

Abstract
Background
Bullous pemphigoid is the most common autoimmune subepidermal blistering disorder with a low incidence in childhood. Combined immunodeficiencies (CIDs) are a group of monogenic inborn errors of immunity (IEIs) characterized by T- and B-cell dysfunction leading to recurrent infections, lymphoproliferation, predisposition to malignancy, and autoimmunity. Here, we report two Afghan siblings with a diagnosis of CID and extremely rare manifestation of diffuse bullous pemphigoid skin lesions.
Case presentation
The older sibling (patient 1) was a 32-month-old male with facial dysmorphism, protracted diarrhea, failure to thrive, recurrent oral candidiasis, recurrent otitis media with tympanic membrane perforation, who had been previously diagnosed with CID. While he was under treatment with intravenous immunoglobulin (IVIg), he developed extensive blistering lesions, which were diagnosed as childhood bullous pemphigoid. Methylprednisolone and azathioprine were added to the regimen, which resulted in a remarkable improvement of the skin lesions and also the feeding condition. However,2 weeks later, he was re-admitted to the intensive care unit (ICU) and eventually died due to fulminant sepsis. Later, his 12-month-old sister (patient 2) with similar facial dysmorphism and a history of developmental delay, food allergy, recurrent oral candidiasis, and respiratory tract infections also developed blistering skin lesions. She was under treatment for occasional eczematous lesions, and had been receiving IVIg for 3 months due to low levels of immunoglobulins. Further immunologic workup showed an underlying CID and thus treatment with IVIg continued, gradually improving her clinical condition. The genetic study of both siblings revealed a novel homozygous mutation in exon 7 of the PGM3 gene, c.845 T > C (p.Val282Ala).
Conclusions
Dermatologic disorders may be the presenting sign in patients with CID and mutated PGM3. This case report further extends the spectrum of skin manifestations that could be observed in PGM3 deficiency and emphasizes the importance of considering CIDs during the assessment of skin disorders, particularly if they are extensive, recurrent, refractory to treatment, and/or associated with other signs of IEIs.
Keywords: PGM3 deficiency, Inborn errors of immunity, Skin, Blister, Case report
Background
Pemphigoid skin diseases are a group of rare autoimmune blistering disorders (AIBD) characterized by the development of autoantibodies that target hemidesmosome proteins involved in the maintenance of the dermo-epidermal integrity [1]. Bullous pemphigoid (BP) is the most prevalent AIBD [2] and classically presents as subepidermal tense blisters arising on a pruritic erythematous or unaffected background [3]. BP usually affects the elderly, and while pediatric cases are rare, they are more commonly associated with widespread lesions and mucosa involvement [4]. Combined immunodeficiencies (CIDs) are monogenic inborn errors of immunity (IEIs) presenting with T- and B-cell dysfunction [5]. Aside from infectious complications, CIDs may be accompanied by a vast spectrum of autoimmune conditions, mainly including endocrinopathies, cytopenias, and enteropathies [6].
Herein, we describe two CID siblings with autoimmune BP lesions who were found to have a novel missense variant in the Phosphoglucomutase 3 (PGM3) gene. This report provides new insights into genotype–phenotype correlation as bullous pemphigoid has never been reported in PGM3 deficiency.
We also aimed to review the existing evidence on pemphigoid skin diseases reported in IEI patients and to investigate PGM3 deficiency in patients with CID and severe CID (SCID) phenotypes.
Method
Medical data of both patients were obtained by direct interview with parents and investigating in- and outpatient medical documents after receiving written informed consent from parents. Demographic data included age, sex, first presentation, age at disease onset and diagnosis, and outcome. The laboratory data included complete cell blood counts, lymphocyte subsets (by flowcytometry), lymphocyte functional assays, serum immunoglobulins (assessed using nephelometry and enzyme-linked immunosorbent assay), antibody titers to vaccinations, Nitroblue tetrazolium (NBT), and skin biopsy. Clinical diagnosis of CID has been established according to the European Society for Immunodeficiencies criteria [7].
Whole exome sequencing (WES) was performed on blood samples of both siblings in the Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria, using the Illumina Nextera DNA Flex Library Exome Kit for library preparation. An Illumina HiSeq3000/4000 instrument was used for 75-bp paired-end sequencing as previously described [8, 9]. Briefly, reads were aligned to the human genome version 19 by means of the Burrows-Wheeler Aligner (BWA). VEP was used for annotating single nucleotide variants (SNVs) and insertions/deletions lists. The obtained list was then filtered according to the presence of variants with a minor allele frequency (MAF) > 0.01 in 1000 Genomes, gnomAD, and dbSNP build 149. After further filtering steps for nonsense, missense, and splice-site variants using the DART software, an internal database was used to filter for recurrent variants. Moreover, variants were prioritized using tools, such as SIFT, Polyphen-2 and the combined annotation dependent depletion (CADD) score [10], that predict the deleteriousness of a present variant. The variant was confirmed through PCR amplification followed by Sanger sequencing in the Watson genetic laboratory, Tehran, Iran.
