

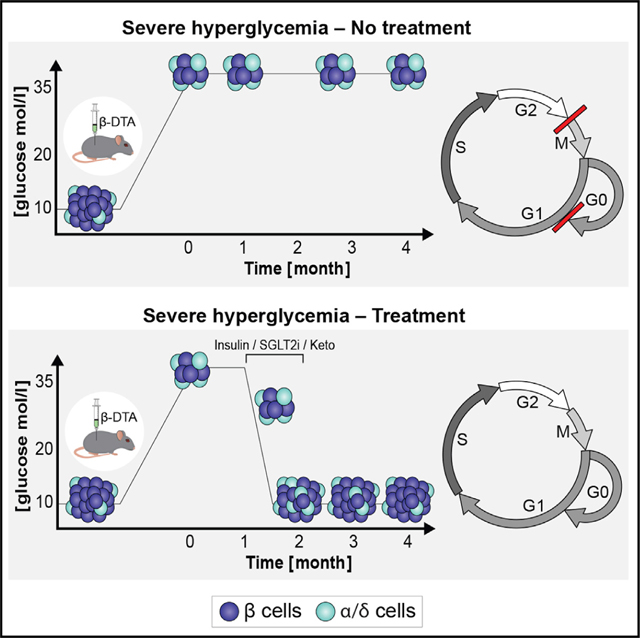
SUMMARY
Diabetogenic ablation of beta cells in mice triggers a regenerative response whereby surviving beta cells proliferate and euglycemia is regained. Here, we identify and characterize heterogeneity in response to beta cell ablation. Efficient beta cell elimination leading to severe hyperglycemia (>28 mmol/L), causes permanent diabetes with failed regeneration despite cell cycle engagement of surviving beta cells. Strikingly, correction of glycemia via insulin, SGLT2 inhibition, or a ketogenic diet for about 3 weeks allows partial regeneration of beta cell mass and recovery from diabetes, demonstrating regenerative potential masked by extreme glucotoxicity. We identify gene expression changes in beta cells exposed to extremely high glucose levels, pointing to metabolic stress and downregulation of key cell cycle genes, suggesting failure of cell cycle completion. These findings reconcile conflicting data on the impact of glucose on beta cell regeneration and identify a glucose threshold converting glycemic load from pro-regenerative to anti-regenerative.
In brief
Furth-Lavi et al. show that massive beta cell ablation resulting in extreme hyperglycemia blocks beta cell replication, preventing recovery of glycemic control by downregulating key cell cycle genes and suppressing cell cycle progression. This blockade is reversed by lowering glucose levels with insulin, an SGLT2 inhibitor, or ketogenic diet.
Graphical Abstract
INTRODUCTION
During persistent hyperglycemia, beta cells are under constant stress due to glucose overloading,1 initiating multiple metabolic or signaling pathways that attempt to dispose of excessive glucose, to minimize damage due to endoplasmatic, oxidative, and genotoxic stress and to increase beta cell proliferation to compensate for increased demand.2,3 However, ultimately, hyperglycemia is toxic to beta cell regeneration, function, and survival. The dual effect of glucose and the mechanisms determining regeneration or compensation versus toxicity and failure remain poorly understood.
Hyperglycemia can be induced in rodents by reduction of beta cell mass using several approaches, including partial pancreatectomy4,5 and the administration of beta cell-selective toxins, such as streptozotocin or alloxan.6 The former results in a simultaneous decrease of non-beta islet cells, such as alpha and delta cells as well as exocrine tissue, and is also associated with significant surgical stress, complicating interpretation of results. Beta cell toxins can result in damage to surviving beta cells and to other organs, also complicating subsequent analyses. To address these shortcomings, we previously developed a transgenic mouse model in which the subunit A of diphtheria toxin is conditionally expressed exclusively in beta cells upon provision of doxycycline (Insulin-rtTA;TET-DTA mice).7 Because of the extreme toxicity of DTA, any beta cell that expresses even a minute amount will succumb. Therefore, unlike other beta cell toxins, which may leave some beta cells alive but permanently damaged, surviving beta cells in this model are considered to be normal and undamaged.
Using this mouse model, we previously demonstrated that partial beta cell ablation, which causes random blood glucose levels to increase to 16.7–33.3 mmol/L, is followed by increased proliferation of surviving beta cells, resulting in most cases in normalization of glucose levels and near-complete recovery of beta cell mass within 10–23 weeks after ablation.7
However, a minority of transgenic mice failed to recover, and remained severely hyperglycemic. In this study we further characterized these mice and investigated the mechanisms underlying the failure to recover, initially hypothesizing that massive destruction resulted in insufficient residual beta cell mass to permit recovery, as demonstrated by Thorel et al. using a similar model of massive beta cell destruction.8 Surprisingly, our findings refuted this hypothesis and, in doing so, we identified and characterized a level of glucotoxicity that is associated with extreme hyperglycemia and results in reversible suppression of beta cell proliferation.
RESULTS
Following massive beta cell destruction, a subgroup of mice remains hyperglycemic
As reported previously, within 1 week of doxycycline treatment Insulin-rtTA;TET-DTA mice reach a peak blood glucose level of 19 to >33 mmol/L (Figure 1A).7 During 2 months of follow-up without doxycycline and on a normal chow diet two distinct subgroups emerged. One subgroup achieved normal or near-normal glycemia (defined here as random glucose levels below 19.4 mmol/L; mean random glucose at 2 months, 11.5 ± 0.5 SE mmol/L), associated with marked improvement of beta cell mass and normalization of islet architecture. The other subgroup of animals failed to recover glycemic control after more than 2 months (mean random glucose at 2 months, 30.8 ± 0.6 mmol/L) (Figures 1A and 1B). Importantly, the two groups showed significantly different random glucose levels 1 day after the end of ablation (average, 26.9 ± 0.8 versus 31.6 ± 0.3 mmol/L, respectively; p < 0.001). In fact, random glucose level at this time point predicted long-term outcome; glucose levels <28.1 mmol/L 1 day after ablation predicted successful recovery in 78.9% of the animals (referred to as the moderate hyperglycemic group [modHG]) and glucose levels >28.1 mmol/L predicted recovery in only 19% of the animals (referred to as the severe hyperglycemic group [sevHG]).
Figure 1. Characteristics of hyperglycemic mice.

(A) One-month-old insulin-rtTA;TET-DTA mice were treated with doxycycline in the drinking water for 1 week and blood glucose levels were monitored for2 months following withdrawal of doxycycline, showing that most insulin-rtTA;TET-DTA mice reduced glucose levels spontaneously (red lines, n = 23 mice), whereas some mice remained hyperglycemic throughout the follow-up period (black lines, n = 43 mice). Blue line represents wild-type (WT) littermates (n = 6 mice). Mean values of each group are shown in dark lines.
(B) Violin plot showing random glucose levels 58 days after doxycycline ablation with black dots representing the animals with severe hyperglycemia, the red dotswith moderate hyperglycemia (see text for definition), and blue dots representing control, WT mice.
(C–G) Panels show glucose (C), insulin (D), insulin/glucose ratios (E), percent Ki67-positive beta cells, n = 6–8 mice per group (F), and beta cell mass (G) in modHG and sevHG mice 1 day after doxycycline treatment, n = 3–4 mice per group.
(H) Representative immunofluorescence images of islets from control, modHG, and sevHG mice co-stained for insulin (green), glucagon (red), and somatostatin (white). Scale bar, 50 μm. In this and subsequent figures, error bars represent standard error of the mean,*p < 0.05, **p < 0.01, ***p < 0.005, ns, p > 0.05.
To further characterize these two groups, we measured random glucose levels, insulin levels, the fraction of proliferating beta cells, and beta cell mass 1 day after ablation in a separate cohort and divided the animals into two groups according to the threshold defined above (modHG = glucose <28.1 mmol/L, mean ± SE glucose 26.6 ± 0.5 mmol/L and sevHG = glucose >28.1 mmol/L, mean ± SE glucose 30.8 ± 0.66 mmol/L; p = 0.0004) (Figures 1C–1G). Whereas absolute insulin levels tended to be lower in the sevHG group (0.99 versus 1.50 ng/mL, p = 0.073), insulin/glucose ratios were significantly lower in this group (0.032 versus 0.056, p = 0.027), suggesting more severe insulin deficiency. The percent of beta cells entering the cell cycle (defined as Ki67 positive on immunostaining) was similar in the two groups (sevHG 4.45% versus modHG 4.81%, p = 0.68) (Figure 1F) and considerably higher than the percentage of Ki67 positivity in beta cells of wild-type (WT) mice of the same age (~2.1%7). However, sevHG mice had significantly lower beta cell mass than modHG mice (0.26 ± 0.03 versus 0.41 ± 0.02 mg, p = 0.014) or WT mice (~1.3 mg)7) (Figure 1G), indicating more efficient beta cell ablation in sevHG mice. Furthermore, immunostaining revealed differences in islet architecture with a more profound decrease in the abundance of beta cells and more alpha and delta cells in the center of the islets of sevHG mice (Figure 1H). We conclude that highly efficient ablation of beta cells, leading to profound disruption of islet architecture and to severe hyperglycemia (>28.1 mmol/L), reduces the likelihood of spontaneous recovery of beta cell mass and glycemia.
