

Graphical abstract
Keywords: Salvia castanea, Plateau adaptation, Underground stem development, Evolution, Secondary metabolites
Highlights
-
•
Salvia castanea evolved the special underground stem with “trunk-branches” developmental pattern.
-
•
Significantly expanded and contracted OGGs, SSGs, and PSGs contribute to morphogenesis and QTP adaptation of S. castanea.
-
•
Tomenta of leaves and periderm of underground stem are like armors protecting S. castanea against QTP environment.
-
•
Secondary metabolites mainly RA and T-IIA realize the functions of armors.
-
•
NAC29 and TGA22 were identified as key TFs involved in regulating T-IIA and RA biosynthesis.
Abstract
Introduction
Salvia castanea, a wild plant species is adapted to extreme Qinghai-Tibetan plateau (QTP) environments. It is also used for medicinal purposes due to high ingredient of tanshinone IIA (T-IIA). Despite its importance to Chinese medicinal industry, the mechanisms associated with secondary metabolites accumulation (i.e. T-IIA and rosmarinic acid (RA)) in this species have not been characterized. Also, the role of special underground tissues in QTP adaptation of S. castanea is still unknown.
Objectives
We explored the phenomenon of periderm-like structure in underground stem center of S. castanea with an aim to unravel the molecular evolutionary mechanisms of QTP adaptation in this species.
Methods
Morphologic observation and full-length transcriptome of S. castanea plants were conducted. Comparative genomic analyses of S. castanea with other 14 representative species were used to reveal its phylogenetic position and molecular evolutionary mechanisms. RNA-seq and WGCNA analyses were applied to understand the mechanisms of high accumulations of T-IIA and RA in S. castanea tissues.
Results
Based on anatomical observations, we proposed a “trunk-branches” developmental model to explain periderm-like structure in the center of underground stem of S. castanea. Our study suggested that S. castanea branched off from cultivated Danshen around 16 million years ago. During the evolutionary process, significantly expanded orthologous gene groups, 24 species-specific and 64 positively selected genes contributed to morphogenesis and QTP adaptation in S. castanea. RNA-seq and WGCNA analyses unraveled underlying mechanisms of high accumulations of T-IIA and RA in S. castanea and identified NAC29 and TGA22 as key transcription factors.
Conclusion
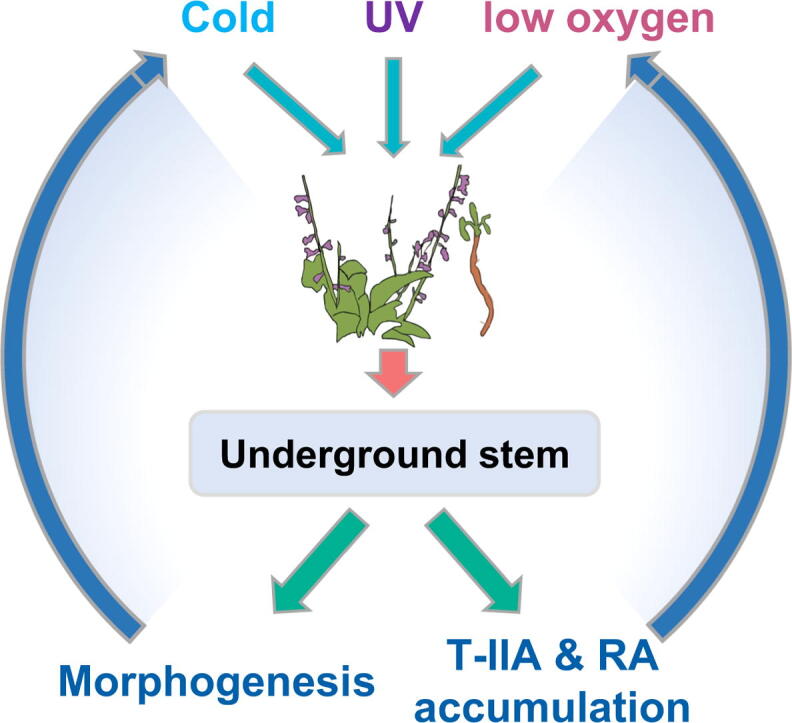
We proposed a “trunk-branches” developmental model for the underground stem in S. castanea. Adaptations to extreme QTP environment in S. castanea are associated with accumulations of high secondary metabolites in this species.
Introduction
The Qinghai-Tibet Plateau (QTP) of China is the highest plateau in the world with an elevation of about 3,000–5,000 m [1], [2], [3]. This region is characterized by extreme climatic conditions, including intense ultraviolet (UV) radiation, low oxygen and low temperature. Salvia castanea (Lamiaceae) is a wild plant species, which primarily grows in QTP regions with no reported artificial cultivation [4], [5]. Although the evolution of Salvia genus has been studied to some extent [6], development of S. castanea and its adaptations to extreme QTP environment have not been reported. Another species in this genus, S. miltiorrhiza (Danshen) is widely cultivated for extracting the active ingredient of tanshinone IIA (T-IIA) from its root tissues [7]. Significantly higher levels of T-IIA and rosmarinic acid (RA) in wild S. castanea make it attractive to medicinal industry [8]. However, accumulation mechanisms of high secondary metabolites (mainly T-IIA and RA) in S. castanea are yet to be explored.
Plant secondary metabolites (PSMs) can be especially affected by environmental factors [9]. Drought could increase the accumulation of secondary metabolites (salvianolic acid B and T-IIA) in S. miltiorrhiza [10]. Dynamic changes in plant secondary metabolites during UV acclimation were reported in Arabidopsis thaliana [11]. Temperature could change the contents of various PSMs [12], as revealed that low-temperature stress could trigger the secondary metabolic pathways of phenylpropanoid and anthocyanin in maize seedlings [13]. Therefore, PSMs could exert long-term effects on plant growth and survival under stressful environments [14]. Plant secondary metabolism is the result of plant adaptation to ecological environment in long-term evolution, and PSMs provide material basis for plants to against adverse environment [15], [16]. In this study, our focused species S. castanea accumulates two kinds of PSMs (Tanshinones and RA), which have relatively clear functions. Tanshinones e.g. dihydrotanshinone I (DT-I), cryptotanshinone (CT), T-I and T-IIA, are lipid-soluble diterpenoid quinone compounds. Plants synthesize these compounds via mevalonate (MVA) pathway in cytoplasm and via 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway in plastids [17], [18]. Tanshinone IIA is a pharmacologically important constituent, which shows anti-hypoxic effect for altitude sickness [19], and prevents myocardial ischemia injury in human [20]. T-IIA also protects cells from oxidative damage by regulating reactive oxygen species (ROS) [21]. Similarly, RA is a natural antioxidant [22], which inhibits lipid peroxidation in situ [23] and protects cells from ionizing radiations [24]. Due to anti-hypoxic, -inflammatory properties of T-IIA and -oxidant character of RA, we propose that S. castanea adapts to extreme QTP environments through high accumulations of these secondary metabolites.
As a main T-IIA synthesis site, it is interesting to understand morphogenesis of underground tissues in S. castanea. Special environment could affect the species-specific morphogenesis such as aerial roots of Ficus microcarpa in tropics or subtropics [25], adventitious roots of lotus in water [26], underground stem of Welwitschia mirabilis in Africa desert [27]. The underlying mechanisms of these specific morphogenesis are complex due to the interactions between environment and endogenous factors. In recent decades, there were some important progresses regarding to plant morphogenesis. Continuous organ initiation and outgrowth in plants rely on proliferation and differentiation of stem cells maintained by CLAVATA (CLV)-WUSCHEL (WUS) negative-feedback loop [28], [29], [30]. Leucine-rich repeat receptor-like protein kinases (LRR-RLKs), including CLV1, RPK2, CLV2 and CRN could be activated by CLV3 signal to repress WUS expression [31], [32], [33]. WUS gene in reverse regulates the expression of CLV3 to form the important CLV-WUS negative-feedback loop and to regulate plant morphogenesis. Further, phytohormones also play crucial roles on plant morphogenesis. For instance, auxin is identified as a trigger for change in plant development [34]. Auxin-dependent pathway is reported to be associated with aerial root initiation, growth, and pattern formation of F. microcarpa [25]. Auxin is regulated by other phytohormones such as ethylene, strigolactone to form complex signaling pathways and realize various plant morphogenesis [35], [36]. Therefore, environment could affect plant morphogenesis by regulating differentiation of stem cells and distribution of phytohormones.
To date, plateau adaptation of S. castanea and high accumulation mechanisms of secondary metabolites (mainly T-IIA and RA) have not been explored. In this study, we used comparative genomics, transcriptomic and weighted gene co-expression network analysis (WGCNA) analyses to understand plateau adaptation of S. castanea from the following aspects, (I) special morphogenesis, (II) species evolution, and (III) high accumulation mechanisms of secondary metabolites (mainly T-IIA and RA). These findings will improve our understandings of plant evolution and survival strategy in response to extreme environment, and provide important candidate genes for genetic improvement of cultivated Chinese medicinal plant Danshen.