The literature search for reported IEI patients with pemphigoid blistering diseases was conducted in PubMed, Web of Science, and Scopus, applying the following keywords: “primary immunodeficiency”, “inborn error of immunity”, “congenital immunodeficiency syndromes”, “inherited immunodeficiency diseases”, in combination with subsequent terminology: “bullous pemphigoid”, “pemphigoid(s)”, “blisters”, and “bullous lesion”. Reference lists of all full-text articles and major reviews were manually searched for additional studies.
Case presentations
Patient 1 was a 32-month-old male born at term to consanguineous Afghan parents living in Iran. At birth, he was noted to have facial dysmorphism with low set ears and hypotelorism but no skeletal abnormalities. The parents reported delay in the separation of umbilical cord by 40 days. He had received routine vaccination with no adverse effect. In his early months of life, he suffered from generalized eczematous lesions and recurrent episodes of oral candidiasis. At the age of 8 months, he developed two prominent erythematous skin lesions on the dorsal surfaces of the hands, which were found to be fungal infections through skin biopsy. He also suffered from protracted non-bloody diarrhea since the age of 14 months leading to hospitalizations. Food allergy, growth failure, and recurrent otitis media with tympanic membrane perforation were among other comorbidities.
When he was 16 months old, laboratory evaluation revealed lymphopenia, neutropenia, thrombocytosis, and increased levels of C-reactive protein (CRP) (Table 1). The serum levels of IgM and IgE were increased but IgG and IgA levels were in the normal range with respect to the age-matched reference values. Flow cytometry showed low CD4+ T cells and CD19+ B cells but normal natural killer (NK) cells. The specific antibody titers to diphtheria and tetanus were not protective. The Nitro blue tetrazolium (NBT) test was normal. The sputum smear and culture were negative for Bacillus Calmette-Guérin (BCG). The quantitative polymerase chain reaction (PCR) for human immunodeficiency viruses (HIV) was negative. According to the European Society for Immunodeficiency (ESID) criteria, the diagnosis of CID was established. He received extended-spectrum antibiotics and amphotericin B, and then prophylaxis with fluconazole and trimethoprim-sulfamethoxazole was initiated. The signs and symptoms were further treated with intravenous immunoglobulin (IVIg) substitution (500 mg/Kg/month) for about 1 year.
Table 1.
Summary of Laboratory Investigations in Two Siblings at the Time of Diagnosis
| Laboratory parameters | Patient 1 (age: 16 months) | Patient 2 (age: 12 months) | Reference value |
|---|---|---|---|
| WBC (cells/mm3) | 5700 | 3900 | 6000–17000 |
| Lymphocyte (cells/mm3) | 1938 | 663 | 3000–9500 |
| Neutrophil (cells/mm3) | 1026 | 2496 | 1500–8500 |
| Eosinophil (cells/mm3) | 114 | 195 | 165–465 |
| IgG (mg/dL) | 849 | 875† | 246–904 |
| IgM (mg/dL) | 256 | 44 | 40–143 |
| IgA (mg/dL) | 47 | 174 | 27–66 |
| IgE (IU/mL) | 475 | 170 | Up to 68 |
| Anti-D IgG (IU/mL) | < 0.01 | 0.006 |
< 0.1: No response 0.1–1: Poor response ˃1: Normal response |
| Anti-T IgG (IU/mL) | 0.05 | 0.39 |
< 0.1: No response 0.1–1: Poor response ˃1: Normal response |
| Plt (× 103 cells/mm3) | 1091 | 738 | 150–450 |
| CD3 + T cells (% of lymphocytes) | 79% | 30% | 50–90 |
| CD4 + T cells (% of lymphocytes) | 12% | 22% | 20–65 |
| CD8 + T cells (% of lymphocytes) | 50% | 2% | 5–40 |
| CD19 + B cells (% of lymphocytes) | 2% | 27.5% | 3–40 |
| CD16 + NK cells (% of lymphocytes) | 15% | 16.7% | 3–15 |
| CD56 + NK cells (% of lymphocytes) | 19% | 21% | 3–15 |
| NBT | 100% | 100% | > 95 |
| CRP | 112 | 3 | < 10 |
| LTT | |||
| PHA | 4.8 | 4.9 | ≥ 3 |
| BCG | 1.1 | 1.4 | ≥ 2.5 |
| Candida | 1.8 | 2.0 | ≥ 2.5 |
Bold items indicate abnormal parameters according to the reference ranges for ages
WBC white blood cell, Ig immunoglobulin, Anti-D anti-diphtheria, Anti-T anti-tetanus, Plt platelet, NK natural killer, CRP C-reactive protein, LTT lymphocyte transformation test, PHA phytohemagglutinin, BCG Bacillus Calmette-Guérin
†While on exogenous immunoglobulin
He was referred to our hospital with a 6-month history of progressive bullous lesions. The lesions involved the face, trunk, palms, and soles, although the mucous membrane was intact (Fig. 1A). The bullae were mostly tense and a few had a thin roof and easily ruptured within 24 h. In the histopathologic examination, spongiotic epidermal reaction and subepidermal blisters associated with perivascular and interstitial infiltration of eosinophils and smaller numbers of neutrophils and lymphocytes were observed (Fig. 2). The immunohistochemistry study showed positive results for the CD1a, S100, and C kit proteins. Using direct immunofluorescence (DIF), linear deposition of IgG and C3 along the dermo-epidermal junction was found, compatible with childhood BP. He also developed hair loss and koilonychia. Methylprednisolone and azathioprine were added to the regimen, which resulted in a remarkable improvement of the skin lesions after 3 weeks. However, 2 weeks later, he was re-admitted to the intensive care unit (ICU) and eventually died due to fulminant sepsis.
