

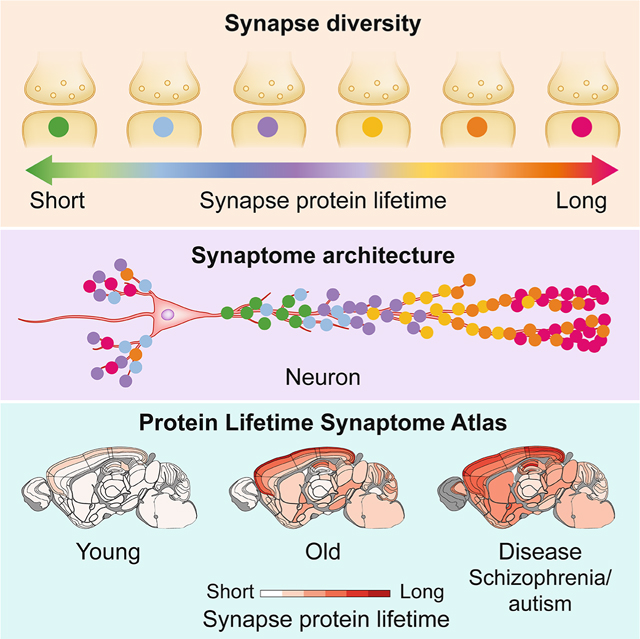
Abstract
The lifetime of proteins in synapses is important for their signaling, maintenance and remodeling, and for memory duration. We quantified the lifetime of endogenous PSD95, an abundant postsynaptic protein in excitatory synapses, at single-synapse resolution across the mouse brain and lifespan, generating the Protein Lifetime Synaptome Atlas. Excitatory synapses have a wide range of PSD95 lifetimes extending from hours to several months, with distinct spatial distributions in dendrites, neurons and brain regions. Synapses with short protein lifetimes are enriched in young animals and in brain regions controlling innate behaviors, whereas synapses with long protein lifetimes accumulate during development, are enriched in the cortex and CA1 where memories are stored, and are preferentially preserved in old age. Synapse protein lifetime increases throughout the brain in a mouse model of autism and schizophrenia. Protein lifetime adds a further layer to synapse diversity and enriches prevailing concepts in brain development, aging and disease.
Keywords: Synapse proteome, postsynaptic density, proteostasis, synaptome, protein turnover, aging, brain development, autism, dendritic spine, pyramidal neuron, HaloTag
eTOC blurb
Protein lifetime in synapses is important for brain development and aging, learning and memory. The authors show that synapses with diverse protein lifetimes are differentially distributed on neurons and in brain regions and that this architecture changes throughout the lifespan and is impacted in a model of autism.
The lifetime of a protein at its subcellular location is a fundamental determinant of its role in homeostatic and adaptive processes. Physiological and pathological events trigger protein modifications, and the subsequent replacement with a newly synthesized protein reinstates cellular functions to their original state, a process known as proteostasis (Kaushik and Cuervo, 2015; Labbadia and Morimoto, 2015). In addition to homeostatic roles, protein turnover is essential for adapting the levels of proteins to developmental and adaptive changes in gene expression; for example, a reduction in an mRNA can only lead to reduced levels of protein if there is ongoing protein removal. The speed of the underlying mechanisms that produce, deliver and remove proteins controls how long it takes for these proteostatic and adaptive processes to complete their task. This in turn is directly related to the lifetime of the protein at its site of action, with a short protein lifetime indicative of rapid homeostasis and adaptation.
In brain synapses, protein turnover controls adaptive processes including synaptic transmission, plasticity and learning (Colledge et al., 2003; Ehlers, 2003; Kato et al., 2005; Patrick et al., 2003). Francis Crick highlighted the importance of synaptic protein lifetime for the duration of memory, reasoning that protein removal erased learning-induced protein modifications (Crick, 1984). An imbalance in the rate of synaptic protein production and removal is thought to underlie learning and other cognitive deficits in genetic forms of autism and intellectual disability (Labbadia and Morimoto, 2015; Louros and Osterweil, 2016). A reduction in the rate of protein removal is also thought to underlie the synaptic and behavioral deficits in old age (Kaushik and Cuervo, 2015; Sabath et al., 2020; Santra et al., 2019) and contribute to the accumulation of toxic protein species, such as aggregates of ß-amyloid and Tau, that contributes to synaptic pathology in Alzheimer’s, Parkinson’s and other neurodegenerative diseases (Helton et al., 2008; Kaushik and Cuervo, 2015; Tai et al., 2012; Vilchez et al., 2014).
Excitatory synapses are highly diverse in molecular composition and morphology (Broadhead et al., 2016; Cizeron et al., 2020; Zhu et al., 2018). The synapse types and subtypes have different spatial distributions on dendrites and between neuron types, and are found in characteristic compositions in brain regions and subregions, forming a highly organized synaptome architecture that changes throughout the lifespan (Cizeron et al., 2020). The spatiotemporal differences in synapse molecular composition raise the possibility that synapses may also differ in their protein lifetime.
The lifetime of endogenous proteins has been measured in bulk brain synapse preparations, which lack information about synapse diversity (Cohen et al., 2013; Dörrbaum et al., 2018; Ehlers, 2003; El-Husseini et al., 2002; Fornasiero et al., 2018; Heo et al., 2018; Price et al., 2010). Measurements of protein lifetime at single-synapse resolution have been limited to small populations of neurons and exogenously expressed proteins (Gray et al., 2006; Steiner et al., 2008; Villa et al., 2016). To overcome these limitations we have adapted HaloTag self-labeling protein tag technology to visualize and quantify the lifetime of endogenous synaptic proteins in individual excitatory synapses in all parts of the mouse brain at any age. HaloTag technology (Los et al., 2008) has been widely used to label different proteins in many cellular and tissue contexts, including studies of protein turnover and lifetime. The system involves two components: the genetically encoded HaloTag protein domain and the HaloTag ligand, a small molecule that binds to the HaloTag domain. The ligand forms a highly stable covalent bond that is irreversible under physiological and non-physiological conditions (Encell et al., 2012; England et al., 2015; Hoelzel and Zhang, 2020; Los et al., 2008; Xue et al., 2015). The binding site for a chloroalkane linker is located within a deep pocket (∼15A) away from the protein surface, protected from the cellular milieu (Los et al., 2008). Coupling the HaloTag ligand to fluorophores enables irreversible labeling and visualization of tagged proteins, and observing the reduction in fluorescent signal over time enables the lifetime of the protein to be measured.
We engineered the mouse genome to express the HaloTag protein domain fused to endogenous PSD95 (PSD95-HaloTag mice) (Fig. 1A, S1A,B). PSD95 (also known as DLG4) is a highly abundant scaffold protein of excitatory synapses that interacts with and organizes more than 100 other postsynaptic proteins, including NMDA and AMPA subtypes of neurotransmitter receptors (Fernandez et al., 2017; Fernandez et al., 2009; Frank et al., 2016; Husi et al., 2000). PSD95 is essential for synaptic plasticity and learning (Carlisle et al., 2008; Fernandez et al., 2017; Fitzgerald et al., 2015; Frank et al., 2016; Migaud et al., 1998; Nithianantharajah et al., 2013). Injecting a fluorescent HaloTag ligand into PSD95-HaloTag mice labels excitatory synapses with a fluorescent date-stamp. The lifetime of PSD95 in a synapse is then measured by visualizing the duration of the labeled protein. We combined this labeling strategy with our synaptome mapping pipeline technology (SYNMAP), which is capable of quantifying the fluorescent signals from billions of individual synapses across the mouse brain (Cizeron et al., 2020; Zhu et al., 2018). We report the creation of a single-synapse resolution atlas (Bulovaite et al., 2021a) of PSD95 protein lifetime across the brain in young, mature and old mice and in a model of neurodevelopmental disorders.
Fig. 1. Visualizing synapse protein lifetime across the mouse brain.
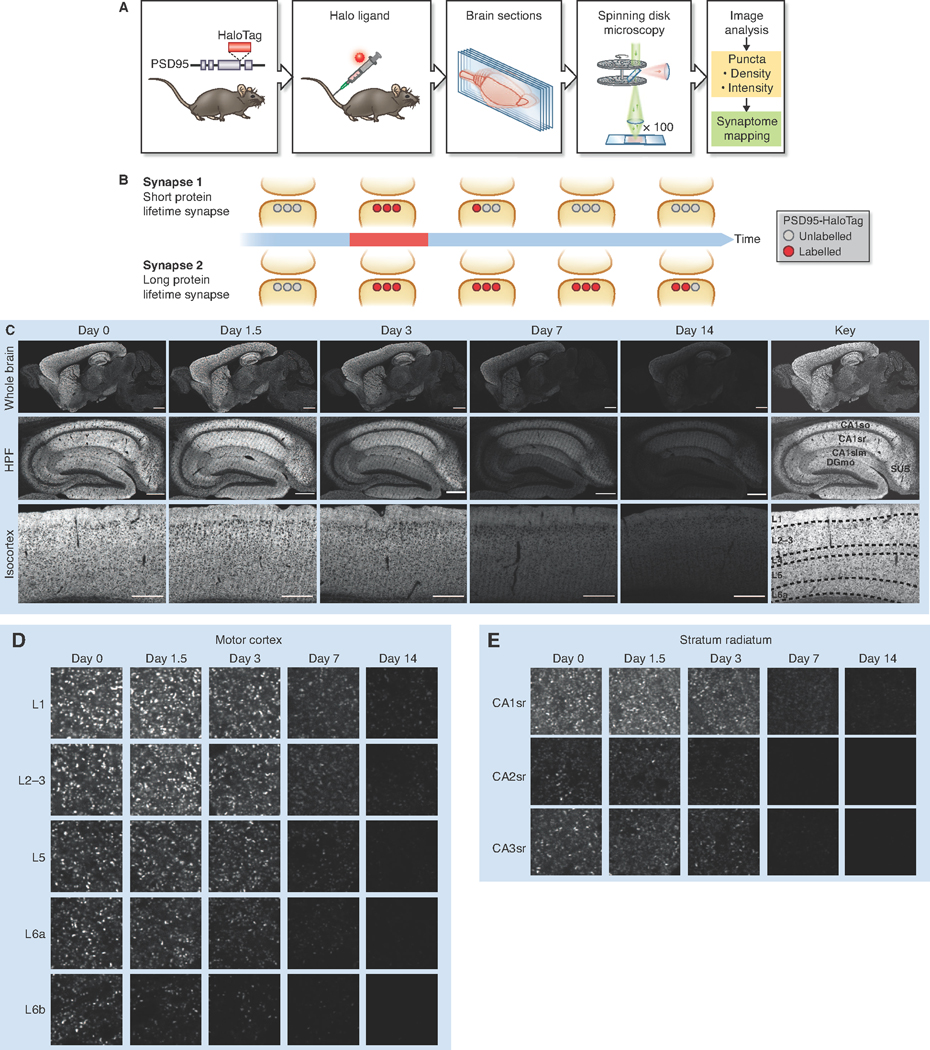
(A) Experimental flow. Homozygous PSD95-HaloTag mice were injected with a Halo ligand, sagittal brain sections imaged using spinning disk confocal microscopy, and synaptic puncta analyzed and mapped. (B) Schematic of PSD95-HaloTag labeling in the postsynaptic terminal of synapses with short or long protein lifetime. (C) SiR-Halo fluorescence labeling at five time points post-injection, showing whole brain, HPF and layers of the isocortex. Key shows regions and layers (Table S1). (D,E) High-magnification representative images of puncta fluorescence decay in layers of the motor cortex (D) and in stratum radiatum of CA1, CA2 and CA3 subregions of the HPF (E). For brain region/subregion abbreviations, see Table S1. Scale bars: 2 mm (C, whole brain), 500 μm (C, HPF and CTX), 5 μm (D,E).
Genetic labeling and visualization of postsynaptic protein lifetime
In PSD95-HaloTag mice, PSD95 expression, protein complex assembly, and the physiology of synaptic transmission and synaptic plasticity are normal (Fig. S1). The bond between PSD95-HaloTag and a fluorescent HaloTag ligand (tetramethyl-rhodamine-Halo, or TMR-Halo) proved to be extremely stable (Fig. S1G), consistent with previous studies (Encell et al., 2012). To irreversibly label and visualize the PSD95-HaloTag in vivo, we injected a cell- and blood brain barrier-permeable fluorescent ligand (silicon-rhodamine-Halo, or SiR-Halo) (Lukinavicius et al., 2013; Masch et al., 2018) into the tail vein of PSD95-HaloTag mice and prepared sections of brain tissue for imaging on a spinning disk confocal microscope (pixel resolution 84 nm and optical resolution, as measured by Full Width at Half Maximum, ∼330 nm in x-y and ∼620 nm in z) (Fig. S2). By examining the persistence of labeling after injection, we could identify the synapses and brain regions with different PSD95 lifetimes (Fig. 1B).
Imaging the brain of 3-month-old PSD95-HaloTag mice 6 hours (day 0) after injection revealed widespread labeling (Fig. 1C, S3A), whereas none was detected in injected wildtype mice (Fig. S3B). 300 nmol of injected SiR-Halo was sufficient to saturate the majority of HaloTag binding sites (Fig. S3C). Higher doses of SiR-Halo were not feasible due to toxic effects of the DMSO solvent (Thackaberry et al., 2014). We confirmed that the SiR-Halo ligand quantitatively labeled all brain regions by injecting it into compound heterozygous Psd95HaloTag/eGFP mice, quantifying the SiR-Halo and eGFP synaptic puncta density in 110 brain regions, and then testing their correlation (R = 0.976, P < 0.0001) (Fig. S3D,E). To better understand how long the SiR-Halo ligand remains in the body after injection, we performed a pharmacokinetic analysis (STAR Methods, Fig S4). The peak concentration of SiR-Halo was detected 1 min after injection (blood, 4.6 μM; brain, 0.08 μM), and by 15 minutes the concentration was 50% of the peak value in both blood and brain. By 24 hours, SiR-Halo was undetectable in blood and brain (< 10 nM detection threshold).