Materials and methods
Plant materials
The wild samples of annual and perennial S. castanea were collected in November 2020, from Jade Dragon Snow (Yulong) Mountain region (100°15′E, 27°9′N, 3052 m) in Lijiang city, Yunnan Province, China (Fig. 1a). Meteorological data from 1979 to 2020 were obtained from http://www.wheata.cn in Yulong Naxi autonomous county (100o25′E, 27o25′N), location close to the sampling site. For the morphological study, root cross and vertical sections were prepared within 1 mm of thickness, and soaked in 70% formaldehyde-acetic acid-ethanol fixative (FAA) for paraffin slicing. Software Caseviewer 2.3 was used for the observation of paraffin section. For the analyses of species evolution and high secondary metabolites accumulation, samples were divided into different tissues (root, stem, leaf and petiole), which were immediately immersed into liquid nitrogen for further PacBio SMRT sequencing analysis. The leaf, periderm, phloem and xylem tissues from both annual and perennial plants, total eight tissues were collected for the next-generation sequencing and active ingredient analyses with three biological replicates. These tissue samples were termed as: annual leaf (A-leaf), annual periderm (A-periderm), annual phloem (A-phloem), annual xylem (A-xylem), perennial leaf (P-leaf), perennial periderm (P-periderm), perennial phloem (P-phloem), and perennial xylem (P-xylem). Annual S. castanea is the plant germinated from the seed. Perennial S. castanea is the plant sprung up from the previous root because S. castanea withers in winter, leaving the roots alive in the ground.
Fig. 1.

Geographic information and morphological characteristics of Salvia castanea. (a) Geographical location of our experimental sample, S. castanea, from Jade Dragon Snow (Yulong) Mountain (100o15′E, 27o9′N, 3,052 m), Lijiang city, Yunnan Province, China. The map was downloaded from the website of http://bzdt.ch.mnr.gov.cn. (b) Morphological characteristics of S. castanea. (c) Features of upper and lower surfaces of S. castanea leaves by microscope.
PacBio and illumina sequencing and data analysis
Total RNAs from four different tissues (root, stem, leaf, and petiole) were extracted using RNAprep Pure Plant Kit DP441 (TIANGEN, Beijing, China) following the manufacturer's procedure. The PacBio single-molecule real-time libraries were constructed in Personal Biotechnology Co., Ltd. Shanghai, China (PacBio Sequel II platform, USA) according to Yue et al. [37]. The longest transcript was selected as unigene after the iterative isoform-clustering (ICE) and CD-HIT software removing redundant sequences. Functional annotations of unigenes were performed in six public databases (NR, Pfam, Eggnog, Swissprot, GO and KEGG). The genes were mainly named according to the annotation of Swissprot database. For more than one gene from S. castanea identified as the homologues of same known gene deposited in Swissprot database, we use ‘1’, ‘2’ and ‘3’ or ‘a’, ‘b’ and ‘c’ to number them. The classification of TF families was defined following the Plant Transcription Factor Database.
RNA sequencing was performed on an Illumina HiSeq 2500 platform (USA), and 150 bp paired-end reads were generated. The raw transcriptome sequencing data have been deposited in the NCBI under the accession number PRJNA800258. After quality control, the clean reads were mapped to a full-length transcriptome using Bowtie2 [38]. To estimate unigene expression levels, the number of mapped clean reads for each gene was determined and normalized for calculating fragments per kilobase transcript length per million fragments mapped (FPKM) value using RNA-Seq with Expectation-Maximization (RSEM) (v1.2.15) [39]. Detailed information of RNA sequencing and mapping is summarized in Tables S1-2. Gene expression profiles for biological replicates were assessed using principal component analysis (PCA) (Fig. S1). Fold change (FC) and binomial tests were used to detect differentially expressed unigenes (DEGs) between samples. DESeq was employed to calculate the false discovery rate (FDR). In this study, P ≤ 0.05 and |log2 FC| ≥ 1 were set as the thresholds to identify the DEGs. We used a hypergeometric test with an FDR correction method using topGO and KEGG pathway for enrichment analysis.
Weighted gene co-expression network analysis (WGCNA) was performed using WGCNA (V1.69) package in R (V3.6.1) [40]. All genes were hierarchically clustered using topological overlap-based dissimilarity measure and a gene dendrogram was generated based on Topological Overlap Matrix (TOM). Gene expression profile of each module identified by gene dendrogram was calculated to test the association with each plant effective component. In co-expression network, the edge weight (ranging from 0 to 1) of any two genes connected was determined based on their topology overlap measure. The networks were visualized using Cytoscape (v.3.8.2).
Phylogenetic tree reconstruction and divergence time estimation
The evolution of S. castanea was analyzed by identifying homologous genes from 15 presentative plant species, including 3 Lamiales, 3 Monocotyledons, 7 Dicotyledons and 2 outgroup speices (Table S3). Unigenes, predicted as protein-coding genes by OrfPredictor [41], were used for further analysis. Predicted polypeptides and proteomes of the above 15 species were clustered into orthologous gene groups (OGGs) using OrthoFinder (v2.2.7) [42]. For species tree reconstruction, we selected those one-to-one ortholog genes among the 15 species. MAFFT (v7.1) [43] was used for multiple alignment of protein sequences of each single-copy homologous gene. Low-quality single-copy regions were removed by TrimAI (v 1.4) [44]. A phylogenetic tree was constructed by using PROTGAMMAJTT model with RAxML (v 8.2) [45]. Maximum likelihood (ML) method was used all the sites for bootstrap analysis with 1,000 replicates for the reliability of interior branches. We set Amborella trichopoda as an outgroup. Internal node time interval of different species was calibrated according to the Timetree database (http://www.timetree.org/) [46], and Bayesian method was used to estimate the divergence time of species by using mcmctree implemented in PAML (v4.9) [47]. CAFÉ (v3.1) [48] was used to accurately estimate the expansion and contraction of OGGs. We further conducted a positive selection analysis for the one-to-one OGGs according to Li et al. [49].
HPLC analysis of active ingredients
Eight tissues including leaf, periderm, phloem and xylem from annual and perennial S. castanea were separately analyzed for active ingredients contents (i.e. T-IIA, T-I, DT-I, CT, RA and salvianolic acid B (SAB)). The samples were dried at 60 °C to constant weight, 20 mg ground powder (passed through 60-mesh sieve) and then mixed with 70% methanol for ultrasonic pretreatment for 1 h, and centrifuged at 8000 rpm min−1 for 10 min. The supernatants were aspirated with a sterile disposable syringe and filtered by 0.22 μm filter membrane for the following HPLC analysis with three biological replicates. Tanshinones and phenoic acids were detected at wavelengths of 270 nm and 288 nm, respectively using waters HPLC system (Milford, MA, USA) according to the methods of Yu et al. [7].
qRT-PCR analysis
The RNAs of S. castanea tissues were returned from RNA-seq experiment. The cDNA was reverse transcribed using TaKaRa PrimeScriptTM RT Master Mix reagent Kit (Perfect Real Time RR036A). The primers used for qRT-PCR were listed in Table S4. The actin gene was selected as a reference. PCRs were conducted using TB Green ® Premix Ex TaqTM II (Tli RNaseH Plus RR420A) (TaKaRa) in QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher) using the following protocol: 95 °C for 30 s, 1 cycle; 95 °C for 5 s and 58 °C for 30 s, 40 cycles according to the method of Xu et al. [50]. The 2−ΔΔCt method with three biological replications was performed for analysis [51].
Statistical analysis
Data for HPLC and qRT-PCR were analyzed using SPSS 22.0 (SPSS, Chicago, IL, USA), fisher least significant difference (LSD) test, two-way analysis of variance (ANOVA), and Duncan's multiple range test were used to determine the significant differences (P < 0.05) between individuals. The results were presented as mean ± standard error (SE).
Results
Morphology and special underground stem developmental pattern
S. castanea species used in this study grew in Yulong Mountain region (Fig. 1a). Meteorological data nearby the sampling site from 1979 to 2020 presented in Table S5. It could be summarized as hypoxia, intense UV light and cold. The oblong ovoid leaves of this species were basal grown, and densely tomentose in the upper and lower surfaces (Fig. 1b, c). During flowering season from July to October, quadrangular stem would bolt from basal leaves and blossom chestnut verticillasters (Fig. 1b). Interestingly, upper lip of flower was also densely villous. All these special characteristics contributed greatly to the adaptation of plateau environment.
Interestingly, underground morphology of this species was special, as multi-branches of tree shrank and buried in the ground. Based on morphological appearance, it was hard to distinguish underground stem from main root (Fig. 2a). However, differentiation of underground stem from bottom up was clearly visible from the cross section of the annual root. The central part of underground stem gradually split to generate periderm-like tissues to surround new generated tissues, leaving the hollow structure in the middle of underground stem. During the development, central part radially separates into several symmetric small “branches” with independent xylem structures (Fig. 2a). The periderm might act as an outer coat or armor closely surrounding the new generated branch-like structures, and it regenerated continuously with the developing underground stem. Paraffin cross-sections unraveled more exquisite numerous branch-like structures, with the differentiation of underground stem from bottom to up (Fig. 2c-h).
Fig. 2.

The underground stem morphological characteristics and developmental progress of Salvia castanea. (a) Annual underground stem of S. castanea, and its different cross-sections from down to up.Fig. 2c-h presented more exquisite paraffin cross-sections, revealing the gradual differentiation to form branches-like structures i.e. 3 branches, 4 branches, 5 branches, 6 branches and so on with the growth of underground stem. Fig. 2i-j were the vertical sections of underground stem, especially Fig. 2j uncovered the bifurcating process of xylem to generate multiple vessel tissues. The core of underground stem was like the results of programmed cell death, and generated the periderm-like structures in the center of underground stem to protect the new generated branches-like structures. (b) Perennial underground stem of S. castanea and cross-sections from down to up.Fig. 2k-l were the paraffin cross-sections of perennial underground stem of S. castanea, with the multiple xylem structures squeezed together. Fig. 2m was the vertical section of S. castanea, presenting multiple xylem vessels with more periderm-like structures in underground stem. Fig. 2n was the paraffin vertical section of perennial underground stem in S. castanea.
