

Abstract
Acute respiratory infection by influenza virus is a persistent and pervasive public health problem. Antiviral innate immunity initiated by type I interferon (IFN) is the first responder to pathogen invasion and provides the first line of defense. We discovered that Axin1, a scaffold protein, was reduced during influenza virus infection. We also found that overexpression of Axin1 and the chemical stabilizer of Axin1, XAV939, reduced influenza virus replication in lung epithelial cells. This effect was also observed with respiratory syncytial virus and vesicular stomatitis virus. Axin1 boosted type I IFN response to influenza virus infection and activated JNK/c-Jun and Smad3 signaling. XAV939 protected mice from influenza virus infection. Thus, our studies provide new mechanistic insights into the regulation of the type I IFN response and present a new potential therapeutic of targeting Axin1 against influenza virus infection.
Keywords: Axin1, influenza virus, interferon, JNK/c-Jun, Smad3
Graphical Abstract

In this study, we have discovered that the host protein Axin1 boosts host immunity in response to influenza virus infection. The finding presents a new potential therapeutic of targeting Axin1 against influenza virus infection.
Introduction
Influenza virus is a member of the Orthomyxoviridae family and is classified into three serotypes: A, B and C according to their internal protein sequences 1. With its global pandemic potential and up to 500,000 annual deaths worldwide during seasonal epidemics, influenza virus is a major public health concern and imposes an enormous economic burden 2. Prevention relying on vaccination has several limitations, including the lag time for vaccine manufacture and the low coverage rate. Considering the increasing level of viral resistance to current anti-influenza drugs targeting the neuraminidase (NA) or M2 channels 3, it is particularly important to develop novel broad spectrum antiviral drugs.
Interferon (IFN) was discovered in 1957 as an agent that inhibits replication of influenza 4. The IFN family of cytokines is the most potent vertebrate-derived signal for mobilizing antimicrobial effector functions against intracellular pathogens 5. Three classes of IFN have been identified and classified according to the receptor complex through which they signal 6. Type I IFNs (IFNβ, 14 IFNα’s, IFNδ, IFNε, IFNκ, IFNo and IFNτ), best known for their antiviral properties, mediate the induction of both the innate immune response and subsequent adaptive immunity to viruses. Type II IFN (IFNγ) stimulates broad immune responses to various pathogens other than viruses. Type III IFNs (3 IFNλs) are also known to regulate antiviral responses and may be ancestral type I IFNs. It is widely accepted that viral attachment and viral dsRNA intermediates accumulating during virus replication are the primary mediators triggering IFN production, ultimately resulting in the expression of thousands of IFN-stimulated genes (ISGs) (OAS1, MX1, and etc.) that limits virus replication 7.
In the early phase of infection, Toll-like receptors, cytosolic RIG-1-like receptors (RIG-1 and MDA5), NOD-like receptors and C-type lectin receptors are major players involved in innate recognition of influenza virus 8. The novel IFN-regulated viral RNA sensor interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) was also identified to have antiviral properties 9. Activation of type I IFN expression by these pattern recognition receptors (PRRs) is tightly controlled by several transcription factors including c-Jun/ATF2 (AP1), interferon regulatory factor 3/7 (IRF3/7), and p50/p65 (NF-κB) 10. Smad3, as a transcription factor, also enhances type I IFN expression by cooperating with IRF7 11. In the later phase of infection, the secreted type I IFN stimulates type I IFN receptor (IFNAR1/2) in an autocrine and paracrine fashion, which leads to the activation of the Janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathway, and finally establishes a cellular antiviral status 12.
Axin, which was identified from analysis of the mouse-Fused locus, is a negative regulator of Axis formation in the development of mouse embryos 13. Axin protein, present in two isoforms (Axin1 and Axin2), acts as an architectural platform for the degradation of β-catenin 14,15. Axin1 has, in fact, emerged as a multidomain scaffolding protein for many other signaling pathways, including the c-Jun-NH2-kinase (JNK) mitogen-activated protein kinase (MAPK) signaling pathway, the p53 signaling pathway, and the transforming growth factor β (TGF-β) signaling pathway 16. Axin1 forms a complex with MEKK1/4 and mediates JNK/c-Jun activation through MKK4/7 17. Axin1 also promotes Smad3 phosphorylation in response to TGF-β 18, and downregulates the negative factor, Smad7, in TGF-β signaling 19. By forming a ternary complex, Axin1 stimulates p53 functions via activation of homeodomain-interacting protein kinase-2 (HIPK2) kinase 20. These intriguing roles of Axin1 in β-catenin-independent signaling open the door to its function in multiple physiological and pathological processes.
In infectious diseases, Axin1 prevents bacterial Salmonella tissue invasion and modulates inflammatory responses during the infection 21. Silencing of Axin1 up-regulates human immunodeficiency virus type I (HIV-1) gene expression and viral replication 22. The compound XAV939, a stabilizer of Axin1, was shown to specifically inhibit poly(ADP-ribose) polymerase tankyrase1/2 (TNK1/2), which induces poly(ADP-ribosyl)ation of Axin1 and in term promotes its proteasome-mediated degradation 23.
In this study, we first discovered that Axin1 was reduced in a mouse model of influenza virus-associated pneumonia. We then demonstrated that Axin1 inhibited replication of influenza virus and other viruses by boosting IFN responses through activation of the JNK/c-Jun and Smad3 signaling pathways. XAV939, a Axin1 stabilizer, protected mice from lethal influenza virus challenge. Our studies, for the first time, demonstrate the role of Axin1, a scaffold protein, in the antiviral network of IFNs.
Materials and Methods
Ethics Statement
The animal studies were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol (VM 15-38) was reviewed and approved by the Institutional Animal Care and Use Committee at Oklahoma State University. All animal studies were performed in a Biosafety Level 2 facility located at Oklahoma State University according to the approved protocol 13-37 & 13-38 following the American Veterinary Medicine Association guidelines on euthanasia. Mice were euthanized if more than 30% of weight was lost. No survival surgery was performed, and all efforts were made to minimize the animal suffering.
Viruses
Influenza virus H1N1 strain A/Puerto Rico/8/1934 (PR/8) was obtained from American Type Culture Collection (ATCC, Manassas, VA). Influenza PR/8-Gaussia luciferase (GLuc) virus 24, and vesicular stomatitis virus with firefly luciferase gene (VSV-Luc) 25 were kind gifts from Dr. Peter Palese (Icahn School of Medicine at Mount Sinai, New York, NY), and from Dr. Sean Whelan (Harvard Medical School, Boston, MA), respectively. GFP-labeled respiratory syncytial virus (RSV) were generated as previously described 26.