Reduction of glycemic load results in beta cell recovery and sustained improvement in glycemic control
We considered various explanations for the failed regeneration of beta cells after severe ablation, proposing the most likely to be cell autonomous. This model predicts that the number of remaining beta cells is insufficient to restore beta cell mass. However, a plausible alternative model suggests that the mechanism of failed recovery is non-cell autonomous, caused by depletion of a necessary trophic factor, such as insulin, or through the direct effect of a disrupted metabolic milieu. We therefore designed experiments to distinguish between these models, starting with the provocative hypothesis that correction of the toxic milieu created by severe insulin deficiency would release the regenerative potential of islets.
To test this hypothesis, 1-month-old Insulin-rtTA;TET-DTA mice were treated with doxycycline in the drinking water for 1 week and were followed for 1 month to exclude mice that recover spontaneously (Figure 2A). Only mice with glucose levels >28.1 nmol/L 1 month after doxycycline were included in the study, which consisted of 1 month treatment with various interventions (see below) followed by an additional month of follow-up without treatment and on a regular laboratory chow diet. In all of the experiments the mice that did not receive active intervention during the month-long treatment period (control mice) failed to demonstrate any improvement in glycemic control, confirming that spontaneous recovery did not occur in these severely ablated mice (Figure 2B).
Figure 2. Reduction of glycemic load results in long-term glucose normalization.

(A) Experimental design. One-month-old insulin-rtTA;TET-DTA mice were treated with doxycycline in the drinking water for 1 week and followed for 1 month to exclude mice that recover spontaneously. Mice with glucose levels >30.5 mmol/L 1 month after doxycycline were treated with insulin, SGLT2, or ketogenic diet (treatment) for 1 month and then followed for an additional month on normal chow diet.
(B) Glucose levels during and after treatment with insulin pellets (n = 14 mice, red), SGLT2i in drinking water (n = 12 mice, green), a ketogenic diet (n = 14 mice, blue), or normal chow diet (controls, n = 32 mice, black) for 1 month (day 0 to day 30) and for an additional month on normal chow. Glucose values for WT mice (n = 5 mice) are shown in gray.
(C) Treated mice from all three treatment cohorts could be divided into two distinct groups (responders, n = 20 mice; and non-responders, n = 20 mice) based on glucose levels 1 month after completion of treatment (see text for details). Dots are colored to represent the treatments given: red, insulin; green, sglt2i; blue, ketogenic diet.
(D) Mean blood glucose levels for responder (solid line) and non-responder (dashed line) mice in each treatment group (color coded) as in (B) (responders, n = 9, 4,and 7; non-responders, n = 5, 8, and 7 mice for insulin, SGLT1i, and KD, respectively). Untreated beta cell ablated (black, n = 32 mice) and WT (gray, n = 5 mice) mice are shown for comparison.
(E) Morphometric assessment of beta cell mass 2 months after beginning of treatment in responders (n = 11 mice) and non-responders (n = 8 mice).
(F) Quantification of beta cell replication 2 months after beginning of treatment in responders and non-responders observed by Ki67 immunostaining. Percentage of proliferation is calculated as the 100 3 number of Ki67+ insulin+ cells/the number of insulin+ cells.
(G) Representative immunofluorescence images of islets from responders and non-responders (n = 3 mice per group) co-stained for insulin (green), glucagon (red), and somatostatin (white). Representative islets from WT and untreated hyperglycemic mice are shown for comparison. Scale bar, 50 μm.
First, we corrected the hyperglycemia with subcutaneous insulin-eluting pellets (~0.5 U insulin/35 g body weight/day), which resulted in an acute decrease in glucose levels to a nadir of 8.8 ± 1.7 mmol/L at day 8. These pellets are calibrated to release insulin over a 30-day period. Indeed, after 20 days, glucose levels increased slowly to a mean of about 16.7 mmol/L measured at day 30. Thereafter, mean glucose levels remained stable at ~18.9 mmol/L until the conclusion of the experiment (Figure 2B). These results indicate that, even after severe beta cell ablation, mice retain the capacity for effective beta cell recovery, but realization of this potential is dependent upon correction of metabolic milieu.
Insulin administration has metabolic effects in addition to lowering glucose, including suppressing lipolysis and ketone body production, while directly stimulating intracellular growth-promoting pathways through insulin receptors expressed on beta cell membranes.9,10 To test whether non-glucose-related effects of insulin contributed to improved recovery, we treated another cohort of mice with the SGLT2 inhibitor dapagliflozin administered in the drinking water. Dapagliflozin inhibits glucose reuptake in the kidney, resulting in reduced glucose levels, without stimulating insulin secretion or suppressing ketone body production.11 Treatment with dapagliflozin resulted in highly significant reduction of glucose levels (nadir 13.6 ± 1 mmol/L at 28 days of treatment). This reduction was maintained for 1 month, until cessation of treatment, after which glucose levels increased to a mean of 21.1 mmol/L (which was significantly lower than glucose levels at the end of beta cell ablation), and were maintained at this level until the conclusion of the experiment (Figure 2B).
Next, we administered a very low carbohydrate, ketogenic diet (KD) to reduce glucose levels by an additional mechanism, substrate deprivation. This diet resulted in a decrease of glucose to a nadir of 16.3 ± 1.5 mmol/L at 21 days. Following cessation of the diet after 30 days and returning to normal laboratory chow, mean glucose levels remained unchanged at 17.4 ± 1.3 mmol/L (Figure 2B).
Three weeks of glycemic control are required to facilitate long-term beta cell recovery
Post-hoc review of individual data revealed that, in each of the three treatment groups, the mice could be divided into two distinct subgroups. Mice that recovered glycemic control and maintained near-normal glucose levels for the duration of the post treatment follow-up (13.4 ± 0.6, 12.1 ± 0.7, 11.3 ± 0.3 mmol/L for insulin, SGLT2i, and KD treatment mice, respectively, referred to as responders), and mice whose glucose levels increased to near-pre-treatment levels and remained there for the duration of the study (26.7 ± 2.9, 28.7 ± 2.9, 23.4 ± 2.6 mmol/L for insulin, SGLT2i, and KD treatment mice, respectively, referred to as non-responders) (Figures 2C, 2D, and S1A). To determine whether this dichotomous response to treatment persists over time, we monitored glucose levels for three additional months in an independent cohort and observed that this clear dichotomy was maintained for an additional 3 months (Figure S1B). The percentage of mice that responded to each treatment ranged from 35% (SGLT2i group) to 50% (insulin- and KD-treated groups). We combined the data from all three interventions, defining two distinct groups, responders (n = 20 mice), with glucose levels of 12.3 ± 0.5 mmol/L at the conclusion of the study, and non-responders (n = 20 mice), with glucose levels of 26.4 ± 1.4 mmol/L at the conclusion of the study (Figure 2C). As predicted, beta cell mass and proliferation index increase was greater in responders compared with non-responders (p < 0.005) (Figures 2E and 2F). Interestingly, beta cell mass in the entire cohort appeared to distribute normally, with responders having a significantly higher mass but not comprising a clearly identifiable sub-population. This suggests that mass per se may not be the only driver of the observed dichotomous response in glucose levels, and a functional component may be playing a role as well. Alternatively, normalization of glucose may require beta cell mass above a certain threshold. Although our data do not show a clear threshold effect, a larger sample size may. In contrast to what was found for beta cell proliferation, beta cell death, measured by TUNEL staining, was detected in a very small fraction of the cells (less than 0.1%) and did not show any significant difference between any of the study groups (Figure S1C). Islet architecture returned to normal in responding mice from all three treatment groups but not in non-responding mice (n = 3 mice from each group) (Figure 2G). We also observed a marked decrease in expression of Nkx6.1, but not Pdx1 (Figures S1F and S1G), as well as the appearance of bihormonal cells expressing glucagon or gastrin12 along with insulin in the untreated severely hyperglycemic mice (Figures S1D and S1E). Improvement of glycemia resulted in normalization of all of these parameters (Figures S1H and S1I).