Spatial diversity in synaptic protein lifetime
Images collected over the course of 2 weeks revealed a loss of labeling in all brain regions, with different regions showing different rates (Fig. 1C, S3A). Synapses with the longest PSD95 protein lifetime were concentrated in the isocortex (neocortex) and hippocampal formation (HPF) (Fig. 1C, S3A). Examination of images at single-synapse resolution revealed that each brain region is composed of populations of synapses with different PSD95 lifetimes (Fig. 1D, 1E, S5), some with long (weeks) and others with short (hours or days) protein lifetimes, which we refer to as LPL and SPL synapses, respectively. These synapse populations were spatially organized. For example, within the laminar organization of the isocortex there was a clear gradient from superficial (layer 1) to deep (layer 6) layers, with synapses with the longest PSD95 lifetime located in layer 1 (Fig. 1C, 1D, S3A, S5). In the HPF, the dendritic fields of pyramidal neurons in the CA1 exhibited synapses with longer PSD95 lifetimes than those in CA2 and CA3 (Fig. 1C, 1E, S3A, S5). This pattern was evident in both basal and apical dendrites, indicating that synaptic protein lifetime is determined, at least in part, by cell-wide mechanisms that differ between pyramidal neuron subtypes (Fig. 1C, S5). Synapses in the dendritic fields of granule cell neurons in the dentate gyrus (DG) and olfactory bulb had short PSD95 protein lifetimes, further suggesting cell-type-specific turnover mechanisms (Fig. 1C, S5). Comparison of the large CA3 thorny excrescence synapses and those in the polymorphic layer of the DG with the smaller CA1 synapses showed that the largest synapses of the HPF (Broadhead et al., 2016; Cizeron et al., 2020; Harris and Weinberg, 2012; Zhu et al., 2018) do not have the longest protein lifetimes (Fig. S5).
A brainwide synaptome atlas of protein lifetime in excitatory synapses
To quantify PSD95 protein lifetime in brain subregions we calculated its half-life using single-synapse resolution data and SYNMAP synaptome mapping technology (Zhu et al., 2018). PSD95-HaloTag mice were injected with SiR-Halo ligand and parasagittal brain sections imaged at post-injection time points (6 h, 1.5, 3, 7, 14 days) (Fig. 1C, S3A). The density and intensity of SiR-Halo-labeled synaptic puncta were quantified in 12 overarching brain areas (isocortex, olfactory areas, HPF, cortical subplate, striatum, pallidum, thalamus, hypothalamus, midbrain, pons, medulla, cerebellum) and 110 subregions registered to the Allen Reference Atlas (Table S1). By fitting a single-phase exponential decay function to the data for every subregion we calculated the average PSD95 puncta density (PSD95 densityt1/2) and intensity (PSD95 intensityt1/2) half-lives and plotted the values in brain maps (Fig. 2A, 2B, S6A,B). PSD95 densityt1/2 is derived from the reduction in the number of SiR-Halo-positive puncta, whereas PSD95 intensityt1/2 is derived from the reduction in SiR-Halo signal within labeled synapses. The two half-life measures were highly correlated (R = 0.9088, P < 0.0001; Fig. S6C). We also tested if SiR-Halo labeling and puncta detection efficiency in brain subregions influenced protein lifetime measurements and found no correlation (Pearson’s correlation, R = 0.1337, P = 0.1638). The half-life density (PSD95 densityt1/2) and intensity (PSD95 intensityt1/2) datasets were used to create the Protein Lifetime Synaptome Atlas (Bulovaite et al., 2021a).
Fig. 2. A brainwide atlas of synapse protein lifetime.

(A) PSD95 puncta half-life across 110 mouse brain subregions (Table S1). The 12 main brain areas are color-coded to the right. N=7–9 Psd95HaloTag/HaloTag animals. (B) PSD95 puncta half-life in HPF, with key elements of the circuitry shown. The pyramidal neurons in entorhinal cortex (EC) layer II project to CA1sl, pyramidal neurons in entorhinal cortex layer III project to CA1sm, and stellate neurons in entorhinal cortex layer II project to DG granule neurons, which in turn project to CA3, which project to CA1 (Marks et al., 2020). (C) Brain subregions ranked according to their half-life (± 95% confidence intervals), color-coded as in (A). (D) Fluorescent puncta decay (mean fraction of puncta remaining at day 7 compared with day 0, ± SD) across apical and basal dendrites of the CA1 pyramidal cells. N=3 Psd95HaloTag/HaloTag animals per time point. Reference points for calculating gradients along the apical dendrite are marked by a–d.
Dendritic and subregional distribution of synapses with different PSD95 lifetimes
We observed a 10-fold difference in PSD95 densityt1/2 between the subregions with the longest (layer 1 of the motor cortex, 12.1 days) and shortest (olfactory bulb glomerular layer, 1.2 days) half-life (Fig. 2A, 2C, S6A, S7A). Comparison of PSD95 densityt1/2 for the 12 overarching brain areas showed that cortical structures are populated by synapses with the longest protein lifetimes, in contrast to subcortical structures which predominantly comprise synapses with the shortest protein lifetimes (Fig. 2A, 2C, S6A,B, S7B-D). All regions of the isocortex exhibited a gradient, with a 2.4-fold range in PSD95 densityt1/2 from layer 1 (10.6 days) to layer 6 (4.5 days) (Fig. 2A, S6A, S7E,F). Similarly, the subregions within the HPF with the shortest PSD95 densityt1/2 were in initial pathways of the trisynaptic circuit (DG, 3.5 days; CA2, 1.9 days; CA3, 3.0 days), whereas those with the longest PSD95 densityt1/2 were in the latter part of the circuit in the CA1 field (5.5 days) and subiculum (6.4 days) (Fig. 2B, S7G,H).
Although dendrites contain protein synthesis and degradation machinery (Biever et al., 2020; Bingol and Schuman, 2006; Ostroff et al., 2002; Steward and Levy, 1982) it is not known whether synapse protein lifetime varies across the dendritic tree. We quantified PSD95 densityt1/2 in a series of parallel windows from the pyramidal neuron soma in the CA1 to the distal basal dendrites in stratum oriens (so) and to the distal apical dendrites in stratum radiatum (sr), stratum lacunosum (sl) and stratum moleculare (sm) (Fig. 2D). In apical dendrites, a gradient of increasing PSD95 protein lifetime was observed (Fig. 2D, a–d: linear regression, R2 = 0.1571, P = 0.007). We next examined segments of the apical dendrite including the sr, sl and sm, which each receive distinct afferents (from CA3, entorhinal cortex layer II and entorhinal cortex layer III, respectively). A positively-sloped gradient in PSD95 lifetime was found in sr (Fig. 2D, a–b: linear regression, R2 = 0.5110, P < 0.0001). There was a significant reduction in PSD95 lifetime between distal sr and the sl (Fig. 2D, b–c: Welch’s t-test, P = 0.0151), which was not due to differences in synapse size (Fig. S8). From the sl to the distal end of the sm we observed a positively-sloped gradient (Fig. 2D, c–d: linear regression, R2 = 0.6234, P = 0.0005). PSD95 puncta density followed a similar gradient to that of PSD95 lifetime, but since half-life measure is independent of absolute puncta numbers at day 0, differences in puncta density cannot affect half-life values (Fig. S8). In the basal dendrites of so there was an initial gradient in PSD95 protein lifetime before levelling and reversing in distal dendrites. These results show that synapse protein lifetime differs within the dendritic tree of neurons and that synapse protein lifetime is influenced by distance from the soma and by afferent inputs.
PSD95 turnover occurs in stable dendritic spines
Dendritic spines can be structurally stable for long or short durations (Grutzendler et al., 2002; Holtmaat et al., 2005; Trachtenberg et al., 2002; Zuo et al., 2005). We asked if protein turnover occurs within stable spines or whether the loss of HaloTag labeling might reflect spine elimination. To visualize dendritic spines housing PSD95 in individual apical dendrites, we dye filled CA1 pyramidal neurons from PSD95-HaloTag mice injected with SiR-Halo (Fig. 3A, S9A). Consistent with previous results that show PSD95 is expressed in a subset of larger, more stable synapses (Cizeron et al., 2020; Fortin et al., 2014; Santuy et al., 2020; Zhu et al., 2018), a subset of spines expressed PSD95 at day 0 and labeled spines were still visible 10 days after SiR-Halo injection. If PSD95 were being turned over in synapses, we reasoned that we could observe both ‘old’ and ‘new’ protein using a two-step labeling procedure. Three days after injecting mice with SiR-Halo we labeled brain sections with TMR-Halo and found that the two labels were colocalized in many synapses (Fig. 3B, S9B).
Fig. 3. PSD95 turnover in dendritic spines.
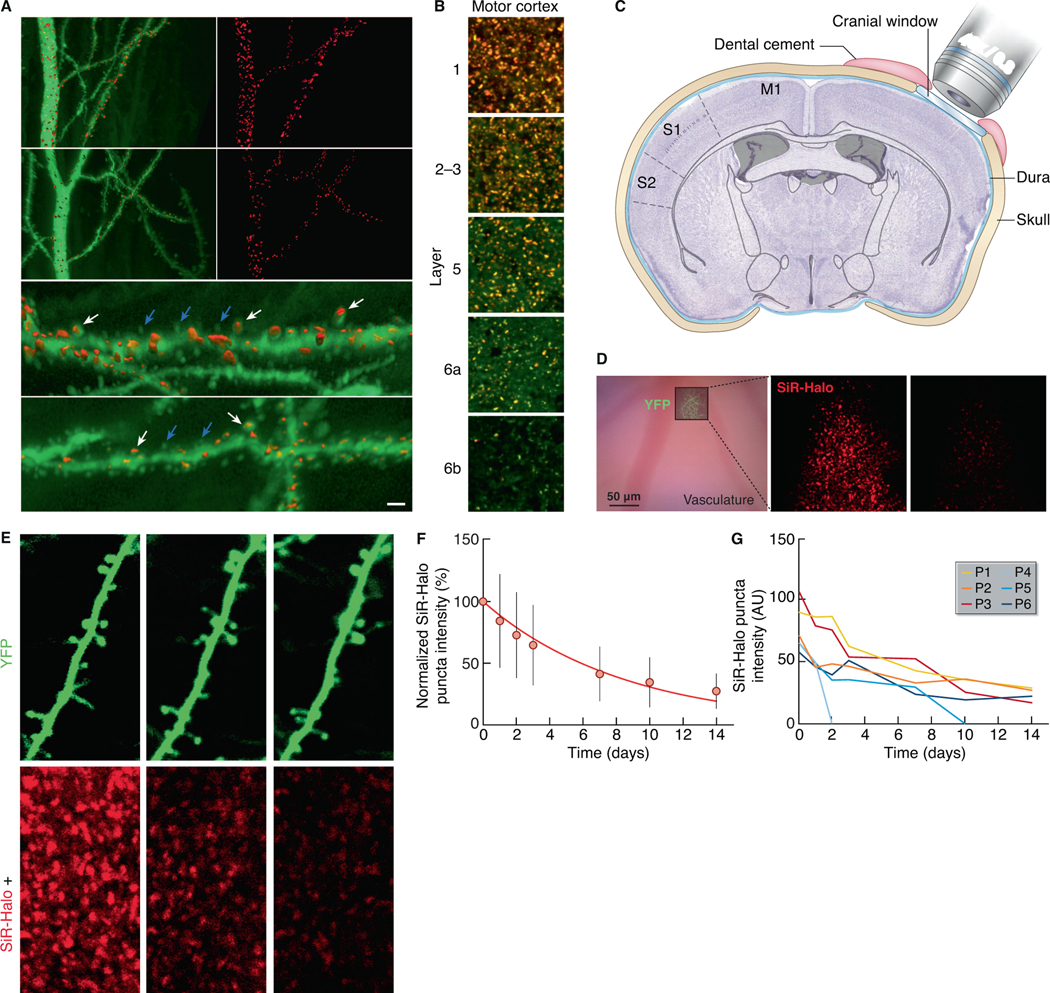
(A) 3D reconstructed dye-filled CA1 neurons and SiR-Halo-positive puncta. (i-iv) CA1 apical dendrites (stratum radiatum) intracellularly injected with Alexa 488 (green) at day 0 (i, ii) and at day 10 post-injection of SiR-Halo (red) (iii, iv). (v, vi) Higher-magnification of (i) and (iii), respectively, showing dendritic spines with SiR-Halo-positive puncta (white arrows) and without SiR-Halo signal (blue arrows). Scale bar: 5 μm (i-iv), 1.5 μm (v, vi). (B) Colocalization (orange) of SiR-Halo (red) injected at day 0 and post-fixation-applied TMR-Halo (green) at day 3 in synapses across layers of the motor cortex. Scale bars: 5 μm. (C) Schematic of the cranial window implant above the somatosensory cortex. (D) Bright-field image (left) of the superficial vasculature and, superposed, a fluorescence image of YFP-labeled axons and dendrites in cortical layer 1 that are used as fiducial points for tracking SiR-Halo ligand puncta over time, as shown for day 0 (center) and day 7 (right). (E) A YFP-labeled dendrite (top) containing SiR-Halo-labeled puncta (bottom), repeatedly imaged over days to weeks. SiR-Halo puncta can be tracked in stable and dynamic spines (e.g. P1-P5) and in the immediate surrounding of the dendrite (e.g. P6). (F) The mean (± SD) fluorescence intensities of individual SiR-Halo puncta (n=295; N=3 Psd95HaloTag/HaloTag mice) normalized to the first imaging time point. Red line, a single exponential fit of the means (λ=0.12/day; t1/2=5.8 days). (G) The measured fluorescence intensity over time of the SiR-Halo puncta examples in (E).
