

Abstract
High-fidelity DNA replication is critical for the faithful transmission of genetic information to daughter cells. Following genotoxic stress, specialized DNA damage tolerance pathways are activated to ensure replication fork progression. These pathways include translesion DNA synthesis, template switching and repriming. In this Review, we describe how DNA damage tolerance pathways impact genome stability, their connection with tumorigenesis and their effects on cancer therapy response. We discuss recent findings that single-strand DNA gap accumulation impacts chemoresponse and explore a growing body of evidence that suggests that different DNA damage tolerance factors, including translesion synthesis polymerases, template switching proteins and enzymes affecting single-stranded DNA gaps, represent useful cancer targets. We further outline how the consequences of DNA damage tolerance mechanisms could inform the discovery of new biomarkers to refine cancer therapies.
High-fidelity DNA replication is constantly challenged by endogenous and exogenous sources of genotoxic stress1 (BOX 1). Endogenous sources of genotoxic stress include abasic sites, improper incorporation of ribonucleotides into replicating DNA, DNA–protein crosslinks, transcription–replication conflicts2, formation of DNA secondary structures, single-stranded DNA (ssDNA) gaps3–8, nucleotide imbalances9,10 and changes in origin firing frequency11. Exogenous sources of genotoxic stress include ionizing radiation and DNA-damaging chemotherapy such as alkylating agents, crosslinking drugs, topoisomerase inhibitors and antimetabolites12 (FIG. 1). The transient slowing or aberrant acceleration of replication forks in response to these challenges is termed ‘replication stress’ and is tightly linked to cancer development3,13–16.
Box 1 |. DNA replication is mediated by a large protein complex known as the replisome that promotes multiple enzymatic activities.
In eukaryotic cells, parental DNA is unwound by the CMG complex, which is composed of cell division control protein 45 (CDC45), mini-chromosome maintenance protein homologues 2–7 (MCM2–7) and the go–ichi–ni–san (GINS) complex. It is then replicated by the leading and lagging strand polymerases Polε and Polδ. The DNA sliding clamp proliferating cellular nuclear antigen (PCNA) is a homotrimer that encircles DNA and is essential for processivity of replicative polymerases. DNA lesions or other sources of DNA replication stress (red triangle) can transiently stall the leading strand polymerase, without affecting the movement of the CMG complex. This process is termed replication fork uncoupling and leads to the accumulation of single-stranded DNA stretches that are promptly coated by the single-stranded DNA-binding protein replication protein A (RPA).
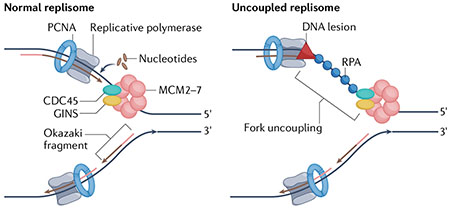
Fig. 1 |. Major mediators of the replication stress response.

Sources of endogenous replication stress include abasic sites, transcription–replication conflicts, the incorporation of ribonucleotides into DNA and protein–DNA crosslinks. Sources of exogenous stress include DNA inter-strand and intra-strand crosslinks induced by DNA crosslinking agents and base damage induced by alkylating drugs. Single-stranded DNA gaps or breaks, secondary DNA structures, including hairpins, and nucleotide imbalances that stall replicative polymerases are also considered sources of DNA replication stress. DNA damage tolerance mechanisms are activated when forks encounter these endogenous or exogenous DNA lesions or roadblocks, which are represented as red triangles. Translesion synthesis (TLS) is a DNA damage tolerance mechanism that involves RAD18-dependent monoubiquitination (mono-Ub) of proliferating cellular nuclear antigen (PCNA), which promotes downstream recruitment of TLS polymerases and DNA synthesis across the DNA lesion. Template switching involves RAD18 and UBC13-mediated polyubiquitination (poly-Ub) of PCNA and the downstream factors RAD51, Bloom syndrome protein (BLM) and Nijmegen breakage syndrome 1 protein (NBS1), allowing use of the complementary base pairs to replicate DNA past the DNA obstacle. Fork reversal, another form of template switching, employs the RAD51, SMARCAL1,ZRANB3, helicase-like transcription factor (HLTF) and F-box DNA helicase 1 (FBH1) enzymes to facilitate replication fork remodelling into a four-way junction structure. Repriming relies on the action of DNA-directed primase/polymerase protein (PRIMPOL), leaving single-stranded DNA (ssDNA) gaps behind the replication forks to be filled post-replicatively. PRIMPOL activity is regulated by ATR and CHK1.
As part of the replication stress response, cancer cells activate various DNA damage tolerance (DDT) pathways17. DDT pathways broadly include translesion DNA synthesis (TLS)18, template switching (TS)19 and repriming20 (FIG. 1). TLS involves specialized polymerases that can replicate through a damaged DNA template21 and is generally regarded as a lower-fidelity form of DDT because the TLS polymerases recruited to stalled replication forks have a high potential for mutagenesis18. TS uses sister chromatid DNA to bypass replication obstacles22 and, as a result, is less likely than TLS to introduce erroneous nucleotides. One version of TS is fork reversal, which promotes the remodelling of replication forks into four-way junction structures upon encountering DNA lesions23. Reversed fork remodelling enables the original lesion to be repositioned ahead of the replication fork junction, facilitating lesion removal before reversed fork restart or lesion bypass through a TS mechanism24. Finally, repriming in human cells involves a specialized polymerase-primase enzyme, DNA-directed primase/polymerase protein (PRIMPOL)25–28, that skips damaged DNA, re-initiating synthesis beyond the lesion and leaving a ssDNA gap between the lesion and the point where synthesis restarts20. The ssDNA gap generated by PRIMPOL-mediated repriming can then be filled post-replicatively through TLS or TS29–32.
The fine-tuning of different DDT mechanisms is an emerging determinant of tumorigenesis and cancer therapy response. Here, we review how the loss of DDT factors can confer an increased cancer risk as DDT proteins are critical for DNA replication in the presence of endogenous and exogenous replication stress. In addition, we describe how the aberrant expression, or indeed the normal function of DDT enzymes upon increased replication stress, can promote the genomic instability that drives cancer development and progression. Many tumours exhibit elevated endogenous replication stress; therefore, we discuss how these pathways can be exploited for cancer cell clearance. Finally, we frame replication stress response mechanisms in the context of current clinical cancer treatments and suggest possible opportunities for biomarker development.
DNA replication stress in tumorigenesis
Translesion DNA synthesis.
Eukaryotic TLS involves polymerases of the Y-family33 — including REV1, Polη, Polι and Poκ — and the B-family (Polζ)34. These Y-family and B-family TLS polymerases lack 3′-to-5′ nucleotide proofreading and exhibit a decreased capacity to distinguish between incoming nucleotides relative to replicative polymerases21. As a result, they are more mutagenic than the polymerases that are part of the core replication complex, with error rates of up to 1 in every 10 nucleotides inserted, compared to errors rates as low as 1 in every 1010 bases for the replicative polymerases Polε and Polδ18,35. Mutagenic events induced by TLS polymerases can contribute to tumorigenesis36 and impact the response of cancer cells to DNA-damaging chemotherapies37, highlighting the importance of these mechanisms in the context of tumour treatment.
The recruitment of TLS polymerases to the DNA is mediated by the ubiquitination of proliferating cell nuclear antigen (PCNA), which is an essential processivity factor for replicative DNA polymerases also known as the DNA sliding clamp. PCNA is monoubiquitinated at lysine 164 by the E3 ligase enzyme RAD18 (REF.38) (FIG. 1), the activity of which is important for the replication of damaged DNA. RAD18 loss can contribute to genomic instability and increased sensitivity to DNA-damaging agents in non-malignant mammalian cells39,40. However, RAD18-dependent PCNA ubiquitination also drives mutagenic TLS41, promoting tumorigenesis. Indeed, the expression of RAD18 is high in a variety of cancer types and correlates with worsened survival outcomes42–44 (TABLE 1). Moreover, RAD18 expression correlates with increased single nucleotide variations in human cancers from The Cancer Genome Atlas, and mutational signatures induced by RAD18 activity in mouse models correlate with mutational landscapes from the COSMIC database41.
Table 1 |.
Replication stress response in cancer and consequences on genome stability
| Replication stress response mechanism | Enzyme | Expression | Activity | Consequences |
|---|---|---|---|---|
| TLS | RAD18 | Moderate or high in most cancersa,42 | PCNA monoubiquitination38 | Mutagenesis leading to elevated TMB and increased neoantigen formation, mutagenesis leading to acquired chemoresistance |
| REV1 | Moderate or high in select livera and lung cancersa,b,37,49 | TLS scaffold45, G>C transversions48 | ||
| Ploζ | Elevated in oesophageal cancersa,57 and gliomasb,55, downregulated in CRCsb, lungb and gastric cancersb,54,58,59 | GC>AA/TT mutations53 | ||
| Polη | High in select basal cell carcinomasa,37 and elevated in head and neck cancersa,63 | Mutations at WA/TW motifs (where W is A or T)64,65 | ||
| Polι | Elevated in breast cancer cellsb,c,72, bladder cancersa,73 and ESCCsa,b,56,74 | T>C, T>A or C>A mutations72 | ||
| Polκ | Elevated in lung cancersb,c,81 | CC:GG interruptions on poly(dA:dT) tracts78 | ||
| TS | UBC13 | Moderate in most cancersa,87 | PCNA polyubiquitination84,85 | SCEs contributing to gross chromosomal aberrations and increased genomic instability, gene amplification leading to acquired chemoresistance |
| HLTF | Low in most cancersa,90 | PCNA polyubiquitination88,89 | ||
| SHPRH | Moderate or high in most cancersa,91 | PCNA polyubiquitination88,89 | ||
| RAD51 | Elevated in pancreatica,129 and breast cancersa,130 | Strand invasion, D-loop formation29,96,97 | ||
| BLM | Moderate or high in most cancersa,100 | D-loop dissolution98,99 | ||
| NBS1 | Moderate or high in most cancersa,101 | Protein recruitment and regulation29 | ||
| Fork reversal/recovery | SMARCAL1 | Moderate in select pancreatic, testis, breast, prostate and thyroid cancersd,117 | Fork reversal108,109 | Fork reversal consequences include replication fork degradation in specific genetic backgrounds, release of degraded or cleaved DNA into the cytosol, and decreased fork repriming Fork recovery consequences include resumption of DNA synthesis and cancer cell survival |
| ZRANB3 | Elevated in testis cancersd,121 | Fork reversal, interaction with polyubiquitinated PCNA111,112 | ||
| HLTF | Low in most cancersa,90 | Fork reversal, PCNA polyubiquitination113,114 | ||
| RAD51 | Elevated in pancreatic129 and breast cancers130 | Fork reversal, fork protection24 | ||
| RECQ1 | Moderate or high in lymphomas, thyroid, head and neck, and carcinoid cancersa,139 | Restart of reversed forks132 | ||
| WRN | Moderate in select testis, thyroid, and head and neck cancersa,138 | Stalled fork processing133 | ||
| DNA2 | Moderate or high in most cancersa,137 | Stalled fork processing133 | ||
| MUS81 | Low in most cancersa,145 | Reversed fork cleavage146,147 | ||
| CSB | Elevated in thyroid and breast cancersd,151 | Stalled fork processing150 | ||
| XRCC1 | High in most cancersa,153 | MMEJ at stalled forks152 | ||
| RNF168 | Downregulated in BRCA1-mutated cancersa,215 | Potential role in break-induced replication-like fork recovery148 | ||
| Fork repriming | PRIMPOL | Moderate in thyroid cancersa,162 | Primase, polymerase25–28 | Increased ssDNA gaps and activation of ssDNA gap filling by TLS or TS, gap processing, and release of DNA into the cytosol |
Replication stress response mechanisms with corresponding enzymes, expression levels in cancer and enzyme activities shown. Consequences of each replication stress response pathway are shown on the right. The Human Protein Atlas uses the terms weak, moderate or high to describe the intensity of immunohistochemistry staining on tumour sections. For our purposes, factors expressed at low levels correspond to weak staining, factors expressed at moderate levels correspond to moderate staining and factors expressed at high levels correspond to strong staining. ‘Elevated’ or ‘downregulated’ refers to increased or decreased protein expression relative to either non-malignant controls or other cancer types. BLM, Bloom syndrome protein; CRC, colorectal cancer; ESCC, oesophageal squamous cell carcinoma; HLTF, helicase-like transcription factor; MMEJ, microhomology-mediated end joining; NBS1, Nijmegen breakage syndrome 1 protein; PCNA, proliferating cellular nuclear antigen; PRIMPOL, DNA-directed primase/polymerase protein; SCE, sister chromatid exchange; SHPRH, SNF2 histone linker PHD ring helicase; ssDNA, single-stranded DNA; TMB, tumour mutational burden; TLS, translesion synthesis; TS, template switching; WRN, Werner syndrome ATP-dependent helicase.
Protein level measured by immunohistochemistry;
RNA measured by quantitative PCR;
Protein level measured by western blot;
RNA measured by RNA sequencing.
In addition to PCNA monoubiquitination, the Y-family TLS polymerase REV1 functions as a scaffold protein to facilitate the downstream recruitment of other TLS polymerases45. In normal cells, REV1 contributes to mitochondrial function46 and somatic hypermutation47. REV1 loss is associated with metabolic dysfunction; a recent study suggested that this dysfunction might be due to the inability of Rev1-knockout mice to appropriately respond to endogenous replication stress46. Interestingly, REV1 can also function in base excision repair, resulting in a mutational signature enriched for C>G transversions48, and is upregulated in hepatocarcinomas and select lung cancers37,49 (TABLE 1).
The B-family polymerase Polζ is involved in the translesion synthesis of DNA adducts that stall replication forks and is composed of the catalytic subunit REV3L and the accessory subunit REV7 (REF.34). Loss of REV3L is associated with chromosomal instability50 and spontaneous tumorigenesis in mouse models51, suggesting that this TLS polymerase is important in DDT in non-malignant cells. Interestingly, the genomic instability observed in REV3L-deficient cells activates an innate immunity-like response involving upregulation of the cGAS–STING pathway and increased micronuclei formation52. When functional, Polζ tends to introduce dinucleotide mutations, with strong preferences for GC>AA or GC>TT mutations53 (TABLE 1). Several single-nucleotide polymorphisms (SNPs) in REV3L have been linked to an increased risk of developing lung cancer in a specific Han Chinese population54. Divergent REV3L expression has been reported across different tumour types: REV3L expression is elevated in gliomas55 and oesophageal squamous cell carcinomas56,57 but appears downregulated in select colorectal, lung and gastric cancer tissues relative to non-cancer controls54,58,59 (TABLE 1).
