

Abstract
Over the past 18 months, the need to perform atomic detail molecular dynamics simulations of the SARS-CoV-2 virion, its spike protein, and other structures related to the viral infection cycle has led biomedical researchers worldwide to urgently seek out all available biomolecular structure information, appropriate molecular modeling and simulation software, and the necessary computing resources to conduct their work. We describe our experiences from several COVID-19 research collaborations and the challenges they presented in terms of our molecular modeling software development and support efforts, our laboratory’s local computing environment, and our scientists’ use of non-traditional HPC hardware platforms such as public clouds for large scale parallel molecular dynamics simulations.
Keywords: Molecular Dynamics Simulation, Remote Visualization, Interactive Computing, COVID-19, SARS-CoV-2
I. Introduction
The COVID-19 pandemic has touched the lives of everyone and demanded a worldwide response. Basic research into the molecular structures and dynamics involved in the SARS-CoV-2 virus that causes COVID-19 aims to elucidate key details of its viral infection cycle, and its unique structural characteristics, to enable and inform more directly clinically oriented research. Our research laboratory at the University of Illinois at Urbana-Champaign and the NIH Center through which we develop and distribute the NAMD [1], [2] and VMD [3] software tools to the biomedical research community, has been in a unique position to contribute directly to the scientific understanding of the SARS-CoV-2 virus, both through its own biomolecular research and through its software development efforts. The COVID-19 pandemic posed many new challenges to conventional molecular simulation practices, due to the need for rapid turnaround, biomedical researchers working remotely during lockdowns, and the need to rapidly adapt existing practices to use new computing platforms and resources. The specific attributes of molecular dynamics studies of SARS-CoV-2 place them at the large end of the spectrum of typical biomolecular simulations. SARS-Cov-2 studies require simulations on the order of one million atoms, e.g., for studies of the spike protein, and up to hundreds of millions of atoms for the full virion and solvent environment [4]. The large simulation sizes pose not only significant computational challenges, but also challenges with respect to the transfer of large all-atom structures and simulation trajectories, and to perform routine visualization and analysis tasks under remote work conditions.
Below, we outline some of the key missions of our laboratory and NIH Center, and we outline some of the key challenges presented by the COVID-19 pandemic and responses developed and undertaken to address them. We describe our approach to providing rapid and responsive software development to serve the needs of the biomolecular research community, to accelerate development and dissemination of our NAMD molecular dynamics simulation software, and enable users of our VMD molecular visualization and analysis software to interact remotely with large data sets, all while working remotely during lockdowns. To help face computing resource challenges, scientific efforts like ours have been supported through generous computer time allocations for COVID-19 related research, most notably through the COVID-19 HPC Consortium and in some cases directly through the NSF supercomputing centers. The availability of diverse computing resources at multiple sites presents its own challenges, due to the need to manage and transfer key data, and due to the desire to achieve optimal performance on many different hardware platforms. We discuss the role of close collaboration with hardware companies to provide higher performance simulation capability on new and emerging hardware platforms, and our approach to making these advances broadly available to the biomedical research community as quickly and responsively as possible. Among the most interesting experiences we describe is the use of public clouds for large scale parallel molecular dynamics simulations with Microsoft Azure [5]. Finally, we conclude with a summary of key lessons learned and opportunities for future research and development.
A. Overview of the NIH Center
The NIH P41 Center for Macromolecular Modeling and Bioinformatics at Illinois has been engaged in developing advanced computational technologies and state-of-the-art software tools [1]–[3], [6], [7] for biomolecular and cell modeling, simulation, and visualization, for more than three decades. Molecular modeling and simulation nowadays constitute an indispensable research component in nearly all areas of molecular sciences. The importance of computational technologies in biological and biomedical research can be best highlighted by the continuing growth in the number of publications in this domain.
Carefully designed and executed biomolecular modeling and simulation of ever larger cellular complexes and membranes yield highly complementary results to experimental structural biology [8] and biophysical techniques [9], [10]. Advanced structural methodologies such as X-ray crystallography and cryo-EM depend largely on computational tools [11], [12], e.g., for data processing and model construction, especially for large supramolecular and cell-scale assemblies [13]. To make the computational technologies easy to use and available to its broad community of biomedical users, the Center has invested substantial time and effort in developing the NAMD and VMD software into highly accessible applications that are currently in use by a very large user base conducting high-impact research. Collectively, these programs currently serve, by very conservative metrics, over 125,000 users. The software programs developed by the Center constitute its main research “instruments,” and their utility by the users defines the principal component of its service to the biomedical community. These instruments enable the researchers, at no cost, to meet their needs for advanced modeling, simulation, and visualization capabilities.
The Center’s software technologies are used extensively to investigate biomolecular and cellular systems and complexes, providing researchers with graphical views and quantitative information at a level of fidelity and accuracy limited only by the extent and quality of available structural information. MD simulations provide valuable atomic-level details about the dynamics of biomolecules when combined with powerful molecular visualization and analysis tools. With their highly complementary capabilities, NAMD and VMD form a unique “Computational Microscope” [14] that enables users to obtain the most detailed, and dynamic, view into molecular events and processes underlying the biochemistry of human health and disease. The comprehensive modeling environment formed by these programs provides biomedical researchers and other molecular scientists with an efficient framework to conduct molecular investigations of their systems and processes of interest, and literally “see” their results.
B. COVID-19 Research Supported by Molecular Dynamics Simulation
Molecular dynamics simulations provide atomic detail views of the structure and dynamics of biomolecular complexes, providing researchers access to temporal and spatial scales inaccessible to experimental imaging methods alone. Preparation of state-of-the-art simulations of biomolecular structures, and particularly new or recently discovered structures, such as those of SARS-CoV-2, relies heavily on modeling approaches that combine structure information from multiple modalities of experimental imaging techniques, typically in concert with homology modeling and other computational structure determination approaches. Molecular dynamics simulation requires the computation and integration of interatomic forces and atom positions over time, for millions of time steps, to achieve biologically relevant simulation time scales. Researchers require access to diverse atomic structure information, powerful visualization and analysis capabilities, tremendous computing resources, and capacious storage infrastructure to study large models, such as the SARS-CoV-2 virus [4], [15].
