

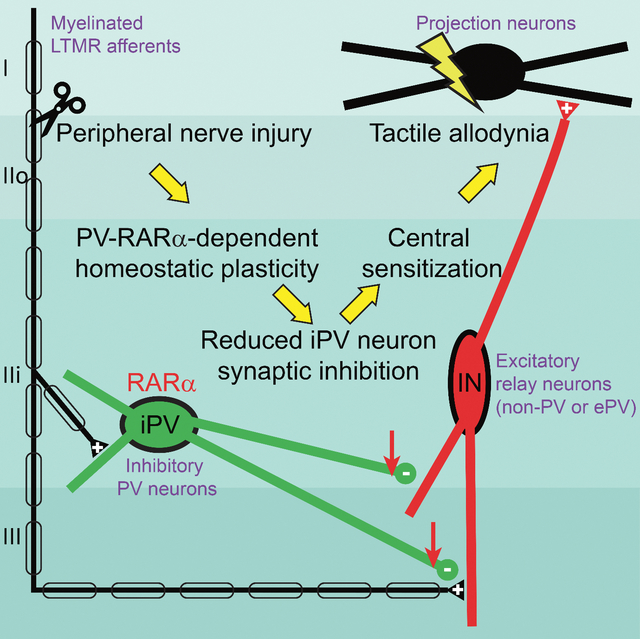
Summary
Central sensitization caused by spinal disinhibition is a key mechanism of mechanical allodynia in neuropathic pain. However, the molecular mechanisms underlying spinal disinhibition after nerve injury remain unclear. Here we show in mice that spared nerve injury (SNI), which induces mechanical hypersensitivity and neuropathic pain, triggers homeostatic reduction of inhibitory outputs from dorsal horn parvalbumin-positive (PV+) interneurons onto both primary afferent terminals and excitatory interneurons. The reduction in inhibitory outputs drives hyperactivation of the spinal cord nociceptive pathway, causing mechanical hypersensitivity. We identified the retinoic acid receptor RARα, a central regulator of homeostatic plasticity, as the key molecular mediator for this synaptic disinhibition. Deletion of RARα in spinal PV+ neurons or application of an RARα antagonist in the spinal cord prevented the development of SNI-induced mechanical hypersensitivity. Our results identify RARα as a crucial molecular effector for neuropathic pain and a potential target for its treatment.
Keywords: homeostatic synaptic plasticity, neuropathic pain, retinoic acid receptor, parvalbumin neurons, synaptic disinhibition
eTOC
Retinoic acid receptor signaling is critically involved in homeostatic synaptic plasticity in many brain circuits. In this study, Cao et al., demonstrated that RARα in spinal cord dorsal horn PV+ neurons is a key mediator for spared-nerve injury-induced homeostatic changes that leads to central sensitization and neuropathic pain.
Graphical Abstract
Introduction
Mechanical allodynia, a pain condition induced by tissue or nerve injury in which an innocuous mechanical stimulus becomes painful, is a prevalent manifestation of chronic pain affecting up to 10% of the population (Bouhassira et al., 2005). Synaptic and non-synaptic changes within the central nervous system that induce and sustain neuropathic pain have been reported in multiple structures involved in pain processing, including in several cortical regions and the dorsal horn of the spinal cord. Among the brain regions involved in chronic pain, synaptic plasticity in the primary somatosensory (S1) and anterior cingulate cortices (ACC) has been extensively studied (Bliss et al., 2016; Kim et al., 2017; Tsuda et al., 2017). Pyramidal neuron hyperactivity in the ACC has been reported in neurogenic pain patients (Santello and Nevian, 2015; Stern et al., 2006) and in rodent models of neuropathic pain (Zhao et al., 2018). Neurophysiology studies further showed that synaptic responses in the ACC are enhanced and excludes further long-term potentiation (LTP) (Li et al., 2010; Wei and Zhuo, 2001). Cortical disinhibition has also been proposed as a potential mechanism underlying neuropathic pain (Blom et al., 2014; Cichon et al., 2017). Although multiple locations in the CNS respond to peripheral nerve injury (Mercer Lindsay et al., 2021; von Hehn et al., 2012), the key circuit responsible for the transformation of normal touch perception into mechanical allodynia is believed to reside within the spinal dorsal horn (Keller et al., 2007; Moehring et al., 2018; Todd, 2017; Torsney and MacDermott, 2006). After peripheral nerve injury, both synaptic and non-synaptic changes in dorsal horn circuitry not only increase the sensitivity of nociceptive projection neurons to noxious stimuli (hyperalgesia), but also change the percept of tactile inputs from touch to pain (mechanical allodynia) (Latremoliere and Woolf, 2009).
Multiple central sensitization mechanisms contributing to hyperalgesia and allodynia have been proposed (Boyle et al., 2019; Finnerup et al., 2021; Gradwell et al., 2020; Ikeda et al., 2003; Polgar et al., 2004; Scholz et al., 2005; Tulleuda et al., 2011; Woolf et al., 1992). One such proposal, the central inhibitory gate theory, places local inhibitory interneurons at key gate-keeping positions of the dorsal horn circuitry (Boyle et al., 2019; Peirs et al., 2020; Petitjean et al., 2015). According to this proposal, reduction in dorsal horn inhibitory tone drives the circuit level changes underlying hyperalgesia and allodynia (Castro-Lopes et al., 1995; Comitato and Bardoni, 2021; Fukuoka et al., 1998; Ibuki et al., 1997; Lever et al., 2003; Lu et al., 2013; Torsney and MacDermott, 2006). Indeed, manipulations that inhibit or enhance GABAergic/glycinergic outputs in central circuits are effective in promoting or alleviating neuropathic pain, respectively (Coull et al., 2003; Eaton et al., 1999; Juarez-Salinas et al., 2019; Knabl et al., 2008; Neumann et al., 2021; Torsney and MacDermott, 2006; Yaksh, 1989; Yamamoto and Yaksh, 1993). Despite the rich literature on circuit changes that underlie the central sensitization leading to allodynic pain, the molecular and cellular mechanisms that produce the change in inhibitory tone after peripheral nerve injury are unclear.
In the present study, we investigated the mechanisms by which dorsal horn synaptic plasticity drives neuropathic pain. Specifically, we tested the hypothesis that peripheral nerve injury induces excessive homeostatic synaptic plasticity at the inhibitory output of the gate-keeping interneurons, resulting in weaker synaptic inhibition, hyperactivation of nociceptive projection neurons, and mechanical hypersensitivity. We demonstrate that injury of the sciatic nerve significantly reduces the synaptic output of inhibitory dorsal horn PV+ interneurons onto both primary afferent terminals and local excitatory interneurons. Moreover, we report that this form of homeostatic plasticity requires the retinoic acid receptor RARα, a key mediator for homeostatic synaptic plasticity at CNS synapses (Aoto et al., 2008; Park et al., 2018; Sarti et al., 2013; Zhong et al., 2018). Genetic deletion of RARα in dorsal horn PV+ neurons or pharmacological inhibition of RARα in the spinal dorsal horn prevented homeostatic downregulation of synaptic inhibition from PV+ neurons, demonstrating the essential role of RARα-dependent homeostatic plasticity in the development of nerve injury-induced mechanical hypersensitivity and chronic pain.
Results
Spared nerve injury (SNI)-induced mechanical hypersensitivity requires RARα in PV+ neurons
We hypothesized that neuropathic pain involving mechanical hypersensitivity may involve homeostatic plasticity, leading us to investigate the functional contribution of RARα as a central regulator of synaptic strength in homeostatic plasticity. We selectively deleted RARα in PV+ neurons by crossing a PV-specific Cre driver line (PvalbCre) with mice bearing a RARα conditional allele (PV-RARα conditional knockout mice). This approach allows deleting RARα from all PV-expressing neurons in the animals. We also included a Cre-dependent YFP reporter allele (Ai3) to enable identification of PV+ cells. Expression fidelity and efficacy of the PvalbCre driver line were verified in three CNS regions (ACC, S1, and spinal cord dorsal horn) known to be involved in pain processing (Figures 1A, 1B). Cre-dependent deletion of RARα was confirmed in spinal cord sections via in situ hybridization (ISH) and single-cell qRT-PCR (Figures 1C–1E).
Figure 1. Deletion of RARα in parvalbumin-expressing (PV+) neurons blocks mechanical hypersensitivity development.
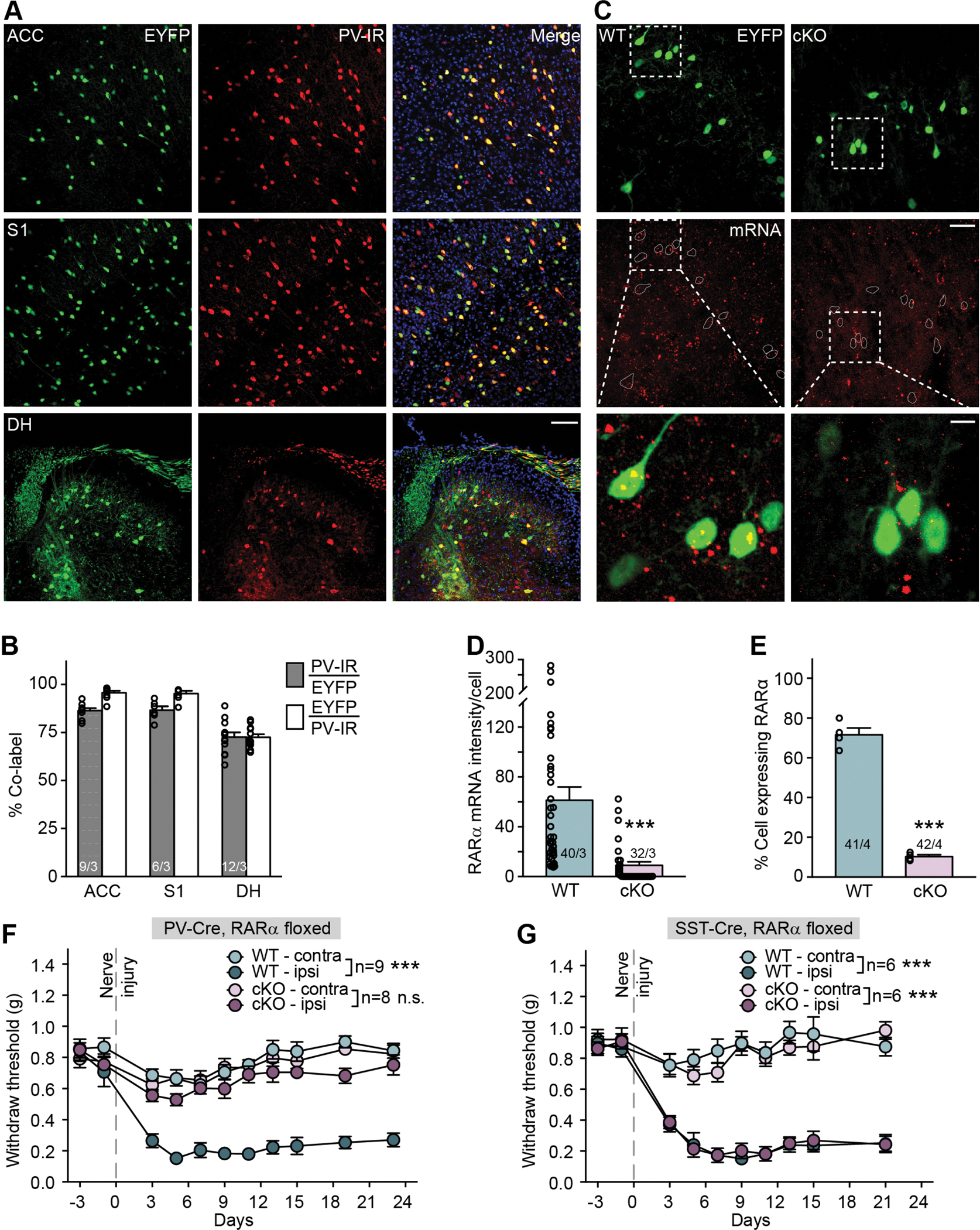
(A) Representative images of anterior cingulate cortex (ACC), primary somatosensory cortex (S1) and spinal cord dorsal horn lumbar region (DH) showing Cre-dependent EYFP reporter (green) and PV-immunoreactivity (PV-IR, red) in PV-Cre::RARα cKO mice. Scale bar: 100 μm. (B) Quantification of PV-Cre driver line expression fidelity (% PV-IR/EYFP) and efficacy (% EYFP/PV-IR) in ACC, S1 and DH. n/N = # of section/# of animals. (C) Representative images of RNA scope in situ hybridization shows RARα mRNA expression (red) in spinal cord sections from WT and PV-RARα cKO mice. PV+ neurons are indicated by EYFP reporter (green). Scale bar: 50 μm. The overlay images of insets (square with white dashed line) are shown at bottom (scale bar: 10 μm). (D) Quantification of RARα mRNA expression with RNAscope in WT (n/N = 40 cells/3 mice) and cKO mice (n/N = 32 cells/3 mice). Mann Whitney test: U = 136, ***, p < 0.001. (E) Quantification of RARα mRNA expression percentage with single-cell qRT-PCR showing percent of RARα+ neurons in PV-expressing spinal dorsal horn neurons (WT: n/N = 41 cells/4 mice; cKO: n/N = 42 cells/4 mice). Two-tailed unpaired t test: t = 17.4, ***, p < 0.001. (F) Mechanical hypersensitivity quantified as paw withdrawal threshold with Von Frey test from hindlimbs ipsilateral and contralateral to the SNI side in WT and PV-RARα cKO mice before and over a period of 3 weeks following SNI surgery. Two-way ANOVA with post-hoc Bonferroni test, WT contra vs. ipsi: interaction (days × SNI), F(10, 160) = 11.21, ***, p < 0.001; SNI factor, F(1, 16) = 121.1, ***, p < 0.001; cKO contra vs. ipsi: interaction, F(10, 140) = 1.22, p = 0.29; SNI factor, F(1, 14) = 2.52, p = 0.13. (G) Mechanical hypersensitivity quantified as paw withdrawal threshold with Von Frey test from hindlimbs ipsilateral and contralateral to the SNI side in WT and SST-RARα cKO mice before and over a period of 3 weeks following SNI surgery. Two-way ANOVA with post-hoc Bonferroni test, WT contra vs. ipsi: interaction (days × SNI), F(9, 90) = 12.62, ***, p < 0.001; SNI factor, F(1, 10) = 125.9, ***, p < 0.001; cKO contra vs. ipsi: interaction, F(9, 90) = 16.83, ***, p < 0.001; SNI factor, F(1, 10) = 99.95, ***, p < 0.001. All graphs represent mean ± SEM.
Given RA and RARα signaling is known to play a central role in homeostatic synaptic plasticity in the cortex (Park et al., 2018; Yee and Chen, 2016; Zhong et al., 2018) and hippocampus (Aoto et al., 2008; Hsu et al., 2019; Sarti et al., 2013), we performed a general behavioral survey. PV-RARα cKO mice exhibited normal working memory, locomotion and anxiety (Figure S1). Based on the essential role of RARα signaling in PV+ neurons in gating inhibitory synaptic outputs during cortical sensory deprivation-induced homeostatic plasticity (Zhong et al., 2018), we asked whether deletion of RARα in PV-expressing neurons may impact the development of neuropathic pain using the SNI model of the sciatic nerve (Decosterd and Woolf, 2000; Shields et al., 2003b) (Figures S2A–S2E). Development of mechanical hypersensitivity, assayed with the von Frey test, was apparent on the ipsilateral hind paw of wildtype (WT) mice after SNI. In PV-RARα cKO mice, development of mechanical hypersensitivity was absent (Figure 1F). We also examined SNI-induced mechanical hypersensitivity in mice with a deletion of RARα specifically in somatostatin (SST) neurons. Like PV+ neurons, SST+ interneurons represent a major class of inhibitory interneurons in brain (Riedemann, 2019). In the spinal cord dorsal horn, however, SST-lineage neurons are excitatory and are thought to be the key component that transmits mechanical pain (Duan et al., 2014). Interestingly, deletion of RARα in SST+ neurons did not prevent the development of mechanical hypersensitivity (Figure 1G). We also examined nocifensive responses to local inflammation induced by subcutaneous injection of complete Freund’s adjuvant (CFA) or 1% formalin, which were indistinguishable between WT and PV-RARα cKO mice (Figures S2F–S2H). Together, these data indicate that RARα expression in PV+ neurons (both in the brain and the spinal cord) is selectively involved in neural circuit modifications contributing to SNI-induced mechanical hypersensitivity, but not in circuit changes mediating inflammatory pain.
