

Abstract
Standard methods to identify microbial contaminants in the environment are slow, laborious, and can require specialized expertise. This study investigated electrochemical detection of microbial contaminants using commercially available, hand-held instruments. Electrochemical assays were developed for a red tide dinoflagellate (Karenia brevis), fecal-indicating bacteria (Enterococcus spp.), markers indicative of human sources of fecal pollution (human cluster Bacteroides and the esp gene of Enterococcus faecium), bacterial pathogens (Escherichia coli 0157:H7, Salmonella spp., Campylobacter jejuni, Staphylococcus aureus), and a viral pathogen (adenovirus). For K. brevis, two assay formats (Rapid PCR-Detect and Hybrid PCR-Detect) were tested and both provided detection limits of 10 genome equivalents for DNA isolated from K. brevis culture and amplified by PCR. Sensitivity with coastal water samples was sufficient to detect K. brevis that was “present” (≤1000 cells/l) without yielding false positive results and the electrochemical signal was significantly different than for samples containing cells at “medium” concentrations (100,000 to < 106 cells/l). Detection of K. brevis RNA was also shown. Multi-target capability was demonstrated with an 8-plex assay for bacterial and viral targets using isolated DNA, natural beach water spiked with human feces, and water and sediments collected from New Orleans, Louisiana following Hurricane Katrina. Furthermore, direct detection of dinoflagellate and bacterial DNA was achieved using lysed cells rather than extracted nucleic acids, allowing streamlining of the process. The methods presented can be used to rapidly (3–5 h) screen environmental water samples for the presence of microbial contaminants and have the potential to be integrated into semi-automated detection platforms.
Keywords: Electrochemical biosensor, Karenia brevis, Pathogen detection, Recreational water quality, Source tracking
1. Introduction
Toxic algal blooms and microbial contaminants impact coastal water quality. As the nation’s coastal areas become more urbanized, poor water quality has increased economic, health, and environmental impacts. Harmful algal blooms (HABs) alone cost the United States 50 million dollars per year (Haugland et al., 2005), and sewage polluted waters adversely affect human health and the economy (Dwight et al., 2005; Leclerc et al., 2002). Environmental managers need rapid and accurate assessments of microbial water quality to restrict human access to contaminated waters and products. Monitoring techniques for HABs that rely on microscopy are time-consuming, labor intensive, error prone (Culverhouse et al., 2003) and require a significant amount of taxonomic expertise (Millie et al., 1997). Standard culture techniques for bacterial indicators of fecal pollution are slow and do not return information on human pathogens or source tracking markers (Bower et al., 2005; Griffin et al., 2001; Scott et al., 2005). As a result, management decisions, ecological study and assessment of control measures are difficult. Biosensors could improve monitoring by providing a means to achieve rapid, on-site identification of microbial contaminants. A robust sensor design is sought, in particular, a sensor that can simultaneously detect multiple targets such as harmful algae, fecal indicators, human pathogens, and source tracking markers.
The electrochemical detection of nucleic acids is qualified to meet the size, cost, and power requirements of on-site water quality testing (Kerman et al., 2004; Wang, 2000). The potential for electrochemical methods to be used as a tool for environmental monitoring depends, in part, on the availability of reproducible biosensors. Wojciechowski and colleagues (Wojciechowski et al., 1999) described electrochemical biosensors that enable sensitive detection of DNA or RNA after PCR or Reverse Transcriptase polymerase chain reaction (Attatippaholkun et al., 2003), respectively. The system, marketed by Alderon Biosciences, Inc. (www.alderonbiosciences.com), uses a hand-held, battery-powered, electrochemical monitor and disposable carbon sensors. One method of detection used with this system, termed “Rapid PCR-Detect,” identifies and quantifies PCR products through biotin and fluorescein labeling during PCR. A second method, “Hybrid PCR-Detect,” provides added specificity by including hybridization to a DNA probe.