Mouse model of influenza viral pneumonia
Male C57BL/6N mice (6-8 weeks of age) were anesthetized with intraperitoneal injection of ketamine (80 mg/kg body weight) and xylazine (10 mg/kg body weight), and inoculated with PR/8 (250 pfu/mouse) intranasally. Mice were weighed daily for the evaluation of loss of body weight and observed for clinical signs such as ruffled fur and respiratory distress. Animals were sacrificed on 0-7 days post infections (dpi). Unlavaged lungs were homogenized in liquid nitrogen by mortar and pestle, aliquoted and stored at −80°C prior to use.
Bronchoalveolar lavages (BAL) analysis
Lungs were lavaged with 1 ml of normal saline three times. BAL fluids were centrifuged at 380 g for 10 min at 4°C. BAL cells were resuspended in normal saline and total cells were counted using a hemocytometer. For differential cell counts, cytospinned cells on the glass slides were stained with Wright-Giemsa.
Histopathology
Unlavaged lungs were instilled with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 20 cm H2O pressure and fixed for 72 h, then embedded in paraffin and sectioned at 4 μm thickness. The sections were stained with hematoxylin and eosin and examined under a light microscope.
Survival study of lethal dose influenza virus infection
Male C57BL/6N mice (6-8 week old) were challenged intranasally with a lethal dose (1,000 pfu) of influenza virus A/PR/8/34 H1N1 under anesthesia. XAV939 (SIGMA, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) and given to the mice orally using metal oral gavage in a dose of 50 mg/kg mixed with 1% methylcellulose (50 μl/mouse) every day, beginning at 1 day before infection (−1 dpi) and continued for 2 or 4 days post infection. Control animals were given vehicle alone in the same formulation. All animals were observed daily for body weight loss and for clinical signs such as ruffled fur, inactivity and difficulty in breathing until 21 days post infection. Animals that showed severe respiratory distress or lost more than 30% body weight were sacrificed.
Cell culture
Human embryonic kidney 293 (HEK293), human lung epithelial A549, and Madin-Darby canine kidney (MDCK) cells (all ATCC, Manassas, VA) were maintained in Earle's Minimal Essential Medium (EMEM) supplemented with 10% fetal bovine serum (FBS). Human epithelial type 2 (HEp2) cells (ATCC, Manassas, VA) were maintained in EMEM supplemented with 10% FBS, glutamine, and non-essential amino acids. Human bronchial/tracheal epithelial cells (HBTEC, FC-0035) were from Lifeline Cell Technology (Frederick, MD) and maintained in BronchiaLife complete medium (LM-0023).
Plasmids and transfection
pCS2-based vectors expressing GFP (15681), Axin1 (21287) and Axin2 (21279) plasmids were obtained from Addgene (Cambridge, MA). All three plasmids had myc-tag. Smad3 signaling and ISRE-luc reporter plasmids were purchased from bpsBioscience (# 60613, San Diego, CA). pRL-TK Renilla luciferase plasmid was obtained from Promega (Madison, WI). HEK293 cells were cultured on 96-well tissue culture plates (for dual luciferase assay) and 12-well tissue culture plates (for all other experiments) until 90% confluent, and then transfected with the appropriate plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Unless otherwise indicated, the plasmid amount used for 12-well plates is 2.5 μg/well for Axin1, Axin2 or GFP and the plasmid amount used for 96-well plates is 100 ng/well for Axin1, Axin2 or GFP, 30 ng/well for the ISRE reporter, 20 ng/well for Smad reporter and 2.5 ng/ml for pRL-TK.
In vitro influenza virus infection
Cells were washed with serum-free complete DMEM and infected with influenza virus at various multiplicity of infection (MOI) in serum-free complete DMEM supplemented with 1 μg/ml L-1-tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (SIGMA, St. Louis, MO) for 1 h. Fresh serum-free complete medium was added, and cells were cultured for an additional 2 to 48 h.
In vitro RSV infection
HEK293 and Hep2 cells were infected with GFP-labeled RSV at a MOI of 0.1 or 1 in complete DMEM for 1 h, as previously described 26. Then cells were changed into fresh complete medium and cultured for an additional 12 to 48 h.
Quantitative real-time PCR
Total RNA was extracted from cells or lung tissues using TRI-Reagent (Molecular Research Center, Cincinnati, OH) and digested with TURBO DNase (Ambion, Austin, TX, USA) to remove the genomic DNA contamination. One μg of RNA was reverse-transcribed into cDNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, CA), random primers, and oligo dT (Promega, Madison, WI). Real-time PCR was carried out on a 7900HT Fast Real-Time PCR System using SYBR Green I detection Master Mix (Eurogentec, Fremont, CA,) as previously described 27. The primers were designed using Primer Express® software (Applied Biosystems, Foster City, CA), and are listed in Table 1.
Table 1.
PCR primer sequences
| Gene | Species | Forward primers | Reverse primers |
|---|---|---|---|
| 18S | Mouse | ATTGCTCAATCTCGGGTGGCTG | CGTTCTTAGTTGGTGGAGCGATTTG |
| IFNβ1 | Mouse | CAGCTCCAAGAAAGGACGAAC | GGCAGTGTAACTCTTCTGCAT |
| MX1 | Mouse | GAAGGCAAGGTCTTGGATG | GCTGACCTCTGCACTTGACT |
| β-actin | Human | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
| IFNβ1 | Human | ATGACCAACAAGTGTCTCCTCC | GGAATCCAAGCAAGTTGTAGCTC |
| OAS1 | Human | TGTCCAAGGTGGTAAAGGGTG | CCGGCGATTTAACTGATCCTG |
| HA | H1N1 influenza | GGCCCAACCACAACACAAAC | AGCCCTCCTTCTCCGTCAGC |
| NP | H1N1 influenza | TGTGTATGGACCTGCCGTAGC | CCATCCACACCAGTTGACTCTTG |
| M1 | H1N1 influenza | CTTCTAACCGAGGTCGAAACGTA | GGTGACAGGATTGGTCTTGTCTTTA |
Western blot
The cells and homogenized lung tissue were lysed in M-PER Mammalian Protein Extraction Reagent containing 1% Halt Protease and Phosphatase Inhibitor Cocktail (Pierce, Rockford, IL) by Dounce homogenizer, followed by sonication and 3 freeze/thaw cycles. The proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. The blots were blocked for 1 h at room temperature with 5% dried milk in Tris-buffered saline TBS-T (10 mM Tris/HCl, 100 mM NaCl and 0.05% Tween, pH 7.5) and incubated overnight at 4°C with anti-Axin1 (1:1000, #20875, Cell Signaling Technology, Danvers, MA), anti-p-JNK (1:1000, #46685, Cell Signaling Technology, Danvers, MA), anti-JNK (1:1000, #92525, Cell Signaling Technology, Danvers, MA), anti-p-STAT1 (Tyr 701) (1:1000, Cell Signaling Technology, Danvers, MA), anti-STAT1 (1:1000, Cell Signaling Technology, Danvers, MA), anti-p-c-Jun (1:1000, #23615, Cell Signaling Technology, Danvers, MA), anti-c-Jun (1:1000, #sc-1694, Santa Cruz Biotechnology, Santa Cruz, CA), anti-p-ATF2 (1:1000, #5112p, Cell Signaling Technology, Danvers, MA), anti-ATF2 (1:1000, #350351, Cell Signaling Technology, Danvers, MA), anti-c-myc (1:2000, DHSB, Iowa City, IO), anti-RSV-M protein serum (1:50, Genscript, Piscataway, NJ), anti-RSV-G protein (1:200, Edward Walsh, University of Rochester School of Medicine, Rochester, NY), anti-NP (1:20, HB-65, ATCC), anti-NS1 (1:1000, #sc-130568, Santa Cruz), anti-β-actin (1:2000, SIGMA, St. Louis, MO) and anti-GAPDH (1:4000, #ab181602, Abcom, Boston, MA) antibodies. The blots were then rinsed in TBS-T, and incubated for 1 h at room temperature with goat anti-rabbit, or goat anti-mouse secondary antibodies, coupled to horseradish peroxidase (1:2000, Jackson Immunoresearch, West Grove, PA). After being washed, the blots were developed by SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). The densities of bands were quantified by ImageJ software (http://rsb.info.nih.gov/ij/).
