

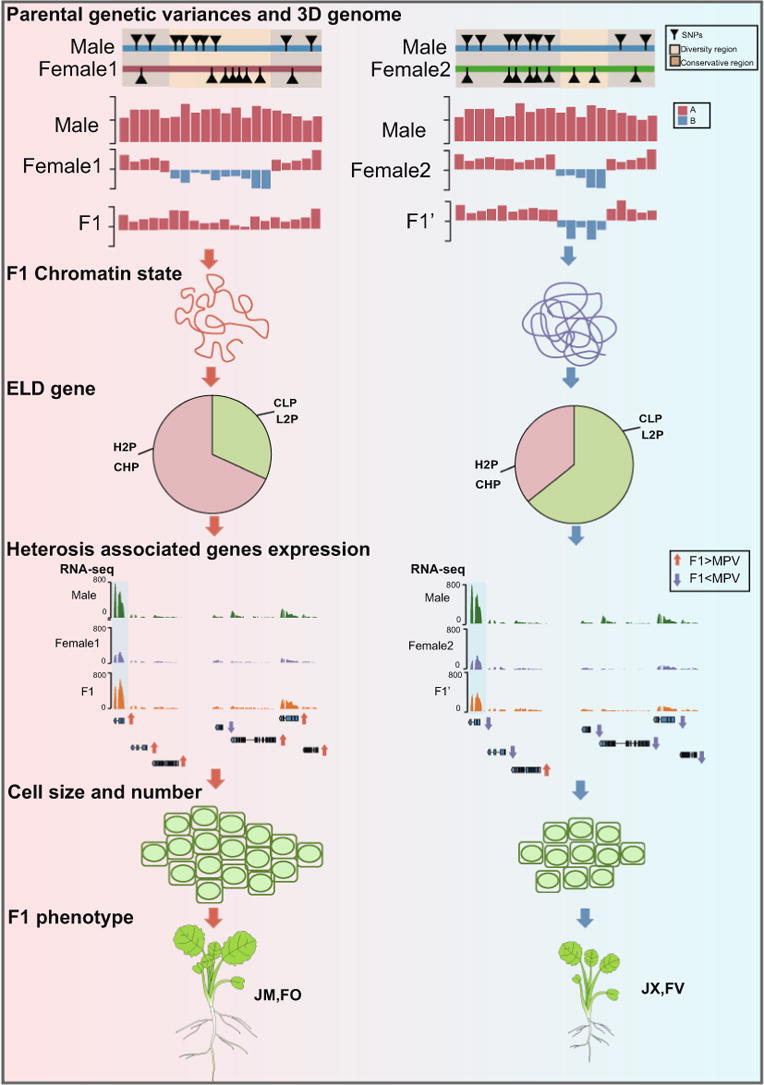
Graphical abstract
Keywords: Brassica napus, Heterosis, 3D chromatin architecture, Genetic variance, Gene expression
Highlights
-
•
The first study to explain the underlying mechanism of heterosis from the perspective of 3D genome in plants.
-
•
Superior heterosis hybrids tend to contain more transcriptionally active chromatin.
-
•
3D chromatin compartments correlate with genetic variance among parents.
-
•
More accessible chromatin accelerates highly expressed ELD (expression level dominance) genes in superior heterosis hybrids.
-
•
Hormone- and cell-cycle-related genes are more up-regulated with changes of 3D genome thereby promoting leaf size.
Abstract
Introduction
Heterosis is the major event driving plant development and promoting crop breeding, but the molecular bases for this phenomenon remain elusive.
Objectives
We aim to explore the effect of three-dimensional (3D) chromatin architecture on the underlying mechanism of heterosis.
Methods
Here, we constructed the North Carolina II (NC-II) population to select superior and inferior heterosis sets by comparing mid-parent heterosis (MPH) in Brassica napus. To decipher the impact of 3D chromatin architecture on the underlying mechanism of heterosis, we combined genetics, transcriptomics and 3D genomics approaches.
Results
We suggest that F1 hybrids with superior heterosis tend to contain more transcriptionally active A compartments compared with F1 hybrids with inferior heterosis, and approximately 19–21% compartment significantly altered in the F1 hybrids relative to the parental lines. Further analyses show that chromatin compartments correlate with genetic variance among parents, which may form the basis for differentially active chromatin compartments. Having more A compartments in F1 hybrids confers a more accessible chromatin circumstance, which promotes a higher proportion of highly expressed ELD (expression level dominance) genes in superior heterosis F1 hybrids (46–64%) compared with inferior heterosis F1 hybrids (22–31%). Moreover, genes related to hormones which affect plant growth, are more up-regulated with changes of 3D genome architecture, and we validate that increased hormone content contributes to cell proliferation and expansion by influencing the key genes of cell cycle thereby promoting leaf size.
Conclusion
Dynamic 3D chromatin architecture correlates with genetic variance among parents and contributes to heterosis in Brassica napus.
Introduction
Heterosis (or hybrid vigor), a biological phenomenon whereby F1 hybrids have superior agronomic traits to their parents, is widespread in both plants and animals. Three classic genetic models have been used to explain heterosis: dominance, overdominance and epistasis [1], [2]. Recent studies have illustrated that these three classic genetic models may contribute to the heterosis performance of offspring collectively, rather than contradictory [3]. In rice, the overall heterozygosity makes little contribution to heterosis, whereas the occurrence of partial dominance and overdominance at the loci contribute to heterotic advantage [4], [5]. These findings indicate that classic models focus more on key heterosis-associated loci and their contributions to heterosis, however, the role of three-dimensional (3D) chromatin architecture remains poorly characterized.
Accumulating evidence obtained using chromatin conformation capture technology indicates that the hierarchical 3D genome architecture in the nucleus is closely related to gene transcription [6]. The hierarchical structures of the 3D genome include compartments, topologically associated domains (TADs) and loops [7]. In general, A compartments are transcriptionally active regions of euchromatin, while B compartments are transcriptionally repressed regions of heterochromatin [7]. The conversion between A and B compartments affects gene expression: the conversion into an A compartment usually increases gene expression, and vice versa [8]. TADs, which are highly conserved among species [7], can spatially constraint the interactions between promoters and remote regulatory elements [9]. Chromatin loops can join cis-regulatory elements and promoters to regulate gene transcription [10].
The 3D architecture of chromatin in plants is increasingly becoming a focus of study [8], [11], [12]. A/B compartments have been found in Arabidopsis, maize, tomato, sorghum, foxtail millet and rice, reflecting their euchromatin and heterochromatin structures [11], [13]. Dynamic 3D genome architecture of plant both exist in the process of evolution and interspecific hybridization. For example, compared to its diploid ancestors, tetraploid cotton exhibits A/B compartment conversion and TAD reorganization, which affect the transcriptional activity of abundant genes [8]. Changes of chromatin compaction and histone have been shown to drive non-additive gene expression in an interspecific Arabidopsis hybrid [12]. Therefore, it is indispensable to explore how 3D genome architecture contributes to plant heterosis mechanism.