When a coronavirus such as SARS-CoV-2 infects a human cell, the first stage is viral entry: so-called spike proteins on the exterior of the virus particle bind to receptors on the host cell, and the membrane of the virus particle fuses with the membrane of the host cell. The membrane fusion allows the release of the viral genome into the host cell cytoplasm and the next stages of infection to proceed. Since the first days of the COVID-19 pandemic, researchers across the molecular modeling field have rapidly stepped forward to perform a great deal of study of the structures and dynamics of SARS-CoV-2 [16], [17], with particular focus on the spike protein and viral entry. One study ongoing in our laboratory examines a part of the SARS-CoV-2 spike protein called the fusion peptide; fusion peptides are suggested to cause membrane curvature during fusion, driving the process forward (Figure 1). Using all-atom molecular dynamics simulations, and following their own related research on characterizing the membrane-bound form of the fusion peptide [18], our laboratory researchers have worked to characterize how multiple fusion peptides form a complex, and how the resulting complex interacts with the human cell membrane. The solvated system size of roughly 600,000 atoms has so far been simulated for a total of 200 ns. Microsoft Azure V100 instances have allowed the researchers to efficiently run GPU-accelerated molecular dynamics simulations using NAMD. Another ongoing study of our laboratory examines the flexibility and dynamics of the SARS-CoV-2 spike protein. In an 11-million atom model system (Figure 2) that captures the crowded environment at the virion surface, multiple full-length spike proteins, complete with the glycan molecules that coat them, are bound to a section of the virion envelope membrane. Concerted motions of three flexible hinge regions of the spike are characterized, in context of the dynamic interactions among the glycans and the interactions between the glycans and the lipids that make up the virus membrane. The functional importance of hinge regions in enhancing viral infection can shed light on the discovery of therapeutics against the virus.
Fig. 1.

A key step in the early stages of SARS-CoV-2 infection is membrane fusion, which allows the release of the viral genome into the host cell. This simulation model, consisting of 600,000 atoms with solvent, is used to study how three fusion peptide monomers — parts of SARS-CoV-2 spike proteins which are suggested to promote fusion by causing membrane curvature — can form a complex which then interacts with the host cell membrane. (Image rendered with VMD.)
Fig. 2.
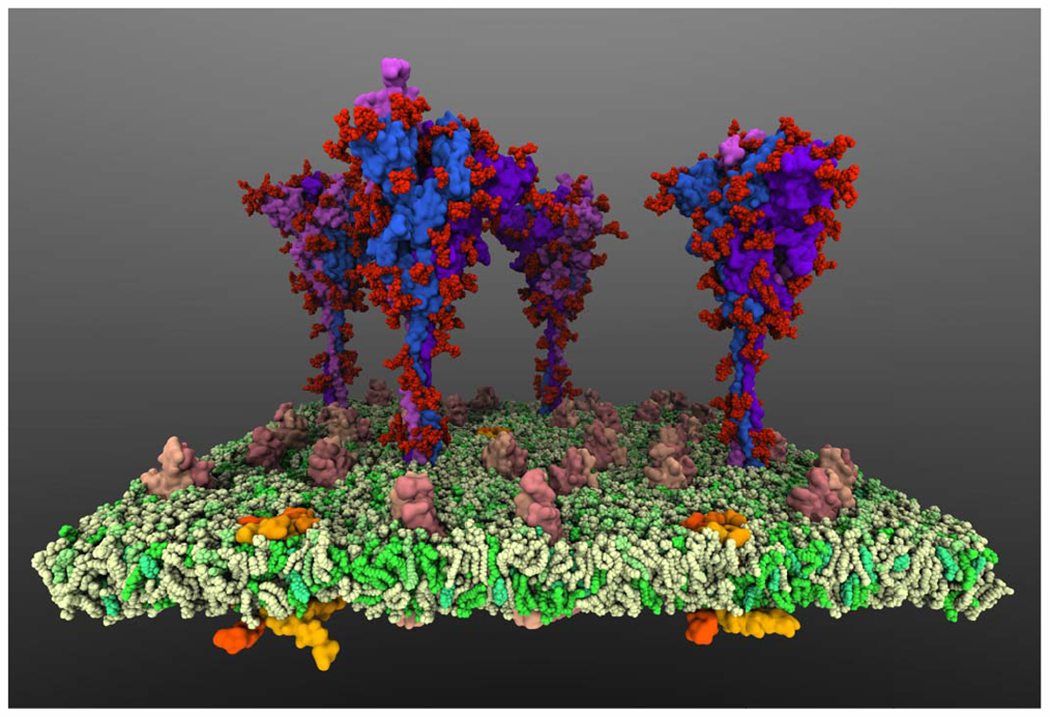
SARS-CoV-2 fuses to and enters human cells upon binding of its spike proteins to human receptors. This simulation model, consisting of 11-million atoms with solvent, captures a realistic representation of multiple, complete spike proteins and their dynamics on the surface of the coronavirus. (Image rendered with VMD.)
II. Responsive Software Development Efforts
During the COVID-19 pandemic, our software team experienced a spike in demands from its user community. As a result of researchers working remotely during so-called “lockdowns,” quarantine periods, or employer-initiated workfrom-home policies, many researchers were faced with the challenge of performing molecular modeling, simulation, and analysis tasks on modest laptop or desktop PC platforms very different from the systems they were accustomed to at their workplace office. These situations led to a wide variety of user requests for improved performance, new features, and quick resolution to software compatibility problems arising from the use of largely unmanaged laptops and PCs.
NAMD and VMD have, during their more than twentyfive year existence, established themselves as trusted, high-quality software applications, supporting all HPC-oriented commodity-based hardware platforms. Both applications are written in C++ and have been CUDA-accelerated since 2007 [19]. For its parallel scaling, NAMD uses the Charm++ parallel programming framework and runtime system, which is also developed at the University of Illinois [20], [21]. Development of these codes has benefited over the years from the consolidation of a variety of available CPU and GPU platforms, not to mention the different flavors of Unix, into predominantly x86 CPUs and NVIDIA GPUs running on Linux. However, recent years have seen the emergence of alternative compute-oriented GPUs, first from AMD and followed by Intel, as well as the need to provide explicit support for CPU vectorization. The need to support emerging platforms during a pandemic, in which the software serves as an important vehicle for biomedical investigation toward understanding and ultimately combating the disease, has sped up the development and release cycles.
A. SARS-CoV-2 Software Challenges
Given their optimized performance and unique capabilities for large biomolecular structures, VMD and NAMD played a key role in computational biophysical studies of SARS-CoV-2. Nevertheless, both programs faced a variety of challenges that required particular attention.