TCID50 assay
MDCK cells in 96-well plates were infected with serial dilutions (10−1 to 10−7) of virus samples in serum-free complete EMEM containing 1 μg/ml TPCK-treated trypsin for 1 h. The cells were cultured with the same medium for 4 days. TCID50 values (median tissue culture infective dose) were calculated by the method of Reed and Muench 28.
Immunofluorescence
Human bronchial/tracheal epithelial cells were cultured on 96-well tissue culture plates. At the end of the experiment, the cells were briefly washed with ice-cold PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. After being washed with PBS again, the cells were permeabilized with 0.3% Triton X-100 for 10 min and blocked with 10% FBS for 1 h at room temperature. After being rinsed, the cells were incubated overnight with anti-NP antibodies (1:20, HB-65, ATCC). Subsequently, cells were washed with PBS and incubated with Alexa 546-conjugated goat anti-mouse IgG (1:200, Invitrogen, Carlsbad, CA) for 1 h. The nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI, 1:1000, Invitrogen, Carlsbad, CA) for 2 min. Images were acquired using a Nikon Eclipse TE-2000 inverted fluorescence microscope.
Statistics analysis
The results were analyzed by one-way ANOVA followed with posthoc Tukey’s test for multiple comparisons of control and treatment groups, or Student’s t-test using GraphPad Prism (version 6). Survival rate between groups was analyzed by Mantel-Cox χ2 test on Kaplan-Meier probability estimates using GraphPad Prism. All results were reported as mean ± SD.
Results
Axin1 is reduced during influenza viral pneumonia
We used a mouse model of viral pneumonia caused by a sub-lethal dose of H1N1 influenza virus infection 29. Histological analysis showed massive infiltration of immune cells (Fig. 1A). The number of neutrophils in BAL significantly increased at Day 3 and persisted until Day 7 (Fig. 1B). While macrophage numbers were increased from Day 5, infiltration of lymphocytes started on Day 6, thus representing the transition from innate immunity to adaptive immunity. To assess viral replication, we measured viral gene expression by measuring hemagglutinin (HA) and nucleoprotein (NP). The mRNA expression of the viral genes reached a peak at Day 5 (Fig. 1C), suggesting that the viral load reached maximum at Day 5. The IFN response was initiated from Day 2 as indicated by an increase in mRNA expression of IFNβ1 and MX1 (Fig. 1D). As a potential antiviral host factor, the expression of Axin1 protein in whole lung tissue was significantly reduced at Day 3 and Day 7 (Fig. 1E). The reduction in Axin1 protein levels was confirmed in the PR/8-infected human bronchial/tracheal epithelial cells (Fig. 1F). The protein levels of Axin1, but not Axin2 were also significantly reduced in THP1-derived human macrophages (Fig. 1G). The results suggest that Axin1 protein is reduced in the lungs at an early stage of influenza viral pneumonia.
Figure 1. Axin1 is reduced during influenza pneumonia.
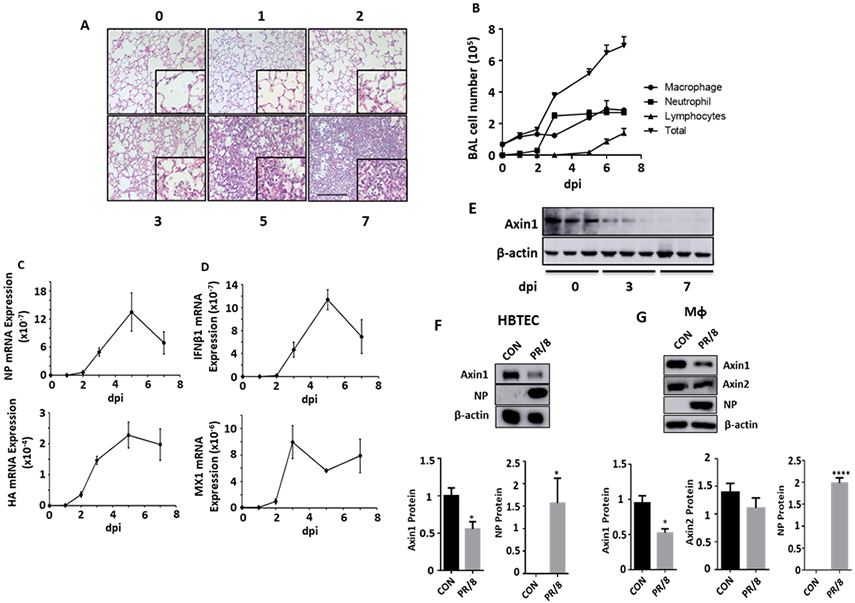
Mice were intranasally inoculated with H1N1 influenza A/PR/8/34 virus (250 pfu/mouse). The lung tissue and bronchoalveolar lavage (BAL) were collected at 1 to 7 days post infection (dpi). (A) H&E staining of paraffin sections of lungs. Scale bar = 100 μm. (B) Differential immune cell counts in BAL. n=3. (C, D) Relative mRNA expression of viral genes (NP and HA) and IFNβ1 and MX1 in whole lung tissues were measured by real-time PCR and normalized to 18S rRNA. n=3. (E) Axin1 protein levels in the lung tissues were measured by Western blot. (F, G) Human bronchial/tracheal epithelial cells (HBTEC) and THP1-derived macrophages (MФ) were infected with H1N1 influenza A/PR/8/34 virus at an MOI of 0.1 for 24 h. Axin1, Axin2, viral NP and β-actin were determined by Western blot. Axin1 and Axin2 protein levels were quantitated to β-actin. Control: CON. n=3. *p < 0.05, ****p < 0.0001 vs CON.