Heterosis is a complex process that is regulated by multiple factors. On the one hand, leaves are the major organ related to plant growth [14]. A key phenotypic difference between parental and hybrid leaves is the degree to which the cell size and cell number increase, resulting in larger leaves, in hybrids [15]. The proper regulation of these two processes is the basis for determining leaf size [16]. Cell proliferation and expansion are inseparable from cell cycle regulation. Numerous cell-cycle-related genes have been identified in plants. The upregulation and/or downregulation of these genes alters cell number and/or cell size during leaf growth [16], [17]. Moreover, auxin, cytokinin (CK), brassinosteroids (BRs) and gibberellin (GA) could control changes in the cell cycle by influencing the expression of cell-cycle-related genes [17]. On the other hand, the phenotypes of offspring also depend on the expression behavior of genes. Expression level dominance (ELD) contributes to heterotic phenotypes, which may be due to the interactions of parental alleles [18], [19]. However, little is known about how 3D chromatin architecture affects gene expression associated with heterosis.
Here, we combined high-throughput chromosome conformation capture (Hi-C), whole-genome sequencing (WGS) and RNA-seq data to explore the influence of the 3D architecture of chromatin on heterosis in B. napus at the seedling stage (Fig. 1A). We revealed dynamic changes of 3D chromatin architecture between parents and F1 hybrids and found that parental genetic variance was associated with parental distinct compartments. In addition, dynamic changes of compartment activities contributed to ELD genes, hormone-related genes and cell-cycle-related genes expression, which were linked to heterosis. This study advances our understanding of heterosis in plants from the perspective of 3D genome.
Fig. 1.

Comparison of the morphology and transcriptomes of parents and hybrids in B. napus. A, The workflow of this study. B, Phenotypes of parents and F1 hybrids at 21 DAS (days after sowing). C, Phenotypes of leaves in hybrids with superior and inferior heterosis. D, Number of upregulated and downregulated genes in F1 hybrids versus the mid-parental value (MPV) of gene expression (Student’s t test, P < 0.05). Data from three biological replicates were combined. E and F, Volcano plots showing genes differentially expressed between FO and FV (E) and between JM and JX (F) (F1 TPM > 1, |log2(fold change)| > 1, Padj < 0.05). Each gene expression level was normalized to TPM (transcripts per million). Red dots, upregulated genes; blue dots, downregulated genes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Methods
Plant materials and growth conditions
The female sterile line, male restorer line, and hybrids of B. napus used in this study were generated at Huazhong Agricultural University [20], [21]. All varieties were grown in a controlled greenhouse (a cycle of 16 h light/8h dark) at 21–23 °C with 30–60% relative humidity under hydroponic conditions with Hoagland nutrient solution in three biologically repeated random block experiments [22]. The parents and combinations with superior heterosis (FO, JM) and inferior heterosis (FV, JX) were selected based on statistical analysis of phenotypes at 30 DAS (days after sowing). O, V, M, and X were the female parents, and F and J were the male parents (Fig. S1). At 21 DAS, 30 similar plants of each accession (10 plants per replicate, three independent experiments with a completely random design) were collected and used to measure phenotypic data, including fresh weight (FW), dry weight (DW), and leaf area (LA). The materials were collected and immediately washed with distilled water, oven-dried at 105 °C for 30 min, and dried further at 80 °C for 60 h. Leaf area was measured using ImageJ (https://imagej.nih.gov/ij/). MPH (mid-parent heterosis) and BPH (better-parent heterosis) were calculated as follows: MPH = (the mean value of hybrid F1 - the average value of both parents)/the average value of both parents × 100%; BPH = (mean value of hybrid F1 - mean value of optimal parent)/mean value of optimal parent × 100%. SPSS Version 18.0 (IBM Corp, Armonk, NY, USA) was used for statistical analysis. Comparisons between different varieties were performed using ANOVA with Tukey’s HSD and two-sided t tests (Fig. S1).
For RNA-seq and Hi-C analyses, the hybrids and parents were grown in the greenhouse under the same conditions. For the whole-genome sequencing, plants were grown in an experimental field under normal environmental conditions (Wuhan; 114.35°E, 30.48°N). Three biological replicates were generated for RNA-seq experiments, two biological replicates for the Hi-C experiment and one biological replicate for WGS experiments.
Hi-C sequencing
For Hi-C sequencing, the plants were grown under the conditions described above and sampled at the same time points. To build a Hi-C library, leaves were ground in liquid nitrogen, and 2 g of homogenous tissue powder was collected. Hi-C sequencing library construction and Illumina sequencing were performed by Novogene Company, China. The experiment was performed using DpnII digestion as previously described [23]. Qualified DNA samples were randomly broken into fragments with a Covaris ultrasonic breaker. The DNA with biotin was captured via adsorption on avidin magnetic beads, and libraries were constructed by performing end repair, adding A tails, adding adaptor, purification and PCR expansion. Following construction, Qubit2.0 was used for preliminary quantification. Subsequently, the insert size of the library was tested using an Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, MA, USA), and the effective concentration of the library was accurately quantified by qPCR to ensure the quality of the library. The final library was sequenced by Illumina HiSeq PE sequencing.
Hi-C read mapping and normalization
Hi-C reads from the superior and inferior hybrid sets were aligned to the newly assembled high-quality ZS11 reference genome [24]. Read mapping and construction of contact maps were performed using Juicer software with default settings [25]. Validated read pairs of B. napus superior and inferior hybrid sets were used to create interaction matrices with a bin size of 50 kb for further analysis. The interaction matrices were normalized using the iced method [26].
Identification of genomic compartments and calculation of ICF and compactness
For compartment analysis, we first removed the centromeric region and the pericentromeric region of each chromosome, as described previously [12], [27]. The locations of the centromere regions of the chromosomes were described previously [24]. PCA was performed separately for each arm with a 50-kb bin size interaction matrix. A or B compartments were determined based on gene density and the sign of the first eigenvector (PC1): A compartments tend to have higher gene densities and B compartments tend to have lower gene densities. ICF (interaction contact fraction) was calculated as described previously [28]. Chromatin compactness was measured as previously described [12].
RNA-seq experiment and sequencings
The second true leaves were collected from 21-day-old seedlings from the parents and hybrids, immediately frozen in liquid nitrogen, and stored at –80 °C for RNA extraction. Three biological replicates were used for each sample, with each replicate consisting of a pool of 8–10 plants.
Library construction and sequencing were carried out by BGI, China. Total RNA was extracted from the samples using the TRIzol method [29]. The RNA was qualified and quantified using a NanoDrop and Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, MA, USA). Following total RNA extraction and DNase treatment, the mRNA was isolated using magnetic beads with Oligo (dT). The mRNA was broken into short fragments in fragmentation buffer. cDNA was synthesized using the mRNA fragments as templates. During the QC (quality control) steps, an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System were used to quantify and qualify the sample library. mRNA-seq libraries were constructed and sequenced on the HiSeq™ 4000 platform following the manufacturer’s instructions.
RNA-seq data analysis
Clean reads were aligned against the ZS11 reference genome using HISAT2 (v2.1.0) with default parameters [30]. The raw read counts for each gene were normalized to TPM (transcripts per million) values with StringTie (v1.3.3b) [31]. Differentially expressed genes (DEGs) in the superior versus inferior hybrids were identified with the DESeq2 package in R [32]. We used the criteria of false discovery rate (FDR) < 0.05 and |log2 fold-change| >1 to determine upregulated and downregulated genes with genes of the F1 lines at TPM > 1. Student’s t test was performed to test the difference between the hybrid and mid-parental value (MPV; P < 0.05).