Germline mutations in the Y-family TLS polymerase Polη predispose carriers to skin tumours as this enzyme is critical for efficient and high-fidelity bypass of UV-induced lesions, including cyclobutane thymine dimers60. Indeed, patients with xeroderma pigmentosum with non-functional Polη are especially prone to malignancies caused by sun exposure61. Polη expression is high in a number of tumour types37,62,63 (TABLE 1), and increased Polη expression in melanomas, chronic lymphocytic leukaemias and germinal centre B cell lymphomas is associated with a mutational signature enriched at WA/TW motifs (where W is A or T), consistent with Polη mutagenic activity64,65.
Polι was originally suggested to serve as a back-up of Polη in UV lesion processing, although its unique structural features suggest an independent, albeit still ill-defined, function for this enzyme in DNA damage bypass66–68. Although Polι deficiencies in normal cells are not linked with pathologies in humans68, several SNPs in POLI (encoding Polι) have been linked to the development of specific tumour types, including melanoma, prostate cancer, lung adenocarcinoma and squamous cell carcinoma69–71. Cell-based studies have shown that Polι activity increases T>C transitions, T>A transversions or C>A transversions in breast cancer cells exposed to UV damage72, and Polι expression is elevated in breast cancer cell lines72, bladder cancer73 and oesophageal squamous cell carcinoma56 (TABLE 1). In vivo studies suggest that Polι expression can contribute to oesophageal squamous cell carcinoma cell migration and invasion74; however, future research will need to determine the molecular links between Polι expression or activity, migration and invasion, and response to chemotherapy.
Polκ is implicated, along with Polη, in the replication of DNA at common fragile sites75 and promotes DNA synthesis when replication fork stalling occurs due to nucleotide deprivation76. Polκ has a propensity to introduce interrupted mutations and undergo polar pausing77; upon hydroxyurea treatment, the mutational signature of Polκ at poly(dA:dT) repeats — sites of fork stalling and collapse in both early and late-replication fragile sites — includes recurrent interruptions of poly(dA:dT) tracts with CC:GG sequences in a mouse cell line model78. In general, mutations in common fragile sites have been associated with genomic instability features that drive tumorigenesis79. Moreover, POLK SNPs are associated with various cancer types, including prostate, breast, lung, melanoma, stomach and large intestine tumours80, and Polκ expression is elevated in lung cancer81 (TABLE 1). It is unclear whether there is a direct relationship between Polκ-dependent mutations within particular sequences and tumorigenesis.
Template switching.
TS has been most extensively studied in yeast and bacteria82,83, and work establishing TS factors and regulators in human cells is limited. In general, TS pathways can lead to genomic instability through genomic rearrangements and sister chromatid exchange (SCE).
TS mechanisms in yeast84,85 and human cells86 are associated with K63-linked polyubiquitination of PCNA by the E2-conjugating enzyme UBC13 following PCNA monoubiquitination by RAD18 (FIG. 1). UBC13, expressed at moderate levels in most tumours87 (TABLE 1), has two E3 ligase partners, helicase-like transcription factor (HLTF) and SNF2 histone linker PHD ring helicase (SHPRH), both of which are implicated in PCNA ubiquitination88,89. HLTF expression is generally low across tumour types90, whereas SHPRH levels appear to be moderate or high in the majority of cancers91 (TABLE 1). Although there is no clear data indicating the molecular mechanism underlying this difference, we speculate that HLTF might be lowly expressed across tumours because of its reported antiproliferative functions92–94 in addition to its roles in the DNA replication stress response95.
Following PCNA polyubiquitination by UBC13, TS might involve molecular steps resembling those of canonical homologous recombination (reviewed in REF.96). The central recombinase factor RAD51 and Nijmegen breakage syndrome 1 protein (NBS1), which is a key component of the MRE11–RAD50–NBS1 (MRN) complex, have been implicated in TS across from abasic sites and benzo[a]pyrene adducts29,97; in this context, RAD51 might potentially mediate TS by promoting strand invasion and branch migration between sister chromatids, whereas the MRN complex might be required to process the stalled replication intermediate. Homologues of the human Bloom syndrome protein (BLM), such as the ATP-dependent helicases SGS1 in Saccharomyces cerevisiae98 and hus2/rqh1 in Schizosaccharomyces pombe99, promote TS by facilitating the dissolution of D-loop structures although there are no mechanistic studies that conclusively demonstrate the function of BLM in TS in human cells. Interestingly, many of these putative TS factors are highly expressed in different cancer types100,101 (TABLE 1).
TS pathways can lead to gross chromosomal rearrangements and gene amplifications102,103, which could in turn affect cancer progression, chemoresponse and clinical survival outcomes102,103. Copy number variations and gene amplifications are likely to occur when replication-associated TS events bypass genomic regions containing a high number of repetitive sequences such as telomeres, tRNA genes and triplet repeats102. Identifying these TS-dependent copy number variations or gene amplifications could uncover targets to improve chemoresponse or reverse resistance resulting from these genomic rearrangements. For example, gene amplification of the HER2 receptor is currently used to inform clinical treatment of specific breast cancers and other HER2-amplified tumours with the HER2-specific antibody trastuzumab104. It is notable that break-induced replication, which is another fork recovery pathway that rescues collapsed or broken replication forks by promoting a TS-like mechanism105, is also an important source of gross chromosomal rearrangements in cancer cells; an in-depth review of the molecular consequences of break-induced replication can be found in REF.105.
Fork reversal.
Replication fork reversal is activated in response to various replication challenges and promotes re-annealing of complementary daughter strands to form a four-way reversed fork structure24.
Several DNA translocases, including Rad5 in budding yeast106 and RAD54 (REF.107), SMARCAL1 (REFS.108,109), FANCM110, ZRANB3 (REFS.111,112) and HLTF113,114 in mammalian cells, can promote fork reversal, although their exact mechanisms are unclear (FIG. 1). Biallelic SMARCAL1 mutations cause Schimke immunoosseous dysplasia, and these clinical phenotypes are linked to the defective replication-associated DNA damage response observed in SMARCAL1-deficient cells115,116. High SMARCAL1 expression has been observed in pancreatic, testis, breast, prostate and thyroid cancer samples117 (TABLE 1). Germline mutations in FANCM, a member of the Fanconi anaemia (FA) complementation group, lead to increased cancer predisposition, consistent with established roles for FA proteins in genome stability118,119. In the context of human malignancy, ZRANB3 variants have been observed in endometrial cancers120 and ZRANB3 RNA expression is highest in testis cancers relative to other tumour types121 (TABLE 1). RAD54 mutations have been detected in a single case of primary lymphoma and a single case of colorectal cancer122, and tumour-associated RAD54 mutations have been linked with genomic instability in cell models123.
The central recombinase factor RAD51 (REF.24), the F-box DNA helicase 1 (FBH1; a helicase and RAD51 ubiquitination regulator)124 and several RAD51 paralogs125,126 have been implicated in reversed fork formation. Germline RAD51 mutations confer an increased cancer risk, particularly for breast and ovarian cancers127, and RAD51 foci formation has been used to assess the homologous recombination proficiency of cancers128. RAD51 protein levels are increased in pancreatic cancer129, breast carcinomas130 and cancer cell lines131 (TABLE 1). As a result, RAD51 inhibitor development127 has emerged as a chemotherapeutic strategy, particularly in combination with targeted therapies such as poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi).
Resolution of reversed fork structures is mediated in humans by the RECQ1 helicase132. The reversed fork restart activity of RECQ1 is regulated by PARP1, which suppresses RECQ1 activity until the damage is repaired132. A second mechanism of reversed-fork processing and restart depends on human DNA replication helicase/nuclease 2 (DNA2) and Werner syndrome ATP-dependent helicase (WRN)133. RECQ1 and WRN, in addition to BLM, RECQ5 and RECQ4, all belong to the RecQ helicase family, and Bloom syndrome, Werner syndrome and Rothmund–Thomson syndrome arise from germline mutations in BLM, WRN and RECQ4, respectively. Hallmarks of these syndromes include chromosomal instability, developmental abnormalities and increased risk of cancer134. Further, a mutation in the zinc-binding domain of RECQ1 that causes a defective DNA replication and DNA damage response following treatment with topoisomerase poisons is associated with RECON syndrome135. Interestingly, a rare RECQ1 mutation has also been correlated with breast cancer susceptibility136, further emphasizing a role for the RecQ helicases in maintaining genome integrity. BLM and DNA2 expression levels tend to be high across various tumour types100,137, whereas WRN seems to be moderately expressed in cancers of the testis, thyroid, head and neck138 (TABLE 1). RECQ1 expression appears moderate or high in lymphomas, thyroid, head and neck, and carcinoid cancers139 (TABLE 1). Targeting RecQ family helicases might increase sensitivity to DNA-damaging chemotherapeutics by preventing their reported functions in DNA repair and replication, with the caveat that the functional inactivation of these enzymes might be toxic to non-malignant cells.
Fork reversal represents a high-fidelity form of DDT and reversed replication forks must be protected from extensive nucleolytic degradation to preserve genome stability. In addition to their critical roles in homologous recombination, the breast cancer susceptibility proteins BRCA1 and BRCA2 act to protect reversed replication forks140–142 and, in their absence, nucleases such as MRE11 and exonuclease 1 (EXO1) target the open DNA end of the reversed fork substrates, leading to extensive fork degradation. Replication fork degradation in cancer is linked to chemosensitivity, whereas restoration of fork protection is associated with drug resistance140,143,144. Interestingly, extensive fork degradation is not a terminal event as BRCA-deficient cells employ specialized fork recovery pathways to rescue degraded forks and withstand DNA damage. In BRCA2-deficient cancer cells, MUS81 (a structure-specific endonuclease that is expressed at low levels across cancer types)145 and DNA polymerase-δ subunit 3 (Polδ3) cooperate to facilitate a break-induced replication-like mechanism of fork restart146,147. Of note, this break-induced replication-like pathway is not employed in BRCA1-deficient backgrounds146, suggesting that BRCA1-deficient cells recover resected forks through a different pathway. Indeed, ectopic expression of the E3 ubiquitin-protein ligase RNF168, together with the DDT enzymes RAD18 and SLF1, contributes to a break-induced replication-like mechanism at stalled replication forks in BRCA1-deficient cells148. It is unclear whether this axis is also active under conditions of endogenous RNF168 expression and whether additional factors are required for fork recovery in BRCA1-deficient cancer cells when RNF168 is not overexpressed. Interestingly, RNF168 loss in BRCA1-heterozygous mice predisposes these animals to tumour development149, suggesting that RNF168 might also mediate a similar replication fork stress response mechanism in non-malignant cells.
Recent work implicates the Cockayne syndrome protein CSB in fork recovery mechanisms in both BRCA1-deficient and BRCA2-deficient cells150. CSB functions in a break-induced replication mechanism of fork restart that depends on MRE11, MUS81 and RAD52 (REF.150). RNA levels expressed from ERCC6 (encoding CSB) are highest in thyroid and breast tumour samples relative to other cancers151 (TABLE 1). DNA repair protein XRCC1, which is involved in ssDNA break repair, is also involved in replication restart in cells lacking BRCA2 (REF.152). XRCC1 is highly expressed in a diverse range of cancers and is synthetically lethal with BRCA2 (REF.153) (TABLE 1). BRCA2-deficient cells activate XRCC1-mediated microhomology-mediated end joining (MMEJ) in collaboration with MRE11 to facilitate recovery of extensively degraded replication forks152. Collectively, these findings underscore the potential of fork recovery mechanisms as possible therapeutic targets.
Repriming and ssDNA gaps.
Repriming is a highly conserved replication stress response that is present across Escherichia coli154, budding yeast155 and human cells25–27. In human cells, repriming is mediated by PRIMPOL25–27 (FIG. 1), which operates in both mitochondria and nuclei156. Although our understanding of mitochondrial repriming is limited, several studies have documented a key role for PRIMPOL during nuclear DNA replication3,4,28,32,97,156–159. PRIMPOL repriming is generally activated in conditions of impaired fork reversal (for example, upon PARPi treatment or loss of SMARCAL1, HLTF or CARM1 expression)3,32,113, increased PRIMPOL expression4,32 or BRCA deficiency160,161. Interestingly, PRIMPOL expression is elevated in thyroid cancers relative to other tumour types162 (TABLE 1) and the point mutant PRIMPOL-Y100H, which alters unique preference of PRIMPOL for dNTPs163, has been identified in lung carcinoma as reported in the COSMIC database164, suggesting that altering PRIMPOL activity could drive tumour formation.
PRIMPOL-dependent repriming introduces ssDNA gaps downstream of the replication obstacle, leaving these gaps to be repaired post-replicatively4,32,97,113,157. Recent studies in cells challenged with cisplatin suggest that there are at least two temporally distinct pathways that repair ssDNA gaps: in G2-phase, a TLS mechanism dependent on RAD18, PCNA monoubiquitination, and REV1 and Polζ promotes gap filling, whereas gap filling is mediated by a TS-like mechanism dependent on UBC13 and RAD51 in S-phase32. The choice and timing of a particular gap-filling pathway likely varies with genetic background and the replication roadblock bypassed during the initial repriming event32,165. Potential risks of these gap-filling mechanisms include mutagenesis in the case of TLS, and SCEs or chromosomal rearrangements that could contribute to genomic instability in TS. Failure to fill the ssDNA gaps leads to persistent ssDNA stretches, which are susceptible to cleavage and nucleolytic processing, potentially contributing to double-stranded DNA break accumulation. Recent studies revealed that accumulation of ssDNA gaps or impaired gap filling increase chemosensitivity upon treatment with PARPi, particularly in BRCA-deficient cancer cells32,160,161,166,167. Recent evidence also implicates the accumulation of ssDNA gaps in contexts where Okazaki fragment maturation and chromatinization is compromised167,168. Thus, factors that promote ssDNA gap generation and the subsequent step of gap filling represent attractive targets to modulate chemotherapy response.
In summary, diverse replication fork stress response mechanisms have different effects on genome stability and tumour development. These findings raise several questions regarding how the differential usage of these pathways affect chemoresponse and clinical outcomes; whether relevant combinatorial treatments could effectively target replication stress response and improve chemosensitivity or combat chemoresistance in a clinical setting; and whether factors involved in replication stress response could serve as useful clinical biomarkers. The next sections outline current research aimed at answering these critical questions.
DNA replication stress in cancer therapy
Targeting TLS and DNA repair polymerases in cancer.