We then analyzed individual mouse data from the three treatment groups to identify factors that predict which mice responded to treatment by maintaining near euglycemia after cessation of treatment (n = 20 mice, responders) and which mice reverted to severe hyperglycemia following treatment cessation (n = 20 mice, non-responders). A glucose level below 16 mmol/L after 3 weeks of treatment was the best predictor of long-term glycemic recovery regardless of which treatment was given, with a chance of recovery of 72% in mice with glucose levels below this threshold compared with 13% in those above this threshold (chi-square with YC = 12.9, p = 0.0003). Interestingly, in the insulin pellet-treated group, the pellet failed within the first 8–14 days in 2 animals, neither of which ultimately recovered. In the 12 remaining animals, only those in which the administered insulin successfully maintained glucose levels below 17 mmol/L for at least 3 weeks recovered (data not shown). This suggests that the failure to lower blood glucose for the full 3-week period was related to treatment failure and not to intrinsic differences between the two groups.
To further characterize those animals that recover, we measured beta cell mass in a separate cohort immediately upon completion of treatment (day 30 shown in Figure 2A). Those animals with blood glucose levels below 16 mmol/L after 3 weeks of treatment had a significantly higher beta cell mass compared with those with glucose levels >16 mmol/L at 3 weeks (0.72 ± 0.08 versus 0.39 ± 0.06 mg, p = 0.026) (Figure S1I).
Taken together, our findings show that severe hyperglycemia results in failure to recover beta cell mass and that this failure can be reversed by reduction of glucose levels by any of several mechanisms to below 16 mmol/L for approximately 3 weeks. To investigate potential mechanisms responsible for this phenomenon, we further characterized the beta cells exposed to severe hyperglycemia in our near-total ablation model.
Prolonged severe hyperglycemia results in failure of beta cell recovery
To determine whether the ability to recover beta cell mass after short-term intensive glucose-lowering treatment is maintained following prolonged hyperglycemia, we induced hyperglycemia using doxycycline at 1 month and then followed the animals untreated for 7 months. The mice tolerated glucose levels >30.5 mmol/L with no apparent untoward effects. At age 8 months, we treated the mice with insulin pellets which normalized their glucose levels for about 1 month, after which glucose levels returned to near pre-treatment levels, confirming the transient function of these pellets while demonstrating an inability to restore islet function and mass after 7 months of severe hyperglycemia (Figure S2B). Plasma insulin levels were dramatically reduced in untreated mice compared with age-matched control mice and did not increase after insulin treatment (Figure S2C). Importantly, the percentage of Ki67-positive beta cells was similar in untreated and treated mice (Figure S2D), suggesting that the ability of beta cells to respond to improved glycemic control with increased proliferation deteriorates after prolonged exposure to severe hyperglycemia. Both treated and untreated mice maintained a small number of islets that were characterized by a low ratio of beta cells to alpha and delta cells. The surviving beta cells lacked detectable levels of key beta cell transcription factors Pdx1 and Nkx6–1, important for maintaining insulin levels, cell identity, and function13,14 (Figure S2E). Double hormone producing cells were not detected. We cannot exclude the possibility that this failure to recovery is related to age per se and not prolonged hyperglycemia, since in 9-month-old mice severe hyperglycemia could not be induced by doxycycline administration, presumably because decreased expression of the transgene at this age. However, previous studies using the same model showed that, even at this age, milder hyperglycemia could be generated following acute beta cell injury and these mice were able to triple proliferation rate and normalize islet architecture.15
Reduction of glycemic load facilitates beta cell regeneration through replication of pre-existing beta cells that survived ablation
Data presented in Figure 2 suggest that the observed increase in beta cell mass following glycemic control may be due to increased proliferation of beta cells that survived ablation, although alternative explanations, that regeneration is based on reprogramming of non-beta islet cells8 or of Ngn3-expressing ductal progenitors16 are also possible. To determine the source of new beta cells in the recovered animals, we performed a lineage tracing experiment. The Insulin-rtTA;TET-DTA mice were crossed with mice carrying an LSL-YFP transgene at the ROSA26 locus and the MIP-CreER transgene, to create quadruple transgenic mice. We permanently pulse-labeled the beta cells of these mice by tamoxifen injection 1 week before their ablation by doxycycline. Animals were sacrificed immediately after ablation (pulse) or after 4 weeks of KD (chase) (Figure 3A) and pancreatic sections were analyzed by immunostaining for YFP and insulin. Importantly, after beta cell mass recovery following the KD intervention, there was no decrease in the percent of labeled beta cells compared with the pulse mice (before treatment), suggesting that there was little or no contribution of non-beta cells (which were by definition unlabeled at the time of tamoxifen injection) to the expansion of beta cell mass (Figures 3B and 3C). These findings are consistent with the findings of Nir et al., who showed that mice spontaneously recovering after 70%–80% beta cell ablation increase beta cell mass by proliferation of pre-existing beta cells.7
Figure 3. New beta cells emerge by replication of preexisting, differentiated beta cells.

(A) Experiment design combining genetic lineage tracing with the DTA-mediated ablation to determine the cellular origin of new beta cells. The beta cells were genetically labeled by administration of two daily injections of tamoxifen (TMX) (arrows) followed by doxycycline for 1 week. Some animals were sacrificed immediately after doxycycline (pulse), whereas others were treated with a ketogenic diet for 1 month and then sacrificed (chase).
(B) The percentage of YFP-positive beta cells was determined at both time points and showed no changes in the fraction of YFP-positive beta cells between thepulse and chase (n = 4 mice pulse; n = 5 mice chase).
(C) Representative immunofluorescence image of islets co-stained for insulin (green) and YFP (red) after KD treatment (chase time point). Scale bar, 50 μm.
Severe hyperglycemia per se is associated with failed beta cell replication in vivo
To better characterize the mechanism by which severe hyperglycemia per se prevents beta cell recovery, and without potential confounding factors associated with beta cell death, we induced moderate or severe hyperglycemia in mice and conducted a BrdU pulse-chase experiment to directly measure beta cell entry into S phase (pulse) and successful replication after 24 h (chase). We implanted 2-month-old ICR mice with osmotic minipumps secreting the insulin receptor antagonist S961 at low (12 nM) and high (24 nM) concentrations for 7 days to induce moderate and severe hyperglycemia, respectively. Random blood glucose levels increased after 1 day and remained at a level of 13.3 ± 1.4 mmol/L on average for the mice who received 12 nM S961 (moderate group) and 30.9 ± 1.6 mmol/L for the mice who received 24 nM S961 (severe group) (Figure 4A). On day 6 we injected 10 mg/mL BrdU, and sacrificed the pulse and chase groups 2 and 24 h after injection, respectively. Entry into the cell cycle at the time of BrdU injection (pulse) was measured by counting the percentage of beta cells staining positive for Ki67, while entry into S phase was measured by counting the percentage of BrdU-positive beta cells (Figures 4B–4D). Successful completion of the cell cycle was determined by comparing the percentage of BrdU-positive beta cells after 24 h to that at 2 h, with the expectation that, if all beta cells entering S phase during the pulse successfully completed the cell cycle, the percentage would double.
Figure 4. Induction of severe hyperglycemia in vivo using a high dose of an insulin receptor blocker inhibits compensatory proliferative response.

(A) Glucose levels following implantation of osmotic pumps containing saline (black, control), low-dose insulin receptor blocker (IRB) S961 (12 nM, moderate, gray) or high-dose IRB (24 mM, severe, red) into 2-month-old ICR mice.
(B) Percent BrdU-positive beta cells at two time points (pulse, 2 h; chase, 24 h) in moderate and severe S961 treatment groups.
(C) Percent Ki67-positive beta cells 2 h after BrdU labeling in moderate and severe S961 treatment groups. n = 3 mice per group.
(D) Representative immunofluorescence image of islets co-stained for insulin (gray), BrdU (red), and Ki67 (green) 2 and 24 h after BruD labeling (n = 3 mice per group). Scale bar, 50 μm.
As demonstrated in Figures 4B and 4C, in mice exposed to moderate hyperglycemia, 4.65% of beta cells were in the cell cycle (Ki67 positive) and 2.9% entered S phase (BrdU positive, 62% of Ki67-positive cells). After 24 h the percentage of BrdU-positive beta cells almost doubled (5.39%; 86% of expected increase), suggesting that the majority of cells entering S phase successfully completed it. In the group exposed to severe hyperglycemia, significantly fewer beta cells (3.03%) were in the cell cycle (p = 0.0251). Of these, 2.11% were in S phase based on BrdU staining (69% of Ki67 positive), and after 24 h the percentage of BrdU-positive beta cells increased to only 2.39% (13% of expected increase), suggesting that only a minority of the beta cells entering S phase successfully completed mitosis.