Axin1 inhibits influenza virus replication
To evaluate the potential role of Axin1 in regulating influenza virus replication, we overexpressed Axin1 or Axin2 in HEK293 cells prior to virus infection. Overexpression of Axin1 or Axin2 had no effects on endogenous Axin2 or Axin1 protein levels (Fig. 2A). However, overexpression of Axin1, but not Axin2, inhibited viral NP, HA and matrix protein (M1) mRNA levels (Fig. 2B). We then knocked down endogenous Axin1 using siRNAs. The knockdown of Axin1 did not affect Axin2 level, but increased viral NP protein levels (Fig. 2C, D). We further tested the effects of Axin1 overexpression on the infection of two nonsegmented negative-sense, single-stranded RNA viruses, VSV and RSV. Using a luciferase reporter virus assay, we found that Axin1 also inhibited VSV replication (Fig. 2E). Similarly, Axin1 drastically reduced RSV viral G and M protein levels at 36 and 48 hours post infection (Fig. 2F). Using GFP-labeled RSV virus, we observed a reduction of GFP signals in Axin1-overexpresing cells, but not in Axin2-overexpressing cells in comparison with blank control without plasmid transfection (Fig. 2G).
Figure 2. Axin1 inhibits virus replication.
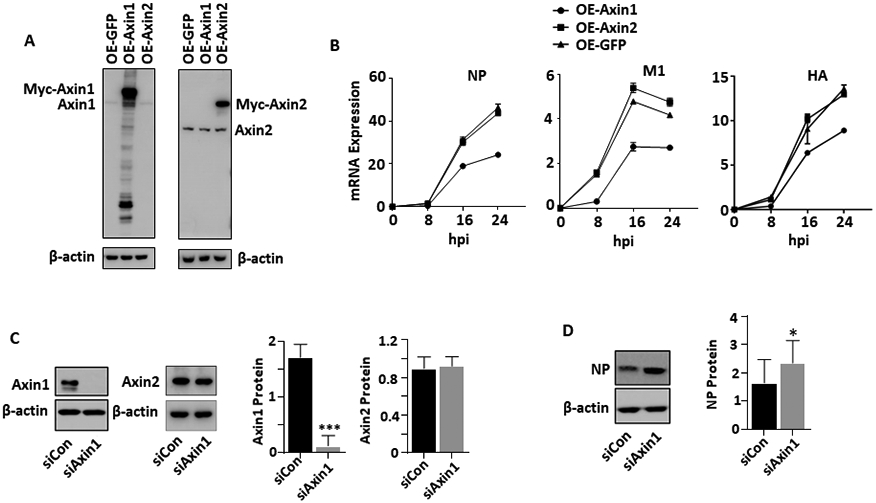
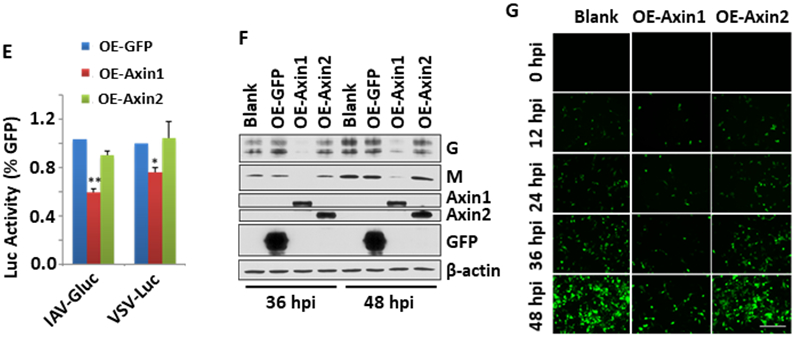
(A, B) HEK293 cells were transfected with GFP, Axin1, or Axin2 plasmid (OE-GFP, OE-Axin1 or OE-Axin2) for 24 h (A) and then infected with H1N1 influenza A/PR/8/34 virus (MOI=2) (B). The cells were collected at 8 to 24 h post infection (hpi). Axin1 and Axin2 protein levels were determined by Western blot and mRNA expression of viral genes (NP, M1 and HA) was analyzed by real-time PCR and normalized to β-actin. n=2. (C, D) A549 cells were transfected with 10 μM of siRNA control or siRNA Axin1 (Dharmacon) for 48 h using Lipfectamine RNAimax (C). The cells were then infected with H1N1 influenza A/PR/8/34 virus (MOI=0.01) for 48 h (D). Axin1, Axin2, β-actin and viral NP levels were determined by Western blot and quantified. n=3. *p < 0.05, ***p < 0.001 v.s. siCon. (E) HEK293 cells were transfected with GFP, Axin1 or Axin2 plasmid and infected with 1 MOI of IAV-Gluc, or VSV-Luc for 16 h. The luciferase activity was determined. n=3. *p <0.05, **P<0.01 v.s. OE-GFP. (F, G) HEK293 cells were transfected with GFP, Axin1, or Axin2 plasmid for 24 h and then infected with GFP-labeled RSV (MOI=1) for indicated times. Viral G and M protein levels were measured by Western blot in lysed cells with β-actin as a loading control. Myc-tagged Axin1, Axin2, and GFP protein levels were also determined using anti-myc antibody (F). GFP images of infected cells were taken (G). Scale bar = 100 μm.
Axin1 boosts IFN response
To determine the possible mechanisms of Axin1-mediated inhibition of influenza virus infection, we examined the effects of Axin1 on IFN responses. Overexpression of Axin1, but not Axin2 markedly increased the mRNA expression of IFNβ1 in the PR/8-infetced cells (Fig. 3A). Accordingly, Axin1 also significantly augmented the mRNA expression of the type I IFN-targeted anti-viral gene, OAS1 in the PR/8-infected cells (Fig. 3B). The phosphorylation of STAT1, the transcription factor essential for turning on ISGs expression 30, was also increased by Axin1 upon PR/8 infection (Fig. 3C). However, these effects were not observed in the uninfected cells (Fig. 3A-C), suggesting that Axin1 only boosts IFN response under the infection conditions and Axin1 alone is not sufficient for inducing IFN response.
Figure 3. Axin1 enhances type I IFN response during influenza virus infection.