One of the challenges posed by SARS-CoV-2 simulations has been the need for scientists to do significant modeling work with hybrid structure information, e.g., from homology modeling (computational) and cryo-electron microscopy (experimental imaging) approaches, owing to the significantly longer time investments required for structures to become available from high resolution techniques such as X-ray crystallography. This has led researchers to employ diverse software tools that interoperate and that can exchange the required modalities of structure information, to permit all-atom SARS-CoV-2 models to be produced, despite of the limitations on the completeness and resolution of any one modality of structure information. The structure refinement and simulation preparation tasks associated with SARS-CoV-2 hybrid modeling efforts were one of the key areas where VMD saw significant demands, in particular for comparing and analyzing structural details [17], and for MDFF structure refinement of cryo-EM images [15], [22], [23]. Since VMD development efforts associated with hybrid structures and MDFF had been an ongoing area of development, making new features and hardware-optimized performance improvements available at a rapid pace was a primary goal.
The large size of SARS-CoV-2 all-atom simulations posed a computing capacity challenge for all researchers. Of particular concern to the NAMD and VMD developers were support for the most broadly used hardware platforms, such as individual GPU-accelerated compute nodes found in desktops, HPC clusters, or the public cloud. For NAMD more specifically, the large scale hardware platforms made available to the scientific community as part of the COVID-19 HPC Consortium were particularly important targets, which included several national supercomputers and public clouds. Key considerations for the COVID-19 HPC consortium platforms were focused on providing builds using optimized low-level networking layers to achieve reduced communication latencies (particularly beneficial for strong scaling of studies of the SARS-CoV-2 spike protein and other components of similar size), and enabling the best combination of hardware-optimized kernels and computing approaches to improve overall computational throughput for all ranges of SARS-CoV-2 simulation sizes, whether performing single- or multiple-replica simulations.
In just over one year, over 350 papers investigating molecular aspects of the novel-coronavirus outbreak have employed the NAMD and VMD software. Among them are scientific breakthroughs that became widely known, for instance, the work led by Prof. Rommie Amaro at UCSD, which was awarded the “Gordon Bell Special Prize for High Performance Computing-Based COVID-19 Research” [4].
B. Rapid, Responsive Software Cycle
In order to make newly developed features, performance enhancements, and hardware support available to the biomedical research community with the least amount of delay, our research laboratory took the unusual step of making multiple feature- and hardware-specific developmental branches of its NAMD software publicly available in addition to standard production releases and nightly builds of the main development branch. By making these additional developmental versions of NAMD publicly available, researchers using hardware platforms such as TACC’s Frontera and Argonne’s Theta-GPU systems were able to run molecular dynamics simulations at performance levels 2× or more that of the production NAMD release, due to integration of algorithmic and hardware-specific improvements. While these developmental branches represent still-unfinished work in terms of full support of all existing NAMD features, optimized performance for all cases, or complete documentation, researchers were, through consultation with the NAMD development team, able to safely make use of the developmental versions for their science projects, using the performance benefits and new features to maximum scientific benefit.
C. Development for Emerging GPU-Dense Platforms
Pre-pandemic, there had already been a concerted effort toward re-architecting NAMD to more fully utilize NVIDIA GPUs by creating a GPU-resident code path that maintains data on the GPU between time steps [1]. The new approach provides significant performance advantages, a 2× to 3× speedup over the earlier GPU-offload approach for computing just the forces on the GPU. The pandemic together with user demand inspired further effort to scale the GPU-resident approach across multiple GPUs on a single node, taking advantage of tightly coupled, NVLinked GPUs. Due to the difficulty scaling the GPU-offload approach on GPU-dense platforms, such as the DGX-series hardware, the GPU-resident version of NAMD can offer 8× or better speedup across an entire node.
Support for AMD and Intel GPUs had already been a priority for the NAMD development team, since the first two U.S. exascale supercomputers will be ORNL Frontier (AMD/HPE) and ANL Aurora (Intel/HPE). NAMD is among the early science applications for both computers, so benefits from the assistance of scientists from ORNL and ANL, as well as dedicated engineering support from AMD and Intel. The porting of NAMD to AMD GPUs is reasonably straightforward, enabled by the similarity of the HIP programming model to CUDA and the HIPify compiler that translates CUDA to HIP, allowing continued maintenance and development from a single CUDA codebase. The porting of NAMD to Intel GPUs is much more involved and is being developed through the Intel oneAPI Center of Excellence formed at the University of Illinois. The new Intel GPUs are best supported through their oneAPI/DPC++ language, which, in spite of being based on the C++-based SYCL programming standard, presents a considerably different programming model from C-based CUDA. Whereas there are already NAMD alpha releases providing AMD GPU support, the first release providing Intel support is not expected until the end of the year.
D. Hardware-Optimized CPU Kernels
The NAMD team’s ongoing collaboration with Intel has also produced CPU kernels optimized for AVX-512. The development is based on the CUDA tiles list kernel for calculating the short-range non-bonded forces, which accounts for the majority of the runtime and up to 90% of the FLOPs required per step. The improvements, released in an alpha version last August, provide NAMD with an almost 2× speedup over the earlier CPU version, and were important to NAMD’s Gordon Bell award-winning COVID-19 research simulations on TACC Frontera [4]. The AVX-512 optimized NAMD kernels have been made publicly available, enabling their use on all applicable computing platforms, in addition to the TACC Frontera system, which is the major NSF-supported supercomputing resource at the time of writing.
III. Interactive Analysis and Visualization
The steps involved in preparing, running, and analyzing molecular dynamics simulations vary by project and nearly always involve the development of a variety of protocols, scripts, and workflows that are somewhat unique to each science project. Although the majority of bulk computer time associated with the work is consumed by the molecular dynamics simulations themselves, much of the “wall clock” project time is associated with a litany of tasks performed by the molecular scientists to conduct and oversee their simulation campaign. In most cases, the best route to making the scientists more productive is to provide them with interactive on-demand access to the data files, short-run computing resources, and visualization and analysis capabilities needed for their project. While the COVID-19 HPC consortium provided large scale computing resources suitable for running molecular dynamics simulations, the major shortcoming associated with these resources was that they generally lacked facilities for high performance remote visualization, necessitating transfer of large data sets for many of the required steps in molecular modeling, simulation preparation, visualization, and analysis of the resulting simulation trajectories. Below we outline some of the steps that were taken to help maximize researcher productivity by maintaining interactive access to the resources normally provided in on-site work, under pandemic lockdown circumstances, with projects distributed to local, cloud, and national computing resources.