Therefore, we proposed a “trunk-branches” developmental model in the underground stem of this species. According to this model, we could easily explain the bifurcating phenomena in Fig. 2e-h, which presented first 3 bifurcations, then it continues to regenerate 4 bifurcations, 5 bifurcations, 6 bifurcations and so on as the underground stem developed. Fig. 2i-j showed the vertical section of underground stem in S. castanea. Interestingly, Fig. 2j revealed the bifurcating process of xylem, which led to generate multiple branch-like structures in underground stem. This xylem trend in longitudinal section further confirmed our hypothesized “trunk-branches” developmental model in the underground stem of S. castanea. This model could also explain why periderm-like structure generated in the center of underground stem. Indeed, it was the newly generated periderm of small bifurcations squeezed together. Considering the basal leaves and special developmental pattern, we confirmed the upper part of main root was indeed underground stem hidden in the soil. However, this branching phenomenon was not found in the roots of cultivated Danshen.
Differentiation of perennial underground stems was more complex than the annuals (Fig. 2b). In perennial underground stems, these branch-like tissues twisted and intertwined to form the perennial underground stem structures, generating more periderm-like tissues in the inner of underground stem in S. castanea (Fig. 2b). The underground stem paraffin cross-sections of perennial S. castanea, showed multiple xylem structures were squeezed together (Fig. 2k-l). However, the outmost periderm was not completely split, and it appeared to be as squeezed branches of a tree without spatial distribution. Vertical section of underground stem in perennial S. castanea (Fig. 2m), and paraffin vertical section (Fig. 2n) further confirmed presence of multiple and complex xylem vessels. Thus, the underground stem of S. castanea is like multiple branches of a tree shrunk, squeezed together and hidden underground.
Phylogenetic relationships and evolution
A total of 40 Gb of clean reads with 18,182 unigenes generated from SMRT sequencing of S. castanea in this study were assessed by BUSCO (Fig. S2a). Most unigenes were distributed in 1,600–1,799 bp, with 45.14% GC and a mean length of 1,678.18 bp (Fig. S2b). Notably, 804 TFs were identified and classified into 52 distinct TF families according to full-length transcriptome of S. castanea (Fig. S2c). The highest number of members was found for bHLH family, followed by ERF and WRKY families (Fig. S2c).
Species-specific genes and phylogenetic tree
Phylogenetic analysis of S. castanea with other 14 representative species was performed to reveal S. castanea evolution (Table S3). Consequently, a total of 17,542 OGGs comprising of 355,262 genes were obtained. Among these, 102,916 genes belonging to 4,555 OGGs were shared by all the 15 species (Fig. S2d). Based on GO enrichment analysis, 8 OGGs including 24 species-specific genes (SSGs) were identified in S. castanea. These SSGs were enriched in QTP adaptation processes such as DNA binding, defense response, programmed cell death (PCD) (Table S6). Especially two PCD genes (NSL1a and NSL1b) might closely related to the formation of bifurcate structure in underground stem of S. castanea to adapt the QTP environment.
Among 4,555 shared OGGs, 212 single-copy OGGs were identified for reconstruction of a species phylogenetic tree (Fig. 3b). It showed that S. castanea was sister to S. miltiorrhiza and branched off from S. miltiorrhiza around 16 million years ago (MYA). Sister lineage of A. paniculata and S. indicum diverged approximately 122 MYA. Three monocotyledons, M. acuminata, O. sativa, and Z. mays, were grouped together, and diverged approximately 387 MYA from the other 10 dicotyledons. A. trichopoda and S. moellendorffii lay at the base of this tree and approximately diverged 1,023 MYA.
Fig. 3.

Salvia castanea phylogeny and its QTP adaptation mechanism from comparative genomic analyses. (a) Positively selected genes closely related to the morphogenesis and QTP adaptation. The genes shown here were mainly named according to the annotation of Swissprot database. (b) Phylogenetic position of S. castanea relative to other 14 representative plant species. The branch lengths of phylogenetic tree were scaled to estimated divergence time, and rhombus nodes indicated the fossil calibration times used for setting the upper and lower bounds of the estimates. Tree topology was supported by posterior probabilities of 1.0 for all nodes. Moreover, 95% credibility intervals were marked to indicate the estimated posterior distributions of divergence time. The numbers of significantly expanded (green) and contracted (red) OGGs were presented on each branch. (c) The number of protein members from significantly expanded OGGs containing ubiquitin, LRR, PPR and some other domains. The domains were identified according to the Pfam database. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Expanded and contracted OGGs
There were 1,013 significantly expanded and 5,750 contracted OGGs identified in S. castanea (Fig. 3b). For the expanded OGGs, KEGG analysis showed that these genes were mainly enriched in spliceosome, ubiquitin mediated proteolysis, cysteine and methionine metabolism pathways (P < 0.05) (Table S7). According to the biological process (BP) category of GO enrichment, the genes were mainly enriched in 33 GO terms regarding development or morphogenesis, especially root system (GO:0022622, P = 9.85 × 10-3) and root development (GO:0048364, P = 9.85 × 10-3). For the molecular function (MF) category, the genes were mainly enriched in GO terms related to QTP adaptation, including 34 genes associated with oxidoreductase activity, 12 with DNA binding, 2 with nucleic acid binding and one with MAP kinase activity (GO:0004707, P = 7.86 × 10-5) (Table S8).
Domain identification revealed that significantly expanded OGGs mainly included ubiquitin, leucine-rich repeat receptor (LRR), pentatricopeptide repeat (PPR) and some other domains (Fig. 3c). There were totally 988 proteins possessing LRR domain, followed by PPR (462 proteins) and ubiquitin (289 proteins). Some other domains included extensin 2 (299 proteins), WD40 (182 proteins), pkinase (117 proteins), and AP2 (42 proteins) (Fig. 3c).
GO analyses for significantly contracted OGGs revealed 37 GO terms were associated with plant development, morphogenesis or histogenesis (Table S9). Noticeably, three SEOB genes were enriched in phloem or xylem histogenesis (GO:0010087, P = 8.99 × 10-3) and phloem development (GO:0010088, P = 8.99 × 10-3). Another gene FPF1 was involved in three GO terms (GO:0048831, GO:0090567 and GO:0048367) regarding shoot system development. Interestingly, following GO terms phloem or xylem histogenesis, phloem development and shoot development could only be observed in contracted, but not in the expanded OGGs. Thus, contracted OGGs might contribute to the specific underground stem architecture in S. castanea.
Positively selected genes
Up to 64 out of 212 single-copy genes were identified as positively selected genes (PSGs) with ω (dn/ds) > 1, and 94 amino acid sites were under positive selection (posterior probability > 0.95) (Table S10). According to annotation, two DNA repair related genes (TYDP1 and SWC4) were PSGs. Ribosome related PSGs (RS6 and RK31), and transporter activity related PSGs (AB7G and AB3F) were involved in the DNA damage response. Moreover, three oxidoreductase activities related PSGs (HBD, DIOX4 and HF101), and three phosphatase activities related PSGs (P2C19, F16P1 and P2C04) were vital in regulating ROS (Fig. 3a, Table S11).
GO annotation showed that PSG PAC was involved in the development of shoot system (GO:0048367), leaf (GO:0048366) and phyllome (GO:0048827). PSG CHLM was involved in chlorophyll processes (GO:0015994, GO:0015995). These results suggested these two PSGs (PAC and CHLM) in S. castanea played vital roles in formation of specific leaves adapted to extreme environments (Fig. 3a, Table S12). As for MF category, 64 PSGs were enriched in ten top GO terms (P < 0.05), including methyltransferase activity (GO:0008168), transferase activity (GO:0016741) and iron-sulfur cluster binding (GO:0051536) etc. (Table S12). KEGG results showed 64 PSGs were mainly enriched in nucleotide excision repair (ko03420), RNA degradation (ko03018) and ribosome (ko03010) (Table S13).
Potential mechanisms of underground stem formation
RNA-seq was conducted to identify the candidate genes regulating underground stem formation in S. castanea. Detected genes could be classified into three categories: plant stem cells related genes (Fig. 4a), phytohormone related genes (Fig. 4b), and branching and root development related genes (Fig. 4c). Auxin was important for plant morphogenesis, and PIN genes were mainly responsible for auxin transportation. In this study, significantly up-regulated PIN1A gene in A-xylem and P-xylem would promote the formation of branch-like structure in underground stem (Fig. 4a). However, some LRR-RLKs genes (BAM1b, BAME2, CLV1) and most of AGL genes were significantly depressed in A-xylem and P-xylem as compared with other tissues. Interestingly, WOX8 gene was stably expressed in all tested tissues (Fig. 4a). Therefore, potential mechanisms of underground stem formation in S. castanea could be concluded as LRR-RLKs, mainly BAM, RPK, CLV and CRN, regulated the stable expression of WOX8, reconstructed its own CLV-WUS negative-feedback loop which was crucial to maintain the stem cell homeostasis (Fig. 4e).
Fig. 4.