To simultaneously visualize PSD95 turnover and spine turnover in the brain of living PSD95-HaloTag mice, we monitored individual synapses in layer 1 of somatosensory cortex through a cranial window (Holtmaat et al., 2009) over weeks after injection of SiR-Halo (Fig. 3C). Viral vectors expressing YFP were used to visualize dendritic spines and estimate spine size (STAR Methods, Fig. 3D, 3E) (Holtmaat et al., 2005). In individual synapses we observed decay profiles that were similar (Fig. 3F) to the population data (Fig. 2). A comparable overall decay of fluorescence was observed in regions imaged only once over the course of the experiment (Fig. S9C), excluding photobleaching as a main factor in driving the fluorescence decay. In line with the dye injection study (Fig. 3A), not all spines expressed PSD95 to a similar extent (Fig. 3E, 3G), although in general SiR-Halo fluorescence intensities correlated well with spine size (Fig. S9C) (Gray et al., 2006). Even within single dendrites, spines variably retained the SiR-Halo label. Label retention was weakly related to spine size (Fig. S9C), and spines observed to disappear often had low levels of PSD95 or rapid loss of labeling (Fig. 3E, 3G), consistent with published observations (Cane et al., 2014; Gray et al., 2006). Together, these data indicate that structurally stable spines differ in their PSD95 content and lifetime, even within the same dendrite.
Synapse classification by protein lifetime
The differences in protein lifetime between individual synapses raise questions about the diversity of synapses that comprise the synaptome. In previous work we used a data-driven approach to classify synapses from across the whole brain and lifespan based on their ‘static’ protein composition (Cizeron et al., 2020; Zhu et al., 2018). Using two markers (PSD95eGFP and SAP102mKO2), each of which assembles a set of postsynaptic proteins into distinct molecular complexes (Frank et al., 2016), we classified synapses into three types: type 1 express PSD95, type 2 express SAP102, and type 3 express both markers. Each of these types was further classified using morphological criteria into a total of 37 subtypes (Zhu et al., 2018). To determine whether the different types and subtypes of synapses have characteristic protein lifetimes, we combined synaptome mapping of PSD95-HaloTag with PSD95eGFP and SAP102mKO2 using triple knock-in mice (Psd95HaloTag/eGFP;Sap102mKO2/y). Six-month-old triple knock-in mice were injected with SiR-Halo and brain sections collected at day 0 and day 7. To test that the estimation of half-life using two time points accurately reflected measurements made with five time points (Fig. 2), we correlated the values obtained in all brain subregions and found a very high correlation (puncta density, R=0.9789, P<0.0001; puncta intensity, R=0.9506, P<0.0001) (Fig. S10A). SiR-Halo-positive synapses were categorized using PSD95eGFP and SAP102mKO2, allowing us to determine the PSD95 lifetime of two synapse types (type 1 and type 3) and 30 of the 37 known subtypes (all except subtypes 12–18, which do not express PSD95) in all brain areas. Ranking the synapse subtypes by their PSD95 protein lifetime from brainwide data (Fig. 4A) and individual regions (Fig. S10B) showed that both type 1 and type 3 synapses contain subtypes with short and long protein lifetimes. Thus, populations of synapses with distinct molecular compositions are further divisible into subtypes with varying protein lifetimes. Synapse subtype 2 maintained the most (96.7%) and subtype 6 the least (23.3%) label after 7 days (Fig. 4A). To visualize the potential duration that any synapse subtype could retain copies of PSD95 we plotted exponential decay functions (Fig. 4B). This revealed that several synapse subtypes in the adult mouse could retain copies of PSD95 for many months and, potentially, as long as the natural lifespan of the mouse (Burt, 1940; Hamilton Jr, 1937).
Fig. 4. PSD95 protein lifetimes differ between synapse subtypes.

(A) Ranked bar plot of synapse subtypes (coded 1–37, Zhu et al, 2018) according to their associated PSD95 lifetimes using whole-brain data. Shown is the percentage of each SiR-Halo-positive subtype remaining at day 7 compared with day 0. (B) Decay curves of PSD95 lifetime in type 1 and type 3 synapse subtypes, shown as percentage of synapses retaining SiR-Halo. Longest-lived subtypes (s) are indicated. (C) Synapse subtype PSD95 protein lifetimes for 110 brain subregions represented as a heatmap. The values in each row are normalized. (D) Self-organizing feature map (SOM) clustering based on subtype densities (STAR Methods). Color key as in Fig. 2A. (E) Percentage of LPL synapses in brain regions and subregions (Table S1). The cortical layers in isocortex are numbered and shown from top to bottom. Days 0 and 7 n=8 Psd95HaloTag/eGFP;Sap102mKO2/y mice.
Building blocks of synaptome architecture
The protein lifetime of each synapse subtype was independent of its location in the brain, with subtype lifetime rankings maintained across brain regions (Fig. 4C, S10B). This indicates that synapse subtypes with different protein lifetimes are general building blocks of whole-brain architecture. Furthermore, these findings suggest that the spatial distribution of synapse subtypes with different protein lifetimes underpins the half-life measurements from individual brain regions. To confirm this, we generated a self-organizing feature map (SOM), which reduces the high dimensionality of synaptome maps of the 37 subtypes to a two-dimensional representation (Fig. 4D, S10C). The SOM shows a systematic organization in which anatomically related regions become closely clustered owing to their similarity in subtype density (Fig. S10C). Consistent with our hypothesis, we observe a systematicity in half-lives when subregion half-life values are shown (Fig. 4D).
LPL synapses are particularly interesting because they could potentially maintain PSD95 modifications for long durations. When grouped together, the five subtypes with longest protein lifetimes (subtypes 2, 34, 3, 35, 5) represented 6.7% of PSD95-positive excitatory synapses in the whole brain; however, there were marked regional and subregional differences (Fig. 4E, S10D), with a 15-fold range across the main brain areas: isocortex (11.9%), striatum (5.9%), cortical subplate (4.4%), cerebellum (3.9%), HPF (3.8%), olfactory areas (3.0%), midbrain (2.4%), hypothalamus (2.2%), medulla (2.2%), pons (2.1%), pallidum (1.8%), thalamus (0.8%). The subregion with the highest percentage of these LPL synapses was layer 2–3 of the somatosensory cortex (17.2%), while the lowest percentage was found in the CA2 stratum lacunosum-moleculare (0.2%). Isocortex regions had the highest percentages of LPL synapses, with superficial layers showing 4- to 5-fold more LPL synapses than the deepest layers. Within the HPF, the subiculum had the highest percentage (9.6%) of LPL synapses of all subregions, followed by CA1sr (5.8%). These results show that the neocortex (isocortex), which is the most recently evolved region of the cortex as compared with the ancient allocortex (olfactory areas and HPF) (Rakic, 2009), contains higher numbers of LPL synapses, with the superficial layers of the neocortex being particularly well populated.
Synapse protein lifetime increases throughout the lifespan
Synapse protein synthesis and turnover are thought to be important during brain development and aging (Hipp et al., 2019; Kaushik and Cuervo, 2015; Labbadia and Morimoto, 2015; Yerbury et al., 2016), yet to our knowledge there have been no studies of synaptic protein turnover comparing mice of different ages. We injected SiR-Halo into PSD95-HaloTag mice at three ages (3 weeks (W), 3 and 18 months (M)) that correspond to childhood, mature adulthood and old age (Cizeron et al., 2020).
These lifespan studies revealed that PSD95 protein lifetime increases in the majority of brain regions from birth to old age (Fig. 5A, 5B, S11A-B). At 3W there was a paucity of subregions showing long protein lifetimes, with only the most superficial layers of the neocortex and CA1 sl/sm showing a PSD95 densityt1/2 above that of other regions (Fig. 5A). Between 3W and 3M, the PSD95 densityt1/2 increased in all subregions, with the greatest increases in subregions of the isocortex and cerebellum granular layer (Fig. 5A, 5B). We also examined age-dependent changes in PSD95 protein lifetime in the dendritic tree of CA1 pyramidal cells (Fig. 5C). At 3W there were nascent gradients in both basal and apical dendrites formed from synapses with short protein lifetimes, and their protein lifetime increased ∼5-fold by 3M. To uncover which synapse subtypes contribute to this increase we measured the effect size (Cohen’s d) for the change in density for all subtypes between 3W and 3M. This identified subtype 2, which is the LPL subtype with the longest protein lifetime, as significantly increased across many brain regions (Fig. S11C i, ii). By contrast, no increase was observed for subtype 34 (a type 3 LPL subtype; Fig. S11C iii), indicating that LPL synapses with different molecular compositions are non-redundant and have distinct developmental trajectories. During development, brain regions differentiate from each other by the acquisition of sets of diverse synapses (Cizeron et al., 2020) and we asked if brain regions differentiate with respect to their synapse protein lifetime compositions. Comparison of the PSD95 densityt1/2 between all subregions showed a high similarity at 3W that dramatically decreased by 3M (Fig. 5D). Together, these data show that from weaning to maturity the mouse brain accumulates synapses with long protein lifetimes, and that brain regions acquire a distinct and thus specialized compositional signature of synapses with different protein lifetimes.
Fig. 5. Synapse protein lifetime changes across the lifespan.

(A) PSD95 puncta half-life across 110 mouse brain subregions in PSD95-HaloTag mice at 3W, 3M and 18M for the whole brain (top row) and HPF (bottom row). N=7–10 Psd95HaloTag/HaloTag mice for each time point and age group. (B) Percentage increase in puncta half-life between 3W and 3M (left brain map) and between 3W and 18M (right brain map). (C) Mean fraction of puncta remaining at day 7 compared with day 0 (± SD) is plotted across basal and apical dendrites of the CA1 dendritic tree at 3W, 3M and 18M (n=3 mice per group). (D) Matrix of half-life similarities between pairs of subregions (rows and columns) for the 3W and 3M age groups. Brain subregions are listed in Table S1. (E) Heatmap showing change (Cohen’s d effect size) in synapse subtype (subtype number shown at top) density across brain subregions between 3M and 18M. Subtypes are ranked with longest lifetime on the left and shortest on the right. *P<0.05 (Benjamini-Hochberg correction). See Fig. S11C for detailed heatmaps.
We next examined how the brain changes with old age. Between 3M and 18M the regional PSD95 densityt1/2 increased in most areas of the brain (Fig. 5A, 5B). By contrast, aging did not influence the spatial organization of synapses on the dendritic tree of CA1 pyramidal neurons (Fig. 5C). Previously, we showed an overall loss of excitatory synapses between 3M and 18M but with a subtype specificity to the retention or loss (Cizeron et al., 2020). Here, we found that the increased regional PSD95 densityt1/2 in old age reflects the preservation of LPL synapses. Although most synapse subtypes decrease between 3M and 18M (Cizeron et al., 2020), the LPL synapses (subtypes 2, 34, 3, 5) were retained (Fig. 5E, S11C iv-vi). Subtype 2 was preferentially retained in neocortex (isocortex), whereas in allocortex (olfactory cortex and HPF) subtypes 34, 3 and 5 were retained, revealing that LPL synapses with different molecular compositions are resilient to aging and populate distinct brain regions.
Increased synaptic protein lifetime in a model of autism and schizophrenia
Mutations in the DLG2 gene cause autism and schizophrenia in humans (Fernandez et al., 2009; Fromer et al., 2014; Ingason et al., 2015; Kirov et al., 2012; Nithianantharajah et al., 2013; Purcell et al., 2014; Reggiani et al., 2017; Ruzzo et al., 2019; Walsh et al., 2008) and the Dlg2 mutant mouse model recapitulates the impairments in attention, learning and cognitive flexibility found in humans (Nithianantharajah et al., 2013). Dlg2 encodes PSD93, a postsynaptic scaffold protein that binds to PSD95, and together these proteins assemble a ternary complex with the NMDA receptor subunit GluN2B (Frank et al., 2016). To test if Dlg2 mutations affect the synaptome map of PSD95 lifetime we bred a cohort of PSD95-HaloTag mice containing heterozygous (Dlg2+/–;Psd95HaloTag/+) or homozygous (Dlg2–/–;Psd95HaloTag/+) null alleles of Dlg2 (Fig. 6, S12). Synaptome maps showed a robust increase in PSD95 protein lifetime across all 110 brain regions of both heterozygous and homozygous Dlg2 mutant mice as compared with PSD95-HaloTag control mice, with a gene-dosage-dependent effect. PSD95 half-life increased up to 210% in heterozygous and 380% in homozygous Dlg2 mutants. Most affected were subregions of the isocortex and HPF CA1, which are areas that show dysfunction in schizophrenia (Bähner and Meyer-Lindenberg, 2017; Heinz et al., 2019), intellectual disability and autism (Carper and Courchesne, 2005; Postema et al., 2019; Weston, 2019).
Fig. 6. Synapse protein lifetime increases in Dlg2 mutant mice.

The percentage increase in PSD95 puncta density half-life in 3M mice carrying heterozygous or homozygous Dlg2 mutations, as compared with control mice. N=5–8 mice for each time point and genotype (animals are heterozygous for PSD95-HaloTag).