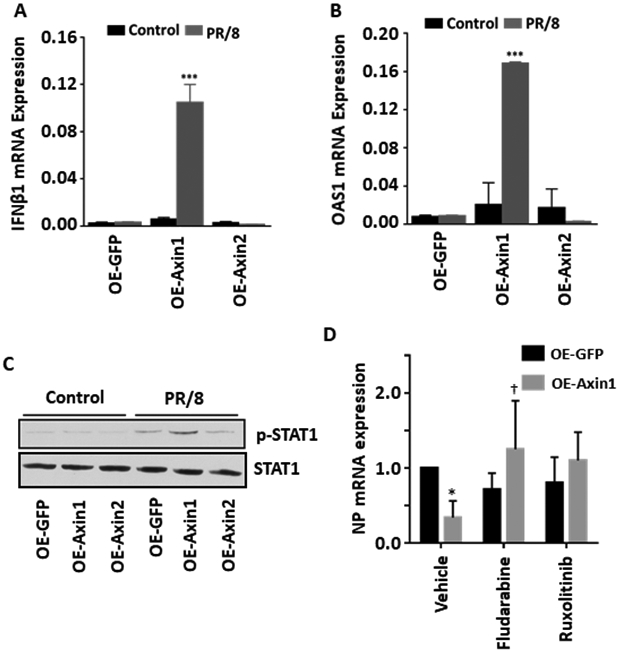
(A, B) HEK293 cells were transfected with GFP, Axin1, or Axin2 plasmid (OE-GFP, OE-Axin1 or OE-Axin2) for 24 h and then infected with H1N1 influenza A/PR/8/34 virus (MOI=2) for 8 h. mRNA expression of IFNβ1 and OAS1 were measured by real-time PCR and were normalized to β-actin. n=3. ***p < 0.001 v.s. OE-GFP. (C) Western blot was carried out to determine the protein expression of p-STAT1 (Tyr 701) and total STAT1 with and without virus infection (2 hpi). (D) HEK293 cells were transfected with GFP or Axin1 and then infected with H1N1 influenza A/PR/8/34 virus (MOI=2) in the presence of vehicle (DMSO), 30 μM fludarabine or 4 μM ruxolinitib for 16 h. NP mRNA levels were determined by real-time PCR and normalized to β-actin. n=3. *p < 0.05 v.s. vehicle-OE-GFP, †p < 0.05 v.s vehicle-Axin1.
To determine whether the Axin1-mediated antiviral effect is via its enhanced IFN response, we blocked IFN response using specific inhibitors of JAK1/2 or STAT1 and measured NP mRNA levels. As shown in Fig. 3D, JAK1/2 inhibitor ruxolitinib or STAT1 inhibitor fludarabine indeed reversed the Axin1-mediated antiviral effects.
JNK/c-Jun and Smad3 mediate Axin1-stimulated IFN response
Both Axin1 and Axin2 inhibit canonical Wnt/β-catenin signaling. However, Axin1, but not Axin2, increased the IFN response and inhibited influenza virus replication, suggesting that Axin1 acts via a different pathway 16. We examined the effects of Axin1 on the JNK/c-Jun pathway. We overexpressed Axin1 or Axin2 in HEK293 cells, infected the cells with PR/8 and examined the phosphorylation of JNK, c-Jun and ATF2. As shown in Fig. 4A and B, overexpression of Axin1, but not Axin2 activated the JNK/c-Jun pathway by increasing the phosphorylation of both JNK and c-Jun without changing ATF2 phosphorylation. Both Axin1 and Axin2 had no effects on the phosphorylation of JNK, c-Jun and ATF2 in the uninfected control cells. Axin1 also specifically triggered the activation of Smad3 signaling as demonstrated by a reporter assay (Fig. 4C). Although Axin1 had a modest effect on Smad3 signaling in the uninfected control cells, Smad3 signaling was further enhanced by PR/8 infection.
Figure 4. Axin1 activates JNK/c-Jun pathway and Smad3 signaling.
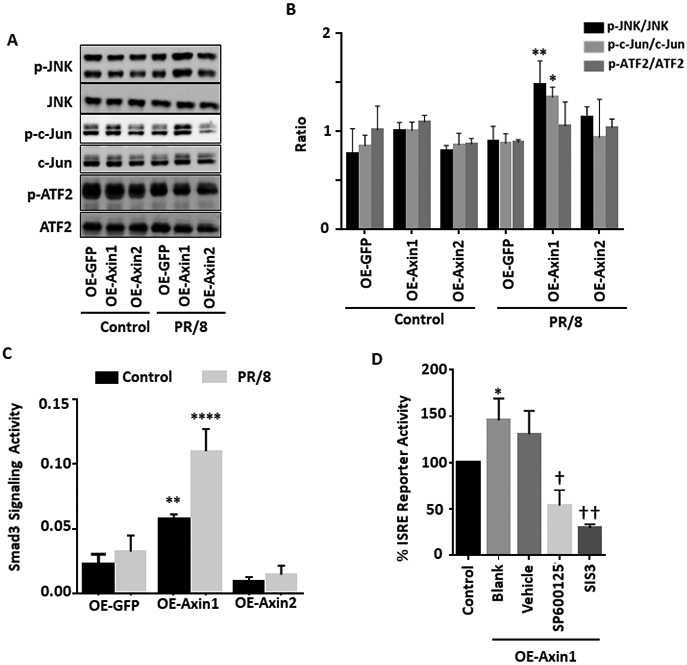
(A, B) HEK293 cells were transfected with GFP, Axin1, or Axin2 plasmid (OE-GFP, OE-Axin1 or OE-Axin2) for 24 h and infected without (Control) or with (PR/8) H1N1 influenza A/PR/8/34 virus (MOI=1) for 2 h. The protein levels of phosphorylated JNK (p-JNK), total JNK (JNK), phosphorylated c-Jun (p-c-Jun), total c-Jun (Jun), phosphorylated ATF2 (p-ATF2), and total ATF2 (ATF2) were determined by Western blot. Relative band intensities of phosphorylated proteins were quantified and normalized to respective total proteins. n=4. *p < 0.05, **p < 0.01 v.s. OE-GFP. (C) Smad3 signaling reporter and pRL-TK plasmids were transfected together with GFP, Axin1, or Axin2 plasmid into HEK293 cells for 24 h and then infected without (Control) or with (PR/8) H1N1 influenza A/PR/8/34 virus (MOI=1) for 8 h. Dual-luciferase activities was performed. The results were expressed as a ratio of Smad3 signaling reporter Firefly luciferase activity to pRL-TK Renilla luciferase activity. n=3. **p < 0.05, ****p < 0.0001 v.s. OE-GFP. (D) HEK293 cells pretreated with 10 μM SP600125 (JNK inhibitor), 10 μM SIS3 (Smad3 inhibitor), or 0.05% DMSO (Vehicle) for 6 h and were transfected with ISRE reporter together with Axin1 plasmid for 24 h. Cells were then infected with H1N1 influenza A/PR/8/34 virus (MOI=2) for 8 h. Dual-luciferase activities were determined and the ISRE_Luc Firefly luciferase activity was normalized to pRL-TK Renilla luciferase activity. n=3. *p < 0.05 v.s. Control. †p < 0.05, ††p < 0.01 v.s. vehicle.
The activity of the interferon-sensitive responsive element (ISRE), primarily responsible for the constitutive expression of ISGs is an indicator of IFN response 31. SP600125, a specific JNK inhibitor 32 and SIS3, a specific Smad3 inhibitor 33, successfully blocked Axin1-stimulated ISRE activity in the PR/8-infected cells (Fig. 4D). The results indicate that both the JNK/c-Jun and Smad3 signaling pathways participated in the Axin1-enhanced IFN response.