Expression profiles of key genes involved in underground stem development and plateau adaptation of Salvia castanea. (a) The different expression genes (DEGs) involved in the auxin transporter PINs, CLAVATA (CLV)-WUSCHEL (WUS) negative-feedback loop and so on in eight tissues of S. castanea. (b) DEGs regarding to three important phytohormones including ethylene, auxin and abscisic acid (ABA). (c) DEGs involved in the strigolactone signal pathway and root development. (d) DEGs regarding cold adaptation and DNA damage repair. (e) Potential mechanisms of underground stem morphogenesis and plateau adaptation of S. castanea. Eight tissues of S. castanea were referring to annual leaf (A-leaf), annual periderm (A-periderm), annual phloem (A-phloem), annual xylem (A-xylem), perennial leaf (P-leaf), perennial periderm (P-periderm), perennial phloem (P-phloem), and perennial xylem (P-xylem). The genes shown here were mainly named according to the annotation of Swissprot database.
Phytohormones such as ethylene, auxin and abscisic acid (ABA) were closely related to the morphogenesis of S. castanea. For example, ethylene biosynthesis related gene ETR2a in A-xylem and P-xylem, and auxin signal related genes AX22D2 in A-leaf, A-xylem, P-leaf and P-xylem were significantly triggered as compared with other tissues. Auxin related gene RAC3 was highly induced in all tested eight samples. ANXD1 gene in ABA signal pathway was induced in all root tissues (Fig. 4b). Strigolactione branching related gene (MAX1) and root development related genes (PMTT and REV1) were highly expressed in A-xylem and P-xylem as compared with other tissues, indicating their crucial functions in regulating underground stem morphogenesis. Moreover, the expressions of DAD2a and DAD2b were significantly enhanced in A-leaf and P-leaf, suggesting their roles in generating multiple basal leaves (Fig. 4c). According to the previous results of Hu et al. (2018) [52], a group of receptor kinases are essential for CLAVATA signalling to maintain stem cell homeostasis. We proposed a complex network to regulate the underground stem morphogenesis in S. castanea as revealed by auxin transport protein PIN, CLV-WUS negative-feedback loop, and phytohormone auxin (Fig. 4e).
Genes involved in QTP adaptation of S. Castanea
Cold adaptation related genes RCI2A1 and RCI2A2 were significantly up-regulated in perennial periderm, phloem and xylem tissues. Cold adaptation in leaf tissues could be regulated by GOLS1, which was significantly triggered in both annual and perennial leaves (Fig. 4d, e). MAPK genes always play vital roles on environment adaptation. Functions of all identified MAPK genes in S. castanea were listed in Table S14. Most of the functions were closely related to QTP adaptation as revealed by resistance to UV, hypoxia, cold and so on. Moreover, most of 47 MAPK genes were significantly induced in all tested tissues of S. castanea (Fig. S3). For instance, MPK3a and MPK3b were highly expressed in all tested 8 tissues, especially in A-leaf and A-xylem. In MAPKK cluster, M2K2a was significantly induced in both annual and perennial leaves as compared to other tissues. However, expression of M2K2b in underground tissues was significantly higher than that of leaves. M2K9 was also significantly induced in all tested tissues, especially in xylem tissues. In MAPKKK cluster, M3K3A was significantly up-regulated in all tested 8 tissues, indicating its important functions in QTP adaptation of S. castanea.
Seven genes related to DNA repair were differentially induced in all tested 8 tissues (Fig. 4d). For instance, DR102 was highly expressed in all tested tissues, especially in A-leaf and P-leaf. However, DNA damage repair related genes DR100a and DR100b were depressed in both A-leaf and P-leaf, implying their vital functions in responding to DNA damage (Fig. 4d, e). Ubiquitin (Ub)-proteasome system (UPS) related genes were highly expressed in most of 49 Ub conjugating enzyme (E2) and 224 Ub ligase (E3) (Fig. S4). Therefore, the aboved results indicated QTP adaptation of S. castanea was regulated by a complex network including MAPK signal transduction, cold adaptation responses, DNA damage repair and ubiquitination process (Fig. 4e).
Genes involved in the biosynthesis of tanshinones
Four tanshinones were mainly distributed in underground tissues (Fig. 5a). Content of each tanshinone was significantly higher in periderm than that in other tissues, no matter in A-periderm or P-periderm. For instance, DT-I, CT, T-I and T-IIA content of A-periderm were 2.97, 2.38, 3.28, 2.07 folds that of A-phloem and A-xylem (sum content of these two tissues), respectively. Each tanshinone content in P-periderm was significantly higher than that in A-periderm except DT-I without significant difference. Interestingly, content of each tanshinone in A-xylem was significantly lower than that in P-xylem, especially for T-IIA which was undetectable in A-xylem tissue. However, the content of each tanshinone in A-phloem was significantly higher than that in P-phloem (Fig. 5a). For example, DT-I, CT, T-I and T-IIA in A-phloem was 2.60, 1.64, 1.77, 1.92 folds that in P-phloem.
Fig. 5.

The mechanisms of high tanshinones accumulation in Salvia castanea. (a) The contents of dihydrotanshinone I (DT-I), cryptotanshinone (CT), tanshinone I (T-I) and T-IIA in eight different tissues including periderm, phloem, xylem and leaf of both annual and perennial S. castanea, respectively. (b) The differential expressions of key genes in MVA and MEP pathways of various tanshinone compounds. (c) Venn diagram regarding to the genes significantly correlated with the contents of CT, DT-I, T-I and T-IIA. (d) Correlation analyses of sixty-three TFs with the contents of four tanshinones. (e) Modules analyses of four tanshinone contents in S. castanea. (f) Networks analyses of genes in the most correlated black module. Data presented in (a) are the means of three replicates (mean ± SE). Values followed by different letters indicated significant differences at P < 0.05 levels.
Majority of genes associated with tanshinone biosynthesis pathways (MVA and MEP) in S. castanea were highly expressed (Fig. 5b). Expressions of 251, 130, 251 and 193 genes were significantly correlated with CT, DT-I, T-I and T-IIA contents, respectively (Fig. 5c). Totally, there were 63 TF genes were identified from 103 shared genes (Fig. 5c,d). WRKY family counted the most member number among all these TFs, all of them were negatively correlated with the contents of four tanshinones. Interestingly, NAC17 and NAC98 had opposite correlations with the contents of four tanshinones.
Totally, 29 distinct modules were identified through correlation analysis with four tanshinones contents by WGCNA analyses (Fig. 5e). Among these modules, black module (454 genes) showed the strongest correlation, followed by darkgreen module (76 genes). Among the black module, five hub genes were associated with tanshinone biosynthesis, including (E)-4-hydroxy-3-methylbut-2-enyl diphosphate synthase, geranylgeranyldiphosphate synthase, cytochrome P450 CYP71D411, 1-deoxy-d-xylulose 5-phosphate reductoisomerase, and cytochrome P450 CYP76AH1. Moreover, five TFs (NAC29, WRK75, HSFB3, MYB36 and AZF2) were identified with top five edge numbers to interact with other genes regulating biosynthesis of tanshinones (Fig. 5f). Additionally, terpenoid backbone biosynthesis pathway was significantly correlated with contents of tanshinones (Table S15), indicating its importance in high accumulation of tanshinones in S. castanea in response to the hypoxia environment of QTP.
Genes involved in the biosynthesis of rosmarinic acid
Rosmarinic acid (RA) contents in annual leaves reached 90.94 mg g−1, which was significantly higher than that of any other tissue (Fig. 6a). Moreover, RA content in each annual tissue was significantly higher than their corresponding perennial tissue. For instance, RA content in A-leaf was 1.76 fold that of P-leaf. The trends of SAB in different tissues were similar to those of RA, except A-xylem, which contained the highest SAB content (5.39 mg g−1) among all the tested tissues. SAB content in perennial leaves was significantly higher than that of each perennial underground tissue (Fig. 6a). For instance, SAB content in P-leaf was 1.29 folds that of P-periderm.
Fig. 6.

The mechanism of high RA accumulation in Salvia castanea. (a) The contents of RA in eight different tissues including periderm, phloem, xylem and leaf of both annual and perennial S. castanea, respectively. Data presented here are the means of three replicates (mean ± SE). Values followed by different letters indicated significant differences at P < 0.05 levels. (b) The differential expressions of key genes in biosynthetic pathway of RA. (c) Phylogenetic relationship of PAL genes in S. castanea and the other 14 species. (d) Venn diagram regarding to the genes significantly correlated with the contents of RA and SAB. (e) Correlation analyses of fifty-five TFs with the accumulation of RA. The genes shown here were mainly named according to the annotation of Swissprot database and the classification of TF families was defined following the Plant Transcription Factor Database.
RNA-seq indicated multiple genes were highly expressed in each step of RA biosynthetic pathway of S. castanea (Fig. 6b). Noticeably, TAT genes were significantly induced both in annual and perennial leaves than in other tissues, which were consistent with the trends of RA contents in different tissues. Moreover, qRT-PCR results for six P450 genes were similar with those in RNA-seq further confirming the validity of transcriptome datasets (Fig. S5).
Phenylalanine ammonia lyase (PAL) is the first enzyme of RA metabolic pathway. To further understand function of PAL family in S. castanea, we conducted a phylogenetic analysis of PAL genes among 15 species (Fig. 6c). Results presented the number of PAL genes in S. castanea were consistent with that in S. miltiorrhiza. All of these PAL genes together with those counterparts from S. indicum and A. paniculata to form two independent clusters (5 and 6) (Fig. 6c). This indicated that the PAL genes of S. miltiorrhiza and S. castanea probably were evolutionarily conserved, and still played a similar role after species divergence.