A. Remote File Transfer
One of the principal problems that arose in association with COVID-19 related research activities was the need to transfer large data sets among computing sites. The data involved in such work includes a diversity of molecular structure information from multiple modalities of imaging, modeling, and simulation, such as cryo-EM density maps, atomic structure models, and molecular dynamics trajectories (time series). Owing to network bandwidth asymmetries and non-uniform performance of network transfers between different sites, our research scientists frequently encountered file transfer performance levels that posed serious problems for accomplishing their work on-time. Traditional HPC centers often provide a wide range of file transfer options, with particular emphasis on HPC-centric tools such as Globus GridFTP. Under normal circumstances when laboratory computer systems are used to perform the majority of computing work, existing installations of the required client software, and the availability of dedicated systems to act as file transfer endpoints, make the use of these tools straightforward. During the COVID-19 pandemic, however, researchers made use of a wide range of non-traditional systems to stand-in for or otherwise augment existing HPC systems, creating many cases where it was not practical to try to use tools like GridFTP or Globus, and as a result, users were left to use rsync, bbcp, and other lightweight file transfer utilities that could be installed on-the-fly by end users.
B. Remote Modeling, Analysis, and Visualization.
The drive to rapidly produce findings in SARS-CoV-2 studies means that researchers performing all-atom molecular modeling can’t afford to wait for structures from highly-curated, high-resolution X-ray crystallography techniques, and they must instead rely on structure information obtainable with a short turnaround time. Structure determination methods such as homology modeling and cryo-electron microscopy have a much shorter development timeline, but must be used in combination with existing X-ray structures and hybrid structure refinement approaches to address their individual limitations in terms of completeness or structure resolution. Researchers studying SARS-CoV-2 therefore typically invest more time into structure and simulation preparation than would otherwise be typical, leading to a much greater reliance on interactive modeling and visualization, to facilitate inspection of complete all-atoms models built from multiple experimental imaging modalities and sources of structure information.
Due to the size of the all-atom structures being modeled, interactive visualization and modeling operations can be challenging. High resolution interactive visualization makes it easier to analyze molecular systems in the million-atom size range. High fidelity interactive ray traced visualizations require molecular scene data to remain entirely memory-resident during rendering, often requiring large memory capacity GPUs [24]–[27]. Traditionally, researchers in our laboratory and others like it work from an on-premises office environment featuring high performance graphical workstations with large-core-count CPUs, hundreds of gigabytes of physical memory, local SSD storage, and 10 GBit networking to petabytes of centralized network file storage.
One of the big challenges facing researchers performing molecular dynamics studies of the SARS-CoV-2 during the COVID-19 pandemic has been the need for remote working arrangements. In 2020 for some SARS-CoV-2 modeling work an increased need for visualization arose just at the time that pandemic conditions limited researchers to working remotely, away from the graphical workstations normally used on-premises. When working remotely, researchers are typically using laptops or PCs with modest performance, memory, and storage capacity. If researchers were limited to using their locally available computing resources, it would be impractical for them to perform the tasks required for simulating COVID-19 due to the size of both the atomic models and resulting simulation trajectories to be analyzed.
To facilitate researchers’ molecular modeling, visualization, and analysis activities working remotely, we deployed 20 remote visualization workstations, configured with large core-count CPUs, large physical memories (128 GB, 512 GB, and two with over 1 TB), and GPU accelerators with large onboard memory (24GB). Although many remote desktop solutions exist, very few permit high resolutions (1920×1080 and higher) at frame rates of 15 frames per second or greater, on low-bandwidth and long-haul network links. Popular remote windowing tools such as VNC use network protocols that perform admirably for conventional graphical interfaces, but harbor severe performance limitations for high resolution 3-D visualization usage. For the purposes of 3-D visualization, hardware-accelerated H.264 and H.265 video streaming provides far better interactivity and acceptable image quality, even at moderate network bit rates. Tan et al. report that HEVC/H.265 video encoding can offer up to a 59% reduction in bit rate as compared with MPEG-4 AVC/H.264 for perceptually comparable video quality, potentially a significant opportunity for increased performance at 4K UHD (3840×2160) and higher resolutions [28]. Developmental versions of VMD have been extended with built-in H.264 and H.265 video streaming using multiple approaches [26], enabling the application to act as its own client/server system, focused on visualization-specific usage. While this mechanism provides a built-in remote visualization solution for VMD itself, it is insufficient for complex molecular modeling workflows involving diverse interactive modeling tools that have been a common element of recent SARS-CoV-2 projects. To facilitate complex interactive modeling workflows, each of the remote visualization servers can also run NICE DCV, a general purpose high performance remote visualization solution that uses a shared library interposing technique to capture and stream windows containing OpenGL-accelerated graphics as compressed H.264 video, for transport over low-bandwidth networks, and decoding at the remote (client) computer, delivering the high-quality rendering at interactive frame rates needed in the daily work of molecular modeling researchers. The key advantage provided by the DCV software is its generality, since it can support all of the commonly used molecular modeling tools, and it provides streaming of not only graphics content via H.264, but provides conventional remote desktop functionality for non-OpenGL application windows.
Several remote work methods that, before the pandemic, were only used by a small number of technology-focused modeling researchers were scaled up to work for many simultaneous users. Expanded services included persistent SSH tunnels used for a range of everyday tasks, remote access to Python Jupyter notebook kernels running on office workstations, and remote file system mounting with macFUSE/sshfs. The associated training and internal documentation were augmented to accommodate the wide range of technical experience of the new users.
C. Re-tasking of Desktop Workstations
During normal operations, our laboratory provides on-site researchers with shared access to several interactive workstations designed to provide high performance for large molecular modeling projects, such as viruses. The shared workstations contain large core-count CPUs, 512GB RAM, and multiple NVIDIA Quadro workstation GPUs, and are well-suited to the entire gamut of molecular modeling tasks, including structure preparation, visualization, simulation, and analysis. During the pandemic, long periods of full-time remote work created a situation in which these powerful workstations, normally made available in a shared laboratory workspace, became idle. In order to better utilize these systems for the duration of fulltime remote work, they were incorporated into the laboratory-wide queuing system, and made available for running both long molecular dynamics simulations and on-demand jobs as described below. In order to facilitate some researchers performing challenging tasks on-site, the workstation windowing system keyboard–mouse activity was used to modulate the queuing system status of the re-tasked workstations to ensure that an on-site user would be prioritized over batch jobs.
