

Abstract
Protein structures are decisive for their activities and interactions with other molecules. Global analysis of protein structures and conformational changes cannot be achieved by commonly used abundance-based proteomics. Here, we integrated cysteine covalent labeling, selective enrichment, and quantitative proteomics to study protein structures and structural changes on a large scale. This method was applied to globally investigate protein structures in HEK293T cells and protein structural changes in the cells with the tunicamycin (Tm)-induced endoplasmic reticulum (ER) stress. We quantified several thousand cysteine residues, which contain unprecedented and valuable information of protein structures. Combining this method with pulsed stable isotope labeling by amino acids in cell culture, we further analyzed the folding state differences between pre-existing and newly synthesized proteins in cells under the Tm treatment. Besides newly synthesized proteins, unexpectedly, many pre-existing proteins were found to become unfolded upon ER stress, especially those related to gene transcription and protein translation. Furthermore, the current results reveal that N-glycosylation plays a more important role in the folding process of the tertiary and quaternary structures than the secondary structures for newly synthesized proteins. Considering the importance of cysteine in protein structures, this method can be extensively applied in the biological and biomedical research fields.
Graphical Abstract
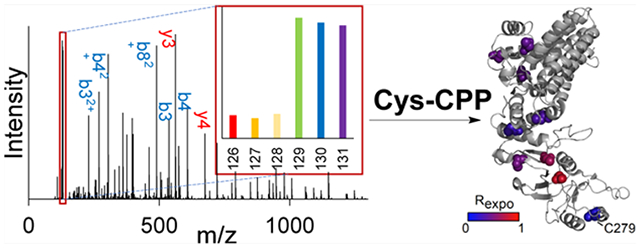
INTRODUCTION
Protein structures and conformational changes are essential to their activities and interactions with other molecules.1,2 In recent years, structural proteomics has attracted much attention because it can provide valuable information about protein structures that is not easily obtained from other methods like nuclear magnetic resonance and cryoEM.3–7 Among the existing structural proteomics approaches, protein footprinting is very attractive.8–10 Typically, covalent protein painting (CPP) using irreversible labeling reagents to quickly target the solvent-accessible amino acid residues in proteins under native conditions is one of the most appealing methods to detect protein structure and conformation changes in different biological environments.11 To date, methionine, tryptophan, and lysine have already served as targets for protein structure analysis at the proteome level.11–14 However, lysine residues are usually protonated under physiological pH and are likely present on the surface of proteins in their well-folded conformations.15 Therefore, most lysine residues may not have significant accessibility differences between the folded and unfolded forms of a protein. Although methionine and tryptophan are relatively more hydrophobic and have higher chances of locating in the protected regions, their frequencies in protein sequence are relatively low,15 and they are less reactive. The cysteine residue is a tempting target because free cysteine is less solvent accessible than other amino acid residues among well-folded proteins in the whole proteome.15,16 Cysteine labeling has been applied to study the structures of purified proteins,17 but is yet to be tested in complex biological environments. In recent years, mass spectrometry (MS)-based strategies have been developed for cysteine reactivity profiling studies.18,19 Through treating the cell lysate with low and high concentrations of semi-reactive thiol probes, the cysteine residues with various reactivities can be quantified on a global scale. However, a highly reactive probe is required for protein structure analysis using CPP, which is different from the cysteine reactivity studies.
In eukaryotic cells, the endoplasmic reticulum (ER) is responsible for the synthesis and maturation of ~40% of the proteins in the proteome.20 The homeostasis inside the ER can be challenged by the alteration of protein synthesis and folding processes, leading to the accumulation of unfolded proteins and the reduced efficiency of protein secretion through the classical secretion pathway.21,22 Over the past decade, the ER stress response has been extensively studied through the comparison of the protein expression differences in cells with and without stress-inducing chemical treatments.23–25 However, the direct consequences of ER stress, that is, protein folding state changes, are not well studied, which cannot be directly analyzed using commonly used abundance-based proteomics approaches. Proteins need to fold into well-defined 3D structures in order to make them functional. Otherwise, their catalytic activities and interactions with other molecules will be negatively impacted. Moreover, studies have shown that proteins outside the ER might also become unfolded during ER stress,26 which is related to the pleiotropic cellular responses to the ER stress,27 Therefore, it is intriguing to profile protein structural changes under ER stress at the proteome level.
In this work, we integrated cysteine covalent labeling, selective enrichment, and quantitative proteomics, called cysteine targeted CPP (Cys-CPP), to investigate protein structures and structural changes on a proteome-wide scale. A probe was designed to target cysteine, which contains a cysteine-reactive group, a biotin moiety for enrichment, and a cleavable linker for generating a small tag for site-specific analysis of cysteine-containing peptides by MS. Cells are lysed under non-denaturing conditions, and the lysate is split into two identical halves. One sample is denatured under a high concentration of chaotropic chemicals along with heating to unfold proteins and achieve maximal cysteine exposure, which serves as an unfolded reference. Then exposed cysteines were covalently modified with the probe in the two samples. After the enrichment of peptides with the modified cysteine, the samples were mixed, followed by fractionation and MS analysis. The ratios of peptides between the native sample and the denatured one provide valuable information about protein structures. Using Cys-CPP, we demonstrate that the ER stress can induce widespread protein unfolding. Furthermore, combining Cys-CPP with pulsed stable isotope labeling by amino acids in cell culture (pSILAC), we studied the folding state differences between pre-existing and newly synthesized proteins under the ER stress and performed a detailed analysis on the effect of protein unfolding caused by ER stress. Overall, the current method can obtain much valuable information about protein structures and their structural changes.
EXPERIMENTAL SECTION
Cell Culture and Lysis.
Baker’s yeast (Saccharomyces cerevisiae) was grown in yeast extract-peptone–dextrose (YPD) (Difco & BBL) medium at 30 °C until OD600 ≈ 1.0. Cells were centrifuged at 3000g for 3 min and washed with ice-cold phosphate-buffered saline (PBS). Cell pellets were lysed by a bead beater four times for 30 s each, and after every time, the tube was rested on ice for 2 min in a lysis buffer [50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH = 7.4, 150 mM NaCl, 20 units/mL benzonase]. The lysate was centrifuged at 16,000g for 10 min, and the protein concentration of the resulting supernatant was determined by the Pierce BCA protein assay and further diluted to 1 mg/mL with the lysis buffer.
HEK293T cells [American type culture collection (ATCC)] were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich) containing 10% fetal bovine serum (FBS) (Thermo) and 100 units/mL penicillin–streptomycin in a humidified incubator with 5.0% CO2 at 37 °C. When cells reached ~80% confluency, they were treated with 0.8 μg/mL tunicamycin or an equal volume of DMSO as a control. Cells were further cultured for 12 h before harvesting. For newly synthesized protein analysis, when HEK293T cells reached ~80% confluency, light media was removed, and cells were washed twice with PBS. Heavy media containing Lys8 and Arg6 and 0.8 μg/mL tunicamycin or an equal volume of DMSO were then added, and cells were incubated for 3 h. The fully heavy-labeled sample was from cells grown in heavy media for five generations.
Cells were then washed with ice-cold PBS and harvested by scraping and centrifugation. Cell pellets were resuspended in the lysis buffer and lysed through frozen-thaw cycles in liquid nitrogen for 3 times. The lysates were clarified at 4600g for 10 min, and the protein concentration was measured and further diluted to 1 mg/mL using the lysis buffer as above.
Tagging the Exposed Cysteine Residues in Proteins and Protein Digestion.
For the condition optimization experiments, the yeast lysate (200 μg proteins each) was used and duplicate experiments were performed. Each lysate was filtered using a 3K MWCO centrifugal filter (VWR) at 14,000g for 15 min and washed twice with 200 μL of the original lysis buffer. The protein concentration of the lysate was then adjusted back to 1 mg/mL and treated with 4.6 M of guanidium hydrochloride (GnHCl) for 15 min. The mixture was further heated at 95 °C for the different times indicated in Figure 1D and then equilibrated at room temperature for 3 min. After protein denaturation, the lysate was treated with different concentrations of the PC biotin–PEG3–NEM for a series of times. The detailed synthesis procedure of the PC biotin–PEG3–NEM was described in the Supporting Information. Excess probe molecules were removed using the methanol–chloroform protein precipitation method. The resulting tagged proteins were digested with trypsin in a digestion buffer (5% ACN, 50 mM HEPES pH = 8.2, and 1.6 M urea) at 37 °C for 16 h. All the procedures mentioned above were performed in the dark.