XAV939 attenuates influenza virus replication in vitro
To further validate the role of Axin1 against virus replication, XAV939, a tankyrase inhibitor 23, was utilized to stabilize Axin1 in lung epithelial A549 cells. XAV939 markedly increases the protein levels of Axin1 in the presence or absence of influenza virus infection (Fig. 5A, B). As a result, XAV939 significantly attenuated the protein expression of influenza viral NP and NS1 (Fig. 5A, C). XAV939 also reduced influenza virus production in a time-dependent manner (Fig. 5D).
Figure 5. XAV939 inhibits virus replication in lung epithelial cells.
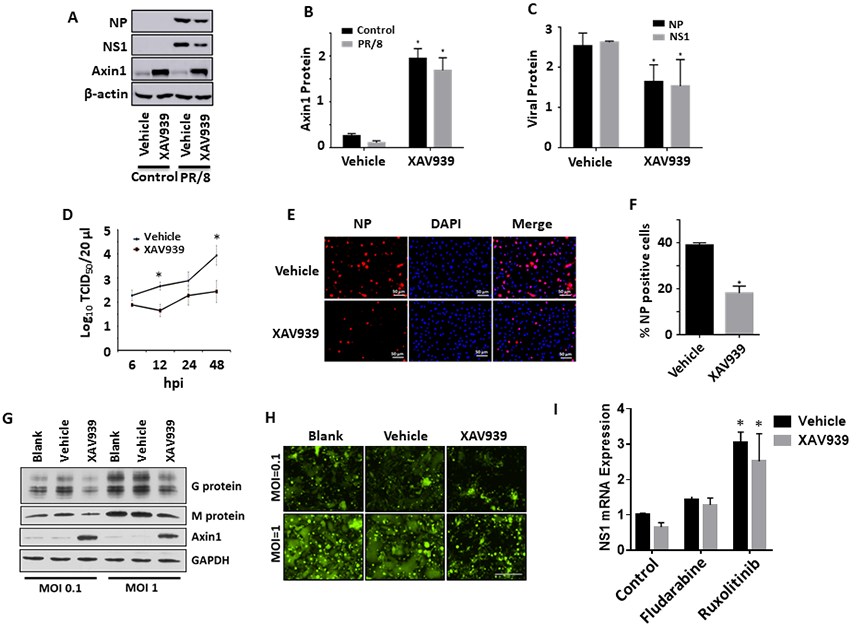
(A-C) A549 cells were pretreated with 10 μM XAV939 or 0.05% DMSO (vehicle) for 24 h. Cells were then infected with H1N1 influenza A/PR/8/34 virus (MOI=0.01) for 48 h. Protein levels of NP, NS1 and Axin1 were determined by western blots and quantitated. n=3. *p < 0.05 v.s vehicle. (D) A549 cells were pretreated with 20 μM XAV939 or 0.05% DMSO (vehicle) for 24 h. Cells were then infected with H1N1 influenza A/PR/8/34 virus (MOI=2) for indicated times. Virus titers in culture media from XAV939-treated A549 cells were determined by TCID50 assay. n=3. *p < 0.05 v.s. vehicle. (E, F) Human bronchial/tracheal epithelial cells were pretreated with 10 μM XAV939 or 0.05% DMSO (vehicle) for 24 h and infected with H1N1 influenza A/PR/8/34 virus (MOI=1) for 8 h. The cells were immunostaining with NP antibodies and NP-positive cells were counted. n=3. *p < 0.05 v.s vehicle. (G, H) Hep2 cells were pretreated with 20 μM XAV939 or 0.05% DMSO (vehicle) for 24 h. Cells were then infected with GFP-labeled RSV (MOI=0.1 or 1) for 48 h. Viral G and M protein expression was measured by Western blot in lysed cells with GAPDH as a loading control. GFP images of infected cells were also taken. Scale bar = 100 μm. (I) A549 cells were treated 10 μM XAV939 or 0.05% DMSO (vehicle) for 24 h and then infected with H1N1 influenza A/PR/8/34 virus (MOI=0.01) in the presence of vehicle (DMSO), 30 μM fludarabine or 4 μM ruxolinitib for 24 h. NP mRNA levels were determined by real-time PCR and normalized to β-actin. n=3. *p < 0.05 v.s. respective control.
As A549 cells are adenocarcinomic human lung epithelial cells, we further examined the effects of XAV939 on influenza virus infection in primary human bronchial/tracheal epithelial cells. XAV939 reduced NP-positive cells in human bronchial/tracheal epithelial cells as determined by immunostaining (Fig. 5E, F).
Finally, XAV939 reduced RSV viral G and M protein levels (Fig. 5G) and virus replication (Fig. 5H) in HEp2 cells.
To determine whether XAV939-mediated inhibition of influenza virus infection is mediated by IFN activation, we examined the effects of IFN signaling inhibitors on influenza virus infection in the absence or presence of XAV939. JAK1/2 inhibitor ruxolitinib or STAT1 inhibitor fludarabine reversed the XAV939-mediated decreases in viral NP and NS1 mRNA expression (Fig. 5I).
XAV939 protects mice from a lethal dose of influenza virus challenge
To further evaluate the therapeutic potential of Axin1 as a novel target for limiting virus infection, XAV939 was further tested as a potential antiviral agent in vivo (Fig. 6A). Oral administration of XAV939 significantly attenuated influenza virus replication in vivo (Fig. 6B) and improved the survival rate of animals challenged with a lethal dose of the virus using two different strategies of XAV939 administration (−1 to 2 and −1 to 4 dpi) (Fig. 6C).
Figure 6. XAV939 protects mice from a lethal influenza virus challenge.
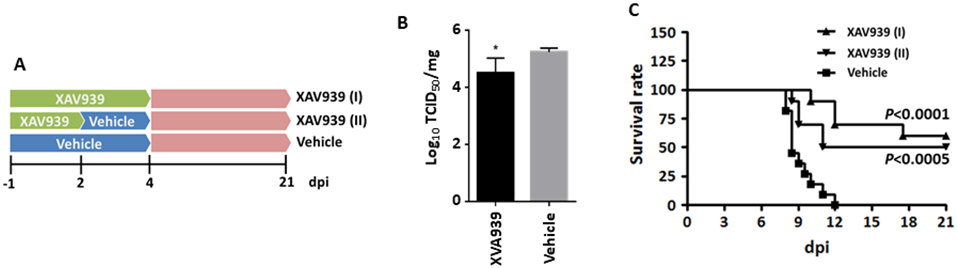
Mice were challenged intranasally with a lethal dose of H1N1 influenza A/PR/8/34 virus. XAV939 treatment was given orally and daily, beginning with one day before infection (−1 dpi) and continued until 2 [XAV939 (I)] or 4 [XAV939 (II)] days post infection. Control animals received vehicle alone. (A) The illumination of experiment design. (B) Virus titer in the homogenized infected lungs at 2 dpi was measured by TCID50 assay in MDCK cells and normalized to total protein amount. n=5. *p < 0.05 v.s. vehicle. (C) Survival rate was monitored throughout the course of the study (10-11 mice/group). Mantel-Cox χ2 test on Kaplan-Meier survival data was used to compare the survival rate between groups.