Numerous approaches for detecting microbial nucleic acids electrochemically have been formulated and tested (Drummond et al., 2003; Kerman et al., 2004; LaGier et al., 2005; Metfies et al., 2005; Zhang et al., 2003). Previous studies using the Alderon system detected single-base mutations in a human gene (Wojciechowski et al., 1999), enterotoxin genes from cultures of the bacterium Staphylococcus aureus (Aitichou et al., 2004), and DNA isolated from algal cultures (Pfiesteria piscicida and Cryptoperidiniopsoid spp.) (Litaker et al., 2001). All of these studies used single-plex reactions to detect nucleic acids isolated from pure culture. Environmental applications require detection in natural samples, with a preference for multi-target detection. Therefore, the use of electrochemical biosensors for environmental microbiology applications needed further testing.
The feasibility of using electrochemical biosensors to detect microbes in environmental samples was tested using Karenia brevis as a model. K. brevis is the causative agent of recurring red tide blooms in the Gulf of Mexico and occasional blooms off the southeastern coast of the United States (Steidinger et al., 1998; Tester and Steidinger, 1997). The lipophilic toxin produced by K. brevis, brevetoxin, can result in massive fish and marine mammal kills (Landsberg, 2002). Aerosolized brevetoxin toxin can cause respiratory distress (Kirkpatrick et al., 2004) when levels of K. brevisexceed 1000 cells/l The harvesting of shellfish is prohibited when cell concentrations reach 5000 K. brevis cells per liter (http://www.floridaaquaculture.com).
The potential to develop multi-target electrochemical detection was tested here using an 8-plex assay of microbes important to microbial water quality monitoring that included fecal-indicating bacteria (Enterococcus spp.), bacterial markers of human fecal pollution (the human-specific HF8 cluster of Bacteroides and the esp gene of Enterococcus faecium), bacterial pathogens (Escherichia coli 0157:H7, Salmonella spp., Campylobacter jejuni and S. aureus), and a viral pathogen (human adenovirus). To address shortfalls associated with the standard culture methods used to monitor recreational water quality, the multi-target assay was designed to return an indication of fecal contamination along with information on whether the contamination was from a human source and if selected human pathogens are present (Griffin et al., 2001).
2. Materials and methods
2.1. Sources of DNA and environmental samples
Algae used in this study were obtained from the Provasoli-Guillard National Center for Culture of Marine Phytoplankton (CCMP) and maintained by the Toxic Algal Culture Core of the Oceans and Human Health Center at the University of Miami, unless otherwise noted (Table 1). Cultures were grown at 22 °C in a light:dark cycle as described by Brand (1990). Cultures were pelleted or filtered onto 5 μm, 47 mm mixed esters of cellulose (MEC) membrane filters (Millipore Corp., Bedford, MA) and DNA was extracted using the FastDNA SPIN Kit for Soil (Qbiogene, Irvine, CA). The quality and amount of extracted DNA was assessed by spectroscopy (Ausubel et al., 1999). Genome equivalents (cell numbers) were estimated from DNA concentrations based on data indicating that a K. brevis cell contains a single DNA genome weighing approximately 100 pg (Lidie et al., 2005; Rizzo et al., 1982).
Table 1.