SNI-induced nociceptive pathway activation is reduced in PV-RARα cKO mice
We next examined SNI-induced neuronal hyperactivation in regions of the central nociceptive pathways from spinal cord to cortical regions. We used FOS expression as an activity indicator because it is primarily expressed in excitatory neurons in the cortex (Li et al., 2020), and neuropathic pain-induced increase in FOS expression almost exclusively occurs in excitatory cortical neurons (Mecca et al., 2021). In the superficial dorsal horn, about 80–86% FOS+ neurons are excitatory (Todd et al., 1994). In our hands, SNI-induced FOS activation occurs mostly in non-PV neurons in the cortical regions and dorsal horn (Figure S3). Taken together, we believe that an increase in FOS-expressing cells typically indicates an elevated neuronal activation in the networks examined. In WT mice, SNI results in increased neuronal activation in both ipsilateral spinal and contralateral supraspinal regions of the ascending nociceptive pathway (Todd, 2017) (Figures 2A and 2B). Specifically, neurons in the ipsilateral dorsal horn, the contralateral lateral parabrachial nucleus (LPBN), ventral posterolateral nucleus of thalamus (VPL), mesencephalic reticular nucleus (mRt), mediodorsal thalamus (MD), primary somatosensory cortex (S1 hind limb region), secondary somatosensory cortex (S2), anterior cingulate cortex (ACC), and medial prefrontal cortex (mPFC) all exhibited elevated FOS immunoreactivity (Figure 2B) (with the exception of the amygdala (Amy) that showed no changes), indicative of a general hyperactivation of nociceptive spinal and brain circuits. Ipsilateral S1, ACC and mPFC also showed significantly higher FOS expression compared to sham-lesion mice, reflecting the bilateral projections of nociceptive and mechanosensory pathways in cortical regions (Figure 2B). The hyperactivation of nociceptive pathway neurons throughout the CNS was abolished by deletion of RARα in PV+ neurons (Figure 2B). In some of the brain regions examined, the RARα deletion in PV+ neurons reduced their basal activity to below the sham lesion level (dashed lines), indicating that the PV-RARα cKO not only prevented the development of hyperactivity in local circuits, but also dampened the basal network activity within these regions (Figure 2B). We also quantified FOS expression in response to acute inflammation induced by formalin injection into the hind paw. As expected from the results of the von Frey test, a comparable level of FOS activation was observed in the ipsilateral spinal cord dorsal horn and in the contralateral brain regions in WT and PV-RARα cKO mice (Figures 2C and 2D). Thus, RARα expression in PV+ neurons in one or multiple loci within the nociceptive pathway is required for development of chronic neuropathic pain, but not for acute inflammatory pain.
Figure 2. PV-RARα deletion reduces SNI-induced neuronal hyperactivity in the ascending nociceptive pathways.
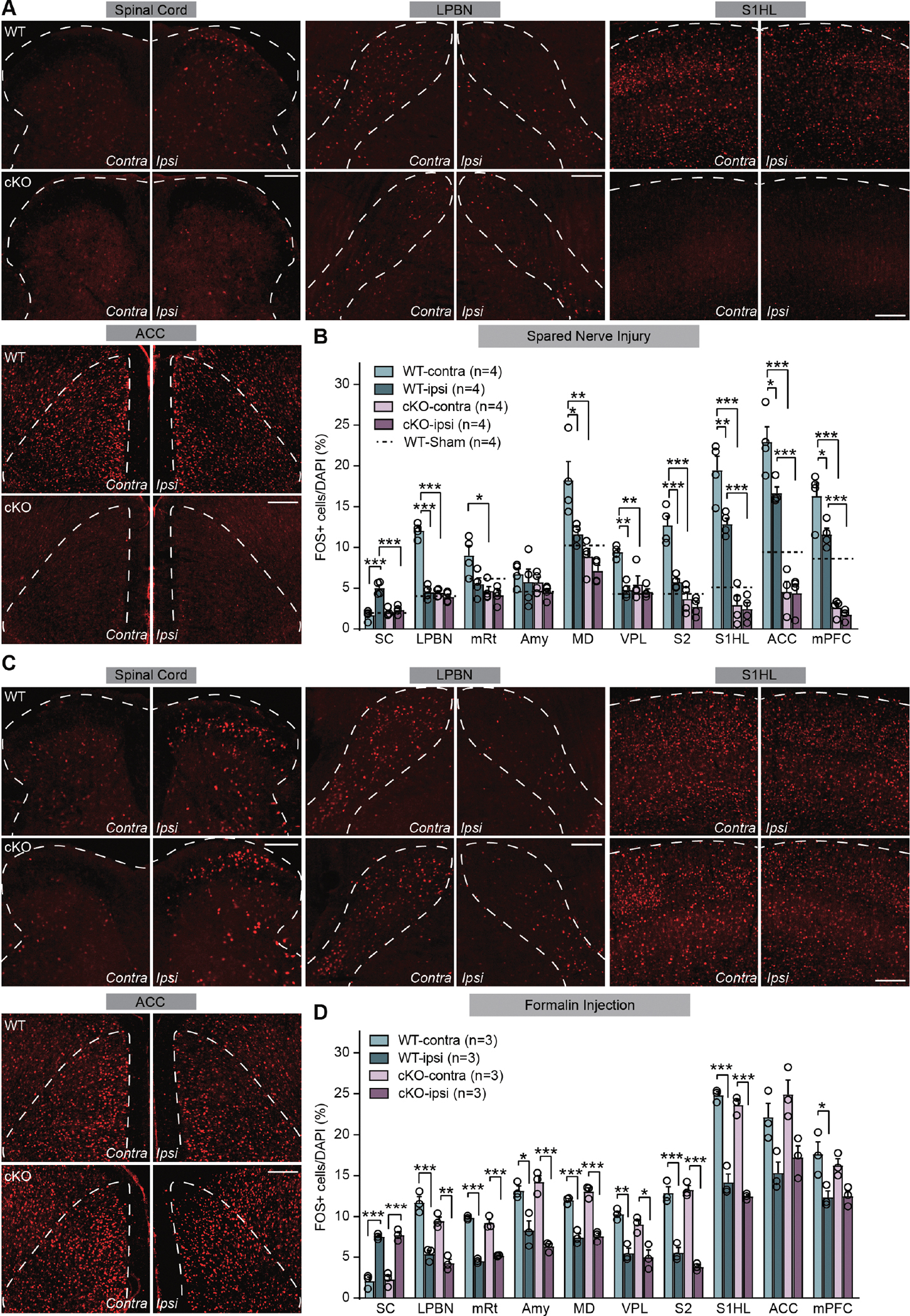
(A) Representative images of FOS immunoreactivity in spinal cord dorsal horn, LPBN (lateral parabrachial nucleus), S1HL (primary somatosensory cortex, hindlimb region) and ACC (anterior cingulate cortex) regions in WT and PV-RARα cKO mice after SNI. Scale bars: 200 μm. (B) Quantification of neuronal activation by SNI in regions of ascending nociceptive pathways. Activation level in sham-lesioned WT mice for each region is indicted by dashed lines. SC: spinal cord; mRt: mesencephalic reticular formation; Amy: Amygdala; MD: mediodorsal thalamus; VPL: central posterolateral thalamic nucleus; S2: secondary somatosensory cortex; mPFC: medial prefrontal cortex. Two-way ANOVA followed by Bonferroni test, interaction (genotype × SNI), SC: F(1, 12) = 24.63, p < 0.001; LPBN: F(1, 12) = 91.46, p < 0.001; mRt: F(1, 12) = 3.51, p = 0.085; Amy: F(1, 12) = 0.024, p = 0.88; MD: F(1, 12) = 3.33, p = 0.093; VPL: F(1, 12) = 8.84, p < 0.05; S2: F(1, 12) = 14.48, p < 0.01; S1HL: F(1, 12) = 6.99, p < 0.05; ACC: F(1, 12) = 5.42, p < 0.05; mPFC: F(1, 12) = 4.11, p = 0.065. SNI factor, SC: F(1, 12) = 35.36, p < 0.001; LPBN: F(1, 12) = 118.8, p < 0.001; mRt: F(1, 12) = 6.28, p < 0.05; Amy: F(1, 12) = 1.36, p = 0.27; MD: F(1, 12) = 10.03, p < 0.01; VPL: F(1, 12) = 20.27, p < 0.001; S2: F(1, 12) = 25.46, p < 0.001; S1HL: F(1, 12) = 9.49, p < 0.01; ACC: F(1, 12) = 6.13, p < 0.05; mPFC: F(1, 12) = 8.77, p < 0.05. Post hoc test, *, p < 0.05; **, p < 0.01; ***, p < 0.001. N = # of mice, (C) Representative images of FOS immunoreactivity in spinal cord dorsal horn, LPBN, S1HL and ACC regions in WT and PV-RARα cKO mice 1.5 h after formalin injection. Scale bars: 200 μm. (D) Quantification of neuronal activation 1.5 hr after formalin injection in regions of ascending nociceptive pathways. Two-way ANOVA followed by Bonferroni test, interaction (genotype × formalin), SC: F(1, 8) = 0.016, p = 0.90; LPBN: F(1, 8) = 1.06, p = 0.33; mRt: F(1, 8) = 6.91, p < 0.05; Amy: F(1, 8) = 3.82, p = 0.086; MD: F(1, 8) = 1.39, p = 0.27; VPL: F(1, 8) = 0.34, p = 0.58; S2: F(1, 8) = 3.46, p = 0.10; S1HL: F(1, 8) = 0.18, p = 0.68; ACC: F(1, 8) = 0.080, p = 0.78; mPFC: F(1, 8) = 0.56, p = 0.48. Formalin factor, SC: F(1, 8) = 154.7, p < 0.001; LPBN: F(1, 8) = 107.0, p < 0.001; mRt: F(1, 8) = 373.2, p < 0.001; Amy: F(1, 8) = 66.97, p < 0.001; MD: F(1, 8) = 161.0, p < 0.001; VPL: F(1, 8) = 49.83, p < 0.001; S2: F(1, 8) = 79.58, p < 0.001; S1HL: F(1, 8) = 283.1, p < 0.001; ACC: F(1, 8) = 20.75, p < 0.01; mPFC: F(1, 8) = 9.01, p < 0.05. Post hoc test, *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data shown as mean ± SEM.
Mechanical allodynia, the hypersensitivity to innocuous tactile stimulation, is a common symptom in neuropathic pain. We thus further quantified SNI-induced changes of spinal neuronal activity in response to light brush stimulation. Compared to sham-lesioned animals, we observed a significantly greater number of FOS+ neurons in the ipsilateral dorsal horn compared to the contralateral side of the WT mice 5–7 days after SNI, indicating heightened sensitivity to innocuous tactile stimulation after injury (Figures 3A and 3B). Importantly, deletion of RARα in PV+ neurons significantly reduced this hypersensitivity (Figure 3B), supporting the conclusion that RARα expression in PV+ neurons is critical for the development of mechanical hypersensitivity. To gain insight into the laminar distribution of light-brush-activated FOS expression in the dorsal horn, we used calcitonin gene-related peptide (CGRP) immunostaining to label lamina I and outer lamina II (IIo) (Duan et al., 2014; Loken et al., 2021; Wang et al., 2018), and locations of PV-EYFP cell bodies to demarcate deeper layers (Hughes et al., 2012). Elevated light brush-induced FOS expression in SNI animals was observed in all laminae (I+IIo, IIi, III-VI) of the WT ipsilateral dorsal horn, and was significantly reduced in PV-RARα cKOs across all laminae of the dorsal horn (Figures 3C and 3D).
Figure 3. PV-RARα deletion reduces light brush stimulation-induced hyperactivation of spinal cord dorsal horn neurons after SNI.
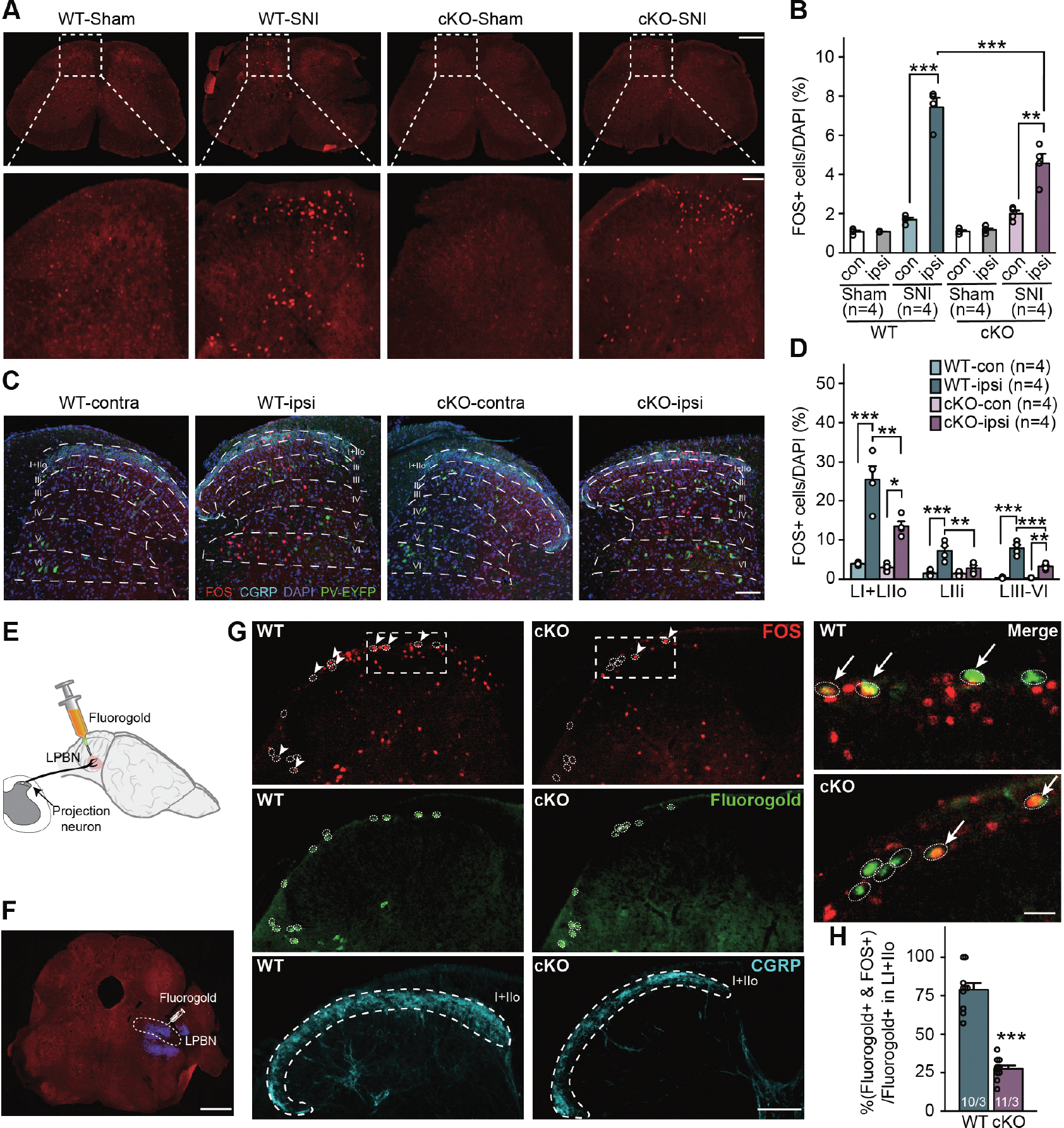
(A) Representative images of spinal cord sections stained with FOS in WT and PV-RARα cKO mice after light brush stimulation to the hind paw receiving SNI. Images of ipsilateral dorsal horn were shown zoomed in at the bottom. Scale bars: 500 μm (top), 100 μm (bottom). (B) Quantification of light brush-activated neurons (FOS+) in WT and PV-RARα cKO mice. Two-way ANOVA followed by Bonferroni test. For SNI animals, WT vs. cKO and ipsi vs. contra, interaction (genotype × SNI), F(1, 12) = 19.73, p < 0.001; SNI factor, F(1, 12) = 136.0, p < 0.001. Post hoc test, **, p < 0.01; ***, p < 0.001. (C) Representative images of dorsal horn sections stained with FOS (red), DAPI (blue), and CGRP (cyan, lamina I+IIo). PV+ cells are indicated by EYFP (Green). Scale bar: 100 μm. (D) Quantification of neuronal activation in different dorsal horn laminae in WT and PV-RARα cKO mice with SNI after light brush stimulation. Two-way ANOVA followed by Bonferroni test, interaction, LI + LIIo: F(1, 12) = 8.29, p < 0.05; LIIi: F(1, 12) = 8.89, p < 0.05; LIII-VI: F(1, 12) = 22.65, p < 0.001. SNI factor, LI + LIIo: F(1, 12) = 70.35, p < 0.001; LIIi: F(1, 12) = 23.42, p < 0.001; LIII-VI: F(1, 12) = 115.8, p < 0.001. Post hoc test, *, p < 0.05; **, p < 0.01; ***, p < 0.001. (E) A schematic drawing depicting retrograde labeling of dorsal horn nociceptive projection neurons via injection of fluorogold into LPBN, where the secondary order neurons receiving projections from dorsal horn are located. (F) A representative image showing the fluorogold injection site (blue) in LPBN. Scale bar: 1 mm. (G) Representative images showing retrogradely Fluorogold-labeled projection neurons (green in white circles) and light brush stimulus-activated FOS+ neurons (red) in the dorsal horn superficial lamina (indicated by CGRP staining in cyan). Higher magnification merged images from LI+IIo of WT and cKO mcie are shown on the right. Scale bars: 100 μm (left) and 20 μm (right). (H) Quantification of Fluorogold+ cells co-labeled with FOS. Statistical analysis was performed using two-sided unpaired t-test. t = 10.81, ***, p < 0.001. n/N = # of sections/# of mice. Data shown as mean ± SEM.