Fig. 1.
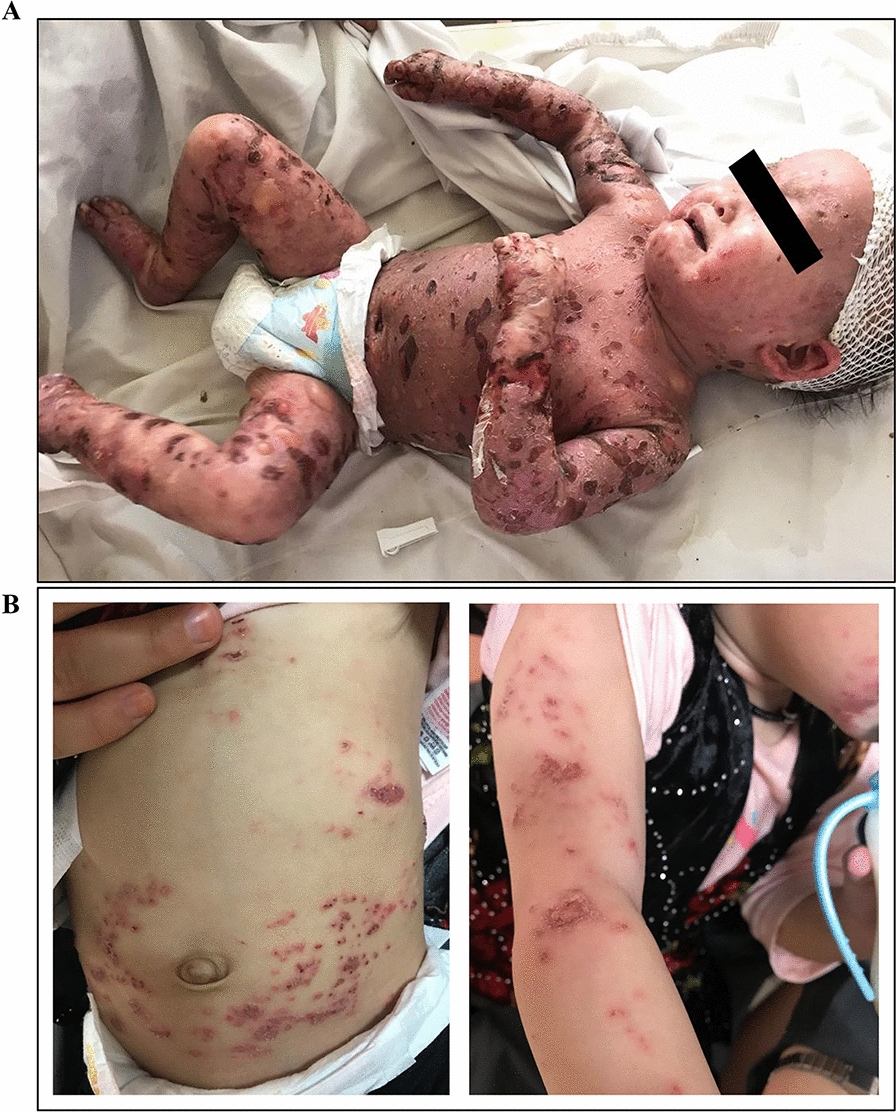
Diffuse bullous lesions in patient 1 A and 2 B
Fig. 2.