Figure 1.
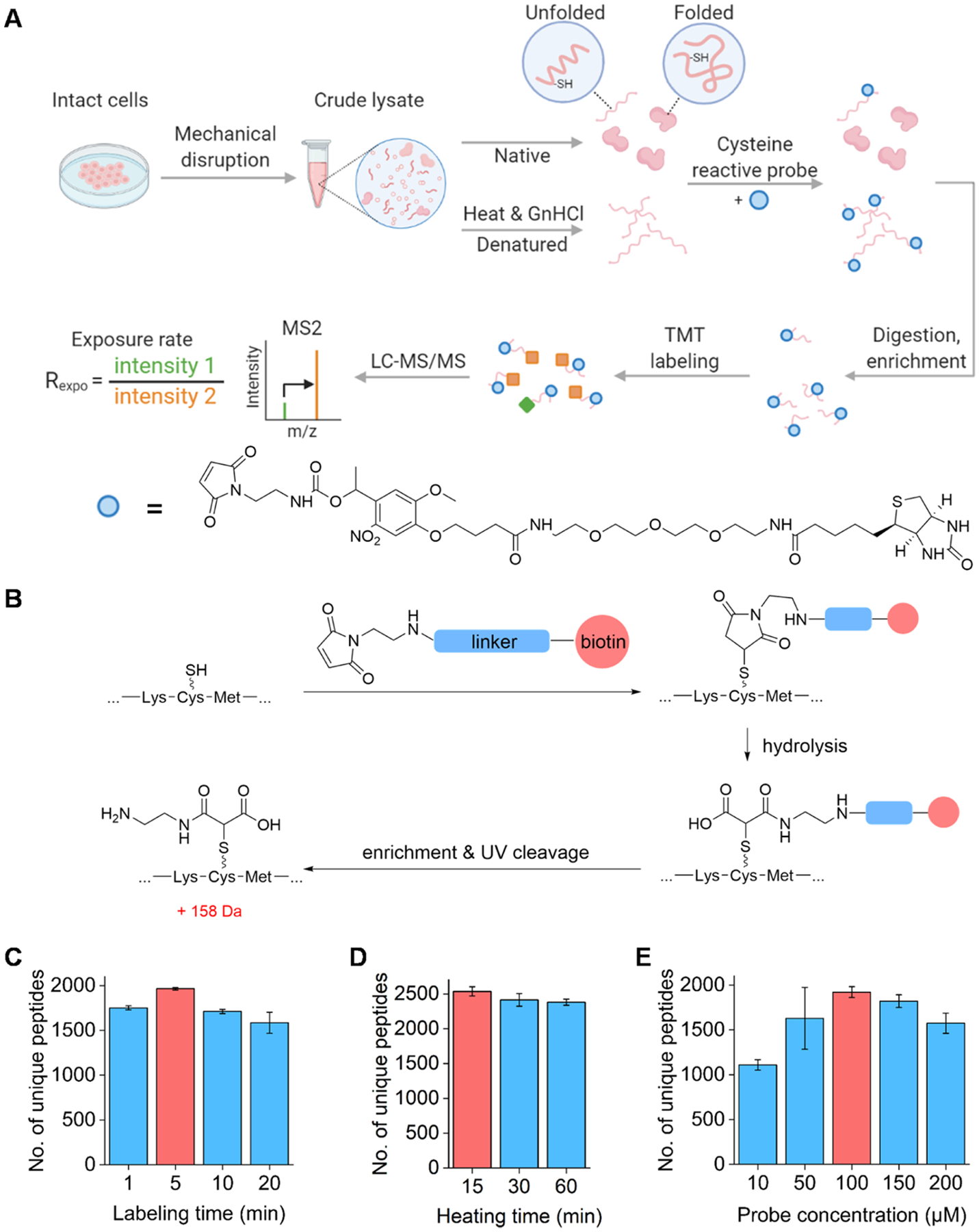
Principle for quantitative analysis of the cysteine exposure rates in the human cell proteome using the cysteine-reactive probe and optimization of experimental conditions. (A) General workflow of Cys-CPP. (B) Reaction between the cysteine residue with the probe. After the enrichment, UV cleavage is performed to leave a small tag on the modified cysteine for site-specific analysis by MS. (C–E) Optimization of the reaction conditions: labeling time (C), heating time (D), and probe concentration (E). The red columns represent the reaction conditions used in the finalized protocol.
For the experiments for studying protein structures, 0.5 mg of proteins for each biological replicate was used. Each lysate was filtered using a 3K MWCO centrifugal filter and washed twice. The protein concentration of the lysate was then adjusted back to 1 mg/mL, and each lysate was evenly separated into two parts, “denatured” and “native”. Proteins in the “denatured” group were treated with 4.6 M GnHCl for 15 min and further heated at 95 °C for 15 min. Those from the “native” group were left on the ice. After the denaturation, the lysate was equilibrated at room temperature for 3 min. Each lysate was then treated with 100 μM PC biotin–PEG3–NEM for 5 min, and excessive probes were removed using the methanol–chloroform protein precipitation method. The tagged proteins were digested as described above.
Labeling of Cysteine-Containing Peptides with TMT, Enrichment, and Fractionation.
After protein digestion, the solution was acidified with TFA to a final pH value of ~2. Peptides were desalted using tC18 Sep-Pak cartridges (Waters) and then lyophilized. The purified peptides were subjected to tandem mass tag (TMT) labeling following the manufacturer’s instructions. The labeled peptide samples were combined, desalted, and lyophilized.
Peptides were then resuspended in 100 mM PBS and incubated with high-capacity neutravidin agarose resins (Thermo) at room temperature for 1 hour. The sample was transferred to a spin column and washed 10 times using 100 mM PBS. The mixture was transferred to a glass vial, and the enriched peptides were then eluted through the cleavage of the photocleavable linker under the UV radiation at 350 nm for 1 hour at room temperature for the first elution and 30 min for the second elution. The eluted peptides were desalted again and lyophilized. For the SILAC experiments, the TMT labeling was performed after the enrichment. Samples were then fractionated using high-pH reversed-phase HPLC (pH = 10). The sample was separated into six fractions for the non-SILAC experiments and 19 fractions for the SILAC experiments using a 4.6 × 250 mm 5 μm particle reversed-phase column (Waters) using a 40 min gradient of 5–50% ACN with 10 mM ammonium acetate. Every fraction was further purified with StageTip before LC–MS/MS.
LC–MS/MS Analysis.
The peptide samples were dissolved in a solvent containing 5% ACN and 4% FA, and 4 μL of the solution was loaded onto a microcapillary column packed with C18 beads (Magic C18AQ, 3 μm, 200 Å, 75 μm × 16 cm, Michrom Bioresources) using a Dionex WPS-3000TPLRS autosampler (UltiMate 3000 thermostatted Rapid Separation Pulled Loop Wellplate Sampler). Peptides were separated by reversed-phase high-performance liquid chromatography using an UltiMate 3000 binary pump with a 120 min gradient of 8–24% ACN (with 0.125% FA). Peptides were detected with a data-dependent Top15 method in a hybrid dual-cell quadrupole linear ion trap—Orbitrap mass spectrometer (LTQ Orbitrap Elite, Thermo Scientific, with Xcalibur 3.0.63 software). For each cycle, one full MS scan (resolution: 60,000) in the Orbitrap cell at the automatic gain control (AGC) target of 1*106 was followed by up to 15 MS/MS recorded in the Orbitrap cell with high mass accuracy and high resolution for the most intense ions. The selected ions were excluded from further analysis for 90 s. Ions with singly or unassigned charges were not fragmented and sequenced. MS/MS scans were activated under HCD at 30% normalized collision energy, and fragments were detected in the Orbitrap cell.
Database Search and Data Filtering.