RAD18 has been investigated as a promising target for cancer treatment owing to its elevated expression across many tumour types and role in initiating TLS (FIG. 2). Indeed, targeting RAD18 with a specific microRNA has been shown to sensitize resistant colorectal carcinoma cells to chemotherapy in vitro169. Recent data reveal that knockout of RAD18 in BRCA1-deficient or BRCA2-deficient cancer cells increases DNA damage and formation of unrepaired ssDNA gaps, leading to cell death161. Further, RAD18-deficient cancer cells are more sensitive than wild type cancer cells to crosslinking agents, including mitomycin C and cisplatin170. The increased sensitivity of RAD18-deficient cells to crosslinking agents could be associated with an additional role of RAD18 in regulating ubiquitination of the FA protein FANCD2 (REFS.170,171), which is a central factor involved in inter-strand crosslink repair172,173. Following these observations, recent studies have screened for chemical inhibitors of the RAD18 pathway by specifically targeting the interaction between RAD18 and its upstream E2-conjugating enzyme partner RAD6 (REF.174), paving the way for future preclinical studies.
Fig. 2 |. The replication stress response in cancer and emerging therapeutic targets.
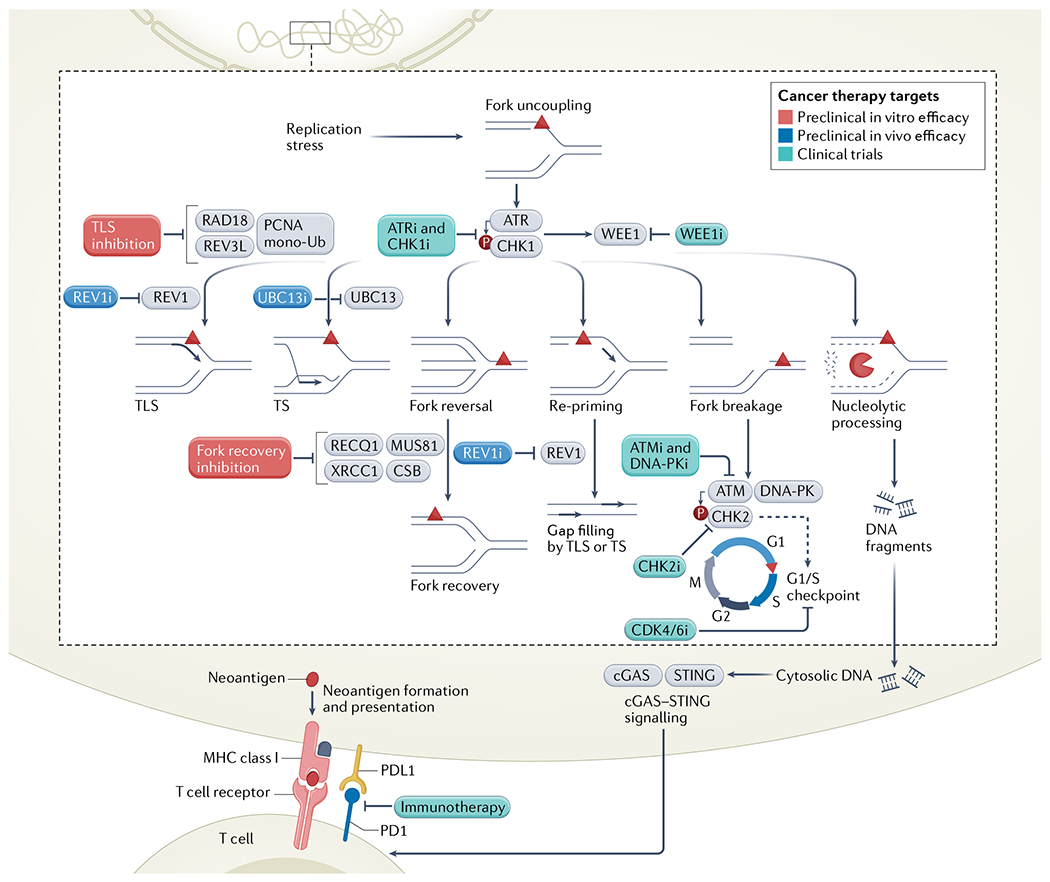
Upon replication fork stalling, single-stranded DNA exposure leads to the activation of the ATR checkpoint and the downstream CHK1 and WEE1 proteins. These signalling pathways can be targeted by ATR, CHK and WEE1 inhibitors, which are being tested in clinical trials. Following ATR induction, cells activate diverse replication stress response pathways, including translesion synthesis (TLS), template switching (TS), fork reversal and repriming. Inhibitors of REV1 and UBC13 have been tested in vivo in several cancer types, and REV1 inhibitors could also be used to target single-stranded DNA gap filling following repriming. Additional TLS enzymes and factors involved in replication fork reversal have also shown promise as therapeutic targets in vitro. Replication-associated breaks activate the ATM, DNA-dependent protein kinase (DNA-PK) and CHK2 kinases. Inhibitors of ATM, DNA-PK and CHK2 are being evaluated for anticancer potential in clinical trials, in addition to CDK4/6 inhibitors, which are associated with cell cycle arrest and antiproliferative effects. Stalled replication forks can also be processed by nucleases, and extensive resection can promote release of DNA fragments into the cytosol, stimulating the cGAS–STINC pathway. cGAS–STINC activation can augment the cancer cell response to immunotherapy, a strategy that is used widely in clinical trials and in the clinic. Inhibitors of DNA replication stress response factors that are already in clinical trial are shown in green, those that have shown preclinical in vivo efficacy are in blue, and those with in vitro efficacy are in red; they are all indicated by the letter i. mono-Ub, monoubiquitination; PCNA, proliferating cellular nuclear antigen.
PCNA monoubiquitination can be directly targeted using the small molecular inhibitor T2-amino alcohol175. Treatment with T2-amino alcohol prevents repair of interstrand DNA crosslinks and increases DNA double-stranded breaks (DSBs) and sensitivity to cisplatin in cell-based assays176. Similarly, preventing PCNA monoubiquitination by mutating lysine 164 to arginine decreases cell proliferation167 and increases sensitivity to UV treatment167,177. Loss of PCNA monoubiquitination increases the response of cells lacking BRCA1 or BRCA2 to PARPi and cisplatin167.
Targeting the deubiquitinase USP1, which removes ubiquitin from monoubiquitinated PCNA, shows promise in exacerbating replication stress and increasing DNA damage in cancer cells178,179. As a result, a clinical trial for advanced solid tumours has recently been developed using a first-in-class USP1 inhibitor, both alone and in combination with PARPi (TABLE 1).
TLS polymerases downstream of RAD18-mediated PCNA ubiquitination have been explored as targets to modulate cancer cell survival and improve therapy response. REV1 expression is associated with the development of chemoresistance to platinum-based drugs in ovarian cancer models180,181 and loss of REV1 in BRCA-deficient cancer cells leads to decreased viability similar to the effects of RAD18 downregulation161. However, the degree of epistasis between these two proteins is untested. A newly developed chemical inhibitor of REV1, JH-RE-06, has been shown to bind to the REV1 C-terminal domain and promote REV1 dimerization182, rendering the enzyme unable to recruit Polζ and initiate TLS. Treatment of mice carrying patient-derived melanoma xenografts with JH-RE-06 in combination with cisplatin substantially reduced tumour burden182. Treatment with a different TLS inhibitor, which also inhibits the interaction between REV1 and TLS enzymes, selectively kills cancer cell lines that rely on TLS for replication, including cells expressing FANCJ-S990A (a mutant copy of the helicase FANCJ that is unable to interact with BRCA1) and cells lacking the negative TLS regulator p21 (REF.166). In these ‘pro-TLS’ backgrounds, treatment with this TLS inhibitor synergizes with other replication stress-inducing agents, including inhibitors of ATR or WEE1 (REF.166), an effect that is attributed to the accumulation of ssDNA gaps. Interestingly, REV1 inhibition preferentially sensitizes BRCA-deficient cancer cells relative to wild type models both in the context of JH-RE-06 monotherapy161 and in combination with cisplatin and PARPi treatment32, which might be a result of the formation of ssDNA gaps161 and decreased gap filling32 in BRCA-deficient backgrounds.
Notably, REV1 loss or inhibition does not sensitize cells to ionizing radiation, possibly owing to the upregulation of autophagy, which is a hallmark of radioresistance183. The complex relationship between autophagy and cancer therapy responses is summarized in REF.184. The impact of TLS polymerase inhibition on autophagy should be considered when targeting these proteins for therapeutic benefit.
Promising results have been demonstrated with in vitro optimization of small-molecule inhibitors that target other TLS enzymes such as Polη and Polκ185–187. Preliminary studies revealed that targeting of Polκ with a small-molecule inhibitor increases sensitivity to the alkylating agent temozolomide in vitro185. Lower Polζ expression is associated with improved response to cisplatin and gemcitabine chemotherapies in head and neck squamous cell carcinomas63 and increased POLN mRNA expression is associated with worsened overall survival in non-small-cell lung cancer (NSCLC)188. Studies also indicate that increased Polζ expression is linked to cisplatin resistance in bladder cancers189 and in ovarian cancer stem cells62. Indeed, Polζ and Polκ have both been shown to facilitate replication past platinum adducts190, suggesting that these TLS polymerases could be targeted to improve platinum-based therapies. A recent study highlighted that Polκ also enables cancer cells to tolerate replication stress resulting from aberrant cyclin-dependent kinase 2 (CDK2) activation191, which can be induced by cyclin E overexpression or WEE1 inhibition191. Consistent with these findings, loss of Polκ or RAD18 sensitizes cancer cells to WEE1 inhibition191.
Polζ might represent a target for improving chemosensitivity and decreasing drug resistance. Increased expression of REV3L — the catalytic subunit of Polζ — is associated with cisplatin resistance in gliomas55 and REV3L loss sensitizes chemoresistant models of NSCLC to platinum-based chemotherapy192. Similarly, loss of REV7 — the accessory subunit of Polζ — improved cisplatin response in a mouse model of NSCLC193, and increased REV7 expression correlates with worsened survival outcomes in patients with diffuse large B cell lymphoma (DLBCL)194. Depletion of REV7 also increases the sensitivity of clear cell ovarian carcinoma to cisplatin195. However, in BRCA1-deficient backgrounds, REV7 loss contributes to PARPi resistance, an effect attributable to its role in non-homologous end joining (NHEJ)196,197. As a result, the impact of REV7 on both TLS and NHEJ must be considered when targeting REV7 across cancers.
Polθ, a member of the A-family of polymerases198 that functions in MMEJ199,200, represents a promising therapeutic target, particularly in BRCA-deficient cancers199–201. Because of its essential role in MMEJ, Polθ loss is synthetically lethal with deficiencies in other DSB repair pathways, including homologous recombination199,200 (FIG. 3), and Polθ inhibition in BRCA-deficient cancer cells increases sensitivity to cisplatin and PARPi199. In addition to its role in MMEJ, Polθ is implicated in the repair of breaks arising from collapsed replication forks, tolerance of G-quadruplex DNA secondary structures and replication stress response upon fork stalling202. Additional work will be critical to define the contributions of each of these mechanisms to chemoresponse as a Polθ inhibitor has recently been combined with PARPi in a clinical trial (TABLE 1). Polθ expression levels and mutational signatures could represent Powerful clinical biomarkers to assess the efficacy of newly developed Polθ inhibitors across a range of tumour types199,203–205.
Fig. 3 |. Chemotherapy and PARPi in BRCA-deficient cancers.
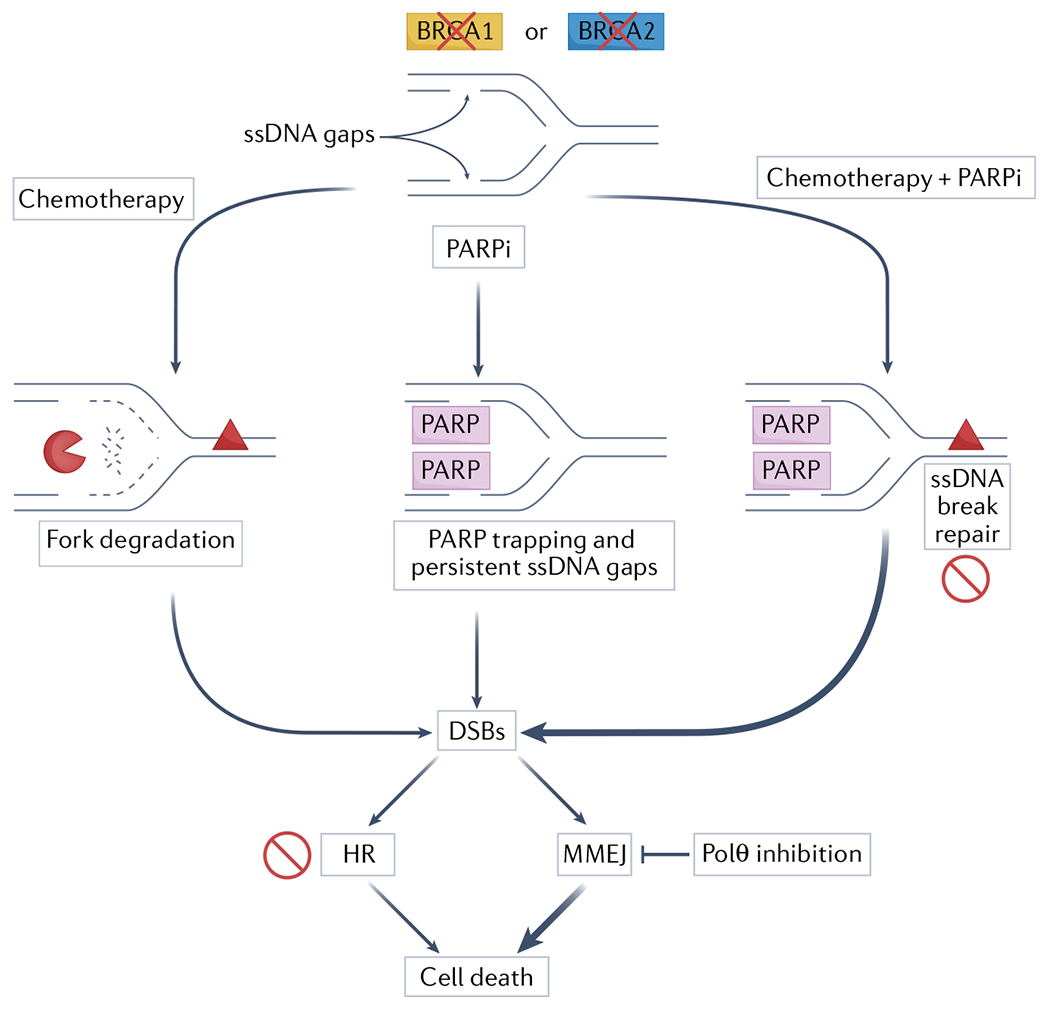
Replication-associated single-stranded DNA (ssDNA) gaps accumulate in BRCA-deficient cancer cells. Upon treatment with DNA-damaging chemotherapy, replication forks are subject to extensive nucleolytic degradation, leading to fork breakage and formation of one-ended double-stranded DNA breaks (DSBs). Degradation mainly originates from reversed forks (not shown). Poly(ADP-ribose) polymerase (PARP) inhibition impairs reversal (not shown), and causes trapping of PARP proteins and persistent ssDNA gaps, which lead to DSBs. Combination of chemotherapy and PARP inhibitor (PARPi) therapy exacerbates DSB formation by blocking ssDNA break repair of chemotherapy-induced lesions in addition to causing ssDNA gap accumulation and PARP trapping. In BRCA-deficient backgrounds, these DSBs cannot be processed by homologous recombination, leading to cell death. These cells also become increasingly reliant on microhomoLogy-mediated end joining (MMEJ) to repair breaks, which provides a rationale for targeting Polθ in BRCA-mutant tumours.