Discussion
We have systematically measured protein lifetime at single-synapse resolution across the brain and lifespan of the mouse using the HaloTag self-labeling system and synaptome mapping technology. We focused on PSD95 because it is a highly abundant protein in the postsynaptic terminal of excitatory synapses that plays a crucial role in assembling glutamate receptors and other proteins into signaling complexes that control synaptic strength, synaptic plasticity and learning (Carlisle et al., 2008; Fernandez et al., 2017; Fitzgerald et al., 2015; Frank et al., 2016; Migaud et al., 1998; Nithianantharajah et al., 2013). Between the ages of 3 and 6 months, when the amounts of PSD95 in individual excitatory synapses remain stable (Cizeron et al., 2020), we found that the vast majority of synapses in all brain areas replaced their PSD95 content within a few days or weeks. This indicates that proteostasis is constantly rebuilding the excitatory synapse proteome and does so many times throughout the life of the mouse in most brain synapses.
Brain synapses differed widely in their PSD95 protein lifetimes and these had distinct spatial distributions. We designated the synapse subtypes with the longest protein lifetimes as LPL synapses and those with the shortest protein lifetime as SPL synapses. It is striking that the neocortex is the brain region most enriched in LPL synapses. Moreover, there is a gradient of enrichment across the layers, with the greatest density of LPL synapses in the superficial layer and the lowest density in the deepest layer. Some LPL synapses in the adult mouse brain showed a potential to retain PSD95 for many months, indicative of a remarkable proteome stability. A progressive lengthening of PSD95 lifetime occurs across the whole brain between 3 weeks and 18 months of age. In the aging mouse brain there is a general loss of synapse number and it was the LPL synapses that are preferentially retained in old age. In a mouse model of autism and schizophrenia there was a brainwide increase in PSD95 lifetime, indicating that germline disease-relevant mutations that cause childhood and young-adult disorders impact on synapse protein lifetime.
Protein composition and lifetime contribute to synapse diversity
Synapse diversity arises by differences in protein composition (Zhu et al., 2018) and here we show that it also arises by differences in protein lifetime. To further understand the relationship between these two parameters we combined synaptome mapping using two markers that inform on protein composition (PSD95eGFP and SAP102mKO2) with the PSD95-HaloTag that allows quantification of protein lifetime. This enabled us to assign a protein lifetime measure to 30 of the previously defined synapse subtypes (Zhu et al., 2018). We found that synapses with different protein compositions (type 1, expressing PSD95 only; type 3, expressing PSD95 and SAP102) included both LPL and SPL synapses with a wide range of PSD95 protein lifetimes. Thus, synapse protein composition and lifetime are two orthogonal parameters that define the synapse proteome, adding another layer to the known diversity of brain synapses. The protein lifetime of synapse subtypes was independent of brain region, strongly supporting the conclusion that synapses with different protein lifetimes are building blocks that can be employed in any area of the brain, where they underpin the synaptome architecture of neurons, regions and, ultimately, the overall brain.
Synaptome architecture arises from the spatial distribution of synapses with different protein lifetimes
LPL and SPL subtypes are distributed in all brain regions at differing abundance in a non-random manner. Within individual dendrites, protein lifetime differed between adjacent synapses, was longer in distal dendrites and appeared to be influenced by synaptic inputs at different locations on the dendritic tree. Synapses within hippocampal subregions and layers showed different protein lifetimes, indicating projection- and cell-type-specific regulation of PSD95 lifetime. It will be of interest to ask if synapse subtypes with different protein lifetimes are in spatial proximity to dendritic translational machinery and proteosomes (Biever et al., 2020; Bingol and Schuman, 2006; Ostroff et al., 2002; Steward and Levy, 1982) and whether the influence of inputs on postsynaptic protein lifetime is controlled by protein interactions across the synaptic cleft or by activity-dependent mechanisms.
A lifespan trajectory of synapse protein lifetime
PSD95 lifetime is shortest in juvenile mice and longest in old mice. This lifespan trajectory of increasing protein lifetime occurs across almost all brain regions. Between 3 and 4 weeks of age there is an increase in the amount of PSD95 in individual synapses (Cizeron et al., 2020), indicating that PSD95 synthesis must exceed its degradation. Yet at this age, synapses have the shortest PSD95 lifetime and the highest rate of protein degradation. This indicates that in juvenile mice, which are at the point of weaning and beginning an independent life, there is a very high rate of synapse proteome construction and remodelling, which is well suited to the adaptive changes programmed by the transcriptome (Skene et al., 2017) and any superimposed activity-dependent changes induced by environmental experience.
We previously showed that specific synapse subtypes are preferentially retained in old age whereas others are lost: ‘age-resilient’ and ‘age-sensitive’ synapse subtypes, respectively (Cizeron et al., 2020). Here we report that the age-resilient synapse subtypes correspond to LPL synapses and the age-sensitive subtypes correspond to SPL synapses. Thus, in old age, synapses with rapid proteostasis and adaptive properties are depleted from the brain, whereas those with slow protein turnover and slow adaptive properties are retained.
Functional implications of synapse protein lifetime diversity
The adaptive and homeostatic properties of synapses have been of intense interest, particularly in the context of how synaptic proteins are modified in response to neuronal activity and environmental experience. Central among these proteins is PSD95 which assembles glutamate receptors with other proteins into supercomplexes (Fernandez et al., 2009; Frank et al., 2016; Frank et al., 2017; Husi et al., 2000). Although these proteins and their modifications are necessary for the initial encoding of memories, the mechanisms controlling the duration of memories and mechanisms of forgetting are less clear. Our discovery of LPL synapses, which could retain copies of PSD95 for many months, challenges Crick’s assumption (Crick, 1984) that synaptic protein lifetime is far shorter than long-term memory duration. Several findings suggest that LPL synapses may be involved with retaining memories for longer durations than SPL synapses. LPL synapses are heavily enriched in the neocortex where long-term memories are stored (Squire and Dede, 2015). Young mice, which have fewer LPL synapses than mature mice, have shorter-lasting memories than adult mice (Akers et al., 2012). The lifetime of PSD95 in CA1, visual and piriform cortex corresponds well with the duration of spatial, visual and olfactory memory representations measured using calcium imaging of individual neurons (Driscoll et al., 2017; Rubin et al., 2015; Schoonover et al., 2021) and with the duration of protein-synthesis-independent memories in dentate gyrus engram cells (Roy et al., 2017). A hallmark of old age is the retention of long-term memories and a reduction in short-term and working memory capacity (Li et al., 2004; Reinert, 1970; Tucker-Drob, 2009), which is paralleled by the preferential retention of LPL but not SPL synapses in old age.
This framework also fits with the anatomical locus of innate behaviors, which are genetically programmed and ‘hard wired’. Basal brain structures that contain circuits mediating innate behaviors are highly enriched in SPL synapses. This would endow these circuits with rapid proteostatic mechanisms capable of erasing and reversing protein modifications and thereby ensuring stable genetically-programmed synaptic functions. In support of the notion that the rapid proteostatic properties of SPL synapses are a mechanism of homeostatic synaptic plasticity, studies in CA1 pyramidal neurons show that synapses closest to the soma, which have the shortest PSD95 lifetime, contribute more to homeostasis than distal synapses (Lee et al., 2013), which have the longest PSD95 lifetime.
Synaptic protein lifetime and mechanisms of disease
Our findings have relevance for brain diseases that arise at different ages. Childhood neurodevelopmental disorders, such as autism, are caused by mutations affecting protein synthesis and degradation (Louros and Osterweil, 2016). In the Dlg2 mutant mouse model of autism and schizophrenia, we have shown that PSD95 lifetime is severely affected in the majority of brain regions. We speculate that the mechanism involves the physical interactions of PSD95 and PSD93 (which is encoded by Dlg2) with NMDA receptors and other proteins in the postsynaptic terminal (Frank et al., 2016). The loss of PSD93 may influence PSD95 interactions with other proteins and its anchoring in the synapse. Many neurodevelopmental disorders are caused by mutations in genes that encode PSD95-interacting proteins (Bayes et al., 2011; Fernandez et al., 2009; Fromer et al., 2014; Husi et al., 2000; Kirov et al., 2012; Purcell et al., 2014; Skene et al., 2017) and it will be of interest to establish whether these affect PSD95 protein lifetime.
The longer PSD95 protein lifetime in synapses of old mice could potentially be explained by the progressive reduction in protein clearance that underpins senescence and longevity (Kelmer Sacramento et al., 2020; Koyuncu et al., 2021). This reduction in protein clearance contributes to the accumulation of toxic and misfolded proteins in neurodegenerative disorders (Labbadia and Morimoto, 2015; Yerbury et al., 2016) and is thought to result from decreased proteasomal activity in aging (Carrard et al., 2003; Dasuri et al., 2009; Hayashi and Goto, 1998; Kelmer Sacramento et al., 2020; Saez and Vilchez, 2014; Vilchez et al., 2014; Zeng et al., 2005). Our findings suggest that SPL synapses would be more efficient at removing toxic proteins than LPL synapses and, consequently, that LPL synapses would be more vulnerable to damage. The accumulation of LPL synapses in old age, together with their enhanced vulnerability to toxic proteins, might at least in part explain the proclivity of Alzheimer’s disease to target synapses in old age and impair memory.
Technical considerations for HaloTag studies of synapse protein lifetimes
Our pharmacodynamic experiments show that the injected SiR-Halo ligand is rapidly cleared from the body, leaving negligible amounts 24 hours after injection. The ligand is therefore only available for labeling the PSD95-HaloTag protein in the brain on the day of injection and does not linger long enough to label protein synthesized on subsequent days. The rapid clearance of ligand from the circulation raises the possibility of using multiple pulses of ligand labeled with different fluorophores to examine PSD95 dynamics with greater temporal precision, exploiting the capacity of super-resolution methods to explore nanoscale lifetime architecture (Masch et al., 2018). Our mice can also be used to examine endogenous PSD95 lifetime in living primary neuronal cultures (Kratschke, 2018) and could be used to study the dwell time of endogenous PSD95 at synapses and any diffusion, trafficking or exchange of PSD95 that occurs between synapses. Endogenous PSD95 can be conditionally tagged for cell-type-specific studies of synapse protein composition and turnover (Zhu et al., 2020). Our biochemical, electrophysiological and anatomical analyses failed to expose an impact of the HaloTag; however, in the absence of a method to measure the lifetime of endogenous PSD95 at single-synapse resolution we cannot exclude the possibility that the tag might have some influence on the lifetime of proteins to which it is attached.
A number of studies have shown that proteins that interact with each other in multiprotein complexes have correlated half-lives (Martin-Perez and Villen, 2017; Mathieson et al., 2018; Price et al., 2010). This implies that PSD95 lifetime could be a ‘sentinel’ for the lifetime of many other postsynaptic proteins that it interacts with. If all proteins in a given dendritic spine had correlated lifetimes this would imply that the whole spine is governed by the same proteostatic mechanisms, such as localized dendritic protein translation and degradation machinery. Our methods could be applied to determine whether pre- and postsynaptic protein lifetimes are correlated.
The Protein Lifetime Synaptome Atlas resource
The atlas provides a versatile reference resource for understanding and modelling memory, molecular and cellular anatomy in health and disease. It provides a framework that can be integrated with anatomical, molecular and phenotypic datasets. Data from the mouse atlas can also be used to generate hypotheses for validation in other species; for example, it will be interesting to ask if the human neocortex is populated with synapses with longer protein lifetimes than those in the mouse. The value of the foundation dataset, which is already linked to the Lifespan Synaptome Atlas (Cizeron et al., 2020), will increase as it is expanded to encompass lifetime studies of further synaptic proteins, both pre- and postsynaptic and inhibitory as well as excitatory. Understanding the spatiotemporal similarities and differences between these proteins will be highly informative as to underlying proteostasis mechanisms. The atlas will take on significant health discovery value as neurological disease models are incorporated and common underlying themes may emerge.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Seth Grant (seth.grant@ed.ac.uk).
Materials availability
Newly generated materials are available from the lead contact upon request and, where appropriate, provision of a materials transfer agreement.
Data and code availability
Synaptome map data have been deposited in Edinburgh DataShare and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Gene targeting and mouse generation
Animal procedures were performed in accordance with UK Home Office regulations and approved by Edinburgh University Director of Biological Services. The genetic targeting strategy adapted from Fernandez et al. (2009) was used to fuse HaloTag protein to the C-terminus of endogenous PSD95. HaloTag coding sequence (Promega) together with a short linker were inserted into the open reading frame of the mouse Psd95 gene, followed by insertion of a loxP floxed PGK-EM7-neo-pA cassette, using recombination in Escherichia coli. E14Tg2a ES cells (from 129P2 ola) were used for gene targeting. Following identification of positive targeting clones using PCR, E3.5 blastocysts from C57BL/6 mice were injected with ES cells containing the target gene. Male chimeras were crossed with C57BL/6 females to produce first generation heterozygous animals. These mice were crossed with CAG Cre recombinase-expressing mice in order to remove the loxP floxed neo cassette in vivo. Animals were then cross-bred to produce a homozygous PSD95-HaloTag colony.
PSD95-HaloTag mice were genotyped by PCR using the following primers (5’−3’): exon F, GTCACATGTCTTTGTGACCTTG; 95UTR F, GATACATGCAGAGAGGAGTGTC; and Halogen F, CTGACTGAAGTCGAGATGGAC. Generation and characterization of the Psd95eGFP/eGFP;Sap102mKO2/mKO2 knock-in mouse line was described previously (Zhu et al., 2018). To establish the Psd95HaloTag/eGFP;Sap102mKO2/y line, double homozygous Psd95eGFP/eGFP;Sap102mKO2/mKO2 mice were crossed with Psd95HaloTag/HaloTag mice. Generation and characterization of the Dlg2 mutant mouse line was described previously (McGee et al., 2001). To establish the Dlg2+/–;Psd95HaloTag/+ line, Dlg2–/– mice were crossed with Psd95HaloTag/HaloTag mice.