Specificity of the Rapid and Hybrid PCR-Detect assays for Karenia brevis when tested against related Dinoflagellates (class Dinophyceae)
| Organism | Strain | Family | Rapida | Hybrida |
|---|---|---|---|---|
| Karenia brevis | CCMP 718 | Gymnodiniaceae | + | + |
| Karenia mikimotoi | NOAA-2b | Gymnodiniaceae | - | - |
| Amphidinium carterae | CCMP 1134 | Gymnodiniaceae | - | - |
| Alexandrium tamarense | CCMP 1493 | Gonyaulacaceae | - | - |
| Gonyaulax cochlea | CCMP 1592 | Gonyaulacaceae | - | |
| Lingulodinium polyedra | CCMP 1738 | Gonyaulacaceae | - | - |
| Protoceratium reticulatum (Gonyaulax grindleyi) | CCMP 1721 | Gonyaulacaceae | - | - |
| Heterocapsa pygmaea | UTEX 2421c | Heterocapsaceae | - | - |
| Katodinium rotundatum (Heterocapsa rotundata) | CCMP 1734 | Heterocapsaceae | - | - |
| Peridinium foliaceum | CCMP 1326 | Peridiniaceae | - | - |
| Prorocentrum hoffmannianum | CCMP 683 | Prorocentraceae | - | - |
| Prorocentrum lima | CCMP 1368 | Prorocentraceae | - | - |
| Symbiodinium species | CCMP 831 | Symbiodiniaceae | - | - |
Samples labeled negative (-) had a mean current value <2x the mean current value of negative controls (no template PCR), while positive samples (+) had a mean current >2x the mean current for negative controls.
Obtained courtesy of S. Morton, NOAA, South Carolina.
UTEX = The culture collection of Algae at the University of Texas.
DNA from E. coli (ATCC #25922) and E. faecalis (ATCC #29212) was isolated from cultures using the FastDNA SPIN Kit for Soil (Qbiogene, Irvine, CA). DNA from S. aureus was obtained from the ATCC (#700699D). DNA from E. coli 0157:H7, C. jejuni, and S. typhi was obtained from the New York State Department of Health courtesy of Dr. Nick Cirino. Adenovirus DNA consisted of a plasmid containing the PCR target (He and Jiang, 2005) and was provided by Dr. Sunny Jiang of the University of California at Irvine. A plasmid containing cloned DNA from the human-specific esp gene of E. faecium (Scott et al., 2005) was provided by Dr. Joan Rose of Michigan State University. The human-specific HF8 cluster of Bacteroides (Bernhard and Field, 2000) was PCR amplified from human fecal DNA and cloned into the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA).
Coastal water samples for analysis of the toxic dinoflagellate K. brevis were obtained from the Rookery Bay National Estuarine Research Reserve (NERR) in Naples, Florida from the sites Caxambas Pass, Goodland, Henderson Creek, Marco Pass, and the 951 Launch Ramp as described in Goodwin et al. (Goodwin et al., 2005). In summary, paired water samples were collected in sterile whirlpacks and shipped overnight to the Florida Fish and Wildlife Research Institute (FWRI; www.floridamarine.org) and to the National Oceanographic and Atmospheric Administration (NOAA) Atlantic Oceanographic and Meteorological Laboratories (AOML). At FWRI, K. brevis was enumerated by microscopy, according to the following eight classifications: not present, present (≤1000 cells/l), very low a (>1000 to < 5000 cells/l), very low b (5000–10,000 cells/l), low a (>10,000 to <50,000 cells/l), low b (50,000 to <100,000 cells/l), medium (100,000 to <106 cells/l), and high (≥106 cells/l). Samples with “medium” concentrations of K. brevis were collected from Caxambas Pass or Marco Pass on 11 February, 2003, 18 February, 2003, 24 March, 2003, and 31 March, 2003. Samples with “present” concentrations were obtained on 11 February, 2003, 18 February, 2003, 26 February, 2003, 24 March, 2003, or 31 March, 2003. Samples in which K. brevis was “not present” were obtained on 11 February, 2003, 26 February, 2003, 11 March, 2003, 24 March, 2003, and 31 March, 2003. Samples shipped to AOML were vacuum-filtered onto MEC filters as described above and frozen at -20 °C until use. Each filter was aseptically cut in two pieces, and DNA was extracted from each filter half. In addition to the electrochemical assessment described here, the samples were also evaluated by a DNA hybridization assay in microplate format and by DNA sequencing (Goodwin et al., 2005).