Given the complexity of spinal cord dorsal horn circuits and heterogeneity of neuronal types, we next specifically examined SNI-induced hyperactivation of nociceptive projection neurons, which is one of the key spinal neuron-types responsible for enhanced pain perception. Activity of these neurons is influenced by nociceptive input, but also can be modulated by descending projections from the brain (Mason, 2009). We identified nociceptive projection neurons using retrograde labeling by unilateral fluorogold microinjections into the contralateral lateral parabrachial nucleus (LPBN) (Todd, 2010; Wang et al., 2018) (Figures 3E–H). Labeling efficiency, indicated by the number of fluorogold-labelled neurons in lamina I+IIo, was comparable between WT and PV-RARα cKO mice (WT: 83 vs cKO: 87, n = 3 mice/group). Remarkably, although a majority of these projection neurons was activated by light-brush stimulation in WT mice (78.9% ± 4.3%) as assessed with FOS expression, only a small fraction of projection neurons was activated in PV-RARα cKO mice (27.6% ± 2.1%) (Figure 3H). Thus, SNI caused hyperactivation of nociceptive projecting neurons by a light tactile stimulus, and the RARα deletion in PV+ neurons prevented this form of hyperactivation.
RARα is not required for SNI-induced reduction in PV+ neuron excitability
Our results thus far show that global deletion of RARα in PV+ neurons prevents mechanical hypersensitivity and reduces neuronal activity throughout central nociceptive circuits. As one of the major inhibitory interneuron types in the dorsal horn, PV+ neurons gate signals from low-threshold mechanoreceptive (LTMR) afferents to postsynaptic excitatory interneurons during tactile perception (Petitjean et al., 2015). A reduction in intrinsic membrane excitability has been described in spinal cord dorsal horn PV+ neurons after SNI and is thought to contribute to SNI-induced disinhibition (Boyle et al., 2019). Given that at this point, we could not rule out contributions of RARα expression in PV+ neurons in the brain, we decided to examine PV+ neuron membrane properties both in the dorsal horn and in the cortex.
In the spinal cord slices of mice with SNI, we recorded from neurons in laminae III-V of the central to lateral half of the dorsal horn region. Although we cannot formally rule out the possibility that some of the neurons recorded from this region do not receive direct input from the spared sural nerve, anatomical studies from both rats and mice suggest that the sural nerve terminals innervate the central to lateral portion of the spinal cord dorsal horn in the lumbar 3–5 region (Boyle et al., 2019; Laedermann et al., 2014; Shields et al., 2003a; Swett and Woolf, 1985; Woolf and Fitzgerald, 1986). PV+ neurons were identified by their EYFP reporter expression, and verified via their characteristic tonic fast-spiking firing patterns in response to current injections (Boyle et al., 2019; Hughes et al., 2012; Petitjean et al., 2015) (Figures S4A and S4B). We did not observe any changes in passive membrane properties (resting membrane potential, membrane capacitance and input resistance) resulting from SNI or the RARα deletion (Figures S4E–S4G). In agreement with previous findings (Boyle et al., 2019), SNI reduced the membrane excitability and increased the action-potential rheobase of PV+ neurons on the ipsilateral, but not the contralateral side (Figures S4C and S4D). We observed similar changes in membrane excitability in RARα cKO PV+ neurons (Figures S4C and S4D), suggesting that RARα is not required for SNI-induced changes in spinal cord PV+ neuron membrane properties. Consistent with these findings in dorsal horn PV+ neurons, ACC PV+ neurons (Figures S5A and S5B) also showed reduced excitability and increased AP rheobase on the contralateral side compared to the ipsilateral side (Figures S5C and S5D), without changes in passive membrane properties (Figures S5E–S5G). Again, deletion of PV-RARα did not block these SNI-induced changes in neuronal excitability (Figures S5C–S5G). Taken together, although changes in PV+ neuron membrane excitability may still contribute to reduced inhibitory tone in the spinal cord dorsal horn and ascending nociceptive pathways, our results rule out these changes as the main mechanism underlying RARα-dependent mechanical hypersensitivity development.
RARα is required in PV+ neurons for SNI-induced disinhibition of primarily afferent inputs
Although our results with RARα deletion in all PV+ neurons do not single out spinal cord PV+ neurons as the only locus for SNI-induced RARα-dependent homeostatic changes, we sought to first investigate potential synaptic mechanisms for SNI-induced hypersensitivity in the spinal cord (Boyle et al., 2019; Petitjean et al., 2015). Two distinct forms of inhibition, presynaptic axoaxonic inhibition on primary sensory afferents and postsynaptic inhibition of excitatory interneurons (including excitatory PV+ neurons, ePV), have been described for dorsal horn inhibitory PV+ neurons (iPV) (Boyle et al., 2019; Gradwell et al., 2020). Dorsal horn iPV+ neurons are thought to gate mechanical pain perception through both forms of synaptic inhibition that target multiple non-PV interneurons in lamina IIi, including SST+ cells (Zhang et al., 2018), PKCγ+ cells (Malmberg et al., 1997; Neumann et al., 2008; Petitjean et al., 2015), vGluT3+ cells (Peirs et al., 2021; Peirs et al., 2015). iPV+ neurons also target other PV+ neurons (both iPV and ePVs) (Gradwell et al., 2020). Thus, reduced iPV+ inhibitory synaptic output could plausibly underlie mechanical hypersensitivity.
We first focused on presynaptic inhibition from iPV+ cells onto primary afferent terminals targeting non-PV cells. To selectively activate PV+ synaptic outputs, we expressed channelrhodopsin (ChR2) in PV+ neurons by crossing a PvalbCre driver line with a floxed Cre-on ChR2 line (Ai32). Although optogenetic activation will activate both iPV and ePV cells, ePV cells are not thought to innervate primary afferents (Gradwell et al., 2021). We then assayed presynaptic inhibition by utilizing a paradoxical property of this synapse, namely GABA-mediated primary afferent depolarization (PAD) that occurs at lower, but not at physiological, temperatures (Boyle et al., 2019; Fink et al., 2014; Rudomin, 2009) (Figure 4A). Specifically, at room temperature (23°C), GABA release from PV+ neuron terminals at the axoaxonic synapses paradoxically induces depolarization of primary afferent terminals that is sufficient to cause glutamate release and generate excitatory postsynaptic currents (EPSCs) (Figure 4A).
Figure 4. Deletion of RARα in PV+ cells prevents SNI-induced presynaptic disinhibition in spinal cord dorsal horn.
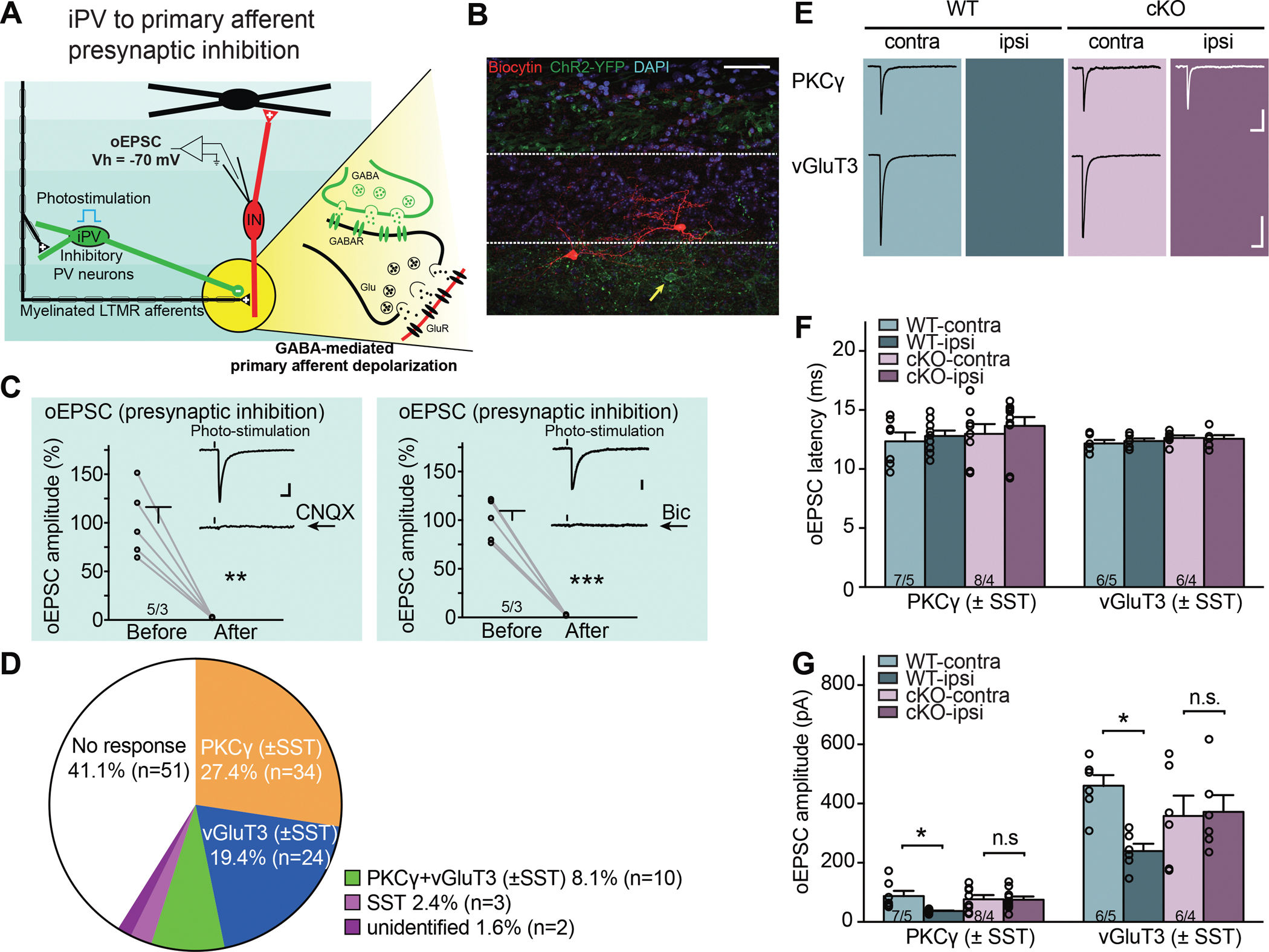
(A) Schematic diagram of local spinal cord dorsal horn circuit pertaining to presynaptic primary afferent inhibition. Inhibitory iPV+ neurons (green) gate primary afferent excitation onto excitatory postsynaptic non-PV interneurons (red) via synaptic inhibition onto primary afferent terminals (presynaptic inhibition). Detailed synaptic organization is depicted in the yellow highlighted part: optogenetic activation of PV+ neurons induces oEPSC in postsynaptic interneurons via primary afferent depolarization (PAD), which triggers glutamate release from afferent terminals at room temperature. oEPSC is recorded with postsynaptic interneuron held at −70 mV (reversal potential of IPSC). (B) A representative image of a recorded non-PV cell (biocytin-labelled, red) in substantia gelatinosa. Yellow arrow indicates a PV-ChR2-YFP cell. Scale bar: 50 μm. (C) oEPSCs, activated by photo-stimulation of PV+ neurons, can be completely blocked by AMPA receptor antagonist CNQX (left, paired t-test, t = 5.97, **, p < 0.01), as well as GABAA receptor antagonist bicuculline (10 μM) (right, paired t-test, t = 9.98, ***, p < 0.001). Scale bars: 50 ms, 20 pA. (D) Single cell qRT-PCR-based categorization of all neurons recorded for PV+ presynaptic inhibition measurement. (E) Samples traces of oEPSCs recorded from PKCγ+ and vGluT3+ postsynaptic neurons bilaterally in WT and cKO mice with SNI. Scale bars: 100 ms, 50 pA (top), 200 pA (bottom). (F) Synaptic latency of oEPSC in both WT and cKO mice. n/N = # of neurons/# of mice. (G) Quantification of oEPSC amplitudes from PKCγ+ and vGluT3+ postsynaptic neurons in response to optogenetic activation of PV+ neurons. Two-way ANOVA followed by Bonferroni test, PKCγ+: interaction (genotype × SNI), F(1, 30) = 4.39, p < 0.05; SNI factor, F(1, 30) = 4.79, p < 0.05; vGluT3+: interaction, F(1, 20) = 5.58, p < 0.05; SNI factor, F(1, 20) = 4.34, p = 0.05. Pos hoc test, *, p < 0.05. n/N = # of neurons/# of mice. Data shown as mean ± SEM.
We recorded from non-PV neurons in laminae IIi-III one week after SNI surgery. To control for variabilities in light penetration and cell depth in slices, we used maximum synaptic responses generated by saturating light intensities (30 mW) for comparison (Figure S6A), and recorded optogenetically evoked PSCs (oPSCs) in response to full-field photo-stimulation of PV-ChR2 cells (Figure 4B, Figures S6B and S6C). oEPSCs were isolated by holding the cells at −70 mV, the reversal potential of IPSCs based on the I-V curve (Figures 4C and S6C). Cells were selected from both ipsilateral and contralateral sides to achieve within-subject controls. Among the 124 recorded neurons (56 in WT and 68 in RARα cKO, Figure 4D), 73 neurons (37 in WT and 36 in RARα cKO) showed clear synaptic responses that could be completely blocked by the AMPA-receptor antagonist CNQX, indicating a comparable level of iPV-target connectivity in both WT and RARα cKO groups. (Figures 4C and 4D). To resolve the molecular identity of the recorded cells (n = 73), we recovered their RNA and performed single-cell qRT-PCR. These experiments revealed that among the postsynaptic target neurons of iPV+ cells in the dorsal horn, PKCγ- and vGluT3-expressing cells are the two major types of excitatory interneurons and represented the majority of the iPV-responsive neurons (~ 80%) we recorded (Figure 4C). The small number of remaining cells were either positive (10) or negative for both PKCγ and vGluT3 (5).
We analyzed the oEPSCs in the PKCγ+ and vGluT3+ neurons, which represent the majority of non-PV neurons recorded in laminae IIi-III. It is worth noting that the oEPSCs recorded in both types of neurons showed similarly long synaptic latencies (Figures 4E and 4F) and could be completely blocked by bicuculline (Gradwell et al., 2021) (Figure 4C), indicative of polysynaptic responses originating from GABA release from iPV+ neurons (Boyle et al., 2019; Gradwell et al., 2021), but not from other excitatory PV-expressing cells in the dorsal horn or the DRG (Niu et al., 2013). The response amplitudes were greater in vGluT3+ neurons than in PKCγ+ neurons (Figures 4E and 4G), suggesting cell type-specific differences in connection strength. In wildtype mice, the amplitude of oEPSC in both PKCγ+ and vGluT3+ neurons on the ipsilateral side were significantly reduced by SNI compared to the contralateral control side, indicating a weakened presynaptic inhibition by PV+ neurons (Figures 4E and 4G). Importantly, this reduction was absent in PV-RARα cKO mice (Figures 4E and 4G). Taken together, these data reveal that PV+ neuron-mediated presynaptic disinhibition contributes, at least in part, the hyperactivity of spinal nociceptive circuits underlying mechanical hypersensitivity, and that this SNI-induced synaptic change requires RARα expression in the PV+ neurons.