The mice were group housed and littermates were randomly assigned to experimental groups. The following groups of mice (m, male; f, female) were used for the study of PSD95 lifetime: 3M animals, day 0 n=9 (5m, 4f), day 1.5 n=8 (4m, 4f), day 3 and day 14 n=8 (3m, 5f), day 7 n=7 (2m, 5f); 3W animals, day 0 n=8 (5m, 3f), day 7 n=8 (6m, 2f); 18M animals, day 0 n=10 (7m, 3f), day 7 n=9 (8m, 1f). For triple colocalization the groups were: day 0 and day 7, n=8 (8f). For studies in Dlg2 mutant animals, 3- to 4-month-old animals were used and the groups were: Dlg2+/+;Psd95HaloTag/+ animals, day 0 n=5 (2m, 3f), day 7 n=6 (2m, 4f); Dlg2+/–;Psd95HaloTag/+ animals, day 0 n=8 (6m, 2f), day 7 n=8 (3m, 5f); Dlg2–/–;Psd95HaloTag/+ animals, day 0 n=7 (3m, 4f), day 7 n=7 (2m, 5f). Sample size was estimated based on Cizeron et al. (2020).
Method details
Western blotting
PSD95 protein expression and abundance were tested in whole-brain synaptosome extracts from wild-type, Psd95+/HaloTag and Psd95HaloTag/HaloTag mice. Three extracts per genotype were loaded onto 4–12% gradient Bis-Tris gels, with 20 μl loaded per well. The gels were subjected to SDS-PAGE and western blotting. The membrane was probed with antibodies for PSD95, PSD95-HaloTag and alpha-Tubulin (loading control) and bands detected using the LI-COR Odyssey imaging system.
SDS-PAGE analysis
Synaptosome preparations from Psd95+/HaloTag and wild-type mice as well as cell lysates from HEK293 cells expressing cytosolic HaloTag protein alongside wild-type controls were incubated with 1 μM tetramethyl-rhodamine HaloTag ligand (TMR-Halo) for 1 hour at 4°C. The preparations were then subjected to SDS-PAGE analysis and imaged using the Li-COR Odyssey imaging system.
Electrophysiological recordings
Hippocampal slices obtained from the dorsal third of the hippocampus were prepared and maintained in vitro using previously described techniques (Babiec et al., 2017) approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles, USA. In experiments using extracellular recording techniques, slices were maintained at 30°C in an interface-slice type recording chamber perfused (2–3 ml/min) with an oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 4 mM KCl, 25 mM NaHCO3, 1 mM NaH2PO4, 2 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose (all Sigma-Aldrich). Field EPSPs (fEPSPs) evoked by Schaffer collateral/commissural fiber stimulation were recorded in stratum radiatum of the CA1 region using low-resistance (5–10 MΩ) glass recording electrodes filled with ACSF (basal stimulation rate = 0.02 Hz). LTP was induced using two, one-second-long trains of 100 Hz stimulation (inter-train interval = 10 seconds). Whole-cell voltage-clamp recordings of evoked excitatory postsynaptic currents (EPSCs) and miniature EPSCs (mEPSCs) were performed using slices maintained in submerged-slice type recording chambers perfused with a modified ACSF containing 2.4 mM KCl, 3.0 mM CaCl2, 2.4 mM MgSO4, 100 μM picrotoxin. In these experiments, recording electrodes were filled with a solution containing 102 mM Cs-gluconate, 20 mM CsCl, 10 mM K-gluconate, 10 mM TEA-Cl, 5 mM QX-314, 0.2 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na-GTP, 20 mM HEPES (pH 7.3, 290 mOsm). Evoked EPSCs (at 0.2 Hz) were recorded at membrane potentials of −80 mV and +40 mV and AMPAR-mediated and NMDAR-mediated components of the EPSCs were estimated by the amplitude of EPSCs 5 and 50 milliseconds after EPSC onset, respectively. Spontaneous mEPSCs were recorded at −80 mV in the presence of 1.0 μM TTX (Alomone Labs). Data collection and initial analysis were performed blind to genotype.
HaloTag ligand generation
SiR-Halo coupling and purification were performed as previously described (Lukinavicius et al., 2013). 50 mg (0.159 mmol) SiR-COOH dye (Spirochrome) and 36 mg (0.161 mmol, 1.5 eq) Amine (O2) HaloTag (Promega) building block were dissolved in 950 μl dimethylformamide (DMF). 90 μl (0.517 mmol, 5 eq) N,N-diisopropylethylamine (DIPEA) were added into the solution. 70 mg (0.135 mmol, 1.3 eq) benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) dissolved in 230 μl DMF was then added to the SiR-COOH and Amine (O2) HaloTag solution. The solution was stirred at room temperature for 2 hours protected from light. Prior to purification, the compound was phase-separated from DMF. The compound was collected with the organic phase (diethyl ether) while the aqueous phase (brine) was discarded. Two to three tablespoons of dried magnesium sulphate were added to the organic phase solution, mixed well and filtered to remove any aqueous remains. Diethyl ether was evaporated, and the crude product was purified using silica gel column chromatography (2% MeOH in dichloromethane) to give the SiR-Halo ligand (45 mg, 63% yield).
In vivo application of HaloTag ligands
SiR-Halo ligand was dissolved in DMSO to a stock concentration of 5 mM. HaloTag ligand solution for injections was prepared as described (Grimm et al., 2017) but at 1.5 mM dye concentration and the total injection volume was adjusted by average weight for the animal group examined. 3W age group received 70 μl (21 μl HaloTag stock solution, 7 μl Pluronic F-127, 42 μl saline), 3M age group received 200 μl (60 μl HaloTag stock solution, 20 μl Pluronic F-127, 120 μl saline) and 18M age group received 300 μl (90 μl HaloTag stock solution, 30 μl Pluronic F-127, 180 μl saline) of HaloTag dye solution. Prior to injection, animals were placed in a heat box for 5–10 minutes to allow the blood vessels of the tail to dilate and become more visible. The mice were placed in a rodent restrainer for the injection. A bolus injection of HaloTag ligand solution was performed into the lateral tail vein. Following injection, the animals were monitored for any adverse effects twice daily for the length of the experiment. The experimenter was blinded to time points and genotypes at the time of injections.
Tissue collection and section preparation
Mice were fully anesthetized by intraperitoneal injection of 0.10–0.20 ml (according to age) pentobarbital (Euthatal). The thorax was opened and an incision made to the right atrium of the heart. A fine needle (26G, 0.45 × 100 mm) was inserted into the left ventricle and the animal was transcardially perfused with 10–15 ml 1xPBS followed by 10–15 ml fixative (4% paraformaldehyde (PFA)) and left at 4°C for 3–4 hours (according to age). Samples were transferred to 30% sucrose solution and incubated for 48–72 hours at 4°C. Brains were then embedded in OCT (embedding matrix for frozen sections) solution (CellPath) inside a plastic mould (Sigma-Aldrich), the moulds placed in beakers containing isopentane (Sigma-Aldrich), and the beakers moved to a container containing liquid nitrogen for freezing. Frozen brains were stored at −80°C for up to 4–5 months. Frozen brain samples were cut at 18 μm thickness using a cryostat (NX70 Thermo Fisher) to obtain sagittal sections referring to 12–13/21 bregma level from the Allen Mouse Brain Atlas (sagittal, https://mouse.brain-map.org/experiment/thumbnails/100042147?image_type=atlas). Cut brain sections were placed on Superfrost Plus glass slides (Thermo Fisher). A drop of 1xPBS was placed on a glass slide prior to picking up the brain sections to ensure the brain tissue lay flat. After cutting, brain sections were left to dry in the dark at room temperature overnight and were then stored at −20°C. Frozen brain sections were placed in a dark chamber and incubated at room temperature for 1 hour prior to mounting the coverslips. Sections were washed with 800 μl 1xPBS to remove any remaining OCT on or around the brain tissue and allowed to dry. A 12 μl drop of MOWIOL solution was applied on top of the brain section. A glass coverslip (18 mm diameter, thickness #1.5, VWR) was carefully lowered on top of the sample to avoid any bubbles forming in between the glass slide and the coverslip. The sections were left to dry in the dark at room temperature overnight and then stored at 4°C for up to 1 week. The experimenter was blinded at the stage of tissue sectioning and section preparation for imaging.
Post-fixation labeling with HaloTag ligands
Brain sections were first washed with 800 μl PBS to remove any remaining OCT and left to dry in the dark. Hydrophobic marker pen was used to draw around sections to contain the solutions. 50 μl 10 μM TMR-Halo in 1xPBS was added to each brain section and samples incubated for 1 hour at room temperature in a wet dark chamber. Brain sections were then washed twice for 10 minutes each with 1xTBS (Tris buffered saline) containing 0.2% Triton X-100 detergent to remove any unbound TMR-Halo ligand and once with 1xTBS for 10 minutes.
Pharmacokinetic analysis
Wild-type C57BL/6 mice (n=21, males and females, ∼2.5–10 months old) were injected with 200 μl 1.5 mM SiR-Halo solution and anaesthetised using isoflurane at different time points post-injection (1, 5, 15, 30 minutes, 1, 6, 24 hours). Additional n=6 mice did not receive an injection of SiR-Halo but were processed as per below and were used as controls. The thorax was opened and a ∼0.5 ml blood sample was drawn from the left ventricle. The animals were then perfused with 12 ml 1xPBS and decapitated. The brains were dissected and snap frozen in liquid nitrogen. The collected blood samples were left to clot for 20 minutes at room temperature and a further 5 minutes on ice and then centrifuged at 10,000 rpm for 10 minutes. The supernatant was collected, and the resulting serum samples as well as brain samples were stored at −80°C until further analysis. Each brain sample was homogenized in 7 ml buffer solution containing 1% sodium deoxycolate, 50 mM Tris pH 9 and 100 mM NaCl. Following ∼20 strokes with a homogenizer, the solution was left on ice for 30 minutes after which another 10 strokes were performed and the solution was incubated for another 30 minutes. The samples were then centrifuged at 50,000 g (20,000 rpm) for 30 minutes at 4°C. The supernatant was collected, aliquoted and stored at −80°C.
The fluorescence of SiR-Halo in blood serum and brain homogenate samples was detected using BioTek CYTATION 3 spectrophotometer (600 nm excitation wavelength). To increase the fluorescence readout (Lukinavicius et al, 2013), blood serum samples were diluted in a ratio of 1:2 in 10% SDS solution (in 1xPBS). Brain homogenates were diluted in a ratio of 1:1 in 10% SDS solution (in 1xPBS). Three mice per time point and a triple replicate per sample were performed.
CA1 pyramidal neuron filling
Mice (n=14) were perfused intracardially with 1xPBS followed by 4% PFA and, after removal, brains were post-fixed in PFA for 24 hours. After washing in PBS, brains were cut in coronal sections (200 μm thick) with a vibratome, and to identify cell nuclei the sections were prelabeled with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma). Then, CA1 pyramidal neurons were individually injected with Alexa Fluor 488 dye solution (Life Technologies) by continuous current until the dendrites were filled completely as previously described (Benavides-Piccione et al., 2013). After injections, sections were mounted in ProLong Gold Antifade mounting medium (Life Technologies). Imaging was performed with a Zeiss LSM 710 confocal microscope. The fluorescence of DAPI, Alexa 488 and 633 (SiR-Halo-positive puncta) was recorded through separate channels. Stacks of images were acquired at high magnification (63x oil immersion; pixel size 0.057 × 0.057 μm; z-step 0.14 μm) and no pixels were saturated within the spines. For intracellular injection experiments, the animals were randomly assigned into groups for different time points for sacrifice.
Spinning disk confocal microscopy
Imaging was performed using an Andor Revolution XDi spinning disk microscope equipped with CSU-X1 (pinhole size 50 μm) and 2x post-magnification lens. Images of 512 × 512 pixels in size and 16-bit depth were obtained using Andor iXon Ultra back-illuminated EMCCD camera and Olympus UPlanSAPO 100x oil-immersion lens (NA 1.4). To cover the whole area of the sagittal brain section, multi-tile single-plane image acquisition was arranged with no overlap between adjacent tiles. Focus in z-plane was achieved by selecting and recording desired z-position for four points on the brain section, which were then used to calculate the intermediate z-positions for in-between image tiles. The optical resolution was estimated based on reconstructed 3D point spread functions (PSFs) and full width at half maximum (FWHM) measurements on images of near-IR FluoSpheres (diameter: 40 nm, excitation/emission wavelength: 660/680 nm; Invitrogen). 100 μl FluoSpheres solution (1:1000 in 1xPBS) was placed on a glass slide with a brain section from a wild-type mouse and incubated for 30 min. After a wash with 1xPBS, the slides were mounted with #1.5 thickness coverslips using MOWIOL as a mounting medium. Z-stack images were acquired with 150 nm step size and 15–20 image planes were obtained for each bead sample. 3D PSFs from 392 beads were reconstructed to obtain average FWHM values.
Imaging parameters
High-magnification (x100) images at single-synapse resolution covering the whole brain section were obtained using 250 EM gain, 2-frame averaging and 5000 millisecond acquisition speed. SiR-Halo was excited at 640 nm, TMR-Halo at 561 nm, eGFP at 488 nm and mKO2 at 561 nm. Emitted light was filtered with a QUAD filter (BP 440/40, BP 521/21, BP 607/34 and BP 700/45).