Definition of ELD gene expression patterns
We divided the genes into additive genes and non-additive genes based on their expression levels. A Student’s t test was conducted to test the difference between the values of hybrids and the MPV. If the values of hybrids were significantly (P < 0.05) different from the MPV, the genes were defined as non-additive genes; if there was no significant difference between the values of hybrids and the MPV, the genes were defined as additive genes. We used non-additive genes to identify ELD genes. The ELD genes were divided into five groups: H2P (higher than both parents), CHP (close to higher parents), CLP (close to lower parents), L2P (lower than both parents) and B2P (between the two parents). If F1 > Paternal = Maternal, F1 > Paternal > Maternal or F1 > Maternal > Paternal, the genes were defined as higher than both parents (H2P); if F1 = Paternal > Maternal or F1 = Maternal > Paternal, the genes were defined as close to higher parent (CHP); if Paternal > F1 > Maternal or Maternal > F1 > Paternal, the genes were defined as between two parents (B2P); if F1 = Paternal < Maternal or F1 = Maternal < Paternal, the genes were defined as close to lower parent (CLP); if F1 < Paternal = Maternal, F1 < Paternal < Maternal or F1 < Maternal < Paternal, the genes were defined as lower than both parents (L2P), where “=” means statistically similar and “>” and “<” mean significantly higher and lower, respectively [33].
WGS experiment and sequencing
Young leaves were collected from the hybrids and parents and stored at −80 °C for DNA isolation. Genomic DNA was extracted from the samples using a Tiangen Genomic DNA Extraction Kit (DP305). Sequencing libraries were prepared according to Illumina’s standard protocol. DNA concentration and integrity were measured using a Qubit Fluorometer and Agilent 2100 bioanalyzer. The matched terminal library with an inserted fragment size of ∼350 bp was sequenced on the Illumina HiSeq PE150 platform. The DNA libraries were constructed and sequenced by Novogene.
WGS data analysis
High-quality SNPs were identified using the Sentieon pipeline [34]. First, BAM files were obtained by Burrows-Wheeler Alignment (BWA) mem algorithm mapping to the current standard reference genome ZS11 [35]. SAMtools was then used to filter the BAM files with quality of >10 [36]. SNPs were collected from all the samples using the Haplotyper algorithm from Sentieon. The VCF files from all samples were merged using the Sentieon GVCFtyper algorithm. To obtain high-quality SNPs, the raw SNPs were filtered using the GATK VariantFiltration module with the parameter --filterExpression ‘QUAL < 30.0 || MQ < 50.0 || QD < 2′ --clusterSize 3 --clusterWindowSize 10. Next, the paternal SNPs were compared with maternal SNPs to identify different SNPs of the parents with a bin size of 50 kb. Subsequently, to identify SVs in the parents, VCF files were used by paragraph with default settings [37]. Finally, paternal SVs were compared with maternal SVs to identify different SVs of the parents with a bin size of 50 kb.
Plant hormone measurements
To measure plant hormone levels, the second true leaves were collected from the parents and hybrids grown under hydroponic conditions at 21 DAS (three biological replicates per variety, approximately seven seedlings per replicate) and rapidly frozen in liquid nitrogen. Freeze-dried leaves were homogenized and crushed in a tissue grinder (400 MM, Retsch; 60 Hz, 60 s) with zirconium beads. A 100 mg sample of dry powder was extracted overnight at 4 °C with 1.0 mL 70% methanol, and acyclovir at a final concentration of 0.1 mg/L was used as an internal standard to measure hormone metabolites [38]. MS2T libraries were constructed using stepwise multiple ions while monitoring enhanced product ions and analyzed by liquid chromatography-electrospray ionization (LC-ESI). Subsequently, the multiple reaction detection method (sMRM) was used to simultaneously perform quantitative analysis of six plant hormones, including CKs, GAs, SAs, auxins, ABA and ACC (biosynthetic precursor of ETH) as previously described using analyst 1.5 software [39]; the MRM detection window was 90 s and the target scanning time was 1.0 s.
Identification of plant hormone, photosynthesis, photorespiration and cell cycle genes in B. napus
The sequences of Arabidopsis plant hormone, photosynthesis and photorespiration genes were obtained from the TAIR database (https://www.arabidopsis.org/). The deduced amino acid sequences of these genes were mapped to the ZS11 proteome using the BLAST pipeline with the parameter setting ‘-max_target_seqs 1 -evalue 1e-5′. The sequences of cell cycle genes and genes that positively regulate the cell cycle obtained from previous reports were used for data analysis as described above [16], [40], [41].
Measuring individual leaves and palisade mesophyll (PM) cells
The second leaves of at least 10 seedlings per plant line were harvested at 21 DAS from plants grown under hydroponic conditions. Four point samples were collected from the second true leaf from the center to the edge of the leaf at the same position with a circular punch. To count PM cells, leaves from plants at the indicated stages were fixed and cleared of chlorophyll with 70% ethanol. The samples were then cleared in chloral hydrate solution (8 g of chloral hydrate: 2 mL of deionized water: 1 mL of glycerol) and photographed under a Nikon ECLIPSE 80i compound microscope with a 10 × ocular and 20 × objective. The cells were counted using the cell counter plugin (http://rsbweb.nih.gov/ij/plugins/cell-counter.html) in ImageJ. Cell sizes and total cell numbers were calculated based on the cell numbers in the image, the area of the image and the overall area of the leaf. Leaf area was determined using ImageJ, and PM cells were counted with the cell counter plugin.
Quantitative reverse-transcription PCR
For qRT-PCR validation experiments, three replicates of 10 seedlings from the parents and hybrids were harvested at the same time at 21 DAS. Total RNA was extracted from 0.1 g FW of various materials using an RNAprep Plant Kit containing DNase I treatment reagent (DP441; Tiangen, Beijing, China), and 2 µg of total RNA was used for cDNA synthesis using a Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (K1622; Thermo, America) according to the manufacturer’s instructions. qRT-PCR was performed using SYBR Green Real-Time PCR Master Mix (Toyobo, Japan) and the CFX96 Real-Time system (Bio-Rad, USA). The data were analyzed with CFX Manager software as described previously [42]. ACTIN7, UBC9 and UBC10 were used as housekeeping genes to normalize the expression data. At least three biological replicates were performed per experiment.
Data availability
All sequencing data generated for this study have been submitted to the NCBI Sequence Read Archive under accession number PRJNA700832, PRJNA701132, and PRJNA701133.
Results
Phenotypic and transcriptomic changes in hybrids
We constructed the North Carolina II (NC-II) population with 132 intraspecific hybrids based on 11 maternal lines (sterile) and 12 paternal lines (fertile), by comparing the mid-parent heterosis (MPH) of these hybrids to select superior and inferior heterosis sets (Table S1). Meanwhile, we combined three major phenotypes fresh weight, dry weight and total leaf area to obtain two superior heterosis sets (F-O-FO, J-M−JM) and two inferior heterosis sets (F-V-FV, J-X-JX) comprehensively (Fig. 1B, C; Table S2; Fig. S1). To explore the influence of the 3D genome architecture on heterosis in B. napus, we combined the phenotypic, transcriptomic, WGS, and Hi-C data (Fig. 1A). First, we examined the transcriptomes of parents and F1 hybrids by RNA-seq analysis. The average mapping rates of all accessions were ∼90% (Fig. S2; Table S3). Compared with MPV (mid-parental value) of gene expression, there were more upregulated genes than downregulated genes in JM and FO, whereas there were more downregulated genes than upregulated genes in JX and FV (P < 0.05) (Fig. 1D; Tables S4–S7). Therefore, according to phenotypic measurements, JM and FO were ‘better’ at being expressed than JX and FV respectively, and the transcriptome levels were consistent with these results. Compared with FV, we identified 5,720 upregulated genes and 4,233 downregulated genes in FO. In addition, compared with JX, we identified 4,228 upregulated and 3,888 downregulated genes in JM (Fig. 1E, F; Tables S8 and S9). These findings suggest that hybrids with superior heterosis include more upregulated genes, altered gene expression patterns may influence their heterotic phenotypes.
