

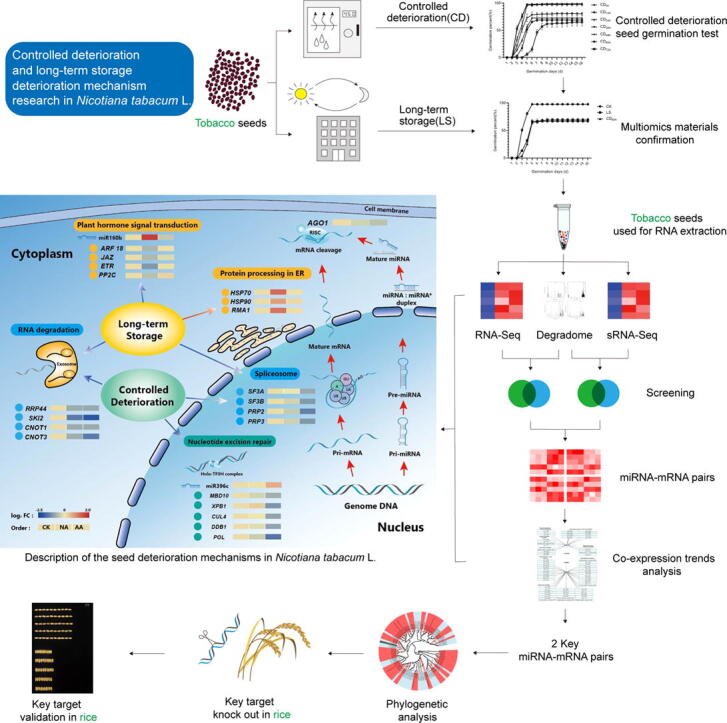
Graphical abstract
Keywords: Controlled deterioration, Long-term storage, Small RNA, Transcriptome, Nicotiana tabacum L., Oryza sativa L.
Highlights
-
•
Transcriptional mechanism of tobacco seed deterioration was revealed by small RNA and transcriptome sequencing.
-
•
The cleavage relationship between tobacco miRNA and mRNA was validated by degradome sequencing.
-
•
Coherent miRNA-mRNA pairs of seed deterioration were screened through combined analysis.
-
•
Rice homologous target genes found that OsARF18 and OsMBD707 were functional in seed deterioration.
-
•
Transcriptional multiomics provides substantial transcriptional data regarding seed deterioration for further study.
Abstract
Introduction
Mature seeds deteriorate gradually and die eventually during long-term storage. Controlled deterioration is often used to accelerate the seed deterioration rate to assess the seed vigor and physiological quality of seed lots.
Objectives
Although it is well known that the process of seed deterioration produced by controlled deterioration is distinct from that caused by long-term storage, the differences in transcriptional levels have not been reported. Clarifying the mechanism of seed deterioration is critical for identifying, conserving and utilizing germplasm resources.
Methods
Tobacco (Nicotiana tabacum L.) seeds were studied thoroughly using transcriptome, small RNA, and degradome sequencing after long-term storage (LS) and controlled deterioration (CD). Co-expression trend analysis identified transcripts involved in tobacco seed deterioration, while phylogenetic analysis helped to uncover comparable targets in rice (Oryza sativa L.) for further verification and utilization.
Results
In LS and CD, a total of 2,112 genes and 164 miRNAs were differentially expressed, including 20 interaction miRNA-mRNA pairs with contrasting expression. Transcriptional multiomics found that the main causes of LS were plant hormone signal transduction and protein processing in the endoplasmic reticulum, whereas the primary cause of CD was nucleotide excision repair dysfunction. The homeostatic balance of RNA degradation and the spliceosome occurred in both modes of seed deterioration. Additionally, co-expression trend analysis identified two coherent pairs, nta-miR160b-NtARF18 and nta-miR396c-NtMBD10, as being significant in LS and CD, respectively. For utilization, rice homologous targets OsARF18 and OsMBD707 were verified to play similar roles in LS and CD, respectively.
Conclusion
This study demonstrated the transcriptional mechanism of tobacco and key genes in seed deterioration. And the application of key genes in rice also verified the feasibility of the multiomics method, guiding the identification of candidate genes to precisely delay seed deterioration in other species of seed research.
Introduction
Seed germination is the beginning of the growth of a new living individual and is crucial to agricultural production [1]. It is inevitable for seeds to degenerate in morphology, physiological function and biochemistry during long-term storage (LS). This will reduce seed vigor and eventually lead to a loss of seed viability and failure to germinate [2], [3].
Seed deterioration occurs over time. The preservation conditions of orthodox seed need low temperatures and low humidity in order to retain seed viability and extend the lifespan of seed [4]. For fast evaluation of the seed storability, various methods have been used to accelerate seed deterioration [5]. Controlled deterioration (CD) is a method of simulating stress conditions that will result in increasing respiration rates and depletion of reserves, culminating in degenerative changes in seed metabolism and eventual deterioration [6]. A systematic proteomic investigation of Arabidopsis seed ageing demonstrates that CD is a suitable way for predicting seed storability and deterioration, and reveals comparable molecular processes in both CD and naturally aged seeds [5]. However, CD reduced seed vigor mainly by altering the expression of seed-stored and newly synthesized transcription factors, and by reducing antioxidant enzyme levels [7], [8]. So far, it is still uncertain which transcripts are affected by CD and where the similarities and differences exist between CD and LS.
Recent advances in sequencing technology have allowed us to get a better understanding of plant physiological systems. Small non-coding RNAs and transcriptome sequencing, which enable post-transcriptional regulation, transcript identification, and quantification of gene expression, have been widely used to gain a better understanding of not only the transcriptional dynamics in plants, but also regulatory networks affecting development and responses to biotic and abiotic stresses [9]. Degradome sequencing acts as a link between miRNA and mRNA, validating miRNA-mediated cleavage of target genes and enabling the discovery of additional targets [10]. The combined analysis of the transcriptome and small RNAs identified a variety of potential affected pathways in biotic and abiotic responses, offering novel insights into the miRNA-mRNA interaction adjustment mechanism [11], [12], [13].
In the present study, both LS and CD seeds of tobacco (Nicotiana tabacum L.) were used to unravel differentially expressed genes, miRNAs and their cleavage connections, as well as seed deterioration mechanisms by means of transcriptional multiomics. Through the extended study of rice homologous target genes, the potential role of key genes in seed deterioration was verified, so as to provide guidance for precise improvement of seed traits in other species.
Materials and methods
Plant materials
Tobacco (Nicotiana tabacum L.) cultivar K326 seeds from 2013 (as a control, CK) and 2004 (as long-term storage, LS) were used as materials. They were stored with an initial 7% seed moisture content and a 100% germination percentage in a 4 °C germplasm resource bank. Rice (Oryza sativa L.) cultivar ZH11 was utilized as a CRISPR background to verify the potential roles of target genes related to seed deterioration.
Controlled deterioration treatment of tobacco seeds
After removing CK and LS from the germplasm resource bank to a laboratory environment (25 °C ± 2 °C and 60% relative humidity) for 24 h, controlled deterioration was performed on CK with the goal of producing a fast deterioration material (coded as controlled deterioration, CD) that is equivalent to the germination, physiological, and enzyme activities of LS. The controlled deterioration procedure was as follows [14]:
Step 1: Taking a portion of CK seeds and adjusting the seed moisture content to 20%.
Step 2: Sealing 1 g of the seeds in an aluminum foil compound bag.
Step 3: The aluminum foil compound bags were immersed in a thermostatic waterbath filling water with 45 °C for 72 h and the bag sample were taken out every 12 h coded as CD0h, CD12h, CD24h, CD36h, CD48h, CD60h and CD72h, meaning controlled deterioration treatment for 0 h, 12 h, 24 h, 36 h, 48 h, 60 h and 72 h, respectively. 3 replicates (3 bags) were used at each sampling time. There was a total of 21 bags of the seeds.