Long-term imaging in vivo
5-month-old male (n=3) and female (n=1) homozygous PSD95-HaloTag mice were used in accordance with the guidelines of the Federal Food Safety and Veterinary Office of Switzerland and in agreement with the veterinary office of the Canton of Geneva (license number GE12219A). To generate sparse expression of YFP in the supragranular layers of cortex, a mix of AAV9.CamKII.Cre.SV40 (2.4×1013 GC/ml; Addgene #105558) and AAV2.Ef1α.DIO.eYFP (4.6×1012 GC/ml; UNC GTC vector #42585) (100 nl in a ratio 1:1500) was injected in the primary somatosensory cortex. Cranial windows were implanted as described previously (Holtmaat et al., 2009) using a circular (3 mm diameter) coverglass pressed down onto the intact dura and glued to the skull using dental acrylic cement. 2–3 weeks after window implantation, SiR-Halo ligand was injected in the tail vein, as described above. Images were acquired in head-fixed mice under anesthesia (0.1 mg/kg Medetomidin and 5 mg/kg Midazolam, i.p.) using a custom-built, two-photon laser-scanning microscope (2PLSM) (Holtmaat et al., 2009) controlled by custom software written in MATLAB (Scanimage, Vidrio Technologies) (Pologruto et al., 2003). A tunable Ti:sapphire laser (Chameleon ultra II, Coherent) was used as a light source, tuned to λ=840 nm for simultaneous excitation of YFP and SiR-Halo ligand, or λ=910 nm for YFP alone. Excitation power was kept constant over time. Fluorescent images were collected using a 20x, 0.95 numerical aperture water-immersion objective (Olympus) and GaAsP photomultiplier tubes (10770PB-40, Hamamatsu). Emitted light was spectrally separated using a 565 nm dichroic mirror (Chroma) and two band-pass emission filters (ET510/50 nm, Chroma; 675/67 nm, Semrock). Field-of-views (FOVs) with YFP-labeled dendrites were first imaged at 6 hours post-injection, and then re-imaged after 1, 2, 3, 7, 10 and 14 days. Additional FOVs were imaged only once over the course of the experiment to measure population fluorescence decay similarly to the in situ experiments (Fig. S25). For each FOV, two separate image stacks (typically 10–30 planes, separated by 1 μm) were acquired at 2 milliseconds/line (image size, 1024 × 1024 pixels; pixel size, 0.06 × 0.06 μm), one at excitation λ=840 nm and one at λ=910 nm. The dim YFP signal that was collected at λ=840 nm served to align the dendrites in both image stacks.
Quantification and statistical analysis
Analysis of electrophysiological data
Acquisition and analysis of electrophysiological data were performed using pCLAMP software (Molecular Devices). Results are reported as mean ± SEM.
Detection of synaptic puncta
Synaptic puncta detection from fluorescence images was performed using a machine learning-based ensemble method developed in-house by Dr Zhen Qiu (Cizeron et al., 2020; Zhu et al., 2018). The classifiers/detectors of PSD95eGFP and SAP102mKO2 were trained in a previous study (Zhu et al., 2018). The training of the image detector for PSD95-HaloTag was required. We first collected a training set of 249 images (10.8 × 10.8 μm) by randomly sampling across 12 main brain regions using bootstrapping. All puncta in the training set were manually located by three independent individuals of varying scientific expertise. Manual annotation of each punctum was then weighted for each individual. The ground truth was generated by weighted averaging over all individuals; an average weight greater than 0.7 was considered as true puncta. The ensemble learning was applied for training and a K-folder method was applied to validate and test the trained detector.
Measurement of synaptic parameters
Upon detection and localization, synaptic puncta were segmented by applying an intensity threshold. The threshold for each detected punctum was set adaptively at 10% of the height of its fluorescence intensity profile. A set of parameters were then quantified for each segmented punctum, including mean pixel intensity, size, skewness, kurtosis, circularity and aspect ratio (Zhu et al., 2018). Additionally, quantification of puncta density per unit area was performed. For calculations of PSD95 lifetime, primarily the measurements of puncta density and total fluorescence intensity content (punctum size x punctum mean intensity) were used.
Comparison of SiR-Halo and eGFP puncta populations
Owing to different imaging and puncta detection parameters, fewer SiR-Halo puncta were detected compared with eGFP (Fig. S8). To assess whether SiR-Halo labeled a representative sample of the diverse population of PSD95-positive synapses, total fluorescence intensity distributions for SiR-Halo and eGFP from injected Psd95eGFP/HaloTag mice were compared. A scaling factor of 0.65 was applied to eGFP puncta distribution to overlay the outlines of the two frequency distributions. The outlines closely overlapped indicating that eGFP and SiR-Halo labeled comparable populations of synaptic puncta, including comparable proportions of puncta of different intensities. These results demonstrate that injection of a fluorescent ligand into PSD95-HaloTag mice efficiently and quantitatively labels PSD95 across all regions of the brain.
SiR-Halo-positive puncta 3D reconstruction
The z-stacks of SiR-Halo and dye-filled neuron images were analyzed with 3D image processing software Imaris 7.6.5 (Bitplane). SiR-Halo-positive puncta were manually reconstructed along apical dendrites (stratum radiatum). For detailed information regarding 3D reconstruction of SiR-Halo-positive puncta, see Figure S23.
Analysis of long-term imaging
The images from λ=840 nm and λ=910 nm acquisitions were overlaid using the Fiji pairwise stitching algorithm (BigStitcher) (Hörl et al., 2019). The images at different time points were aligned using the ‘Correct_3D_Drift’ plugin in Fiji (Parslow et al., 2014). To correct for slight variations in excitation efficiency and image quality at different time points, the images were normalized using the YFP fluorescence signal from the dendrites, under the assumption that YFP levels did not substantially change over time, and background subtracted using the blood vessel interior as the baseline signal. For single SiR-Halo punctum decay analysis, the total fluorescence of each punctum at every time point was measured from the plane that yielded the brightest signal. For spine SiR-Halo puncta, the spines were selected in the YFP channel. YFP and SiR-Halo fluorescence intensity (from 910 nm and 840 nm excitation, respectively) was integrated in the spine head. To correct for bleaching due to repeated imaging of the puncta, on the first day of imaging a series of seven images were taken of a control FOV in rapid succession (within 5 minutes). For each point in the series the level of bleaching was estimated (∼3%) and applied as a correction factor to the corresponding time point in the long-term imaging series.
Colocalization of synaptic puncta
Object-based colocalization method was employed to assess double (between SiR-Halo and eGFP) and triple (between SiR-Halo, eGFP and mKO2) colocalization of markers of synaptic puncta. The double colocalization between two channels (SiR-Halo and eGFP) was obtained by minimizing the summation of the total centroid distance between all eGFP and SiR-Halo using linear programming (Zhu et al., 2018). The triple localization is computationally much more complex and an NP-hard problem, thus it cannot be resolved efficiently using linear programming. New multiple-channel localization algorithms were developed by adapting the multiple hypothesis tracking algorithm (Chenouard et al., 2013) to the particle tracking problem: the image channel of different markers were considered as temporal image frames in tracking, but more image matching between different markers was added because there was no temporal sequence in multilocalization. A distance threshold of 400 nm was applied to identify markers that were colocalized or triple localized. 400 nm is within the range of the previously measured diameters of postsynaptic density as observed by electron microscopy (Harris et al., 1992).
Classification of synaptic puncta
Using classifiers trained and built in a previous study (Cizeron et al., 2020), Psd95eGFP/+;Sap102mKO2/y synapses were classified into 37 subtypes based on the parameters measured in the previous steps. A small percentage of puncta (∼0.002%) that did not get classified into any subtype group were denoted ‘other subtype’. The classification results were then combined with the triple localization result of PSD95-HaloTag to quantify the lifetime of different synapse subtypes over all brain regions/subregions at time points post SiR-Halo injection.
Segmentation of brain regions and subregions
An overview montage of each imaged sagittal brain section was stitched from ∼45,000–50,000 image tiles using a custom MATLAB script and downsized by a factor of 16. Quantification of PSD95-Halo labeling was performed on anatomical brain regions as defined by the Allen Mouse Brain Atlas. A combination of manual and semi-automated delineation methods was employed for defining 110 anatomical regions in each sagittal brain image. Manual delineations of subcortical brain regions (subregions of the olfactory areas, cortical subplate, striatum, pallidum, hypothalamus, thalamus, midbrain, cerebellum, pons, medulla) were performed using a polygon selection tool in Fiji/ImageJ (Schindelin et al., 2012; Schneider et al., 2012). Semi-automated delineations of isocortex and hippocampal formation were performed with in-house custom-built software. The program provides a user interface for a freeform deformation of a surface on which we map a specially prepared image that contains a multitude of delineated regions (Sederberg and Parry, 1986). The specially prepared images were, in most cases, delineation templates obtained from the Allen Mouse Brain Atlas. The delineation templates mapped on a digital surface are subdivided into a uniform grid so that each cell contains a subdivision of the delineation template. The software allows the user to manipulate the grid points, which results in the deformation of the grid cells and in turn leads to the deformation of the delineation template (Catmull, 1974). The software superimposes the delineation template on top of the brain image being delineated. The user can deform the delineation template to ‘fit’ the underlying montage image of the brain section. The software has a function allowing the user to paint over any damaged areas on the tissue to exclude them from the finalized delineation. The final delineation is saved as a set of .roi files representing individual brain regions, which are used in the later steps of the mapping pipeline.
Decay rate estimation
PSD95 decay rate was estimated by two alternative methods: (a) puncta count-based estimation and (b) total punctum intensity-based estimation. Method (a) relies purely on the presence or absence of fluorescent synaptic puncta, whereas method (b) considers the fluorescence content within each punctum. For both methods, the mean at day 0 was considered a reference point to which all the subsequent values were normalized. Single-phase exponential decay function was fitted to fractions of puncta/synapse fluorescence remaining:
where N(t) denotes the puncta density/fluorescence intensity at time t, N0 = N(0) is quantity at t = 0 and λ is the decay rate constant.
Puncta count-based estimation
The detected number of fluorescent puncta per 100 μm2 (puncta density) was estimated for each ROI from every brain examined using the SYNMAP pipeline. Normalized puncta densities (y-axis) for each animal were plotted against time (x-axis) and an exponential decay function was fitted to estimate half-life and ± 95% confidence interval.
Total puncta intensity-based estimation
For each detected punctum within an ROI, mean fluorescence intensity was multiplied by punctum size to obtain the total fluorescence content for each punctum. The resulting fluorescence intensities were summed to obtain the total synaptic fluorescence content within a given brain region. Normalized fluorescence intensities (y-axis) for each animal were plotted against time (x-axis) and an exponential decay function was fitted to estimate half-life and ± 95% confidence interval.
PSD95 half-life calculations
PSD95 half-life was defined as the time required for the PSD95-Halo puncta density/fluorescence intensity to fall to half its initial value. Half-life was calculated based on the decay constant obtained from exponential curve fitting via the following formula:
PSD95 fraction remaining calculation
In order to compare the decay of puncta number/fluorescence intensity between two time points, for each ROI we estimated the fraction of puncta/fluorescence intensity remaining at day 7 compared with day 0. The mean values for fraction remaining were calculated using:
where A represents the mean of day 7 measurements and B is the mean of day 0 measurements. The error in the measurement of fraction remaining was estimated by following the statistical rules for propagation of uncertainty. The formula used for estimating the standard deviation of a ratio of means was:
where σf is a standard deviation of the function f (ratio of mean (day 7)/mean (day 0)), σA is the standard deviation of A and σB is the standard deviation of B.
Statistical analysis
Covariance of pairs of synaptic parameters (density, half-life) was assessed using Pearson’s correlation function in MATLAB (Figs. S8 and S13). Correlation coefficient (R) values are presented alongside P-values that determine the statistical significance (P < 0.05) of the correlation between the two variables.
Bayesian analysis
Changes in subtype densities between 3M and18M were established in a previous study (Fig. S13 in Cizeron et al, 2020) and the heatmaps (Figs. 5E and S32) were then permutated column-wisely based on the ranking order of the subtype lifetime in Fig. 4A. The changes in subtype densities (Figs. 5E and S32) were tested using Bayesian estimation (Kruschke, 2013). Bayesian testing was also used to compare PSD95 lifetime between the Dlg2 mouse mutant and control (Fig. S33). The input data were modelled assuming a t-distribution, and a Markov chain Monte Carlo algorithm was then performed to estimate the posterior distribution of the changes. P-values were then calculated for the distribution to complete the test. Each data point in each subregion was modelled and tested separately. The results were finally corrected over all subregions using the Benjamini-Hochberg procedure.
Cohen’s d effect size
The difference between two groups was measured by calculating a Cohen’s d effect size (Figs. 5E, S32 and S33), which is based on the difference between two means divided by their pooled standard deviation:
The pooled standard deviation was calculated as follows:
Self-organizing feature maps
Self-organizing feature maps (SOMs) (Kohonen, 1982) were constructed based on the subtype density of 110 subregions. SOMs preserve the local neighborhood so that subregions which are similar (neighbors) in the 37-dimensional subtype space become neighbors in the 2-dimensional output space. Network nodes were connected on a 2-dimensional rectangular grid of size 35 × 35 nodes. Input consisted of the 37 subtype densities of a subregion, where each subtype was normalized to [0, 1]. Number of training iterations was set to 45 and starting neighborhood size was set to 15 and reduced by a factor 0.9 for each iteration and weights were initialized to [0, 1]. After training, the strongest responding node was identified for each subtype and the position of this node was used for positioning the name of the subregion (Fig. S28A) (half-height of left-hand side of first letter of subregion name) as well as the position of the disc (disc center) representing the subregion (Fig. 4D, S28B). Radius of discs was proportional to the intensity half-life (PSD95 intensityt1/2) of the subregion represented by the disc. Colors of subregions used for presentations were the same as in Figure 2A.