RARα in PV+ neurons is required for SNI-induced disinhibition of excitatory interneurons
To assay the direct postsynaptic inhibition of non-PV neurons by PV+ cells, we recorded oIPSCs in non-PV cells in acute spinal cord slices in the presence of CNQX and APV, which blocks all excitatory synaptic transmission (Figure 5A). We verified that all oIPSCs were completely blocked by bicuculline and strychnine (Figure S6D). We recorded from a total of 106 non-PV cells (50 WT and 56 cKO). 64 of these neurons, majority of which were PKCγ+ (25) or vGluT3+ (24), showed responses (Figure 5B). Unlike presynaptic inhibition-induced oEPSCs, direct postsynaptic oIPSCs exhibited a shorter monosynaptic response latency, which was comparable across all groups (Figure 5D). oIPSCs in vGluT3+ neurons had a greater amplitude compared PKCγ+ neurons, and both types of neurons showed reduced oIPSCs after SNI on the ipsilateral side (Figures 5C and 5E). Deletion of RARα in PV+ neurons did not alter oIPSC amplitudes in control neurons, but completely prevented the SNI-induced reduction in postsynaptic inhibition (Figure 5E), indicating that SNI-induced PV+ neuron-mediated postsynaptic disinhibition is also RARα-dependent.
Figure 5. Deletion of RARα in PV+ cells prevents the SNI-induced postsynaptic disinhibition onto non-PV cells in spinal cord dorsal horn.
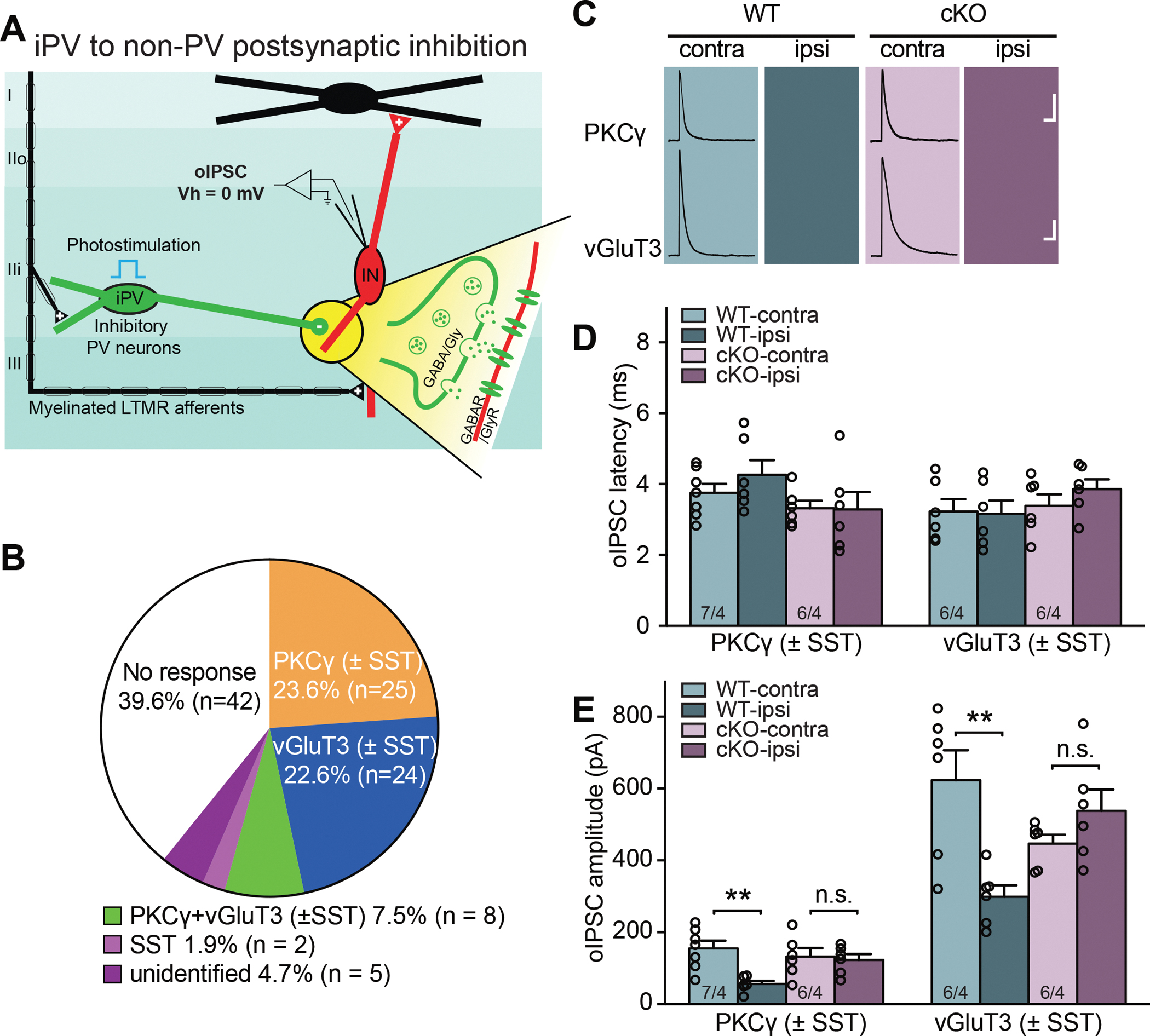
(A) Schematic diagram of local spinal dorsal horn circuit pertaining to postsynaptic inhibition from iPV+ neurons to non-PV excitatory interneurons. Detailed synaptic organization is depicted in the yellow highlighted part. oIPSCs were measured in postsynaptic interneurons (held at 0 mV) with optogenetic activation of PV+ cells in the presence of CNQX and APV (to block PAD-induced oEPSCs). (B) Single cell qRT-PCR-based categorization of all neurons recorded for PV+ postsynaptic inhibition measurement. (C) Sample traces of oIPSCs recorded from PKCγ+ and vGluT3+ postsynaptic neurons bilaterally in WT and cKO mice with SNI. Scale bars: 100 ms, 50 pA (top), 100 pA (bottom). (D) Synaptic latency of oIPSC in both WT and cKO mice. n/N = # of neurons/# of mice. (E) Quantification of oIPSC amplitudes from PKCγ+ and vGluT3+ postsynaptic neurons in response to optogenetic activation of PV+ neurons. Two-way ANOVA followed by Bonferroni test, PKCγ+: interaction (genotype × SNI), F(1, 21) = 5.78, p < 0.05; SNI factor, F(1, 21) = 8.30, p < 0.01; vGluT3: interaction, F(1, 20) = 14.32, p < 0.01; SNI factor, F(1, 20) = 4.48, p < 0.05. Pos hoc test, **, p < 0.01. n/N = # of neurons/# of mice. Data shown as mean ± SEM.
Lastly, we examined another synaptic output from inhibitory PV+ neurons (iPVs), which targets both excitatory and inhibitory PV+ neurons (ePVs and iPVs) in the dorsal horn and presumably provides feedforward inhibition and disinhibition, respectively (Gradwell et al., 2021) (Figure 6A). The recordings were conducted in the presence of CNQX and APV to block all excitatory transmission. Light stimulation evoked a large current at holding potential of 0 mV, which is a combination of ChR2-mediated photo current and oIPSC (Figure 6B). Subtraction of the isolated photocurrent (evoked response after the addition of bicuculline and strychnine) from the total current yielded pure oIPSCs for quantification (Figure 6B). After each recording, total mRNA was recovered and single-cell qRT-PCR was performed to verify the expressing of PV, GAD1 (for iPVs) and vGluT2 (for ePVs). We recorded 48 PV+ neurons (24 in WT and 24 in cKO). qPCR results showed no overlap between GAD1+ and vGluT2+ populations. 12 of 24 neurons in WT and 12 of 24 in cKO were ePVs, while the remaining neurons were iPVs. Both ePVs and iPVs receive significant and comparable synaptic inhibition from iPVs (Figures 6C and 6E) (Gradwell et al., 2021). Importantly, similar to synaptic inhibition onto non-PV neurons, SNI significantly reduced iPV inhibition onto both ePV and iPV neurons ipsilateral to the injured side, and PV-specific RARα deletion completely blocked this reduction in inhibition (Figures 6A and 6F).
Figure 6. Deletion of RARα in PV+ cells prevents the SNI-induced postsynaptic disinhibition onto PV+ cells in spinal cord dorsal horn.
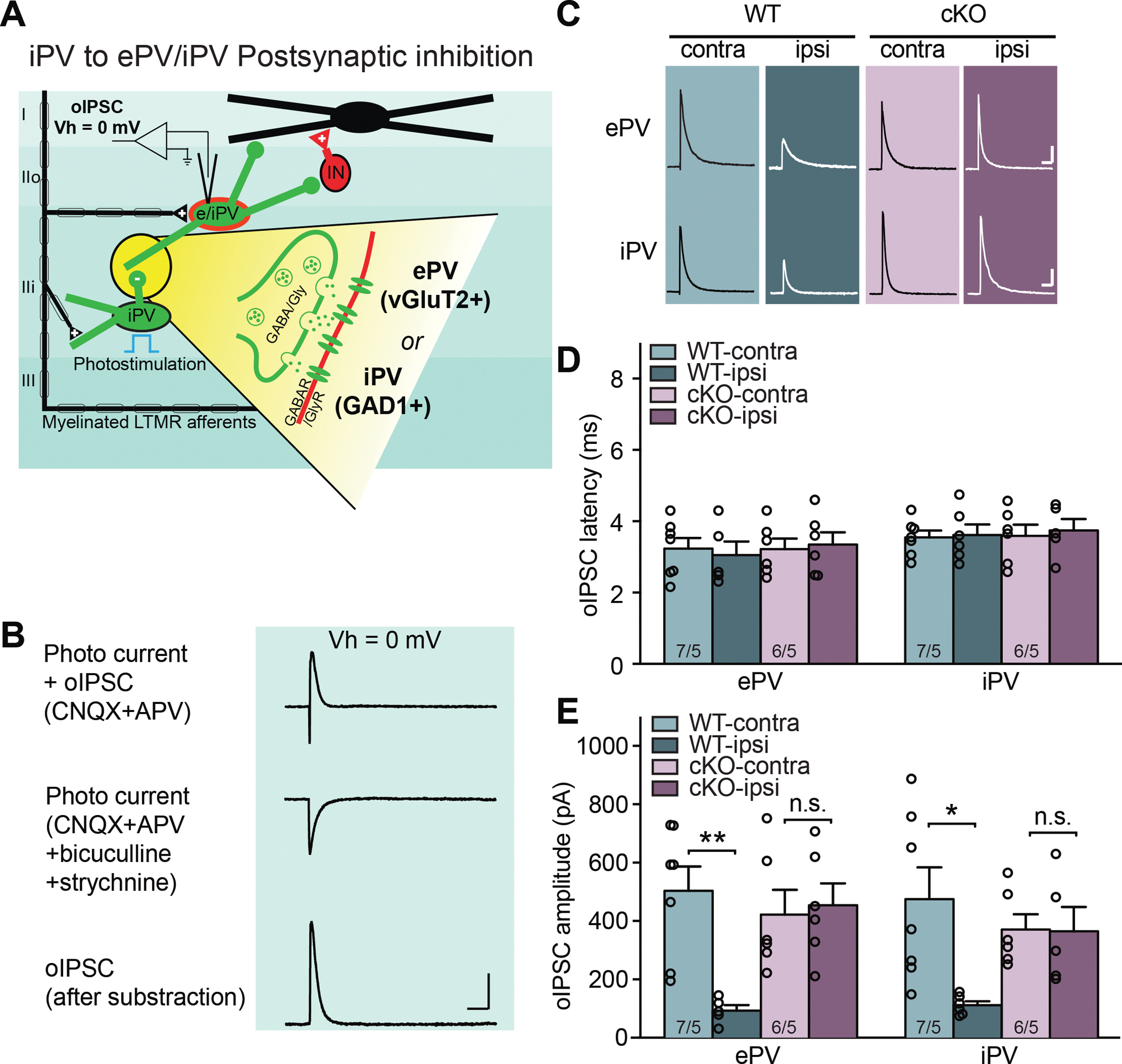
(A) Schematic diagram of local spinal dorsal horn circuit pertaining to postsynaptic inhibition from iPV+ neurons to other PV+ (ePV and iPV) interneurons. Detailed synaptic organization is depicted in the yellow highlighted part. oIPSCs were measured in postsynaptic interneurons (held at 0 mV) with optogenetic activation of PV+ cells in the presence of CNQX and APV (to block PAD-induced oEPSCs). (B) Example traces of optogenetically evoked currents in a PV+ neuron recorded at 0 mV holding potential. Subtraction of isolated photocurrent (middle) from total current (top) yields pure oIPSCs (bottom) from PV+ neurons. (C) Sample traces of oIPSCs recorded from ePV and iPV postsynaptic neurons bilaterally in WT and cKO mice with SNI. Scale bars: 100 ms, 100 pA. (D) Synaptic latency of oIPSC in both WT and cKO mice. n/N = # of neurons/# of mice. (E) Quantification of oIPSC amplitudes from ePV and iPV postsynaptic neurons in response to optogenetic activation of PV+ neurons. Two-way ANOVA followed by Bonferroni test, ePV: interaction (genotype × SNI), F(1, 20) = 8.43, p < 0.01; SNI factor, F(1, 20) = 6.16, p < 0.05; iPV: interaction, F(1, 20) = 5.23, p < 0.05; SNI factor, F(1, 20) = 5.57, p < 0.05. Pos hoc test, *, p < 0.05; **, p < 0.01. n/N = # of neurons/# of mice. Data shown as mean ± SEM.
Together, our synaptic response analysis provides strong evidence that concomitant pre- and postsynaptic disinhibition of the iPV+ output onto specific postsynaptic excitatory neurons in the spinal dorsal horn circuit underlies the SNI-induced central sensitization. Importantly, these synaptic changes resemble homeostatic plasticity found in other inhibitory synapses in the brain and share the same molecular mechanism (Zhong et al., 2018), relying on RARα expression in PV+ neurons. The observation that iPV to iPV inhibition is also reduced by SNI may suggest a paradoxical decrease in feedforward disinhibition, although the functional outcome of such changes is yet to be explored. Given that the vast majority of postsynaptic cells are excitatory interneurons (non-PV neurons and ePVs), and that in vivo photostimulation of ePVs leads to significant activation of nociceptive processing neurons in the superficial lamina (Gradwell et al., 2021), we propose that the overall effect of disinhibition of spinal cord dorsal horn iPV outputs under SNI is central sensitization.
Selective deletion of RARα in spinal cord dorsal horn PV+ neurons prevents mechanical hypersensitivity
Our synaptic physiology in the dorsal horn of the spinal cord supports the notion that SNI modifies local dorsal horn circuits by homeostatically reducing inhibitory synaptic outputs from PV+ neurons upon sensory deafferentation, thereby causing a misdirection of tactile signals to nociceptive pathways. Deletion of RARα from PV+ neurons prevented such changes in dorsal horn circuits and blocked the development of mechanical hypersensitivity. However, since RARα is deleted from all PV+ neurons throughout the CNS, we cannot exclude the possibility that deletion of RARα in PV+ neurons in cortical and subcortical brain regions alters cortical outputs, which provide an essential descending feedback signal that blocks spinal cord hyperactivity and may have mediated the effect of the RARα deletion (Liu et al., 2018; Ossipov et al., 2014). To distinguish between these two possibilities, we injected AAVs encoding Flp-dependent Cre and tdTomato into the dorsal horn of PV-FlpO:RARαfl/fl double-mutant mice to selectively delete RARα in dorsal horn PV+ neurons (Figures 7A–7C). Three weeks after viral infection, we performed SNI surgery.
Figure 7. Spinal cord-specific deletion of PV-RARα prevents the development of mechanical hypersensitivity.