D. On-Demand Interactive Job Scheduling
The ability to immediately schedule interactive or high-priority rapid turnaround jobs is of critical importance to many molecular modeling and simulation preparation/debugging activities. The HPC clusters operated by our laboratory are configured such that each resource (compute node) has two queues associated with it: a standard (long running job) queue, and a short (interactive, debugging, or other rapid-turnaround job) queue. The queuing system is configured such that the given resource’s short queue is prioritized and will immediately preempt any existing job running in the standard (long running) queue, putting the existing job to sleep and waking upon completion of the preempting job, via Unix job control signals (SIGSTOP and SIGCONT). The job preemption feature of the queuing system permits interactive and high-priority jobs to be run on-demand, normally within 5 seconds. An end user can either submit a batch job to the special queue, or the qrsh utility can be used to open an interactive shell on the computing system, for up to the wall clock time limit set in the queuing system configuration.
E. Hardware Maintenance Challenges During Lockdowns
The running of hardware diagnostics was also impacted by pandemic conditions; for example, our NVIDIA DGX-2 required expertise beyond our vaccinated system administration team members, necessitating remote audio and video communication, which is difficult to accomplish in a loud machine room when wearing masks. Individual headsets used with cellphone audio/video conference software helped communication with remote technicians significantly, even more so when multiple workers on site in a loud machine room needed to both work with a remote technician and communicate in-room with each other while following social distancing guidelines. Remote collaboration was also needed for hardware maintenance of various laboratory workstations and was challenging in situations such as new machine installation of “bleeding-edge” OSes and drivers, in which screen sharing is impossible.
IV. Using Diverse HPC Resources
Traditional HPC resources, including the NSF resources at supercomputing centers such as TACC, PSC, and NCSA, and the DOE resources at national laboratories such as ORNL, ANL, and LLNL, are available to just a limited number of researchers, primarily through the NSF award programs XSEDE and LRAC, and the DOE award programs such as INCITE. Many more researchers are supported mainly through university or departmental computing resources or, if fortunate enough, through self-maintained resources. The growing demand for HPC resources by researchers has led to the rise of HPC-as-a-Service (HPCaaS), made available through companies such as Microsoft, Amazon, Google, and Oracle. For example, much of the computing needs at the NIH are met by Microsoft Azure cloud computing services. These services can be advantageous to researchers when the time required to compete for an XSEDE award is too costly or the cost of supporting internal resources is too prohibitive, with the added attraction of gaining access to some of the latest computing technologies while paying for only the computing time used.
In response to the pandemic, key players from government, industry, and academia joined together to form the COVID-19 HPC Consortium, making their collective computing resources available through a special-purpose rapid-turnaround proposal mechanism. Our laboratory was granted computing resources for COVID-19 research on both traditional (ORNL Summit, LLNL Lassen, TACC Frontera and Stampede2) and non-traditional public cloud (Microsoft Azure) resources. Ongoing research projects include virus spike simulations, systematic construction of the entire viral envelope, and dynamics of the Delta variant spike, so far with one publication [18] and others to follow.
A. Exploiting Traditional HPC Resources
To facilitate COVID-19 research teams using traditional supercomputing resources as assigned by the COVID-19 HPC Consortium, Center staff worked with researchers to ensure they had access to site-optimized builds of existing and newly-developed versions of NAMD. Although pre-built NAMD binaries are made available for download, parallel builds of NAMD for HPC resources seldom work out-of-the-box. Separate builds of NAMD need to be maintained on site for the various platforms. The NAMD development team actively maintains builds of NAMD on several commonly used resources, including ORNL Summit (IBM POWER9, NVIDIA V100), TACC Frontera (Intel Xeon), and TACC Stampede2 (Intel KNL). When a previously unsupported computing resource is provided, such as LLNL Lassen (IBM POWER9, NVIDIA V100), NAMD must be built on site. Even though Lassen has a similar base architecture as Summit, the 4-GPU per node Lassen configuration differs enough from the 6-GPU per node configuration to require revised launch scripts in order to correctly exploit the POWER9 CPU cores together with the GPUs on each machine.
B. Exploiting HPC-as-a-Service on Public Clouds
Microsoft Azure has been especially generous, providing ongoing resources to our laboratory researchers, including both NVIDIA GPU-accelerated compute nodes and also AMD Rome Infiniband clusters, with computing time approaching a retail cost of $5.5 million USD. Part of this effort has also included collaboration with the Microsoft Azure team to test the scalability limits of NAMD across their HBv2 (AMD Rome) and HBv3 (AMD Milan) clusters and test NAMD’s recent multi-GPU single-node scalability on NDv2 (NVIDIA V100) and NDv4 (NVIDIA A100) nodes.
Much of our laboratory’s Azure computing has utilized their NVIDIA V100 resources. For ease of use, the Azure staff was kind enough to ease the use of their GPU-based resources by partitioning their 8-way GPU nodes to be exposed as individual single-GPU nodes through a Slurm queuing system, noting that this was initially done in the early summer of 2020 before the fast multi-GPU single-node support was yet available in NAMD. We were able to build NAMD in-house, periodically updating the installation when new features were added and bugs were fixed. Efficient performance, comparable to what has already been reported [1], was seen for molecular dynamics simulation protocols, including simple equilibration, applied electric fields, loosely coupled replica exchange, and ligand flooding, with system sizes from 100,000 to 600,000 atoms, and with simulated timescales of up to 15μs.
The use of the Azure AMD Rome cluster has posed greater difficulties. We acknowledge that the quality of resources is remarkable, in which an unprecedented result was shown last year in the scaling of NAMD to 86,400 cores on a public cloud [5]. Our best benchmarking results show that the Azure HBv2 cluster can achieve supercomputer-like performance, as shown in Figure 3, with nodes consisting of dual AMD EPYC 7002-series CPUs (128 cores total) linked using 200 Gb/sec HDR Infiniband; only 120 cores are exposed per node, with the remaining 8 running the cluster hypervisor software. By contrast, the TACC Frontera nodes consist of dual Intel Xeon Platinum 8280 CPUs (56 cores total) with HDR-100 Inifiniband linking the nodes. The plot shows that the Azure performance almost matches Frontera, except for difficulties scaling the smaller system beyond 128 nodes. The synthetic benchmark systems are constructed from the STMV (satellite tobacco mosaic virus) system, a cube of about 1.068M atoms, which is replicated in an array; for example, the 210 STMV benchmark is formed by an array of 7 × 6 × 5 STMV. Although the best performance from Azure HBv2 is almost as good as Frontera, the average performance of Azure is somewhat less, with a higher variability between runs as the number of atoms per core drops below 1000, as shown in Figure 4, which is indicative of latency issues, possibly due to the hypervisor. In practice, we have had difficulties in using the Azure AMD Rome resources for our science research, including for our 11-million atom SARS-CoV-2 spike simulations, plagued by lower-than-expected performance due to bad nodes and possible networking latency and bandwidth issues. We have gone so far as to writing our own ping-pong tests to locate and avoid scheduling jobs to bad or slow nodes in the cluster. Although there is a great deal of freedom in having a dedicated (virtual) cluster, it requires greater oversight of the resources than would be required for running at a traditional HPC center.