WGCNA analyses identified 261 genes were significantly correlated with the accumulation of RA, among which 55 genes were TF gene (Fig. 6d,e). However, only 61 genes were significantly correlated to SAB accumulation (Fig. 6d). Further, majority of the identified TFs such as bHLH, bZIP and ERF were positively correlated with RA accumulation (Fig. 6e). Interestingly, NAC17 gene was positively and NAC98 was negatively correlated with RA accumulation. Among the identified TF genes, SRM1 belongs to MYB family and LBD37 belongs to LBD family had the highest positive and negative correlation coefficient, respectively.
Co-expression networks resulted in 32 distinct modules, among which brown module (779 genes) had a significantly positive correlation with both RA and SAB contents (Fig. 7a). Further, in brown module, hub gene with the highest edge number (658 edges) was skp1, which was involved in root growth and auxin response [53]. Among the identified TF hub genes, TGA22, WRK33 and WRK46 were listed to be with top 3, most edge numbers (Fig. 7b). Four other modules were also positively related with RA contents, including turquoise (3,946 genes), black (330 genes), tan (111 genes) and midnightblue (89 genes) modules. Four modules were negatively related with RA contents, including red (556 genes), purple (135 genes), salmon (99 genes) and lightcyan (85 genes) modules. Moreover, two modules yellow (777 genes) and cyan (91 genes) were positively related with SAB contents (Fig. 7a).
Fig. 7.

WGCNA analyses of RA and SAB in Salvia castanea (a) Modules analyses of RA and SAB contents in S. castanea. (b) Networks analyses of genes in brown module which was positively correlated to the contents of both RA and SAB. (c) Networks analyses of genes in turquoise module, which was most positively related to the RA contents and with the most genes of 3,946. (d) Genes in positively correlated turquoise module were enriched in 19 pathways according to KEGG analyses. (e) The important biological processes enriched by the genes from turquoise module. (f) TF numbers identified from turquoise module genes. The genes shown here were mainly named according to the annotation of Swissprot database and the classification of TF families was defined following the Plant Transcription Factor Database.
Turquoise module, with most genes and most positively related to RA accumulation, was selected for further co-expression network. Twelve TFs including WRK24, TGA21, GBF4a etc. were identified to be with most interactions to other genes. Among these TFs, WRK24 contained most edge number (3,778) (Fig. 7c). Genes in turquoise module could be classified into 10 categories with 19 pathways, which were closely related to QTP adaptation (Fig. 7d). Noticeably, mRNA surveillance pathway (ko03015) and nucleotide excision repair (ko03420) were enriched in this module (Fig. 7d, Table S16). The genes from this module were involved in several important biological processes closely related to the QTP adaptation, mainly including terpenoid metabolic, oxidation-reduction, response to ROS, cellular response to oxidative stress, response to superoxide (Fig. 7e). In addition, total 215 TFs were identified from this module genes, among which WRKY, NAC, ERF and TALE families were accounted for the top four number of genes (Fig. 7f).
Discussion
Environment enforce the adaptive evolution
Environment variations played a key role in plant morphogenesis and adaptive evolution. This has been observed in several plant species i.e. Welwitschia mirabilis in Africa deserts[27], Crucihimalaya himalaica in QTP [54] and Hordeum spontaneum in QTP [55]. Living organisms evolve and adapt to the harsh environment through positive selection. For example, W. mirabilis has evolved an underground stem, however, inner structure of this stem is yet to be explored. In this study, we studied underground stem structure and proposed the “truck-branches” development model in S. castanea for adapting to harsh QTP environment (Fig. 1, Fig. 2, Table S7). Our study suggested that S. castanea branched off from cultivated S. miltiorrhiza around 16 MYA (Neogene, Fig. 3), when QTP could receive more rains from the Indian ocean monsoon. With the uplift of this plateau, the environment of QTP became extreme [1], [2], [5], and S. castanea began to evolve specific underground stem structure.
These severe environmental stresses have also triggered a cascade of events now visible in S. castanea, such as induction of species-specific genes, expanded and contracted OGGs, and PSGs. Different species had their own strategies in response to different environment even if on the same QTP [49], [54]. One important strategy was inducing the MAPK pathway in plants in response to various environmental stresses. In our studied S. castanea, functions of induced MAPK genes were closely related to QTP adaptation. For instance, MPK3a and MPK3b genes had the clear functions in response to UV-B, cold, and freezing [56], [57], [58]. M2K2a and M2K2b genes were with oomycete pathogen resistance [59] and cold acclimation [60], M2K9 could give cellular response to hypoxia [61]. M3K3A and M3K5G could resist to both bacterial and fungal pathogens [62], and adapt to the QTP wild environment. These MAPK genes might be generated by alternative splicing, and have important functions in response to the harsh environment. Therefore, our findings would provide insights into the evolution of S. castanea, and improve our understandings of survival strategy in response to extreme environment.
PCD genes and PSGs contribute to the morphogenesis and QTP adaptation
Programmed cell death (PCD) genes have been reported to play crucial roles in plant development [63], [64]. PCD process accompanied by DNA degradation has been studied in different plant development processes such as endosperm development [65], leaf morphogenesis [66], and vessel differentiation [67], [68]. Therefore, PCD was vital in plant cell fate, organ differentiation and further morphogical establishment. In our study, two PCD genes (NSL1a and NSL1b) were included in 24 species-specific genes among studied 15 species (Fig. S8). This indicated these two PCD genes might contribute to formation of branch-like structures in the underground stem of S. castanea. Our hypothesized “trunk-branches” developmental model indicated the phenomenon of bifurcations in the underground stem might be controlled by the program, implying the crucial function of these two PCD genes. This model also easily explained why some periderm-like structures were found in the center of underground stem. That was indeed the new generated periderm surrounding the new bifurcations, and inner multiple xylem vessels were squeezed together forming the special underground stem structure in S. castanea.
As we know, PSGs were evolved under driving force of environment, therefore these genes always play important roles in adaptation of specific environment. Among the 64 identified PSGs of S. castanea (Fig. 3a, Table S12), DIOX4 was found to be 2-oxoglutarate-dependent dioxygenase gene, which has been reported critical for chilling stress tolerance [69]. Thus, this PSG DIOX4 in S. castanea might contribute greatly to QTP adaptation in response to cold winter. Another PSG PAC has been linked with leaf and chloroplast development in A. thaliana [70], [71]. The molecular function PAC protein has been further investigated to facilitate biogenesis of 50S ribosomal subunit [71], implying its important functions in regulating morphogenesis of S. castanea, especially for densely tomentose leaves. Iron-sulfur cluster has been reported it could act as sensor of gases i.e. oxygen and nitric oxide, catalyze oxidation-reduction, and conduct DNA repair [72], [73]. In this study, 64 PSGs enriched in iron-sulfur cluster binding GO term, indicating these PSGs were functional to deal with oxidative or DNA damage induced by QTP stresses. Moreover, nucleotide excision repair pathway reported in ectothermic snakes and C. himalaica in response to QTP stresses [49], [54], was also enriched in our results. Antioxidative enzymes could be triggered to confront ROS induced by environment stresses [20], [50], [74]. Thus, our identified oxidoreductase activity related PSGs (HBD, DIOX4, HF101) play crucial roles in scavenging unregulated ROS in S. castanea. Therefore, 24 species-specific genes, especially two PCD genes, combined with 64 PSGs contributed greatly to the QTP adaptation in S. castanea. Two PCD genes (NSL1a and NSL1b), some PSGs i.e. DIOX4, PAC, HBD, DIOX4, HF101 might be the important candidate genes responsible for survival of the plant in the harsh climatic condition.
Expanded OGGs contributed to morphogenesis and QTP adaptation
Proteins from significantly expanded OGGs contained LRR domain (Fig. 3c), which is an important member of CLV/WUS negative-feedback loop and regulates organ initiation, species morphogenesis and stem cell homeostasis [25], [28], [29], [52]. LRR could regulate the cell fate, plant development and evolution innovation [25], [33], [75]. Therefore, these LRR domain in S. castanea might contribute greatly to formation of special underground stem. CLV3 is an important feedback signal gene in CLV/WUS loop to depress the expression of WOX gene [29]. However, CLV3 was undetectable in S. castanea. It might be due to lack of high-quality genome information, or related to, maintain stable expressions of WOX8 gene for homeostasis of stem cells in different tissues. Synergism of WOX8, auxin transport protein PIN and some important phytohormone realized the morphogenesis of special underground stem in S. castanea. This speculated mechanism has been proved in other species such as aerial roots in Ficus microcarpa [25], and lotus adventitious roots (ARs) [26]. Therefore, WOX8 gene was essential in S. castanea, and could be a candidate gene responsible to special morphogenesis and QTP adaptation of this species.