These results demonstrate that, in contrast to short-term severe hyperglycemia (Figure 1F), extended exposure (6 days) to extreme hyperglycemia has a negative effect on beta cell entry into the cell cycle. Furthermore, our data suggest that prolonged severe hyperglycemia inhibits beta cells at the G2/M checkpoint, preventing them from completing the cell cycle.
In depth investigation of the molecular mechanisms responsible for the in vivo observations described in the Insulin-rtTA;TET-DTA mouse model was not technically feasible as we were unable to isolate intact islets from these animals. We therefore performed ex vivo and in vitro experiments on WT ICR mouse islets and human beta cell line EndoC-betaH2, respectively, mimicking the in vivo conditions.
First, we incubated WT mouse islets in 5, 20, or 35 mM glucose for 3 days and, using whole-islet confocal microscopy (see STAR Methods), determined the percentage of beta cells that take up EthDIII (indicating cell death) or stained for Ki67 (indicating cycling cells). As shown in Figure 5A, apoptosis was unchanged at 20 mM glucose but appeared to increase, at least in some islet preparations, when exposed to 35 mM glucose (p = 0.17).
Figure 5. Exposure of mouse islets or human EndoC-BH2 cells to severe hyperglycemia results in suppression of proliferation-related gene sets.

(A and B) WT mouse islets were incubated for 3 days in 11.8, 20, or 35 mM glucose and whole islets were immunostained (see STAR Methods) for insulin and Ki67 to assess beta cell proliferation (A) or incubated with EthDIII to measure apoptosis (B). Each dot represents an islet, n = 4–9 islets per group.
(C) Gene set enrichment analysis indicating biological pathways enriched in genes up- or downregulated in mouse islets incubated in 20 mM (gray bar) or 35 mM(black bar) glucose. p < 0.05 was considered as a significant enrichment (n = 3 mice replicates).
(D) Heatmap displaying the relative expression levels (as median log2 reads) of G1/S and G2/M transition genes in mouse islets incubated in normal (11.8 mM), moderate (20 mM), and extreme (35 mM) glucose concentrations. Green, low; black, intermediate; and red, high expression levels.
(E) Gene set enrichment analysis indicating biological pathways that are enriched in genes up- or downregulated in EndoC-betaH2 human beta cell line incubated under 20 mM (gray bar) or 35 mM (black bar) glucose (n = 3 replicates). p < 0.05 was considered as a significant enrichment.
(F) Heatmap displaying the relative expression levels (as median log2 reads) of replication genes in EndoC-betaH2 cells incubated in 5 mM, 20 mM, or 35 mM glucose concentrations. Green, low; black, intermediate; and red, high expression levels.
Importantly, Ki67 positivity increased markedly at 20 mM glucose, but failed to increase above baseline levels when exposed to 35 mM glucose (Figure 5B). When compared with the findings described above, this observation supports the notion that the negative effect of glucose levels on cell cycle entry is dependent on both glucose levels and duration of exposure. One day after completion of doxycycline treatment, Ki67 positivity was similarly increased in mice with moderate and severe hyperglycemia (Figure 1F), whereas a 6-day in vivo exposure to 30.9 ± 1.6 mmol/L partially prevented hyperglycemia-induced entry into the cell cycle (Figure 4B), and 3 days of 35 mmol/L glucose ex vivo completely eliminated the effect (Figure 5B). The more dramatic effect ex vivo could be related to the fact that in the in vivo model, glucose levels varied between mice, and over the 6 days, modulating the effect.
To determine the overall effect of extreme hyperglycemia on the islet cell expression program, we incubated WT mouse islets or ENDO-betaH2 cells for 3 days in basal glucose levels (11.8 mM for mouse islets, 5.5 mM for EndoC-betaH2 cells), 20 or 35 mM glucose, simulating normoglycemic, hyperglycemic, and extreme hyperglycemic conditions, respectively, and subjected the islets and cells to transcriptome analysis. Differential expression analysis of mouse islets treated with 20 mM glucose (simulating moderate hyperglycemia) compared with 11.8 mM glucose revealed 349 upregulated and 228 downregulated genes out of a total of 12,272 detected genes, whereas comparing 35 mM glucose to 11.8 mM glucose identified 446 upregulated and 333 downregulated genes. Over representation analysis17 and gene set enrichment analysis, genomica, (http://genomica.weizmann.ac.il/), revealed that incubation with both 20 and 35 mM glucose increase the expression of beta cell function genes (Figure 5C and Table S1). These included genes, such as Iapp (islet amyloid polypeptide), G6pc2 (Glucose-6-Phosphatase Catalytic Subunit 2), and Pcsk1 (proprotein convertase subtilisin), as well as genes related to glycolysis and gluconeogenesis, including Mdh1 (malate dehydrogenase 1), Hax1 (Hcls1-associated protein X-1), Pdhb (pyruvate dehydrogenase E1 subunit beta), Akr1a1 (aldo-keto reductase), and AldoB (aldolaseB). Among the genes similarly upregulated in both 20 and 35 mM glucose, we detected genes associated with enhanced respiration, ER stress, and oxidative stress.
In contrast, cell cycle genes showed markedly different responses to moderate and severe hyperglycemia. These genes were upregulated in cells exposed to 20 mM glucose, consistent with our findings above (Figures 2D–2F) and supported by previous observations that glucose acts as a beta cell mitogen in both rodents and humans at 20 mM glucose18 (Figure 5D). However, the same genes show an opposite trend when cells were exposed to 35 mM glucose, reducing in expression below the level in islets incubated in 11.8 mM glucose. Many cell cycle genes downregulated at 35 mM glucose act specifically during G1/S and G2/M checkpoints, among them the DNA replication licensing factors (Mcm3, Mcm5, Mcm6, and Mcm8), members of the pre-replication complex, and cyclin E2, which is required for progression through S phase19 as well as the G2/M-related members of the centromere-associated proteins Cenpl, Cenph, and Cenpo, kinetochore localized astrin binding protein (Knstrn), spindle and kinetochore-associated protein 1 (Ska1), a microtubule-binding subcomplex of the outer kinetochore that is essential for proper chromosome segregation20 and structural maintenance of chromosome 4 (Smc4). Moreover, the downregulation of the mitotic kinase Pbk in islets treated with extreme glucose concentrations further support cell arrest at G1, as was previously demonstrated in transformed T cells.21 Interestingly, both Cdk1 and cyclin B, which phosphorylates Pbk, are also downregulated under extreme glucose concentrations. The reduced levels of cell cycle genes further support our in vivo and ex vivo findings (Figures 4B–4D and 5B), demonstrating that prolonged exposure to severe hyperglycemia inhibits cell cycle entry and progression.
To determine whether these changes are limited to rodent beta cells, we repeated the experiment on the human beta cell line (EndoC-betaH2).22 Importantly, we observed similar changes to the transcriptome, including enrichment of respiration, stress, and apoptosis pathways in cells incubated with 20 mM glucose for 3 days that were further enhanced by 35 mM glucose (Figure 5E). In this model, since the cells are proliferating at their maximum capacity, increasing glucose concentration from 5 to 20 mM was not expected to stimulate cell cycle entry, as was observed for mouse islets treated with 20 mM glucose. Importantly, however, and consistent with the findings in mouse islets, exposure to 35 mM glucose did result in downregulation of key cell cycle regulators of both G1/S and G2/M checkpoints, such as ATM23 and EP300 (the inhibition of which was shown to cause accumulation of cells in G1 state24) as well as TOP2A, BRCA2, and PLK425–27 (Figure 5F).
These molecular profiles, together with our in vivo analyses, identify a mechanism for the failure of compensatory regeneration of mouse and human beta cells in which prolonged severely elevated glucose levels block cell cycle entry and progression, impairing beta cell compensatory proliferation and beta cell mass recovery. Importantly, in mice exposed to severe hyperglycemia for 1 month or less, this blockage can be reversed by lowering glucose levels below a defined threshold for a sufficient period of time.
DISCUSSION
Glucose levels are thought to play a key role in determining the beta cell’s response to metabolic stress, initially boosting insulin secretion and beta cell replication but later fueling a vicious cycle that precipitates beta cell failure and hyperglycemia.28–31 Beta cell failure is thought to be caused by a number of mechanisms collectively termed glucotoxicity, which has been shown to involve oxidative stress, endoplasmic stress, and more recently the DNA damage response.2,3,32–34 Most studies of glucotoxicity focus on the effect of mild or moderate hyperglycemia on beta cell proliferation, apoptosis, dedifferentiation, and cell-autonomous function, usually quantified as glucose-stimulated insulin secretion.