Targeting TS and replication fork recovery in cancer.
Linking differential expression levels of UBC13, RAD51, BLM and NBS1 to defects in TS and to cancer chemotherapy response is complicated by the lack of direct methods for investigating homology-mediated TS mechanisms that do not necessarily involve strand transfer. In addition, these proteins have multiple cellular roles: UBC13 promotes DSB signalling and ubiquitinates cytosolic NF-κB pathway targets206; RAD51 plays multiple roles in replication fork stability125; and RAD51, BLM and NBS1 function in homologous recombination96. Consequently, we note that any potential chemotherapeutic benefit associated with targeting these factors cannot be absolutely associated with changes in TS efficiency.
UBC13 upregulation promotes breast and colorectal cancer cell metastasis through JNK and MAP kinase activation207,208 and melanoma growth through MEK signalling209 although another study proposes that UBC13 is downregulated in paclitaxel-resistant ovarian cancer cells, with lower expression contributing to worsened outcomes210. Interestingly, UBC13 inhibition with a small molecular inhibitor, NSC697923, has been shown to kill neuroblastoma cells211, DLBCL cells212 and melanoma cells in vitro209. Differences across studies might point to tumour-specific impacts of UBC13 expression and activity and indicate a need to evaluate any off-target effects of UBC13 inhibitors that could contribute to observed cell-killing phenotypes. In addition to UBC13, downstream TS factors might also constitute potential clinical targets and biomarkers.
Loss of the fork recovery factor MUS81 is associated with increased hydroxyurea sensitivity in BRCA2-deficient cancer cells146 and, similarly, CSB downregulation increases hydroxyurea sensitivity in BRCA1-deficient and BRCA2-deficient backgrounds150. Moreover, MUS81 promotes progression of serous ovarian carcinoma213 and knockdown increases sensitivity of epithelial ovarian cancer to PARPi214. Both overexpression and downregulation of RNF168 decrease viability in BRCA1-deficient cancers215. These data suggest that the relationship between RNF168 expression and cell survival is distinct from the roles of other recovery factors such as MUS81 and CSB, whose targeting can increase sensitivity to replication stress inducers. Future research should focus on defining whether, in addition to its role in fork recovery148, the roles of RNF168 in chromatin ubiquitination216 and DNA damage signalling149 should be considered when targeting this factor to improve cancer cell chemoresponse.
Leveraging replication stress in PARPi cancer therapy.
The development of PARPi therapies has significantly improved survival outcomes in homologous recombination-deficient cancers. Therapies using PARPi and chemotherapy are proposed to kill homologous recombination-deficient tumours, such as those harbouring BRCA1 or BRCA2 mutations, through synthetic lethality217,218. PARPi leads to trapping of PARP proteins on DNA and causes an increase in ssDNA breaks, which are converted into irreparable DSBs during replication in BRCA-deficient tumours218–220 and lead to cell death (FIG. 3). Toxic DSBs might also originate from the degradation and collapse of stalled replication forks upon treatment with DNA-damaging chemotherapy that cannot be adequately protected in the absence of BRCA proteins140–142 (FIG. 3). Interestingly, PARPi has also shown promise in targeting homologous recombination-proficient cancer cells as loss of RNase H2, which is involved in removal of erroneous ribonucleotides from the DNA, sensitizes BRCA-proficient cells to olaparib221.
PARPi efficacy is hampered by the development of resistance222,223 and several regulators of PARP trapping have recently emerged as key modulators of PARPi224–226. Reported mechanisms of PARPi resistance (reviewed in REF.227) include BRCA reversion mutations that restore homologous recombination in these tumours; upregulation of efflux pumps that clear PARPi from cancer cells; restoration of homologous recombination through the downregulation of NHEJ factors, including tumour suppressor P53-binding protein 1 (53BP1) and the Shieldin complex; diminished PARP trapping via PARP mutations; and rescued PARylation and decreased binding of PARP to DNA through loss of poly-ADP ribose glycohydrolase (PARG)228, which opposes PARP activity. Another emerging mechanism of chemoresistance is the restoration of replication fork stability in BRCA-deficient cancer cells, independent of the re-establishment of homologous recombination function in these genetic backgrounds140,229.
ssDNA gaps are frequently formed as a consequence of replication stress and several studies propose that the accumulation of ssDNA gaps in BRCA-deficient cancer cells, exacerbated by treatment with PARPi, modulates cancer cell survival and drug sensitivity8,32,160,161,166,167,230 (FIG. 3). Therefore, PRIMPOL, which generates ssDNA gaps during repriming and is regulated by the ATR4 and CHK1 (REF.231) kinases (discussed below), might represent a key regulator of cancer response to PARPi or emerging ATR and CHK1 inhibitors.
While PRIMPOL activity is typically associated with leading-strand ssDNA gaps, recent evidence suggests that aberrant Okazaki fragment processing (OFP) could lead to ssDNA gaps on the lagging DNA strand5,6,167,168. Defects in the canonical OFP pathway232, which involves flap endonuclease 1 (FEN1) and DNA ligase I, or a backup OFP mechanism that relies on PARP, XRCC1 and DNA ligase 3 (REF.6) have been implicated in increased sensitivity to cancer therapies, including to PARPi. Restoration of efficient OFP or upregulation of OFP pathways may also contribute to chemoresistance in certain genetic contexts, including BRCA1-deficient cancer cells8. Moreover, models of PARPi resistance in BRCA-deficient cancer cells can be re-sensitized to PARPi through depletion of DNA ligase 3 (REF.233), which could be explained by an increased reliance of BRCA-deficient cells on DNA ligase 3-mediated OFP or base excision repair. The ssDNA gap accumulation model of chemoresponse234,235 raises the important possibility that gap-filling mechanisms can be targeted to sensitize BRCA-deficient tumours to PARPi and other DNA-damaging chemotherapy to overcome chemoresistance in these cancers.
There are distinctions to be made between in vitro, in vivo and clinical models of PARPi resistance. Patient data has revealed cases of PARPi resistance caused by reversion mutations in BRCA1 and BRCA2 (REF.227) as well as by diminished PARP trapping via a PARP1 mutation in a single instance227. Preclinical in vivo model data support PARPi resistance through decreased 53BP1 and Shieldin expression in patient-derived xenografts236. However, it remains to be shown whether restoration of replication fork stability or changes in ssDNA gap formation and repair impact clinical PARPi resistance. Translation of findings from in vitro models of chemotherapy resistance to clinical models of disease is complicated by the fact that multiple chemoresistance mechanisms can be activated in the same cell and across cells within the same tumour. In vitro studies must be expanded to other BRCA-mutated and wild type tumour types and potentially combine parallel assessments of different mechanisms of chemoresistance found in the clinical setting.
Exacerbating replication stress in cancer with cell cycle-checkpoint inhibitors.
DDT mechanisms are temporally regulated throughout the cell cycle19 and checkpoint inhibitors are emerging as promising drugs for cancer treatment as they affect the ability of specific DTT pathways to repair or bypass a lesion in S-phase before cells enter G2-phase or reach mitosis.
ATR kinase orchestrates different cellular pathways in response to replication stress, including the enforcement of an S/G2 checkpoint237, regulation of intracellular dNTP levels and origin firing. ATR might also play a role in replication fork reversal but its contribution is unclear238–240. The ATR signalling cascade, involving CHK1 phosphorylation, is activated upon exposure of stretches of ssDNA that form during replication fork stalling and uncoupling241. Based on the role of ATR in preserving replication fork stability and enforcing an appropriate cell-checkpoint response, inhibition of ATR kinase and of its downstream CHK1 substrate are relevant strategies to improve cancer chemoresponse (FIG. 2). Preclinical data show that ATR inhibitors and CHK1 inhibitors can re-sensitize PARPi-resistant, BRCA1-deficient cancer cells to PARPi, making the ATR–CHK1 pathway an attractive therapeutic target in settings of drug resistance229,242. CHK1 inhibition also decreases tumour growth in mouse models lacking activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1), which has been uncovered as a key regulator of the cell cycle243–245. Multiple clinical trials have used preclinical mechanistic insight to inform combination therapies with ATR inhibitors and either PARPi, platinum-based chemotherapy, antimetabolites or radiotherapy (TABLE 2). Similarly, CHK1/2 inhibitors have been included in clinical trials as a monotherapy or in combination with PARPi, gemcitabine and even PDL1 blockade (TABLE 2). Importantly, the inhibition of ATR appears to have lower toxicity than inhibition of CHK1, which could be related to the non-specificity of some CHK1 inhibitors that target both CHK1 and CHK2 (TABLE 2). Activation of ATR and CHK1 might be a useful biomarker to predict tumour response to emerging targeted and combinatorial therapies that induce replication stress. For a comprehensive review of ATR kinase and its functions at replication forks, we direct readers to REFS.1,241.
Table 2 |.
Clinical trials with emerging targeted therapies
| Inhibitor | Cancers | Combination treatments | Clinical trial numbera |
|---|---|---|---|
| ATR inhibitors | |||
| ART0380 | Advanced cancer or metastatic cancer, ovarian cancer, primary peritoneal cancer, fallopian tube cancer | Gemcitabine (antimetabolite) and irinotecan (topoisomerase inhibitor) | NCT04657068 |
| AZD4547 | NSCLC, squamous cell carcinoma, adenocarcinoma | Durvalumab (PDL1 blockade) | NCT02664935 |
| AZD6738 | High-grade serous carcinoma | Olaparib (PARPi) | NCT03462342 |
| Advanced solid tumours, advanced pancreatic adenocarcinoma | Gemcitabine (antimetabolite) | NCT03669601 | |
| CCRCC, locally advanced pancreatic cancer, metastatic renal cell carcinoma, metastatic urothelial carcinoma, metastatic pancreatic cancer | Olaparib (PARPi) | NCT03682289 | |
| Breast cancers | Olaparib (PARPi) and durvalumab (PDL1 blockade) | NCT03740893 | |
| Prostate cancer | Olaparib (PARPi) | NCT03787680 | |
| Gynaecological cancers | Olaparib (PARPi) | NCT04065269 | |
| BAY1895344 | Advanced solid tumours | Pembrolizumab (PDL1 blockade) | NCT04095273 |
| Advanced solid tumours (excluding prostate cancer) and ovarian cancer | Niraparib (PARPi) | NCT04267939 | |
| Berzosertib (also known as M6620 and VX-970) | Ovarian serous tumour, recurrent fallopian tube carcinoma, recurrent ovarian carcinoma, recurrent primary peritoneal carcinoma | Gemcitabine (antimetabolite) | NCT02595892 |
| Metastatic colorectal, lung, small-cell lung and pancreatic carcinomas; refractory colorectal, small-cell lung and pancreatic carcinomas; unresectable colorectal, small-cell lung and pancreatic carcinomas | Irinotecan (topoisomerase inhibitor) | NCT02595931 | |
| SCLC, high-grade neuroendocrine cancers | Lurbinectedin (transcription inhibitor) | NCT04802174 | |
| Adult leiomyosarcoma | Gemcitabine (antimetabolite) | NCT04807816 | |
| Homologous recombination-deficient cancer, SCLC and advanced solid tumours | Sacituzumab govitecan (topoisomerase inhibitor antibody conjugate) | NCT04826341 | |
| NSCLC, SCLC, ovarian cancers, uterine cervical cancers, neuroendocrine carcinoma, extrapulmonary small cell cancer | Topotecan (topoisomerase inhibitor) | NCT02487095 | |
| Advanced stage solid tumours | Monotherapy or in combination with carboplatin and paclitaxel | NCT03309150 | |
| Oesophageal adenocarcinoma, squamous cell carcinoma, solid tumours | Cisplatin, capecitabine (antimetabolite) and radiation | NCT03641547 | |
| Advanced solid tumours | Topotecan (topoisomerase inhibitor) | NCT05246111 | |
| Refractory solid tumours | Veliparib (PARPi) and cisplatin (crosslinking agent) | NCT02723864 | |
| Ceralasertib | Advanced solid tumours, head and neck squamous cell carcinoma, NSCLC, gastric cancer, breast cancer and ovarian cancer | Carboplatin (crosslinking agent) | NCT02264678 |
| Head and neck squamous cell carcinoma | Olaparib (PARPi) | NCT03022409 | |
| Metastatic triple-negative breast cancer | Olaparib (PARPi) | NCT03330847 | |
| Elimusertib | Advanced cancers of the gastrointestinal system | Fluorouracil, irinotecan (topoisomerase inhibitor) and leucovorin (enhances fluorouracil) | NCT04535401 |
| Various metastatic and unresectable cancers | Irinotecan and topotecan (topoisomerase inhibitors) | NCT04514497 | |
| HPV-mediated oropharyngeal carcinoma | Pembrolizumab (PDL1 blockade) and radiation | NCT04576091 | |
| IMP9064 | Advanced adult solid tumours | Senaparib (PARPi) | NCT05269316 |
| M1774 | Metastatic or locally advanced unresectable solid tumours | Niraparib (PARPi) | NCT04170153 |
| M4344 | PARPi-resistant recurrent ovarian cancer | Niraparib (PARPi) | NCT04149145 |
| RP-3500 | Advanced solid tumours | Talazoparib (PARPi) and gemcitabine (antimetabolite) | NCT04497116 |
| Advanced solid tumours | Niraparib or olaparib (PARPi) | NCT04972110 | |
| VX-970 | Refractory solid tumours | Veliparib (PARPi) and cisplatin (crosslinking agent) | NCT02723864 |