Similarity matrices
Each row/column in the matrix represents one delineated brain subregion (Fig. 5D). Elements in the matrix are the synaptome similarities between two subregions quantified by differences in the PSD95 lifetime. A Gaussian kernel function was applied to convert the differences into similarities (Zhu et al., 2018).
Protein Lifetime Synaptome Atlas website
The Protein Lifetime Synaptome Atlas website (Bulovaite et al., 2021a) provides users with detailed information on PSD95 lifetime in regions and subregions of the mouse brain across the lifespan. The main panel shows the heatmap of protein half-life for the selected punctum parameter (density, intensity) and age or age comparison. The brain region navigator provides a collapsible tree structure that allows the user to pinpoint any subregion, its half-life values, and linked punctum parameter and synapse type/subtype prevalence data from the Lifespan Synaptome Atlas (Cizeron et al., 2020). Half-life data for further synaptic proteins will be added as they become available.
Additional resources
All brain maps generated for the study are freely available at (Bulovaite et al., 2021).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HaloTag | Promega | Cat# 9211 RRID: AB_2688011 |
| Mouse anti-PSD95 (clone 16/PSD-95) | BD Biosciences | Cat# 610496 RRID: AB_397862 |
| Bacterial and virus strains | ||
| AAV9.CamKII.Cre.SV40 | Addgene | Cat# 105558 |
| AAV2.Ef1α.DIO.eYFP | UNC GTC vector | Cat# 42585 |
| Chemicals, peptides, and recombinant proteins | ||
| 4’,6 diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | Cat# D9542 |
| Alexa Fluor 488 dye solution | Life Technologies | Cat# A10436 |
| Pluronic F-127 (20% in DMSO) | Thermo Fisher | Cat# P3000MP |
| ProLong Gold Antifade mounting medium | Life Technologies | Cat# P36930 |
| OCT embedding matrix | CellPath | Cat# KMA-0100-00A |
| TMR-Halo | Promega | Cat# G8251 |
| SiR-COOH | Spirochrome | Cat# SC004 |
| Amine (O2) HaloTag | Promega | Cat# P6711 |
| Deposited data | ||
| Allen mouse brain reference atlas | Allen Brain Institute | https://mouse.brain-map.org/static/atlas |
| Synaptome data, including data for time points, individual animals and half-life values | This paper | https://doi.org/10.7488/ds/3070 |
| Experimental models: Cell lines | ||
| Embryonic stem cell line used for gene targeting: E14Tg2a | 08021401 | |
| Experimental models: Organisms/strains | ||
| Mouse: PSD95HaloTag/HaloTag | This paper | N/A |
| Mouse: PSD95eGFP/eGFP | Zhu et al, 2018 | N/A |
| Mouse: SAP102mKO2/mKO2 | Zhu et al, 2018 | N/A |
| Mouse: Dlg2−/− | McGee et al, 2001 | N/A |
| Oligonucleotides | ||
| Mouse genotyping primers PSD95-HaloTag: exon F, GTCACATGTCTTTGTGACCTTG | This paper | N/A |
| Mouse genotyping primers PSD95-HaloTag: 95UTR F, GATACATGCAGAGAGGAGTGTC | This paper | N/A |
| Mouse genotyping primers PSD95-HaloTag: Halogen F, CTGACTGAAGTCGAGATGGAC | This paper | N/A |
| Recombinant DNA | ||
| HaloTag coding sequence | Promega | https://www.promega.co.uk/resources/technologies/halotag/ |
| Software and algorithms | ||
| MATLAB | MathWorks | https://uk.mathworks.com/products/matlab.html |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Python | Python Software Foundation | https://www.python.org/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Imaris 7.6.5 | Bitplane | https://imaris.oxinst.com/ |
| ScanImage | Vidrio Technologies | http://scanimage.vidriotechnologies.com/display/SIH/ScanImage+Home |
| Correct_3D_Drift (Fiji plugin) | Parslow et al, 2014 | https://imagej.net/plugins/correct-3d-drift |
| pCLAMP 10.7 | Molecular Devices | https://support.moleculardevices.com/s/article/Axon-pCLAMP-10-Electrophysiology-Data-Acquisition-Analysis-Software-Download-Page |
| Code for fitting of exponential decay function and Pearson’s correlation test | This paper | https://github.com/EditaBu/Synapse_Protein_Lifetime_code_repository.git |
| Code for Bayesian test | This paper | https://github.com/NilsWinter/matlab-bayesian-estimation |
| Other | ||
| PSD95 protein lifetime maps: resource website | This paper | http://brain-synaptome.org/Protein_Lifetime/ |
Highlights.
Brain excitatory synapses differ widely in postsynaptic protein lifetime
Synapses with diverse protein lifetimes are spatiotemporally distributed
Age-resilient synapses have long protein lifetimes
Synapse protein lifetime increases in a model of autism and schizophrenia
Acknowledgments:
General:
Sarah Lempriere, Cathy McLaughlin, Emma Sigfridsson, Rand Dahan, Gabor Varga, Emily Robson, Theresa Wong, Bev Notman, Ian Hawes, Dimitra Koukaroudi, William Mungall, and Fabio De Moliner for technical assistance. Colin Davey for editing. Debbie Maizels for artwork. Trevor Robbins, Szu-Han Wang, Tim Bussey for comments on the manuscript.
Funding:
SGNG: The European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (695568 SYNNOVATE), Simons Foundation Autism Research Initiative (529085), and the Wellcome Trust (Technology Development Grant 202932). JD: Interdisciplinary Platform Cajal Blue Brain (CSIC, Spain). PMS: Spanish Ministerio de Ciencia e Innovación (IJCI-2016-27658). TJO: National Institute of Mental Health Grant (R01MH060919-15). AH: Swiss National Science Foundation (Grant 31003A_173125), the Swiss National Centre Competence in Research (NCCR) Synapsy (Grant 51NF40-185897), and a gift from a private foundation with public interest through the International Foundation for Research in Paraplegia (chair Alain Rossier). For the purpose of open access the author has applied a CC-BY public copyright licence to any author accepted manuscript version arising from this submission.
Footnotes
Competing interests:
Authors declare they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and materials availability:
Data are available at the Protein Lifetime Synaptome Atlas website (Bulovaite et al.,2021a) and Edinburgh DataShare (Bulovaite et al., 2021b).
References and notes:
- Akers KG, Arruda-Carvalho M, Josselyn SA, and Frankland PW. (2012). Ontogeny of contextual fear memory formation, specificity, and persistenece in mice. Learning and Memory. 19, 598–604. 10.1101/lm.027581.112 [DOI] [PubMed] [Google Scholar]
- Babiec WE, Jami SA, Guglietta R, Chen PB, and O’Dell TJ. (2017). Differential regulation of NMDA receptor-mediated transmission by SK channels underlies dorsal-ventral differences in dynamics of schaffer collateral synaptic function. J. Neurosci. 37, 1950–1964. 10.1523/JNEUROSCI.3196-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähner F, and Meyer-Lindenberg A. (2017). Hippocampal–prefrontal connectivity as a translational phenotype for schizophrenia. Eur. Neuropsychopharmacol. 27, 93–106. 10.1016/j.euroneuro.2016.12.007 [DOI] [PubMed] [Google Scholar]
- Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, and Grant SG. (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 14, 19–21. 10.1038/nn.2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides-Piccione R., Fernaud-Espinosa I., Robles V., Yuste R., and DeFelipe J. (2013). Age-based comparison of human dendritic spine structure using complete three-dimensional reconstructions. Cereb. Cortex 23, 1798–1810. 10.1093/cercor/bhs154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biever A, Glock C, Tushev G, Ciirdaeva E, Dalmay T, Langer JD, and Schuman EM. (2020). Monosomes actively translate synaptic mRNAs in neuronal processes. Science 367, eaay4991. 10.1126/science.aay4991 [DOI] [PubMed] [Google Scholar]
- Bingol B, and Schuman EM. (2006). Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441, 1144–1148. 10.1038/nature04769 [DOI] [PubMed] [Google Scholar]
- Broadhead MJ, Horrocks MH, Zhu F, Muresan L, Benavides-Piccione R, DeFelipe J, Fricker D, Kopanitsa MV, Duncan RR, and Klenerman D, et al. (2016). PSD95 nanoclusters are postsynaptic building blocks in hippocampus circuits. Sci. Rep. 6, 24626. 10.1038/srep24626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulovaite E, Qiu Z, Gokhale R, Fransen E, and Grant SG. (2021a). Protein Lifetime Synaptome Atlas. http://brain-synaptome.org/protein_lifetime
- Bulovaite E, Qiu Z, and Grant SG. (2021b). Edinburgh Datashare. 10.7488/ds/3070 [DOI]
- Burt WH. (1940). Territorial behavior and populations of some small mammals in southern Michigan. University of Michigan Press. [Google Scholar]
- Cane M, Maco B, Knott G, and Holtmaat A. (2014). The relationship between PSD-95 clustering and spine stability in vivo. J. Neurosci. 34, 2075–2086. 10.1523/JNEUROSCI.3353-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, and O’Dell TJ. (2008). Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J. Physiol. 586, 58855900. 10.1113/jphysiol.2008.163469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carper RA, and Courchesne E. (2005). Localized enlargement of the frontal cortex in early autism. Biol. Psychiatry 57, 126–133. 10.1016/j.biopsych.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Carrard G, Dieu M, Raes M, Toussaint O, and Friguet B. (2003). Impact of ageing on proteasome structure and function in human lymphocytes. Int. J. Biochem. Cell Biol. 35, 728–739. 10.1016/S1357-2725(02)00356-4 [DOI] [PubMed] [Google Scholar]
- Catmull EE. (1974). A subdivision algorithm for computer display of curved surfaces. PhD thesis, University of Utah. [Google Scholar]
- Chenouard N, Bloch I, and Olivo-Marin J. (2013). Multiple hypothesis tracking for cluttered biological image sequences. IEEE Trans. Pattern Anal. Mach. Intell. 35, 2736–3750. 10.1109/TPAMI.2013.97 [DOI] [PubMed] [Google Scholar]
- Cizeron M, Qiu Z, Koniaris B, Gokhale R, Komiyama NH, Fransen E, and Grant SGN. (2020). A brainwide atlas of synapses across the mouse life span. Science 369, 270–275. 10.1126/science.aba3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LD., Zuchman R., Sorokina O., Muller A., Dieterich DC., Armstrong JD., Ziv T., and Ziv NE. (2013). Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One 8, e63191. 10.1371/journal.pone.0063191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, and Scott JD. (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40, 595–607. 10.1016/S0896-6273(03)00687-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. (1984). Memory and molecular turnover. Nature 312, 101. 10.1038/312101a0 [DOI] [PubMed] [Google Scholar]
- Dasuri K, Zhang L, Ebenezer P, Liu Y, Fernandez-Kim SO, and Keller JN. (2009). Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mech. Ageing. Dev. 130, 777–783. 10.1016/j.mad.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörrbaum AR, Kochen L, Langer JD, and Schuman EM. (2018). Local and global influences on protein turnover in neurons and glia. Elife 7, e34202. 10.7554/eLife.34202.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll LN, Pettit NL, Minderer M, Chettih SN, and Harvey CD. (2017). Dynamic reorganization of neuronal activity patterns in parietal cortex. Cell 170, 986–999.e916. 10.1016/j.cell.2017.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242. 10.1038/nn1013 [DOI] [PubMed] [Google Scholar]
- El-Husseini AE-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, GauthierCampbell C, Aguilera-Moreno A, Nicoll RA, and Bredt DS. (2002). Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863. 10.1016/S00928674(02)00683-9 [DOI] [PubMed] [Google Scholar]
- Encell LP, Friedman Ohana R, Zimmerman K, Otto P, Vidugiris G, Wood MG, Los GV, McDougall MG, Zimprich C, Karassina N, et al. (2012). Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr. Chem. Genomics 6, 55–71. 10.2174/1875397301206010055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- England CG, Luo H, and Cai W. (2015). HaloTag technology: a versatile platform for biomedical applications. Bioconjug. Chem. 26, 975–986. 10.1021/acs.bioconjchem.5b00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, Collins MO, Frank RAW, Zhu F, Kopanitsa MV, Nithianantharajah J, Lempriere SA, Fricker D, Elsegood KA, McLaughlin CL, et al. (2017). Arc requires PSD95 for assembly into postsynaptic complexes involved with neural dysfunction and intelligence. Cell Rep. 21, 679–691. 10.1016/j.celrep.2017.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E., Collins MO., Uren RT., Kopanitsa MV., Komiyama NH., Croning MD., Zografos L., Armstrong JD., Choudhary JS., and Grant SG. (2009). Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 5, 269. 10.1038/msb.2009.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ, Pinard CR, Camp MC, Feyder M, Sah A, Bergstrom HC, Graybeal C, Liu Y, Schluter OM, Grant SG, et al. (2015). Durable fear memories require PSD-95. Mol. Psychiatry 20, 901–912. 10.1038/mp.2014.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornasiero EF, Mandad S, Wildhagen H, Alevra M, Rammner B, Keihani S, Opazo F, Urban I, Ischebeck T, Sakib MS, et al. (2018). Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 9, 4230. 10.1038/s41467-018-06519-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Tillo SE, Yang G, Rah J-C, Melander JB, Bai S, Soler-Cedeño O, Qin M, Zemelman BV, and Guo C. (2014). Live imaging of endogenous PSD-95 using ENABLED: a conditional strategy to fluorescently label endogenous proteins. J. Neurosci. 34, 16698–16712. 10.1523/JNEUROSCI.3888-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RA, Komiyama NH, Ryan TJ, Zhu F, O’Dell TJ, and Grant SG. (2016). NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun. 7, 11264. 10.1038/ncomms11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RAW, Zhu F, Komiyama NH, and Grant SG. (2017). Hierarchical organization and genetically separable subfamilies of PSD95 postsynaptic supercomplexes. J. Neurochem. 142, 504–511. 10.1111/jnc.14056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, and Svoboda K. (2006). Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 4, e370. 10.1371/journal.pbio.0040370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. 10.1038/nmeth.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, and Gan WB. (2002). Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Hamilton WJ Jr (1937). Growth and life span of the field mouse. The American Naturalist 71, 500–507. 10.1086/280735 [DOI] [Google Scholar]