3D chromatin architecture changes dynamically in B. napus hybrids
To investigate the dynamics of 3D genome organization during B. napus hybridization, we performed Hi-C experiments using DnpII for the heterosis groups, generating a total of 101 billion sequencing read pairs. We mapped the clean Hi-C data against the high-quality ZS11 reference genome [24] and obtained 34 billion usable valid interaction read pairs for subsequent comparative 3D structural analysis (Table S10). We constructed chromatin interactive heat maps and identified chromatin compartments at a resolution of 50 kb based on high-quality Hi-C data in all groups (Figs. S3 and S4). We identified A/B compartments, which corresponding to euchromatin and heterochromatin, based on principal component analysis (Fig. 2A; Table S11). Particularly, hybrids with superior heterosis tend to contain more A compartments while hybrids with inferior heterosis tend to contain more B compartments, which is congruent with their gene expressions and implies 3D chromatin architecture may influence phenotype of heterosis (Fig. 2B). Compared with the activity of parental compartments, approximately 19–21% compartments in F1 hybrids were obviously changed, which suggested that 3D chromatin architecture is dynamic among parents and hybrids (Fig. 2C). Furthermore, analysis of these changed compartments, under the condition of different compartment activity existed in parents, hybrids with superior heterosis (FO, JM) occupied a higher ratios of A compartments than B compartments, whereas the opposite situation was observed in FV and JX (Fig. S5A).
Fig. 2.

Comparison of dynamic changes in the 3D structure of chromatin between parents and hybrids. A, Distribution of compartments in superior and inferior hybrid sets. Red and blue histograms represent A and B compartments, respectively. B, Percentages of different compartments in superior and inferior hybrids. C, Percentages of compartment activity during B. napus hybridization. The order of compartment activity is paternal-maternal-F1. D, Distribution of chromatin interactions in the whole genome of the F-O-FO and J-M−JM groups. E and F, Relationship of chromatin compactness between parents and hybrids between FO and F (E) and FO and O (F). rho, Spearman’s rank correlation coefficient. G, Chromatin interactions in B. napus represented by the C04 chromosome of F-O-FO. The upper track shows the distribution of chromatin compartments in the 3D structure. Red histograms represent A compartments; blue histograms represent B compartments; gray columns indicate masked regions represented as centromere and pericentromeric regions. The lower track shows chromatin interactions at 50-kb resolution. In each heat map, strong interactions are indicated in red and weak interactions in blue, and the dark blue area in the middle represents the pericentromeric regions. H, Features of compartments represented by the first eigenvector from principal component analysis (PCA). Red and blue histograms represent A and B compartments, respectively. I, Dynamic activities of compartments contribute to gene expression in F1 hybrids. The expression level of each gene was normalized to transcripts per million (TPM). *P < 0.05; **P < 0.01; ***P < 0.001 (Wilcoxon rank-sum test). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To gain more insight into the detailed dynamic 3D genome organization between the parents and hybrids, we introduced the interaction contact fraction (ICF) to describe the interactions of chromatin and compactness to display the status of chromatin, respectively [12], [28]. We identified the interactions of hybrids chromatin were greatly altered compared to their parents (Fig. 2D; Fig. S5B, C). In addition, the chromatin compactness of an F1 hybrid was generally quite different from that of either parent, but more likely to resemble that of the maternal than the paternal chromatin (Fig. 2E, F; Fig. S6). Altogether, at the chromosomal level, not only chromatin interactions but also the distribution of compartments changed during hybridization (Fig. 2G; Figs. S7 and S8). Two representative cases showed that FO and JM harbored more A compartments than FV and JX, respectively (Fig. 2H; Fig. S9). Next, we sought to test whether compartment switching contributes to gene expression, which has been observed in plants [8], [11]. As expected, compared with distinct compartments in parents, the genes of hybrids were detected in A compartments with higher gene expression than those of in B compartments (Fig. 2I). Therefore, the hierarchical 3D chromatin architecture changed dynamically among parents and hybrids, and the compartment activity differed between superior and inferior hybrid sets, which strongly affected the regulation of gene expression.
Chromatin compartments correlate with genetic variance among parents
Genetic diversity between parents could be used to further improve yields [43]. F1 hybrid heterosis shows preferential association with the parents’ individual performance [44], [45]. We speculated that there is a relationship between the genetic variance and parental distinct compartments. To verify this hypothesis, we defined regions of chromatin compartments with different activity between parents as ‘diversity regions’ and those with similar activity between parents as ‘conservative regions’ and introduced the analysis of the WGS data between parents. By comparing single-nucleotide polymorphism (SNP) density between parents, we found superior heterosis parents with more SNPs than those in inferior heterosis parents (Fig. S10). Observing the location of parental distinct compartments, we identified approximately 81% parental distinct compartments show preferential association with genetic variance regions between parents (Fig. 3A). Remarkably, throughout the genome, the SNP density was higher in diversity regions, whereas in conservative regions (within 2 Mb at upstream and downstream of diversity regions), the SNP density was lower (Fig. 3B). Moreover, the SNP density between parents showed the same trend (Fig. 3C). Next, we divided SNP density into four groups based on quantiles with different orders between parents. As the SNP density between parents increased, the proportion of different compartment activities between parents (which we refer to as diversity regions) increased (Fig. 3D; Fig. S11). Although a small portion of diversity regions located in non-genetic variance regions between parents, we speculated that may because of the parental epigenetic difference, the most majority diversity regions link to parental genetic variance regions. In one case, in the F-O-FO superior heterosis set, SNP density was higher in diversity regions than in the surrounding conservative regions (Fig. 3E). In another case, the same was true in the J-M−JM set (Fig. 3F).
Fig. 3.

Dynamic changes in compartments correlate with genetic variance among parents. A, Distribution of parental distinct compartments among parental genetic variance regions. B, SNP density levels between parents around diversity region borders. Lines show average values across 50-kb bins. C, Distribution of SNP density between diversity regions and conservative regions. ***P < 0.001 (Wilcoxon rank-sum test.). D, Percentages of F-O-FO diversity regions and conservative regions with SNP density classifications. According to the analysis of SNP density order between parents, quartile 1 is smaller and quartile 4 is larger. ***P < 0.001; NS, no significant difference (Fisher’s exact test). E and F, Example of the relationship between diversity/conservative regions and SNP density in parents. The two upper tracks show the features of compartments in the parents: red, A; blue, B. The middle track shows the distribution of diversity regions and conservative regions. The two lower tracks show the distribution of different SNPs between parents and the number of SNPs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We also examined the different structural variations (SVs) between parents. Diversity regions contained more SVs than the surrounding conservative regions (Fig. S12A, B). Moreover, in parents with more different SVs, these SVs tended to be located in diversity regions; the opposite was true for parents with no different SV regions (Fig. S12C, D). Therefore, we inferred that genetic variance among parents is closely related to chromatin hierarchical structures, perhaps forming the basis for their differentially active chromatin compartments.