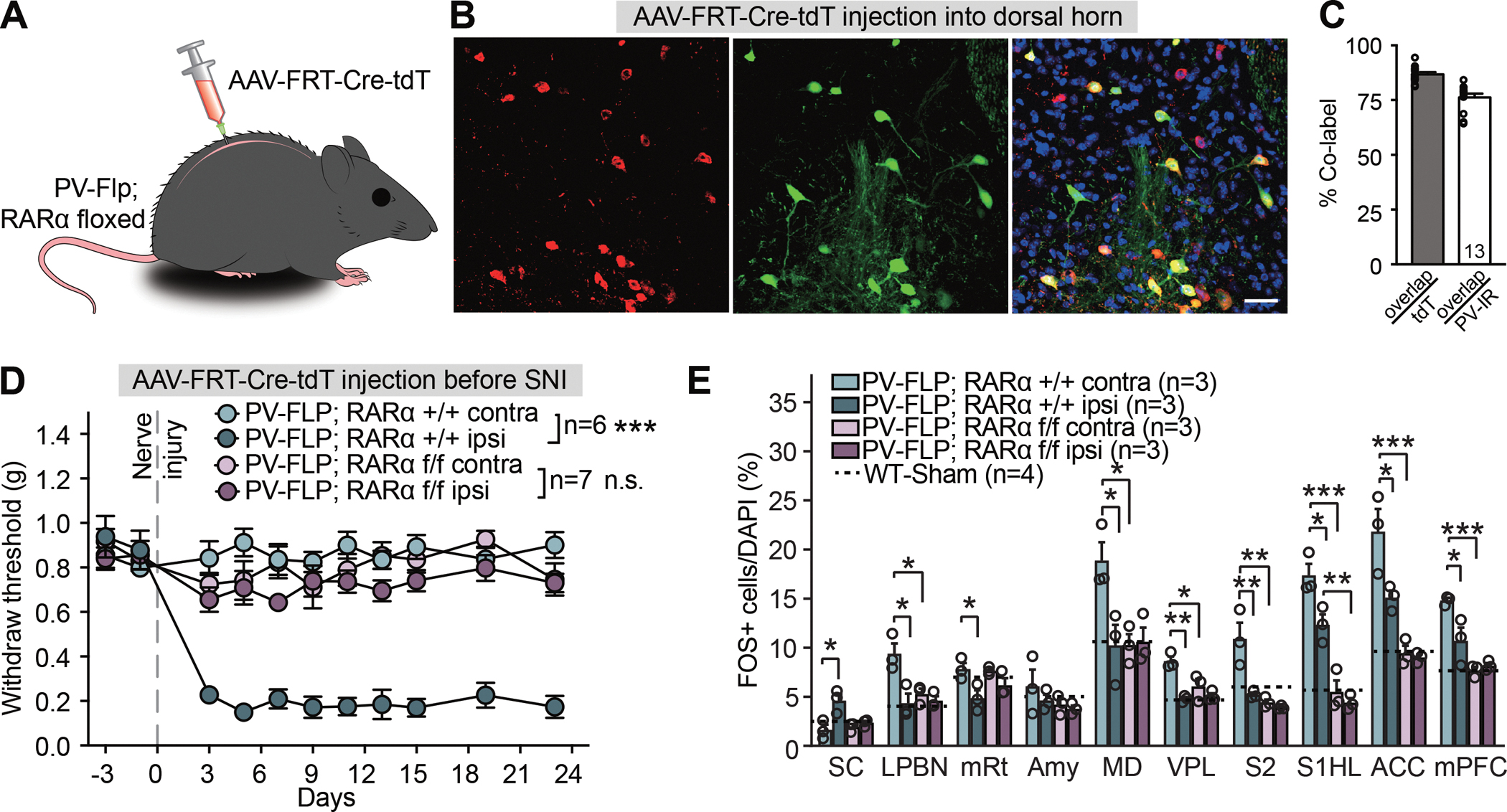
(A) Experimental design to obtain RARα deletion in dorsal horn PV+ neurons. (B) Representative images showing FRT-Cre-tdTomato expression in PV-immunoreactive neurons in spinal dorsal horn. Scale bar: 20 μm. (C) Quantification of PV-Flp driver line expression fidelity (% PV-IR/tdTomato) and efficacy (% tdTomato/PV-IR) in spinal dorsal horn. (D) Mechanical hypersensitivity quantified as paw withdrawal threshold with von Frey filaments in WT and spinal-specific PV-RARα cKO mice before and over a period of three weeks after SNI. Two-way ANOVA with Bonferroni test, PV-FLP; RARα +/+ contra vs. ipsi: interaction (days × SNI), F(10, 100) = 15.96, ***, p < 0.001; SNI factor, F(1, 10) = 337.4, ***, p < 0.001; PV-FLP; RARα f/f contra vs. ipsi: interaction, F(10, 120) = 0.74, p = 0.68; SNI factor, F(1, 12) = 2.85, p = 0.12. (E) Quantification of neuronal activation by SNI in regions of the ascending nociceptive pathways. Activation level in sham-treated WT mice for each region is indicted by dashed lines. SC: spinal cord; LPBN: lateral parabrachial nucleus; mRt: mesencephalic reticular formation; Amy: Amygdala; MD: mediodorsal thalamus; VPL: ventral posterolateral thalamic nuclei; S2: secondary somatosensory cortex; S1HL: Primary somatosensory cortex, hindlimb region; ACC: anterior cingulate cortex; mPFC: medial prefrontal cortex. Two-way ANOVA followed by Bonferroni test, interaction (genotype × SNI), SC: F(1, 8) = 7.53, p < 0.05; LPBN: F(1, 8) = 7.41, p < 0.05; mRt: F(1, 8) = 1.54, p = 0.25; Amy: F(1, 8) = 0.28, p = 0.61; MD: F(1, 8) = 8.32, p < 0.05; VPL: F(1, 8) = 8.46, p < 0.05; S2: F(1, 8) = 9.51, p < 0.05; S1HL: F(1, 8) = 3.90, p = 0.084; ACC: F(1, 8) = 6.10, p < 0.05; mPFC: F(1, 8) = 10.01, p < 0.05. SNI factor, SC: F(1, 8) = 12.91, p < 0.01; LPBN: F(1, 8) = 12.8, p < 0.01; mRt: F(1, 8) = 13.7, p < 0.01; Amy: F(1, 8) = 1.04, p = 0.34; MD: F(1, 8) = 7.19, p < 0.05; VPL: F(1, 8) = 23.49, p < 0.01; S2: F(1, 8) = 13.82, p < 0.01; S1HL: F(1, 8) = 9.31, p < 0.05; ACC: F(1, 8) = 7.94, p < 0.05; mPFC: F(1, 8) = 6.28, p < 0.05. Post hoc test, *, p < 0.05; **, p < 0.01; ***, p < 0.001.N = # of mice. Data shown as mean ± SEM.
Remarkably, mechanical hypersensitivity failed to develop in mice with the spinal cord PV-RARα cKO (Figure 7D). By contrast, the significantly increased sensitivity to cold temperature as a form of thermal allodynia that was also evident after SNI (Brenner et al., 2012; Francois et al., 2017) was not blocked by the PV-RARα deletion in the spinal cord (Figures S7A and S7B). These results suggest a critical and highly specific involvement of RARα expression in dorsal horn PV+ neurons in the development of mechanical neuropathic pain. Additionally, SNI-induced neuronal hyperactivity in ascending nociceptive pathways (with the exception of amygdala not showing hypersensitivity) was also prevented by the spinal PV-RARα cKO (Figure 7E). Importantly, unlike the global PV-RARα deletion, the spinal cord-specific RARα deletion did not affect the basal activity of neurons in any of the regions examined, as assayed by FOS expression (Figure 7E). This result indicates that a reduced basal activity in brain regions in the global PV-RARα cKO is not the main cause of blocked hyperactivity in the brain regions involved in neuropathic pain. Interestingly, injection of AAVs encoding Flp-dependent Cre into these mice one week after SNI surgery, when mechanical hypersensitivity is fully developed, failed to reverse mechanical hypersensitivity (Figure S7C), revealing that RARα is critically involved in the induction of the dorsal horn circuit plasticity that establishes SNI-induced mechanical hypersensitivity, but is not required for its maintenance.
RARα antagonist infusion in spinal cord prevents mechanical hypersensitivity
Given the essential function of RARα in dorsal horn PV+ neurons in the development of mechanical hypersensitivity, we next asked whether pharmacological inhibition of RARα could prevent neuropathic pain development. We are fully aware of the limitation of this approach as the drug will inhibit RARα in all cell types. Nevertheless, we believe this is an attractive alternative to Cre-dependent deletion of RARα because of its reversibility – once the pump runs empty, RARα function will be restored, which allows us to probe its contribution to the maintenance phase of mechanical hypersensitivity.
We intrathecally delivered the selective RARα antagonist Ro41–5253 for seven days with an osmotic pump placed at the lumbar vertebrae L3–5 (Hu et al., 2020; Ke et al., 2019) (Figure 8A). SNI surgery was performed two days after pump implantation. SNI-induced mechanical hypersensitivity was detected in the vehicle infusion group for at least two weeks after surgery, indicating that the pump implantation surgery does not interfere with hypersensitivity development (Figure 8A). Infusion of Ro41–5253 for two days before the SNI surgery did not affect tactile perception, as evidenced by the unchanged withdrawal threshold in response to an innocuous mechanical stimulation (Figure 7B). Additionally, recordings from dorsal horn non-PV neurons in acute spinal cord slices acute showed no acute effect of Ro41–5253 on basic cell membrane properties or synaptic responses (Figure S8). Continuous infusion of Ro41–5253 for an additional five days prevented the development of SNI-induced mechanical hypersensitivity during this period (Figure 8B, first infusion), indicating that locally blocking spinal RARα before nerve injury effectively prevents SNI-induced mechanical hypersensitivity. To further test the potential impact of the RARα antagonist on existing hypersensitivity, we allowed neuropathic pain to develop after the first pump was emptied for three more weeks, and infused Ro41–5253 for a second time. Consistent with the results obtained with AAV-mediated RARα deletion after SNI, pharmacologically blocking RARα function cannot reverse the existing hypersensitivity (Figure 8B, second infusion).
Figure 8. Region-specifical deletion of PV-RARα prevents the development of neuropathic pain.
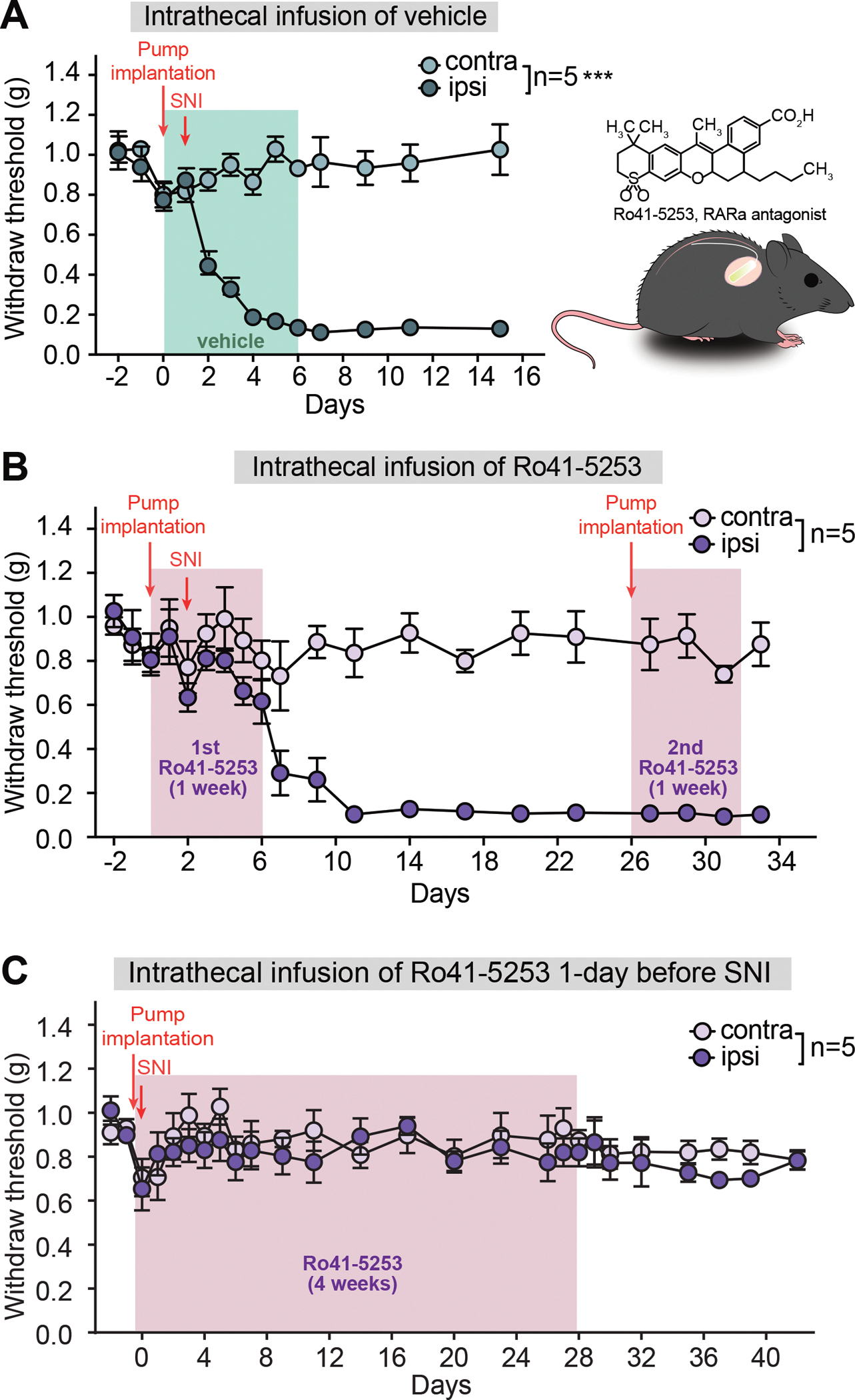
(A) Mechanical hypersensitivity quantified as paw withdrawal threshold in animals receiving vehicle infusion and SNI (performed one day after pump implantation. Two-way ANOVA with Bonferroni test, interaction (days × SNI), F(12, 96) = 17.62, ***, p < 0.001; SNI factor, F(1, 8) = 333.7, ***, p < 0.001. (B) Paw withdrawal threshold in animals receiving two infusions of a RARα antagonist Ro41–5253 (6–7 days each) and SNI. First infusion started 2 days before SNI and second infusion started 24 days after SNI when mechanical hypersensitivity was evident. Two-way ANOVA followed by Bonferroni test, first infusion: interaction, F(6, 48) = 0.60, p = 0.73; SNI factor, F(1, 8) = 1.736, p = 0.22; second infusion: interaction, F(2, 16) = 0.82, p = 0.46; SNI factor, F(1, 8) = 179.8, ***, p < 0.001, Data shown as mean ± s.e.m. (C) Paw withdrawal threshold in animals receiving 4-week of a RARα antagonist Ro41–5253 infusion and SNI (performed one day after pump implantation). Two-way ANOVA followed by Bonferroni test, interaction, F(16, 128) = 0.50, p = 0.94; SNI factor, F(1, 8) = 0.83, p = 0.39 (0–28 days); interaction, F(6, 48) = 0.33, p = 0.92; SNI factor, F(1, 8) = 3.05, p = 0.12 (29–42 days). Data shown as mean ± SEM.
Since short-term infusion of Ro41–5253 only temporarily blocked mechanical hypersensitivity development, we were curious whether the development of mechanical hypersensitivity may be more permanently prevented by a prolonged blockade of RARα function. We therefore implanted one day before SNI surgery an osmotic pump enabling the delivery of Ro41–5253 for four weeks. This approach prevented the development of SNI-induced mechanical hypersensitivity for the four weeks when RARα function was blocked (Figure 8C). Interestingly, unlike the results from short-term blockade (Figure 8B), no further mechanical hypersensitivity was evident weeks after the pump became empty (Figure 8C), suggesting the existence of a critical period for RARα-mediated homeostatic modulation of dorsal horn circuit function after the peripheral nerve injury. This result is particularly significant in that it highlights a critical time window during which spinal synaptic plasticity may be actively engaged in driving central sensitization. This window may also be of significance for effective therapeutic intervention after nerve injury.
Discussion
Homeostatic synaptic plasticity has been studied extensively in the context of maintaining neural network stability through counteracting and preventing runaway Hebbian plasticity (Monday et al., 2018; Turrigiano, 2012; Yee et al., 2017). The physiological role and pathological implications of homeostatic plasticity, however, are incompletely understood. We now show that peripheral nerve injury, which leads to a chronic aberration of afferent input signals to spinal cord circuits, triggers homeostatic synaptic compensation in local spinal dorsal horn circuits, and thereby as an unwanted by-product with vast pathophysiological consequences, causes the redirection of tactile sensory signals into spinal nociceptive circuits, thus inducing allodynia and neuropathic pain.