| WEE1 inhibitors | |||
| Adavosertib (also known as AZD1775 and MK-1775) | Glioblastoma and recurrent glioblastoma | Temozolomide (alkylating agent) and radiation | NCT01849146 |
| Cancers of the nervous system | Irinotecan (topoisomerase inhibitor) | NCT02095132 | |
| Ovarian tumours, recurrent fallopian tube carcinoma, recurrent ovarian carcinoma and recurrent primary peritoneal carcinoma | Gemcitabine (antimetabolite) | NCT02101775 | |
| Metastatic or unresectable pancreatic adenocarcinoma | Gemcitabine (antimetabolite) and paclitaxel (antimicrotubule) | NCT02194829 | |
| Metastatic triple-negative breast cancer | Ceralasertib (ATR inhibitor) and olaparib (PARPi) | NCT03330847 | |
| Cancers of the female reproductive system | Cisplatin (crosslinking agent) and radiation | NCT03345784 | |
| Prostate cancer | Monotherapy | NCT03385655 | |
| Advanced solid tumours | Carboplatin and paclitaxel | NCT02341456 | |
| Head and neck squamous cell carcinoma | Cisplatin and radiation | NCT02585973 | |
| Leukaemias and myelodysplastic syndromes/cancers | Cytarabine (antimetabolite) | NCT02666950 | |
| Metastatic colorectal cancer | Irinotecan (topoisomerase inhibitor) | NCT02906059 | |
| Triple-negative metastatic breast cancer | Cisplatin (crosslinking agent) | NCT03012477 | |
| Hypopharynx squamous cell carcinoma, oral cavity squamous cell carcinoma and larynx cancer | Cisplatin and radiation | NCT03028766 | |
| Epithelial ovarian cancer | Carboplatin (crosslinking agent) | NCT01164995 | |
| Pancreatic adenocarcinoma | Gemcitabine (antimetabolite) and radiation | NCT02037230 | |
| Head and neck squamous cell carcinoma | Cisplatin and docetaxel (antimicrotubule) | NCT02508246 | |
| CHK1 inhibitors | |||
| LY2603618 | NSCLC | Pemetrexed (antimetabolite) and cisplatin | NCT01139775 |
| LY2880070 | Ewing sarcoma and Ewing-like sarcoma | Gemcitabine (antimetabolite) | NCT05275426 |
| Prexasertibb | Ovarian cancer, breast cancer, prostate cancer | Monotherapy | NCT02203513 |
| Advanced cancers | Monotherapy | NCT02873975 | |
| Advanced solid tumours | Olaparib (PARPi) | NCT03057145 | |
| Advanced solid tumours | LY3300054 (PDL1 blockade) | NCT03495323 | |
| Recurrent, refractory, or paediatric brain tumours, medulloblastoma and CNS tumours | Cyclophosphamide (alkylating agent) and filgrastim (biologic agent) | NCT04023669 | |
| SCH 900776 | Adult leukaemias | Cytarabine (antimetabolite) | NCT01870596 |
| SRA737 | Advanced solid tumours | Gemcitabine (antimetabolite) and cisplatin | NCT02797977 |
| ATM inhibitors | |||
| AZD0156 | Advanced solid tumours | Olaparib (PARPi), irinotecan (topoisomerase inhibitor) and fluorouracil (antimetabolite) | NCT02588105 |
| AZD1390 | Glioblastoma, brain cancers and leptomeningeal disease | Radiation | NCT03423628 |
| CHK2 inhibitors | |||
| PHI-101 | Platinum-refractory or resistant ovarian carcinoma, platinum-resistant fallopian tube carcinoma and platinum-resistant primary peritoneal carcinoma | Monotherapy | NCT04678102 |
| DNA-PK inhibitors | |||
| AZD7648 | Advanced cancers | Pegylated liposomal doxorubicin (topoisomerase inhibitor) | NCT03907969 |
| Peposertib (also known as nedisertib and M3814) | Advanced solid tumours | Cisplatin and radiation | NCT02516813 |
| Solid tumours | Avelumab (immune-checkpoint blockade) and palliative radiation | NCT03724890 | |
| Glioblastoma and gliosarcoma | Temozolomide (alkylating agent) and radiation | NCT05002140 | |
| Locally advanced rectal cancer | Capecitabine (antimetabolite) and radiation | NCT03770689 | |
| Inhibitors of both ATM and DNA-PK | |||
| XRD-0394 | Metastasis, locally advanced solid tumours and recurrent cancer | Palliative radiation | NCT05002140 |
| Polθ inhibitors | |||
| ART4215 | Advanced or metastatic cancer and breast cancer | Talazoparib (PARPi) | NCT04991480 |
| USP1 inhibitors | |||
| KSQ-4279 | Advanced solid tumours | PARPi | NCT05240898 |
CCRCC, clear cell renal cell carcinoma; CNS, central nervous system; DNA-PK, DNA-dependent protein kinase; NSCLC, non-small-cell lung carcinoma; PARPi, poly(ADP-ribose) polymerase inhibitor; SCLC, small cell lung carcinoma.
NCT numbers, along with cancer types and drugs, were compiled from completed, recruiting and active trials from ClinicalTrials.gov.
Prexasertib has also shown inhibitory activity against CHK2; however, the biological antitumour activity of prexasertib has been linked to CHK1 inhibition and not CHK2 inhibition.
The G2 checkpoint kinase WEE1, which is downstream of ATR and CHK1, shows promise as a therapeutic target (FIG. 2 and TABLE 2). WEE1 inhibitors reduce tumour growth in combination with ATR inhibitors in mouse models of DLBCL246, are synergistic with CHK inhibitors in acute lymphoblastic leukaemia247 and improve PARPi response in triple-negative breast cancer248,249. WEE1 inhibition contributes to replication fork stress by disrupting nucleotide pools, leading to replication fork collapse and DSBs250, and by promoting replication fork degradation251. In addition, treatment of ex vivo models of ovarian cancer with CHK1 inhibitors or with both CHK1 and WEE1 inhibitors increases sensitivity to PARG inhibition, which induces replication fork catastrophe and increased DNA damage252. These data provide mechanistic insight into the potential clinical efficacy of combining CHK1 and WEE1 inhibitors with replication stress-inducing agents such as PARPi and PARG inhibitors.
In the absence of ATR, additional kinases, such as ATM and DNA-dependent protein kinase (DNA-PK), can phosphorylate CHK1 upon replication stress253. ATM is also involved in sensing DSBs, including those resulting from collapsed replication forks, and phosphorylates CHK2 as part of the DSB signalling cascade254. As a result, ATM and CHK2 inhibitors have entered clinical trials in combination with chemotherapeutic agents that induce replication stress, including PARPi (TABLE 1). Drugs that specifically target CHK2 have been investigated in fewer clinical trials than those that target CHK1, which suggests that they might be less effective as antineoplastics. Indeed, this difference might be connected to CHK2 being non-essential in cells, whereas CHK1 is an essential gene255. ATM-deficient tumours have been targeted with a wide range of drugs, including platinum-based agents, ATR inhibitors, PARPi and CHK1/2 inhibition strategies256,257. In addition to ATR and ATM, DNA-PK plays critical roles both in DSB repair through NHEJ and in the replication stress response. Upon replication stress induction, DNA-PK facilitates ATR–CHK1 checkpoint activation258, and concurrent ATR and DNA-PK inhibition increases radio-sensitivity in colon and head and neck squamous cell carcinoma cell lines259. In vitro data show that DNA-PK inhibition can improve cancer cell response to PARPi, doxorubicin and radiation treatment, and ATM-deficient cancer cells are also highly sensitive to combined DNA-PK inhibition and PARPi treatment260. As a result, DNA-PK inhibitors are in clinical development for the treatment of solid malignancies (TABLE 2).
CDK inhibitors that impair cell cycle progression are increasingly used in the clinic, including as second-line therapy for breast, prostate and ovarian cancer261. However, resistance to CDK inhibitors — and particularly to CDK4/6 inhibitors — is seen frequently and the pleotropic effects of many CDK inhibitors make it difficult to pinpoint mechanisms that underlie differential sensitivity262,263. CDK inhibitors typically arrest cells in G1/S phase (FIG. 2), which can decrease replication-associated toxicity induced by DNA-damaging chemotherapy or radiotherapy264,265. As a result, the blunting of chemotherapy by CDK inhibitors is a relevant concern266. Despite these concerns, recent preclinical findings show that CDK4/6 inhibitors have synergistic effects with PARPi267. Pre-treatment with the CDK4/6 inhibitor palbociclib sensitizes cancer cells to a wide range of genotoxic agents, such as aphidicolin, camptothecin and doxorubicin, and is associated with prolonged replication stress268 (FIG. 2). Post-treatment with palbociclib following incubation with DNA-damaging agents, such as gemcitabine, cisplatin and topoisomerase poisons, also enhances cancer cell killing269. Although most CDK inhibitors are used as monotherapies, a number of clinical trials have tested combinations of CDK inhibitors, particularly CDK4/6 inhibitors, with platinum-based agents, epirubicin and gemcitabine; however, toxicity with these therapies remains a challenge270.
Bolstering immunotherapy response in cancer with replication stress induction.
Recent studies have explored a connection between replication fork perturbations, inflammatory signalling and cancer immunotherapy response. The finding that STING inflammatory signalling is upregulated in response to replication stress271–273 suggests that replication stress might cause the release of DNA fragments from the nucleus into the cytosol, causing cGAS–STING pathway activation. Further studies have implicated the processing of stalled replication forks271,274,275 and micronuclei276,277 as sources of these DNA fragments. Upregulation of an interferon-like response by cGAS–STING leads to T cell priming and recruitment and can boost the efficacy of immunotherapies278, suggesting that combinatorial treatments exploiting STING-inducing replication stress with immune-checkpoint blockade represent a promising therapeutic strategy (FIG. 2).
Increased expression of specific TGFβ-responsive genes is associated with immunotherapy resistance in gynaecological cancers279, and additional work has shown that blocking TGFβ signalling restrains tumour growth in a breast cancer mouse model that is resistant to immune-checkpoint blockade280. One study identified that loss of Mediator complex subunit 12 (MED12; a coactivator that functions in transcription) contributes to chemoresistance by upregulating TGFβ signalling and restoring replication fork stability in BRCA-deficient cancer cells281. In BRCA-competent cells, BRCA1, which plays roles in both DSB repair and replication fork stability, is downregulated by TGFβ through its interactions with miR-182 (REF.282); upon stimulation by TGFβ, this microRNA decreases BRCA1 protein levels in mouse and human cells282,283. These data highlight MED12 and the TGFβ signalling axis as promising therapeutic targets for combatting immune therapy resistance and improving drug sensitivity, particularly through promoting replication stress and decreasing DNA-damage repair capacity. TGFβ preserves genomic stability more broadly by mediating ATM and p53 checkpoint activation and promotion of the DNA damage response and DSB repair through SMAD proteins284–286. To refine this preliminary evidence, additional studies must define the contribution of the TGFβ pathway across cancer cell types, especially in cancer stem cell populations in which TGFβ expression has been suggested to both suppress287 and promote cancer stem cell features in different contexts288,289.
Several publications broadly suggest that tumour mutational burden (TMB) is linked to immunotherapy response290,291. While the impact of TMB on therapy response is not straightforward in every cancer subtype nor with every drug regimen, TMB is generally proposed to correlate with increased neoantigen formation292. These neoantigens are recognized by T cells293 and could boost efficacy of immune-checkpoint blockade and improve tumour killing. To date, and based on the preclinical data described above, numerous clinical trials have combined immune-checkpoint blockade with a range of chemotherapeutics294,295 (FIG. 2). One study also uncovered a “replication stress response gene expression signature”, which was predictive of immune-checkpoint blockade response in preclinical cancer models296, suggesting that replication stress-linked TMB or neoantigen formation could be useful as biomarkers. These data emphasize the importance of further mechanistic investigation to exploit the link between replication stress pathways, DNA damage response and immune system activation.
DNA replication stress and clinical biomarkers.
DDT pathways could be useful for the development of new clinical biomarkers (FIG. 4). TLS enzymes are generally elevated across select tumour types37,56,57,62,63,72,73, possibly because of their contribution to mutagenesis, which drives carcinogenesis and also promotes tumour evolution and resistance to chemotherapy. Increased RAD18, PCNA ubiquitination and TLS polymerase expression relative to normal tissue controls or over time within the same tumour might represent novel biomarkers to predict therapy response and clinical outcomes43,44,57,69–71,74,297. Similarly, TS proteins, fork reversal factors and fork recovery enzymes are elevated in a variety of tumours87,91,100,101,117,121,137–139,145,151,153. The successful development of these DDT factors as biomarkers will require large-scale studies in a wide range of cancers to assess the tumour specificity of these proteins and establish relevant expression thresholds across cancer types. It should be noted that upregulation of selected DTT factors could be specific to the DNA-damaging agents that tumour cells are exposed to during treatment, which is an important consideration for the clinical applicability of these factors as biomarkers. Further, the replication stress-independent roles of these factors will also need to be explored to determine whether they contribute to cancer cell survival or response to DNA-damaging agents.
Fig. 4 |. The replication stress response in cancer and implications for cancer biomarkers.