Discussion
The mortality rate of pneumonia and influenza virus infection continually declined throughout the 20th century because of the improvement of medical strategies that prevented secondary lung infections 2,34. However, acute pulmonary infection remains a substantial concern as acute lower respiratory infections still cause the most deaths and are the largest economic burden among all infectious diseases worldwide. Unlike respiratory syncytial virus (RSV), which is the major cause of respiratory illness (bronchiolitis or pneumonia) in young children 35, influenza virus sickens patients of all ages 36. In our study, we discovered the protective effect of Axin1 and its stabilizer XAV939 on influenza virus and RSV infection. Axin1 boosts the antiviral type I IFN response by augmenting JNK/c-Jun and Smad3 signaling. Targeting Axin1 to regulate the IFN response is therefore a potential broad-spectrum antiviral therapeutic approach.
A unique feature in signaling transduction is that several different pathways depend on a group of crucial regulators referred to as scaffolds, which simultaneously bind several components in the same or different signaling pathways and augment specificity and efficacy during signal transduction. By serving as a multidomain scaffold, Axin1 coordinates several different protein complexes involved in regulating TGF-β/Smad, JNK/c-Jun, and p53 signaling 16. In this study, we found that both transcription factors, c-Jun and Smad3, were activated by Axin1.
Axin2 did not amplify the type I IFN response and affect virus replication. A similar effect was also observed in bacterial Salmonella infection 21. Considering that both Axin1 and Axin2 inhibited Wnt/catenin signaling 14, the enhancement of type I IFN synthesis by Axin1 is probably not through manipulation of Wnt/catenin signaling. Actually, Wnt3a has been reported to amplify IFN response 37 potentially through interaction between β-Catenin and LRRFIP1b 38. This implies that enhancement of type I IFN synthesis by Axin1 through JNK/c-Jun and Smad3 signaling is intensive enough to override the potential negative effect of suppressed Wnt/catenin signaling on ISREs responses. Considering the additional effect of type I IFN sensitizing the host to secondary bacterial pneumonia post influenza infection 39 and promoting cell death 40 as Axin1 is normally involved 41, more detailed studies will need to be carried out.
Axin1 has been reported to be the rate limiting factor for β-catenin destruction complex assembly due to its low basal expression 42. This unique feature and its antiviral properties make it a perfect host factor for use against virus infection. In this study, we used XAV939 to stabilize Axin1 and evaluated its potential antiviral activity against influenza virus in vitro and in vivo. XAV939 has previously been reported to inhibit herpes simplex virus replication 43. Oral administration of XAV939 stabilizes Axin1 in the lung as previously reported 44. Our current studies indicate that XAV939 successfully reduces influenza virus replication in vitro and in vivo, and protects the mice from lethal influenza virus infection.
IFN exerts significant protection by limiting respiratory virus infection including highly pathogenic H5N1 influenza A virus infection in animals 45,46. However, viruses can block nearly all aspects of the IFN regulatory pathway through intimate interplay to avoid compromising virus replication 47. In our study, we found that Axin1, as an antiviral mediator, was reduced in influenza viral pneumonia. This could be due to the complicated systemic response to virus infection, sophisticated paracrine signaling, and an adequate set of protein degradation machinery. Several other components of the ubiquitin-proteasome pathway (USP34 and RNF146) and small ubiquitin-related modifier (SUMO) are also involved in regulating homeostasis of Axin1 48-50. Targeting these molecules to stabilize Axin1 could also be potentially utilized against virus replication.
Exploitation by influenza virus of host cellular signaling to support viral replication is very effective. However, this dependency might also be utilized to develop novel antiviral approaches. Our work here is an example of translating this information into potential therapeutics. We discovered a novel function of Axin1 in limiting virus infection and incorporated this scaffold protein into the antiviral network of interferon.
Acknowledgements
This work was supported by National Institutes of Health grants AI121591, AI152004, HL135152, GM103648, the Oklahoma Center for Adult Stem Cell Research-A Program of Tobacco Settlement Endowment Trust (TSET), the Oklahoma Center for the Advancement of Science and Technology (HR-20-050) and the Lundberg-Kienlen Endowment fund (to LL). SL is supported by NIH grant AI126360 and GM103648. JPM is supported by NIH grants AI62629 and GM103648 and the Merit Review Program of the Department of Veterans Affairs, (BX001937). We thank Dr. Peter Palese (Icahn School of Medicine at Mount Sinai) and Dr. Sean Whelan (Harvard Medical School) for providing influenza PR/8-GLuc virus and VSV-Luc, respectively. GFP plasmid constructed by Dr. Frank Costantini (Columbia University) and Axin1/2 plasmids generated by Dr. Michael Klymkowsky (University of Colorado) were obtained from Addgene. We also appreciate Dr. Kevin Tan and Dr. Harold A. Chapman (UCSF) for providing the XAV939 formulation used in animal study.