The resulting raw files with MS/MS spectra were converted into mzXML files and then searched against the baker’s yeast (S. cerevisiae) or human (Homo sapiens) protein database (downloaded from Uniprot) using the SEQUEST algorithm (version 28).28 The following parameters were used during the search: 10 ppm precursor mass tolerance; 0.025 Da product ion mass tolerance; two missed cleavages; variable modifications: oxidation of methionine (+15.9949 Da) and modification on the cysteine residue (+158.0691); static modifications: TMT (+229.1629) on the lysine residue and the peptide N-terminus. For the newly synthesized protein experiment, cysteine modification was set as +387.2321 because free amine on the modified cysteine was labeled with the TMT reagent as well, and heavy lysine (+8.0142) and heavy arginine (+6.0201) were added as differential modifications. The false discovery rates (FDR) of cysteine-containing peptide and protein identifications were evaluated and controlled by the target-decoy method.29,30 Each protein sequence was listed in both forward and reversed orders. Linear discriminant analysis (LDA) was employed to control the quality of peptide identifications.31 Peptides shorter than seven amino acid residues in length were discarded. Furthermore, peptide spectral matches were filtered to <1%. The data set was restricted to cysteine-containing peptides when determining FDRs. In order to evaluate the site localization confidence, a ModScore was calculated for each labeled cysteine site,32 which applies a probabilistic algorithm that considers all cysteines in a peptide and uses the presence of MS/MS fragment ions unique to each cysteine site to evaluate the confidence of localization when the best site match is compared with the next best one. Sites with ModScore >13 (p < 0.05) were considered to be confidently localized. For quantification, only peptides with at least one confidently localized site were used. Because cysteine is a relatively rare amino acid, most peptides have only one site identified.
Calculation of the Cysteine Exposure Rate.
The TMT reporter ion intensities were used to quantify peptides. The isotopic information provided by Thermo was utilized to calibrate the ion intensities. Intensity normalization was performed in the replicate experiments to correct for the differences in protein loading. For the non-SILAC experiments, only peptides with the sum signal-to-noise ratio (S/N) greater than 30 and with the reporter ions found in all six channels were further analyzed for quantification. For sites that were detected more than once, the mean S/N was calculated from all peptides with the same sites. For each site, the S/N from the “native” channels was divided by S/N from the “denatured” channels of the same site to calculate the exposure rate. Three biological replicates were used to calculate the exposure rate significance, and we considered sites with a student’s t-test p-value < 0.05 as confidently quantified. For the SILAC experiments, only peptides with the sum S/N greater than 30 and with the reporter ions found at all four channels were further analyzed for quantification. For sites that were detected more than once, the mean S/N was calculated from all peptides with the same site. Peptides with high variation (CV > 0.25) between the replicate native or denatured channels were removed. For each site, the S/N from the “native” group channels was divided by the S/N from the “denatured” group channels of the same site to calculate the exposure rate.
Proteomics Data Analysis.
All analyses were performed in Excel and OriginLab 2020 unless mentioned otherwise. Protein property prediction was performed on Predict_Property standalone package (v1.01) downloaded from the Raptox server, which includes secondary structure, solvent accessibility, and predicted site disorderness,33–35 and protein sequences for property calculation were downloaded from Uniport. GO terms and pathway analysis were performed using the database for annotation, visualization, and integrated discovery (DAVID),36 where all identified proteins in the experiments served as background. Protein subcellular locations were determined by their GO cellular compartment annotation. All boxplots were generated using OriginLab 2020. The error bar with the column plot represents the absolute error of the data. The bold line within the box indicates the median value; the box borders represent the first and third quartiles; and the whiskers mean the minimum and maximum values are within 1.5 times the interquartile range.
RESULTS
Studying Protein Structures through Quantification of the Cysteine Exposure Rate Using Cys-CPP.
It is well known that cysteine plays a crucial role in protein structures. In this work, we studied protein structures by measuring the exposure rate of the cysteine residue in native proteins using Cys-CPP (Figure 1A). Cys-CPP enables us to quantify the accessibility alteration of the cysteine residues with the rationale that the structure and conformation change of proteins will result in the alteration of the surface accessibility of free thiols. Hence, we comprehensively studied protein structures by labeling surface-exposed cysteines. To achieve this, we synthesized a cysteine-reactive probe containing (1) a maleimide-based cysteine targeting warhead, (2) biotin, and (3) a UV-cleavable linker to label the cysteine residues and then effectively enrich cysteine-containing peptides (Figure S1). After the enrichment, the linker was cleaved under UV radiation, and a small tag remained on the peptide (Figure 1B).
To obtain the protein structural information under conditions similar to their native environment, we mechanically broke cells to collect the lysates without detergents. Small molecules were filtered out to avoid interferences from non-protein thiols (such as glutathione), and the lysates were then evenly split into two portions. One portion was in native conditions, while the other was treated with heat and a high concentration of the chemical denaturant, guanidium hydrochloride (GnHCl), to reach maximal denaturation of proteins,37 The cysteine residues of proteins are thus exposed as much as possible in the denatured sample. The cysteine-reactive probe was then added to both denatured and native protein samples, and the reaction occurred under the optimized conditions. The optimization results for the reaction conditions are shown in Figure 1C–E. After protein digestion, labeled cysteine-containing peptides were enriched through the biotin–avidin interactions. The cleavable linker in the probe can generate a small tag on the labeled cysteine (+158 Da) under UV radiation for site-specific analysis by MS, as shown in Figure 1B. This small tag containing a free amine group can also facilitate peptide ionization. The ratio of each peptide was then used to calculate the cysteine exposure rate, Rexpo. It should be noted that the protein abundance differences across the samples were normalized by using the exact same amount of the original cell lysates for the native and denatured samples.
We first applied this method to study the cysteine exposure rates of proteins in HEK293T cells under normal conditions. We performed the biological triplicate experiments, and the results are highly reproducible (Figure S2A,B). In this experiment, the exposure rates of 4391 unique cysteine sites were quantified (Figure S2C and Table S1). An example of peptide identification and quantification is shown in Figure 2A. The peptide VAHALAEGLGVIAC#IGEK (# refers to the labeling site) from triosephosphate isomerase was confidently identified with an Xcorr of 6.94. Another example is protein CCT3, which belongs to the chaperonin-containing T-complex assisting in protein folding and refolding in the cytosol. Nine out of ten cysteines in CCT3 were quantified (Figures 2B and S3). The intensities of the TMT38 reporter ions allowed us to accurately quantify the exposure rates of different cysteine sites, for instance, 0.16 for C279. This exposure rate agrees with its local environment in the crystal structure of the protein, where it is located between two helices.
Figure 2.