Dynamic compartments influence ELD gene expression
Expression level dominance (ELD) genes, which could potentially be responsible for generating the heterosis phenotypes [46]. Higher than both parents (H2P) and close to higher parent (CHP) type ELD genes are expressed at higher levels in F1 hybrids than in both parents and at levels similar to highly expressed genes in one parent, respectively. Lower than both parents (L2P) and close to lower parent (CLP) type ELD genes are expressed at lower levels in F1 hybrids than in both parents and at similar levels to genes expressed at lower levels in one parent, respectively. Finally, between two parents (B2P) type ELD genes are those whose expression in the F1 hybrid is between two parents [33]. The emergence of ELD genes has been detected in allopolyploids [18], [46].
The concept of ELD gene expression prompted us to ask whether the dynamic changes in chromatin compartment activity are associated with ELD genes. Compared with hybrids (JX, FV with 27–39%) with inferior heterosis, hybrids (JM, FO with 43–60%) with superior heterosis contain more percentages of highly expressed H2P and CHP genes. Conversely, hybrids (JX, FV with 61–73%) with inferior heterosis contain more percentages of L2P and CLP genes than those of hybrids (JM, FO with 40–57%) with superior heterosis (Fig. S13; Tables S12–S15). As mentioned above, hybrids with superior heterosis cover more A compartments in diversity regions, we further explored the correlation of compartment activity and ELD genes. Among ELD genes, H2P and CHP genes were more highly expressed than CLP and L2P genes in diversity regions (Fig. 4A). Consistent with the dynamic changes of the compartment characteristics in the offspring (Fig. 2B), the H2P and CHP genes described above accounted for a large proportion of ELD genes in JM (64%) and FO (46%), whereas L2P and CLP genes accounted for a large proportion of these genes in JX (60%) and FV (74%) (Fig. 4B). These results suggest that changes in compartment activity in F1 hybrids might contribute to the differential expression of ELD genes. Next, we analyzed the distribution of the ELD genes in the dynamic compartments regions, indicating that ELD genes of hybrids with superior heterosis tend to lie in more transcriptionally active A compartments, whereas those of in hybrids with inferior heterosis tend to lie in more transcriptionally in-active B compartments (FV) or not significant existed in A/B compartments (JX) (Fig. 4C). In addition, we examined folding status of chromatin, observing that the chromatin compactness of FV was higher than that of FO, and the chromatin compactness of JX was higher than that of JM (Fig. 4D). These findings suggest that 3D chromatin architectures provide a more accessible chromatin basis for ELD gene expression in superior heterosis hybrids. On the basis of these results, we propose that the chromatin activity of different compartments affects the expression of ELD genes: ELD genes in F1 hybrids with A compartments tend to be highly expressed, whereas ELD genes in F1 hybrids with B compartments tend to be expressed at lower levels (Fig. 4E, F).
Fig. 4.

Dynamic 3D chromatin architecture contributes to ELD gene expression. A, Distribution of ELD gene expression in hybrids compared with MPV. The dotted line indicates a slope of 1, meaning that the MPV is equivalent to F1 gene expression. Above the dotted line are H2P (higher than both parents) and CHP (close to higher parents) group genes; below the dotted line are CLP (close to lower parents), L2P (lower than both parents) and B2P (between two parents) group genes. B, Percentages of ELD genes in diversity regions. C, Relationship between ELD genes and compartment activity in F1 hybrids. D, Distribution of compactness in F1 chromatin located in diversity regions. P values were determined using the Wilcoxon rank-sum test. E and F, Examples of the relationship between dynamic 3D chromatin compartments and ELD genes. The upper track shows the feature of the compartments: red, A compartments; blue, B compartments. The middle track shows the genes in the chosen region: genes shown in red are ELD genes, and genes shown in black are additive genes. The lower track shows the expression patterns of ELD genes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Enhanced expression of hormone-related and cell-cycle-related genes affected by dynamic 3D chromatin architecture
Hormones are crucial for mediating leaf growth and have an impact on cell cycle control [17]. These observations prompted us to investigate whether dynamic changes in chromatin structure are linked to the expression of hormone- and cell cycle-related genes. Auxin, cytokinins (CKs), brassinosteroids (BRs) and gibberellin (GA) contribute to plant growth, whereas salicylic acid (SA), ethylene (ETH), and abscisic acid (ABA) do not directly influence plant growth but are involved in stress adaptation (Fig. 5A) [41]. First, we investigated the expression of auxin-, CK-, BR-, and GA-related genes in B. napus. We found that there were more upregulated than downregulated genes in these categories in the superior hybrid sets compared to the MPV (Student’s t test, P < 0.05) (Fig. S14; Tables S16–S19).
Fig. 5.

Effects of dynamic 3D architecture on hormone-related genes, cell proliferation and cell expansion. A, A diagram of hormones closely associated with plant growth. Red and gray arrows represent hormones beneficial and disadvantageous to plant growth, respectively. Hormones: CK, cytokinins; GA gibberellin; BR, brassinosteroids; SA, salicylic acid; ABA, abscisic acid; ETH, ethylene. B, Number of upregulated and downregulated hormone-related genes in F1 hybrids versus the MPV (Student’s t test, P < 0.05) related to auxin, CK, GA and BR. C, Comparison of hybrid gene expression and the MPV. The dotted line indicates a slope of 1, meaning that the MPV is equivalent to F1 gene expression. Red and gray dots indicate the genes that are more highly and more weakly expressed in the F1 hybrids versus the MPV, respectively. D, Examples of the relationship between 3D chromatin structure and plant-hormone-related gene expression. The first track shows the features of the Hi-C chromatin interaction. Strong interactions are shown in red and weak interactions are shown in blue. The second track shows the distribution of compartments: red, A; blue, B. The third track shows RNA-seq read coverage in the chosen region. The fourth track shows the genes in the chosen region. Red denotes hormone-related genes; red arrows represent genes that were more highly expressed in the F1 hybrids versus the MPV; purple arrows represent genes that were expressed at lower levels in the F1 hybrids versus the MPV. E, Depiction of the regulatory network of auxin and auxin-related genes. Heat map of auxin-related gene expression: dark red and dark blue represent high and low gene expression levels, respectively. Orange and blue triangles around auxin represent relatively high hormone levels in superior hybrids and relatively low hormone levels in inferior hybrids, respectively. The bottom arrow indicates the four phases of the cell cycle. F, The second leaf areas of parents and hybrids. G, Paradermal optical sections of the palisade mesophyll (PM) layer showing the larger cells of superior heterosis sets. H and I, Measurements of PM cell size (H) and estimated cell number (I) in the hybrids. (F, H and I) The percentage above the bar is the MPH (mid-parent heterosis) ratio in the corresponding F1 hybrid. Red asterisks indicate significant differences from the MPV, and black asterisks indicate values above the BPV (better parent value). Statistical comparisons among the J-M−X−JM−JX (uppercase blue letters) and F-O-V-FO-FV (uppercase red letters) are shown, and categories with different letters are significantly different from the others (Student’s t test, P < 0.01). All error bars are S.E.M. n = 10–14. J, Relative contributions of the increases in cell size and cell number to the larger leaves of the hybrids. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We then focused on the relationship between 3D chromatin architecture and hormone-related gene expression. Consistently, superior hybrids tended to have more upregulated genes in diversity regions (Student’s t test, P < 0.05) (Fig. 5B). We then investigated gene expression in diversity regions of F1 hybrids compared to the MPV. There was a higher proportion of highly expressed hormone-related genes (versus the MPV) in JM and FO and a lower proportion of these genes in JX and FV (Fig. 5C). Furthermore, in superior hybrids with A compartments, most hormone-related genes (which support plant growth) were upregulated in diversity regions, as were most surrounding genes, whereas in inferior hybrids with B compartments, most of these genes were downregulated (Fig. 5D; Fig. S15). These findings suggest that 3D chromatin architecture regulates the expression of hormone-related genes. To verify this notion, we measured the levels of these hormones, except for BR, due to experimental technical limitations (Fig. 5A). Auxin, CK, and GA levels were significantly higher in the superior hybrid sets, whereas SA, ETH, and ABA levels were significantly higher in the inferior hybrid sets (Figs. S16–S18).