Here, we identified a mechanism of beta cell glucotoxicity, associated with extremely high glucose levels (>28 mmol/L), which in vivo appears to cause a distinct phenotype of reversible failure of compensatory beta cell proliferation. A similar dichotomous response was shown in rats following near-total pancreatectomy, with severe hyperglycemia persisting for >14 weeks in a subgroup of animals.35 In an effort to characterize this extreme level of glucotoxicity we made the following observations.
Reversible failure of beta cell compensatory proliferation in the presence of severe hyperglycemia
Mice with severe hyperglycemia (>28 mmol/L) had marked decrease in beta cell mass with disrupted islet architecture and, as opposed to mice with less severe hyperglycemia, failed to spontaneously recover glycemic control. Improved glycemic control induced by any of three independent treatments (insulin, SGLT2 inhibitor, or KD) released the replication potential of remaining beta cells, resulting in increased beta cell mass and sustained glycemic control. These three treatment modalities affect the metabolic milieu differently, the only common factor being the lowering of glucose levels, suggesting the presence of toxic effects of severe hyperglycemia per se on beta cell replication that were not present at less severely elevated glucose levels. Interestingly, glucokinase reaches maximum catalytic activity (Vmax) at approximately 20–25 mmol/L glucose,36 raising the question of how the beta cell senses glucose levels above this threshold. Our findings do not negate possible additional negative effects by other metabolic abnormalities seen in diabetes, such as dyslipidemia and ketonemia (in the case of diabetic ketoacidosis), but focus solely on the toxic effect of severely elevated glucose levels. Importantly, whereas these effects were reversible after exposure to severe hyperglycemia in vivo for 1 month, after exposure to severe hyperglycemia for 7 months, short-term glycemic control failed to induce any measurable recovery (Figure S2B).
Reduction of ambient glucose levels below 16 mmol/L for 3 weeks was the best predictor of long-term recovery of beta cell functional mass, indicating that the mechanism preventing recovery involves metabolic or structural changes that take time to reverse. We found that increased beta cell mass at the end of the treatment period distinguished mice that achieved glycemic recovery from those that reverted to hyperglycemia after completion of the treatment period. The beta cell mass of both groups exceeded that of mice with similar initial glucose levels prior to any treatment, suggesting that reduction of hyperglycemia releases the regenerative capabilities of the beta cell and that approximately 3 weeks is required to achieve a beta cell mass sufficient to maintain glycemic control in most animals. As the degree of ablation in this experimental model is variable, a shorter treatment period may be sufficient for some mice, whereas a longer treatment period may be required for mice with more severe ablation.
Severe hyperglycemia prevents beta cell recovery by blocking proliferation
Failure of beta cell recovery in the presence of severe hyperglycemia could be due to either increased cell death (apoptosis) or failure of proliferation. We were unable to identify apoptosis in vivo using TUNEL staining, but the apoptotic process can be very rapid and this method may not be sufficiently sensitive to measure a low level of chronic apoptosis in pancreatic tissue sections.37 Some increase in apoptosis was suggested in our in vitro whole-islet model (Figure 5A) but this effect was modest at most and it did not reach statistical significance.
Lineage tracing studies showed that beta cell mass recovery was driven by proliferation of beta cells existing at the time of ablation. This is similar to the finding of Nir et al.7 in the same model exposed to lower levels of hyperglycemia but in contrast to the findings of Thorel et al.,8 discrepancies that can be explained by differences in the experimental models used. Based on our histologic findings, the extent of beta cell ablation seen in our most severely hyperglycemic animals did not reach the level of ablation (>99%) reported by Thorel et al. This is further supported by the finding that the mice in the Thorel et al. study reached lethal levels of insulin insufficiency, whereas even the mice with the most severe hyperglycemia in our study survived without treatment. Perhaps most importantly, the mice in the Thorel et al. study were only treated with insulin when glucose levels exceeded 20 mmol/L and the treatment regimen did not achieve the threshold needed to permit recovery as identified in our study. Furthermore, the mechanism of recovery identified in the Thorel et al. study, alpha to beta cell transdifferentiation, was much slower and less efficient, resulting in only 17% increase of beta cell mass at 10 months of follow-up compared with the approximate doubling of beta cell mass after 1 month of treatment and 1 month of follow-up in our model.
The cell cycle is a complex multistep process regulated at a number of key checkpoints. The protein Ki67 is expressed in cells throughout the cell cycle, whereas BrdU is permanently incorporated into DNA only during S phase. When comparing Ki67 positivity in beta cells exposed to extreme levels of glucose (>30–35 mmol/L) with those exposed to moderate levels of glucose (20–25 mmol/L) we observed a decreased percentage of Ki67-positive beta cells (Figures 4C and 5B). Using the BrdU pulse/chase protocol we were able to determine that severely elevated glucose significantly affected cell cycle entry and progression, suggesting blockage at G1/S and G2/M checkpoints. This finding was further supported by the transcriptome analysis of mouse islets and EndoC-betaH2 cells exposed to 3 days of severe glucose concentrations (35 mM), which revealed downregulation of cell cycle genes involved in both G1/S and G2/M checkpoints. The transcriptome data obtained from this analysis provide an important resource for future studies aiming to understand the mechanistic link between the effect of enhanced glucose metabolism and glucotoxicity on cell cycle arrest in beta cells, a topic that has recently gained increased attention.38,39
Taken together, these findings support the hypothesis that whereas moderate and temporary hyperglycemia induces beta cells to proliferate, extreme chronic hyperglycemia has a detrimental effect on beta cell regeneration. In a paper published while our study was under review, Xi et al. demonstrated that glucagon receptor blockade resulted in beta cell regeneration in the same TET-DTA mouse model used in our study.40 Whereas Xi et al. interpreted their data to indicate a specific alpha cell-related process driving proliferation, their findings are consistent with ours, that glycemic control itself is unmasking compensatory proliferation. Although they demonstrate evidence for alpha to beta cell transdifferentiation, only a small percentage of the observed increase in beta cell mass can be explained by transdifferentiation. Their observation that FGF21 administration resulted in improved glycemia without improvement in beta cell mass could be due to the previously reported anti-proliferative effect of FGF21 on beta cells.41
Potential relevance to human diabetes
In describing the functional changes associated with the development of diabetes, Weir et al. propose that progression from mild to severe hyperglycemia can be triggered by a relatively small additional decrease in beta cell functional mass, presumably activating a vicious cycle of glucotoxicity. Initially, this marked decrease can be reversible.42 We and others reported previously that patients with type 2 diabetes (T2D) and very poor glycemic control improve their beta cell function after 2 weeks of intensive treatment.43,44 Notwithstanding the differences between humans and mice, our studies suggest that longer periods of intensive control may further improve long-term glycemic control in patients with poorly controlled T2D. Thus, although the absolute glucose levels and durations may be different, the concept of beta cell recovery following extended intensive glycemic control is relevant to human T2D and possibly T1D, making detailed mechanistic studies in animal models potentially relevant to the development of therapeutic strategies to prevent progression, and perhaps even reverse diabetogenic changes.
In conclusion, we demonstrate a mechanism of beta cell toxicity, caused by severe hyperglycemia and mediated by a reversible inhibition of cell cycle gene expression and thus compensatory beta cell proliferation. Further study is needed to determine the significance of this mechanism of glucotoxicity in the pathogenesis and progression of clinical diabetes.
Limitations of the study
We measured blood glucose levels using the ACCU-Chek Performa Glucometer (Roche), which can measure glucose levels up to 33.3 mmol/L. Since on many occasions measured glucose levels reached this limit, it is highly likely that the mice with severe hyperglycemia had glucose levels significantly higher than those reported here.
To predict spontaneous recovery following ablation (Figures 1A and 1B) we used random glucose levels as a surrogate for degree of beta cell ablation. Other criteria, such as plasma insulin levels, pancreatic insulin content, or beta cell mass, may be better predictors; however, these data were not available on a cohort followed long term to determine recovery potential. We do not believe that this limitation significantly influenced our findings.
This study focuses on the effect of extreme hyperglycemia on beta cell proliferation, which may be of limited capacity in humans. Improved beta cell function following glycemic control has been demonstrated in T2D43–45 and even in new-onset T1D following hematopoietic stem cell transplantation.46 While the former is unlikely to be related to proliferation, the latter could be. In addition, it is possible that extreme hyperglycemia negatively affects different aspects of beta cell biology in different species, for example, leading to cell cycle arrest in mice and to cell death or dysfunction in human beta cells.
Using lineage tracing we demonstrated that the bulk of the observed regeneration could be explained by proliferation of previously existing beta cells. Our findings cannot exclude a minor contribution from other sources, such as transdifferentiation from non-beta cells40 or neogenesis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin Glaser (ben.glaser@mail.huji.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession numbers for the RNA sequencing data reported in this paper are NCBI GEO: GSE209751 for mouse islets and GSE212749 for EndoC-betaH2 human beta cell line.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Genotypes used in this study
Sex and age
Both males and female mice were used for all experiments. Age of the mice varies between experiments and is indicated in the text and figure legends.