The other identified significantly expanded OGGs in S. castanea (Fig. 3c) were also closely related to QTP adaptation. Pentatricopeptide repeat (PPR) domain has been reported to regulate editing of RNA to ensure an accurate translation of functional proteins [76]. PPR protein (WSL5) was essential for chloroplast biogenesis in rice under cold stress [77]. PPR gene TCD10 was needed for chloroplast development in rice under cold stresses [78]. PPR protein SOAR1 was reported to play crucial roles in Arabidopsis in response to drought, salt and cold stresses [79]. Thus, we suggested a crucial role of PPRs for QTP adaptation of S. castanea, especially for the cold environment. Moreover, PPR proteins were not only important for RNA editing and stress response, but also for plant growth and development [80]. PPR was also one kind of peptide, might undertake similar function of CLV3 peptide in S. castanea. Therefore, our studied species S. castanea might possess its own CLV/WUS negative-feedback loop involved PPR. PPR might contribute to not only QTP adaptation, but also special morphogenesis of S. castanea. Other identified domains from significantly expanded OGGs were also important (Fig. 3c). Extensin 2 domain has been reported to have important roles in signaling cascade in response to forthcoming challenge on QTP [81]. WD40 could form MYB-bHLH-WD40 (MBW) complex to regulate secondary metabolism [82]. Pkinase domains is regarding to leaf morphogenesis or seedling development [83], [84]. AP2/ERF is key regulator connecting important phytohormonal signals to regulate plant growth and development, defense response, metabolism [85].
Moreover, Ub-proteasome system (UPS) plays vital roles in degradation of proteins, which had suffered irrecoverable injuries from QTP environment [49], [54]. Our analysis presented Ub domain contained OGGs underwent a significant expansion (Fig. 3c), and this was consistent with the findings of Zhang et al. [54] who reported the adaptation mechanism of a high-altitude plant Crucihimalaya himalaica. Furthermore, most of our detected genes in UPS were highly expressed and proved the crucial roles of Ub-mediated proteolysis in QTP adaptation of S. castanea. In addition, expanded OGGs of S. castanea were enriched in phosphatidylinositol signaling system, which was critical for plant developmental processes including seed development, growth and reproduction, aging and cell responses to environmental stress [86], [87]. Therefore, significantly expanded OGGs of S. castanea might play vital roles on QTP adaptation or morphogenesis of this species. It was worth to note that much more significantly contracted OGGs were observed in S. castanea as compared to other sequenced species, which may result from tissue-specific expression and lack of reference genome of our studied species S. castanea (Fig. 3b). We may infer that the actual contracted OGGs would be much fewer. Interestingly, phloem and shoot developments related OGGs were significantly contracted, indicating they were closely related to form this special underground stem structure in S. castanea.
Secondary metabolites helped to realize the QTP adaptation
Secondary metabolites were not absolutely required for plant survival, but for cell signaling, defense and antioxidant in response to environment stresses [88]. T-IIA has definite functions of anti-hypoxia, anti-oxidant and anti-inflammatory [19], [20], [21], therefore, high contents of T-IIA in S. castanea (Fig. 5a), especially in periderm could promote adaptation to hypoxic environment and protect from pathogen infection. We speculated that continuous generating branch-like structures surrounded by periderm in underground stem might be due to guaranteeing the transportation of T-IIA from periderm to inner phloem or xylem, because T-IIA could protect the plant cells against hypoxia, pathogen and so on. RA was highly antioxidative [22], [23] and against UV [24], thus high accumulations of RA in leaves of S. castanea (Fig. 6a) could scavenge ROS induced by intense UV on QTP. Therefore, periderm of underground stem and tomenta of leaves were like armor protecting S. castanea against harsh environment through accumulations of specific secondary metabolites (mainly T-IIA and RA).
Our results revealed terpenoid backbone biosynthesis pathway was significantly enriched in T-IIA biosynthesis, which was consistent with the results in S. miltiorrhiza [89]. However, terpenoid backbone biosynthesis pathway was also significantly enriched in RA biosynthesis in S. castanea, indicating this might be an important pathway crossing with other metabolic pathways and regulated biosynthesis of these two metabolites. TFs regulated secondary metabolisms had been reported in various species i.e. S. miltiorrhiza [7], [90], Crocus sativus [37], Arabidopsis thaliana [91] and so on. Thus, our identified TFs NAC29 and WRK75 in T-IIA biosynthesis pathway, WRKY33, WRKY24, TGA22 etc. in RA biosynthesis pathway might play crucial roles in biosynthesis of these two secondary metabolites (Fig. 5, Fig. 6). Overall, the special underground stem structure and secondary metabolism of S. castanea provide insights into the mechanisms of QTP adaptation, and further provided some candidate genes for genetic improvement of cultivated crops, especially for Chinese medicinal plant S. miltiorrhiza.
Conclusion
S. castanea is a wild resource that grows on QTP. Here, we assembled a high-quality full-length transcriptome of this species and revealed it branched off from a Chinese model medicinal plant S. miltiorrhiza around 16 Mya. We proposed a “trunk-branches” developmental model to explain periderm-like structures found inner underground stem of S. castanea. Significantly expanded OGGs in CLV/WUS negative-feedback loop, and contracted OGGs with the function of shoot development might lead to specific underground stem structure of S. castanea. Moreover, 24 species-specific genes especially two programmed cell death genes (NSL1a and NSL1b), and 64 positively selected genes also contributed greatly to the morphogenesis and QTP adaptation of this species. High accumulations of two secondary metabolites (T-IIA and RA) could protect S. miltiorrhiza against hypoxia and intense UV radiation stresses on QTP. Continuous generating branch-like structures in underground stems might be due to guaranteeing the transportation of T-IIA from periderm to inner phloem and xylem. WGCNA analyses revealed that some important TFs such as NAC29 and TGA22 were involved in regulating T-IIA and RA biosynthesis. Overall, the special underground stem architecture and secondary metabolism of S. castanea provided insights into the mechanisms of QTP adaptation, and further provided some candidate genes for genetic improvement of cultivated S. miltiorrhiza.
CRediT authorship contribution statement
Ling Xu: Conceptualization, Methodology, Funding acquisition, Writing – original draft. Mengting Cao: Methodology, Resources, Writing – original draft. Qichao Wang: Methodology, Formal analysis, Visualization. Jiahao Xu: Methodology, Formal analysis, Writing – review & editing. Chenglin Liu: Formal analysis, Visualization. Najeeb Ullah: Formal analysis, Writing – review & editing. Juanjuan Li: Formal analysis, Writing – review & editing. Zhuoni Hou: Investigation. Zongsuo Liang: Supervision, Project administration. Weijun Zhou: Writing – review & editing, Funding acquisition. Ake Liu: Conceptualization, Formal analysis, Visualization, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31871694, 31800255), Basic Research Foundation for Young Scientists of Shanxi Province (20210302124145), Shanxi Scholarship Council of China (2021-153), and Collaborative Innovation Center for Modern Crop Production co-sponsored by Province and Ministry (CIC-MCP). We also thank the graduate student Xin Li from Zhejiang Sci-Tech University for her help during experiment.
Compliance with Ethics Requirements
All experiments in this article do not include human or animal related research.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.02.004.
Contributor Information
Zongsuo Liang, Email: liangzs@zstu.edu.cn.
Weijun Zhou, Email: wjzhou@zju.edu.cn.