Standard germination test of tobacco seeds
Four replications of 100 seeds each for CK, LS and all batches of CD were used. Each of the 100-seeds was placed in a 12 cm diameter petri dish containing three layers of water-saturated germination paper. Then, seeds were kept in a growth chamber (DGX-800E, Safe Experiment Instrument Factory, China) with 250 µmol m−2 s−1 light intensity and an alternate cycle of 8 h of light at 30 °C and 16 h of darkness at 20 °C, and the standard seed germination test protocol strictly complies with International Rules for Seed Testing (ISTA) [15]. The germinated seeds were counted daily, and on the 7th and 14th days, respectively, the germination energy (GE) and germination percentage (GP) were calculated [15]. Seedling length with 10 randomly chosen seedlings in each replication was manually measured after 14 days of germination [16]. The germination index (GI), mean germination time (MGT) and vigor index (VI) were calculated using formulae based on the daily number of germinated seeds.
where Gt is number of seed newly germinated at time Dt; Dt is days from when set to germinate; S is seedling length (cm) [17].GraphPad Prism 8.01 (GraphPad Software, San Diego, CA 92108, USA) was used to display the germination trends line and histograms.
Measurement of antioxidant enzyme activity
CD60h was initially used as the CD sample to test the antioxidant enzyme activity. Samples (0.1 g) of tobacco seedlings after standard germination were standardized in 0.5 ml of 50 mM potassium phosphate buffer (i.e., pH 7.0, comprising 1 mM EDTANa2 in addition to 0.5% PVP, w/v) on ice. Accordingly, centrifugation of the homogenate was conducted for 20 min at 12,000 × g at 4 °C. Peroxidase (POD), catalase (CAT), superoxide dismutase (SOD), and glutathione reductase (GR) were measured in the supernatant according to Chance et al [18], Aebi [19], Giannopolitis et al [20] and Yang et al [21], respectively.
RNA extraction
CD60h was finally selected as the CD sample for the multiomics study, which had similar performance as LS in seed germination performance and antioxidant enzyme activities. All seeds of CK, LS and CD were balanced in a laboratory environment (25 °C ± 2 °C, and 20% relative humidity) for 7 days prior to RNA extraction, and total RNA was extracted three times in each sample using 0.2 g of materials. To guarantee the quality of de novo transcriptome and small RNA sequencing, the purity, concentration, and integrity of RNA samples were determined. Degradome sequencing was performed on whole mixed RNA samples.
Transcriptome sequencing
The NEB Next UltraTM RNA Library Prep Kit for Illumina platform (Illumina Inc., San Diego, CA) 240 bp paired-end strategy was used to generate the transcriptome library from the high-quality total RNA. Transcriptome sequencing was performed on a total of nine samples. The data sets were purified of adaptor sequences and low-quality sequencing reads. Following data processing, raw sequences were converted to clean reads. The Hisat2 tool was used to map these clean reads to the reference genome sequence [22].
Differential gene expression analysis
The FPKM (fragments per kilobase of transcript per million fragments mapped) method was used to assess gene expression levels, and the genes identified in the assembly were then examined for their expression patterns across all samples using the DESeq2 method [23]. Compared with CK, genes in LS and CD with P value < 0.01 and fold change ≥ 1.5 detected by DESeq2 were assigned as differentially expressed. A KEGG enrichment analysis of DEGs was carried out by KOBAS software [24]. The heat map was generated using normalized (separate z-score computed per gene) expression values in units of FPKM.
Small RNA sequencing
The Illumina TruSeq Small RNA Library Prep Kit was used to create small RNA libraries from nine small RNA samples (Illumina Inc., San Diego, CA). Reads comprising adaptor, ploy-N, low-quality, and sequences less than 18 nt or longer than 30 nt were removed from the raw data to produce clean reads. The clean reads were compared with the Silva, GtRNAdb, Rfam, and Repbase databases to filter ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and other ncRNA and repetitions using the Bowtie tools [25]. By comparing the genome with known miRNAs from miRBase, the remaining reads were utilized to identify existing miRNAs and predict new miRNAs. New miRNA secondary structures were predicted with the help of the Randfold tools [26].
Differential miRNA expression analysis
The transcripts per kilobase of exon model per million mapped reads (TPM) were used to indicate miRNA expression. The R-based DESeq2 package was used to undertake differential expression analysis of miRNAs with fold change ≥ 1.5 and P value < 0.05 in LS and CD compared with CK [23]. Normalized (separate z-score calculated per gene) expression data in TPM was used to create the heat map.
Target validation by degradome
To create a degradome library for degradome sequencing, equal volumes of nine RNA samples were mixed together. The Illumina HiSeq 2500 sequencing platform was used to sequence all of the samples. Low-quality reads, reads containing 'N's, and any reads with adaptor and primer contamination were removed from the retrieved sequencing reads obtained by degradome sequencing. Cleaveland v4.5 was used to predict the cleavage sites of known and new miRNAs [27]. Cleavage sites at the 10th position relative to the aligned miRNA were deemed significant if the P value < 0.05. Based on the read abundance at the 10th position, the discovered sites were divided into five groups (0–4): At the cleavage site, categories 0–3 have many reads mapped, whereas category 4 contains just one read. With category 0 (highest confidence) and category 4 (lowest confidence), these distinct categories show the amount of forecast confidence (minimum confidence).
Coherent miRNA-mRNA pair screening and DEGs’ co-expression trend analysis
For transcriptome, small RNA sequencing, and degradome, the Jvenn tool was utilized to screen differentially expressed miRNAs and mRNAs [28]. DEGs’ co-expression pattern analysis tools on the platform BMKCloud (http://www.biocloud.net) were used to analyze DEGs’ co-expression (http://www.biocloud.net).
Phylogenetic analysis and validation of the rice homologous genes OsARF18 and OsMBD707
Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [29] and Evolview V3.0 (https://www.evolgenius.info/evolview/) [30] were used to do the phylogenetic analysis. CRISPR/Cas9 mutants of Osarf18 and Osmbd707 were generated in the Oryza sativa Japonica cv. ZH11 background [31]. The 1000 grain weight was obtained by weighing 1000 seeds of 10 randomly selected rice plants per line [32]. Seed length, width, and thickness were calculated using ten 10-seed replications.
The standard germination test of rice seeds after long-term storage and controlled deterioration
The seeds of ZH11, Osarf18 and Osmbd707 mutants, stored at 25 °C room temperature, were used to verify the characteristics of OsARF18 and OsMBD707. The standard germination test was respectively performed on rice seeds stored for 4 months and 8 months, and seeds with 15% moisture content subjected to controlled deterioration for 72 h under 45 °C with 3 replicates of 50 seeds for each material [15]. GE and GP were measured on the 5th and 14th days, respectively. GI, VI and the fold change of GI and VI were also calculated. For each iteration, ten normal rice seedlings were chosen randomly.
Tetrazolium testing of rice seeds
Tetrazolium testing was used for the rapid evaluation of seed viability as follows [15]: Three replicates of 30 seeds were randomly selected, stripped off their palea and lemma, and immersed in water for 12 h at room temperature. Afterwards, the whole seed was cut longitudinally and immersed in a 0.5% tetrazolium solution in a dark incubator for 3 h at 30 °C. Finally, the embryo stained red was identified as a viable seed according to the standard [15].
Subcellular localization of OsARF18 and OsMBD707 proteins in rice protoplasts
Rice protoplasts were used to determine the subcellular localization of the OsARF18 and OsMBD707 proteins. Rice plasticity 1 (OsRPL1) is nuclear localized [33], and the OsRPL1-CFP (35S: OSRPL1-CFP) is defined as a nuclear localization marker. OsARF18 and OsMBD707 were fluorescently fused to the pCAM1300 (YFP) vector, respectively. As localization signals for targets, OsARF18-YFP (35S: OsARF18-YFP) and OsMBD707-YFP (35S: OsMBD707-YFP) were produced. The findings of subcellular localization were obtained using an Olympus FV3000 laser confocal microscopy system (Olympus Co., Ltd. Tokyo, Japan).