Maintenance and care
PCR genotyping was performed on DNA extracted from ear punch biopsies, using the primers listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| guinea pig anti insulin 1:5 | Dako | Cat# IR002; RRID:AB_2800361 |
| Goat anti-Pdx1 1:2500 | Generous gift from Chris Wright | |
| Rabbit anti-Nkx6.1 1:100 | Abcam | Cat# Ab221549; RRID:AB_2754979 |
| Rabbit monoclonal anti Ki-67 | Thermo Fisher Scientific | Cat# RM-9106; RRID:AB_2341197 |
| Mouse anti-glucagon | Abcam | Cat# Ab10988; RRID:AB_297642 |
| Mouse monoclonal anti BrdU (5-bromo-2-deoxyuridine) | GE Healthcare Life Biosciences | Cat# RPN202; RRID:AB_2314032 |
| Goat anti-GFP 1:400 | Abcam | Cat# AB6673; RRID:AB_305643 |
| Rabbit anti Somatostatin 1:200 | Abcam | Cat# Ab108456; RRID:AB_11158517 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 711–545; RRID:AB_2313584 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Goat IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 705–545; RRID:AB_2336933 |
| Alexa Fluor 488-AffiniPure Donkey Anti-Guinea Pig IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 706–545; RRID:AB_2340472 |
| Cy3-AffiniPure Donkey Anti-Mouse IgM, μ Chain Specific | Jackson ImmunoResearch Labs | Cat# 715–165; RRID:AB_2340812 |
| Alexa Fluor 647-AffiniPure Donkey Anti-Mouse IgM, μ Chain Specific | Jackson ImmunoResearch Labs | Cat# 715–605; RRID:AB_2340863 |
| Cy3-AffiniPure Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 711–165; RRID:AB_2307443 |
| Cy5-AffiniPure Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 711–175; RRID:AB_2340607 |
|
| ||
| Biological samples | ||
|
| ||
| Pancreatic islets isolated from WT ICR mice for ex vivo experiments | Envigo, Israel | Cat# HSD:ICR MICE - 260 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Ethidium Homodimer III- Dead cell stain 1:500 | Biotum | Cat# 40050 |
| DAPI | Roche | Cat# 10236276001 |
| CAS-Block™ | Thermo Fisher Scientific | Cat# 008120 |
| Tamoxifen (20mg/mL in corn oil) | Sigma | Cat# T5648 |
| Doxycycline hydrochloride, (200μg/mL, 2% w/v sucrose) | Proteogenix | Cat# A600889–0100 |
| Insulin pellets | LinBit implants LinShin, Scarborough ON, Canada | Cat# Pr-1-B |
| Dapagliflozin 25mg/kg/day in drinking water | AstraZeneca | |
| Ketogenic diet | Envigo, Israel | Cat# TD. 96355 |
|
| ||
| Critical commercial assays | ||
|
| ||
| In Situ Cell Death Detection Kit, Fluorescein (TUNEL) | Roche | Cat# 11684795910 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| RNeasy Mini Kit | QIAGEN | Cat# 74106 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-Seq dataset of WT mouse islets exposed to 11.8, 20 and 35mM glucose for 72 h | This paper | GSE209751 |
| RNA-Seq dataset of EndoC-BetaH2 exposed to 5, 20 and 35mM glucose for 72 h | This paper | GSE212749 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| EndoC-betaH2 – human pancreatic beta cells | provided by Endocell and Raphael Scharfmann | https://doi.org/10.1172/JCI72674 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Insulin-rtTA; TET-DTA | https://www.zotero.org/google-docs/?NmVJ8n | N/A |
| MipCreER | https://www.zotero.org/google-docs/?ePSfVp | N/A |
| Rosa26-LSL-YFP | https://www.zotero.org/google-docs/?VgU7i3 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Insulin-CreER_F 5′-TGCCACGACCAAGTGACAGC-3′ | Dor et al.47 | N/A |
| Insulin-CreER_R 5′-CCAGGTTACGGATATAGTTCATG-3′ | Dor et al.47 | N/A |
| ROSA26_F 5′-AAAGTCGCTCTGAGTTGTTAT-3′ | Soriano et al.48 | N/A |
| WT 5′-GAAAGACCGCGAAGAGTTTG-3′ | Soriano et al.48 | N/A |
| ROSA26_R 5′-TAAGCCTGCCCAGAAGACTC-3′ | Soriano et al.48 | N/A |
| TET-DTA_F 5′-TTTTGACCTCCATAGAAGAC-3′ | Lee et al.49 | N/A |
| TET-DTA_R 5′-GGCATTATCCACTTTTAGTGC-3′ | Lee et al.49 | N/A |
| Insulin-rtTA_F 5′-TAGATGTGCTTTACTAAGTCATCGCG-3′ | Milo-Landesman et al.50 | N/A |
| Insulin-rtTA_R 5′-AGATCGAGCAGGCCCTCGATGGTAG-3′ | Milo-Landesman et al.50 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| NIS-Elements | Nikon | RRID:SCR_014329 |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Adobe Photoshop CC | Adobe | http://www.adobe.com/es/products/photoshop.html |
All animals were housed in the Hebrew University Specific Pathogen Free (SPF) small animal facility and fed standard laboratory mouse chow unless otherwise noted.
The joint ethics committee (IACUC) of the Hebrew University and Hadassah Medical Center approved the study protocol for animal welfare. The Hebrew University is an AAALAC international accredited institute.
Islet culture
5–6 weeks old mice were sacrificed under general anesthesia; the pancreas was exposed and injected with the enzyme solution (Collagenase P, Roche); 1.5mg/mL, 5mL per mice) through the main bile duct until full distension was achieved. The pancreatic tissue was then surgically removed and incubated at 37°C for 5 min. Enzyme kinetics was slowed by addition of cold RPMI (Biological industries). Mechanical disruption of the digested pancreatic tissue was achieved by repeated passages through pipette until complete release of free islets was observed under the microscope. Isolated islets used for in vitro experiments were cultured overnight at 37°C, 5% CO2, in RPMI-1066 medium (Biological industries) supplemented with 10% fetal bovine serum, 2 mmol/l L-glutamine, 100 U/mL penicillin and 100 mg/mL streptomycin.
Cell lines
EndoC-betaH2 human beta cell line were provided by Endocell and Raphael Scharfmann and cultured and propagated as described previously (http://www.endocells.com/products-and-services/.22 Briefly, EndoC cells were seeded at a density of 70K cells/cm2 on ECM coated plates prepared 1–24 h before seeding. Coating medium ingredients are DMEM 4.5g/L glucose, Penicillin/Streptomycin 1%, Fibronectin 2mg/mL, ECM 1%. Culture medium contained DMEM low glucose (1g/L), 2% Albumin from bovine serum fraction V, 50μM 2-mercaptoethanol, 10mM nicotinamide, 5.5 μg/mL transferrin, 6.7 ng/mL sodium selenite and Penicillin (100 units/mL)/Streptomycin (100 μg/mL). cells were subculture for propagation once a week at a ratio of 1:2.
METHOD DETAILS
Activation of transgenes
To activate Cre recombinase in transgenic mice carrying the MIP-CreER transgene, tamoxifen (20mg/mL in corn oil; Sigma) was injected subcutaneously (2 injections of 8 mg each, at day 0 and day 2). To activate the diphtheria toxin in Insulin-rtTA; TET-DTA mice, doxycycline (Dexon) was administered in the drinking water (200μg/mL doxycycline, 2% w/v sucrose). Water was changed every 5 days. Four-week-old mice were treated with doxycycline for 7 days and then allowed to recover, in the absence of doxycycline.
Blood glucose measurements
Blood glucose measurements were performed on blood collected following amputation of the distal tail using an ACCU-Chek Performa Glucometer (Roche).
In vivo treatments
The following treatments were given as described in the results section: Insulin pellets (LinBit implants; LinShin, Scarborough, ON, Canada), which were implanted subcutaneously to hyperglycemic Insulin-rtTA; TET-DTA mice, two implants for the first 20g body weight and another pellet for each additional 5g); dapagliflozin (SGLT2 inhibitor) 25mg/kg/day in the drinking water; ketogenic diet (Teklad Custom Diet TD. 96355, Envigo) containing 90.5% fat, 9.2% protein, 0.3% carbohydrate (% of total caloric content).
Pulse chase experiment on WT ICR hyperglycemic mice
WT ICR mice were implanted with osmotic minipumps secreting the insulin receptor antagonist S961 at low (12nM) and high (24nM) concentrations for 7 days to induce moderate and severe hyperglycemia respectively. On day 6 we injected 10mg/mL BrdU (dissolved in PBS) intraperitoneally and sacrificed the pulse and chase groups two and 24 h post injection respectively.