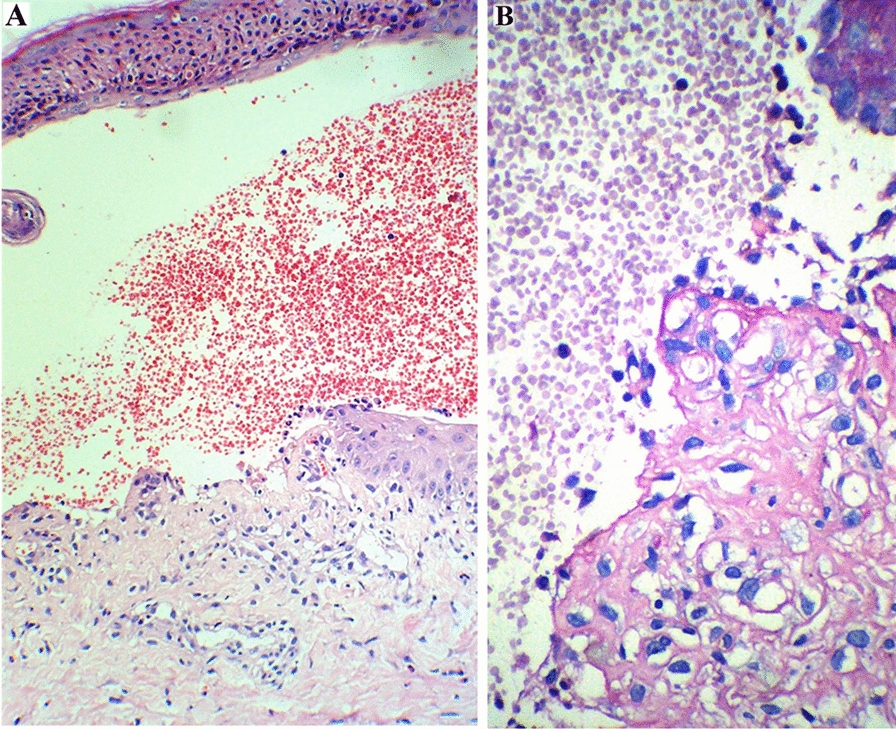
The histopathologic findings of skin lesions. a Subepidermal blister which appears intraepidermal located at the periphery as a result of epithelial regeneration. The blister is filled with many red blood cells and small number of neutrophils, eosinophils and lymphocytes, H&E × 200. b Periodic acid Schiff (PAS) positive basement membrane is focally presented at the base of the subepidermal blister, PAS × 400
One week after he passed away, patient 2 (his sister) presented at 1-year-old with severe eczematous skin lesions, which progressed to the forms of papulopustular and bullous rashes on the entire trunk and limbs and suffered from lesions similar to her sibling’s (Fig. 1B). She had facial dysmorphism comparable to her brother’s, had long been under treatment for occasional eczematous lesions, and had been receiving IVIg for 3 months due to low levels of immunoglobulins. No post-vaccination complication was reported. She also suffered from delayed umbilical cord separation (by 30 days), developmental delay, allergy to cow’s milk, recurrent oral candidiasis, and episodes of respiratory tract infections since infancy. Eventually, based on the basic immunologic workup, an underlying CID was suspected and treatment with IVIg continued. She is now well and in a relatively stable health condition. Later, the genetic study on blood samples of both siblings by WES revealed a novel homozygous ENST00000506587.5:c.845 T > C, p.Val282Ala variant in exon 7 of the PGM3 gene [SIFT: deleterious, PolyPhen: probably damaging, CADD score: 25].
Both parents were shown to be heterozygous for the variant (Fig. 3).
Fig. 3.

The family pedigree and chromatograms. Sanger sequencing confirmed a homozygous missense variant (c.845 T > C, p.Val282Ala) in exon 7 of the PGM3 gene in the index patient A. The mother B and father C were heterozygous for the variant. Squares, male; circle, female; solid symbols, affected subjects; slashed symbol, deceased subject
Discussion
Cutaneous lesions may be the first or predominant presentation in patients with IEIs. The most commonly reported skin disorders include eczema, mucocutaneous candidiasis, skin abscess, granulomas, erythroderma, warts, molluscum contagiosum, alopecia, and vitiligo (for a detailed review, refer to [11]). Among others, pemphigoid skin diseases are extremely rare dermatologic manifestations in IEIs and there have been few reports of only 14 patients with these complications.
Pemphigoid skin diseases are autoimmune subepidermal bullous disorders encompassing different subtypes, namely BP, mucous membrane pemphigoid, epidermolysis bullosa acquisita, gestational pemphigoid, and anti-p200 pemphigoid, with BP being the most prevalent [1]. Although rare, the diagnosis of childhood BP is considered in young patients (≤ 18 years old) with tense subepidermal bullae and dermal infiltration, predominantly by eosinophils. Nonetheless, the definite diagnosis is ascertained by DIF showing linear deposition of IgG and/or C3 alongside the basement membrane zone or by the detection of circulating IgG autoantibodies against the basement membrane through indirect immunofluorescence [12].