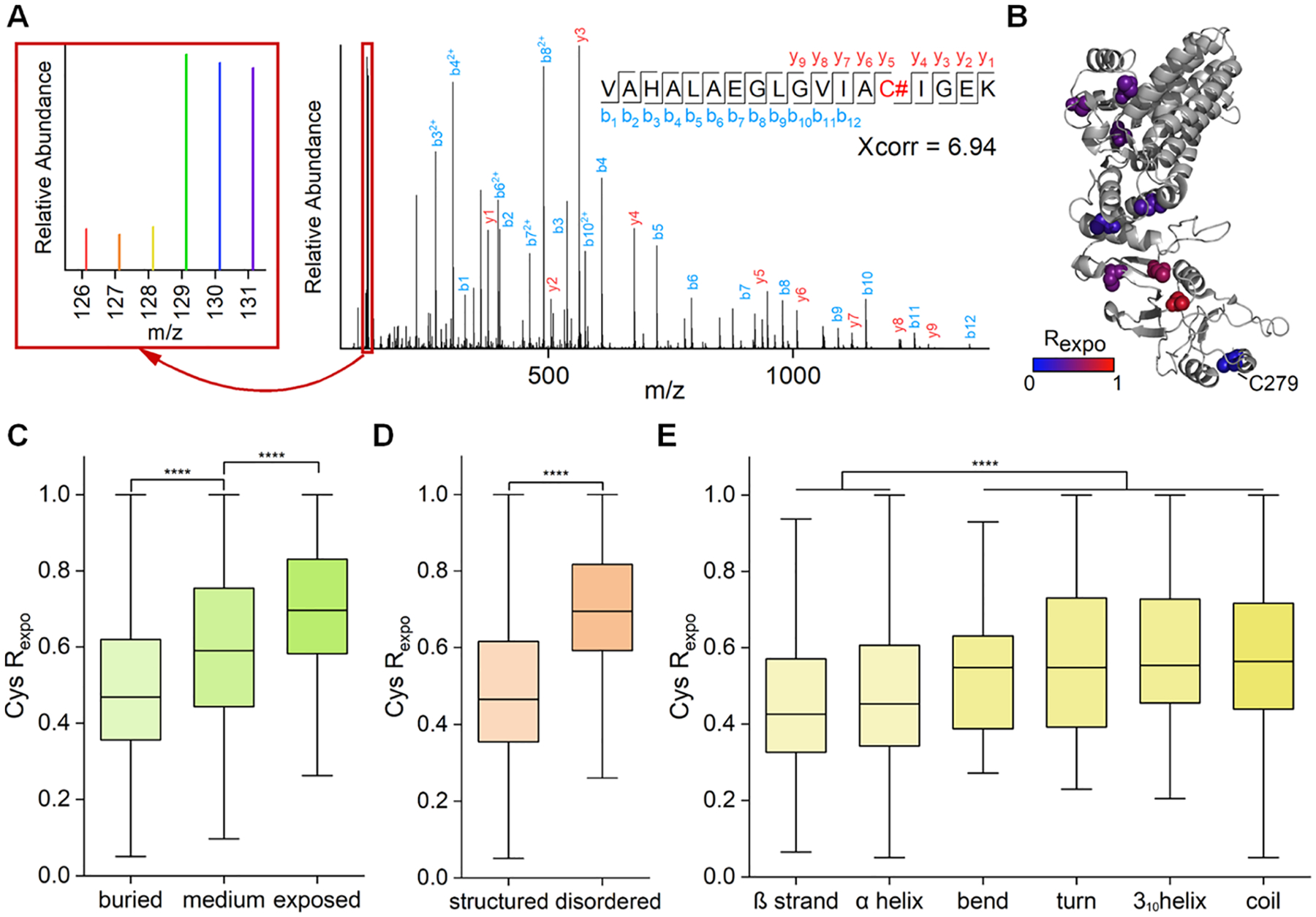
Analysis of the cysteine exposure rates in HEK293T cells. (A) Example tandem mass spectrum of VAHALAEGLGVIAC#IGEK (# refers to the labeling site). The intensities of the reporter ions allowed us to accurately quantify the exposure rate of a cysteine (an enlarged view of the reporter ion intensities is in the left box). (B) Demonstration of the localizations and the exposure rates of quantified cysteines of CCT3 (PDB code: 6qb8). (C–E) Comparison of the exposure rates of cysteines with different solvent accessibility (C), predicted disorderness (D), and secondary structures (E) (****p < 0.0001, Kolmogorov–Smirnov test).
We compared the exposure rates of the cysteine residues with their computationally predicted solvent accessibilities.33,34 The “buried,” “medium,” and “exposed” features of cysteine in the manuscript were predicted, and thus they may not be in total agreement with the actual solvent accessibility of each cysteine. Therefore, some disagreement could be from the prediction errors. Overall, the results showed a substantial positive correlation (Figure 2C). We then studied the effect of neighboring amino acids in the protein sequence on the cysteine exposure rates (Figure S4). Cysteines next to polar amino acid residues like serine and threonine have relatively higher exposure rates, which is consistent with the common belief that hydrophilic residues are more likely to be on the protein surface. Moreover, higher exposure rates were observed for the cysteine residues near the glycine and proline residues. This trend can be rationalized by the fact that the glycyl and prolyl residues frequently occur in turns and loops, which result in less sterically rigid hindrance to nearby cysteines.39,40 According to previous studies, electrostatic modulation caused by proximally charged residues can affect the cysteine reactivity because positive charges would stabilize the more reactive, nucleophilic thiolate form of free thiols.41 However, this has a minimal effect on protein structure analysis in this work because the primary sequence is the same in both the native and denatured forms of each protein. As expected, no apparent difference was observed among the exposure rates of the cysteines near positively charged, negatively charged, or uncharged polar residues (Figure S4). As expected, the measured cysteine exposure rates provide valuable information about protein structures. Analysis of the correlation between the cysteine exposure rates and their region disorderness (Figure 2D) or located secondary structures (Figure 2E) shows that the lower exposure rates are correlated well with more stable and structured local environments. Together, these data demonstrate that the current method can accurately measure the cysteine exposure rates and provide valuable information about protein structures.
Subsequently, the cysteine exposure rates are divided into quintile segments, with Q1 (0–20%) representing the sites in the most structured regions and Q5 (80–100%) standing for those in the least structured regions. Enrichment analysis based on biological process (BP) for proteins containing the sites in each segment shows that the majority of processes are enriched among proteins with the sites in the well-structured region (i.e., proteins with the sites in Q1) (Figure S5 and Table S2). Interestingly, proteins responsible for tRNA aminoacylation are enriched in all five segments, which indicates that those proteins have variable structures. It was reported that many eukaryotic cytoplasmic aminoacyl-tRNA synthetases acquired new domains compared with their archaeal or bacterial counterparts, and those new domains or the linker regions are more structurally disordered, which are essential for activating and regulating the functions of aminoacyl-tRNA synthetases.42
Analysis of Protein Unfolding Induced by Tunicamycin.
Protein N-glycosylation often occurs co-translationally, and it plays a vital role in the regulation of protein folding. It is speculated that the inhibition of protein N-glycosylation will result in misfolding of many newly synthesized proteins in the ER. Correspondingly, they will be trapped in the ER and generate enormous stress to cells, resulting in pleiotropic consequences beyond the ER stress responses. The current method provides an excellent opportunity to study protein structural changes in cells under the inhibition of protein N-glycosylation at the proteome-wide level.
Tm is most commonly used to inhibit protein N-glycosylation due to its high efficiency toward the integral membrane enzyme of GlcNAc-1-P-transferase, which catalyzes the first step of the N-glycan formation.43 After cells were treated with a high concentration of Tm for 12 h, the exposure rates of 3227 unique cysteine sites were quantified (Figure S6 and Table S3). Among those, 2561 unique cysteines from 1484 proteins were commonly quantified in both treated and untreated samples. The exposure rate of each cysteine upon the Tm treatment was compared with the corresponding one in the normal sample (Figure 3A). As expected, many cysteines shifted to higher exposure rates because inhibiting N-glycosylation significantly interrupted the protein folding in the ER.44,45 To further investigate the impact of protein structural changes, we compared the exposure rate of each cysteine with the corresponding one without the treatment (Figure S7). The results indicate that Tm has a more unfolding effect on the structurally unstable regions in proteins. The subsequent analysis of secondary structures and site disorderness also supports this conclusion (Figure S8).
Figure 3.