A spiking approach was used to mimic beach water contaminated with a human fecal source. Beach water was obtained in a sterile bottle from Hobie Beach, FL (Shibata et al., 2004) at knee-deep depth in May 2006. A contaminated sample was prepared by spiking 100 ml of water with 0.001% (w/v) human feces collected from a healthy volunteer. This sample and 100 ml of natural beach water were filtered onto 0.40 μm polycarbonate filters (#K04CP04700, GE Osmonics, Minnetonka, MN). A crude lysate of DNA (Haugland et al., 2005) was prepared by placing the filters (sample side-in) into 2.0-ml screw-cap tubes containing 0.3 g sterile glass beads (#G-1277 Sigma, St. Louis, MS) and 600 μl of AE buffer (Qiagen, Valencia, CA) and bead-beating for 1 min at the maximum setting (6.5) on a FastPrep instrument (QBiogene, Irvine, CA). Filter debris and glass beads were pelleted by centrifugation for 5 min at 14,000g. The supernatant was removed and an aliquot of the supernatant was diluted 1:5 in AE buffer for use as a working stock. The crude lysates were stored at -20 °C until use.
Environmental samples were collected from flooded regions in New Orleans, Louisiana following Hurricane Katrina as described in Sinigalliano et al. (Sinigalliano et al., submitted for publication). Briefly, the three samples tested here were collected on 12 November, 2005 (FDP-25 and FDP-26) and 25 March, 2006 (YD3). Water samples (FDP-25 and FDP-26) were collected from a canal adjacent to the 17th street water-pumping station south of Lake Pontchartrain (N29°59.251, W90°07.446) before and after a pumping event. The sample taken before the pumping event (FDP-25) contained 115 enterococci/100 ml water, while the sample taken after the pumping event contained 1481 enterococci/100 ml water. Visible pieces of feces were noted during collection of FDP-26. For both samples, DNA was extracted from 100 ml canal water concentrated onto 0.45 μm polycarbonate filters, using the FastDNA SPIN Kit for Soil (QBiogene, Irvine, CA). A sediment sample (YD3), shown to contain 1039 Enterococci/g soil, was collected from a region flooded by Lake Pontchartrain and an associated industrial canal break (N30°01.414, W90 °01.222). Specifically, 500 mg of soil from site YD3 was collected from surface soil sediments (top 3 cm) and DNA was extracted using the FastDNA SPIN Kit for Soil (QBiogene, Irvine, CA) (Sinigalliano et al., submitted for publication).
2.2. K. brevis PCR primers and DNA probes
PCR primers and probes (Table 2) were designed in silico to hybridize sequences within the D1/D2 region of the ribosomal RNA large subunit (LSU) of K. brevis (Goodwin et al., 2005). Forward primers were labeled with biotin and reverse primers were labeled with fluoroscein (FITC). Both assays used the same forward primer (Brevis-1-PCR-F). However, the Rapid PCR-Detect assay used a reverse primer specific for K. brevis (Karenia-1-PCR-1); whereas, the Hybrid PCR-Detect used a general eukaryotic reverse primer (Goodwin et al., 2005; Kiesling et al., 2002) labeled with free phosphate at the 5′ end (R635-1-PCR-R). The Hybrid PCR-Detect assay employed the Karenia-1-PCR-1 sequence as a probe. All primers and probes were synthesized by Sigma-Genosys (www.sigmagenosys.com). Probe and primer sequences were aligned with Karenia LSU rRNA sequences available from Gen-Bank (http://www.nlm.nih.gov) using the default settings of the AlignX module of Vector NTI software (Invitrogen, Calrsbad, CA).
Table 2.