The first report documenting childhood pemphigoid in a setting of immunodeficiency was made about two decades ago when Bloomfield et al. [13] reported an 8-month-old girl with thymic hypoplasia, autoimmune hemolytic anemia, T cell lymphopenia, and juvenile pemphigoid. She failed to respond to steroids and, despite adding sulphapyridine to the regimen, the bullae became extensive and she eventually died due to pulmonary edema. After that, clinically similar patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) [14–16] and IPEX-like syndrome [17, 18], hyper IgE syndrome (HIES) [19, 20], zeta-chain-associated protein kinase 70 (ZAP70) deficiency [21], and common variable immunodeficiency (CVID) [22] were reported to suffer from pemphigoid blistering conditions, including BP, pemphigoid nodularis, and mucous membrane pemphigoid.
A summary of the demographic and clinical features of IEI patients who had pemphigoid blistering disorder complications are provided in Table 2. It is worth noting that the focus of this review is on BP disorders and associated variants. There have also been reports of other similar childhood blistering disorders in IEI patients such as chronic granulomatous disease (CGD) with linear IgA dermatosis [23], auto-inflammation and phospholipase Cγ2 (PLCγ2)-associated antibody deficiency and immune dysregulation (APLAID) syndrome with early-onset blistering lesions [24], and SERPING1 mutation with bullous lesions at the site of angioedema [25] that are not discussed here.
Table 2.
A summary of the demographic and clinical features of IEI patients with pemphigoid blistering diseases
| Patient | Type of PIDs (Mutated gene) | Type of blistering disorder | Age (years) | Sex | Parental consanguinity | Origin | The onset of skin disorder (years) | Blister involvement | DIF | Other comorbidities | Treatment | D/A | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CID (PGM3) | Bullous pemphigoid | 2.7 | M | Yes | Afghanistan | 2.2 | Face, trunk, extremities | IgG, C3 | FTT, enteropathy, allergy, recurrent otitis media with tympanic membrane perforation, recurrent oral candidiasis, hair loss, koilonychia | Prednisolone, azathioprine | D | This report, 2022 |
| 2 | CID (PGM3) | Bullous pemphigoid | 1.0 | F | Yes | Afghanistan | NA | Face, trunk, extremities | IgG, C3 | Food allergy, eczema, FTT, Recurrent oral candidiasis, RTI, recurrent oral candidiasis | IVIg | A | This report, 2022 |
| 3 | IPEX (FOXP3) | Bullous pemphigoid | 7.0 | M | No | Denmark | 2.0 | Face, extremities, gluteal region | IgG | FTT, Lymphoid interstitial pneumonia | Prednisolone, Azathioprine, MMF, CsA | A | Anderson et al. 2021 |
| 4 | IPEX (FOXP3) | Pemphigoid nodularis | 23.0 | NA | NA | USA | NA | NA | NA | Recurrent RTI, Enteropathy, Eczema/psoriasis, Thyroiditis, Nephropathy, Hypogammaglobulinemia | Immunosuppressive, IVIg, HSCT | NA | Rosenberg et al. 2018 [13] |
| 5 | CVID (NA) | Mucous membrane pemphigoid | 73.0 | F | NA | USA | 72.2 | Oral mucosa | IgG, C3 | No | Topical dexamethasone | A | Doll et al., 2017 [21] |
| 6 | ZAP70 Deficiency (ZAP70) | Bullous pemphigoid | 2.0 | M | No | USA | 1.6 | Face, trunk, extremities, oral mucosa | IgG | Inflammatory colitis, Hemophilia, Nephrotic Syndrome | HSCT | A | Chan et al., 2016 [20] |
| 7 | ZAP70 Deficiency (ZAP70) | Bullous pemphigoid | 0.2 | F | No | USA | 0.1 | Face, trunk, extremities | IgG | Growth failure, Inflammatory colitis, Proteinuria, autoimmune thyroiditis | HSCT | A | Chan et al., 2016 [20] |
| 8 | HIES (STAT3) | Bullous pemphigoid | 30.0 | F | NA | Indonesia | 4.0 | Entire body | NA | Esophagus stricture, Recurrent RTI, Lung TB, Alopecia, Hair loss, Anonychia, Anemia, Malnutrition | Prednisolone, Topical antibiotics | A | Budiyani et al. 2016 [18] |
| 9 | IPEX-like syndrome (CD25/IL2RA) | Bullous pemphigoid | 8.0 | F | Yes | Italy | 1.0 | NA | NA | Autoimmune enteropathy, CMV infection, Diffuse eczema, Autoimmune thyroiditis, Alopecia universalis, Lymphadenopathies | Plasmapheresis | A | Goudy et al. 2013 [16] |