Fig. 3.
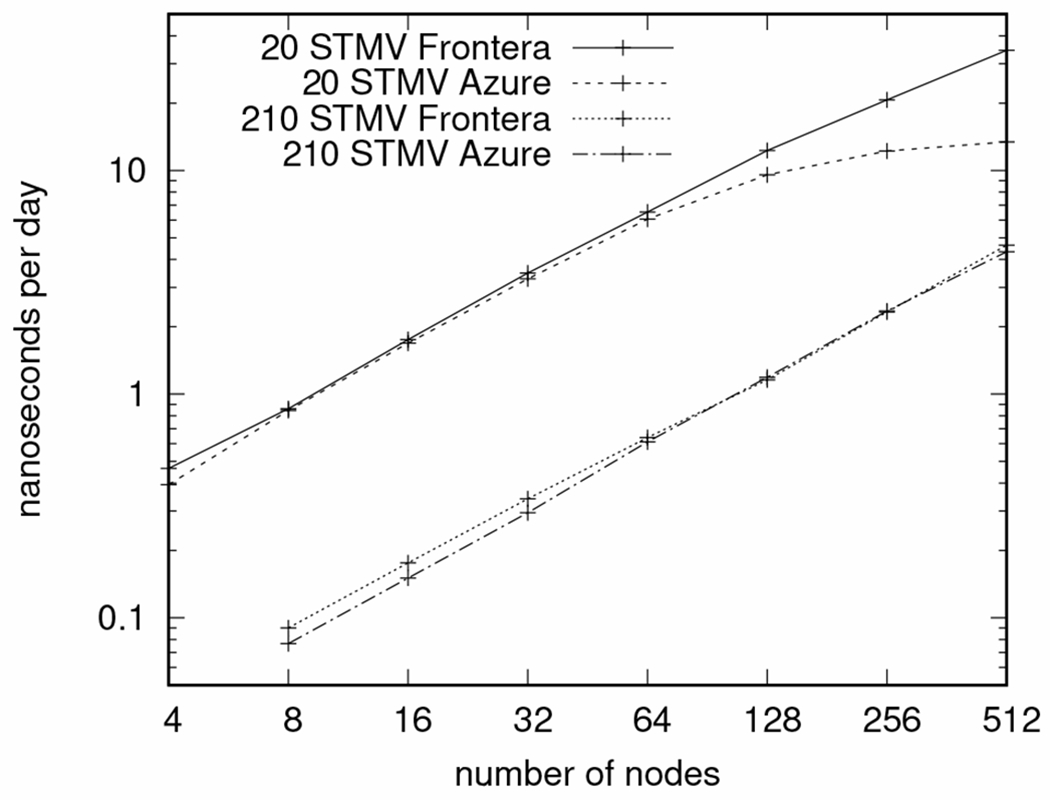
Comparing the best performance of Azure HBv2 cluster to TACC Frontera, scaling the STMV synthetic benchmarks. The smaller system size has about 21M atoms, and the larger system size has about 224M atoms. Azure is running the standard x86 build of NAMD enhanced with autovectorization compiler options for AVX2 and FMA support. Frontera is running the AVX-512 (tiles kernel) optimized version of NAMD. The Azure performance almost matches Frontera except when scaling the smaller system beyond 128 nodes.
Fig. 4.

Showing the average performance, with error bars, of Azure HBv2 cluster, scaling the STMV synthetic benchmarks. The smallest sized system of 10 STMV has about 10.5M atoms. Not only does the scaling plateau as the number of atoms per core drops below 1000, but there is also greater variability in performance of consecutive runs of each benchmark.
C. Overcoming the Challenges of Running on Public Clouds
One key difference between traditional HPC centers versus HPCaaS on public clouds, is that the traditional HPC centers have full-time staff with HPC-specific expertise dedicated to their specific computers to provide high reliability. Typical HPCaaS resources are partitioned for specific use among a pool of similar resources, where in many cases it is up to the users of the resource to manage their own queuing system and various other system administration related tasks with less overall involvement by support staff. Molecular dynamics simulation provides an extreme test case for parallel scaling due to overall performance being very sensitive to communication latency. Scaling issues related to hardware configuration states relevant to cloud HPC that are not emphasized in non-HPC cloud offerings — aspects of intra- and inter-server connectivity, and locality of file systems — may need to be addressed with iterative rounds of testing and reconfiguration, in collaboration between HPC users and cloud support staff. This is similar to repeated rounds of HPC acceptance testing or early HPC “friendly user time,” but mixed with large-scale production computing. Software management is oftentimes eased through the use of pre-built container images, which places burden back on the software developer to produce and maintain working container images.
HPCaaS offers the possibility of automatically making use of lower-priced, low-priority, evictable cloud instances — for example Azure “Spot Instances” — to work piecewise through lower-urgency larger compute projects that checkpoint with low overhead, and can be moved with acceptable cost to run in a compute location that has lower demand on its resources. Molecular dynamics simulations fall into this category, but simulations which require large node counts would require robust automatic checking to ensure such jobs do not fall prey to compromised performance brought on by slow nodes or latency problems.
V. HPC Collaboration Challenges
Beyond the laboratory’s activities to develop and distribute molecular modeling tools, and conduct its own simulations, its staff were also directly involved in close collaborations with external science teams performing SARS-CoV-2 simulations in response to COVID-19. Due to the timeliness, complexity, and scope of SARS-CoV-2 research, large multi-institutional collaborations enabled results to be achieved with much shorter turnaround than is typically the case, in large part due to the ability of collaborators to collectively bring their unique or specialized expertise, data, computing resources, and software advances to bear on the problem [4]. As one might imagine, some of the principal challenges associated with such a large multi-institutional collaboration are related to conducting large project meetings entirely virtually, on-demand transfer of very large data sets between multiple national HPC centers and the home institutions of collaborators, and marshalling diverse computational resources at multiple sites to perform the individual tasks in a large science workflow.