Rice hull fungal pathogen isolation and propagation
In a 100 ml conical flask, add 20 rice seeds of each sample, 20 ml of sterilized water, and 1 drop of Tween 20. A 220 RPM shaker was then used to vibrate the conical container for 2 h after the cotton stopper was inserted. Take 20 μL from the conical flask and distribute it uniformly onto a sterile PDA plate, and then place it in an incubator at 28 °C, 12 h light/12 h dark for 2 days. The blank control was sterile water, and 4 replicates were set for each sample. Nikon Eclipse Ni (Nikon Co., Ltd. Tokyo, Japan) was used to photograph the seed hull. ABI 3730xl sequencing platform was used for ITS sequencing of fungus which was isolated and purified from the seed hull (Zhejiang Sunya Biotechnology Co., Ltd, Hangzhou, China), and the primer sequence was TCCTCCGCTTATTGATATGC.
Sequencing data validation by qRT-PCR
To validate the gene and miRNA expression data acquired from high-throughput sequencing, qRT-PCR was conducted on randomly chosen 20 genes and 6 miRNAs. The principle of miRNA primers was designed by Kramer [34] (Supplementary table 1). Primer Premier 6.0 (PREMIER Biosoft International, San Francisco, USA) was used to construct the gene-specific primers. The qRT-PCR assays were done using the LightCycler 96 System (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany) with three biological replicates and two technical replicates using the endogenous control L25 ribosomal protein (L18908) [35]. The heat map was created using normalized (separate z-score calculated per gene) qRT-PCR expression data.
Statistical analysis
SAS software was used to analyze data using one-way analysis of variance (ANOVA). The least significant difference of 0.05 was used for multiple comparisons (LSD, p < 0.05). Before statistical comparison, the percentage data were arcsintrans generated using ŷ=arcsin [sqrt (x/100)].
Results
Controlled deterioration treatment of tobacco seeds
The standard germination tests on all controlled deterioration materials (Fig. 1A) revealed that germination energy (Fig. 1B), germination percentage (Fig. 1C), mean germination time (Fig. 1D), and vigor index (Fig. 1E) all showed a significant decreasing trend with the extension of controlled deterioration time. Seeds treated with CD for 60 h (CD60h) performed similarly in germination performance as long-term storage (LS) seeds (Fig. 1F) in comparison with CK (Table 1). CD60h also showed no difference in POD (Fig. 1G), CAT (Fig. 1H), SOD (Fig. 1I) and GR (Fig. 1J) activities as compared with LS. Therefore, CD60h was eventually used as the CD material in transcriptional multiomics.
Fig. 1.

Tobacco seed deterioration and screening by controlled deterioration. (A) germination progress curve of K326 tobacco seeds with different controlled deterioration degrees. controlled deterioration condition: adjust the seeds water content to 20% and seal them with aluminum foil compound bags. Each bag contains 1 g seeds. A total of 21 aluminum foil bags. All the bags were placed at 45℃ constant temperature, and three bags (three repetitions of a sample) were taken out every 12 h for germination test. CD0h, CD12h, CD24h, CD36h, CD48h, CD60h, and CD72h respectively represent controlled deterioration treatment for 0 h, 12 h, 24 h, 36 h, 48 h, 60 h and 72 h. (B) Germination energy. (C) Germination percentage. (D) Mean germination time. (E) Vigor index. (F) Germination progress curve of K326 (CK) tobacco seeds with long-term storage (LS) and controlled deterioration for 60 h. (G) Peroxidase (POD) activity; (H) Catalase (CAT) activity; (I) Superoxide dismutase (SOD) activity; (J) Glutathione reductase (GR) activity. LSD (α = 0.01) method was used for multiple comparison.
Table 1.
Germination and vigor traits of tobacco seeds.
| Sample | GE (%) | GP (%) | GI | VI | MGT(d) |
|---|---|---|---|---|---|
| CK | 97.67 A | 97.67 A | 27.79 A | 0.231 A | 3.66 A |
| LS | 68.00B | 69.00B | 16.33B | 0.124B | 4.43B |
| CD | 66.67B | 66.67B | 14.80B | 0.111B | 4.62B |
GE: germination energy; GP: germination percentage; GI: germination index; MGT: mean germination time; VI: vigor index.
CK: a control; LS: long-term storage; CD: controlled deterioration.
LSD (α = 0.01) method was used for multiple comparison.
Transcriptome sequencing and differential gene expression analysis
CK, LS and CD each had 3 biological replicates, and a total of 9 samples were sequenced. A total of 59.24 Gb of clean data was acquired, with at least 5.96 Gb of data for each sample. Following the strict quality filters, 198,141,321 clean reads with at least 93.94 percent of bases scoring Q30 were produced. These reads were mapped to the reference genome (Nicotiana tabacum L., Ntab-K326, https://www.ncbi.nlm.nih.gov/assembly/GCA_000715075.1), with the mapping ratio varying from 93.80% to 95.34%. Mapping results identified 10,720 novel genes, 8,137 of which were functionally annotated. Table 2 summarizes the Illumina transcriptome sequencing results for tobacco.
Table 2.
Summary of Illumina sequencing for tobacco.
| Sample ID | CK1 | CK2 | CK3 | LS1 | LS2 | LS3 | CD1 | CD2 | CD3 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Transcriptome | Total Reads | 42,622,840 | 49,732,294 | 42,573,646 | 41,434,632 | 43,519,600 | 44,031,206 | 44,240,984 | 39,863,606 | 48,263,834 |
| Mapped Reads | 39,982,315 (93.80%) |
47,183,484 (94.87%) |
40,087,241 (94.16%) |
39,198,865 (94.60%) |
41,147,772 (94.55%) |
41,401,820 (94.03%) |
42,179,969 (95.34%) |
37,736,471 (94.66%) |
45,696,348 (94.68%) |
|
| Unique Mapped Reads | 39,027,372 (91.56%) |
45,629,269 (91.75%) |
39,102,164 (91.85%) |
37,815,101 (91.26%) |
39,503,497 (90.77%) |
39,249,961 (89.14%) |
40,429,785 (91.39%) |
36,363,694 (91.22%) |
44,031,747 (91.23%) |
|
| Multiple Map Reads | 954,943 (2.24%) |
1,554,215 (3.13%) |
985,077 (2.31%) |
1,383,764 (3.34%) |
1,644,275 (3.78%) |
2,151,859 (4.89%) |
1,750,184 (3.96%) |
1,372,777 (3.44%) |
1,664,601 (3.45%) |
|
| Reads Map to '+' | 19,900,201 (46.69%) |
23,289,454 (46.83%) |
19,956,917 (46.88%) |
19,315,647 (46.62%) |
20,163,271 (46.33%) |
20,045,365 (45.53%) |
20,615,689 (46.60%) |
18,568,401 (46.58%) |
22,450,587 (46.52%) | |
| Reads Map to '-' | 19,853,058 (46.58%) |
23,316,832 (46.88%) |
19,915,107 (46.78%) |
19,361,005 (46.73%) |
20,243,653 (46.52%) |
20,209,634 (45.90%) |
20,705,257 (46.80%) |
18,613,951 (46.69%) |
22,527,705 (46.68%) |
|
| GC (%) | 44.14 | 44.31 | 44.13 | 44.42 | 44.74 | 44.93 | 44.75 | 44.76 | 44.66 | |
| Q30 (%) | 93.94 | 94.62 | 94.77 | 94.48 | 94.8 | 94.84 | 94.92 | 94.36 | 94.62 | |
| Small RNA sequencing | Raw reads | 10,477,149 | 10,953,865 | 9,708,645 | 10,782,588 | 10,385,697 | 10,194,116 | 10,026,146 | 11,192,222 | 10,952,757 |
| Length < 18 | 45,683 | 47,414 | 46,735 | 54,943 | 54,524 | 62,959 | 38,954 | 47,982 | 44,626 | |
| Length > 30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Clean reads | 10,431,466 | 10,906,451 | 9,661,910 | 10,727,645 | 10,331,173 | 10,131,157 | 9,987,192 | 11,144,240 | 10,908,131 | |
| Q30 (%) | 95.39 | 97.2 | 97.44 | 96.2 | 97.07 | 97.39 | 96.62 | 97.18 | 97.1 | |
| Unannotated Reads | 5,642,352 | 5,950,969 | 5,004,153 | 4,898,855 | 4,691,841 | 4,354,346 | 5,323,006 | 5,620,048 | 6,237,320 | |
| Mapped Reads | 2,425,447 (42.99%) |
2,903,460 (48.79%) |
2,494,010 (49.84%) |
2,906,795 (59.34%) |
2,949,585 (62.87%) |
2,761,009 (63.41%) |
2,972,714 (55.85%) |
3,147,683 (56.01%) |
3,578,622 (57.37%) |
|
| Mapped reads ‘+’ | 1,717,946 (30.45%) |
2,057,964 (34.58%) |
1,769,847 (35.37%) |
2,055,999 (41.97%) |