| 10 | IPEX-like syndrome (NA) | Bullous pemphigoid | 7.0 | NA | NA | Italy | NA | NA | NA | Arthritis, AIHA, Autoimmune hepatitis, Enteropathy | Steroids, Rituximab, MMF, cyclophosphamide | NA | Barzaghi et al. 2012 [17] |
| 11 | HIES (NA) | Bullous pemphigoid | 0.5 | M | No | Turkey | 0.2 | Face, trunk, extremities, oral mucosa | IgG | Recurrent RTI, Recurrent oral thrush, Otitis, Severe eczema | Prednisolone, Antibiotics | A | Erbagci et al. 2008 [19] |
| 12 | CVID | IgA mucous membrane pemphigoid | 46.0 | F | NA | Canada | 45.3 | Conjunctiva | IgA | Eye disorder, meningioma, atrial myxoma | Dapsone, IVIG | Suwattee et al. 2004 | |
| 13 | IPEX (FOXP3) | Bullous pemphigoid, pemphigoid nodularis | 14.0 | M | No | USA | 0.1 | Face, trunk, extremities | IgG, C3 | Autoimmune enteropathy, Recurrent RTI, Abscess, Asthma, growth failure, VZV and EBV infection | Prednisolone, CsA, Dapsone, Azathioprine, MTX, Hydroxyzine, SSRI, Topical tacrolimus, IVIg, Antibiotics, Rituximab | A | Ferguson et al. 2000 [14], McGinnes et al. 2006 [15] |
| 14 | NA | Juvenile pemphigoid | 0.8 | F | NA | UK | 0.7 | Face, trunk, extremities | IgG | Thymic hypoplasia, AIHA, T lymphopenia | Prednisolone, sulphapyridine | D | Bloomfield et al. 1982 [12] |
A alive, AIHA autoimmune hemolytic anemia, CGD chronic granulomatous disease, CID combined immunodeficiency, CMV cytomegalovirus, CsA cyclosporine A, CVID common variable immunodeficiency, D dead, DIF direct immunofluorescence, EBV epstein-barr virus, F female, FOXP3 forkhead box protein P3, HIES hyper IgE syndrome, HSCT hematopoietic stem cell transplantation, Ig immunoglobulin, IVIg intravenous immunoglobulin, IL2RA interleukin-2 receptor alpha chain, IPEX immune dysregulation, polyendocrinopathy, enteropathy, X-linked, M male, MMF Mycophenolate mofetil, MTX methotrexate, NA not available, PID primary immunodeficiency disorder, RTI respiratory tract infection, STAT signal transducer and activator of transcription, TB tuberculosis, VZV Varicella zoster virus, ZAP70 zeta-chain-associated protein kinase 70
Intriguingly, five out of 14 reported IEI patients with BP disorders were diagnosed with IPEX or IPEX-like syndrome. This association may result from a low regulatory T cell (T reg) count [26], which is reported in 68% and 50% of patients with IPEX and IPEX-like syndrome, respectively [27]. In fact, recent studies have demonstrated that T reg cells play an important role in preventing the production of autoantibodies against BP180 and BP230 in human and mice models [28].
The protein encoded by PGM3 is required for the reversible conversion of N-acetylglucosamine-6-phosphate (GlcNAc-6-P) to N-acetylglucosamine-1-phosphate (GlcNAc-1-P) during the synthesis of UDP-GlcNAc, with an intra- and extracellular structural role, as well as a role in cell signaling [29]. Variants in PGM3 were first assumed to be responsible for autosomal recessive forms of hyper IgE syndrome, patients with CID/SCID phenotype and mutated PGM3 are reported in the literature [30–33], mainly manifesting as facial dysmorphisms, skeletal abnormalities, neurologic disorders, renal disorders, and gastrointestinal complications, and less frequently congenital heart disorders, and recurrent respiratory tract infections. Most patients suffered from skin disorders mainly in the form of eczema or as a consequence of infections but none of them had BP as the patients depicted here. Therefore, underlying PGM3 mutation should be suspected in (S)CID patients with facial and skeletal abnormalities.
To our knowledge, this is the first report of childhood bullous pemphigoid in the setting of PGM3 deficiency. Dermatologic disorders may be the presenting sign in patients with CID. In a cohort of 696 CID patients, about 11% of non-syndromic CID patients primarily presented with generalized eczema, skin infections, and abscesses, and almost half of the patients with syndromic CID developed dermatologic abnormalities during the course of the disease [34]. These findings emphasize the importance of considering CIDs during the assessment of skin disorders, particularly if they are extensive, recurrent, refractory to treatment, and/or associated with other signs of IEIs [11].