Ake Liu, Email: akeliu@126.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Yin A.n., Harrison T.M. Geologic Evolution of the Himalayan-Tibetan Orogen. Annu Rev Earth Pl Sc. 2000;28(1):211–280. [Google Scholar]
- 2.Xing Y., Ree R.H. Uplift-driven diversification in the Hengduan mountains, a temperate biodiversity hotspot. Proc Natl Acad Sci U S A. 2017;114(17):E3444–E3451. doi: 10.1073/pnas.1616063114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu XiaoDong, Dong BuWen. Influence of the Tibetan Plateau uplift on the Asian monsoon-arid environment evolution. Chinese Sci Bull. 2013;58(34):4277–4291. [Google Scholar]
- 4.Cheviron Z.A., Brumfield R.T. Genomic insights into adaptation to high-altitude environments. Heredity. 2012;108(4):354–361. doi: 10.1038/hdy.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiao Q., Wang Q., Han X.i., Guan Y., Sun H., Zhong Y., et al. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci Rep. 2016;6(1) doi: 10.1038/srep21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H., Ma P.F., Li H.T., Hu G.X., Li D.Z. Comparative plastomic analysis and insights into the phylogeny of Salvia (Lamiaceae) Plant Diversity. 2021;43(1):15–26. doi: 10.1016/j.pld.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu H., Guo W., Yang D., Hou Z., Liang Z. Transcriptional profiles of SmWRKY family genes and their putative roles in the biosynthesis of tanshinone and phenolic acids in Salvia miltiorrhiza. Int J Mol Sci. 2018;19:1593–1610. doi: 10.3390/ijms19061593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang D., Ma P., Liang X., Liang Z., Zhang M., Shen S., et al. Metabolic Profiles and cDNA-AFLP Analysis of Salvia miltiorrhiza and Salvia castanea Diel f. tomentosa Stib. PLoS One. 2012;7:e29678. doi: 10.1371/journal.pone.0029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y., Kong D., Fu Y., Sussman M.R., Wu H. The effect of developmental and environmental factors on secondary metabolites in medicinal plants. Plant Physiol Biochem. 2020;148:80–89. doi: 10.1016/j.plaphy.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Liu H., Wang X., Wang D., Zou Z., Liang Z. Effect of drought stress on growth and accumulation of active constituents in Salvia miltiorrhiza Bunge. Ind Crops Prod. 2011;33(1):84–88. [Google Scholar]
- 11.Hectors K., Oevelen S.V., Geuns J., Guisez Y., Jansen M.A.K., Prinsen E. Dynamic changes in plant secondary metabolites during UV acclimation in Arabidopsis thaliana. Physiol Plant. 2014;152(2):219–230. doi: 10.1111/ppl.12168. [DOI] [PubMed] [Google Scholar]
- 12.Yang L., Wen K.S., Ruan X., Zhao Y.X., Wei F., Wang Q. Response of Plant Secondary Metabolites to Environmental Factors. Molecules. 2018;23(4):762. doi: 10.3390/molecules23040762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christie P.J., Alfenito M.R., Walbot V. Impact of low-temperature stress on general phenylpropanoid and anthocyanin pathways: Enhancement of transcript abundance and anthocyanin pigmentation in maize seedlings. Planta. 1994;194(4):541–549. [Google Scholar]
- 14.Kurepin L.V., Ivanov A.G., Zaman M., Pharis R.P., Hurry V., Hüner N.P.A. In: Photosynthesis: Structures, Mechanisms, and Applications. Hou H.J.M., Najafpour M.M., Moore G.F., Allakhverdiev S.I., editors. Springer International Publishing; Cham: 2017. Interaction of Glycine betaine and plant hormones: protection of the photosynthetic apparatus during abiotic stress; pp. 185–202. [DOI] [Google Scholar]
- 15.Moore B.D., Andrew R.L., Külheim C., Foley W.J. Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytol. 2014;201(3):733–750. doi: 10.1111/nph.12526. [DOI] [PubMed] [Google Scholar]
- 16.Speed M.P., Fenton A., Jones M.G., Ruxton G.D., Brockhurst M.A. Coevolution can explain defensive secondary metabolite diversity in plants. New Phytol. 2015;208(4):1251–1263. doi: 10.1111/nph.13560. [DOI] [PubMed] [Google Scholar]
- 17.Kai G., Liao P., Xu H., Wang J., Zhou C., Zhou W., et al. Molecular mechanism of elicitor-induced tanshinone accumulation in Salvia miltiorrhiza hairy root cultures. Acta Physiol Plant. 2012;34(4):1421–1433. [Google Scholar]
- 18.Vranová E., Coman D., Gruissem W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol. 2013;64(1):665–700. doi: 10.1146/annurev-arplant-050312-120116. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y., Duo D., Yan Y., He R., Wang S., Wang A., et al. Extract of Salvia przewalskii repair tissue damage in chronic hypoxia maybe through the RhoA–ROCK signalling pathway. Biol Pharm Bull. 2020;43:432–439. doi: 10.1248/bpb.b19-00775. [DOI] [PubMed] [Google Scholar]
- 20.Xu W., Yang J., Wu L. Cardioprotective effects of tanshinone IIA on myocardial ischemia injury in rats. Pharmazie. 2009;64:332–336. [PubMed] [Google Scholar]
- 21.Liu T., Jin H., Sun Q.-R., Xu J.-H., Hu H.-T. The neuroprotective effects of tanshinone IIA on β-amyloid-induced toxicity in rat cortical neurons. Neuropharmacology. 2010;59(7-8):595–604. doi: 10.1016/j.neuropharm.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 22.Alagawany M., Abd El-Hack M.E., Farag M.R., Gopi M., Karthik K., Malik Y.S., et al. Rosmarinic acid: modes of action, medicinal values and health benefits. Anim Health Res Rev. 2017;18(2):167–176. doi: 10.1017/S1466252317000081. [DOI] [PubMed] [Google Scholar]
- 23.Fadel O., El Kirat K., Morandat S. The natural antioxidant rosmarinic acid spontaneously penetrates membranes to inhibit lipid peroxidation in situ. BBA-Biomembranes. 2011;1808(12):2973–2980. doi: 10.1016/j.bbamem.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Sánchez-Campillo M., Gabaldon J.A., Castillo J., Benavente-García O., Del Baño M.J., Alcaraz M., et al. Rosmarinic acid, a photo-protective agent against UV and other ionizing radiations. Food Chem Toxicol. 2009;47(2):386–392. doi: 10.1016/j.fct.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X., Wang G., Zhang S., Chen S., Wang Y., Wen P., et al. Genomes of the banyan tree and pollinator wasp provide insights into gig-wasp coevolution. Cell. 2020;183:875–889. doi: 10.1016/j.cell.2020.09.043. [DOI] [PubMed] [Google Scholar]
- 26.Geiss G., Gutierrez L., Bellini C. Adventitious root formation: new insights and perspectives. Annu Plant Rev. 2009;37:127–156. [Google Scholar]
- 27.Wan T., Liu Z., Leitch I.J., Xin H., Maggs-Kölling G., Gong Y., et al. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat commun. 2021;12:4247. doi: 10.1038/s41467-021-24528-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fletcher J.C., Brand U., Running M.P., Simon Rüdiger, Meyerowitz E.M. Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science. 1999;283(5409):1911–1914. doi: 10.1126/science.283.5409.1911. [DOI] [PubMed] [Google Scholar]
- 29.Brand U., Fletcher J.C., Hobe M., Meyerowitz E.M., Simon Rüdiger. Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science. 2000;289(5479):617–619. doi: 10.1126/science.289.5479.617. [DOI] [PubMed] [Google Scholar]
- 30.Schoof H., Lenhard M., Haecker A., Mayer K.F.X., Jürgens G., Laux T. The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell. 2000;100(6):635–644. doi: 10.1016/s0092-8674(00)80700-x. [DOI] [PubMed] [Google Scholar]
- 31.Clark S.E., Williams R.W., Meyerowitz E.M. The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis. Cell. 1997;89(4):575–585. doi: 10.1016/s0092-8674(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 32.Kayes J.M., Clark S.E. CLAVATA2, a regulator of meristem and organ development in Arabidopsis. Development. 1998;125:3843–3851. doi: 10.1242/dev.125.19.3843. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa M., Shinohara H., Sakagami Y., Matsubayashi Y. Arabidopsis CLV3 peptide directly binds CLV1 ectodomain. Science. 2008;319(5861) doi: 10.1126/science.1150083. [DOI] [PubMed] [Google Scholar]
- 34.Vanneste S., Friml J. Auxin: A trigger for change in plant development. Cell. 2009;136(6):1005–1016. doi: 10.1016/j.cell.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Qin H., Huang R. Auxin controlled by ethylene steers root development. Int J Mol Sci. 2018;19(11):3656. doi: 10.3390/ijms19113656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agusti J., Herold S., Schwarz M., Sanchez P., Ljung K., Dun E.A., et al. Strigolactone signaling is required for auxin-dependent stimulation of secondary growth in plants. Proc Natl Acad Sci U S A. 2011;108(50):20242–20247. doi: 10.1073/pnas.1111902108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue J., Wang R., Ma X., Liu J., Lu X., Balaso Thakar S., et al. Full-length transcriptome sequencing provides insights into the evolution of apocarotenoid biosynthesis in Crocus sativus. Comput Struct Biotec. 2020;18:774–783. doi: 10.1016/j.csbj.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Min X.J., Butler G., Storms R., Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005;33(Web Server):W677–W680. doi: 10.1093/nar/gki394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S., Stecher G., Suleski M., Hedges S.B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34(7):1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- 47.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics. 1997;13(5):555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 48.De Bie T., Cristianini N., Demuth J.P., Hahn M.W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 49.Li J.-T., Gao Y.-D., Xie L., Deng C., Shi P., Guan M.-L., et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc Natl Acad Sci U S A. 2018;115(33):8406–8411. doi: 10.1073/pnas.1805348115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu L., Li J., Najeeb U., Li X., Pan J., Huang Q., et al. Synergistic effects of EDDS and ALA on phytoextraction of cadmium as revealed by biochemical and ultrastructural changes in sunflower (Helianthus annuus L.) tissues. J Hazard Mater. 2021;407:124764. doi: 10.1016/j.jhazmat.2020.124764. [DOI] [PubMed] [Google Scholar]