PCR primers and probes used in the Rapid and Hybrid PCR-Detect assays developed for Karenia brevis
| Primer or probea | Nucleotide sequence (5′ → 3′) | Tm (°C) | Use |
|---|---|---|---|
| Brevis-1-PCR-F | Biotin-TGTTGTCTAAGGTGATAGCTTGC | 62.1 | Forward primer for Rapid and Hybrid |
| Karenia-1-PCR-R | FITC-GAAGCAAATTACCATGTCCCTAG | 62.4 | Reverse primer for Rapid and probe for Hybrid |
| R635-1-PCR-R | Phosphate-GGTCCGTGTTTCAAGACGG | 65.5 | Reverse primer for Hybrid |
Sequences previously published in Goodwin et al., 2005; FITC = fluorescein.
2.3. PCR amplification of algal DNA
DNA was amplified from algal cultures and coastal water samples by standard PCR on an Eppendorf Master-cycler using K. brevis primers as summarized in Table 2. For the Rapid PCR-Detect assay, the PCR contained 5 μl of 10× DyNAzyme II buffer (contains 1.5 mM MgCl2), 0.2 mM dNTP, 10 pmol forward primer, 10 pmol reverse primer, 1 μl DNA, 1 U DyNAzyme II DNA polymerase (Finnzymes, www.finnzymes.fi), and nuclease-free water for a total reaction volume of 50 μl. PCRs were similar for the Hybrid PCR-Detect assay except that 50 pmol of each primer was used.
B2 Thermal cycling for the Rapid PCR-Detect assay consisted of one cycle at 94 °C for 10 min, 30 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; followed by a final extension step at 70 °C for 8 min. Thermal cycling for the Hybrid PCR-Detect assay consisted of one cycle at 94 °C for 10 min, 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min; followed by one final extension step at 70 °C for 8 min. Amplicons used for electrochemical analysis were confirmed by agarose gel electrophoresis (Ausubel et al., 1999). The amplicon sizes for the Rapid and Hybrid PCR-Detect assays were 138 and 322 base pairs, respectively.
2.4. Reverse transcriptase PCR of K. brevis
Total RNA was isolated from K. brevis using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA quality and quantity was assessed by agarose gel electrophoresis and spectroscopy prior to using RT-PCR to detect the LSU rRNA of K. brevis. Initially, total isolated RNA was synthesized into complementary DNA using the protocol included with the Access RT-PCR System (Promega, Madison, WI). Each 25 μl reaction contained 1 μl (10 ng) K. brevis total RNA, 12.5 μl of AccessQuick 2 × Master Mix, 0.5 μl of AMV Reverse Transcriptase, 10 pmol of each primer (Table 2), and nuclease-free water. For both the Rapid and Hybrid PCR-Detect assays, the first-strand cDNA synthesis was carried out at 45 °C for 45 min in an Eppendorf Mastercycler prior to PCR. PCR thermal cycling for the Rapid PCR-Detect assay consisted of one cycle at 94 °C for 10 min, 30 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; followed by a final extension step at 70 °C for 8 min. Thermal cycling for the Hybrid PCR-Detect assay consisted of one cycle at 94 °C for 10 min, 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min; followed by one final extension step at 70 °C for 8 min. Successful amplification was confirmed by agarose gel electrophoresis.
2.5. Filter PCR of K. brevis
K. brevis cells grown in culture were counted, serially diluted into 100 ml of natural seawater culture media (Andersen et al., 1997), and concentrated onto replicate 25 mm membrane filters (0.45 μm Supor-450; Pall Corp., East Hills, NY). For PCR amplification, a 5 mm diameter circular section of each filter was removed from the center of each membrane using a standard paper hole puncher and placed into a 500 μl PCR tube using sterile forceps (Kirchman et al., 2001). To prevent contamination between PCRs, the hole puncher was treated with DNA AWAY (Molecular BioProducts, San Diego, CA), 100% ethanol and flamed between samples. The PCR and cycling conditions were identical to the Rapid PCR-Detect protocol except a final reaction volume of 100 μl was used and 15 μg of bovine serum albumin (10 mg/ml stock; A7030; Sigma, St. Louis, MS) was added to each reaction. Electrochemical detection of PCR products was carried out using the standard Rapid PCR-Detect protocol.