In the spinal cord dorsal horn, nociceptive and tactile mechanosensory information are transmitted to separate circuits comprising projection neurons and local interneurons (Choi et al., 2020; Peirs and Seal, 2016; Todd, 2010). Under normal conditions, an innocuous touch activates mechanosensory inputs via myelinated Aδ and Aβ fibers as well as unmyelinated C fibers, thereby exciting mechanosensory projection neurons and generating a touch sensation (Handler and Ginty, 2021; Koch et al., 2018). Normally, the flow of sensory information from Aβ fibers to nociceptive circuits is blocked by local inhibitory interneurons that potently inhibit excitatory interneurons, such as somatostatin-expressing interneurons, and additionally suppress presynaptic sensory terminals, thus preventing activation of nociceptive projection neurons and ascending nociceptive pathway by tactile sensory inputs under normal conditions (Duan et al., 2014; Hughes and Todd, 2020; Peirs et al., 2015; Petitjean et al., 2015; Takazawa and MacDermott, 2010; Zholudeva et al., 2021). In this study, we show that the loss of afferent inputs reduces synaptic inhibition onto both primary afferent axonal terminals (presynaptic inhibition) and postsynaptic excitatory relay interneurons (postsynaptic inhibition) by a homeostatic mechanism requiring RARα in PV-expressing inhibitory interneurons. We demonstrate that blocking homeostatic synaptic plasticity in spinal PV+ neurons by deletion of RARα prevents the reduction of synaptic inhibition by SNI and the development of mechanical hypersensitivity. These results not only show that homeostatic synaptic plasticity may be a key mechanism driving allodynia, but also suggest that the molecular pathway mediating homeostatic plasticity could be a therapeutic target for neuropathic pain.
Several types of spinal cord dorsal horn interneuron have been implicated in central sensitization and mechanical allodynia, including interneurons that express somatostatin (Duan et al., 2014), dynorphin (Duan et al., 2014), calretinin (Peirs et al., 2015), vesicular glutamate transporter vGluT3 (Peirs et al., 2015), and PV (Petitjean et al., 2015). In the brain, PV-expressing GABAergic interneurons effectively control the spike patterns of pyramidal neurons by synapsing onto their axon initial segment (PV-expressing chandelier cells) or onto their soma and proximal dendrites (basket cells) (Cobb et al., 1995; Cobb et al., 1997; Lewis and Lund, 1990; Miles et al., 1996; Pawelzik et al., 2002). In the spinal cord dorsal horn, PV+ neurons (up to 75% of which express GABA and glycine as neurotransmitters (Abraira et al., 2017; Gradwell et al., 2021)) primarily localize to the inner lamina II and lamina III (Hughes et al., 2012), where they provide an additional mode of synaptic inhibition termed presynaptic inhibition (Comitato and Bardoni, 2021). By forming axo-axonic synapses onto primary afferent terminals, iPV+ neurons activate GABAARs present in the afferent terminals and curtail glutamate release from primary afferent inputs through shunting inhibition (Hughes et al., 2012). Spinal iPV+ neurons normally tightly control the flow of tactile sensory information from peripheral inputs to ascending nociceptive pathways via this form of presynaptic inhibition and via their postsynaptic inhibition of excitatory vGluT3+ or PKCγ+ interneurons (Braz et al., 2014; Hughes and Todd, 2020). Here, we found that the inhibition of PKCγ+, vGluT3+, and iPV+/ePV+ interneurons by iPV+ neurons is significantly reduced after peripheral nerve injury, illustrating the widespread impact of postsynaptic inhibition in the context of neuropathic pain. Furthermore, we demonstrate that SNI reduces presynaptic inhibition onto primary afferents, which synapse onto both PKCγ+ and vGluT3+ interneurons. PKCγ+ and vGluT3+ interneurons, the major excitatory interneuron subtypes in dorsal horn laminae II and III that relay Aβ afferent input to lamina I nociceptive projection neurons, have both been implicated in neuropathic pain (Cheng et al., 2017; Malmberg et al., 1997; Peirs et al., 2021; Petitjean et al., 2015; Seal et al., 2009). The two concomitantly occurring effects of inhibitory synaptic plasticity onto PKCγ+ and vGluT3+ neurons described in our study demonstrate that the spinal cord dorsal horn circuit adjusted itself homeostatically to compensate for the loss of peripheral input due to the nerve injury during SNI, leading to hypersensitivity of the ascending nociceptive pathway and neuropathic pain.
While the circuit level remodeling in the spinal dorsal horn during neuropathic pain is being mapped out by many studies, the molecular players enabling synaptic plasticity underlying these circuit remodeling is less understood. In dissecting the molecular mechanisms underlying the homeostatic plasticity that induces disinhibition of sensory gating by PV+ neurons, we examined the role of RARα because of its central contribution to homeostatic synaptic plasticity (Hsu et al., 2019; Park et al., 2018; Sarti et al., 2013; Yee and Chen, 2016; Zhong et al., 2018). In mature neurons, RARα translocates from the nucleus to the cytosol to act as a retinoic acid-dependent translational regulator in homeostatic synaptic plasticity (Maghsoodi et al., 2008; Poon and Chen, 2008). Synaptic RA/RARα signaling is essential for homeostatic synaptic plasticity at both excitatory and inhibitory synapses in the hippocampus and neocortex (Aoto et al., 2008; Park et al., 2018; Sarti et al., 2013; Yee and Chen, 2016; Zhong et al., 2018). Of particular interest is RARα’s involvement in homeostatic synaptic plasticity in the primary visual cortex (V1). Prolonged visual deprivation induces homeostatic synaptic plasticity at both V1 excitatory and inhibitory cortical synapses toward increased synaptic excitation/inhibition balance (Maffei and Turrigiano, 2008; Miska et al., 2018; Zhong et al., 2018). Among the synapses involved, synaptic inhibition from PV+ neurons to L2/3 pyramidal neurons shows a profound reduction after loss of visual inputs, a form of homeostatic plasticity that requires RARα expression in PV+ neurons. In the current study, we demonstrate that a similar form of homeostatic synaptic plasticity occurs in spinal dorsal horn iPV+ neurons, with RARα as the key effector. Peripheral nerve injury drives homeostatic changes that lead to a loss of inhibitory output from iPV+ neurons, which causes primary afferent inputs to erroneously drive superficial lamina nociceptive projecting neuron and ascending nociceptive pathway activation. Upon the PV-specific genetic deletion of RARα, this form of homeostatic synaptic plasticity is inhibited and the gate remains closed, thus preventing the development of mechanical hypersensitivity. These results indicate that the basic molecular mechanisms underlying homeostatic synaptic plasticity may be conserved across CNS circuits. Additional work is needed to understand the signaling mechanisms of the critical window during which homeostatic synaptic plasticity leading to disinhibition of the dorsal horn circuit occurs, including the involvement of microglial activation (Chen et al., 2018; Denk et al., 2016; Guan et al., 2016; Peng et al., 2016). Pathological neuronal activity resulting from homeostatic plasticity after nerve injury or cochlear trauma is a common theme in many types of chronic pain or in tinnitus (Eggermont and Roberts, 2004; Flor et al., 1995; Moller, 2007). Thus, identifying key molecular players as potential druggable targets will be the first step towards identifying effective remedies for treating these debilitating conditions.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lu Chen, at luchen1@stanford.edu.
Materials availability
This study did not generate new unique reagents or mouse lines.
Data and code availability
All primary data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animals were housed and used in experiments following Stanford University APLAC guidelines. Postnatal day 35–56 male and female littermates were used for this study. Mice were group-housed with littermates and maintained under a 12 h light/dark cycle. The RARαfl/fl mice (C57BL/6 background) were originally obtained as a generous gift from Dr. Pierre Chambon and Norbert Ghyselinck (IGBNC, Stasbourg, France)(Chapellier et al., 2002). The a floxed ChR2 line (Ai32, stock # 012569) and C57BL/6 wild-type mice were obtained from The Jackson Laboratory (Bar Harbor, ME).
Cell type-specific RARα cKO mice were generated by crossing the RARαfl/fl line with cell type-specific driver lines (PV-Cre, stock #008069; SST-Cre, stock #013044, The Jackson Laboratory, Bar Harbor, ME) and an EYFP reporter line (Stock #007903, The Jackson Laboratory, Bar Harbor, ME). Litters were genotyped by PCR using the protocol described previously.
For regional ablation of RARα, RARαfl/fl mice were crossed to PV-2A-FlpO line (stock #022730, The Jackson Laboratory, Bar Harbor, ME). WT mice crossed to PV-2A-FlpO served as controls. Litters were genotyped for flox and FlpO.
METHOD DETAILS
In situ hybridization
In situ hybridization with RNAscope Technology(Wang et al., 2018) (Advanced Cell Diagnostics Bioscience) was used to detect the expression of RARα mRNA. P35-P42 WT and RARα KO mice were deeply anesthetized with ketamine-xylazine followed by perfusion with 0.1M PBS and fixed with 4% PFA. Spinal cord L3-L5 regions were dissected, cryoprotected in 30% sucrose overnight, sectioned at 14 μm and maintained at −80 °C. On the day of ISH, tissue was thawed from −80 °C, washed with PBS and processed according to the manufacturer’s protocol. Briefly, the tissue was permeabilized, incubated with protease for 30 min, and hybridized with probe(s) for 2 hr at 40 °C. After amplification of mRNA signals, the slides were mounted with DAPI. A scanning microscope (BX61VS; OLYMPUS) and a confocal microscope (Nikon Instruments) were used to obtain whole-spinal cord slice images and higher-resolution images, respectively.
Single-cell qRT-PCR
mRNAs extracted from single cells were amplified using the protocol previously described (Zhong et al., 2018). Acute slices were obtained from P35-P42 mice and single cell contents were extracted from the substantia gelatinosa in the spinal cord. mRNAs from single cell extracts were amplified use Superscript III One-Step RT-PCR System with Platinum Taq High Fidelity DNA polymerase (Invitrogen, cat 12574035). RT-qPCR was then performed using TaqMan Gene Expression Master Mix (Applied Biosystems, cat 4369016) with Taqman primers from Life Technologies; RARα (Mm00436262_m1), Actinβ (Mm02619580_g1), GFAP (Mm01253033_m1), PV (Mm00443100_m1), SST (Mm.PT.53a.7678291), PKCγ (Mm.PT.58.45983184), VGluT3 (Mm.PT.58.17676720), GAD1 (Mm.PT.58.10501737), VGluT2 (Mm.PT.58.10363705). Actinβ was used as the endogenous control. Cells expressing GFAP, a marker for astrocytes, were excluded from the sample.
Spared nerve injury surgery
The spared nerve injury (SNI) surgery was performed based on previously established methods(Decosterd and Woolf, 2000). Briefly, mice were anesthetized with a mixture of ketamine and xylazine (0.1 ml/20g, intraperitoneal injection). The sciatic nerve with three branches was exposed, and the common peroneal and tibial branches were ligated with 8–0 nylon suture, sectioned distal to the ligation, removing 2–4 mm of the distal nerve stump. Care was taken to avoid any stretching or contact with the spared sural nerve. Sham operation without axotomy or contralateral intact hindlimb responses were served as controls.
Inflammatory pain model of complete Freund’s adjuvant (CFA) injection
CFA (Sigma, F5881) was diluted 1:1 in saline. Mice received a single subcutaneous injection of 20 μl CFA in the plantar surface of a single hind paw delivered with a 27-G needle after a baseline of von Frey filaments test. The contralateral hindlimb without injections was served as control.
Osmotic pump implantation
P35-P42 mice were anesthetized with 2.5% isoflurane, placed into a stereotaxic frame and a polyethylene catheter (Mouse Intrathecal Catheter MIT-02) was inserted into the spinal subarachnoid space of each mouse so as to place its tip 1–1.5 mm rostral to the dural incision at the L3–5 intervertebral space. The other end of the catheter was attached to an osmotic pump (Alzet 1007D) that infused the solution at a flow rate of 0.5 μl/h for 7 days. The osmotic pump was filled with Ro41–5253 (1.25 μg/h) or vehicle (aCSF). All animals had free access to food and water from the time of surgery until they were sacrificed. Each mouse was examined every day for behavioral changes.
AAV Vector construction, AAV preparation and Stereotaxic injections
The AAV-Frt-Cre-tdTomato vector was constructed using a synapsin promoter that drives the expression of an inverted Cre-IRES-tdTomato sequence, which is flanked by two FRT F545 sites. The AAV Frt-Cre-tdTomato was packaged with AAV-DJ capsids for high efficiency in vivo neuronal infection. AAVs were prepared as described(Zolotukhin et al., 1999). For intraspinal injections, a midline incision along the left lumbar vertebrae was carefully performed until the spinal cord was visible from the intervertebral spaces. No laminectomy was performed to maximally avoid trauma. Two stereotaxic injections of 250 nl AAVs on each side 400 μM lateral to the posterior spinal arteries and 400 μm from the dura were made in L3–5. The lassimus dorsi were sutured to protect the spinal cord and the skin was sealed with silk sutures(Peirs et al., 2015). For intracortical injections, 400 nL AAVs were injected into the anterior cingulate cortex (in mm: AP 0.90/1.40, ML ± 0.25, DV 1.50) or somatosensory cortex hindlimb region (in mm: AP −0.30/−0.80, ML ±1.50, DV 1.00)(Cetin et al., 2006; Park et al., 2018).
Retrograde labeling
7 days after SNI surgery, WT or PV-Cre RARα KO mice were anaesthetized with isoflurane and 300 nl of fluorogold (FG, 2% in sterilized water) were injected unilaterally with a glass micropipette into the lateral parabrachial nucleus (AP 5.0 mm; ML 1.4 mm; DV −2.3 mm from brain surface). 5–7 days following injection, spinal cord and brain tissues was collected for histological analysis.
Immunohistochemistry
Innocuous mechanical stimulation for FOS induction
Five to seven days after SNI surgery, animals were lightly restrained using a rodent restraint bag and both hind paws were stroked lightly with a #5 painting brush. Each 2-second stroke was applied from the middle of the foot to the distal foot pad along the peripheral side to cover the sural nerve territory. These strokes were applied once every 4 s for 10 min(Liu et al., 2018). This touch stimulus does not elicit a flexion reflex in normal mice. Animals were perfused 1.5 hr later with 4% PFA and spinal cord and brain tissues were postfixed in 30% sucrose and 4% PFA for 2 additional days. 30 μm coronal sections of whole brain and cross sections of spinal cord L3-L5 were obtained and stored in PBS at 4 °C. After permeabilization and blocking, slices were subsequently incubated with a FOS antibody (ABE457, Millipore) overnight at 4 °C, followed by incubation with an Alexa Fluor 546 donkey anti-rabbit secondary anti-body (ThermoFisher, cat#A10040) for 1 h at room temperature. The slices were then washed and mounted on slides with Vectashield-containing DAPI (H-1500, Vector Laboratories). Images were taken from 2 or 3 slices per animal. A minimal of 3 animals were analyzed for each group. The number of FOS+ cells was quantified in a fixed area within sections.
Electrophysiology
Acute spinal cord slice preparation: 5–7-week-old mice (5–7 days after SNI) were anesthetized with isoflurane, decapitated, and the vertebral column was rapidly removed and placed in oxygenated ice-cold dissection solution containing (in mM): 250 sucrose, 2.5 KCl, 25 NaHCO3, 1 NaH2PO4, 25 glucose, 6 MgCl2, 0.5 CaCl2, and 5 kynurenic acid (pH = 7.4, 320 mOsm). The lumbar spinal cord was isolated, embedded in a 3% low melting point agarose block and transverse of parasagittal slices (250 μm thick) with dorsal roots attached were made using a vibrating microtome (Leica VT1200). Slices were incubated for 10–15 min in oxygenated recovery solution containing (in mM): 92 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, HEPES 20, Glucose 25, 5 Sodium Ascorbate, 2 thiourea, 3 Sodium Pyruvate, 10 MgSO4, 0.5 CaCl2 (pH = 7.3~7.4, 300~310 mOsm) then transferred to aCSF containing (in mM): 125 NaCl, 2.5 KCl, 1.0 NaH2PO4, 25 NaHCO3, 25 glucose, 1 MgCl2, and 2 CaCl2 (pH = 7.4, 320 mOsm). For Acute brain slices preparation: Following isoflurane anesthesia, mice were decapitated and brains were quickly removed and transferred into ice-cold high sucrose aCSF. Coronal slices of 300 μm were made and allowed to recover at 32 °C – 34 °C for 30 min before being moved to ACSF at room temperature (Zhong et al., 2018).
Patch-clamp recordings of PV+ neurons were performed at room temperature. PV+ neurons located within or closely juxtaposed to the substantia gelatinosa in the spinal cord, or within layer II/III of the anterior cingulate cortex were identified with a Cre-dependent YFP reporter. For spinal cord recordings, glass pipettes (5–6 MΩ tip resistance) were filled with an internal solution containing (in mM): 120 K-methyl-sulfonate, 10 NaCl, 10 EGTA, 1 CaCl2, 10 HEPES, 0.5 Na3GTP, 5 MgATP (pH = 7.3, 310–320 mOsm). For cortical recordings, recording pipettes (3–4 MΩ) were filled with an internal solution containing (in mM): 130 K-gluconate, 10 KCl, 10 HEPES, 5 MgATP, 0.3 Na3GTP, and 0.2 EGTA (pH = 7.3, 300~310 mOsm). Membrane excitability experiments were performed in current-clamp mode with injections of incremental DC current steps (800 ms, −50 to +250 pA with a step interval of 20 pA). The numbers of action potentials elicited by injected currents were counted (Zhong et al., 2018). Data were acquired using a Multiclamp 700B amplifier and pClamp10 software (Molecular Devices, USA). Sampling rate was 10 kHz.