2,081,665 (44.37%) |
1,953,601 (44.87%) |
2,107,669 (39.60%) |
2,228,967 (39.66%) |
2,550,516 (40.89%) |
|
| Mapped reads ‘-’ | 707,501 (12.54%) |
845,496 (14.21%) |
724,163 (14.47%) |
850,796 (17.37%) |
867,920 (18.50%) |
807,408 (18.54%) |
865,045 (16.25%) |
918,716 (16.35%) |
1,028,106 (16.48%) |
|
| Known miRNAs | 101 | 111 | 110 | 112 | 115 | 107 | 96 | 92 | 103 | |
| Novel miRNAs | 306 | 302 | 306 | 305 | 299 | 305 | 302 | 293 | 300 | |
| Total miRNAs | 407 | 413 | 416 | 417 | 414 | 412 | 398 | 385 | 403 | |
| Degradome | Data summary | Noncoding RNA annotation | Noncoding RNA unique tags | |||||||
| Clean Data | Q30 (%) | Unannotated Reads | Mapped | Pefect map | rRNA | tRNA | snoRNA | snRNA | Other | |
| 20,467,151 | 94.59 | 5,048,084 | 3,707,641 | 3,201,096 | 13,549 | 199 | 408 | 2 | 1,364 | |
The differentially expressed genes (DEGs) in LS and CD were discovered by comparing LS and CD with CK. A total of 2112 genes (fold change ≥ 1.5; P value < 0.01) (Fig. 2A) were expressed differentially after normalized read counts FPKM were calculated for each gene. There were 374 up-regulated genes and 748 down-regulated genes in LS, whereas 112 up-regulated genes and 1243 down-regulated genes were found in CD (Fig. 2B). KEGG enrichment was used to classify all of the DEGs in LS and CD. Protein processing in the endoplasmic reticulum (ko04141) pathway was found to be the most critical element in LS (Fig. 2C), while spliceosome (ko03040), ribosome biogenesis in eukaryotes (ko03008), RNA degradation (ko03018), phosphatidylinositol signaling system (ko04070) and RNA transport (ko03013) pathways were the most important factors due to CD (Fig. 2D). In addition, 365 (347 + 17 + 1, Fig. 2B) genes commonly involved in LS and CD regulation were annotated, and the results revealed that spliceosome (ko03040) was the most prevalent cause of seed deterioration (Fig. 2E).
Fig. 2.

Transcriptome analysis and KEGG pathway enrichment of tobacco seeds. (A) Heat map of enriched genes (the ratio of total; fold change > 1.5; P < 0.01) in three groups. Each group contained three biologically independent replicates (n = 3). (B) Statistics of DEG number. (C) KEGG pathway enrichment of DEGs in LS; (D) KEGG pathway enrichment of DEGs in CD; Each circle represents a KEGG pathway, as shown by the left legend. It shows the ratio of “x/y” (DEGs in the route vs. all DEGs in all pathways), and “y” (genes in all pathways). The rich factor denotes the pathway's importance. The color of the circle signified the q value, which is a multiple hypothesis test adjusted p value. (E) KEGG pathway enrichment of common DEGs in both LS and CD. The ordinate represents the name of KEGG pathway, and the abscissa represents the proportion of DEGs in the pathway with all genes in all pathways.
Small RNA sequencing and differential miRNA expression analysis
By constructing and sequencing nine small RNA libraries, miRNAs linked with seed deterioration were identified. Raw reads totaling 94.67 million with an average of 10.51 million reads per sample were obtained. 443,820 reads with a length < 18 nt or > 30 nt were first discarded, followed by the removal of rRNA, tRNA, snRNA, snoRNA, other ncRNA and reads mapping to repeats (Table 2). A total of 126 known miRNAs and 306 novel miRNAs were identified, in which hairpin energy ranged from −165.3 to −19.9 kcal/mol based on Randfold (Supplementary table 2). The total number of miRNAs in each sample ranged from 385 to 417. The length distribution of the small RNA reads indicated that 24 nt (42.82%) was the most abundant small RNA class, followed by 21 nt (26.85%), 22 nt (12.73%), and 20 nt small RNAs (9.72%) (Supplementary Fig. 1A). There was no significant base bias shown in each position of the miRNA nucleotide (Supplementary Fig. 1B-E).
Additionally, TPM was utilized to assess differentially expressed miRNAs (DEMs), and 164 miRNAs (fold change ≥ 1.5; P value < 0.05) were obtained in total, including 52 known and 112 novel miRNAs (Fig. 3A). In comparison to CK, LS detected 73 DEMs, whereas CD identified 120 DEMs (Fig. 3B). 29 DEMs were found to be functional in both LS and CD, but nta-miR6145d, nta-miR6146a, nta-miR6146b, nta-miR6149a, nta-miR6149b, nta-miR6153, and nta-miR6157 were expressed in completely opposite forms in LS and CD.
Fig. 3.

Small RNA, degradome sequencing and coherent miRNA-mRNA pairs searching in tobacco seeds. (A) Heat map of enriched miRNAs (the ratio of total; fold change > 1.5; P < 0.05) in three groups. Each group contained three biologically independent replicates (n = 3) used for the heat map. The heat map was generated using normalized (separate z-score computed per gene) expression values in units of TPM. (B) Statistics of DEM number. (C) nta-miR160b-NtARF18 alignments validated by degradome sequencing. (D) nta-miR396c-NtMBD10 alignments validated by degradome sequencing. The red dots represent the cleavage nucleotide positions on the target genes. (E) coherent miRNA-mRNA pairs searching using venn analysis. Degradome validated miRNAs and DEMs were gathered to find degradome validated DEMs, which were then used to find their target genes; Degradome validated genes and DEGs were gathered to find degradome validated DEGs, which were then used to find miRNAs targeting DEGs. (F) A combined view of the expression level of coherent pairs of differentially expressed miRNAs and their target genes. Heat map of coherent miRNAs and genes in two groups. Each group contained three biologically independent replicates (n = 3) used for the heat map. The heat map was generated using normalized (separate z-score computed per gene) expression values in units of TPM and FPKM respectively in miRNAs and genes. The abscissa is the sample name, and the ordinate is the miRNA or gene name with its NR annotation.
Target validation by degradome and coherent miRNA-mRNA pair screening
Degradome sequencing was used to confirm target genes with perfect or near-perfect matching. Through degradome sequencing, 20,467,151 of clean data (Q30 > 94.59%) was collected (Table 2), and a total of 263 targets were found for 51 known nta-miRNAs and 71 novel miRNAs, resulting in 267 target associations (P value < 0.05, category ≤ 4) (Supplementary table 3). Gene 1261 (auxin response factor 18, NtARF18) was cleaved by nta-miR160b (Fig. 3C) and Gene 12,019 (methyl-CpG-binding domain-containing protein 10, NtMBD10) by nta-miR396c (Fig. 3D).
The degradome-validated DEMs and DEGs were utilized to search for their partners (Fig. 3E), and a total of 54 validated DEMs and 15 validated DEGs were discovered. We summarized 20 coherent pairings with opposing regulatory interactions that were verified using degradome, including ten pairs belonging to LS and ten pairs belonging to CD (Fig. 3F). It showed that nta-miR160b, nta-miRNA167d/e, miR171a, nta-miR394, nta-miR6155, and 4 novel miRNAs played the core roles in LS, while nta-miR156a/e/f, nta-miR396c, and 6 novel miRNAs were involved in CD. As for their targets, they showed a completely opposite regulatory relationship. In LS, NtARF18, NtARF8, NtTPR4, NteIF3, Calcineurin subunit B, 60 s ribosomal protein L37a, CBSPB1, and Ribulose-phosphate 3-epimerase were down-regulated, while Senescence-associated protein and Galactinol synthase 1 were up-regulated. In CD, NtSPL6, NtSPL12, Tropinone reductase, F-box protein 7 and Acetylornithine deacetylase were up-regulated, while NtMBD10, protein phosphatase type 2c, CBSPB1, 4-alpha-glucanotransferase DEP2, and Anther-specific myb-related protein 1 were down-regulated.