Histology and immunohistochemistry
Tissue processing was performed as previously described.7 Briefly, 5 μm thick pancreatic paraffin sections were rehydrated, and antigen retrieval was performed using a PickCell pressure cooker. Antibodies used are listed in the key resources table. All fluorescent images were taken using a Nikon 90i C1 confocal microscope or an Olympus FV1000 confocal microscope. For beta cell mass determination, consecutive paraffin sections, 75 μ m apart, spanning the entire pancreas, were stained for insulin and hematoxylin. Digital 4x images were obtained, stitched together using NIS-Elements software, and the fraction of tissue stained for insulin was determined.
Lineage tracing study
The Insulin-rtTA; TET-DTA mice were crossed with mice carrying a LSL-YFP transgene at the ROSA26 locus and the MIP-CreER transgene to make Insulin-rtTA; TET-DTA/MIP-CreER; YFP quadruple transgenic mice for a beta cell lineage tracing induced by tamoxifen injection one week prior to their ablation by doxycycline. Doxycycline was added to drinking water for 1 week and the animals were sacrificed immediately after ablation (pulse) or after four weeks of ketogenic diet (chase) and pancreatic sections were analyzed by immunostaining for YFP and insulin.
TUNEL staining
TUNEL staining was performed using In Situ Cell Death AP (Roche) according to manufacturer instructions. Slides were pre-treated with 1:1000 proteinase K (Roche) for 10 min. Fluorescent images were taken on a Nikon C1 confocal microscope at 40x magnification.
Whole mount staining
Whole mount staining of islets was done as previously described.53 Islets were washed with PBS, fixed with paraformaldehyde, permeabilized and then stained using anti Ki67, anti Pdx1 and Ethidium Homodimer III as described in the key resources table. Images were captured on Olympus FV1000 confocal microscope.
RNA sequencing and data processing and analysis
To determine the effect of elevated glucose levels on the beta cell expression profile, mouse islets and EndoC-BH2 human beta cells were incubated for 3 days with normal, moderately elevated (20mM) and severely elevated (35mM) glucose levels. At the end of the experiment, we extracted RNA with RNeasy kit (Qiagen). For mouse islet samples, libraries were prepared using CEL-Seq2 protocol, as published by Hashimshony et al.54 The libraries were sequenced on the Illumina Hiseq 2500 sequencer (Illumina) rapid mode, 15 bases for read 1 and 52 bases for read 2. Demultiplexing was performed in two steps. First, Illumina demultiplexing was performed using bcl2fastq Illumina software with the following parameters: barcode-mismatches = 1, minimum-trimmed-read-length = 0, and mask-short-adapter-reads = 0. Second, Cell-seq demultiplexing using the pipeline described in Hashimshony et al. (https://github.com/yanailab/CEL-Seq-pipeline) was executed with the following parameters: min_bc_quality = 15, bc_length = 6, umi_length = 6, and cut_length = 50. RNA measurements, library preparation and sequencing were performed by the Technion Genomics Center, Technion, Israel. For the following steps, a modified version of the CELSeq2 version 1.16.0 pipeline was applied. Raw reads were processed with cutadapt, removing from the 3′ end low quality bases, adapter sequences and poly-A stretches. Processed reads were aligned to the mouse genome with TopHat. The genome version was GRCm38, with gene annotations from the Ensembl database, supplemented with the spike-in ERCC data. Alignment allowed up to 2 mismatches per read. Raw counts were calculated with htseq-count, ignoring the UMI information. Raw counts were normalized and differential expression was calculated with DESeq2, after removing genes with a mean normalized expression less than 2. Default parameters were used except for disabling the search for outlier genes (setting cooksCutoff = FALSE). For human EndC-BH2, libraries were prepared with Illumina TruSeq stranded mRNA library kit and sequenced on the Illumina NovaSeq S1 with 100 bp single read and depth of 24.86 (MR) +/− 1.62 range. We aligned and quantified the reads using RUM and the human genome release hg19.
To characterize the expression program of beta cells exposed to severe glucose levels, we performed differential expression analysis between the transcriptomes of mouse islets exposed to normal (11.8mM), moderate (20mM) and severe (35mM) glucose levels and EndoC-betaH2 human beta cell line exposed to normal (5mM), moderate (20mM) and severe (35mM) glucose levels and conducted gene set enrichment analysis (GSEA) using a hypergeometric test by Genomica software (http://genomica.weizmann.ac.il/) considering gene sets with a p value < 0.01 and an FDR <0.05 to be significantly enriched. In addition, we performed an over-representation test (geneset enriched in upregulated or downregulated genes).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using a 2-tailed Student’s t-test except where indicated otherwise. Data are presented as mean ± SE. The number of mice used in each experiment is stated in the figure legends as “n = ”. In all statistical analyses * represents p < 0.05; ** represents p < 0.01; *** represents p < 0.005, and NS represents p > 0.05.
Supplementary Material
Highlights.
Massive beta cell ablation in mice results in persistent, severe hyperglycemia
Extremely high glucose levels suppress beta cell replication and cell cycle progression
Short-term glycemic control unmasks regenerative potential of beta cells
ACKNOWLEDGMENTS
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (UC4DK104119 to B.G.); BIRAX Regenerative Medicine Initiative (14BX14NHBG to B.G.); Israel Science Foundation (ISF, 1782/18 to B.G.); ISF-Juvenile Diabetes Research Foundation Joint Program in Type 1 Diabetes Research (2982/20 to B.G.); The United States-Israel Binational Science Foundation (BSF, 2019314 to B.G.); Deutsche Forschungsgemeinschaft (NI 1495/5-1 to Y.D.); The Ernest and Bonnie Beutler Research Program of Excellence in Genomic Medicine (to Y.D.), The Alex U Soyka Pancreatic Cancer Fund (to Y.D.), The Israel Science Foundation, the Waldholtz/Pakula Family (to Y.D.), and the Robert M. and Marilyn Sternberg Family Charitable Foundation (to Y.D.). Y.D. holds the Walter and Greta Stiel Chair and Research grant in heart studies.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111719.