Given the roles of hormone-related genes in the cell cycle, we asked whether a regulatory network might accelerate cell proliferation and cell expansion due to dynamic changes in 3D chromatin architecture. Notably, two key genes in the auxin pathway, KUA1/MPYH and TOR, were upregulated in FO and JM but downregulated (or expressed at similar levels to the MPV) in FV and JX in diversity regions (Figs. S19 and S20). KUA1/MPYH and TOR genes play roles in the auxin biosynthesis pathway in Arabidopsis [47], [48]. Overexpressing KUA1/MPYH upregulates the phytochrome-interacting factor genes PIF4/PIF5 in Arabidopsis, leading to the upregulation of auxin biosynthesis genes YUCC8 and SAUR thereby enhancing auxin accumulation and plant growth [47]. Here, we found these genes were upregulated in FO, likely leading to the accumulation of auxin, whereas they were downregulated in FV, leading to relatively low auxin contents (Fig. 5E; Fig. S21; Table S20).
Auxin accumulation facilitates the upregulation of PRZ1, which in turn promotes the expression of the core cell cycle gene CYCB1 [41]. Gene expression analysis showed that PRZ1 and CYCB1 were highly expressed in FO and weakly expressed in FV. The upregulation of CYCB1 could promote the G2-to-M phase transition and accelerate the cell cycle (Fig. 5E; Fig. S21; Table S20) [16], [41]. ROP2 expression increases in response to auxin accumulation, thereby promoting TOR gene expression. This in turn leads to increased expression of E2FA and/or E2FB, which are core genes of the cell cycle [41], [48]. Indeed, E2FA were upregulated in JM, accelerating the transition of the cell cycle from the G1 to the S phase but downregulated in JX (Fig. 5E; Fig. S21; Table S20) [41]. In addition, KUA1 and TOR also affect cell expansion [49], [50].
To verify the influence of this putative gene regulatory network on cell proliferation and cell expansion in the hybrids, we performed cytological experiments (Fig. S22). The size of the second leaf was greater (a mark of superior hybrid status) than the MPV in JM and FO, and was slightly larger than the MPV in JX and FV (Fig. 5F). Cell number and cell size showed the same trends (Fig. 5G–I). Moreover, genes that positively regulate the cell cycle were much more likely to be upregulated in superior hybrids (Fig. S23; Tables S21–S29). Finally, by evaluating the contribution of cell number and cell size to the leaf area, we determined that both contribute to leaf size, but to different degrees (Fig. 5J). In general, it appears that changes in the gene regulatory network related to KUA1/MPYH and TOR due to dynamic changes in 3D chromatin structure and to a greater extent promote cell proliferation and cell expansion thus promoting leaf size in the superior heterosis hybrids.
Discussion
The mechanism of heterosis is an intriguing issue in the field of plant biology. To date, most studies of heterosis have focused on the differences in traits due to differences in DNA sequence, leading to the classic hypotheses. However, classic models ignore the role of chromatin in the 3D space and focus more on differences caused by changes in DNA sequence in one dimension. Thus, how dynamic 3D genome architecture affects hybrid heterosis remains elusive.
Different compartments between parents were enriched in parental genetic diversity areas
The genetic variance among parents strongly affects the offspring’s growth. In agricultural breeding, offspring are usually selected based on the large genetic differences between parents [43], [51]. Here, we found that the chromatin compartment correlates with genetic variance among parents. Recent studies have combined WGS and 3D genome analysis to detect the relationship of SVs and chromatin structure to the regulation of gene expression in humans [52]. By disrupting higher-order chromatin organization, SVs have the potential to alter the gene dosage, thereby regulating gene expression [52]. In particular, we believe that not all genetic loci contribute to heterosis. A weak positive correlation was previously detected between parental SNP density and biomass, indicating that there are still some limitations in the process of heterosis [53].
Compared to model organisms such as humans and mice, analysis of 3D chromatin structure in plants, especially higher plants, is still at a preliminary stage. Detecting chromatin interactions at the gene level in higher plants is still quite difficult [10]. Therefore, based on the current data and the current knowledge of the 3D chromatin structure of polyploid plants, we only determined that genetic variants such as SNPs and SVs are associated with the dynamic changes in 3D chromatin structure in F1 hybrids, but we could not determine which specific variants cause differences in their 3D chromatin structures. More in-depth research on this topic is needed in the future.
Chromatin compartments influence expression of ELD genes and plant-hormone-related genes
A/B compartment switching is closely related to gene expression: the B-to-A switch tends to lead to the upregulation of genes, and the A-to-B switch tends to lead to their downregulation, which has been found in rice and cotton [8], [54]. We found that dynamic 3D chromatin architecture contributes to ELD gene expression. F1 hybrids with superior heterosis harbored a larger proportion of highly expressed ELD genes, whereas lower expression levels of ELD genes existed in inferior heterosis F1. High-parental-ELD genes might play a vital role in B. napus hybrids [46]. In addition, dynamic 3D chromatin structure contributes to plant-hormone-related gene expression of the offspring, which is associated with plant growth. The hormone levels in the plants varied, leading to enhanced cell proliferation and cell expansion in hybrids with superior heterosis and thus enhanced leaf growth (Fig. 5). Similar findings have also suggested that early establishment of auxin biosynthesis, promoting cell proliferation and cell expansion to plant development in early biomass heterosis in B. napus (canola) hybrids [55], but we provided the evidence from dynamic 3D chromatin structure perspective. In addition to the growth-related plant hormones examined in this study, other hormones, such as ABA, SA and ETH, also affect the cell cycle [16], [40], [41].
However, we believe that not all changes of dynamic 3D chromatin structure contribute to heterosis, and we focus attention on the beneficial parts. The phenomena presented by the 3D chromatin structure changes of B. napus in different periods may also be different, but our study mainly observes the seedling stage. In addition, the contribution of the parental 3D structure to F1 hybrids is difficult to judge and will require further investigation.