Footnotes
Conflict of Interest:
The authors have no conflict of interest to declare
References
- 1.Medina RA, and García-Sastre A (2011). Influenza A viruses: new research developments. Nature Reviews Microbiology 9, 590–603. 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizgerd JP (2012). Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med 186, 824–829. 10.1164/rccm.201206-1063PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Müller KH, Kakkola L, Nagaraj AS, Cheltsov AV, Anastasina M, and Kainov DE (2012). Emerging cellular targets for influenza antiviral agents. Trends Pharmacol Sci 33, 89–99. 10.1016/j.tips.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs A, and Lindenmann J (1957). Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147, 258–267. 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 5.McNab F, Mayer-Barber K, Sher A, Wack A, and O'Garra A (2015). Type I interferons in infectious disease. Nature Reviews Immunology 15, 87–103. 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walter MR (2020). The Role of Structure in the Biology of Interferon Signaling. Frontiers in Immunology 11. 10.3389/fimmu.2020.606489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowie AG, and Unterholzner L (2008). Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8, 911–922. 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbalat R, Ewald SE, Mouchess ML, and Barton GM (2011). Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29, 185–214. 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 9.Pichlmair A, Lassnig C, Eberle CA, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, et al. (2011). IFIT1 is an antiviral protein that recognizes 5'-triphosphate RNA. Nat Immunol 12, 624–630. 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 10.Honda K, Takaoka A, and Taniguchi T (2006). Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Qing J, Liu C, Choy L, Wu RY, Pagano JS, and Derynck R (2004). Transforming growth factor beta/Smad3 signaling regulates IRF-7 function and transcriptional activation of the beta interferon promoter. Mol Cell Biol 24, 1411–1425. 10.1128/mcb.24.3.1411-1425.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai T, and Akira S (2006). Innate immune recognition of viral infection. Nat Immunol 7, 131–137. 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 13.Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL 3rd, Lee JJ, Tilghman SM, Gumbiner BM, and Costantini F (1997). The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90, 181–192. 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 14.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, and Costantini F (2002). Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22, 1172–1183. 10.1128/mcb.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, and Clevers H (2012). Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256. 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Cortese MS, Uversky VN, and Dunker AK (2008). Intrinsic disorder in scaffold proteins: getting more from less. Prog Biophys Mol Biol 98, 85–106. 10.1016/j.pbiomolbio.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Neo SY, Wang X, Han J, and Lin SC (1999). Axin forms a complex with MEKK1 and activates c-Jun NH(2)-terminal kinase/stress-activated protein kinase through domains distinct from Wnt signaling. J Biol Chem 274, 35247–35254. 10.1074/jbc.274.49.35247. [DOI] [PubMed] [Google Scholar]
- 18.Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, and Kato M (2001). Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol 21, 5132–5141. 10.1128/mcb.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Rui H, Wang J, Lin S, He Y, Chen M, Li Q, Ye Z, Zhang S, Chan SC, et al. (2006). Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. Embo j 25, 1646–1658. 10.1038/sj.emboj.7601057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rui Y, Xu Z, Lin S, Li Q, Rui H, Luo W, Zhou HM, Cheung PY, Wu Z, Ye Z, et al. (2004). Axin stimulates p53 functions by activation of HIPK2 kinase through multimeric complex formation. Embo j 23, 4583–4594. 10.1038/sj.emboj.7600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang YG, Wu S, Xia Y, Chen D, Petrof EO, Claud EC, Hsu W, and Sun J (2012). Axin1 prevents Salmonella invasiveness and inflammatory response in intestinal epithelial cells. PLoS One 7, e34942. 10.1371/journal.pone.0034942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kameoka M, Kameoka Y, Utachee P, Kurosu T, Sawanpanyalert P, Ikuta K, and Auwanit W (2009). Short communication: RNA interference directed against Axin1 upregulates human immunodeficiency virus type 1 gene expression by activating the Wnt signaling pathway in HeLa-derived J111 cells. AIDS Res Hum Retroviruses 25, 1005–1011. 10.1089/aid.2008.0284. [DOI] [PubMed] [Google Scholar]
- 23.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620. 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 24.Heaton NS, Leyva-Grado VH, Tan GS, Eggink D, Hai R, and Palese P (2013). In vivo bioluminescent imaging of influenza a virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol 87, 8272–8281. 10.1128/jvi.00969-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, and Whelan SP (2009). Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5, e1000394. 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitra R, Baviskar P, Duncan-Decocq RR, Patel D, and Oomens AG (2012). The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J Virol 86, 4432–4443. 10.1128/jvi.06744-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng T, Gao L, Bhaskaran M, Guo Y, Gou D, Narayanaperumal J, Chintagari NR, Zhang K, and Liu L (2009). Pleiotrophin Regulates Lung Epithelial Cell Proliferation and Differentiation during Fetal Lung Development via β-Catenin and Dlk1. Journal of Biological Chemistry 284, 28021–28032. 10.1074/jbc.M109.052530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.REED LJ, and MUENCH H (1938). A SIMPLE METHOD OF ESTIMATING FIFTY PER CENT ENDPOINTS12. American Journal of Epidemiology 27, 493–497. 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 29.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, and Rosen H (2011). Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146, 980–991. 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy DE, and Darnell JE Jr. (2002). Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3, 651–662. 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 31.Reid LE, Brasnett AH, Gilbert CS, Porter AC, Gewert DR, Stark GR, and Kerr IM (1989). A single DNA response element can confer inducibility by both alpha- and gamma-interferons. Proc Natl Acad Sci U S A 86, 840–844. 10.1073/pnas.86.3.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A 98, 13681–13686. 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jinnin M, Ihn H, and Tamaki K (2006). Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol 69, 597–607. 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
- 34.Fauci AS, and Morens DM (2012). The perpetual challenge of infectious diseases. N Engl J Med 366, 454–461. 10.1056/NEJMra1108296. [DOI] [PubMed] [Google Scholar]
- 35.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, and Fukuda K (2003). Mortality associated with influenza and respiratory syncytial virus in the United States. Jama 289, 179–186. 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 36.Webster RG, Bean WJ, Gorman OT, Chambers TM, and Kawaoka Y (1992). Evolution and ecology of influenza A viruses. Microbiol Rev 56, 152–179. 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. (2009). A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139, 1255–1267. 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, and Cao X (2010). The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat Immunol 11, 487–494. 10.1038/ni.1876. [DOI] [PubMed] [Google Scholar]
- 39.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, and Deng JC (2009). Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119, 1910–1920. 10.1172/jci35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K, and Yagita H (1999). Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J Exp Med 189, 1451–1460. 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, Wang X, Wu X, Rui Y, Liu W, Wang J, Wang X, Liou YC, Ye Z, and Lin SC (2007). Daxx cooperates with the Axin/HIPK2/p53 complex to induce cell death. Cancer Res 67, 66–74. 10.1158/0008-5472.Can-06-1671. [DOI] [PubMed] [Google Scholar]
- 42.Lee E, Salic A, Krüger R, Heinrich R, and Kirschner MW (2003). The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol 1, E10. 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Yamauchi Y, Kamakura M, Murayama T, Goshima F, Kimura H, and Nishiyama Y (2012). Herpes simplex virus requires poly(ADP-ribose) polymerase activity for efficient replication and induces extracellular signal-related kinase-dependent phosphorylation and ICP0-dependent nuclear localization of tankyrase 1. J Virol 86, 492–503. 10.1128/jvi.05897-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ulsamer A, Wei Y, Kim KK, Tan K, Wheeler S, Xi Y, Thies RS, and Chapman HA (2012). Axin pathway activity regulates in vivo pY654-β-catenin accumulation and pulmonary fibrosis. J Biol Chem 287, 5164–5172. 10.1074/jbc.M111.322123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, and Durbin JE (1998). The role of interferon in influenza virus tissue tropism. J Virol 72, 8550–8558. 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price GE, Gaszewska-Mastarlarz A, and Moskophidis D (2000). The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol 74, 3996–4003. 10.1128/jvi.74.9.3996-4003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katze MG, He Y, and Gale M Jr. (2002). Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2, 675–687. 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 48.Lui TT, Lacroix C, Ahmed SM, Goldenberg SJ, Leach CA, Daulat AM, and Angers S (2011). The ubiquitin-specific protease USP34 regulates axin stability and Wnt/β-catenin signaling. Mol Cell Biol 31, 2053–2065. 10.1128/mcb.01094-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, et al. (2011). RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13, 623–629. 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 50.Kim MJ, Chia IV, and Costantini F (2008). SUMOylation target sites at the C terminus protect Axin from ubiquitination and confer protein stability. Faseb j 22, 3785–3794. 10.1096/fj.08-113910. [DOI] [PMC free article] [PubMed] [Google Scholar]