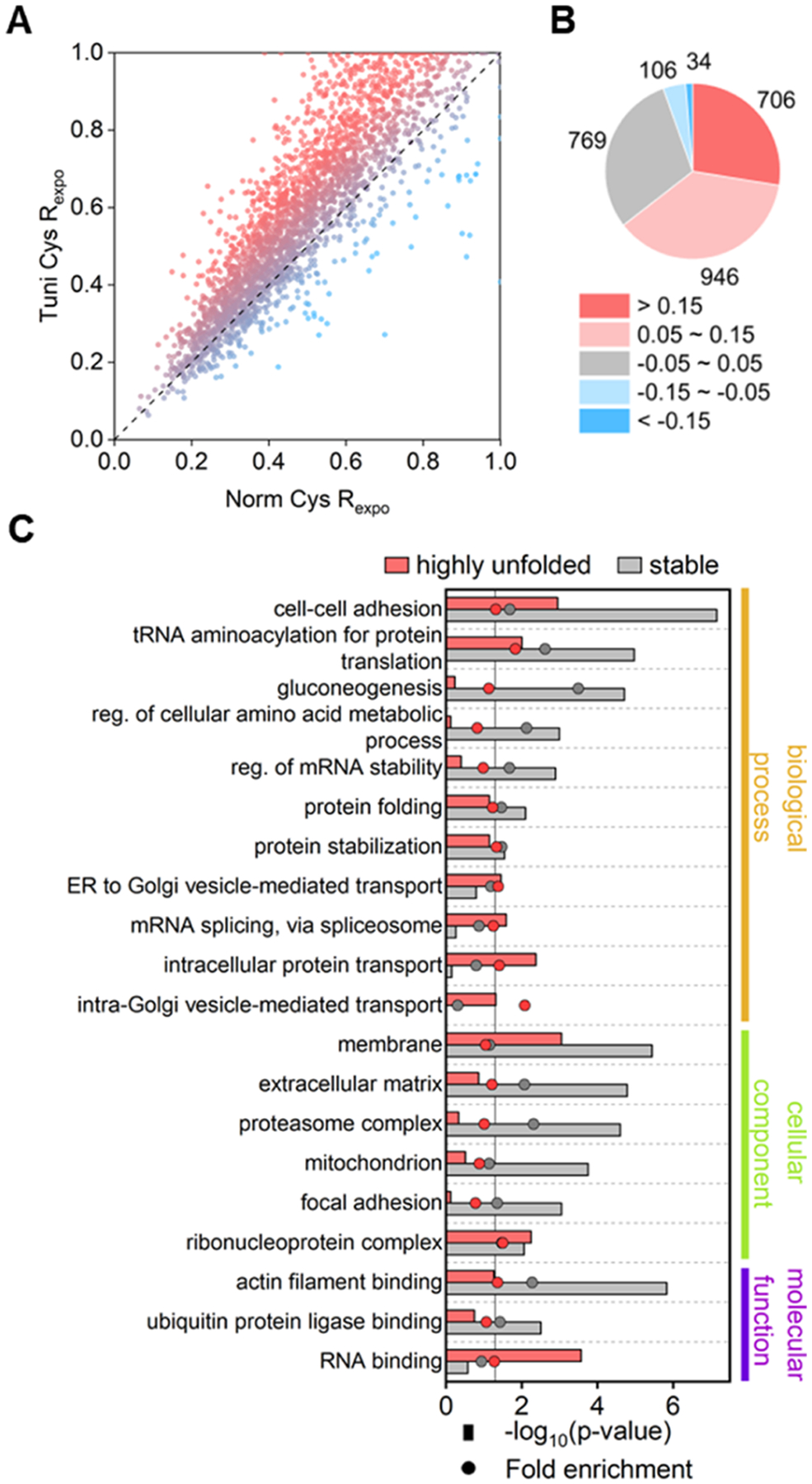
Systematic study of protein folding affected by the inhibition of protein N-glycosylation using Tm. (A) Comparison of the cysteine exposure rates between the Tm-treated and control samples. (B) Number of proteins within different ranges of the exposure rate changes. (C) GO enrichment analysis among proteins with cysteine in highly unfolded or stable regions. Fold enrichment values were calculated by comparing the frequency of total proteins annotated to that term in all identified proteins to the frequency representing the number of proteins in the highly damaged or stable group that fall under the same term.
To find the subproteome with significantly increased unfolded copies, we assigned the sites with the differences of >0.15 between the cysteine exposure rates with and without the Tm treatment as becoming highly exposed (segment 1, S1), which approximately stands for >30% change in unfolding stoichiometry for most surrounding regions (see Supporting Information for the derivation). Sites with the exposure rate differences that fall within −0.05 to 0.05 were considered to have no obvious changes in their surrounding (segment 3, S3), with their approximately unfolding stoichiometry changes of <10%. On this basis, all sites were divided into five segments (Figure 3B). Nearly one-third of the cysteine-located regions showed no significant structural change under the Tm treatment, while 28% of all sites became highly exposed.
We then performed gene ontology (GO) enrichment analysis among proteins with sites belonging to S1 or S3 (Figure 3C and Table S4). Proteins related to cell–cell adhesion and protein translation were enriched in the two groups. This is also supported by the exposure rate analysis results obtained from the experiment under the untreated condition (Figure S5). Terms enriched in the S3, such as gluconeogenesis and protein folding, contain proteins with well-structured regions (Figure S5). Specific terms related to vesicle trafficking, such as ER to Golgi vesicle-mediated transport and intracellular protein transport, were enriched exclusively in S1, suggesting the vital role of N-glycosylation in the folding and trafficking of secretory proteins.46 Analysis of all cysteine exposure rates of proteins in different organelles based on cellular components (Figure S9 and Table S5) also shows that proteins in the ER and the Golgi are more unfolded, indicating that the inhibition of N-glycosylation primarily impacts proteins in the classical secretory pathway, as expected.
Folding State Changes of Newly Synthesized Proteins under the Tunicamycin Treatment.
In cells, nascent peptides are glycosylated inside the ER before reaching their mature and functional conformations.47 Therefore, the inhibition of N-glycosylation may affect the folding of newly synthesized proteins, especially those that are N-glycosylated. Detecting the exposure rates in the whole proteome may thus underestimate the impact of the Tm treatment on protein folding because pre-existing and well-folded proteins are expected to be much less affected, but they were quantified simultaneously. Therefore, an analysis focusing on newly synthesized proteins can provide more accurate information about the direct impact of Tm on protein structural changes. Furthermore, investigation of the structural changes of pre-existing proteins allows us to obtain the indirect effect of Tm in cells.
By coupling the current method with pSILAC, we investigated the effects of Tm on newly synthesized proteins and pre-existing ones, respectively. The experimental design is displayed in Figure 4A. A fully heavy isotope-labeled sample served as a boosting source to assist in the identification of low-abundant newly synthesized proteins.48,49 About 3000 pre-existing proteins were quantified with or without the Tm treatment, while 1783 newly synthesized proteins were quantified in the control samples and 879 proteins in the Tm-treated samples (Figures 4B, S10, and Table S6). Fewer proteins quantified with the inhibition of N-glycosylation were expected because the decreased overall protein synthesis and the enhanced protein degradation are common cellular responses to relieve the protein unfolding stress. In total, 896 unique cysteine sites were quantified in all four groups, and their distributions are plotted in Figure 4C. Despite the fact that those newly synthesized proteins were generally more unfolded, inhibiting N-glycosylation resulted in a more significant unfolding impact on newly synthesized ones than those pre-existing proteins.
Figure 4.

Investigation of the effect of N-glycosylation inhibition on the folding states of newly synthesized and pre-existing proteins, respectively. (A) Experimental setup for quantifying the folding state changes of newly synthesized proteins and pre-existing ones. (B) Overlap of newly synthesized and pre-existing proteins quantified in cells with the tunicamycin treatment (Tuni) or without the treatment (Norm). Protein number in each group is in the brackets. (C) Distributions of the exposure rates in different groups of proteins (***p < 0.001, ****p < 0.0001, Kolmogorov–Smirnov test). (D) Distributions of the cysteine exposure rate differences in different secondary structures between newly synthesized and pre-existing proteins in cells with or without the Tm treatment (*p < 0.05, ***p < 0.001, Kolmogorov–Smirnov test).
Protein folding is an error-prone process in nature.50,51 To differentiate the unfolding effects of the Tm treatment from intrinsic errors in the natural folding process, we subsequently compared these exposure rate differences in different secondary structures between the newly synthesized and pre-existing copies of proteins (Figure 4D). The ordered regions (helix and strand) have more remarkable structural changes than the disordered regions (coil) in both treated and untreated samples, suggesting that intrinsic folding errors primarily impair the formation of protein secondary structures. The Tm treatment resulted in a greater unfolding effect in the coil regions but not in the helix and strand regions. This trend indicates that N-glycosylation majorly impacts the folding process of the protein tertiary or quaternary structures rather than their secondary structures. It has long been proposed that bulky and hydrophilic N-glycans play a vital role in the hydrophobic collapse-based protein folding process, but those studies focused on a small number of proteins or relied on computational simulation.52,53 The current systematic investigation reveals that N-glycosylation plays a more critical role in the formation of protein tertiary or quaternary structures besides regulating the secondary structures.
Profiling Proteins with Unstable Regions under the Tunicamycin Stress.
Proteins rely on proper conformations to perform their biological activities and even partially unfolded ones may not function effectively. Some proteins may be much easier to lose their structures under stress conditions, while others may be the opposite. Identifying the unstable regions can provide insights into cellular stress responses that abundance-based protein analysis cannot offer. A recent study profiled stress-induced protein aggregates to find misfolding-prone proteins, and about 10% of the proteome was reported to become less soluble in at least one case of cell stresses.54 However, misfolded proteins are not guaranteed to form aggregates.55 Hence, direct detection of protein unfolding allowed us to identify those with structures less resistant to stress.