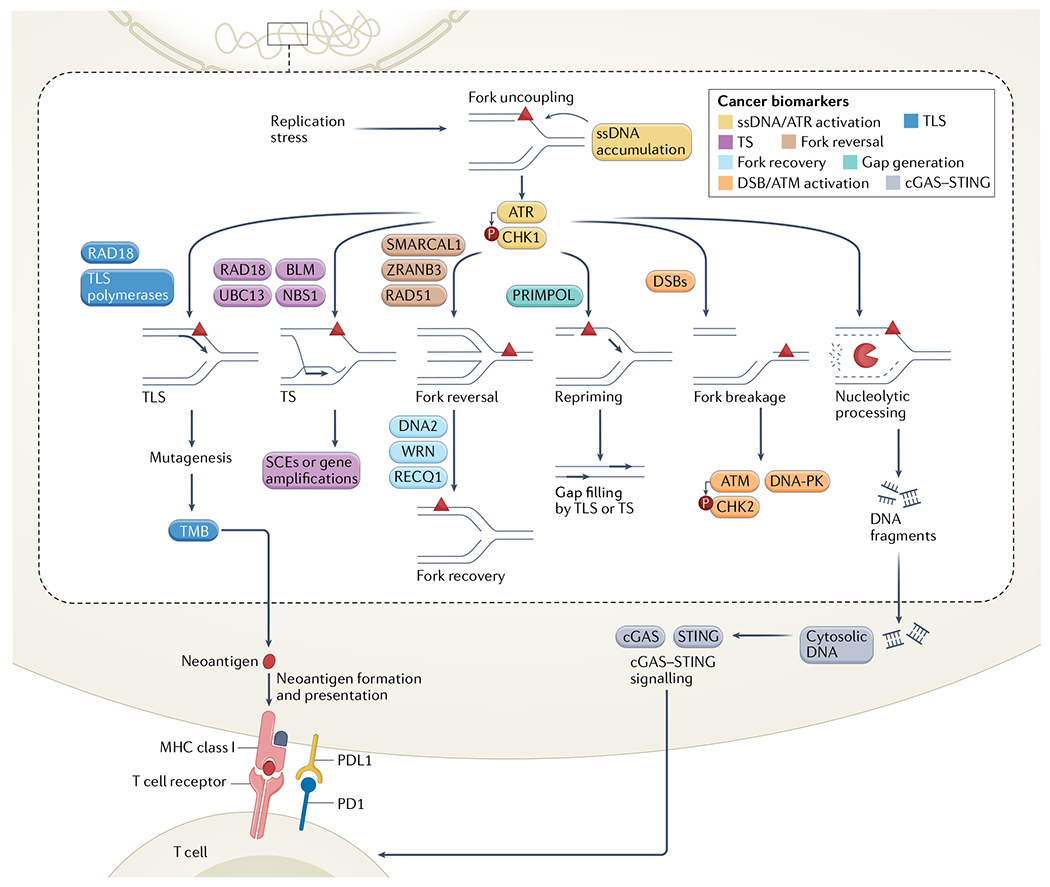
Potential biomarkers are highlighted within the context of different replication stress response pathways. Biomarkers associated with accumulation of single-stranded DNA (ssDNA) and ATR–CHK1 activation in yellow; translesion synthesis (TLS) enzyme expression or activity and TLS-mediated mutagenesis in blue; template switching (TS) enzyme expression or activity and TS-dependent sister chromatid exchange (SCE) or gene amplifications in purple; fork recovery protein expression in light blue; fork reversal protein expression in brown; DNA-directed primase/polymerase protein (PRIMPOL) expression in green and downstream gap filling by TS or TLS-based mechanisms; DNA double strand break (DSB) and ATM activation in orange; and cGAS–STING activation in grey. BLM, Bloom syndrome protein; NBS1, Nijmegen breakage syndrome 1 protein; TMB, tumour mutational burden; WRN, Werner syndrome ATP-dependent helicase.
Functional assays to assess replication stress activities in tumour samples could be used as biomarkers. For example, single-molecule DNA fibre assays have been used in preclinical studies to monitor replication fork stability in high-grade serous ovarian cancers organoids298. TS-dependent SCE or gene amplifications and TLS-mediated mutagenesis could also provide useful readouts to assess possible response to chemotherapy. In addition, increased accumulation of ssDNA and ATR–CHK1 activation could serve as biomarkers to predict response to drugs that induce replication stress (FIG. 4), including PARPi. Chromatin-bound RPA might be a useful readout of ssDNA gaps in cancer cells299, although two major challenges are currently associated with this approach: replication-associated ssDNA gaps need to be distinguished from regions of ssDNA generated in other phases of the cell cycle, and background levels of chromatin-bound RPA need to be studied and baseline thresholds established across diverse tumour types.
Recent data have shown that PARP trapping levels could also be used as a biomarker as they are indicative of PARPi sensitivity in cancer cells300. Finally, accumulation of cytosolic DNA and activation of the cGAS–STING pathway might also represent novel biomarkers to evaluate the combinatorial benefit of replication stress-inducing chemotherapy with immunotherapy or with other agents that increase TMB and neoantigen formation.
Conclusion
Precision medicine continues to revolutionize cancer care, informing the strengths and limitations of therapeutic strategies and uncovering emerging resistance mechanisms. In parallel, the field continues to identify foundational pathways of replication stress and DNA damage response using genome-wide screens and sequencing techniques301,302. These tools and the data they uncover are transforming the way we understand tumours at the molecular level and open new strategies to improve clinical cancer care. Future work needs to identify the tumour or cancer cell-type specificity of these emerging pathways and assess the adequacy of in vitro and in vivo cancer models that are used to elucidate replication stress and DNA repair mechanisms. Further, the feasibility and scalability of possible biomarkers and targets outlined in this Review must be explored given that multiple mechanisms of chemosensitivity and resistance can be activated within a single tumour or within a single cell. These studies are essential to solidify new findings on replication stress that are actively shaping clinical medicine, including the link between replication stress and immunotherapy, which is emerging as a promising direction for cancer treatment303,304. We predict that new cross-disciplinary studies will continue to inform the complex interplay of replication stress response mechanisms, DNA damage repair signalling and the tumour microenvironment, better predicting and improving response to therapy.
Acknowledgements
The authors would like to thank L. Zou, P. Verma and J. Eissenberg for their careful reading of this manuscript and for their insightful feedback, and A. Meroni for comments on the figures. This work was supported by the National Cancer Institute (NCI) grants F30CA254215 to E.C. and R01CA237263 and R01CA248526 to A.V.; the US Department of Defense (DOD) Breast Cancer Research Program (BRCP) Expansion Award BC191374 to A.V.; the Alvin J. Siteman Cancer Center Siteman Investment Program (supported by The Foundation for Barnes-Jewish Hospital, Cancer Frontier Fund) to A.V.; and the Barnard Foundation to A.V.
Glossary
- DNA lesions
Modifications introduced On the DNA helix by different genotoxic agents.
- Xeroderma pigmentosum
An autosomal recessive genetic disease caused by biallelic mutations of specific proteins that are involved in molecular mechanisms required to cope with UV-induced DNA lesions, including Polη.
- Polar pausing
Transient pausing of the replication fork in response to a unidirectional barrier that only inhibits replication fork progression in one direction.
- Schimke immuno-osseous dysplasia
A multi-system autosomal recessive genetic disease caused by inheritance of biallelic SMARCAL1 mutations, with renal disease being a major cause of mortality in patients with this disease.
- RECON syndrome
An autosomal recessive genetic disease caused by biallelic mutations in the RECQL1 DNA helicase, which functions in the DNA damage response.
- Microhomology-mediated end joining
(MMEJ). One of the DNA double-strand break repair pathways, along with homologous recombination and non-homologous end joining, which relies on microhomology sequences (1–16 nucleotides) to anneal and align double-strand break ends for repair.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Zeman MK & Cimprich KA Causes and consequences of replication stress. Nat. Cell Biol 16, 2–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamperl S, Bocek MJ, Saldivar JC, Swigut T & Cimprich KA Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genois MM et al. CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol. Cell 81, 784–800.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinet A et al. PRIMPOL-mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Mol. Cell 77, 461–474.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates how the balance between repriming and fork reversal governs the adaptive response of BRCA1-deficient cells to cisplatin.
- 5.Hanzlikova H et al. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. Cell 71, 319–331.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaitsiankova A et al. PARP inhibition impedes the maturation of nascent DNA strands during DNA replication. Nat. Struct. Mol. Biol 29, 329–338 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work uncovers the mechanism by which PARPi promotes the formation of ssDNA gaps, which are an emerging determinant of PARPi response in BRCA-deficient cancers.
- 7.van Wietmarschen N & Nussenzweig A Mechanism for synthetic lethality in BRCA-deficient cancers: no longer lagging behind. Mol. Cell 71, 877–878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong K et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 81, 3227 (2021). [DOI] [PubMed] [Google Scholar]; This publication links ssDNA gap accumulation and defective Okazaki fragment processing with PARPi sensitivity in BRCA1-deficient cells, revealing new potential targets in BRCA-deficient cancers.
- 9.Donne R et al. Replication stress triggered by nucleotide pool imbalance drives DNA damage and cGAS-STING pathway activation in NAFLD. Dev. Cell 57, 1728–1741.e6 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Koç A, Wheeler LJ, Mathews CK & Merrill GF Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem 279, 223–230 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Flach J et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ubhi T & Brown GW Exploiting DNA replication stress for cancer treatment. Cancer Res. 79, 1730–1739 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Bartkova J et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Gorgoulis VG et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Maya-Mendoza A et al. High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Quinet A & Vindigni A Superfast DNA replication causes damage in cancer cells. Nature 559, 186–187 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Sale JE Competition, collaboration and coordination–determining how cells bypass DNA damage. J. Cell Sci 125, 1633–1643 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Sale JE Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol 5, a012708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branzei D & Szakal B DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44, 68–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinet A, Tirman S, Cybulla E, Meroni A & Vindigni A To skip or not to skip: choosing repriming to tolerate DNA damage. Mol. Cell 81, 649–658 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waters LS et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev 73, 134–154 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Branzei D Ubiquitin family modifications and template switching. FEBS Lett. 585, 2810–2817 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Neelsen KJ & Lopes M Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol 16, 207–220 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Zellweger R et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bianchi J et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 52, 566–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Gomez S et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 52, 541–553 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mouron S et al. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol 20, 1383–1389 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Wan L et al. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 14, 1104–1112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adar S, Izhar L, Hendel A, Geacintov N & Livneh Z Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 37, 5737–5748 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edmunds CE, Simpson LJ & Sale JE PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 30, 519–529 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Jansen JG et al. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair 8, 1444–1451 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Tirman S et al. Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol. Cell 81, 4026–4040.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes previously uncharacterized ssDNA gap-filling pathways in human cells and shows that these pathways can be targeted to increase cancer cell genomic instability and sensitivity to PARPi and cisplatin treatment.
- 33.Sale JE, Lehmann AR & Woodgate R Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol 13, 141–152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gan GN, Wittschieben JP, Wittschieben BO & Wood RD DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 18, 174–183 (2008). [DOI] [PubMed] [Google Scholar]
- 35.McCulloch SD & Kunkel TA The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18, 148–161 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y. et al. Diverse roles of RAD18 and Y-family DNA polymerases in tumorigenesis. Cell Cycle 17, 833–843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamanaka K, Chatterjee N, Hemann MT & Walker GC Inhibition of mutagenic translesion synthesis: a possible strategy for improving chemotherapy? PLoS Genet. 13, e1006842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe K. et al. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tateishi S. et al. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol. Cell Biol 23, 474–481 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon JH, Prakash S & Prakash L Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc. Natl Acad. Sci. USA 109, 7799–7804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lou J. et al. Rad18 mediates specific mutational signatures and shapes the genomic landscape of carcinogen-induced tumors in vivo. NAR Cancer 3, zcaa037 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The Human Protein Atlas. RAD18. Protein Atlas https://www.proteinatlas.org/ENSG00000070950-RAD18 (2022).
- 43.Baatar S. et al. High RAD18 expression is associated with disease progression and poor prognosis in patients with gastric cancer. Ann. Surg. Oncol 27, 4360–4368 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Wu B. et al. High expression of RAD18 in glioma induces radiotherapy resistance via down-regulating P53 expression. Biomed. Pharmacother 112, 108555 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Kikuchi S, Hara K, Shimizu T, Sato M & Hashimoto H Structural basis of recruitment of DNA polymerase zeta by interaction between REV1 and REV7 proteins. J. Biol. Chem 287, 33847–33852 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.In Het Panhuis W. et al. Rev1 deficiency induces replication stress to cause metabolic dysfunction differently in males and females. Am. J. Physiol. Endocrinol. Metab 322, E319–E329 (2022). [DOI] [PubMed] [Google Scholar]
- 47.Jansen JG et al. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J. Exp. Med 203, 319–323 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts SA & Gordenin DA Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 14, 786–800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dumstorf CA, Mukhopadhyay S, Krishnan E, Haribabu B & McGregor WG REV1 is implicated in the development of carcinogen-induced lung cancer. Mol. Cancer Res 7, 247–254 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wittschieben JP, Reshmi SC, Gollin SM & Wood RD Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res. 66, 134–142 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Wittschieben JP et al. Loss of DNA polymerase zeta enhances spontaneous tumorigenesis. Cancer Res. 70, 2770–2778 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin SK, Tomida J & Wood RD Disruption of DNA polymerase zeta engages an innate immune response. Cell Rep. 34, 108775 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seplyarskiy VB, Bazykin GA & Soldatov RA Polymerase zeta activity is linked to replication timing in humans: evidence from mutational signatures. Mol. Biol. Evol 32, 3158–3172 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Zhang S. et al. REV3L 3’UTR 460T>C polymorphism in microRNA target sites contributes to lung cancer susceptibility. Oncogene 32, 242–250 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Wang H. et al. REV3L confers chemoresistance to cisplatin in human gliomas: the potential of its RNAi for synergistic therapy. Neuro-Oncol. 11, 790–802 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou J. et al. Overexpression of DNA polymerase iota (Poliota) in esophageal squamous cell carcinoma. Cancer Sci. 103, 1574–1579 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu X. et al. REV3L, the catalytic subunit of DNA polymerase zeta, is involved in the progression and chemoresistance of esophageal squamous cell carcinoma. Oncol. Rep 35, 1664–1670 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Brondello JM et al. Novel evidences for a tumor suppressor role of Rev3, the catalytic subunit of Pol zeta. Oncogene 27, 6093–6101 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Pan Q, Fang Y, Xu Y, Zhang K & Hu X Down-regulation of DNA polymerases kappa, eta, iota, and zeta in human lung, stomach, and colorectal cancers. Cancer Lett. 217, 139–147 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Yoon JH, Prakash L & Prakash S Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl Acad. Sci. USA 106, 18219–18224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masutani C. et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399, 700–704 (1999). [DOI] [PubMed] [Google Scholar]
- 62.Srivastava AK et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl Acad. Sci. USA 112, 4411–4416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou W. et al. Expression of DNA translesion synthesis polymerase eta in head and neck squamous cell cancer predicts resistance to gemcitabine and cisplatin-based chemotherapy. PLoS ONE 8, e83978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexandrov LB & Stratton MR Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev 24, 52–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogozin IB et al. DNA polymerase eta mutational signatures are found in a variety of different types of cancer. Cell Cycle 17, 348–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jansen JG et al. Redundancy of mammalian Y family DNA polymerases in cellular responses to genomic DNA lesions induced by ultraviolet light. Nucleic Acids Res. 42, 11071–11082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sertic S. et al. Coordinated activity of Y family TLS polymerases and EXO1 protects non-S phase cells from UV-induced cytotoxic lesions. Mol. Cell 70, 34–47.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 68.McIntyre J. Polymerase iota - an odd sibling among Y family polymerases. DNA Repair 86, 102753 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Luedeke M. et al. Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol. Biomark. Prev 18, 3030–3035 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Silvestrov P, Maier SJ, Fang M & Cisneros GA DNArCdb: a database of cancer biomarkers in DNA repair genes that includes variants related to multiple cancer phenotypes. DNA Repair 70, 10–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakiyama T. et al. Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int. J. Cancer 114, 730–737 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Yang J, Chen Z, Liu Y, Hickey RJ & Malkas LH Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 64, 5597–5607 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Yuan F. et al. Overexpressed DNA polymerase iota regulated by JNK/c-Jun contributes to hypermutagenesis in bladder cancer. PLoS ONE 8, e69317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zou S. et al. DNA polymerase iota (Pol iota) promotes invasion and metastasis of esophageal squamous cell carcinoma. Oncotarget 7, 32274–32285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Twayana S. et al. Translesion polymerase eta both facilitates DNA replication and promotes increased human genetic variation at common fragile sites. Proc. Natl Acad. Sci. USA 118, e2106477118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tonzi P, Yin Y, Lee CWT, Rothenberg E & Huang TT Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. eLife 7, e41426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hile SE & Eckert KA DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 36, 688–696 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tubbs A. et al. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma K. et al. Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int. J. Mol. Sci 13, 11974–11999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stern HR, Sefcikova J, Chaparro VE & Beuning PJ Mammalian DNA polymerase kappa activity and specificity. Molecules 24, 2805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Q-Wang J. et al. DNA polymerase kappa, implicated in spontaneous and DNA damage-induced mutagenesis, is overexpressed in lung cancer. Cancer Res. 61, 5366–5369 (2001). [PubMed] [Google Scholar]
- 82.Lovett ST Template-switching during replication fork repair in bacteria. DNA Repair 56, 118–128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ulrich HD Timing and spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett. 585, 2861–2867 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Branzei D, Seki M & Enomoto T Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cell 9, 1031–1042 (2004). [DOI] [PubMed] [Google Scholar]
- 85.Takahashi TS, Wollscheid HP, Lowther J & Ulrich HD Effects of chain length and geometry on the activation of DNA damage bypass by polyubiquitylated PCNA. Nucleic Acids Res. 48, 3042–3052 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chiu RK et al. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2, e116 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.The Human Protein Atlas. UBE2N. Protein Atlas https://www.proteinatlas.org/ENSG00000177889-UBE2N (2022).