2.6. Amplification of bacterial and viral targets
Monoplex PCR amplification for bacterial and viral targets was carried out as described above for the Rapid PCR-Detect protocol for algal DNA except that 5 μl(1 ng) of DNA was used and 15 μg (1.5 μl) of 10 mg/ml stock bovine serum albumin (#A7030; Sigma, St. Louis, MS). For detection of Salmonella, each 50 μl PCR also contained 2% formamide and an additional 1 U of DNA polymerase. Amplification of beach water spiked with human feces was carried out by using 5 μl of crude lysate diluted 1:5 as a template in each 50 μl reaction. PCR products were analyzed using gel electrophoresis. Table 3 summarizes the primers, references, and thermal cycling conditions used for the multi-target assays.
Table 3.
PCR primers and conditions used in the multi-target electrochemical assay for bacterial and viral targets
| Target | Gene | Primer label-name-sequence, 5′ → 3′ (μM per PCR) |
Cyclinga | Reference |
|---|---|---|---|---|
| Human-specific Enterococcus faeciumb |
esp | Biotin-espF-TATGAAAGCACAAGTT (0.3) FITC-espR- ACGTCGAAAGTTCGATTTCC (0.3) |
94 °C 1 min; 58 °C 1 min; 72 °C 1 min; 40 cycles |
Scott et al. (2005) |
| Human-specific Bacteroidesb |
16S, HF8 gene cluster |
Biotin-HF183F- ATCATGAGTTCACATGTCCG (0.4) FITC-Bac708R- CAATCGGAGTTCTTCGTG (0.4) |
94 °C 30 s; 59 °C 30 s; 72 °C 30 s; 40 cycles |
Bernhard and Field (2000) |
| Campylobacter jejuni c | HipO | Biotin-CjF1-TGCTAGTGAGGTTG CAAAAGAATT (0.5) FITC-CjR1- TCATTTCGCAAAAAAATCCAAA (0.5) |
94 °C 30 s; 60 °C 30 s; 72 °C 30 s; 40 cycles |
LaGier et al. (2004) |
| Salmonella spp.c | IpaB | Biotin-IpaBF- GGACTTTTTAAAAGCGGCGG (0.3) FITC-IpaBR- GCCTCTCCCAGAGCCGTCTGG (0.3) |
94 °C 1 min; 62 °C 1 min; 72 °C 1 min; 35 cycles |
Kong et al. (2002) |
| E. coli 0157:H7c | rfb | Biotin-0157PF8- CGTGATGATGTTGAGTTG (1.0) FITC-0157PR8- AGATTGGTTGGCATTACTG (1.0) |
94 °C 30 s; 55 °C 30 s; 72 °C 30 s; 40 cycles |
Maurer et al. (1999) |
| Human Adenovirusc | Hexon | Biotin-AD2F- CCCTGGTAKCCRATRTTGTA (0.3) FITC-AD3R- GACTCYTCWGTSAGYGGCC (0.3) |
94 °C 30 s; 60 °C 30 s; 72 °C 30 s; 40 cycles |
He and Jiang (2005) |
| Enterococcus spp.d | 23S rRNA | Biotin-ECST748F- AGAAATTCCAAACGAACTTG (0.9) FITC-ENC854R- CAGTGGTCTACCTCCATCATT (0.3) |
94 °C 30 s; 60 °C 30 s; 72 °C 30 s; 30 cycles |
Haugland et al. (2005) |
| Staphylococcus aureus c | clfA | Biotin-clfAF-GCAAAATCCAGCACA ACAGGAAACGA (0.1) FITC-clfAR-CTTGATCT CCAGCCATAATTGGTGG (0.1) |
94 °C 1 min; 55 °C 1 min; 72 °C 1 min; 40 cycles |
Mason et al. (2001) |
For all reactions and targets, the PCR thermal cycling was preceded by an initial heat denaturation step (94 °C 10 min) and followed by a final extension step (70 °C 8 min).