Acknowledgements
Authors thanks patients and their family for their participation in this case study.
Abbreviations
- AIBD
Autoimmune blistering disorders
- APLAID
Auto-inflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation
- BP
Bullous pemphigoid
- CGD
Chronic granulomatous disease
- CIDs
Combined immunodeficiencies
- CRP
C-reactive protein
- CVID
Common variable immunodeficiency
- DIF
Direct immunofluorescence
- HIES
Hyper IgE syndrome
- HIV
Human immunodeficiency viruses
- ICU
Intensive care unit
- IEIs
Inborn errors of immunity
- IPEX
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- IVIg
Intravenous immunoglobulin
- MAF
Minor allele frequency
- NBT
Nitro blue tetrazolium
- NK
Natural killer
- PCR
Polymerase chain reaction
- PGM3
Phosphoglucomutase 3
- SNVs
Single nucleotide variants
- WES
Whole exome sequencing
- ZAP70
Zeta-chain-associated protein kinase 70
Author contributions
ZCH and MJ contributed to the conceptualization, data curation, and supervision; MF and MJ wrote the original draft; MeM performed the immunological tests. FA was involved in the diagnosis and treatment of skin lesions; MKh elicited the histopathologic examination. ASR, AF, JD, MK and KB performed the genetic studies. JE, MaM, GE and SF reviewed and edited the final manuscript. All authors read and approved the final manuscript.
Funding
The authors received no specific funding for this research.
Availability of data and materials
The detailed laboratory data of two patients are available at supplementary material.
Declarations
Ethics approval and consent to participate
This case study was accredited by Ethical Committee of Shahid Beheshti University of Medical Sciences. Informed consent was obtained from both patient’s parents.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mahnaz Jamee, Email: mahnaz.jamee@gmail.com.
Zahra Chavoshzadeh, Email: zahra_chavoshzadeh@yahoo.com.
References
- 1.Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. 2018;54(1):26–51. doi: 10.1007/s12016-017-8633-4. [DOI] [PubMed] [Google Scholar]
- 2.Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. 2018;5:296. doi: 10.3389/fmed.2018.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egami S, Yamagami J, Amagai M. Autoimmune bullous skin diseases, pemphigus and pemphigoid. J Allergy Clin Immunol. 2020;145(4):1031–1047. doi: 10.1016/j.jaci.2020.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Chou C-S. Childhood bullous pemphigoid: a case report and literature review. J Clin Exp Dermatol Res. 2014 doi: 10.4172/2155-9554.S6-010. [DOI] [Google Scholar]
- 5.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. 2020;40(1):24–64. doi: 10.1007/s10875-019-00737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azizi G, Ziaee V, Tavakol M, Alinia T, Yazdai R, Mohammadi H, et al. Approach to the management of autoimmunity in primary immunodeficiency. Scand J Immunol. 2017;85(1):13–29. doi: 10.1111/sji.12506. [DOI] [PubMed] [Google Scholar]
- 7.Abinun MAM, Beaussant Cohen S, Buckland M, Bustamante J, Cant AJ, et al. ESID registry - Working definitions for clinical diagnosis of PID. https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria. Accessed 22 Nov 2022.
- 8.Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki ÖY, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol. 2016;17(12):1352–1360. doi: 10.1038/ni.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozen A, Comrie WA, Ardy RC, Domínguez Conde C, Dalgic B, Beser ÖF, et al. CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. N Engl J Med. 2017;377(1):52–61. doi: 10.1056/NEJMoa1615887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma D, Jindal AK, Rawat A, Singh S. Approach to a child with primary immunodeficiency made simple. Indian Dermatol Online J. 2017;8(6):391–405. doi: 10.4103/idoj.IDOJ_189_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemeth AJ, Klein AD, Gould EW, Schachner LA. Childhood bullous pemphigoid Clinical and immunologic features, treatment, and prognosis. Arch Dermatol. 1991;127(3):378–86. doi: 10.1001/archderm.1991.01680030098014. [DOI] [PubMed] [Google Scholar]