REFERENCES
- 1.Yan LJ (2014). Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J. Diabetes Res 2014, 137919. 10.1155/2014/137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poitout V, and Robertson RP (2002). Minireview: secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology 143, 339–342. 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 3.Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T,Schyr-Ben Haroush R, Hija A, Stolovich-Rain M, Furth-Lavi J, Granot Z, et al. (2014). Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in β cells. Cell Metab. 19, 109–121. 10.1016/j.cmet.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Lee SH, Hao E, and Levine F. (2011). beta-Cell replication and islet neogenesis following partial pancreatectomy. Islets 3, 188–195. 10.4161/isl.3.4.16338. [DOI] [PubMed] [Google Scholar]
- 5.Bonner-Weir S, Trent DF, and Weir GC (1983). Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J. Clin. Invest 71, 1544–1553. 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szkudelski T. (2001). The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res 50, 537–546. [PubMed] [Google Scholar]
- 7.Nir T, Melton DA, and Dor Y. (2007). Recovery from diabetes in mice by beta cell regeneration. J. Clin. Invest 117, 2553–2561. 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, and Herrera PL (2010). Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464, 1149–1154. 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothenberg PL, Willison LD, Simon J, and Wolf BA (1995). Glucose-induced insulin receptor tyrosine phosphorylation in insulin-secreting beta-cells. Diabetes 44, 802–809. 10.2337/diab.44.7.802. [DOI] [PubMed] [Google Scholar]
- 10.Harbeck MC, Louie DC, Howland J, Wolf BA, and Rothenberg PL(1996). Expression of insulin receptor mRNA and insulin receptor substrate 1 in pancreatic islet beta-cells. Diabetes 45, 711–717. 10.2337/diab.45.6.711. [DOI] [PubMed] [Google Scholar]
- 11.Al Jobori H, Daniele G, Adams J, Cersosimo E, Triplitt C, DeFronzo RA, and Abdul-Ghani M. (2017). Determinants of the increase in ketone concentration during SGLT2 inhibition in NGT, IFG and T2DM patients. Diabetes Obes. Metab 19, 809–813. 10.1111/dom.12881. [DOI] [PubMed] [Google Scholar]
- 12.Dahan T, Ziv O, Horwitz E, Zemmour H, Lavi J, Swisa A, Leibowitz G, Ashcroft FM, In’t Veld P, Glaser B, and Dor Y. (2017). Pancreatic b-cells express the fetal islet hormone gastrin in rodent and human diabetes. Diabetes 66, 426–436. 10.2337/db16-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T, et al. (2014). Pdx1 maintains β cell identity and function by repressing an a cell program. Cell Metab. 19, 259–271. 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor BL, Liu FF, and Sander M. (2013). Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 4, 1262–1275. 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stolovich-Rain M, Hija A, Grimsby J, Glaser B, and Dor Y. (2012). Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J. Biol. Chem 287, 27407–27414. 10.1074/jbc.M112.350736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gribben C, Lambert C, Messal HA, Hubber E-L, Rackham C, Evans I, Heimberg H, Jones P, Sancho R, and Behrens A. (2021). Ductal Ngn3-expressing progenitors contribute to adult β cell neogenesis in the pancreas. Cell Stem Cell 28, 2000–2008.e4. 10.1016/j.stem.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL,Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levitt HE, Cyphert TJ, Pascoe JL, Hollern DA, Abraham N, Lundell RJ, Rosa T, Romano LC, Zou B, O’Donnell CP, et al. (2011). Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD–severe combined immunodeficiency (SCID) mice. Diabetologia 54, 572–582. 10.1007/s00125-010-1919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, and Lehner CF (1994). Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell 77, 107–120. 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 20.Hanisch A, Silljé HHW, and Nigg EA (2006). Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J. 25, 5504–5515. 10.1038/sj.emboj.7601426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa C, Senba M, and Mori N. (2018). Mitotic kinase PBK/TOPK asa therapeutic target for adult T-cell leukemia/lymphoma. Int. J. Oncol 53, 801–814. 10.3892/ijo.2018.4427. [DOI] [PubMed] [Google Scholar]
- 22.Scharfmann R, Pechberty S, Hazhouz Y, von Bülow M, Bricout-Neveu E, Grenier-Godard M, Guez F, Rachdi L, Lohmann M, Czernichow P, and Ravassard P. (2014). Development of a conditionally immortalized human pancreatic β cell line. J. Clin. Invest 124, 2087–2098. 10.1172/JCI72674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakkenist CJ, and Kastan MB (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Carpizo V, Ruiz-Llorente S, Sarmentero J, Graña-Castro O, Pisano DG, and Barrero MJ (2018). CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC regulatory axis in proliferation. Epigenet. Chromatin 11, 30. 10.1186/s13072-018-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida K, and Miki Y. (2004). Role of BRCA1 and BRCA2 as regulatorsof DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 95, 866–871. 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Press MF, Xie B, Davenport S, Zhou Y, Guzman R, Nolan GP, O’Brien N, Palazzolo M, Mak TW, Brugge JS, and Slamon DJ (2019). Role for polo-like kinase 4 in mediation of cytokinesis. Proc. Natl. Acad. Sci. USA 116, 11309–11318. 10.1073/pnas.1818820116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen CF, Zhang T, Barisic M, Kalitsis P, and Hudson DF (2020). Topoisomerase IIα is essential for maintenance of mitotic chromosome structure. Proc. Natl. Acad. Sci. USA 117, 12131–12142. 10.1073/pnas.2001760117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bensellam M, Laybutt DR, and Jonas JC (2012). The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Mol. Cell. Endocrinol 364, 1–27. 10.1016/j.mce.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Chang-Chen KJ, Mullur R, and Bernal-Mizrachi E. (2008). Beta-cell failure as a complication of diabetes. Rev. Endocr. Metab. Disord 9, 329–343. 10.1007/s11154-008-9101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tornovsky-Babeay S, Weinberg-Corem N, Ben-Haroush Schyr R, Avrahami D, Lavi J, Feleke E, Kaestner KH, Dor Y, and Glaser B. (2021). Biphasic dynamics of beta cell mass in a mouse model of congenital hyperinsulinism: implications for type 2 diabetes. Diabetologia 64, 1133–1143. 10.1007/s00125-021-05390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, and Sharma A(2001). Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 50, S154–S159. 10.2337/diabetes.50.2007.s154. [DOI] [PubMed] [Google Scholar]
- 32.Korsgren O, Jansson L, Sandler S, and Andersson A. (1990). Hyperglycemia-induced B cell toxicity. The fate of pancreatic islets transplanted into diabetic mice is dependent on their genetic background. J. Clin. Invest 86, 2161–2168. 10.1172/JCI114955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, Arvan P, and Alonso LC (2015). Insulin demand regulates β cell number via the unfolded protein response. J. Clin. Invest 125, 3831–3846. 10.1172/JCI79264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma RB, Landa-Galván HV, and Alonso LC (2021). Living dangerously: protective and harmful ER stress responses in pancreatic β-cells. Diabetes 70, 2431–2443. 10.2337/dbi20-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, Bonner-Weir S, and Weir GC (2003). Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J. Biol. Chem 278, 2997–3005. 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- 36.Matschinsky FM, Glaser B, and Magnuson MA (1998). Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes 47, 307–315. 10.2337/diabetes.47.3.307. [DOI] [PubMed] [Google Scholar]
- 37.Elmore S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol 35, 495–516. 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguayo-Mazzucato C, Andle J, Lee TB, Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, and Bonner-Weir S. (2019). Acceleration of b cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 30, 129–142.e4. 10.1016/j.cmet.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tudurí E, Soriano S, Almagro L, Montanya E, Alonso-Magdalena P, Nadal Á, and Quesada I. (2022). The pancreatic β-cell in ageing: implications in age-related diabetes. Ageing Res. Rev 80, 101674. 10.1016/j.arr.2022.101674. [DOI] [PubMed] [Google Scholar]
- 40.Xi Y, Song B, Ngan I, Solloway MJ, Humphrey M, Wang Y, Mondal K, Wu H, Liu W, Lindhout DA, et al. (2022). Glucagon-receptor-antagonism-mediated β-cell regeneration as an effective anti-diabetic therapy. Cell Rep. 39, 110872. 10.1016/j.celrep.2022.110872. [DOI] [PubMed] [Google Scholar]
- 41.So WY, Cheng Q, Xu A, Lam KSL, and Leung PS (2015). Loss of fibroblast growth factor 21 action induces insulin resistance, pancreatic islet hyperplasia and dysfunction in mice. Cell Death Dis. 6, e1707. 10.1038/cddis.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weir GC, and Bonner-Weir S. (2004). Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53, S16–S21. [DOI] [PubMed] [Google Scholar]
- 43.Glaser B, Leibovich G, Nesher R, Hartling S, Binder C, and Cerasi E(1988). Improved beta-cell function after intensive insulin treatment in severe non-insulin-dependent diabetes. Acta Endocrinol. 118, 365–373. 10.1530/acta.0.1180365. [DOI] [PubMed] [Google Scholar]
- 44.Ilkova H, Glaser B, Tunç kale A, Bagriaç ik N, and Cerasi E. (1997). Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Care 20, 1353–1356. 10.2337/diacare.20.9.1353. [DOI] [PubMed] [Google Scholar]
- 45.Retnakaran R, and Zinman B. (2012). Short-term intensified insulin treatment in type 2 diabetes: long-term effects on β-cell function. Diabetes Obes. Metab 14, 161–166. 10.1111/j.1463-1326.2012.01658.x. [DOI] [PubMed] [Google Scholar]
- 46.Couri CEB, Oliveira MCB, Stracieri ABPL, Moraes DA, Pieroni F, Barros GMN, Madeira MIA, Malmegrim KCR, Foss-Freitas MC, Simões BP, et al. (2009). C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 301, 1573–1579. 10.1001/jama.2009.470. [DOI] [PubMed] [Google Scholar]
- 47.Dor Y, Brown J, Martinez OI, and Melton DA (2004). Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46. 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 48.Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet 21, 70–71. 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 49.Lee P, Morley G, Huang Q, Fischer A, Seiler S, Horner JW, Factor S, Vaidya D, Jalife J, and Fishman GI (1998). Conditional lineage ablation to model human diseases. Proc. Natl. Acad. Sci. USA 95, 11371–11376. 10.1073/pnas.95.19.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milo-Landesman D, Surana M, Berkovich I, Compagni A, Christofori G, Fleischer N, and Efrat S. (2001). Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant. 10, 645–650. [PubMed] [Google Scholar]
- 51.Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, et al. (2010). Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59, 3090–3098. 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol 1, 4. 10.1186/1471-213x-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahnfelt-Rønne J, Hald J, Bødker A, Yassin H, Serup P, and Hecksher-Sørensen J. (2007). Preservation of proliferating pancreatic progenitor cells by Delta-Notch signaling in the embryonic chicken pancreas. BMC Dev. Biol 7, 63. 10.1186/1471-213X-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, et al. (2016). CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77. 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA sequencing data reported in this paper are NCBI GEO: GSE209751 for mouse islets and GSE212749 for EndoC-betaH2 human beta cell line.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.