For synaptic inhibition experiments, a floxed ChR2 line (stock # 012569) was crossed with the PV-Cre driver line to allow optogenetic activation of PV+ terminals. Patch-clamp recordings were targeted in PV/ChR2-negative neurons in substantia gelatinosa for both presynaptic inhibition and postsynaptic inhibition assessment. Recording pipettes (3–4 MΩ) were filled with an internal solution containing (in mM): 132 CsMeSO3, 8 CsCl, 10 HEPES, 0.6 EGTA, 4 MgATP, 0.4 Na3GTP, and 10 Na-phosphocreatine (pH = 7.3, 300~310 mOsm). Full-field photostimulation (PS) of PV-ChR2-expressing neurons was achieved using single light pulses to induce maximal responses (450nm wavelength, 1 ms, 30 mW, Laser Diode MDL-III-450), which was collimated and coupled to the epifluorescence path of an Olympus BX51 microscope. For presynaptic inhibition through axo-axonic synapse onto primary afferent terminals, primary afferent depolarization (PAD)-induced optically evoked EPSC was recorded in postsynaptic neurons at the reversal potential of IPSCs (−70 mV) at room temperature(Boyle et al., 2019). For postsynaptic inhibition, glutamate receptor antagonists CNQX (10 μM) and APV (100 μM) were added to the recording aCSF, and optically evoked IPSCs were recorded in the postsynaptic neurons held at 0 mV.
Behavioral tests
Open field test:
Mice were placed in a 40 cm (L) × 40 cm (W) × 40 cm (H) open-field chamber. Locomotor activity was recorded for 30 minutes using an overhead digital camera and tracked using Viewer III tracking system (BIOSERVE, Bonn, Germany). Time spent in the center was measured by a 10 cm by 10 cm area in the center.
Elevated plus maze:
An elevated plus maze (Stoelting Co. IL) is composed of two open arms and two closed arms that extend from a central square area and elevate to a height of 50cm above floor level. The mouse was placed in the center of the maze facing an open arm and left to freely explore the maze for 10 min. The amounts of the time spent in the open vs. closed arms were recorded by Viewer III tracking system (BIOBSERVE).
Y-Maze test:
A plastic y-maze (Stoelting Co. IL) was used to measure spatial working memory. Individual mice were placed in the center of the Y-maze and allowed to freely explore for 5 minutes. The sequences and total numbers of arm entries were recorded and analyzed with the Viewer III tracking system. Visiting all three different arms consecutively was termed a ‘correct’ trial and visiting one arm twice or more than three consecutive entries was termed a ‘wrong’ trial. Spontaneous alternation was calculated as the percentage of the ‘correct’ trial to the total trials.
Hargreaves test:
The Hargreaves test was performed as described (Francois et al., 2017). Mice were placed in a red plastic cylinder on a glass floor maintained at 30 °C. A radiant heat beam was focused onto the hind paw (UGO Basile, model 37370). The latency to hind-paw withdrawal was recorded with at least 3 trials per animals repeated ≥ 5 min apart. Beam intensity was adjusted so that mice displayed a latency of 8–12 s. A cut-off latency of 30 s was set to avoid tissue damage.
Cold plantar assay:
The cold plantar assay was adapted from previous studies (Brenner et al., 2012). To make the cold probe, dry ice was filled into a truncated 3 ml syringe. Mice were placed in a red plastic cylinder with their hind-paw on a 1/4” thick Pyrex borosilicate glass floor. The center of the dry ice was pressed to the glass underneath the hind-paw with light pressure. The withdrawal latency was recorded with at least 3 trials per mice repeated at least 15 min interval. A cut-off latency of 20 s was set to avoid tissue damage.
Formalin test:
Mice received a single subcutaneous injection of 50 μl of 1% formalin in the plantar surface of a single hind paw delivered with a 26-G needle. Spontaneous pain behavior, characterized by increased paw flinching, licking, and elevated paw were scored in the 60 min period after the injection.
Von Frey withdrawal threshold test:
Each mouse was habituated in a small (7.5 × 7.5 × 15 cm) plastic cage for at least 20 minutes before testing. Mechanical sensitivity was determined with a series of von Frey filaments (bending forces: 0.04, 0.07, 0.16, 0.4, 0.6, 1, 1.4, 2 and 4 g) applied within the sciatic nerve territory (lateral part of the hind paw) to assess mechanical withdrawal thresholds. Filaments were applied perpendicular to the hind paw surface with sufficient force to cause a slight bending of the filament. A positive response was characterized by a rapid withdrawal of the paw away from the stimulus fiber within 4 s. The Up-Down method was used to determine the mechanical threshold (50% withdrawal threshold). The 50% threshold was calculated using the formula: 50% threshold (g) = 10(X+kd)/104, where X = the value (in log units) of the final von Frey filament, k = tabular value for the response pattern(Chaplan et al., 1994; Gonzalez-Cano et al., 2018) and d = the average increment (in log units between von Frey filaments).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were conducted with the experimenters being “blind” to the genotypes and treatment parameters. Animals in the same litter were randomly assigned to different treatment groups and blinded to experimenters. Sample sizes (n number of independent biological replicates) were first determined based on similar experiments performed and published by our lab and others in the same field(Hsu et al., 2019; Liu et al., 2018; Wang et al., 2018). To ensure reproducibility, each experiment was performed in at least three independent litters (N number), with multiple animals per litter. For non-behavioral experiments (e.g. histology), a minimum of 3 technical duplicates were analyzed and results averaged to generate a single data point for that particular animal. All samples in each group were analyzed without exclusions with one exception: if the injection site was found inaccurate during end-point histological verification, all data from that particular animal were excluded. The n/N (number of cells/number of mice) for each experiment are indicated in figure legends.
All results are presented as mean ± SEM, and statistical analysis were performed using GraphPad Prism 9 software (GraphPad Software, San Diego, CA). The distribution of data in each set of experiments was tested for normality using Shapiro-Wilk normality test. Two-tailed unpaired or paired t test (for parametric test) or Mann-Whitney U tests (for nonparametric test) was applied for two groups comparisons. For multiple groups comparisons, one-way ANOVA or two-way ANOVA (for parametric test) were applied, followed by appropriate post hoc tests (as indicated in figure legends).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal anti-parvalbumin (1:500) | Millipore Sigma | Cat#P3088, RRID: AB_477329 |
| Rabbit Polyclonal anti-c-Fos (1:500) | Millipore Sigma | Cat#ABE457, RRID: AB_2631318 |
| Mouse Monoclonal anti-CGRP (1:500) | Abcam | Cat#Ab81887, RRID: AB_1659411 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, Alexa Fluor 488 (1:200) | Abcam | Cat#Ab150113,RRID: AB_2576208 |
| Donkey anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 546 (1:200) | Thermo Fisher Scientific | Cat#A-10040, RRID: AB_2534016 |
| Oligonucleotides | ||
| Primers for Cre: Forward 5’-CACCCTGTTACGTATAGCCG-3’; Reverse 5’-GAGTCATCCTTAGCGCCGTA-3’ |
Zhong et al., 2018 | N/A |
| Primers for flox site for RARα: Forward 5’-GTGTGTGTGTGTATTCGCGTGC-3’; Reverse 5’-ACAAAGCAAGGCTTGTAGATGC-3’ |
Sarti et al., 2012 | N/A |
| Primers for EYFP: WT Forward: 5’-AAGGGAGCTGCAGTGGAGTA-3’ WT Reverse: 5’-CCGAAAATCTGTGGGAAGTC-3’; Mutant Forward: 5’-ACATGGTCCTGCTGGAGTTC-3’; Mu Reverse: 5’-GGCATTAAAGCAGCGTATCC-3’ |
The Jackson Laboratory | Primer: oIMR9020 oIMR9021 oIMR9102 oIMR9103 |
| Primers for ChR2: WT Forward: 5’-AAGGGA GCTGCAGTGGAGTA-3’; WT Reverse: 5’-CCGAAAATCTGTGGGAAGTC-3’; Mu Forward: 5’-CATTGGTGGCACTGAGATTG-3’; Mu Reverse: 5’-GAACTTCAGGGTCAGCTTGC-3’ |
The Jackson Laboratory | Primer: oIMR9020 oIMR9021 22166 oIMR9402 |
| Primers for FLpO: WT forward: 5’-GGATGCTTGCCGAAGATAAG-3’; Mu forward: 5’-CTGAGCAGCTACATCAACAGG-3’; common reverse: 5’-TGTTTCTCCAGCATTTCCAG-3’ |
The Jackson Laboratory | Primer: 17564 17566 16211 |
| qRT-PCR primer for RARα | ThermoFisher Scientific | Mm00436262_m1 |
| qRT-PCR primer for Actinβ | ThermoFisher Scientific | Mm02619580_g1 |
| qRT-PCR primer for GFAP | ThermoFisher Scientific | Mm01253033_m1 |
| qRT-PCR primer for PV | ThermoFisher Scientific | Mm00443100_m1 |
| qRT-PCR primer for SST | Integrated DNA Technologies | Mm.PT.53a.7678291 |
| qRT-PCR primer for PKCγ | Integrated DNA Technologies | Mm.PT.58.45983184 |
| qPT-PCR primer for Gad1 | Integrated DNA Technologies | Mm.PT.58.10501737 |
| qRT-PCR primer for VGluT2 | Integrated DNA Technologies | Mm.PT.58.10363705 |
| qRT-PCR primer for VGluT3 | Integrated DNA Technologies | Mm.PT.58.17676720 |
| Critical commercial assays | ||
| SuperScript III One-Step RT-PCR System with Platinum Taq High Fidelity DNA Polymerase | ThermoFisher Scientific | Cat#12574035 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat#4369016 |
| RNAscope Probe-Mm-Rara | Advanced Cell Diagnostics | Cat#463801 |
| Vectashield Hardset Antifade mounting medium with DPAI | Vector laboratories | Cat#H-1500–10 |
| Bacterial and Virus Strains | ||
| AAV-Frt-Cre-IRES-tdTomato | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formaldehyde solution | Millipore Sigma | Cat#252549, CAS: 50–00-0 |
| Complete Freund’s Adjuvant | Millipore Sigma | Cat# F5881 |
| Ro41–5253 | Abcam | Cat# Ab141629 CAS: 144092–31-9 |
| Fluorogold | Thermo Fisher Scientific | Cat#NC1363013 |
| Biocytin | Millipore Sigma | Cat#B4261 CAS: 576–19-2 |
| Streptavidin, Alexa Fluor 555 conjugate (1:1000) | Thermo Fisher Scientific | Cat#S21381 |
| CNQX | Tocris Biosciences | Cat#0190 CAS: 115066–14-3 |
| D-APV | Tocris Biosciences | Cat#0106 CAS: 79055–68-8 |
| Bicuculline | Tocris Biosciences | Cat#0131 CAS: 38641–83-7 |
| Strychine | Millipore Sigma | Cat#S0532 CAS: 57–24-9 |
| Experimental Models: Organisms/Strains | ||
| Mouse: PVcre: B6;129P2-Pvalbtm1(cre)Arbr/J | The Jackson Laboratory | JAX: 008069 RRID: IMSR_JAX:008069 |
| Mouse: SSTcre: Ssttm2.1(cre)Zjh/J | The Jackson Laboratory | JAX: 013044 RRID: IMSR_JAX:013044 |
| Mouse: RARαfl/fl: C57BL/6 | Ref (Zhong et al., 2018) | N/A |
| Mouse: Ai3(RCL-EYFP): B6.Cg-Gt(ROSA)26Sortm3(CAG-EYFP)Hze/J | The Jackson Laboratory | JAX: 007903 RRID: IMSR_JAX:007903 |
| Mouse: Ai32(RCL-ChR2(H134R)/EYFP): B6;129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J | The Jackson Laboratory | JAX: 012569 RRID: IMSR_JAX:012569 |
| Mouse: Pvalb-T2A-FlpO-D: B6.Cg-Pvalbtm4.1(flpo)Hze/J | The Jackson Laboratory | JAX: 022730 RRID: IMSR_JAX:022730 |
| Software and Algorithms | ||
| Viewer III tracking system | Biobserve | http://www.biobserve.com/behavioralresearch/products/viewer/ |
| ImageJ 1.53 | NIH | https://imagej.nih.gov/ij/ |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| OlyVIA V3.3 | Olympus | https://www.olympus-lifescience.com/en/support/downloads/ |
| Axon pCLAMP 10 | Molecular Devices | https://support.moleculardevices.com/s/article/Axon-pCLAMP-10-Electrophysiology-Data-Acquisition-Analysis-Software-Download-Page |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Spared nerve injury (SNI) induces homeostatic changes in spinal cord circuit
SNI results in reduced synaptic inhibitory outputs from dorsal horn PV+ neurons
RARα mediates SNI-induced reduction in PV+ neuron inhibition
Blocking RARα function in spinal cord blocks mechanical allodynia after SNI
ACKNOWLEDGMENTS
We thank Drs. Dong Wang, Dan Berg and Chelsie L. Brewer for technical assistances, and Drs. Kristin Arendt and Omid Miry for critical reading of the manuscript. Special thanks to Dr. David Hughes (University of Glasgow) for critical discussions of the study. The work was supported by NIH grants MH086403 (L.C.), NS11566001 (L.C.), HD104458 (L.C.), and a Stanford School of Medicine Dean’s postdoctoral fellowship (B.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures, which can be found with this article online at <URL>.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraira VE, Kuehn ED, Chirila AM, Springel MW, Toliver AA, Zimmerman AL, Orefice LL, Boyle KA, Bai L, Song BJ, et al. (2017). The Cellular and Synaptic Architecture of the Mechanosensory Dorsal Horn. Cell 168, 295–310 e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, and Chen L (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Kaang BK, and Zhuo M (2016). Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nature reviews Neuroscience 17, 485–496. [DOI] [PubMed] [Google Scholar]
- Blom SM, Pfister JP, Santello M, Senn W, and Nevian T (2014). Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 5754–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, Cunin G, Fermanian J, Ginies P, Grun-Overdyking A, et al. (2005). Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 114, 29–36. [DOI] [PubMed] [Google Scholar]
- Boyle KA, Gradwell MA, Yasaka T, Dickie LC, Polgar E, Ganley RP, Orr DPH, Watanabe M, Abraira VE, Kuehn ED, et al. (2019). Defining a Spinal Microcircuit that Gates Myelinated Afferent Input: Implications for Tactile Allodynia. Cell Rep 28, 526–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz J, Solorzano C, Wang X, and Basbaum AI (2014). Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 82, 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, and Gereau R.W.t. (2012). A novel behavioral assay for measuring cold sensation in mice. Plos One 7, e39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Lopes JM, Malcangio M, Pan BH, and Bowery NG (1995). Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Res 679, 289–297. [DOI] [PubMed] [Google Scholar]
- Cetin A, Komai S, Eliava M, Seeburg PH, and Osten P (2006). Stereotaxic gene delivery in the rodent brain. Nat Protoc 1, 3166–3173. [DOI] [PubMed] [Google Scholar]
- Chapellier B, Mark M, Garnier JM, LeMeur M, Chambon P, and Ghyselinck NB (2002). A conditional floxed (loxP-flanked) allele for the retinoic acid receptor alpha (RARalpha) gene. Genesis 32, 87–90. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Chen G, Zhang YQ, Qadri YJ, Serhan CN, and Ji RR (2018). Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 100, 1292–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Duan B, Huang T, Zhang Y, Chen Y, Britz O, Garcia-Campmany L, Ren X, Vong L, Lowell BB, et al. (2017). Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat Neurosci 20, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Hachisuka J, Brett MA, Magee AR, Omori Y, Iqbal NU, Zhang D, DeLisle MM, Wolfson RL, Bai L, et al. (2020). Parallel ascending spinal pathways for affective touch and pain. Nature 587, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, Blanck TJJ, Gan WB, and Yang G (2017). Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat Neurosci 20, 1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, and Somogyi P (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Halasy K, Vida I, Nyiri G, Tamas G, Buhl EH, and Somogyi P (1997). Synaptic effects of identified interneurons innervating both interneurons and pyramidal cells in the rat hippocampus. Neuroscience 79, 629–648. [DOI] [PubMed] [Google Scholar]
- Comitato A, and Bardoni R (2021). Presynaptic Inhibition of Pain and Touch in the Spinal Cord: From Receptors to Circuits. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, and De Koninck Y (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942. [DOI] [PubMed] [Google Scholar]
- Decosterd I, and Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. [DOI] [PubMed] [Google Scholar]
- Denk F, Crow M, Didangelos A, Lopes DM, and McMahon SB (2016). Persistent Alterations in Microglial Enhancers in a Model of Chronic Pain. Cell Rep 15, 1771–1781. [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng LZ, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren XY, et al. (2014). Identification of Spinal Circuits Transmitting and Gating Mechanical Pain. Cell 159, 1417–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton MJ, Martinez MA, and Karmally S (1999). A single intrathecal injection of GABA permanently reverses neuropathic pain after nerve injury. Brain Res 835, 334–339. [DOI] [PubMed] [Google Scholar]
- Eggermont JJ, and Roberts LE (2004). The neuroscience of tinnitus. Trends Neurosci 27, 676–682. [DOI] [PubMed] [Google Scholar]
- Fink AJ, Croce KR, Huang ZJ, Abbott LF, Jessell TM, and Azim E (2014). Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Kuner R, and Jensen TS (2021). Neuropathic Pain: From Mechanisms to Treatment. Physiol Rev 101, 259–301. [DOI] [PubMed] [Google Scholar]
- Flor H, Elbert T, Knecht S, Wienbruch C, Pantev C, Birbaumer N, Larbig W, and Taub E (1995). Phantom-limb pain as a perceptual correlate of cortical reorganization following arm amputation. Nature 375, 482–484. [DOI] [PubMed] [Google Scholar]