- 88.Lin JR, Zeman MK, Chen JY, Yee MC & Cimprich KA SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 42, 237–249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Motegi A et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl Acad. Sci. USA 105, 12411–12416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.The Human Protein Atlas. HLTF. Protein Atlas https://www.proteinatlas.org/ENSG00000071794-HLTF (2022). [Google Scholar]
- 91.The Human Protein Atlas. SHPRH. Protein Atlas https://www.proteinatlas.org/ENSG00000146414-SHPRH (2022). [Google Scholar]
- 92.Liu L et al. HLTF suppresses the migration and invasion of colorectal cancer cells via TGF-β/SMAD signaling in vitro. Int. J. Oncol 53, 2780–2788 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Moinova HR et al. HLTF gene silencing in human colon cancer. Proc. Natl Acad. Sci. USA 99, 4562–4567 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sandhu S et al. Loss of HLTF function promotes intestinal carcinogenesis. Mol. Cancer 11, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dhont L, Mascaux C & Belayew A The helicase-like transcription factor (HLTF) in cancer: loss of function or oncomorphic conversion of a tumor suppressor? Cell Mol. Life Sci 73, 129–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jasin M & Rothstein R Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol 5, a012740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Piberger AL et al. PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat. Commun 11, 5863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liberi G et al. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 19, 339–350 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pietrobon V et al. The chromatin assembly factor 1 promotes Rad51-dependent template switches at replication forks by counteracting D-loop disassembly by the RecQ-type helicase Rqh1. PLoS Biol. 12, e1001968 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.The Human Protein Atlas. BLM. Protein Atlas https://www.proteinatlas.org/ENSG00000197299-BLM (2022).
- 101.The Human Protein Atlas. NBN. Protein Atlas https://www.proteinatlas.org/ENSG00000104320-NBN (2022).
- 102.Hasty P & Montagna C Chromosomal rearrangements in cancer: detection and potential causal mechanisms. Mol. Cell. Oncol 1, e29904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tanaka H & Watanabe T Mechanisms underlying recurrent genomic amplification in human cancers. Trends Cancer 6, 462–477 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vicario R et al. Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS ONE 10, e0129876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sakofsky CJ & Malkova A Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol 52, 395–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blastyak A et al. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 28, 167–175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bugreev DV, Rossi MJ & Mazin AV Cooperation of RAD51 and RAD54 in regression of a model replication fork. Nucleic Acids Res. 39, 2153–2164 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Betous R et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 26, 151–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kolinjivadi AM et al. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and Stable Rad51 nucleofilaments. Mol. Cell 67, 867–881.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gari K, Decaillet C, Stasiak AZ, Stasiak A & Constantinou A The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 29, 141–148 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Vujanovic M et al. Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell 67, 882–890.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yusufzai T & Kadonaga JT Annealing helicase 2 (AH2), a DNA-rewinding motor with an HNH motif. Proc. Natl Acad. Sci. USA 107, 20970–20973 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bai G et al. HLTF promotes fork reversal, limiting replication stress resistance and preventing multiple mechanisms of unrestrained DNA synthesis. Mol. Cell 78, 1237–1251.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blastyak A, Hajdu I, Unk I & Haracska L Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell Biol 30, 684–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bansbach CE, Boerkoel CF & Cortez D SMARCAL1 and replication stress: an explanation for SIOD? Nucleus 1, 245–248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elizondo LI et al. Schimke immuno-osseous dysplasia: SMARCAL1 loss-of-function and phenotypic correlation. J. Med. Genet 46, 49–59 (2009). [DOI] [PubMed] [Google Scholar]
- 117.The Human Protein Atlas. SMARCAL1 . Protein Atlas https://www.proteinatlas.org/ENSG00000138375-SMARCAL1 (2022).
- 118.Bogliolo M et al. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med 20, 458–463 (2018). [DOI] [PubMed] [Google Scholar]
- 119.Catucci I et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med 20, 452–457 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Jones S et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat. Commun 5, 5006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.The Human Protein Atlas. ZRANB3. Protein Atlas https://www.proteinatlas.org/ENSG00000121988-ZRANB3 (2022).
- 122.Hiramoto T et al. Mutations of a novel human RAD54 homologue, RAD54B, in primary cancer. Oncogene 18, 3422–3426 (1999). [DOI] [PubMed] [Google Scholar]
- 123.Smirnova M, Van Komen S, Sung P & Klein HL Effects of tumor-associated mutations on Rad54 functions. J. Biol. Chem 279, 24081–24088 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Fugger K et al. FBH1 catalyzes regression of stalled replication forks. Cell Rep. 10, 1749–1757 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Berti M et al. Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat. Commun 11, 3531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rein HL, Bernstein KA & Baldock RA RAD51 paralog function in replicative DNA damage and tolerance. Curr. Opin. Genet. Dev 71, 86–91 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grundy MK, Buckanovich RJ & Bernstein KA Regulation and pharmacological targeting of RAD51 in cancer. NAR Cancer 2, zcaa024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cruz C et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol 29, 1203–1210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maacke H et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 19, 2791–2795 (2000). [DOI] [PubMed] [Google Scholar]
- 130.Maacke H et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 88, 907–913 (2000). [DOI] [PubMed] [Google Scholar]
- 131.Raderschall E et al. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 62, 219–225 (2002). [PubMed] [Google Scholar]
- 132.Berti M et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol 20, 347–354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thangavel S et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chu WK & Hickson ID RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Abu-Libdeh B et al. RECON syndrome is a genome instability disorder caused by mutations in the DNA helicase RECQL1. J. Clin. Invest 132, e147301 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cybulski C et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet 47, 643–646 (2015). [DOI] [PubMed] [Google Scholar]
- 137.The Human Protein Atlas. DNA2. Protein Atlas https://www.proteinatlas.org/ENSG00000138346-DNA2 (2022).
- 138.The Human Protein Atlas. WRN. Protein Atlas https://www.proteinatlas.org/ENSG00000165392-WRN (2022).
- 139.The Human Protein Atlas. RECQL. Protein Atlas https://www.proteinatlas.org/ENSG00000004700-RECQL (2022). [Google Scholar]
- 140.Ray Chaudhuri A et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication was one of the first to explore how replication fork stability is associated with chemoresponse in BRCA-deficient cancer cells.
- 141.Schlacher K et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ying S, Hamdy FC & Helleday T Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 72, 2814–2821 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Dungrawala H et al. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell 67, 374–386.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guillemette S et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 29, 489–494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.The Human Protein Atlas. MUS81. Protein Atlas https://www.proteinatlas.org/ENSG00000172732-MUS81 (2022).
- 146.Lemacon D et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rondinelli B et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol 19, 1371–1378 (2017). [DOI] [PubMed] [Google Scholar]
- 148.Krais JJ & Johnson N Ectopic RNF168 expression promotes break-induced replication-like DNA synthesis at stalled replication forks. Nucleic Acids Res. 48, 4298–4308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zong D et al. BRCA1 haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Mol. Cell 73, 1267–1281.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Batenburg NL et al. Cockayne syndrome group B protein regulates fork restart, fork progression and MRE11-dependent fork degradation in BRCA1/2-deficient cells. Nucleic Acids Res. 49, 12836–12854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.The Human Protein Atlas. ERCC6. Protein Atlas https://www.proteinatlas.org/ENSG00000225830-ERCC6 (2022).
- 152.Eckelmann BJ et al. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2, zcaa013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.The Human Protein Atlas. XRCC1. Protein Atlas https://www.proteinatlas.org/ENSG00000073050-XRCC1 (2022).
- 154.Heller RC & Marians KJ Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439, 557–562 (2006). [DOI] [PubMed] [Google Scholar]
- 155.Fumasoni M, Zwicky K, Vanoli F, Lopes M & Branzei D Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 complex. Mol. Cell 57, 812–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bailey LJ, Bianchi J & Doherty AJ PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 47, 4026–4038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.González-Acosta D et al. PrimPol-mediated repriming facilitates replication traverse of DNA interstrand crosslinks. EMBO J. 40, e106355 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Keen BA, Bailey LJ, Jozwiakowski SK & Doherty AJ Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res. 42, 12102–12111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kobayashi K et al. Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15, 1997–2008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Panzarino NJ et al. Replication gaps underlie BRCA deficiency and therapy response. Cancer Res. 81 , 1388–1397 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work links replication-associated ssDNA gaps with therapy response in BRCA-deficient cancer cells and proposes a gap-centred framework for understanding ‘BRCAness’ phenotypes.
- 161.Taglialatela A et al. REV1-Polζ maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol. Cell 81, 4008–4025.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights the dependence of BRCA-deficient cells on ssDNA gap filling to promote cell survival, supporting the use of novel TLS inhibitors in combination with other cancer therapies.
- 162.The Human Protein Atlas. PRIMPOL. Protein Atlas https://www.proteinatlas.org/ENSG00000164306-PRIMPOL (2022).
- 163.Diaz-Talavera A et al. A cancer-associated point mutation disables the steric gate of human PrimPol. Sci. Rep 9, 1121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bamford S et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M & Ulrich HD Processing of DNA polymerase-blocking lesions during genome replication is spatially and temporally segregated from replication forks. Mol. Cell 77, 3–16.e14 (2020). [DOI] [PubMed] [Google Scholar]
- 166.Nayak S et al. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv 6, eaaz7808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thakar T et al. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun 11, 2147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Thakar T et al. Lagging strand gap suppression connects BRCA-mediated fork protection to nucleosome assembly by ensuring PCNA-dependent CAF-1 recycling. Nat. Commun 13, 5323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu RL, Dong Y, Deng YZ, Wang WJ & Li WD Tumor suppressor miR-145 reverses drug resistance by directly targeting DNA damage-related gene RAD18 in colorectal cancer. Tumour Biol. 36, 5011–5019 (2015). [DOI] [PubMed] [Google Scholar]
- 170.Williams SA, Longerich S, Sung P, Vaziri C & Kupfer GM The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood 117, 5078–5087 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Geng L, Huntoon CJ & Karnitz LM RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol 191 , 249–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lopez-Martinez D, Liang CC & Cohn MA Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol. Life Sci 73, 3097–3114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kim H & D’Andrea AD Regulation of DNA crosslink repair by the Fanconi anemia/BRCA pathway. Genes. Dev 26, 1393–1408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fenteany G et al. A series of xanthenes inhibiting Rad6 function and Rad6-Rad18 interaction in the PCNA ubiquitination cascade. iScience 25, 104053 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Punchihewa C et al. Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication. J. Biol. Chem 287, 14289–14300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Inoue A et al. A small molecule inhibitor of monoubiquitinated proliferating cell nuclear antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem 289, 7109–7120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Qin Z et al. DNA-damage tolerance mediated by PCNA*Ub fusions in human cells is dependent on Rev1 but not Pokη. Nucleic Acids Res. 41, 7356–7369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Coleman KE et al. USP1-trapping lesions as a source of DNA replication stress and genomic instability. Nat. Commun 13, 1740 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lim KS et al. USP1 is required for replication fork protection in BRCA1-deficient tumors. Mol. Cell 72, 925–941.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lin X, Okuda T, Trang J & Howell SB Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol 69, 1748–1754 (2006). [DOI] [PubMed] [Google Scholar]
- 181.Okuda T, Lin X, Trang J & Howell SB Suppression of hREV1 expression reduces the rate at which human ovarian carcinoma cells acquire resistance to cisplatin. Mol. Pharmacol 67, 1852–1860 (2005). [DOI] [PubMed] [Google Scholar]
- 182.Wojtaszek JL et al. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178, 152–159.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ikeh KE et al. REV1 inhibition enhances radioresistance and autophagy. Cancers 13, 5290 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mulcahy Levy JM & Thorburn A Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 27, 843–857 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ketkar A et al. Inhibition of human DNA polymerases eta and kappa by indole-derived molecules occurs through distinct mechanisms. ACS Chem. Biol 14, 1337–1351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Coggins GE et al. N-Aroyl indole thiobarbituric acids as inhibitors of DNA repair and replication stress response polymerases. ACS Chem. Biol 8, 1722–1729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zafar MK et al. A small-molecule inhibitor of human DNA polymerase η potentiates the effects of cisplatin in tumor cells. Biochemistry 57, 1262–1273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ceppi P et al. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res 15, 1039–1045 (2009). [DOI] [PubMed] [Google Scholar]
- 189.Zhang J et al. A PolH transcript with a short 3’UTR enhances PolH expression and mediates cisplatin resistance. Cancer Res. 79, 3714–3724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jha V & Ling H Structural basis for human DNA polymerase kappa to bypass cisplatin intrastrand cross-link (Pt-GG) lesion as an efficient and accurate extender. J. Mol. Biol 430, 1577–1589 (2018). [DOI] [PubMed] [Google Scholar]
- 191.Yang Y et al. DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress. J. Cell Biol 216, 3097–3115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Doles J et al. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl Acad. Sci. USA 107, 20786–20791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vassel FM, Bian K, Walker GC & Hemann MT Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc. Natl Acad. Sci. USA 117, 28922–28924 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Okina S et al. High expression of REV7 is an independent prognostic indicator in patients with diffuse large B-cell lymphoma treated with rituximab. Int. J. Hematol 102, 662–669 (2015). [DOI] [PubMed] [Google Scholar]
- 195.Niimi K et al. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci. 105, 545–552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ghezraoui H et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xu G et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that REV7 loss but not loss of the REV1 or REV3L TLS polymerases represents a BRCA1-independent mechanism of HR restoration that contributes to PARPi resistance.