DEGs’ co-expression trends and phylogenetic analyses
A DEGs’ co-expression trend analysis was carried out to find the target genes that are co-expressed with those in key differential pathways. It showed that nta-miR160b-NtARF18 played an important role in LS, which was co-expressed with those genes involved in plant hormone signal transduction (ko04075) and ribosome biogenesis in eukaryotes (ko03008), while nta-miR396c-NtMBD10 was effective in CD, which was co-expressed with those genes in nucleotide excision repair (ko03420), RNA degradation (ko03018), and spliceosome (ko03040) pathways (Fig. 4A).
Fig. 4.

DEGs’ co-expression trends and phylogenetic analyses in tobacco and rice. (A) DEGs’ co-expression trends analysis. (B) Phylogenetic analysis. All the ARF and MBD family genes in rice and tobacco were used to portray the evolution tree based on neighbor-joining method [78].
NtARF18, NtMBD10, and the other ARF and MBD family genes in tobacco and rice were annotated and clustered together to depict the phylogenetic tree to investigate the probable roles and similarities of target genes in seed deterioration (Fig. 4B). The homologous genes of NtARF18 found in rice were LOC_Os06g47150 (OsARF18), LOC_Os10g33940 (OsARF22), LOC_Os02g41800 (OsARF8), and LOC_Os04g43910 (OsARF10). LOC_Os12g42550 (OsMBD707) was the rice gene that corresponded to NtMBD10.
Validation and utilization of rice homologous target genes
The homologous genes LOC_Os06g47150 (OsARF18) and LOC_Os12g42550 (OsMBD707) were chosen to verify the target genes found by multiomics and phylogenetic methods in rice. Both OsARF18 (Supplementary Fig. 2A) and OsMBD707 (Supplementary Fig. 2B) are located in the nucleus, where they influence gene expression as transcription factors and methylation-binding proteins, respectively. Using the CRISPR/Cas9 technique, Osarf18 and Osmbd707 mutants with two lines were produced from Oryza sativa Japonica cv. ZH11 (Fig. 5A) [31].
Fig. 5.

Validation of rice homologous genes OsARF18 and OsMBD707. (A) The background of Osarf18 and Osmbd707 mutants. Mutants were created in the Oryza sativa Japonica cv. ZH11 background by CRISPR/Cas9 method [31]. (B-C) The rice seedlings after 14 days of a standard germination test, in which ZH11, Osarf18-1, Osarf18-2, Osmbd707-1 and Osmbd707-2 have been stored for 8 months at room temperature. The bar represents 50 mm. (D-F) Standard germination test of ZH11, Osarf18 and Osmbd707 stored for 4 and 8 months. (D) Germination percentage; (E) Germination index; (F) Vigor index. (G) The fold change of germination index and vigor index, which is represented by the percentage of difference between 4 months of storage and 8 months of storage. (H-I) Rice seedlings after 14 days of a standard germination test, in which ZH11, Osarf18-1, Osarf18-2, Osmbd707-1 and Osmbd707-2 have been treated for 72 h of controlled deterioration. The bar represents 50 mm. (J-L) Standard germination test of ZH11, Osarf18 and Osmbd707 before and after 72 h of controlled deterioration. (J) Germination percentage; (K) Germination index; (L) Vigor index. (M) The fold change of germination index and vigor index, which is represented by the percentage of difference before and after 72 h of controlled deterioration. LSD (α = 0.01) method was used for multiple comparison.
For Osarf18, Osmbd707, and ZH11, standard germination tests were conducted. After 8 months of storage at room temperature, the OsARF18 knockout resulted in rice seedling growth retardation (Fig. 5B), but the OsMBD707 knockout resulted in seedling growth acceleration (Fig. 5C). Compared with seed germination data stored for 4 months and 8 months, the GP (Fig. 5D), GI (Fig. 5E), and VI (Fig. 5F) of ZH11 and Osarf18 mutants declined dramatically, with the GI and VI of Osarf18 mutants decreasing by approximately 50% and more (Fig. 5G). However, Osmbd707 mutants still maintained 100% GP and the highest GI and VI among these materials (Fig. 5D–F). Tetrazolium testing results showed that the seed viability of Osarf18 was indeed significantly lower than that of ZH11 and Osmbd707 after 8 months of storage (Supplementary Fig. 3A). Short-term CD treatment greatly reduced the GI (Fig. 5K) and VI (Fig. 5L) of Osarf18, Osmbd707 and wild type ZH11, notably in Osmbd707 mutants (Fig. 5M). However, it did not considerably lower the GP (Fig. 5J) of any material, and the Tetrazolium testing also validated this result (Supplementary Fig. 3B).
The characteristics of seeds were also investigated (Supplementary Fig. 3C). The 1000 grain weight of Osarf18 mutants was considerably increased (Supplementary Fig. 3D) due to the full-scale growth of seed width (Supplementary Fig. 3E), thickness (Supplementary Fig. 3F), and length (Supplementary Fig. 3G). On the other hand, Osmbd707-1 and Osmbd707-2 seeds were much smaller than ZH11 and Osarf18-1 and Osarf18-2 seeds (Supplementary Fig. 3C-G). Furthermore, there were many black spots on the hull of Osarf18 mutants. It was identified as Talaromyces pinophilus, a highly cellulase-producing filamentous fungus (Supplementary Fig. 4).
Description of potential mechanism in seed deterioration
The mechanisms of seed deterioration in LS and CD varied at the transcriptional level (Fig. 6). Jasmonate-zim domain (JAZ), Ethylene receptor (ETR) PP2C, and ARF18, which belong to the plant hormone signal transduction pathway, were down-regulated in LS. Heat shock protein 70 (HSP70), Heat shock protein 90 (HSP90) and RING membrane-anchor 1(RMA1) were up-regulated in LS, and these proteins were implicated in protein processing in the endoplasmic reticulum (ER) pathway. Xeroderma pigmentosum type B (XPB1), Cullin 4A(CUL4), MBD10, Damage Specific DNA Binding Protein (DDB1) and DNA polymerase delta subunit 1 (POL1) were dramatically down-regulated in CD, which was related to the nucleotide excision repair pathway. At the same time, miR160b and miR396c played vital roles in LS and CD, respectively. Furthermore, the similarities between LS and CD were the down-regulation of Splicing Factor 3A (SF3A), Splicing Factor 3B (SF3B), Proline-rich protein 2 (PRP2), Proline-rich protein 3 (PRP3) in the spliceosome pathway, and Ribonuclease II/R family protein (RRP44), SKI2, CCR4-NOT Transcription Complex Subunit 1(cNOT1), and CCR4-NOT Transcription Complex Subunit 3 (cNOT3) in the RNA degradation pathway.
Fig. 6.

Graphical summary of mainly genes, miRNAs, and pathways involved in seed deterioration in tobacco. Each group contained three biologically independent replicates (n = 3) and their mean value was used for the heat map. The heat map was generated using normalized (separate z-score computed per gene) expression values in units of TPM and FPKM respectively representing miRNAs and genes.
Sequencing data validation by qRT-PCR
A quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis was used to confirm gene and miRNA expression patterns. qRT-PCR was used to verify the expression of 20 randomly chosen genes and 6 miRNAs (Supplementary Fig. 5). For the majority of the samples, qRT-PCR analysis revealed similar gene expression patterns (up-regulation or down-regulation) as high-throughput sequencing. These findings indicated a high degree of concordance between the results of high-throughput sequencing and qRT-PCR.