The underlying mechanism of heterosis is caused by many factors
Heterosis is a complicated process. Here, we examined the possible impact of 3D chromatin structure on this process. Besides those suggested by our findings, other factors may also lead to heterosis in the offspring, such as photosynthesis, photorespiration, plant hormone transport, gene interactions and so on [56], [57], [58]. We also verified the changes in expression of genes related to photosynthesis and photorespiration in F1 hybrids (Fig. S24A), suggesting that the modes of photosynthesis of superior and inferior heterosis sets may be different. Some offspring accumulate organic matter by reducing photorespiration, whereas others do so by increasing photosynthesis, but the specific mechanism remains to be investigated (Fig. S24B). The specific period of plant growth could also affect the underlying mechanism of heterosis. Above all, the complex process of heterosis is based on multiple factors.
Conclusions
In summary, we found chromatin compartments correlated with parental genetic variance areas. F1 with superior heterosis tend to contain more active A compartments, which contributes to ELD genes and hormone genes highly expressed, thereby promoting leaf size. Our findings enhance the understanding of the underlying mechanism of heterosis from a 3D chromatin structure perspective and provide new insights for exploring the mechanism of plant heterosis.
Ethical statement
This article does not contain any studies with human or animal subjects.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
This work was funded by the National Key Research and Development Program of China (2021YFF1000100), Hubei Provincial Science and Technology Major Project (2021ABA011), National Natural Science Foundation of China (32070559), Hubei Provincial Natural Science Foundation of China (2019CFA014), and the Fundamental Research Funds for the Central University HZAU (2662018PY068).
Author contributions
Yue Hu: analyzed the RNA-seq, Hi-C, WGS data and wrote the manuscript draft. Jie Xiong: contributions to experiments of RNA-seq, Hi-C, WGS, qRT-PCR, cytological experiment and plant hormone measurements, and manuscript drafting. Nesma Shalby: contributions to experiments of RNA-seq, Hi-C. Chenjian Zhuo: organized phenotype photos. Yupeng Jia: contributions to figure preparation, and manuscript preparation. Jinxing Tu and Qing-Yong Yang: conceived and designed the project and revised the manuscript.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.01.001.
Contributor Information
Qing-Yong Yang, Email: yqy@mail.hzau.edu.cn.
Jinxing Tu, Email: tujx@mail.hzau.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Li L., Lu K., Chen Z., Mu T., Hu Z., Li X. Dominance, overdominance and epistasis condition the heterosis in two heterotic rice hybrids. Genetics. 2008;180(3):1725–1742. doi: 10.1534/genetics.108.091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnable P.S., Springer N.M. Progress toward understanding heterosis in crop plants. Annu Rev Plant Biol. 2013;64(1):71–88. doi: 10.1146/annurev-arplant-042110-103827. [DOI] [PubMed] [Google Scholar]
- 3.Liu J., Li M., Zhang Q.i., Wei X., Huang X. Exploring the molecular basis of heterosis for plant breeding. J Integr Plant Biol. 2020;62(3):287–298. doi: 10.1111/jipb.12804. [DOI] [PubMed] [Google Scholar]
- 4.Huang X., Yang S., Gong J., Zhao Y., Feng Q.i., Gong H., et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat Commun. 2015;6(1) doi: 10.1038/ncomms7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X., Yang S., Gong J., Zhao Q., Feng Q.i., Zhan Q., et al. Genomic architecture of heterosis for yield traits in rice. Nature. 2016;537(7622):629–633. doi: 10.1038/nature19760. [DOI] [PubMed] [Google Scholar]
- 6.Pei L., Li G., Lindsey K., Zhang X., Wang M. Plant 3D genomics: the exploration and application of chromatin organization. New Phytol. 2021;230(5):1772–1786. doi: 10.1111/nph.17262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doğan E.S., Liu C. Three-dimensional chromatin packing and positioning of plant genomes. Nat Plants. 2018;4(8):521–529. doi: 10.1038/s41477-018-0199-5. [DOI] [PubMed] [Google Scholar]
- 8.Wang M., Wang P., Lin M., Ye Z., Li G., Tu L., et al. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat Plants. 2018;4(2):90–97. doi: 10.1038/s41477-017-0096-3. [DOI] [PubMed] [Google Scholar]
- 9.Oudelaar A.M., Higgs D.R. The relationship between genome structure and function. Nat Rev Genet. 2021;22(3):154–168. doi: 10.1038/s41576-020-00303-x. [DOI] [PubMed] [Google Scholar]
- 10.Sun Y., Dong L., Zhang Y., Lin D.a., Xu W., Ke C., et al. 3D genome architecture coordinates trans and cis regulation of differentially expressed ear and tassel genes in maize. Genome Biol. 2020;21(1) doi: 10.1186/s13059-020-02063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong P., Tu X., Chu P.-Y., Lü P., Zhu N., Grierson D., et al. 3D chromatin architecture of large plant genomes determined by local A/B compartments. Mol Plant. 2017;10(12):1497–1509. doi: 10.1016/j.molp.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W., Hu B.o., Becker C., Doğan E.S., Berendzen K.W., Weigel D., et al. Altered chromatin compaction and histone methylation drive non-additive gene expression in an interspecific Arabidopsis hybrid. Genome Biol. 2017;18(1) doi: 10.1186/s13059-017-1281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C., Cheng Y.-J., Wang J.-W., Weigel D. Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat Plants. 2017;3(9):742–748. doi: 10.1038/s41477-017-0005-9. [DOI] [PubMed] [Google Scholar]
- 14.Du F., Guan C., Jiao Y. Molecular mechanisms of leaf morphogenesis. Mol Plant. 2018;11(9):1117–1134. doi: 10.1016/j.molp.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Groszmann M., Gonzalez-Bayon R., Greaves I.K., Wang L., Huen A.K., Peacock W.J., et al. Intraspecific Arabidopsis hybrids show different patterns of heterosis despite the close relatedness of the parental genomes. Plant Physiol. 2014;166(1):265–280. doi: 10.1104/pp.114.243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vercruysse J., Baekelandt A., Gonzalez N., Inzé D. Molecular networks regulating cell division during Arabidopsis leaf growth. J Exp Bot. 2020;71(8):2365–2378. doi: 10.1093/jxb/erz522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimotohno A., Aki S.S., Takahashi N., Umeda M. Regulation of the plant cell cycle in response to hormones and the environment. Annu Rev Plant Biol. 2021;72(1):273–296. doi: 10.1146/annurev-arplant-080720-103739. [DOI] [PubMed] [Google Scholar]
