol 2020; 332: 113388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacKay CI, Bick D, Prokop JW, et al. Expanding the phenotype of the CDKL5 deficiency disorder: are seizures mandatory? Am J Med Genet A 2020; 182: 1217–22. [DOI] [PubMed] [Google Scholar]
- 32.Muñoz IM, Morgan ME, Peltier J, et al. Phosphoproteomic screening identifies physiological substrates of the CDKL5 kinase. EMBO J 2018; 37: e99559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard H, Junaid M, Wong K, Aimetti AA, Pestana Knight E, Downs J. Influences on the trajectory and subsequent outcomes in CDKL5 deficiency disorder. Epilepsia 2022; 63: 352–63. [DOI] [PubMed] [Google Scholar]
- 34.Sakki HEA, Dale NJ, Sargent J, Perez-Roche T, Bowman R. Is there consensus in defining childhood cerebral visual impairment? A systematic review of terminology and definitions. Br J Ophthalmol 2018; 102: 424–32. [DOI] [PubMed] [Google Scholar]
- 35.Heussler HS, Hiscock H. Sleep in children with neurodevelopmental difficulties. J Paediatr Child Health 2018; 54: 1142–47. [DOI] [PubMed] [Google Scholar]
- 36.Lo Martire V, Alvente S, Bastianini S, et al. CDKL5 deficiency entails sleep apneas in mice. J Sleep Res 2017; 26: 495–97. [DOI] [PubMed] [Google Scholar]
- 37.Mazurek MO, Sohl K. Sleep and behavioral problems in children with autism spectrum disorder. J Autism Dev Disord 2016; 46: 1906–15. [DOI] [PubMed] [Google Scholar]
- 38.Robertson J, Baines S, Emerson E, Hatton C. Prevalence of constipation in people with intellectual disability: a systematic review. J Intellect Dev Disabil 2018; 43: 392–406. [DOI] [PubMed] [Google Scholar]
- 39.Liang JS, Huang H, Wang JS, Lu JF. Phenotypic manifestations between male and female children with CDKL5 mutations. Brain Dev 2019; 41: 783–89. [DOI] [PubMed] [Google Scholar]
- 40.Mingorance A, Jaksha A, Smart T, Sherriff L, Valentine J. The voice of the patient report: CDKL5 deficiency disorder (CDD). June 17, 2020. https://www.cdkl5.com/wp-content/uploads/2020/06/CDD-VoP-REPORT.pdf (accessed Dec 10, 2020).
- 41.Olson HE, Kelly M, LaCoursiere CM, et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol 2017; 81: 419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Y, Wang ZI, Sarwar S, et al. Brain morphological abnormalities in children with cyclin-dependent kinase-like 5 deficiency disorder. Eur J Paediatr Neurol 2021; 31: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gan L, Cookson MR, Petrucelli L, La Spada AR. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci 2018; 21: 1300–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu YC, Xiong ZQ. Molecular and synaptic bases of CDKL5 disorder. Dev Neurobiol 2019; 79: 8–19. [DOI] [PubMed] [Google Scholar]
- 45.Hector RD, Kalscheuer VM, Hennig F, et al. CDKL5 variants: improving our understanding of a rare neurologic disorder. Neurol Genet 2017; 3: e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertani I, Rusconi L, Bolognese F, et al. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J Biol Chem 2006; 281: 32048–56. [DOI] [PubMed] [Google Scholar]
- 47.Demarest S, Pestana-Knight EM, Olson HE, et al. Severity assessment in CDKL5 deficiency disorder. Pediatr Neurol 2019; 97: 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baltussen LL, Negraes PD, Silvestre M, et al. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J 2018; 37: e99763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eyers PA. A new consensus for evaluating CDKL5/STK9-dependent signalling mechanisms. EMBO J 2018; 37: e100848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonard H, Junaid M, Wong K, Demarest S, Downs J. Exploring quality of life in individuals with a severe developmental and epileptic encephalopathy, CDKL5 deficiency disorder. Epilepsy Res 2021; 169: 106521. [DOI] [PubMed] [Google Scholar]
- 51.Stosser MB, Lindy AS, Butler E, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med 2018; 20: 403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mori Y, Downs J, Wong K, Anderson B, Epstein A, Leonard H. Impacts of caring for a child with the CDKL5 disorder on parental wellbeing and family quality of life. Orphanet J Rare Dis 2017; 12: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mori Y, Downs J, Wong K, Heyworth J, Leonard H. Comparing parental well-being and its determinants across three different genetic disorders causing intellectual disability. J Autism Dev Disord 2018; 48: 1651–65. [DOI] [PubMed] [Google Scholar]
- 54.Tangarorang J, Leonard H, Epstein A, Downs J. A framework for understanding quality of life domains in individuals with the CDKL5 deficiency disorder. Am J Med Genet A 2019; 179: 249–56. [DOI] [PubMed] [Google Scholar]