VI. Conclusions and Future Work
Throughout the COVID-19 pandemic, changes to work environment, the use of both conventional and non-traditional computing platforms, and the need for rapid access to new structure information, optimized and updated software tools, and collaboration among geographically dispersed research teams have remained challenges. We have reported some of our experiences during this time, and outlined solutions that have assisted our research staff, software developers, and collaborative teams to remain productive under adverse circumstances. Here we summarize a few of our observations and suggest opportunities for future work related to our experiences to date.
Due to our research laboratory being theoretical and conducting strictly computer-based experimentation in-house, the entire lab was able to continue successful operation remotely, supported by regular meetings via video conferencing and group messaging software, and general productivity achieved typical levels once some of our internal computing infrastructure was re-tasked to support improved remote access. There are key advantages to continuing remote work — for example, no time required for commuting to the office — and the logistics of scheduling external guest speakers for the laboratory’s ongoing weekly seminar series is much simpler and cheaper, with the added bonus that a number of distant colleagues now routinely remotely attend the seminar series and join discussions. Our experiences with the re-tasking of desktop computing resources in laboratory-wide queuing systems as additional compute nodes proved very successful, as did the deployment of a larger number of remote visualization systems. These efforts, in combination with configuring the queuing system with support for easy-to-use on-demand job launching helped maximize the productivity of laboratory researchers on the large number of human-centric tasks required for successful molecular dynamics simulation campaigns.
The researchers in our laboratory, as well as many others, have benefited from the computer time made available through the COVID-19 HPC Consortium. Our software development has benefited from continued collaboration with industry partners NVIDIA, Intel, and AMD providing dedicated software engineering resources to help maintain and improve our software on their most recent hardware. Much of our success in addressing virus-scale problems has been due to our own preparedness — a great deal of past development had already gone into NAMD and VMD to handle cell-sized systems of up to two billion atoms, and work had already been underway to optimize NAMD’s GPU performance.
The concept of HPC-as-a-Service (HPCaaS) shows great promise as a powerful tool to make tremendous computing resources available with rapid turnaround, not only in terms of the current COVID-19 pandemic, but more generally as a responsive route to bringing computation to bear on other similarly important and time-critical problems. Existing HPCaaS approaches are still somewhat incomplete and fall short of conventional HPC centers in terms of ease of deployment of large scale jobs, and ensuring performance in the kinds of dynamic runtime environments found in public clouds. There are opportunities for HPCaaS to influence the design of public cloud software and hardware infrastructure, and for the HPC community to develop software layers, libraries, and other techniques to achieve reliably high performance, even on public clouds, when a large node allocation includes a few underperforming systems or network links. Such reliable large-node allocations would allow another opportunity, to automatically make use of low-priority evictable instances, taking advantage of the low-overhead checkpointing offererd by much simulation software. Another unrealized opportunity for HPCaaS would be to facilitate both remote visualization and coordination of persistent filesystems on public clouds, with easy-to-use interfaces for launching and configuring resources. Although NAMD and VMD have been made available in hardware-optimized container images for multiple platforms, and within cloud virtual machine images, the user interfaces provided by existing public clouds are presently very complex and pose a barrier to adoption by biomedical researchers.
The approaches we have described here have helped researchers in our laboratory and users of our NIH Center-developed software remain productive during the COVID-19 pandemic, as evidenced by over 350 citations of NAMD and VMD from biophysical studies of SARS-CoV-2 during 2020. We expect that ongoing collaborations with hardware vendors, public cloud providers, and the HPC community at large will continue to improve upon the results reported here, to better address the pandemic. While our focus has been COVID-19 research specifically, we feel that many of the lessons learned are of general interest, and would be directly applicable to many other domains of science and engineering under similar circumstances of urgent need for computational studies to inform science, policy, or decision making.
Acknowledgments
Supported by NIH grant P41-GM104601.
Acknowledgments
This work was supported by NIH grant P41-GM104601 and COVID-19 HPC Consortium projects MCB200137 and MCB200183, including a Microsoft Azure Sponsorship. Thanks to NIH P41 Center members Defne Gorgun, Tianle Chen, Muyun Lihan, Karan Kapoor, and Sepehr Dehghanighahnaviyeh for valuable discussion of science projects and to Ronak Buch and Paritosh Garg for important technical advice and assistance.
Contributor Information
David J. Hardy, Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign Urbana, Illinois, USA
Barry Isralewitz, Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign Urbana, Illinois, USA.
John E. Stone, Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign Urbana, Illinois, USA
Emad Tajkhorshid, Beckman Institute for Advanced Science and Technology University of Illinois at Urbana-Champaign Urbana, Illinois, USA.