- 13.Bloomfield S, Stockdill G, Barnetson RS. Thymic hypoplasia, auto-immune haemolytic anaemia and juvenile pemphigoid in an infant. Br J Dermatol. 1982;106(3):353–355. doi: 10.1111/j.1365-2133.1982.tb01735.x. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg JM, Maccari ME, Barzaghi F, Allenspach EJ, Pignata C, Weber G, et al. Neutralizing anti-cytokine autoantibodies against interferon-α in immunodysregulation polyendocrinopathy enteropathy X-linked. Front Immunol. 2018;9:544. doi: 10.3389/fimmu.2018.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson PJ, Blanton SH, Saulsbury FT, McDuffie MJ, Lemahieu V, Gastier JM, et al. Manifestations and linkage analysis in X-linked autoimmunity-immunodeficiency syndrome. Am J Med Genet. 2000;90(5):390–397. doi: 10.1002/(SICI)1096-8628(20000228)90:5<390::AID-AJMG9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 16.McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55(1):143–148. doi: 10.1016/j.jaad.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 17.Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146(3):248–261. doi: 10.1016/j.clim.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barzaghi F, Passerini L, Gambineri E, Ciullini Mannurita S, Cornu T, Kang ES, et al. Demethylation analysis of the FOXP3 locus shows quantitative defects of regulatory T cells in IPEX-like syndrome. J Autoimmun. 2012;38(1):49–58. doi: 10.1016/j.jaut.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budiyani L, Idayu A, Wijaya I, Widhani A. Sindrom Hiper IgE dengan manifestasi bullous pemphigoid dan striktur esofagus. J Penyakit Dalam Indonesia. 2016;3:100. doi: 10.7454/jpdi.v3i2.96. [DOI] [Google Scholar]
- 20.Erbagci Z. Childhood bullous pemphigoid in association with hyperimmunoglobulin E syndrome. Pediatr Dermatol. 2008;25(1):28–33. doi: 10.1111/j.1525-1470.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 21.Chan AY, Punwani D, Kadlecek TA, Cowan MJ, Olson JL, Mathes EF, et al. A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J Exp Med. 2016;213(2):155–165. doi: 10.1084/jem.20150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doll R, Johnson J, Tcheurekdjian H, Hostoffer R. Common variable immune deficiency associated with pemphigoid. Ann Allergy Asthma Immunol. 2017;118(1):122. doi: 10.1016/j.anai.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Sillevis Smitt JH, Leusen JH, Stas HG, Teeuw AH, Weening RS. Chronic bullous disease of childhood and a paecilomyces lung infection in chronic granulomatous disease. Arch Dis Child. 1997;77(2):150–152. doi: 10.1136/adc.77.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neves JF, Doffinger R, Barcena-Morales G, Martins C, Papapietro O, Plagnol V, et al. Novel PLCG2 mutation in a patient with APLAID and cutis laxa. Front Immunol. 2018;9:2863. doi: 10.3389/fimmu.2018.02863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serpa FS, Veronez CL, Campinhos FL, Moyses TR, Pesquero JB. SERPING1 mutation in a rare hereditary angioedema with skin blisters. Ann Allergy Asthma Immunol. 2019;122(3):340–341. doi: 10.1016/j.anai.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 26.Schultz B, Hook K. Bullous diseases in children: a review of clinical features and treatment options. Paediatr Drugs. 2019;21(5):345–356. doi: 10.1007/s40272-019-00349-3. [DOI] [PubMed] [Google Scholar]
- 27.Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like syndrome. J Allergy Clin Immunol Pract. 2020 doi: 10.1016/j.jaip.2020.04.070. [DOI] [PubMed] [Google Scholar]
- 28.Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. 2018;142(6):1818–30.e6. doi: 10.1016/j.jaci.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Fliegauf M, Grimbacher B. Hyper-IgE syndromes: reviewing PGM3 deficiency. Curr Opin Pediatr. 2014;26(6):697–703. doi: 10.1097/MOP.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 30.Stray-Pedersen A, Backe PH, Sorte HS, Mørkrid L, Chokshi NY, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95(1):96–107. doi: 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernth-Jensen JM, Holm M, Christiansen M. Neonatal-onset T(-)B(-)NK(+) severe combined immunodeficiency and neutropenia caused by mutated phosphoglucomutase 3. J Allergy Clin Immunol. 2016;137(1):321–324. doi: 10.1016/j.jaci.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 32.Pacheco-Cuéllar G, Gauthier J, Désilets V, Lachance C, Lemire-Girard M, Rypens F, et al. A novel PGM3 mutation is associated with a severe phenotype of bone marrow failure, severe combined immunodeficiency, skeletal dysplasia, and congenital malformations. J Bone Mineral Res. 2017;32(9):1853–1859. doi: 10.1002/jbmr.3173. [DOI] [PubMed] [Google Scholar]
- 33.Fusaro M, Vincent A, Castelle M, Rosain J, Fournier B, Veiga-da-Cunha M, et al. Two novel homozygous mutations in phosphoglucomutase 3 leading to severe combined immunodeficiency, skeletal dysplasia, and malformations. J Clin Immunol. 2021 doi: 10.1007/s10875-021-00985-w. [DOI] [PubMed] [Google Scholar]
- 34.Abolhassani H, Chou J, Bainter W, Platt CD, Tavassoli M, Momen T, et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J Allergy Clin Immunol. 2018;141(4):1450–1458. doi: 10.1016/j.jaci.2017.06.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The detailed laboratory data of two patients are available at supplementary material.