- 55.Downs J, Jacoby P, Leonard H, et al. Psychometric properties of the Quality of Life Inventory-Disability (QI-Disability) measure. Qual Life Res 2019; 28: 783–94. [DOI] [PubMed] [Google Scholar]
- 56.Alexander HB, Broshek DK, Quigg M. Quality of life in adults with epilepsy is associated with anticonvulsant polypharmacy independent of seizure status. Epilepsy Behav 2018; 78: 96–99. [DOI] [PubMed] [Google Scholar]
- 57.Williams K, Jacoby P, Whitehouse A, et al. Functioning, participation, and quality of life in children with intellectual disability: an observational study. Dev Med Child Neurol 2021; 63: 89–96. [DOI] [PubMed] [Google Scholar]
- 58.Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol 2020; 24: 15–23. [DOI] [PubMed] [Google Scholar]
- 59.Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018; 59: 1062–71. [DOI] [PubMed] [Google Scholar]
- 60.Kothur K, Holman K, Farnsworth E, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 2018; 59: 132–40. [DOI] [PubMed] [Google Scholar]
- 61.Helbig I, Riggs ER, Barry CA, et al. The ClinGen Epilepsy Gene Curation Expert Panel—bridging the divide between clinical domain knowledge and formal gene curation criteria. Hum Mutat 2018; 39: 1476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Axeen EJT, Olson HE. Neonatal epilepsy genetics. Semin Fetal Neonatal Med 2018; 23: 197–203. [DOI] [PubMed] [Google Scholar]
- 63.Muir AM, Myers CT, Nguyen NT, et al. Genetic heterogeneity in infantile spasms. Epilepsy Res 2019; 156: 106181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gardella E, Marini C, Trivisano M, et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018; 91: e1112–24. [DOI] [PubMed] [Google Scholar]
- 65.Greene C, Daniels C, Love-Nichols J, et al. Clinical features of CDKL5 deficiency disorder compared to other infantile onset genetic epilepsies. American Epilepsy Society 74th Annual Meeting; Dec 4–8, 2020 (abstr 1030). [Google Scholar]
- 66.Jian L, Nagarajan L, de Klerk N, et al. Predictors of seizure onset in Rett syndrome. J Pediatr 2006; 149: 542–47. [DOI] [PubMed] [Google Scholar]
- 67.Cutri-French C, Armstrong D, Saby J, et al. Comparison of core features in four developmental encephalopathies in the Rett natural history study. Ann Neurol 2020; 88: 396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Müller A, Helbig I, Jansen C, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol 2016; 20: 147–51. [DOI] [PubMed] [Google Scholar]
- 69.Aledo-Serrano Á, Gómez-Iglesias P, Toledano R, et al. Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: findings from a multicenter cohort. Epilepsy Behav 2021; 118: 107946. [DOI] [PubMed] [Google Scholar]
- 70.Dale T, Downs J, Wong K, Leonard H. The perceived effects of cannabis products in the management of seizures in CDKL5 deficiency disorder. Epilepsy Behav 2021; 122: 108152. [DOI] [PubMed] [Google Scholar]
- 71.Olson H, Demarest S, Pestana-Knight E, et al. Infantile spasms in CDKL5 deficiency disorder respond poorly to first line treatments. The 145th Annual Meeting of the American Neurological Association; Oct 3, 2020 (abstr K-593). [Google Scholar]
- 72.Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: experience of >100 patients. Epilepsia 2017; 58: 1415–22. [DOI] [PubMed] [Google Scholar]
- 73.Lim Z, Wong K, Downs J, Bebbington K, Demarest S, Leonard H. Vagus nerve stimulation for the treatment of refractory epilepsy in the CDKL5 deficiency disorder. Epilepsy Res 2018; 146: 36–40. [DOI] [PubMed] [Google Scholar]
- 74.Specchio N, Chez M, Tarquinio D, et al. Ganaxolone in children with CDKL5 gene-related epileptic encephalopathy: preliminary analysis from an open-label trial. Epilepsia 2021; 58: s163–64. [Google Scholar]
- 75.Pestana Knight EM, Amin S, Bahi-Buisson, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2022; 21: 417–27. [DOI] [PubMed] [Google Scholar]
- 76.Devinsky O, King L, Bluvstein J, Friedman D. Ataluren for drug-resistant epilepsy in nonsense variant-mediated Dravet syndrome and CDKL5 deficiency disorder. Ann Clin Transl Neurol 2021; 8: 639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Demarest S, Jeste S, Agarwal N, et al. Efficacy, safety and tolerability of soticlestat (TAK-935/OV935) as an adjunctive therapy in patients with 15q duplication syndrome (Dup15q) or cyclin-dependent kinase-like 5 deficiency disorder (CDD) in a signal-finding phase 2 study (ARCADE) (4096). Neurology 2021; 96 (suppl 15): 4096. [Google Scholar]