References
- [1].Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Hénin J, Jiang W, McGreevy R, Melo MCR, Radak B, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev M, Luthey-Schulten Z, Kalé LV, Schulten K, Chipot C, and Tajkhorshid E, “Scalable molecular dynamics on CPU and GPU architectures with NAMD,” Journal of Chemical Physics, vol. 153, p. 044130, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K, “Scalable molecular dynamics with NAMD,” Journal of Computational Chemistry, vol. 26, pp. 1781–1802, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Humphrey W, Dalke A, and Schulten K, “VMD: visual molecular dynamics,” Journal of Molecular Graphics, vol. 14, no. 1, pp. 33–38, 1996. [DOI] [PubMed] [Google Scholar]
- [4].Casalino L, Dommer AC, Gaieb Z, Barros EP, Sztain T, Ahn S-H, Trifan A, Brace A, Bogetti AT, Clyde A, Ma H, Lee H, Turilli M, Khalid S, Chong LT, Simmerling C, Hardy DJ, Maia JD, Phillips JC, Kurth T, Stern AC, Huang L, McCalpin JD, Tatineni M, Gibbs T, Stone JE, Jha S, Ramanathan A, and Amaro RE, “AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics,” The international Journal of High Performance Computing Applications, p. 10943420211006452, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Peck O, “Azure scaled to record 86,400 cores For molecular dynamics,” HPCwire, Oct 2020. [Online]. Available: https://www.hpcwire.com/2020/11/20/azure-scaled-to-record-86400-cores-for-molecular-dynamics/
- [6].Melo MCR, Bernardi RC, Rudack T, Scheurer M, Riplinger C, Phillips JC, Maia JDC, Rocha GB, Ribeiro JV, Stone JE, Nesse F, Schulten K, and Luthey-Schulten Z, “NAMD goes quantum: An integrative suite for hybrid simulations,” Nature Methods, vol. 15, pp. 351–354, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Trabuco LG, Villa E, Mitra K, Frank J, and Schulten K, “Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics,” Structure, vol. 16, pp. 673–683, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, and Zhang P, “Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics,” Nature, vol. 497, pp. 643–646, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Milles LF, Schulten K, Gaub HE, and Bernardi RC, “Molecular mechanism of extreme mechanostability in a pathogen adhesin,” Science, vol. 359, pp. 1527–1533, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Goh BC, Hadden JA, Bernardi RC, Singharoy A, McGreevy R, Rudack T, Cassidy CK, and Schulten K, “Computational methodologies for real-space structural refinement of large macromolecular complexes,” Annual Review of Biophysics, vol. 45, pp. 253–278, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Singharoy A, Venkatakrishnan B, Liu Y, Mayne CG, Chen C-H, Zlotnick A, Schulten K, and Flood AH, “Macromolecular crystallography for synthetic abiological molecules: Combining xMDFF and PHENIX for structure determination of cyanostar macrocycles,” Journal of the American Chemical Society, vol. 137, pp. 8810–8818, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guo Q, Lehmer C, Martínez-Sánchez A, Rudack T, Beck F, Hartmann H, Pérez-Berlanga M, Frottin F, Hipp MS, Hard FU, Edbauer D, Baumeister W, and Fernández-Busnadiego R, “In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment,” Cell, vol. 172, pp. 696–705, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Singharoy A, Maffeo C, Delgado-Magnero KH, Swainsbury DJ, Sener M, Kleinekathöfer U, Vant JW, Nguyen J, Hitchcock A, Isralewitz B et al. , “Atoms to phenotypes: Molecular design principles of cellular energy metabolism,” Cell, vol. 179, no. 5, pp. 1098–1111, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lees AJ, Hardy J, and Revesz T, “Parkinson’s disease,” Lancet, vol. 373, pp. 13–19, 2009. [DOI] [PubMed] [Google Scholar]
- [15].Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, Zhang J, Weng T, Zhang Z, Wu Z, et al. , “Molecular architecture of the SARS-CoV-2 virus,” Cell, vol. 183, no. 3, pp. 730–738, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Padhi AK, Rath SL, and Tripathi T, “Accelerating COVID-19 research using molecular dynamics simulation,” Journal of Physical Chemistry B, vol. 125, no. 32, pp. 9078–9091, 2021. [DOI] [PubMed] [Google Scholar]
- [17].Henderson R, Edwards RJ, Mansouri K, Janowska K, Stalls V, Gobeil SMC, Kopp M, Li D, Parks R, Hsu AL, and et al. , “Controlling the SARS-CoV-2 spike glycoprotein conformation,” Nature Structural & Molecular Biology, vol. 27, no. 10, pp. 925–933, Oct 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gorgun D, Lihan M, Kapoor K, and Tajkhorshid E, “Binding mode of SARS-CoV2 fusion peptide to human cellular membrane,” Biophysical Journal, vol. 120, no. 3, p. 191a, 2021, published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stone JE, Phillips JC, Freddolino PL, Hardy DJ, Trabuco LG, and Schulten K, “Accelerating molecular modeling applications with graphics processors,” Journal of Computational Chemistry, vol. 28, pp. 2618–2640, 2007. [DOI] [PubMed] [Google Scholar]
- [20].Kalé LV and Krishnan S, “Charm++: Parallel programming with message-driven objects,” in Parallel Programming using C++, Wilson GV and Lu P, Eds. MIT Press, 1996, pp. 175–213. [Google Scholar]
- [21].Acun B, Buch R, Kale L, and Phillips JC, “NAMD: Scalable molecular dynamics based on the Charm++ parallel runtime system,” in Exascale Scientific Applications: Scalability and Performance Portability, Straatsma TP, Antypas KB, and Williams TJ, Eds. CRC Press, 2017, ch. 5. [Google Scholar]
- [22].Stone JE, McGreevy R, Isralewitz B, and Schulten K, “GPU-accelerated analysis and visualization of large structures solved by molecular dynamics flexible fitting,” Faraday Discussions, vol. 169, pp. 265–283, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Singharoy A, Teo I, McGreevy R, Stone JE, Zhao J, and Schulten K, “Molecular dynamics-based refinement and validation for sub-5Åcryo-electron microscopy maps,” eLife, vol. 10.7554/eLife.16105, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stone JE, Vandivort KL, and Schulten K, “GPU-accelerated molecular visualization on petascale supercomputing platforms,” in Proceedings of the 8th international Workshop on Ultrascale Visualization, ser. UltraVis ’13. New York, NY, USA: ACM, 2013, pp. 6:1–6:8. [Google Scholar]
- [25].Stone JE, Sener M, Vandivort KL, Barragan A, Singharoy A, Teo I, Ribeiro JV, Isralewitz B, Liu B, Goh BC, Phillips JC, MacGregor-Chatwin C, Johnson MP, Kourkoutis LF, Hunter CN, and Schulten K, “Atomic detail visualization of photosynthetic membranes with GPU-accelerated ray tracing,” Parallel Computing, vol. 55, pp. 17–27, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stone JE, Sherman WR, and Schulten K, “Immersive molecular visualization with omnidirectional stereoscopic ray tracing and remote rendering,” 2016 IEEE international Parallel and Distributed Processing Symposium Workshop (IPDPSW), pp. 1048–1057, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sener M, Levy S, Stone JE, Christensen A, Isralewitz B, Patterson R, Borkiewicz K, Carpenter J, Hunter CN, Luthey-Schulten Z, and Cox D, “Multiscale modeling and cinematic visualization of photosynthetic energy conversion processes from electronic to cell scales,” Parallel Computing, 2021, published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tan T, Weerakkody R, Mrak M, Ramzan N, Baroncini V, Ohm J, and Sullivan G, “Video quality evaluation methodology and verification testing of HEVC compression performance,” Circuits and Systems for Video Technology, IEEE Transactions on, vol. 26, no. 1, pp. 76–90, Jan 2016. [Google Scholar]