Discussion
Seed deterioration by controlled deterioration
Seeds can maintain their vitality at low moisture content and low temperatures for a long time. However, they still undergo inevitable deterioration, and the rate of deterioration dictates the seed longevity [36]. CD is a commonly accelerated ageing method used in practices to access the quality of seeds or to better understand the mechanism of seed deterioration. When seeds were treated by CD for a certain period of time, generally about 24 h, seed vigor would decrease significantly. However, short-term high temperature and high humidity treatments could improve seed vigor to a certain extent, which might be due to high moisture and high temperature in CD promoting the enhancement of seed imbibition and metabolism during this period [14]. A similar phenomenon was also found in our study. It indicated that there were some essential differences between CD and the deterioration caused by LS.
Characteristics of seed deterioration at the transcriptional level
Transcriptome analysis showed that there were some different genes involved in the protein processing of ER pathway (ko04141) in LS seeds, such as HSP90, HSP70 and so on. Increasing evidence indicates that ER plays a part in plant defense [37]. HSP90, along with HSP70 and co-chaperones, is crucial for DNA repair [38]. It is well known that the ability of the embryo’s genomic DNA repair to reverse DNA damage affects seed vigor and viability [39], [40]. The up-regulated expression of HSP90 and HSP70 indicated that the LS seeds were attempting to repair damaged genes and fighting against senescence in order to survive longer. Another feature of LS seed was the alteration of the plant hormone signal transduction (ko04075). The genes GH3, JAZ, ETR, and PP2C, which were respectively related to auxin, jasmonic acid, ethylene and abscisic acid, were all down-regulated in LS, and they all participated in the plant hormone signal transduction system [41], [42], [43].
Controlled deterioration cussed a particular change in nucleotide excision repair (ko03420), making the genomic response more susceptible to plant alterations [44]. CUL4 and DDB1 are essential components of the Cul4/DDB1 (XPE) and Cul4-CSA complexes, which are involved in global genome repair and transcription-coupled repair, respectively [45]. In the universal transcription factor TFIIH complex, XPB is the largest subunit necessary for both transcription and nucleotide excision repair [46], [47], [48]. The POL1 gene encodes the catalytic and proofreading subunits of DNA polymerase-delta, which is responsible for DNA synthesis of the lagging strand during DNA replication [49]. All of these changes suggested that CD led to failure of nucleotide excision repair, thereby reducing seed vigor and viability.
The common causes of LS and CD were spliceosome (ko03040) and RNA degradation (ko03018) pathways. Spliceosome is a multi-megadalton ribonucleoprotein complex made up of five snRNPs and other proteins that catalyzes pre-mRNA splicing by removing noncoding sections (introns) from a precursor messenger RNA (pre-mRNA) [50]. RNA degradation, whether as a monitoring mechanism to eliminate aberrant mRNAs or during RNA processing to create mature transcripts, plays a critical role in maintaining cellular homeostasis [51]. Changes in the primary components of the spliceosome and exosome severely limit the generation of mature transcripts and the removal of aberrant mRNAs, resulting in a decrease in seed vigor.
Seed deterioration characters at the small RNA level and utilization of key targets
The role of miRNA in the regulation of seed deterioration was further analyzed by small RNA and degradome sequencing technologies, which provided abundant information for the study of seed vigor mechanisms.
In LS seeds, up-regulated nta-miR160b and nta-miRNA167e were expressed contrary to their targets, NtARF18 and NtARF8, respectively. MiR167 is a conserved miRNA family that plays a role in growth and development control [52]. In Arabidopsis, over-expression of miR167c inhibits somatic embryo development by silencing its targets ARF6 and ARF8 [53], [54]. Plant growth and development have been shown to be required for MiR160 family members [55]. Transgenic rice plant expressing an miR160-resistant form of ARF18 exhibited pleiotropic defects, including small seeds and dwarf stature [56], [57]. Knockout of miR160 in Arabidopsis caused significantly smaller grain size and weight [58], while over-expression of miR160 decreases the sensitivity of Arabidopsis to ABA during seed germination [56]. ARFs are transcription factors that regulate the expression of early auxin response genes [59]. ARF10, ARF16 and ARF17 are targeted by miR160 in Arabidopsis thaliana [55]. Nta-miR160b cleaves NtARF18, and its rice homolog OsARF18 could be cleaved by Osa-miR160 [57]. In our research, knockout of OsARF18 increased 1000 grain weight, seed length, width and thickness, which had positive significance for the improvement of grain yield. However, the seeds of the Osarf18 mutant were more sensitive to LS, and seed viability declined faster at room temperature. Meanwhile, Osarf18 mutant seeds were more sensitive to a filamentous fungus called Talaromyces pinophilus, which infested the hull of rice seeds and produced excessive amounts of cellulase, causing rice grain blast. The sensitivity of the Osarf18 mutant to LS and fungi might be due to the decreased ABA sensitivity leading to the decline of overall plant adaptation, which still needs further study.
In CD seeds, the nta-miR156 family was down-regulated, while nta-miR396c was up-regulated. MiR156 is a highly conserved miRNA family [60]. Over-expression of miR156 might increase the level of DELLA and GA-decomposing enzymes, and eliminate the mutational background of miR156-non-targeting SPL, resulting in a more robust phenotype [61], [62]. MiR396 was previously shown to target a collection of transcription factor GRF (growth regulating factor) genes and to play a vital function in the response to biotic and abiotic stress [63], [64]. Blocking miR396 increased rice yield and conferred broad resistance to necrotrophic and hemibiotrophic fungal pathogens in Arabidopsis [64], [65], while over-expression of miR396 decreased rice tolerance to salt and alkali stress [66], as well as causing obvious floral organ defects in rice, Arabidopsis and tobacco [67], [68], [69]. NtMBD10 was cleaved by nta-miR396c, and has been shown to play a vital role in CD. The MBD protein may play a role in seed longevity, DNA damage repair and genome stability maintenance through decoding the genetic information encoded by methylated DNA [70], [71], [72], [73], [74], [75]. In Arabidopsis, AtMBD10 collaborated with AtMBD6, suppressing ribosomal DNA loci in nucleolar dominance [76]. Overexpression of the homologous gene OsMBD707 results in increased tiller angles and decreased photoperiod sensitivity [77]. In our work, the rice homolog OsMBD707 of NtMBD10 was used to verify its role in seed CD. Knockout of OsMBD707 did lead to an accelerated decline of seed vigor during CD, but significantly increased seed tolerance to LS compared with wide type and Osarf18, which might be a result of methylation-demethylation imbalance. Furthermore, it proved again that LS and CD did have some essential difference in seed deterioration.
Conclusion
Transcriptional multiomics revealed the mechanism of slow deterioration during long-term storage (LS) and fast deterioration under controlled deterioration (CD) in Nicotiana tabacum L. seeds. Simultaneously, a large number of transcripts associated with seed deterioration were discovered. The genes NtARF18 and NtMBD10 related to LS and CD in tobacco seeds were identified by multiomics analysis, and the homologous genes OsARF18 and OsMBD707 in rice were preliminarily verified to play similar roles in LS and CD, respectively. This study verifies the feasibility of multiomics in studying seed deterioration and also provides a novel way to identify seed deterioration genes in other species, thus providing guidance for precisely delaying seed deterioration.
Data available
Sequencing data has been submitted in NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) with the BioProject ID PRJNA695068.