- 198.Yousefzadeh MJ & Wood RD DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair 12, 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ceccaldi R et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mateos-Gomez PA et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhou J et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2, 598–610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, along with Ceccaldi et al. (2015) and Mateos-Gomez et al. (2015), establishes Polθ as a target in BRCA-deficient cancers and provides critical mechanistic insight for utility of Polθ inhibitors, one of which is now being used in a clinical trial.
- 202.Schrempf A, Slyskova J & Loizou JI Targeting the DNA repair enzyme polymerase theta in cancer therapy. Trends Cancer 7, 98–111 (2021). [DOI] [PubMed] [Google Scholar]
- 203.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a comprehensive landscape of tumour mutational signatures and links replication stress response mechanisms with several of these mutational patterns in cancers.
- 204.Higgins GS et al. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 1, 175–184 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lemée F et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl Acad. Sci. USA 107, 13390–13395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hodge CD, Spyracopoulos L & Glover JN Ubc13: the Lys63 ubiquitin chain building machine. Oncotarget 7, 64471–64504 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wu Z, Shen S, Zhang Z, Zhang W & Xiao W Ubiquitin-conjugating enzyme complex Uev1A-Ubc13 promotes breast cancer metastasis through nuclear factor-small ka, CyrillicB mediated matrix metalloproteinase-1 gene regulation. Breast Cancer Res. 16, R75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wu Z, Neufeld H, Torlakovic E & Xiao W Uev1A-Ubc13 promotes colorectal cancer metastasis through regulating CXCL1 expression via NF-small ka, CyrillicB activation. Oncotarget 9, 15952–15967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dikshit A et al. UBE2N promotes melanoma growth via MEK/FRA1/SOX10 signaling. Cancer Res. 78, 6462–6472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang X et al. The inhibition of UBC13 expression and blockage of the DNMT1-CHFR-Aurora A pathway contribute to paclitaxel resistance in ovarian cancer. Cell Death Dis. 9, 93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Cheng J et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis. 5, e1079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pulvino M et al. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 120, 1668–1677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lu R et al. MUS81 participates in the progression of serous ovarian cancer associated with dysfunctional DNA repair system. Front. Oncol 9, 1189 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhong A et al. MUS81 inhibition increases the sensitivity to therapy effect in epithelial ovarian cancer via regulating CyclinB pathway. J. Cancer 10, 2276–2287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Krais JJ et al. RNF168-mediated ubiquitin signaling inhibits the viability of BRCA1-null cancers. Cancer Res. 80, 2848–2860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Luijsterburg MS et al. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife 6, e20922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bryant HE et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 218.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 219.Fong PC et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med 361, 123–134 (2009). [DOI] [PubMed] [Google Scholar]
- 220.Turner N, Tutt A & Ashworth A Targeting the DNA repair defect of BRCA tumours. Curr. Opin. Pharmacol 5, 388–393 (2005). [DOI] [PubMed] [Google Scholar]
- 221.Zimmermann M et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.D’Andrea AD Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 71, 172–176 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Janysek DC, Kim J, Duijf PHG & Dray E Clinical use and mechanisms of resistance for PARP inhibitors in homologous recombination-deficient cancers. Transl. Oncol 14, 101012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fugger K, Hewitt G, West SC & Boulton SJ Tackling PARP inhibitor resistance. Trends Cancer 7, 1102–1118 (2021). [DOI] [PubMed] [Google Scholar]
- 225.Gatti M, Imhof R, Huang Q, Baudis M & Altmeyer M The ubiquitin Ligase TRIP12 limits PARP1 trapping and constrains PARP inhibitor efficiency. Cell Rep. 32, 107985 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Krastev DB et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nat. Cell Biol 24, 62–73 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Noordermeer SM & van Attikum H PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol. 29, 820–834 (2019). [DOI] [PubMed] [Google Scholar]
- 228.Gogola E et al. Selective loss of PARG restores parylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 33, 1078–1093.e12 (2018). [DOI] [PubMed] [Google Scholar]
- 229.Yazinski SA et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 31, 318–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Simoneau A, Xiong R & Zou L The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 35, 1271–1289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mehta KPM et al. CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci. Adv 8, eabm0314 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Guo E et al. FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proc. Natl Acad. Sci. USA 117, 19415–19424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Paes Dias M et al. Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1/53BP1 double-deficient cells by exposing ssDNA gaps. Mol. Cell 81, 4692–4708.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication describes a mechanism of PARPi resistance mediated by LIG3 loss and accumulation of ssDNA in BRCA1/53BP1-deficient cells, pointing towards the clinical promise of targeting ssDNA gaps therapeutically to address chemoresistance.
- 234.Cantor SB Revisiting the BRCA-pathway through the lens of replication gap suppression: “Gaps determine therapy response in BRCA mutant cancer”. DNA Repair 107, 103209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cong K & Cantor SB Exploiting replication gaps for cancer therapy. Mol. Cell 82, 2363–2369 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dev H et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol 20, 954–965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Saldivar JC et al. An intrinsic S/G. Science 361, 806–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Couch FB et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dibitetto D et al. Fork slowing and reversal as an adaptive response to chronic ATR inhibition. Preprint at bioRxriv 10.1101/2021.05.18.444697 (2021). [DOI] [Google Scholar]
- 240.Mutreja K et al. ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links. Cell Rep. 24, 2629–2642.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yazinski SA & Zou L Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu. Rev. Genet 50, 155–173 (2016). [DOI] [PubMed] [Google Scholar]
- 242.Parmar K et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res 25, 6127–6140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chaikovsky AC et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 592, 794–798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Maiani E et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 592, 799–803 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Simoneschi D et al. CRL4. Nature 592, 789–793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Young LA et al. Differential activity of ATR and WEE1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 79, 3762–3775 (2019). [DOI] [PubMed] [Google Scholar]
- 247.Ghelli Luserna Di Rora A et al. Synergism through WEE1 and CHK1 inhibition in acute lymphoblastic leukemia. Cancers 11, 1654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chen X et al. Targeting replicative stress and DNA repair by combining PARP and Wee1 kinase inhibitors is synergistic in triple negative breast cancers with cyclin E or BRCA1 alteration. Cancers 13, 1656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ha DH et al. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci. Rep 10, 9930 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Beck H et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell Biol 32, 4226–4236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Elbaek CR et al. WEE1 kinase protects the stability of stalled DNA replication forks by limiting CDK2 activity. Cell Rep. 38, 110261 (2022). [DOI] [PubMed] [Google Scholar]
- 252.Pillay N et al. DNA replication vulnerabilities render ovarian cancer cells sensitive to poly(ADP-Ribose) glycohydrolase inhibitors. Cancer Cell 35, 519–533.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is the first to address how PARG inhibition impacts replication stress across a variety of ovarian cancer models and to test combinatorial treatment of PARG inhibitors with those of CHK1 and WEE1 in preclinical models.
- 253.Buisson R, Boisvert JL, Benes CH & Zou L Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol. Cell 59, 1011–1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Balmus G et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun 10, 87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bartek J & Lukas J Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429 (2003). [DOI] [PubMed] [Google Scholar]
- 256.Choi M, Kipps T & Kurzrock R ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther 15, 1781–1791 (2016). [DOI] [PubMed] [Google Scholar]
- 257.Lavin MF & Yeo AJ Clinical potential of ATM inhibitors. Mutat. Res 821, 111695 (2020). [DOI] [PubMed] [Google Scholar]
- 258.Lin YF et al. PIDD mediates the association of DNA-PKcs and ATR at stalled replication forks to facilitate the ATR signaling pathway. Nucleic Acids Res. 46, 1847–1859 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hafsi H et al. Combined ATR and DNA-PK inhibition radiosensitizes tumor cells independently of their p53 status. Front. Oncol 8, 245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Fok JHL et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun 10, 5065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zhang M et al. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 11, 1913–1935 (2021). [PMC free article] [PubMed] [Google Scholar]
- 262.Alvarez-Fernandez M & Malumbres M Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 37, 514–529 (2020). [DOI] [PubMed] [Google Scholar]
- 263.Asghar US, Kanani R, Roylance R & Mittnacht S Systematic review of molecular biomarkers predictive of resistance to CDK4/6 inhibition in metastatic breast cancer. JCO Precis. Oncol 6, e2100002 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dean JL, McClendon AK & Knudsen ES Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem 287, 29075–29087 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Johnson SM et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J. Clin. Invest 120, 2528–2536 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Roberts PJ et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl Cancer Inst 104, 476–487 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li S et al. Pan-cancer analysis reveals synergistic effects of CDK4/6i and PARPi combination treatment in RB-proficient and RB-deficient breast cancer cells. Cell Death Dis. 11, 219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Crozier L et al. CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J. 41, e108599 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Salvador-Barbero B et al. CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell 38, 584 (2020). [DOI] [PubMed] [Google Scholar]; This study provides mechanistic evidence to support the emerging treatment paradigm of CDK4/6 inhibition, which is used clinically in combination with DNA-damaging therapies.
- 270.Panagiotou E, Gomatou G, Trontzas IP, Syrigos N & Kotteas E Cyclin-dependent kinase (CDK) inhibitors in solid tumors: a review of clinical trials. Clin. Transl. Oncol 24, 161–192 (2022). [DOI] [PubMed] [Google Scholar]
- 271.Coquel F et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018). [DOI] [PubMed] [Google Scholar]; This work provides a mechanistic link between replication fork stress, nucleolytic processing and accumulation of cytosolic DNA that activates an innate immune-like response.
- 272.Kreienkamp R et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. 22, 2006–2015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Orvain C et al. Hair follicle stem cell replication stress drives IFI16/STING-dependent inflammation in hidradenitis suppurativa. J. Clin. Invest 130, 3777–3790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Shen YJ et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 11, 460–473 (2015). [DOI] [PubMed] [Google Scholar]
- 275.Técher H & Pasero P The replication stress response on a narrow path between genomic instability and inflammation. Front. Cell Dev. Biol 9, 702584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ragu S, Matos-Rodrigues G & Lopez BS Replication stress, DNA damage, inflammatory cytokines and innate immune response. Genes 11, 409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Ni Y et al. High TGF-beta signature predicts immunotherapy resistance in gynecologic cancer patients treated with immune checkpoint inhibition. NPJ Precis. Oncol 5, 101 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Li S et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in TH cells. Nature 587, 121–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Jackson LM et al. Loss of MED12 activates the TGFbeta pathway to promote chemoresistance and replication fork stability in BRCA-deficient cells. Nucleic Acids Res. 49, 12855–12869 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Moskwa P et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 41, 210–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Martinez-Ruiz H et al. A TGFbeta-miR-182-BRCA1 axis controls the mammary differentiation hierarchy. Sci. Signal. 9, ra118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kirshner J et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 66, 10861–10869 (2006). [DOI] [PubMed] [Google Scholar]
- 285.Wang M et al. Novel Smad proteins localize to IR-induced double-strand breaks: interplay between TGFβ and ATM pathways. Nucleic Acids Res. 41, 933–942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wiegman EM, Blaese MA, Loeffler H, Coppes RP & Rodemann HP TGFbeta-1 dependent fast stimulation of ATM and p53 phosphorylation following exposure to ionizing radiation does not involve TGFbeta-receptor I signalling. Radiother. Oncol 83, 289–295 (2007). [DOI] [PubMed] [Google Scholar]
- 287.Tang B et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 67, 8643–8652 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Wen H et al. Inhibiting of self-renewal, migration and invasion of ovarian cancer stem cells by blocking TGF-beta pathway. PLoS ONE 15, e0230230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Yan G et al. TGFbeta/cyclin D1/Smad-mediated inhibition of BMP4 promotes breast cancer stem cell self-renewal activity. Oncogenesis 10, 21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chan TA et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol 30, 44–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Strickler JH, Hanks BA & Khasraw M Tumor mutational burden as a predictor of immunotherapy response: is more always better? Clin. Cancer Res 27, 1236–1241 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wang S, Xie K & Liu T Cancer immunotherapies: from efficacy to resistance mechanisms - not only checkpoint matters. Front. Immunol 12, 690112 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sim MJW & Sun PD T cell recognition of tumor neoantigens and insights into T cell immunotherapy. Front. Immunol 13, 833017 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Salas-Benito D et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discov. 11, 1353–1367 (2021). [DOI] [PubMed] [Google Scholar]
- 295.Zhang JY, Yan YY, Li JJ, Adhikari R & Fu LW PD-1/PD-L1 based combinational cancer therapy: icing on the cake. Front. Pharmacol 11, 722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.McGrail DJ et al. Replication stress response defects are associated with response to immune checkpoint blockade in nonhypermutated cancers. Sci. Transl Med 13, eabe6201 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Zou S et al. RAD18 promotes the migration and invasion of esophageal squamous cell cancer via the JNK-MMPs pathway. Cancer Lett. 417, 65–74 (2018). [DOI] [PubMed] [Google Scholar]
- 298.Hill SJ et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 8, 1404–1421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work provides evidence that DNA replication fork dynamics could be used to predict therapy sensitivity in patient-derived tumour samples, building on previous findings that connected replication fork degradation phenotypes with drug response in cell-based models.
- 299.Maréchal A & Zou L RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 25, 9–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Michelena J et al. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun 9, 2678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Hussmann JA et al. Mapping the genetic landscape of DNA double-strand break repair. Cell 184, 5653–5669.e25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Olivieri M et al. A genetic map of the response to DNA damage in human cells. Cell 182, 481–496.e21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication uses a powerful genome-wide screening to demonstrate the breadth of DNA damage response pathways essential for coping with a variety of replication stress inducers and DNA-damaging agents.
- 303.Chen M, Linstra R & van Vugt MATM Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim. Biophys. Acta Rev. Cancer 1877, 188661 (2022). [DOI] [PubMed] [Google Scholar]
- 304.Chabanon RM et al. Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat. Rev. Cancer 21, 701–717 (2021). [DOI] [PubMed] [Google Scholar]