- Francois A, Low SA, Sypek EI, Christensen AJ, Sotoudeh C, Beier KT, Ramakrishnan C, Ritola KD, Sharif-Naeini R, Deisseroth K, et al. (2017). A Brainstem-Spinal Cord Inhibitory Circuit for Mechanical Pain Modulation by GABA and Enkephalins. Neuron 93, 822–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Tokunaga A, Kondo E, Miki K, Tachibana T, and Noguchi K (1998). Change in mRNAs for neuropeptides and the GABA(A) receptor in dorsal root ganglion neurons in a rat experimental neuropathic pain model. Pain 78, 13–26. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cano R, Boivin B, Bullock D, Cornelissen L, Andrews N, and Costigan M (2018). Up-Down Reader: An Open Source Program for Efficiently Processing 50% von Frey Thresholds. Frontiers in Pharmacology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwell MA, Boyle KA, Browne TJ, Bell AM, Leonardo J, Peralta Reyes FS, Dickie AC, Smith KM, Callister RJ, Dayas CV, et al. (2021). Diversity of inhibitory and excitatory parvalbumin interneuron circuits in the dorsal horn. Pain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwell MA, Callister RJ, and Graham BA (2020). Reviewing the case for compromised spinal inhibition in neuropathic pain. Journal of Neural Transmission 127, 481–503. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, et al. (2016). Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler A, and Ginty DD (2021). The mechanosensory neurons of touch and their mechanisms of activation. Nat Rev Neurosci 22, 521–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Li J, Wu D, Sudhof TC, and Chen L (2019). Synaptic retinoic acid receptor signaling mediates mTOR-dependent metaplasticity that controls hippocampal learning. Proc Natl Acad Sci U S A 116, 7113–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, van Dam AM, Wang Y, Lucassen PJ, and Zhou JN (2020). Retinoic acid and depressive disorders: Evidence and possible neurobiological mechanisms. Neuroscience and Biobehavioral Reviews 112, 376–391. [DOI] [PubMed] [Google Scholar]
- Hughes DI, Sikander S, Kinnon CM, Boyle KA, Watanabe M, Callister RJ, and Graham BA (2012). Morphological, neurochemical and electrophysiological features of parvalbumin-expressing cells: a likely source of axo-axonic inputs in the mouse spinal dorsal horn. Journal of Physiology-London 590, 3927–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, and Todd AJ (2020). Central Nervous System Targets: Inhibitory Interneurons in the Spinal Cord. Neurotherapeutics 17, 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibuki T, Hama AT, Wang XT, Pappas GD, and Sagen J (1997). Loss of GABA-immunoreactivity in the spinal dorsal horn of rats with peripheral nerve injury and promotion of recovery by adrenal medullary grafts. Neuroscience 76, 845–858. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, and Sandkuhler J (2003). Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 299, 1237–1240. [DOI] [PubMed] [Google Scholar]
- Juarez-Salinas DL, Braz JM, Etlin A, Gee S, Sohal V, and Basbaum AI (2019). GABAergic cell transplants in the anterior cingulate cortex reduce neuropathic pain aversiveness. Brain : a journal of neurology 142, 2655–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Q, Li R, Cai L, Wu SD, and Li CM (2019). Ro41–5253, a selective antagonist of retinoic acid receptor alpha, ameliorates chronic unpredictable mild stress-induced depressive-like behaviors in rats: Involvement of regulating HPA axis and improving hippocampal neuronal deficits. Brain Research Bulletin 146, 302–309. [DOI] [PubMed] [Google Scholar]
- Keller AF, Beggs S, Salter MW, and De Koninck Y (2007). Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Molecular pain 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Kim SK, and Nabekura J (2017). Functional and structural plasticity in the primary somatosensory cortex associated with chronic pain. J Neurochem 141, 499–506. [DOI] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, Brockhaus J, Sergejeva M, Hess A, Brune K, et al. (2008). Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451, 330–334. [DOI] [PubMed] [Google Scholar]
- Koch SC, Acton D, and Goulding M (2018). Spinal Circuits for Touch, Pain, and Itch. Annu Rev Physiol 80, 189–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann CJ, Pertin M, Suter MR, and Decosterd I (2014). Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol Pain 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, and Woolf CJ (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain 10, 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever I, Cunningham J, Grist J, Yip PK, and Malcangio M (2003). Release of BDNF and GABA in the dorsal horn of neuropathic rats. Eur J Neurosci 18, 1169–1174. [DOI] [PubMed] [Google Scholar]
- Lewis DA, and Lund JS (1990). Heterogeneity of chandelier neurons in monkey neocortex: corticotropin-releasing factor- and parvalbumin-immunoreactive populations. The Journal of comparative neurology 293, 599–615. [DOI] [PubMed] [Google Scholar]
- Li J, Jiang RY, Arendt KL, Hsu YT, Zhai SR, and Chen L (2020). Defective memory engram reactivation underlies impaired fear memory recall in Fragile X syndrome. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, et al. (2010). Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 330, 1400–1404. [DOI] [PubMed] [Google Scholar]
- Liu Y, Latremoliere A, Li X, Zhang Z, Chen M, Wang X, Fang C, Zhu J, Alexandre C, Gao Z, et al. (2018). Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loken LS, Braz JM, Etlin A, Sadeghi M, Bernstein M, Jewell M, Steyert M, Kuhn J, Hamel K, Llewellyn-Smith IJ, et al. (2021). Contribution of dorsal horn CGRP-expressing interneurons to mechanical sensitivity. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Dong H, Gao Y, Gong Y, Ren Y, Gu N, Zhou S, Xia N, Sun YY, Ji RR, et al. (2013). A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J Clin Invest 123, 4050–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, and Turrigiano GG (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, and Chen L (2008). Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci U S A 105, 16015–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Chen C, Tonegawa S, and Basbaum AI (1997). Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science 278, 279–283. [DOI] [PubMed] [Google Scholar]
- Mason P (2009). Descending Modulation of Nociception. In Encyclopedia of Neuroscience, Binder MD, Hirokawa N, and Windhorst U, eds. (Berlin, Heidelberg: Springer Berlin Heidelberg; ), pp. 944–946. [Google Scholar]
- Mecca CM, Chao D, Yu G, Feng Y, Segel I, Zhang Z, Rodriguez-Garcia DM, Pawela CP, Hillard CJ, Hogan QH, et al. (2021). Dynamic Change of Endocannabinoid Signaling in the Medial Prefrontal Cortex Controls the Development of Depression After Neuropathic Pain. J Neurosci 41, 7492–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer Lindsay N, Chen C, Gilam G, Mackey S, and Scherrer G (2021). Brain circuits for pain and its treatment. Science translational medicine 13, eabj7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Toth K, Gulyas AI, Hajos N, and Freund TF (1996). Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16, 815–823. [DOI] [PubMed] [Google Scholar]
- Miska NJ, Richter LM, Cary BA, Gjorgjieva J, and Turrigiano GG (2018). Sensory experience inversely regulates feedforward and feedback excitation-inhibition ratio in rodent visual cortex. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring F, Halder P, Seal RP, and Stucky CL (2018). Uncovering the Cells and Circuits of Touch in Normal and Pathological Settings. Neuron 100, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller AR (2007). Tinnitus and pain. Prog Brain Res 166, 47–53. [DOI] [PubMed] [Google Scholar]
- Monday HR, Younts TJ, and Castillo PE (2018). Long-Term Plasticity of Neurotransmitter Release: Emerging Mechanisms and Contributions to Brain Function and Disease. Annual Review of Neuroscience, Vol 41 41, 299–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann E, Kupfer L, and Zeilhofer HU (2021). The alpha2/alpha3GABAA receptor modulator TPA023B alleviates not only the sensory but also the tonic affective component of chronic pain in mice. Pain 162, 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Braz JM, Skinner K, Llewellyn-Smith IJ, and Basbaum AI (2008). Innocuous, not noxious, input activates PKCgamma interneurons of the spinal dorsal horn via myelinated afferent fibers. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 7936–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Ding L, Li JJ, Kim H, Liu J, Li H, Moberly A, Badea TC, Duncan ID, Son YJ, et al. (2013). Modality-based organization of ascending somatosensory axons in the direct dorsal column pathway. J Neurosci 33, 17691–17709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Morimura K, and Porreca F (2014). Descending pain modulation and chronification of pain. Current Opinion in Supportive and Palliative Care 8, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Tjia M, Zuo Y, and Chen L (2018). Postnatal Ablation of Synaptic Retinoic Acid Signaling Impairs Cortical Information Processing and Sensory Discrimination in Mice. J Neurosci 38, 5277–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelzik H, Hughes DI, and Thomson AM (2002). Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. The Journal of comparative neurology 443, 346–367. [DOI] [PubMed] [Google Scholar]
- Peirs C, Dallel R, and Todd AJ (2020). Recent advances in our understanding of the organization of dorsal horn neuron populations and their contribution to cutaneous mechanical allodynia. Journal of neural transmission 127, 505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, and Seal RP (2016). Neural circuits for pain: Recent advances and current views. Science 354, 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Williams SPG, Zhao XY, Arokiaraj CM, Ferreira DW, Noh MC, Smith KM, Halder P, Corrigan KA, Gedeon JY, et al. (2021). Mechanical Allodynia Circuitry in the Dorsal Horn Is Defined by the Nature of the Injury. Neuron 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Williams SPG, Zhao XY, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, et al. (2015). Dorsal Horn Circuits for Persistent Mechanical Pain. Neuron 87, 797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Gu N, Zhou L, U, B.E., Murugan M, Gan WB, and Wu LJ (2016). Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun 7, 12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean H, Pawlowski SA, Fraine SL, Sharif B, Hamad D, Fatima T, Berg J, Brown CM, Jan LY, Ribeiro-da-Silva A, et al. (2015). Dorsal Horn Parvalbumin Neurons Are Gate-Keepers of Touch-Evoked Pain after Nerve Injury. Cell Rep 13, 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar E, Gray S, Riddell JS, and Todd AJ (2004). Lack of evidence for significant neuronal loss in laminae I-III of the spinal dorsal horn of the rat in the chronic constriction injury model. Pain 111, 144–150. [DOI] [PubMed] [Google Scholar]
- Poon MM, and Chen L (2008). Retinoic acid-gated sequence-specific translational control by RARalpha. Proc Natl Acad Sci U S A 105, 20303–20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedemann T (2019). Diversity and Function of Somatostatin-Expressing Interneurons in the Cerebral Cortex. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudomin P (2009). In search of lost presynaptic inhibition. Exp Brain Res 196, 139–151. [DOI] [PubMed] [Google Scholar]
- Santello M, and Nevian T (2015). Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron 86, 233–246. [DOI] [PubMed] [Google Scholar]
- Sarti F, Zhang Z, Schroeder J, and Chen L (2013). Rapid suppression of inhibitory synaptic transmission by retinoic acid. J Neurosci 33, 11440–11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, and Woolf CJ (2005). Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 25, 7317–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, and Edwards RH (2009). Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature 462, 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Eckert WA 3rd, and Basbaum AI (2003a). Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain 4, 465–470. [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, and Basbaum AI (2003b). Spared nerve injury model of neuropathic pain in the mouse: A behavioral and anatomic analysis. J Pain 4, 465–470. [DOI] [PubMed] [Google Scholar]
- Stern J, Jeanmonod D, and Sarnthein J (2006). Persistent EEG overactivation in the cortical pain matrix of neurogenic pain patients. Neuroimage 31, 721–731. [DOI] [PubMed] [Google Scholar]
- Swett JE, and Woolf CJ (1985). The somatotopic organization of primary afferent terminals in the superficial laminae of the dorsal horn of the rat spinal cord. J Comp Neurol 231, 66–77. [DOI] [PubMed] [Google Scholar]
- Takazawa T, and MacDermott AB (2010). Synaptic pathways and inhibitory gates in the spinal cord dorsal horn. Ann N Y Acad Sci 1198, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ (2010). Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ (2017). Identifying functional populations among the interneurons in laminae I-III of the spinal dorsal horn. Molecular pain 13, 1744806917693003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Brodbelt AR, Price RF, and Shehab SA (1994). Some inhibitory neurons in the spinal cord develop c-fos-immunoreactivity after noxious stimulation. Neuroscience 63, 805–816. [DOI] [PubMed] [Google Scholar]
- Torsney C, and MacDermott AB (2006). Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci 26, 1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Koga K, Chen T, and Zhuo M (2017). Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J Neurochem 141, 486–498. [DOI] [PubMed] [Google Scholar]
- Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, and Gasull X (2011). TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Molecular pain 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor perspectives in biology 4, a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, and Woolf CJ (2012). Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Tawfik VL, Corder G, Low SA, Francois A, Basbaum AI, and Scherrer G (2018). Functional Divergence of Delta and Mu Opioid Receptor Organization in CNS Pain Circuits. Neuron 98, 90–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, and Zhuo M (2001). Potentiation of sensory responses in the anterior cingulate cortex following digit amputation in the anaesthetised rat. J Physiol 532, 823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, and Fitzgerald M (1986). Somatotopic organization of cutaneous afferent terminals and dorsal horn neuronal receptive fields in the superficial and deep laminae of the rat lumbar spinal cord. J Comp Neurol 251, 517–531. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Shortland P, and Coggeshall RE (1992). Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 355, 75–78. [DOI] [PubMed] [Google Scholar]
- Yaksh TL (1989). Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 37, 111–123. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, and Yaksh TL (1993). Effects of intrathecal strychnine and bicuculline on nerve compression-induced thermal hyperalgesia and selective antagonism by MK-801. Pain 54, 79–84. [DOI] [PubMed] [Google Scholar]
- Yee AX, and Chen L (2016). Differential regulation of spontaneous and evoked inhibitory synaptic transmission in somatosensory cortex by retinoic acid. Synapse 70, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee AX, Hsu YT, and Chen L (2017). A metaplasticity view of the interaction between homeostatic and Hebbian plasticity. Philosophical transactions of the Royal Society of London Series B, Biological sciences 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Zhang YQ, Goulding M, Wang YQ, and Ma Q (2018). Timing Mechanisms Underlying Gate Control by Feedforward Inhibition. Neuron 99, 941–955 e944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Zhou H, Huang L, Xie Z, Wang J, Gan WB, and Yang G (2018). Neuropathic Pain Causes Pyramidal Neuronal Hyperactivity in the Anterior Cingulate Cortex. Front Cell Neurosci 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zholudeva LV, Abraira VE, Satkunendrarajah K, McDevitt TC, Goulding MD, Magnuson DSK, and Lane MA (2021). Spinal Interneurons as Gatekeepers to Neuroplasticity after Injury or Disease. J Neurosci 41, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong LR, Chen X, Park E, Sudhof TC, and Chen L (2018). Retinoic Acid Receptor RARalpha-Dependent Synaptic Signaling Mediates Homeostatic Synaptic Plasticity at the Inhibitory Synapses of Mouse Visual Cortex. J Neurosci 38, 10454–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, and Muzyczka N (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6, 973–985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primary data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.