Compliance with Ethics Requirements
This research meets all Ethics Requirements.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was supported by National Natural Science Foundation of China No. 32072127, Natural Science Foundation of Zhejiang Province No. LY21C130006, Dabeinong Funds for Discipline Development and Talent Training, and Collaborative Innovation Center for Modern Crop Production co-sponsored by Province and Ministry (CIC-MCP).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.03.009.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Bewley J.D. Seed Germination and Dormancy. Plant Cell. 1997;9(7):1055–1066. doi: 10.1105/tpc.9.7.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammond S.T., Brown J.H., Burger J.R., Flanagan T.P., Fristoe T.S., Mercado-Silva N., et al. Food Spoilage, Storage, and Transport: Implications for a Sustainable Future. Bioscience. 2015;65(8):758–768. [Google Scholar]
- 3.Ventura L., Donà M., Macovei A., Carbonera D., Buttafava A., Mondoni A., et al. Understanding the molecular pathways associated with seed vigor. Plant Physiol Bioch. 2012;60:196–206. doi: 10.1016/j.plaphy.2012.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Walters C., Wheeler L., Stanwood P.C. Longevity of cryogenically stored seeds. Cryobiology. 2004;48(3):229–244. doi: 10.1016/j.cryobiol.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Rajjou L., Lovigny Y., Groot S.P.C., Belghaz M., Job C., Job D. Proteome-wide characterization of seed aging in Arabidopsis: A comparison between artificial and natural aging protocols. Plant Physiol. 2008;148(1):620–641. doi: 10.1104/pp.108.123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fantazzini T.B., da Rosa S.D.V.F., Pereira C.C., Pereira D.D., Cirillo M.A., Ossani P.C. Association between the artificial aging test and the natural storage of coffee seeds(1) J Seed Sci. 2018;40(2):164–172. [Google Scholar]
- 7.Kimura M., Nambara E. Stored and neosynthesized mRNA in Arabidopsis seeds: effects of cycloheximide and controlled deterioration treatment on the resumption of transcription during imbibition. Plant Mol Biol. 2010;73(1-2):119–129. doi: 10.1007/s11103-010-9603-x. [DOI] [PubMed] [Google Scholar]
- 8.Min C.W., Lee S.H., Cheon Y.E., Han W.Y., Ko J.M., Kang H.W., et al. In-depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. J Proteomics. 2017;169:125–135. doi: 10.1016/j.jprot.2017.06.022. [DOI] [PubMed] [Google Scholar]
- 9.Garg V., Khan A.W., Kudapa H., Kale S.M., Chitikineni A., Sun Q.W., et al. Integrated transcriptome, small RNA and degradome sequencing approaches provide insights into Ascochyta blight resistance in chickpea. Plant Biotechnol J. 2019;17(5):914–931. doi: 10.1111/pbi.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S.S., Chen Y., Lu M.J. Degradome Sequencing in Plants. Methods Mol Biol. 2019;1932:197–213. doi: 10.1007/978-1-4939-9042-9_15. [DOI] [PubMed] [Google Scholar]
- 11.Han X., Yin H., Song X., Zhang Y., Liu M., Sang J., et al. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol J. 2016;14(6):1470–1483. doi: 10.1111/pbi.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y., Xu Z., Mo Q., Zou C., Li W., Xu Y., et al. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann Bot-London. 2013;112(3):633–642. doi: 10.1093/aob/mct133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y., Li L., Tang S., Liu J., Zhang H., Zhi H., et al. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. Bmc Genet. 2016;17(1) doi: 10.1186/s12863-016-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma W.-G., Zhang Z.-H., Zheng Y.-Y., Pan W., Qiu T., Guan Y.-J., et al. Determination of tobacco (Nicotiana tabacum) seed vigour using controlled deterioration followed by a conductivity test. Seed Sci Technol. 2020;48(1):1–10. [Google Scholar]
- 15.ISTA. International Rules for Seed Testing (2010 edn). Bassersdorf, Switzerland: International Seed Testing Association; 2010.
- 16.Xu S., Hu J., Li Y., Ma W., Zheng Y., Zhu S. Chilling tolerance in Nicotiana tabacum induced by seed priming with putrescine. Plant Growth Regul. 2011;63(3):279–290. [Google Scholar]
- 17.Luo Y., Guan Y.J., Huang Y.T., Li J., Li Z., Hu J. Single counts of radicle emergence provides an alternative method to test seed vigour in sweet corn. Seed Sci Technol. 2015;43(3):519–525. [Google Scholar]
- 18.Chance B, Maehly A. [136] Assay of catalases and peroxidases. 1955. [DOI] [PubMed]
- 19.Aebi H. [13] Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 20.Giannopolitis C.N., Ries S.K. Superoxide Dismutases. 1. Occurrence in Higher-Plants. Plant Physiol. 1977;59(2):309–314. doi: 10.1104/pp.59.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang S.u., Ulhassan Z., Shah A.M., Khan A.R., Azhar W., Hamid Y., et al. Salicylic acid underpins silicon in ameliorating chromium toxicity in rice by modulating antioxidant defense, ion homeostasis and cellular ultrastructure. Plant Physiol Bioch. 2021;166:1001–1013. doi: 10.1016/j.plaphy.2021.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12) doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mao X., Cai T., Olyarchuk J.G., Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 25.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedlander M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Addo-Quaye C., Miller W., Axtell M.J. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25(1):130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C. jvenn: an interactive Venn diagram viewer. Bmc Bioinformatics. 2014;15(1) doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madeira F., Park Y.m., Lee J., Buso N., Gur T., Madhusoodanan N., et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47(W1):W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian B., Gao S., Lercher M.J., Hu S., Chen W.H. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019;47(W1):W270–W275. doi: 10.1093/nar/gkz357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma X., Zhang Q., Zhu Q., Liu W., Chen Y., Qiu R., et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol Plant. 2015;8(8):1274–1284. doi: 10.1016/j.molp.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Kalpanadevi C., Singh V., Subramanian R. Impact of physicochemical properties on duration and head rice yield during abrasive and friction milling of rice. J Food Sci Tech Mys. 2019;56(8):3900–3909. doi: 10.1007/s13197-019-03861-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C.C., Yuan W.-Y., Zhang Q.F. RPL1, a Gene Involved in Epigenetic Processes Regulates Phenotypic Plasticity in Rice. Mol Plant. 2012;5(2):482–493. doi: 10.1093/mp/ssr091. [DOI] [PubMed] [Google Scholar]
- 34.Kramer M.F. Stem‐Loop RT‐qPCR for miRNAs. Curr Protocols Mol Biol. 2011;95(1) doi: 10.1002/0471142727.mb1510s95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt G.W., Delaney S.K. Stable internal reference genes for normalization of real-time RT-PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol Genet Genomics. 2010;283(3):233–241. doi: 10.1007/s00438-010-0511-1. [DOI] [PubMed] [Google Scholar]
- 36.Walters C., Ballesteros D., Vertucci V.A. Structural mechanics of seed deterioration: Standing the test of time. Plant Sci. 2010;179(6):565–573. [Google Scholar]
- 37.Ai G., Zhu H., Fu X., Liu J., Li T., Cheng Y., et al. Phytophthora infection signals-induced translocation of NAC089 is required for endoplasmic reticulum stress response-mediated plant immunity. Plant J. 2021;108(1):67–80. doi: 10.1111/tpj.15425. [DOI] [PubMed] [Google Scholar]
- 38.Niu P., Liu L., Gong Z., Tan H., Wang F., Yuan J., et al. Overexpressed heat shock protein 70 protects cells against DNA damage caused by ultraviolet C in a dose-dependent manner. Cell Stress Chaperon. 2006;11(2):162. doi: 10.1379/CSC-175R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterworth W.M., Bray C.M., West C.E. Seeds and the Art of Genome Maintenance. Frontiers. Plant Sci. 2019;10 doi: 10.3389/fpls.2019.00706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugliani M., Rajjou L., Clerkx E.J.M., Koornneef M., Soppe W.J.J. Natural modifiers of seed longevity in the Arabidopsis mutants abscisic acid insensitive3-5 (abi3-5) and leafy cotyledon1-3 (lec1-3) New Phytol. 2009;184(4):898–908. doi: 10.1111/j.1469-8137.2009.03023.x. [DOI] [PubMed] [Google Scholar]