All sites quantified with or without the treatment were categorized into five segments based on their exposure rate changes, as described above. Compared with the whole proteome analysis, fewer cysteines became highly exposed in this experiment (Figures 3B and 5A). We reason that the short Tm treatment time in the pSILAC experiment caused fewer unfolded proteins to accumulate in the cells. Interestingly, despite that 16.4% of the cysteines became highly exposed in the newly synthesized group (174 out of 1059 sites) under the Tm treatment, 13.9% of the sites in the pre-existing group (710 out of 5093 sites) did the same (Figure 5A). Considering the S1 and S2 segments together, the pre-existing group has more cysteines being more exposed than the newly synthesized one. This observation indicates that a large portion of the well-folded pre-existing proteins became extensively unfolded under ER stress. The proteasome may serve as an example. For the core subunits (20S), newly synthesized copies became more unfolded than their pre-existing counterparts under the Tm treatment (Figure S11A). Conversely, the unfolding stress has a greater impact on the pre-existing regulatory subunits (19S). The possible explanation is that the regulatory subunits are more flexible in the proteasome complex and have a greater possibility to interact with other unfolded proteins, while the core subunits are protected by the regulatory subunits in the proteasome. Thus, the pre-existing regulatory subunits are more prone to damage by the unfolding stress. According to a study focused on the degradation of newly synthesized proteins, the newly synthesized regulatory subunits had much more extended residence periods than the core subunits due to the higher usage rates of the regulatory subunits.56 The current results suggest that the relatively unstable nature of the regulatory subunits could be a possible reason why the newly synthesized regulatory subunits are used more frequently.
Figure 5.
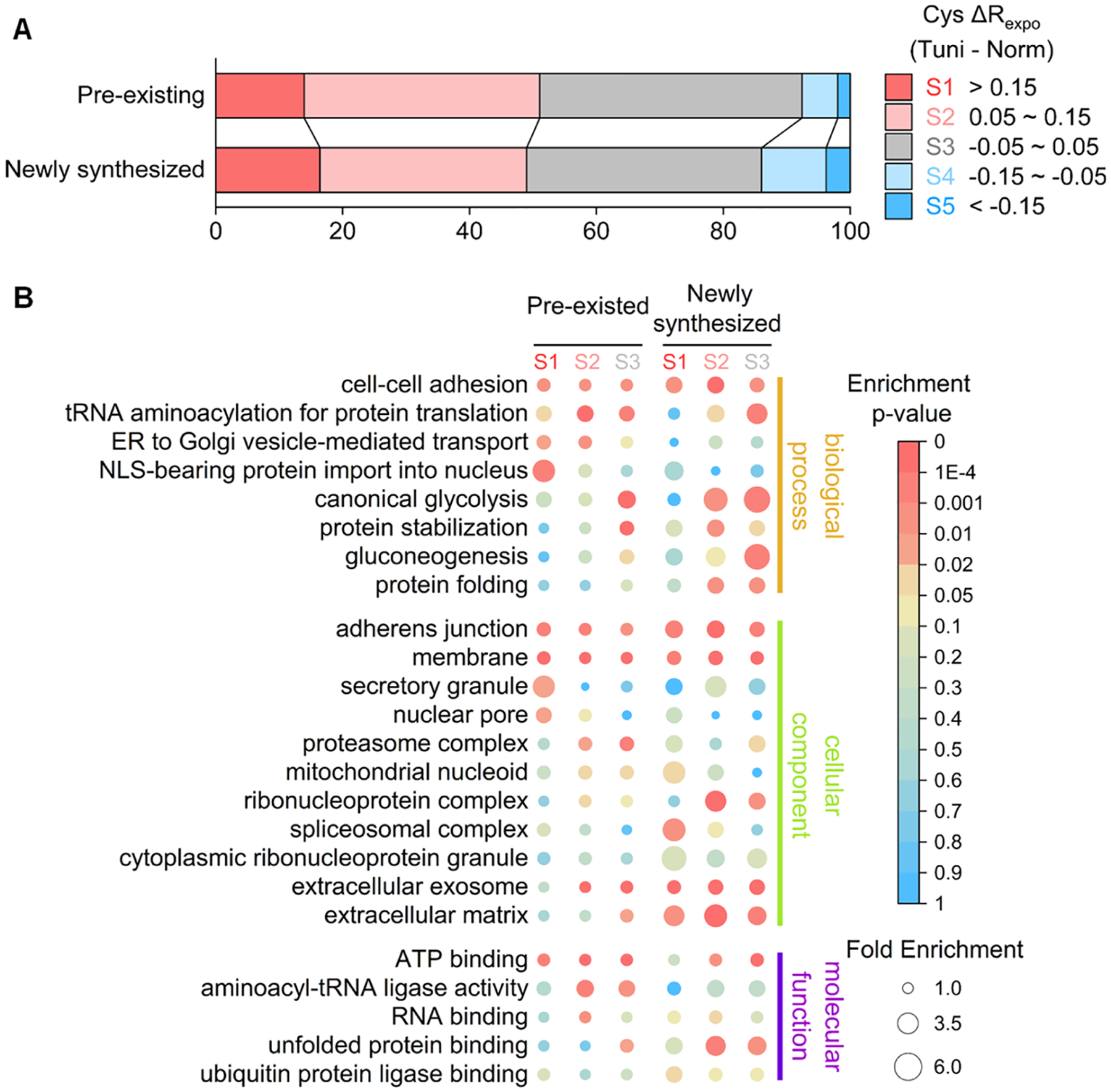
Analysis of the folding states of pre-existing and newly synthesized proteins affected by the N-glycosylation inhibition using Tm. (a) Protein numbers within each segment of the exposure rate changes in pre-existing and newly synthesized proteins (Information about each segment is in the text). (B) GO enrichment analysis among proteins with the cysteine exposure rate belonging to each segment.
Subsequent GO enrichment analysis among the proteins with the sites belonging to S1–S3 was performed to understand protein unfolding (Figure 5B and Table S7). We primarily focus on terms related to protein folding and degradation, which are critical for cells to cope with ER stress. Proteins related to protein folding, protein stabilization, and unfolded protein binding were enriched in the S2 segment of the newly synthesized group. In contrast, they were not enriched in the S1 and S2 segments of the pre-existing group, indicating that significant portions of the newly synthesized stress-responsive copies were not folded well. Similarly, ubiquitin ligase binding-related proteins were overrepresented in the newly synthesized S1 segment but not in the pre-existing group. Although proteins for the proteostasis regulation were reported to be significantly upregulated under unfolding stresses,24,57 the current results suggest that the enhancement of the ability to reduce unfolded protein loads was compromised in cells because many newly synthesized proteins related to proteostasis regulation are not fully functional.
The abovementioned results demonstrate that proteins related to secretion were enriched in highly unfolded segments (Figure 3C). Further analysis reveals that proteins related to ER to Golgi vesicle-mediated transport and secretory granule were enriched in the S1 or S2 segment of the pre-existing group, indicating that unfolded protein loads in the secretory pathway mainly hampered the established vesicle transportation system. However, for the extracellular matrix (ECM) proteins, their pre-existing copies were stable while the newly synthesized counterparts were not well-folded under the Tm treatment. ECM proteins are often heavily glycosylated, and many of them are highly structured scaffold proteins.58,59 The results demonstrate that those well-structured pre-existing proteins maintained their structures under ER stress. Nevertheless, a great portion of the newly synthesized ECM proteins could not fold well because of the inhibition of N-glycosylation.
Key Pathways Impaired by Tm-Induced Protein Unfolding.
To evaluate the effect of the Tm treatment on cellular activities, we conducted the KEGG pathway analysis for the unfolded newly synthesized and pre-existing proteins. Some enriched pathways were found in both protein groups, including RNA transport and endocytosis (Figure S12A). The pathways enriched in the newly synthesized group are mainly related to metabolism processes, while the overrepresented pathways in the pre-existing group cover various aspects from DNA replication, ubiquitin-mediated proteolysis to RNA degradation, indicating that the secondary effects of the unfolding stress have a more profound influence on cell activities.