- 51.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Hu C., Zhu Y., Cui Y., Cheng K., Liang W., Wei Z., et al. A group of receptor kinases are essential for CLAVATA signalling to maintain stem cell homeostasis. Nat Plants. 2018;4(4):205–211. doi: 10.1038/s41477-018-0123-z. [DOI] [PubMed] [Google Scholar]
- 53.del Pozo J.C., Dharmasiri S., Hellmann H., Walker L., Gray W.M., Estelle M. AXR1-ECR1-dependent conjugation of RUB1 to the Arabidopsis Cullin AtCUL1 is required for auxin response. Plant Cell. 2002;14(2):421–433. doi: 10.1105/tpc.010282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang T., Qiao Q., Novikova P.Y., Wang Q., Yue J., Guan Y., et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. P Natl A Sci. 2019;116(14):7137–7146. doi: 10.1073/pnas.1817580116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen Q., Fu L., Dai F., Jiang L., Zhang G., Wu D. Multi-omics analysis reveals molecular mechanisms of shoot adaption to salt stress in Tibetan wild barley. BMC Genomics. 2016;17:889. doi: 10.1186/s12864-016-3242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Casadevall R., Rodriguez R.E., Debernardi J.M., Palatnik J.F., Casati P. Repression of growth regulating factors by the microRNA396 inhibits cell proliferation by UV-B radiation in Arabidopsis leaves. Plant Cell. 2013;25(9):3570–3583. doi: 10.1105/tpc.113.117473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lang J., Genot B., Bigeard J., Colcombet J. MAP Kinases 3 and 6 control Salicylic Acid signaling by upregulating NLR receptors during Pattern- and Effector-Triggered Immunity. J Exp Bot. 2022 doi: 10.1093/jxb/erab544. erab544. [DOI] [PubMed] [Google Scholar]
- 58.Li H., Ding Y., Shi Y., Zhang X., Zhang S., Gong Z., et al. MPK3- and MPK6-mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev Cell. 2017;43(5):630–642.e4. doi: 10.1016/j.devcel.2017.09.025. [DOI] [PubMed] [Google Scholar]
- 59.Li J., Deng F., Wang H., Qiang X., Meng Y., Shan W. The Raf-like kinase Raf36 negatively regulates plant resistance against the oomycete pathogen Phytophthora parasitica by targeting MKK2. Mol Plant Pathol. 2021 doi: 10.1111/mpp.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furuya T., Matsuoka D., Nanmori T. Membrane rigidification functions upstream of the MEKK1-MKK2-MPK4 cascade during cold acclimation in Arabidopsis thaliana. FEBS Lett. 2014;588(11):2025–2030. doi: 10.1016/j.febslet.2014.04.032. [DOI] [PubMed] [Google Scholar]
- 61.Licausi F., Perata P. Chapter 4: Low oxygen signaling and tolerance in plants. Adv Bot Res. 2009;50:139–198. [Google Scholar]
- 62.Sun T., Nitta Y., Zhang Q., Wu D., Tian H., Lee J.S., et al. Antagonistic interactions between two MAP kinase cascades in plant development and immune signaling. EMBO Rep. 2018;19(7) doi: 10.15252/embr.201745324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ranganath R.M., Nagashree N.R. Role of programmed cell death in development. Int Rev Cytol. 2001;202:159–242. doi: 10.1016/s0074-7696(01)02005-8. [DOI] [PubMed] [Google Scholar]
- 64.Bai M., Liang M., Huai B., Gao H., Tong P., Shen R., et al. Ca2+-dependent nuclease is involved in DNA degradation during the formation of the secretory cavity by programmed cell death in fruit of Citrus grandis 'Tomentosa'. J Exp Bot. 2020;71(16):4812–4827. doi: 10.1093/jxb/eraa199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bozhkov P.V., Filonova L.H., Suarez M.F., Helmersson A., Smertenko A.P., Zhivotovsky B., et al. VEIDase is a principal caspase-like activity involved in plant programmed cell death and essential for embryonic pattern formation. Cell Death Differ. 2004;11(2):175–182. doi: 10.1038/sj.cdd.4401330. [DOI] [PubMed] [Google Scholar]
- 66.Gunawardena A.H.L.A.N. Programmed cell death and tissue remodelling in plants. J Exp Bot. 2008;59:445–451. doi: 10.1093/jxb/erm189. [DOI] [PubMed] [Google Scholar]
- 67.Fukuda H. Programmed cell death of tracheary elements as a paradigm in plants. Plant Mol Biol. 2000;44:245–253. doi: 10.1023/a:1026532223173. [DOI] [PubMed] [Google Scholar]
- 68.Chen H.-M., Pang Y.u., Zeng J., Ding Q.i., Yin S.-Y., Liu C., et al. The Ca2+-dependent DNases are involved in secondary xylem development in Eucommia ulmoides. J Integ Plant Biol. 2012;54(7):456–470. doi: 10.1111/j.1744-7909.2012.01134.x. [DOI] [PubMed] [Google Scholar]
- 69.Hu T., Wang Y., Wang Q., Dang N., Wang L., Liu C., et al. The tomato 2-oxoglutarate-dependent dioxygenase gene SlF3HL is critical for chilling stress tolerance. Hortic Res. 2019;6(1) doi: 10.1038/s41438-019-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reiter R.S., Coomber S.A., Bourett T.M., Bartley G.E., Scolnik P.A. Control of leaf and chloroplast development by the Arabidopsis gene pale cress. Plant Cell. 1994;6:1253–1264. doi: 10.1105/tpc.6.9.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meurer J., Schmid L.-M., Stoppel R., Leister D., Brachmann A., Manavski N. PALE CRESS binds to plastid RNAs and facilitates the biogenesis of the 50S ribosomal subunit. Plant J. 2017;92(3):400–413. doi: 10.1111/tpj.13662. [DOI] [PubMed] [Google Scholar]
- 72.Grazina R., Pauleta S.R., Moura J.J.G., Moura I. Iron–sulfur centers: new roles for ancient metal sites. Compr Inorg Chem II (Second Edition) 2013;3:103–148. [Google Scholar]
- 73.Jagannathan B., Golbeck J.H. FX, FA, and FB iron-sulfur clusters in type I photosynthetic reaction centers. Encyclopedia of Biological Chemistry. 2013:335–342. [Google Scholar]
- 74.Gill R.A., Zang L., Ali B., Farooq M.A., Cui P., Yang S., et al. Chromium-induced physio-chemical and ultrastructural changes in four cultivars of Brassica napus L. Chemosphere. 2015;120:154–164. doi: 10.1016/j.chemosphere.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 75.Ratnaparkhe M.B., Wang X., Li J., Compton R.O., Rainville L.K., Lemke C., et al. Comparative analysis of peanut NBS-LRR gene clusters suggests evolutionary innovation among duplicated domains and erosion of gene microsynteny. New Phytol. 2011;192(1):164–178. doi: 10.1111/j.1469-8137.2011.03800.x. [DOI] [PubMed] [Google Scholar]
- 76.Cheng S., Gutmann B., Zhong X., Ye Y., Fisher M.F., Ba F., et al. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 2016;85:532–547. doi: 10.1111/tpj.13121. [DOI] [PubMed] [Google Scholar]
- 77.Liu X., Lan J., Huang Y., Cao P., Zhou C., Ren Y., et al. WSL5, a pentatricopeptide repeat protein, is essential for chloroplast biogenesis in rice under cold stress. J Exp Bot. 2018;69(18):4495. doi: 10.1093/jxb/ery259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu L., Wu J., Liu Y., Gong X., Xu J., Lin D., et al. The rice pentatricopeptide repeat gene TCD10 is needed for chloroplast development under cold stress. Rice (N Y). 2016;9(1) doi: 10.1186/s12284-016-0134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang S.-C., Mei C., Liang S., Yu Y.-T., Lu K., Wu Z., et al. Crucial roles of the pentatricopeptide repeat protein SOAR1 in Arabidopsis response to drought, salt and cold stresses. Plant Mol Biol. 2015;88(4-5):369–385. doi: 10.1007/s11103-015-0327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qin T., Zhao P., Sun J., Zhao Y., Zhang Y., Yang Q., et al. Research progress of PPR proteins in RNA editing, stress response, plant growth and development. Front Genet. 2021;12 doi: 10.3389/fgene.2021.765580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Borassi C., Sede A.R., Mecchia M.A., Salgado Salter J.D., Marzol E., Muschietti J.P., et al. An update on cell surface proteins containing extensin-motifs. J Exp Bot. 2016;67(2):477–487. doi: 10.1093/jxb/erv455. [DOI] [PubMed] [Google Scholar]
- 82.Zhao M., Li J., Zhu L., Chang P., Li L., Zhang L. Identification and characterization of MYB-bHLH-WD40 regulatory complex members controlling anthocyanidin biosynthesis in blueberry fruits development. Genes (Basel) 2019;10:496. doi: 10.3390/genes10070496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ye M., Zhu X., Gao P., Jiang L., Wu R. Identification of quantitative trait loci for altitude adaptation of tree leaf shape with Populus szechuanica in the Qinghai-Tibetan plateau. Front Plant Sci. 2020;11 doi: 10.3389/fpls.2020.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.López-Hernández F., Cortés A.J. Last-Generation Genome-Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.) Front Genet. 2019;10:954. doi: 10.3389/fgene.2019.00954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gu C., Guo Z., Hao P., Wang G., Jin Z., Zhang S. Multiple regulatory roles of AP2/ERF transcription factor in angiosperm. Bot Stud. 2017;58:6. doi: 10.1186/s40529-016-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dove S.K., Cooke F.T., Douglas M.R., Sayers L.G., Parker P.J., Michell R.H. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390(6656):187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- 87.Stephens L.R., Hughes K.T., Irvine R.F. Pathway of phosphatidylinositol (3,4,5)-trisphosphate synthesis in activated neutrophils. Nature. 1991;351(6321):33–39. doi: 10.1038/351033a0. [DOI] [PubMed] [Google Scholar]
- 88.Pichersky E., Gang D.R. Genetics and biochemistry of secondary metabolites in plants: an evolutionary perspective. Trends Plant Sci. 2000;5(10):439–445. doi: 10.1016/s1360-1385(00)01741-6. [DOI] [PubMed] [Google Scholar]
- 89.Wenping H., Yuan Z., Jie S., Lijun Z., Zhezhi W. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics. 2011;98(4):272–279. doi: 10.1016/j.ygeno.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 90.Hao X., Pu Z., Cao G., You D., Zhou Y., Deng C., et al. Tanshinone and salvianolic acid biosynthesis are regulated by SmMYB98 in Salvia miltiorrhiza hairy roots. J Adv Res. 2020;23:1–12. doi: 10.1016/j.jare.2020.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Teng S., Keurentjes J., Bentsink Leónie, Koornneef M., Smeekens S. Sucrose-specific induction of anthocyanin biosynthesis in Arabidopsis requires the MYB75/PAP1 gene. Plant Physiol. 2005;139(4):1840–1852. doi: 10.1104/pp.105.066688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.