- 41.Natarajan B., Kalsi H.S., Godbole P., Malankar N., Thiagarayaselvam A., Siddappa S., et al. MiRNA160 is associated with local defense and systemic acquired resistance against Phytophthora infestans infection in potato. J Exp Bot. 2018;69(8):2023–2036. doi: 10.1093/jxb/ery025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huot B., Yao J., Montgomery B.L., He S.Y. Growth-Defense Tradeoffs in Plants: A Balancing Act to Optimize Fitness. Mol Plant. 2014;7(8):1267–1287. doi: 10.1093/mp/ssu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miransari M., Smith D.L. Plant hormones and seed germination. Environ Exp Bot. 2014;99:110–121. [Google Scholar]
- 44.Chien L.-C., Wu Y.-H., Ho T.-N., Huang Y.-Y., Hsu T. Heat stress modulates nucleotide excision repair capacity in zebrafish (Danio rerio) early and mid-early embryos via distinct mechanisms. Chemosphere. 2020;238 doi: 10.1016/j.chemosphere.2019.124653. [DOI] [PubMed] [Google Scholar]
- 45.Gao J., Buckley S.M., Cimmino L., Guillamot M., Strikoudis A., Cang Y., et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. Elife. 2015;4 doi: 10.7554/eLife.07539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo J., Cimermancic P., Viswanath S., Ebmeier C., Kim B., Dehecq M., et al. Architecture of the Human and Yeast General Transcription and DNA Repair Factor TFIIH. Mol Cell. 2015;59(5):794–806. doi: 10.1016/j.molcel.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rimel J.K., Taatjes D.J. The essential and multifunctional TFIIH complex. Protein Sci. 2018;27(6):1018–1037. doi: 10.1002/pro.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greber B.J., Nguyen T.H.D., Fang J., Afonine P.V., Adams P.D., Nogales E. The cryo-electron microscopy structure of human transcription factor IIH. Nature. 2017;549(7672):414–417. doi: 10.1038/nature23903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palles C., Cazier J.B., Howarth K.M., Domingo E., Jones A.M., Broderick P., et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genet. 2013;45(6):713. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Will C.L., Luhrmann R. Spliceosome Structure and Function. Csh Perspect Biol. 2011;3(7) doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sokhi U.K., Das S.K., Dasgupta S., Emdad L., Shiang R., DeSalle R., et al. Human Polynucleotide Phosphorylase (hPNPaseold-35): Should I Eat You or Not-That Is the Question? Adv Cancer Res. 2013;119:161–190. doi: 10.1016/B978-0-12-407190-2.00005-8. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Z.-X., Feng Q., Cao X.-L., Zhu Y., Wang H.e., Chandran V., et al. Osa-miR167d facilitates infection of Magnaporthe oryzae in rice. J Integr Plant Biol. 2020;62(5):702–715. doi: 10.1111/jipb.12816. [DOI] [PubMed] [Google Scholar]
- 53.Su Y.H., Liu Y.B., Zhou C., Li X.M., Zhang X.S. The microRNA167 controls somatic embryogenesis in Arabidopsis through regulating its target genes ARF6 and ARF8. Plant Cell Tiss Org. 2016;124(2):405–417. [Google Scholar]
- 54.Gutierrez L., Mongelard G., Floková K., Păcurar D.I., Novák O., Staswick P., et al. Auxin Controls Arabidopsis Adventitious Root Initiation by Regulating Jasmonic Acid Homeostasis. Plant Cell. 2012;24(6):2515–2527. doi: 10.1105/tpc.112.099119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X., Huang J., Wang Y., Khanna K., Xie Z., Owen H.A., et al. The role of floral organs in carpels, an Arabidopsis loss-of-function mutation in MicroRNA160a, in organogenesis and the mechanism regulating its expression. Plant J. 2010;62(3):416–428. doi: 10.1111/j.1365-313X.2010.04164.x. [DOI] [PubMed] [Google Scholar]
- 56.Liu P.P., Montgomery T.A., Fahlgren N., Kasschau K.D., Nonogaki H., Carrington J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007;52(1):133–146. doi: 10.1111/j.1365-313X.2007.03218.x. [DOI] [PubMed] [Google Scholar]
- 57.Huang J., Li Z., Zhao D. Deregulation of the OsmiR160 Target Gene OsARF18 Causes Growth and Developmental Defects with an Alteration of Auxin Signaling in Rice. Sci Rep. 2016;6(1) doi: 10.1038/srep29938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y., Shi C., Yang T., Zhao L., Chen J., Zhang N., et al. High-throughput sequencing revealed that microRNAs were involved in the development of superior and inferior grains in bread wheat. Sci Rep. 2018;8(1) doi: 10.1038/s41598-018-31870-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagen G., Guilfoyle T. Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol. 2002;49(3–4):373–385. [PubMed] [Google Scholar]
- 60.Jerome Jeyakumar J.M., Ali A., Wang W.-M., Thiruvengadam M. Characterizing the Role of the miR156-SPL Network in Plant Development and Stress Response. Plants-Basel. 2020;9(9):1206. doi: 10.3390/plants9091206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xing S.P., Salinas M., Hohmann S., Berndtgen R., Huijser P. miR156-Targeted and Nontargeted SBP-Box Transcription Factors Act in Concert to Secure Male Fertility in Arabidopsis. Plant Cell. 2010;22(12):3935–3950. doi: 10.1105/tpc.110.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun T.-P. The Molecular Mechanism and Evolution of the GA-GID1-DELLA Signaling Module in Plants. Curr Biol. 2011;21(9):R338–R345. doi: 10.1016/j.cub.2011.02.036. [DOI] [PubMed] [Google Scholar]
- 63.Yuan S., Li Z., Yuan N., Hu Q., Zhou M., Zhao J., et al. MiR396 is involved in plant response to vernalization and flower development in Agrostis stolonifera. Hortic Res-England. 2020;7(1) doi: 10.1038/s41438-020-00394-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soto-Suárez M., Baldrich P., Weigel D., Rubio-Somoza I., San Segundo B. The Arabidopsis miR396 mediates pathogen-associated molecular pattern-triggered immune responses against fungal pathogens. Sci Rep. 2017;7(1) doi: 10.1038/srep44898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao F., Wang K., Liu Y., Chen Y., Chen P., Shi Z., et al. Blocking miR396 increases rice yield by shaping inflorescence architecture. Nat Plants. 2016;2(1) doi: 10.1038/nplants.2015.196. [DOI] [PubMed] [Google Scholar]
- 66.Gao P., Bai X.i., Yang L., Lv D., Li Y., Cai H., et al. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta. 2010;231(5):991–1001. doi: 10.1007/s00425-010-1104-2. [DOI] [PubMed] [Google Scholar]
- 67.Liu H., Guo S., Xu Y., Li C., Zhang Z., Zhang D., et al. OsmiR396d-Regulated OsGRFs Function in Floral Organogenesis in Rice through Binding to Their Targets OsJMJ706 and OsCR4. Plant Physiol. 2014;165(1):160–174. doi: 10.1104/pp.114.235564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liang G., He H., Li Y., Wang F., Yu D. Molecular Mechanism of microRNA396 Mediating Pistil Development in Arabidopsis. Plant Physiol. 2014;164(1):249–258. doi: 10.1104/pp.113.225144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baucher M., Moussawi J., Vandeputte O.M., Monteyne D., Mol A., Pérez-Morga D., et al. A role for the miR396/GRF network in specification of organ type during flower development, as supported by ectopic expression of Populus trichocarpa miR396c in transgenic tobacco. Plant Biol. 2013;15(5):892–898. doi: 10.1111/j.1438-8677.2012.00696.x. [DOI] [PubMed] [Google Scholar]
- 70.Jin B.L., Robertson K.D. DNA Methyltransferases, DNA Damage Repair, and Cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore L.D., Le T., Fan G. DNA Methylation and Its Basic Function. Neuropsychopharmacol. 2013;38(1):23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lombard D.B., Chua K.F., Mostoslavsky R., Franco S., Gostissa M., Alt F.W. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 73.Zou X.Q., Ma W., Solov'yov I.A., Chipot C., Schulten K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012;40(6):2747–2758. doi: 10.1093/nar/gkr1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Law J.A., Jacobsen S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sen P., Shah P.P., Nativio R., Berger S.L. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166(4):822–839. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Preuss S.B., Costa-Nunes P., Tucker S., Pontes O., Lawrence R.J., Mosher R., et al. Multimegabase Silencing in Nucleolar Dominance Involves siRNA-Directed DNA Methylation and Specific Methylcytosine-Binding Proteins. Mol Cell. 2008;32(5):673–684. doi: 10.1016/j.molcel.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qu M., Zhang Z., Liang T., Niu P., Wu M., Chi W., et al. Overexpression of a methyl-CpG-binding protein gene OsMBD707 leads to larger tiller angles and reduced photoperiod sensitivity in rice. Bmc. Plant Biol. 2021;21(1) doi: 10.1186/s12870-021-02880-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saitou N., Nei M. The Neighbor-Joining Method - a New Method for Reconstructing Phylogenetic Trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.