For instance, the RNA transport pathway was highly enriched in both groups. Detailed analysis shows that many essential protein complexes were structurally changed under the unfolding stress (Figure S12B). Typically, nucleocytoplasmic transport-related proteins, including the nuclear pore complex (NPC) and the nuclear transport complex, are generally unfolded under the Tm treatment. NPC transports macromolecules between the cytoplasm and the nucleus.60 Here, many NPC proteins became highly unfolded under the ER stress, especially in the pre-existing group. Proteins related to the nuclear pore were exclusively enriched in the pre-existing S1 segment as well (Figure 5B). It should be noted that the cytoplasmic barrier of the NPC, the cytoplasmic fibrils proteins, was significantly more damaged than other NPC proteins (Figure S11B), which are reported to have relatively poorly structured regions.61 This result emphasizes that the flexible structures tend to become more unfolded under the ER stress. The impairment of the nucleocytoplasmic transport process was reported to be tightly related to the unfolding stress and neurodegeneration diseases.62 The current results suggest that the structurally impaired NPC proteins may be the reason behind this, and this intriguing finding needs more follow-up studies.
DISCUSSION
Extensive abundance-based proteomics analyses were performed to study cellular responses to ER stress,21,22,57 but they cannot provide direct information about protein structure changes. Alterations in protein structures caused by ER stress can conceivably influence enzyme activities and cellular events beyond what we have already known. In particular, the pleiotropic effects of the Tm-induced protein unfolding stress need to be further explored.
In this work, we measured protein structural alterations using a newly developed method called Cys-CPP. A cysteine-targeting probe was synthesized to label solvent-exposed free thiols in native proteins. It should be noted that heat and GnHCl treatments were used to denature the lysate in the reference sample, which may hamper the reactivity of thiols through oxidation or electrostatic effects and then could cause over-represented exposure rates. However, these impacts were minimum because only 5% of quantified cysteines had their exposure rates greater than 1.05, which could be partially caused by some experimental errors. First, this method was applied to study protein structures in HEK293T cells. As expected, the results demonstrated that the cysteine exposure rates correlated very well with the local structures of proteins. Furthermore, we employed this method to investigate protein structural changes in cells with the ER stress caused by Tm. Stable protein regions like helix and strand had significantly less structural changes compared with unstable regions under the unfolding stress.
It is well-known that cells treated with Tm, which is a potent protein N-glycosylation inhibitor and commonly used as a stress-inducing compound, have pleiotropic consequences beyond the ER stress responses and may not be able to mimic the actual physiological ER stress.27,63 Under the Tm treatment, hundreds of genes were regulated, but the vast majority were not directly related to the ER functions and the classical protein secretory pathway.27,64 We studied newly synthesized and pre-existing proteins separately to investigate the primary and secondary effects of Tm on protein structures. The current results suggest that N-glycosylation impacts the tertiary and quaternary structures of proteins more than the secondary structures. Previously, N-glycans were reported to most influence the solvation of polypeptides, affecting the polypeptide hydrophobic collapse process.47,53 The formation of secondary structures, on the other hand, is predominately decided by the local sequence65 and generally happens after the hydrophobic collapse.66 The current results reveal that N-glycosylation of polypeptides is often not decisive to the formation of protein secondary structures.
From this study, the Tm toxicity at least partially attributes to the unfolding of some pre-existing proteins, which is less relevant to the primary effect of Tm. Damaged proteins were involved in multiple BPs, including intracellular protein transport and ER to Golgi vesicle-mediated transport, most relevant to the accumulation of unfolded proteins within the ER. In contrast, nucleocytoplasmic transport and aminoacyl-tRNA biosynthesis are more likely to be a common consequence of the unfolding stress caused by a limited capacity for proteostasis regulation, as reported previously.24,26,54 Among pre-existing proteins with dramatic structure changes, they are involved in different pathways from DNA replication and ubiquitin-mediated proteolysis to RNA degradation, indicating that the secondary effects of the protein unfolding stress have a more profound influence on cell activities. The new approach can comprehensively investigate protein structure changes under different kinds of stresses in the future.
CONCLUSIONS
In this work, we systematically studied protein structures and structural changes by quantifying the exposure rates of the cysteine residues. A probe was designed to target cysteine, which contains the cysteine reaction group, biotin for enrichment, and a cleavable linker for generating a small tag for site-specific analysis of cysteine-containing peptides by MS. Using the current method, we demonstrate that the ER stress can induce widespread protein unfolding. Furthermore, by combining Cys-CPP with pSILAC, we studied the structure change differences in both pre-existing and newly synthesized proteins under the ER stress and performed a detailed analysis of the effect of protein unfolding caused by the ER stress. The folding of newly synthesized proteins was more affected by Tm. Unexpectedly, the results also reveal that many pre-existing proteins related to the transcription, translation, and nucleocytoplasmic transport pathways became unfolded upon the ER stress. Additionally, the systematic investigation found that N-glycosylation plays a more important role in the folding process of the tertiary or quaternary structures than in the secondary structures of newly synthesized proteins. This study can clearly demonstrate that the Tm-induced unfolding stress is not restricted to proteins in the classical secretory pathway. An in-depth and accurate understanding of the protein structural changes under the ER stress provides insights into protein functions and cellular activities.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R01GM127711 and R01GM118803).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c03076.
Detailed experimental procedures; synthesis of the cysteine reactive probe; reproducibility of data from untreated HEK293T lysate; the exposure rates of cysteines in CCT3; cysteine exposure rates with different neighboring residues; GO enrichment analysis of proteins with differentially exposed cysteine; reproducibility of data from Tm-treated HEK293T lysate; cysteine exposure rate changes with original exposure rate; comparison of the exposure rates changes and local protein structure; reproducibility of data from SILAC experiments; exposure rates of selected protein complexes; and KEGG pathway analysis (PDF)
Quantified exposure rates in normal HEK293T cells using Cys-CPP (XLSX)
GO enrichment analysis of proteins with differentially exposed cysteines (XLSX)
Quantified exposure rates in Tm-treated HEK293T cells using Cys-CPP (XLSX)
Comparison of proteins with stable and highly exposed cysteine (XLSX)
Cysteine exposure rate changes in different cellular compartments under tunicamycin treatment (XLSX)
Quantified newly synthesized proteins exposure rates in normal HEK293T cells using Cys-CPP and pSILAC (XLSX)
GO enrichment analysis comparing differentially unfolded pre-existing and newly synthesized proteins (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.2c03076
The authors declare no competing financial interest.
Raw files are available via ProteomeXchange with identifier PXD028864.
Contributor Information
Kejun Yin, School of Chemistry and Biochemistry and the Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Ming Tong, School of Chemistry and Biochemistry and the Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States; Present Address: Novo Nordisk research centre, Beijing, 100102, China.
Fangxu Sun, School of Chemistry and Biochemistry and the Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States; Present Address: Biogen, Cambridge, 02142, MA, USA..
Ronghu Wu, School of Chemistry and Biochemistry and the Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States;.