- 18.Yoo M.-J., Szadkowski E., Wendel J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity. 2013;110(2):171–180. doi: 10.1038/hdy.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M., Wang R., Wu X., Wang J. Homoeolog expression bias and expression level dominance (ELD) in four tissues of natural allotetraploid Brassica napus. BMC Genomics. 2020;21(1):330. doi: 10.1186/s12864-020-6747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bu S.H., Xinwang Z., Yi C., Wen J., Jinxing T.u., Zhang Y.M., et al. Interacted QTL mapping in partial NCII design provides evidences for breeding by design. PLoS ONE. 2015;10(3):e0121034. doi: 10.1371/journal.pone.0121034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao X., Li B., Zhang K.a., Hu K., Yi B., Wen J., et al. Breeding signature of combining ability improvement revealed by a genomic variation map from recurrent selection population in Brassica napus. Sci Rep. 2016;6(1) doi: 10.1038/srep29553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tocquin P., Corbesier L., Havelange A., Pieltain A., Kurtem E., Bernier G., et al. A novel high efficiency, low maintenance, hydroponic system for synchronous growth and flowering of Arabidopsis thaliana. BMC Plant Biol. 2003;3(1):2. doi: 10.1186/1471-2229-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Berkum N.L., Lieberman-Aiden E., Williams L., Imakaev M., Gnirke A., Mirny L.A., et al. Hi-C: a method to study the three-dimensional architecture of genomes. JoVE-J Vis Exp. 2010;(39) doi: 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song J.-M., Guan Z., Hu J., Guo C., Yang Z., Wang S., et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat Plants. 2020;6(1):34–45. doi: 10.1038/s41477-019-0577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durand N.C., Shamim M.S., Machol I., Rao S.S.P., Huntley M.H., Lander E.S., et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3(1):95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9(10):999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie T., Zhang F.-G., Zhang H.-Y., Wang X.-T., Hu J.-H., Wu X.-M. Biased gene retention during diploidization in Brassica linked to three-dimensional genome organization. Nat Plants. 2019;5(8):822–832. doi: 10.1038/s41477-019-0479-8. [DOI] [PubMed] [Google Scholar]
- 28.Heinz S., Texari L., Hayes M.G.B., Urbanowski M., Chang M.W., Givarkes N., et al. Transcription elongation can affect genome 3D structure. Cell. 2018;174(6):1522–1536.e22. doi: 10.1016/j.cell.2018.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ChomczzDski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15(532–4):6–7. [PubMed] [Google Scholar]
- 30.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y.-J., Gao S.-Q., Tang Y.-M., Gong J., Zhang X., Wang Y.-b., et al. Transcriptome analysis of wheat seedling and spike tissues in the hybrid Jingmai 8 uncovered genes involved in heterosis. Planta. 2018;247(6):1307–1321. doi: 10.1007/s00425-018-2848-3. [DOI] [PubMed] [Google Scholar]
- 34.Kendig K.I., Baheti S., Bockol M.A., Drucker T.M., Hart S.N., Heldenbrand J.R., et al. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front Genet. 2019;10 doi: 10.3389/fgene.2019.00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S., Krusche P., Dolzhenko E., Sherman R.M., Petrovski R., Schlesinger F., et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019;20(1) doi: 10.1186/s13059-019-1909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W., Gong L., Guo Z., Wang W., Zhang H., Liu X., et al. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol Plant. 2013;6(6):1769–1780. doi: 10.1093/mp/sst080. [DOI] [PubMed] [Google Scholar]
- 39.Chen W., Gao Y., Xie W., Gong L., Lu K., Wang W., et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 2014;46(7):714–721. doi: 10.1038/ng.3007. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez C. The Arabidopsis cell division cycle. The Arabidopsis Book. 2009;7:e0120. doi: 10.1199/tab.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.del Pozo J.C., Lopez-Matas M.A., Ramirez-Parra E., Gutierrez C. Hormonal control of the plant cell cycle. Physiol Plantarum. 2005;123(2):173–183. [Google Scholar]
- 42.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 43.Hegstad J.M., Nelson R.L., Renny-Byfield S., Feng L., Chaky J.M. Introgression of novel genetic diversity to improve soybean yield. Theor Appl Genet. 2019;132(9):2541–2552. doi: 10.1007/s00122-019-03369-2. [DOI] [PubMed] [Google Scholar]
- 44.Geng X., Qu Y., Jia Y., He S., Pan Z., Wang L., et al. Assessment of heterosis based on parental genetic distance estimated with SSR and SNP markers in upland cotton (Gossypium hirsutum L.) BMC Genomics. 2021;22(1) doi: 10.1186/s12864-021-07431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu D., Gu X., Zhang S., Dong S., Miao H., Gebretsadik K., et al. Molecular basis of heterosis and related breeding strategies reveal its importance in vegetable breeding. Hortic Res-England. 2021;8(1) doi: 10.1038/s41438-021-00552-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen Y., Sun S., Hua S., Shen E., Ye C.-Y., Cai D., et al. Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 2017;91(5):874–893. doi: 10.1111/tpj.13605. [DOI] [PubMed] [Google Scholar]
- 47.Kwon Y., Kim J.H., Nguyen H.N., Jikumaru Y., Kamiya Y., Hong S.-W., et al. A novel Arabidopsis MYB-like transcription factor, MYBH, regulates hypocotyl elongation by enhancing auxin accumulation. J Exp Bot. 2013;64(12):3911–3922. doi: 10.1093/jxb/ert223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X., Cai W., Liu Y., Li H., Fu L., Liu Z., et al. Differential TOR activation and cell proliferation in Arabidopsis root and shoot apexes. Proc Natl Acad Sci USA. 2017;114(10):2765–2770. doi: 10.1073/pnas.1618782114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu D., Wang T., Persson S., Mueller-Roeber B., Schippers J.H.M. Transcriptional control of ROS homeostasis by KUODA1 regulates cell expansion during leaf development. Nat Commun. 2014;5(1):3767. doi: 10.1038/ncomms4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Vleesschauwer D., Filipe O., Hoffman G., Seifi H.S., Haeck A., Canlas P., et al. Target of rapamycin signaling orchestrates growth-defense trade-offs in plants. New Phytol. 2018;217(1):305–319. doi: 10.1111/nph.14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hochholdinger F., Baldauf J.A. Heterosis in plants. Curr Biol. 2018;28(18):R1089–R1092. doi: 10.1016/j.cub.2018.06.041. [DOI] [PubMed] [Google Scholar]
- 52.Spielmann M., Lupiáñez D.G., Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018;19(7):453–467. doi: 10.1038/s41576-018-0007-0. [DOI] [PubMed] [Google Scholar]
- 53.Yang M., Wang X., Ren D., Huang H., Xu M., He G., et al. Genomic architecture of biomass heterosis in Arabidopsis. Proc Natl Acad Sci USA. 2017;114(30):8101–8106. doi: 10.1073/pnas.1705423114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao L., Wang S., Cao Z., Ouyang W., Zhang Q., Xie L., et al. Chromatin loops associated with active genes and heterochromatin shape rice genome architecture for transcriptional regulation. Nat Commun. 2019;10(1) doi: 10.1038/s41467-019-11535-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu A., Wang A., Zhang Y., Dennis E.S., Peacock W.J., Greaves A.I.K. Early establishment of photosynthesis and auxin biosynthesis plays a key role in early biomass heterosis in Brassica napus (canola) hybrids. Plant Cell Physiol. 2020;61(6):1134–1143. doi: 10.1093/pcp/pcaa038. [DOI] [PubMed] [Google Scholar]
- 56.Chen Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat Rev Genet. 2013;14(7):471–482. doi: 10.1038/nrg3503. [DOI] [PubMed] [Google Scholar]
- 57.Huot B., Yao J., Montgomery B.L., He S.Y. Growth-defense tradeoffs in plants: a balancing act to optimize fitness. Mol Plant. 2014;7(8):1267–1287. doi: 10.1093/mp/ssu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinoshita A., Vayssières A., Richter R., Sang Q., Roggen A., Van Driel A.D., et al. Regulation of shoot meristem shape by photoperiodic signaling and phytohormones during floral induction of Arabidopsis. eLife. 2020;9:e60661. doi: 10.7554/eLife.60661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated for this study have been submitted to the NCBI Sequence Read Archive under accession number PRJNA700832, PRJNA701132, and PRJNA701133.