- 78.Devinsky O, King L, Schwartz D, Conway E, Price D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia 2021; 62: e98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yawno T, Miller SL, Bennet L, et al. Ganaxolone: a new treatment for neonatal seizures. Front Cell Neurosci 2017; 11: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fazzari M, Frasca A, Bifari F, Landsberger N. Aminoglycoside drugs induce efficient read-through of CDKL5 nonsense mutations, slightly restoring its kinase activity. RNA Biol 2019; 16: 1414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sullivan J, Scheffer IE, Lagae L, et al. Fenfluramine HCl (Fintepla) provides long-term clinically meaningful reduction in seizure frequency: analysis of an ongoing open-label extension study. Epilepsia 2020; 61: 2396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Terzic B, Cui Y, Edmondson AC, et al. X-linked cellular mosaicism underlies age-dependent occurrence of seizure-like events in mouse models of CDKL5 deficiency disorder. Neurobiol Dis 2021; 148: 105176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang HT, Zhu ZA, Li YY, et al. CDKL5 deficiency in forebrain glutamatergic neurons results in recurrent spontaneous seizures. Epilepsia 2021; 62: 517–28. [DOI] [PubMed] [Google Scholar]
- 84.Fuchs C, Fustini N, Trazzi S, Gennaccaro L, Rimondini R, Ciani E. Treatment with the GSK3-beta inhibitor tideglusib improves hippocampal development and memory performance in juvenile, but not adult, Cdkl5 knockout mice. Eur J Neurosci 2018; 47: 1054–66. [DOI] [PubMed] [Google Scholar]
- 85.Trazzi S, De Franceschi M, Fuchs C, et al. CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum Mol Genet 2018; 27: 1572–92. [DOI] [PubMed] [Google Scholar]
- 86.Tang S, Terzic B, Wang IJ, et al. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat Commun 2019; 10: 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yennawar M, White RS, Jensen FE. AMPA receptor dysregulation and therapeutic interventions in a mouse model of CDKL5 deficiency disorder. J Neurosci 2019; 39: 4814–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fuchs C, Gennaccaro L, Ren E, et al. Pharmacotherapy with sertraline rescues brain development and behavior in a mouse model of CDKL5 deficiency disorder. Neuropharmacology 2020; 167: 107746. [DOI] [PubMed] [Google Scholar]
- 89.Gao Y, Irvine EE, Eleftheriadou I, et al. Gene replacement ameliorates deficits in mouse and human models of cyclin-dependent kinase-like 5 disorder. Brain 2020; 143: 811–32. [DOI] [PubMed] [Google Scholar]
- 90.Trovò L, Fuchs C, De Rosa R, et al. The green tea polyphenol epigallocatechin-3-gallate (EGCG) restores CDKL5-dependent synaptic defects in vitro and in vivo. Neurobiol Dis 2020; 138: 104791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Patnaik A, Spiombi E, Frasca A, Landsberger N, Zagrebelsky M, Korte M. Fingolimod modulates dendritic architecture in a BDNF-dependent manner. Int J Mol Sci 2020; 21: E3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barbiero I, Peroni D, Siniscalchi P, et al. Pregnenolone and pregnenolone-methyl-ether rescue neuronal defects caused by dysfunctional CLIP170 in a neuronal model of CDKL5 deficiency disorder. Neuropharmacology 2020; 164: 107897. [DOI] [PubMed] [Google Scholar]
- 93.Balestra D, Giorgio D, Bizzotto M, et al. Splicing mutations impairing CDKL5 expression and activity can be efficiently rescued by U1snRNA-based therapy. Int J Mol Sci 2019; 20: E4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Halmai JANM, Deng P, Gonzalez CE, et al. Artificial escape from XCI by DNA methylation editing of the CDKL5 gene. Nucleic Acids Res 2020; 48: 2372–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fallah MS, Eubanks JH. Seizures in mouse models of rare neurodevelopmental disorders. Neuroscience 2020; 445: 50–68. [DOI] [PubMed] [Google Scholar]
- 96.Berg AT, Palac H, Wilkening G, Zelko F, Schust Meyer L. SCN2A-developmental and epileptic encephalopathies: challenges to trial-readiness for non-seizure outcomes. Epilepsia 2021; 62: 258–68. [DOI] [PubMed] [Google Scholar]
- 97.Saldaris J, Weisenberg J, Pestana-Knight E, et al. Content validation of clinician-reported items for a severity measure for CDKL5 deficiency disorder. J Child Neurol 2021; 36: 998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Benke TA, Kind PC. Proof-of-concept for a gene replacement approach to CDKL5 deficiency disorder. Brain 2020; 143: 716–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barbiero I, De Rosa R, Kilstrup-Nielsen C. Microtubules: a key to understand and correct neuronal defects in CDKL5 deficiency disorder? Int J Mol Sci 2019; 20: E4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.