REFERENCES
- (1).Pegram LM; Liddle JC; Xiao Y; Hoh M; Rudolph J; Iverson DB; Vigers GP; Smith D; Zhang H; Wang W; Moffat JG; Ahn NG Proc. Natl. Acad. Sci. U.S.A 2019, 116, 15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wheat A; Yu C; Wang X; Burke AM; Chemmama IE; Kaake RM; Baker P; Rychnovsky SD; Yang J; Huang L Proc. Natl. Acad. Sci. U.S.A 2021, 118, No. e2023360118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kaur U; Meng H; Lui F; Ma R; Ogburn RN; Johnson JHR; Fitzgerald MC; Jones LM J. Proteome Res 2018, 17, 3614–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lössl P; Waterbeemd M; Heck AJ EMBO J. 2016, 35, 2634–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).de Souza N; Picotti P Curr. Opin. Struct. Biol 2020, 60, 57–65. [DOI] [PubMed] [Google Scholar]
- (6).Hoofnagle AN; Resing KA; Ahn NG Annu. Rev. Biophys. Biomol. Struct 2003, 32, 1–25. [DOI] [PubMed] [Google Scholar]
- (7).Chakrabarty JK; Bugarin A; Chowdhury SM J. Proteomics 2020, 225, 103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu XR; Zhang MM; Gross ML Chem. Rev 2020, 120, 4355–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Guan JQ; Chance MR Trends Biochem. Sci 2005, 30, 583–592. [DOI] [PubMed] [Google Scholar]
- (10).Johnson DT; Punshon-Smith B; Espino JA; Gershenson A; Jones LM Anal. Chem 2020, 92, 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bamberger C; Pankow S; Martínez-Bartolomé S; Ma M; Diedrich J; Rissman RA; Yates JR 3rd J. Proteome Res 2021, 20, 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Walker EJ; Bettinger JQ; Welle KA; Hryhorenko JR; Ghaemmaghami S Proc. Natl. Acad. Sci. U.S.A 2019, 116, 6081–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu Y; Strickland EC; Fitzgerald MC Anal. Chem 2014, 86, 7041–7048. [DOI] [PubMed] [Google Scholar]
- (14).Yu KW; Niu MM; Wang H; Li YX; Wu ZP; Zhang B; Haroutunian V; Peng JM J. Am. Soc. Mass Spectrom 2021, 32, 936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baud F; Karlin S Proc. Natl. Acad. Sci. U.S.A 1999, 96, 12494–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Marino SM; Gladyshev VN J. Mol. Biol 2010, 404, 902–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Potter ZE; Lau HT; Chakraborty S; Fang L; Guttman M; Ong SE; Fowler DM; Maly DJ Cell Chem. Biol 2020, 27, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MB; Bachovchin DA; Mowen K; Baker D; Cravatt BF Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kuljanin M; Mitchell DC; Schweppe DK; Gikandi AS; Nusinow DP; Bulloch NJ; Vinogradova EV; Wilson DL; Kool ET; Mancias JD; Cravatt BF; Gygi SP Nat. Biotechnol 2021, 39, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Uhlén M; Fagerberg L; Hallström BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjöstedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Pontén F Science 2015, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- (21).Hetz C; Chevet E; Oakes SA Nat. Cell Biol 2015, 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Taylor RC; Berendzen KM; Dillin A Nat. Rev. Mol. Cell Biol 2014, 15, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Travers KJ; Patil CK; Wodicka L; Lockhart DJ; Weissman JS; Walter P Cell 2000, 101, 249–258. [DOI] [PubMed] [Google Scholar]
- (24).Itzhak DN; Sacco F; Nagaraj N; Tyanova S; Mann M; Murgia M Dis. Models Mech 2019, 12, dmm040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kudelko M; Chan CW; Sharma R; Yao Q; Lau E; Chu IK; Cheah KS; Tanner JA; Chan DJ Proteome Res. 2016, 15, 86–99. [DOI] [PubMed] [Google Scholar]
- (26).Owyong TC; Subedi P; Deng J; Hinde E; Paxman JJ; White JM; Chen W; Heras B; Wong WWH; Hong Y Angew. Chem., Int. Ed. Engl 2020, 59, 10129–10135. [DOI] [PubMed] [Google Scholar]
- (27).Bergmann TJ; Fregno I; Fumagalli F; Rinaldi A; Bertoni F; Boersema PJ; Picotti P; Molinari MJ Biol. Chem 2018, 293, 5600–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Eng JK; McCormack AL; Yates JR J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- (29).Elias JE; Gygi SP Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (30).Peng JM; Elias JE; Thoreen CC; Licklider LJ; Gygi SP J. Proteome Res 2003, 2, 43–50. [DOI] [PubMed] [Google Scholar]
- (31).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villén J; Haas W; Sowa ME; Gygi SP Cell 2010, 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Beausoleil SA; Villén J; Gerber SA; Rush J; Gygi SP Nat. Biotechnol 2006, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- (33).Wang S; Ma J; Xu J Bioinformatics 2016, 32, i672–i679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang S; Li W; Liu S; Xu J Nucleic Acids Res. 2016, 44, W430–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang S; Peng J; Ma J; Xu J Sci. Rep 2016, 6, 18962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Huang W; Sherman BT; Lempicki RA Nat. Protoc 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- (37).West GM; Tang L; Fitzgerald MC Anal. Chem 2008, 80, 4175–4185. [DOI] [PubMed] [Google Scholar]
- (38).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon R; Mohammed AK; Hamon C Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (39).Richardson JS Adv. Protein Chem 1981, 34, 167–339. [DOI] [PubMed] [Google Scholar]
- (40).Wilmot CM; Thornton JM J. Mol. Biol 1988, 203, 221–232. [DOI] [PubMed] [Google Scholar]
- (41).Lutolf MP; Tirelli N; Cerritelli S; Cavalli L; Hubbell JA Bioconjugate Chem. 2001, 12, 1051–1056. [DOI] [PubMed] [Google Scholar]
- (42).Yang XL Chem. Biol 2013, 20, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yoo J; Mashalidis EH; Kuk ACY; Yamamoto K; Kaeser B; Ichikawa S; Lee SY Nat. Struct. Mol. Biol 2018, 25, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xu C; Ng DT Nat. Rev. Mol. Cell Biol 2015, 16, 742–752. [DOI] [PubMed] [Google Scholar]
- (45).Roth J Chem. Rev 2002, 102, 285–304. [DOI] [PubMed] [Google Scholar]
- (46).Pfeffer SR Mol. Biol. Cell 2017, 28, 712–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Imperiali B; O’Connor SE Curr. Opin. Chem. Biol 1999, 3, 643–649. [DOI] [PubMed] [Google Scholar]
- (48).Suttapitugsakul S; Tong M; Sun F; Wu R Anal. Chem 2021, 93, 2694–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yi L; Tsai CF; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian WJ Anal. Chem 2019, 91, 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Braselmann E; Chaney JL; Clark PL Trends Biochem. Sci 2013, 38, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Anfinsen CB Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- (52).Cheng S; Edwards SA; Jiang Y; Gräter F Chemphyschem 2010, 11, 2367–2374. [DOI] [PubMed] [Google Scholar]
- (53).Lu D; Yang C; Liu ZJ Phys. Chem. B 2012, 116, 390–400. [DOI] [PubMed] [Google Scholar]
- (54).Sui X; Pires DEV; Ormsby AR; Cox D; Nie S; Vecchi G; Vendruscolo M; Ascher DB; Reid GE; Hatters DM Proc. Natl. Acad. Sci. U.S.A 2020, 117, 2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chaturvedi SK; Siddiqi MK; Alam P; Khan RH Process Biochem. 2016, 51, 1183–1192. [Google Scholar]
- (56).Tong M; Smeekens JM; Xiao H; Wu R Chem. Sci 2020, 11, 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bull VH; Thiede B Electrophoresis 2012, 33, 1814–1823. [DOI] [PubMed] [Google Scholar]
- (58).Vogel V Annu. Rev. Biophys. Biomol. Struct 2006, 35, 459–488. [DOI] [PubMed] [Google Scholar]
- (59).Lynch M; Barallobre-Barreiro J; Jahangiri M; Mayr MJ Intern. Med 2016, 280, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Strambio-De-Castillia C; Niepel M; Rout MP Nat. Rev. Mol. Cell Biol 2010, 11, 490–501. [DOI] [PubMed] [Google Scholar]
- (61).Hampoelz B; Andres-Pons A; Kastritis P; Beck M Annu. Rev. Biophys 2019, 48, 515–536. [DOI] [PubMed] [Google Scholar]
- (62).Zhang K; Daigle JG; Cunningham KM; Coyne AN; Ruan K; Grima JC; Bowen KE; Wadhwa H; Yang P; Rigo F; Taylor JP; Gitler AD; Rothstein JD; Lloyd TE Cell 2018, 173, 958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Fribley A; Zhang K; Kaufman RJ Methods Mol. Biol 2009, 559, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Thibault G; Ismail N; Ng DT Proc. Natl. Acad. Sci. U.S.A 2011, 108, 20597–20602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Srinivasan R; Rose GD Proc. Natl. Acad. Sci. U.S.A 1999, 96, 14258–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sadqi M; Lapidus LJ; Muñoz V Proc. Natl. Acad. Sci. U.S.A 2003, 100, 12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.