- Harris KM, and Weinberg RJ. (2012). Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 4, a005587. 10.1101/cshperspect.a005587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, and Tsao B. (1992). Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci. 12, 2685–2705. 10.1523/JNEUROSCI.12-07-02685.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T., and Goto S. (1998). Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech. Ageing. Dev. 102, 55–66. 10.1016/S00476374(98)00011-6 [DOI] [PubMed] [Google Scholar]
- Heinz A, Murray GK, Schlagenhauf F, Sterzer P, Grace AA, and Waltz JA. (2019). Towards a unifying cognitive, neurophysiological, and computational neuroscience account of schizophrenia. Schizophr. Bull. 45, 1092–1100. 10.1093/schbul/sby154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Otsuka T, Lee M-C, Mu Y, and Ehlers MD. (2008). Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc. Natl. Acad. Sci. USA 105, 19492–19497. 10.1073/pnas.0802280105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo S, Diering GH, Na CH, Nirujogi RS, Bachman JL, Pandey A, and Huganir RL. (2018). Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 115, E3827–E3836. 10.1073/pnas.1720956115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, and Hartl FU. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. 10.1038/s41580-019-0101-y [DOI] [PubMed] [Google Scholar]
- Hoelzel CA, and Zhang X. (2020). Visualizing and manipulating biological processes using HaloTag and SNAP‐Tag technologies. ChemBioChem 21, 1935–1946. 10.1002/cbic.202000037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee W-CA, et al. (2009). Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 4, 1128–1144. 10.1038/nprot.2009.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, and Svoboda K. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. 10.1016/j.neuron.2005.01.003 [DOI] [PubMed] [Google Scholar]
- Hörl D, Rojas Rusak F, Preusser F, Tillberg P, Randel N, Chhetri RK, Cardona A, Keller PJ, Harz H, Leonhardt H, et al. (2019). BigStitcher: reconstructing high-resolution image datasets of cleared and expanded samples. Nat. Methods 16, 870–874. 10.1038/s41592-019-0501-0 [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, and Grant SG. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669. 10.1038/76615 [DOI] [PubMed] [Google Scholar]
- Ingason A., Giegling I., Hartmann AM., Genius J., Konte B., Friedl M., Schizophrenia Working Group of the Psychiatric Genomics, Ripke C, Sullivan S, F. P., St Clair D, et al. (2015). Expression analysis in a rat psychosis model identifies novel candidate genes validated in a large case-control sample of schizophrenia. Transl. Psychiatry 5, e656. 10.1038/tp.2015.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Rouach N, Nicoll RA, and Bredt DS. (2005). Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc. Natl. Acad. Sci. USA 102, 5600–5605. 10.1073/pnas.0501769102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, and Cuervo AM. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. 10.1038/nm.4001 [DOI] [PubMed] [Google Scholar]
- Kelmer Sacramento E, Kirkpatrick JM, Mazzetto M, Baumgart M, Bartolome A, Di Sanzo S, Caterino C, Sanguanini M, Papaevgeniou N, and Lefaki M, et al. (2020). Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol. Syst. Biol. 16, e9596. 10.15252/msb.20209596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153. 10.1038/mp.2011.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T. (1982). Self-organized formation of topologically correct feature maps. Biol. Cybern. 43, 59–69. 10.1007/BF00337288 [DOI] [Google Scholar]
- Koyuncu S, Loureiro R, Lee HJ, Wagle P, Krueger M, and Vilchez D. (2021). Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature 596, 285–290. 10.1038/s41586-021-03781-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratschke M. (2018). Investigating PSD-95 turnover at the synapse using the HaloTag technology. [Google Scholar]
- Kruschke JK. (2013). Bayesian estimation supersedes the t test. J. Exp. Psychol. Gen. 142, 573–603. 10.1037/a0029146 [DOI] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. 10.1146/annurev-biochem-060614-033955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S, and Pak DT. (2013). Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron 77, 99–114. 10.1016/j.neuron.2012.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Lindenberger U, Hommel B, Aschersleben G, Prinz W, and Baltes PB. (2004). Transformations in the couplings among intellectual abilities and constituent cognitive processes across the life span. Psychol. Sci. 15, 155–163. 10.1111/j.09567976.2004.01503003.x [DOI] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Louros SR, and Osterweil EK. (2016). Perturbed proteostasis in autism spectrum disorders. J. Neurochem. 139, 1081–1092. 10.1111/jnc.13723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G., Umezawa K., Olivier N., Honigmann A., Yang G., Plass T., Mueller V., Reymond L., Correa IR Jr., Luo ZG., et al. (2013). A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 5, 132–139. 10.1038/nchem.1546 [DOI] [PubMed] [Google Scholar]
- Marks WD, Yamamoto N, and Kitamura T. (2020). Complementary roles of differential medial entorhinal cortex inputs to the hippocampus for the formation and integration of temporal and contextual memory (Systems Neuroscience). Eur. J. Neurosci. 54, 6762–6779. 10.1111/ejn.14737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Perez M, and Villen J. (2017). Determinants and Regulation of Protein Turnover in Yeast. Cell Syst. 5, 283–294. 10.1016/j.cels.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masch JM, Steffens H, Fischer J, Engelhardt J, Hubrich J, Keller-Findeisen J, D’Este E, Urban NT, Grant SGN, Sahl SJ, et al. (2018). Robust nanoscopy of a synaptic protein in living mice by organic-fluorophore labeling. Proc. Natl. Acad. Sci. USA 115, E8047–E8056. 10.1073/pnas.1807104115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, and Becher I. (2018). Systematic analysis of protein turnover in primary cells. Nat. Commun. 9, 1–10. 10.1038/s41467-018-03106-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, and Prehoda KE. (2001). Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol. Cell 8, 1291–1301. 10.1016/S10972765(01)00411-7 [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. (1998). Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439. 10.1038/24790 [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Komiyama NH, McKechanie A, Johnstone M, Blackwood DH, St Clair D, Emes RD, van de Lagemaat LN, Saksida LM, Bussey TJ, et al. (2013). Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nat. Neurosci. 16, 16–24. 10.1038/nn.3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, and Harris KM. (2002). Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35, 535–545. 10.1016/S0896-6273(02)00785-7 [DOI] [PubMed] [Google Scholar]
- Parslow A, Cardona A, and Bryson-Richardson RJ. (2014). Sample drift correction following 4D confocal time-lapse imaging. J. Vis. Exp. 86, 51086. 10.3791/51086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, and Schuman EM. (2003). Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr. Biol. 13, 2073–2081. 10.1016/j.cub.2003.10.028 [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, and Svoboda K. (2003). ScanImage: Flexible software for operating laser scanning microscopes. Biomed. Eng. Online 2, 13. 10.1186/1475-925X-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postema MC., van Rooij D., Anagnostou E., Arango C., Auzias G., Behrmann M., Busatto Filho G., Calderoni S., Calvo R., and Daly E. (2019). Altered structural brain asymmetry in autism spectrum disorder in a study of 54 datasets. Nat. Commun. 10, 4958. 10.1038/s41467-019-13005-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, and Ghaemmaghami S. (2010). Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 107, 14508–14513. 10.1073/pnas.1006551107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. 10.1038/nrn2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiani C, Coppens S, Sekhara T, Dimov I, Pichon B, Lufin N, Addor MC, Belligni EF, Digilio MC, Faletra F, et al. (2017). Novel promoters and coding first exons in DLG2 linked to developmental disorders and intellectual disability. Genome Med. 9, 67. 10.1186/s13073-017-0452-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert G. (1970). Comparative factor analytic studies of intelligence throughout the human life-span. In Life-span Developmental Psychology (Elsevier), pp. 467–484. 10.1016/B978-0-12-293850-4.50023-6 [DOI] [Google Scholar]
- Roy DS, Muralidhar S, Smith LM, and Tonegawa S. (2017). Silent memory engrams as the basis for retrograde amnesia. Proc. Natl. Acad. Sci. USA 114, E9972–E9979. 10.1073/pnas.1714248114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin A, Geva N, Sheintuch L, and Ziv Y. (2015). Hippocampal ensemble dynamics timestamp events in long-term memory. Elife 4, e12247. 10.7554/eLife.12247.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzo EK, Perez-Cano L, Jung JY, Wang LK, Kashef-Haghighi D, Hartl C, Singh C, Xu J, Hoekstra JN, Leventhal O, et al. (2019). Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866.e26. 10.1016/j.cell.2019.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath N, Levy-Adam F, Younis A, Rozales K, Meller A, Hadar S, Soueid-Baumgarten S, and Shalgi R. (2020). Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 117, 31902–31913. 10.1073/pnas.2018138117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez I, and Vilchez D. (2014). The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genomics 15, 38–51. 10.2174/138920291501140306113344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M, Dill KA, and de Graff AM. (2019). Proteostasis collapse is a driver of cell aging and death. Proc. Natl. Acad. Sci. USA 116, 22173–22178. 10.1073/pnas.1906592116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuy A., Tomás-Roca L., Rodríguez J-R., González-Soriano J., Zhu F., Qiu Z., Grant SG., DeFelipe J., and Merchan-Perez A. (2020). Estimation of the number of synapses in the hippocampus and brain-wide by volume electron microscopy and genetic labeling. Sci. Rep. 10, 14014. 10.1038/s41598-020-70859-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonover CE, Ohashi SN, Axel R, and Fink AJ. (2021). Representational drift in primary olfactory cortex. Nature 594, 541–546. 10.1038/s41586-021-03628-7 [DOI] [PubMed] [Google Scholar]
- Sederberg TW, and Parry SR. (1986). Free-form deformation of solid geometric models. SIGGRAPH Comput. Graph. 20, 151–160. 10.1145/15886.15903 [DOI] [Google Scholar]
- Skene NG, Roy M, and Grant SG. (2017). A genomic lifespan program that reorganises the young adult brain is targeted in schizophrenia. Elife 6, e17915. 10.7554/eLife.17915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, and Dede AJ. (2015). Conscious and unconscious memory systems. Cold Spring Harb. Perspect. Biol. 7, a021667. 10.1101/cshperspect.a021667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, and Sabatini BL. (2008). Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 60, 788–802. 10.1016/j.neuron.2008.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, and Levy WB. (1982). Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J. Neurosci. 2, 284–291. 10.1523/JNEUROSCI.02-03-00284.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai H-C, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, and Hyman BT. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181, 1426–1435. 10.1016/j.ajpath.2012.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackaberry EA, Wang X, Schweiger M, Messick K, Valle N, Dean B, Sambrone A, Bowman T, and Xie M. (2014). Solvent-based formulations for intravenous mouse pharmacokinetic studies: tolerability and recommended solvent dose limits. Xenobiotica 44, 235–241. 10.3109/00498254.2013.845706 [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, and Svoboda K. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- Tucker-Drob EM. (2009). Differentiation of cognitive abilities across the life span. Dev. Psychol. 45, 1097–1118. 10.1037/a0015864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Saez I, and Dillin A. (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659. 10.1038/ncomms6659 [DOI] [PubMed] [Google Scholar]
- Villa KL, Berry KP, Subramanian J, Cha JW, Oh WC, Kwon H-B, Kubota Y, So PT, and Nedivi E. (2016). Inhibitory synapses are repeatedly assembled and removed at persistent sites in vivo. Neuron 89, 756–769. 10.1016/j.neuron.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543. 10.1126/science.1155174 [DOI] [PubMed] [Google Scholar]
- Weston CS. (2019). Four social brain regions, their dysfunctions, and sequelae, extensively explain autism spectrum disorder symptomatology. Brain Sci. 9, 130. 10.3390/brainsci9060130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L., Karpenko IA., Hiblot J., and Johnsson K. (2015). Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol. 11, 917–923. 10.1038/nchembio.1959 [DOI] [PubMed] [Google Scholar]
- Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, Cashman NR, Wilson MR, and Ecroyd H. (2016). Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505. 10.1111/jnc.13575 [DOI] [PubMed] [Google Scholar]
- Zeng BY, Medhurst AD, Jackson M, Rose S, and Jenner P. (2005). Proteasomal activity in brain differs between species and brain regions and changes with age. Mech. Ageing Dev. 126, 760–766. 10.1016/j.mad.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Zhu F, Cizeron M, Qiu Z, Benavides-Piccione R, Kopanitsa MV, Skene NG, Koniaris B, DeFelipe J, Fransen E, Komiyama NH, et al. (2018). Architecture of the mouse brain synaptome. Neuron 99, 781–799.e710. 10.1016/j.neuron.2018.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Collins MO, Harmse J, Choudhary JS, Grant SGN, and Komiyama NH. (2020). Cell-type-specific visualisation and biochemical isolation of endogenous synaptic proteins in mice. Eur. J. Neurosci. 51, 793–805. 10.1111/ejn.14597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, and Gan WB. (2005). Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46, 181–189. 10.1016/j.neuron.2005.04.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Synaptome map data have been deposited in Edinburgh DataShare and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Data are available at the Protein Lifetime Synaptome Atlas website (Bulovaite et al.,2021a) and Edinburgh DataShare (Bulovaite et al., 2021b).