Human fecal marker.
Waterborne pathogen.
Fecal indicator; FITC = fluorescein.
2.7. Electrochemical detection of K. brevis
A single-target format was used for electrochemical detection of K. brevis. Disposable carbon sensors coated with NeutrAvidin™ were obtained from Alderon Biosciences (Durham, NC). The screen printed sensors incorporate a conventional three-electrode configuration comprised of a circular (5 mm diameter) working electrode, a counter electrode, and a silver (Ag/AgCl) reference electrode, all printed on a polycarbonate substrate (4.5 cm × 1.5 cm). Both working and counter electrodes were made of heat-cured carbon composite inks. A ring-shaped layer printed around the three electrodes comprised the reservoir of the electrochemical cell, which can accommodate a volume of up to 50 μl.
During PCR, amplicons were labeled on one end with biotin to allow capture onto the NeutrAvidin™-coated electrodes (i.e., biosensors). Amplicons were also labeled with fluorescein during PCR (Rapid PCR-Detect) or during hybridization to a probe post λ-exonuclease digestion (Hybrid PCR-Detect). The amount of DNA bound to the sensor was proportional to the electrochemical current generated via horseradish peroxidase (HRP) chemistry and intermittent pulse amperometry (IPA). The HRP was conjugated to the amplicon via fluorescein, oxidized by hydrogen peroxide (Eq. (1)), and regenerated upon donation of an electron to 3,3′,5,5′-tetramethylbenzidine (TMB) (Eq. (2)). The sensor reader generated a charged electrode surface by applying millisecond pulses of -100 mV potential and the TMB was regenerated (Eq. (3)) upon transferring an electron to the carbon sensor surface, thus completing the catalytic cycle.
| (1) |
| (2) |
| (3) |
With the Rapid PCR-Detect assay, amplicons were labeled with biotin on one end and fluorescein on the other during PCR (Table 2). Reagents supplied with the Rapid PCR-Detect Reagent Kit (Alderon Biosciences, Durham, NC) were used. Sensors were pre-washed with 100 μlof 1× wash solution for 10 min at room temperature. The buffer was removed just prior to addition of sample to ensure that the sensors were not dry. Following PCR, 1 μl of amplicon was diluted 1:100 in 100 μl of nuclease-free water. A drop (40 μl) of diluted amplicon was placed on a pre-washed sensor and allowed to bind for 5 min at room temperature. Using a squirt bottle, sensors were washed twice with 1× wash solution to remove unbound constituents. The wash solution was removed and the sensor was incubated for 5 min with 50 μl of horseradish peroxidase (HRP)-conjugated anti-fluorescein antibody (0.75 U/μl) at room temperature. Sensors were washed twice with 1x wash solution and inserted into an AndCare 100 Sensor reader (Alderon Biosciences, Durham, NC). The electro-chemical reaction was initiated by adding 50 μl of HRP substrate (0.4 g/μl of TMB containing 0.02% hydrogen peroxidase). A reading was obtained in approximately 30 s. The sensor reader was set to perform IPA analysis according to the following parameters: 15 s delay, -100 mV pulse potential (versus Ag/AgCl reference electrode), 10 ms pulse time, 10 s measurement time, 10 μA current range, and 5 Hz frequency. A total of 50 current measurements were collected from each sensor during the IPA analysis. Each reported current value was the mean of the last five measurements collected. The mean value from each sensor well was displayed on the instrument following IPA.
With Hybrid PCR-Detect, PCR amplicons were labeled with biotin and a free phosphate group (Table 2). Addition of λ-exonuclease was then used to convert the double stranded PCR product into single-stranded DNA molecules labeled at the 5′ end with biotin. Reagents supplied with the Hybrid PCR-Detect Reagent Kit (Alderon Biosciences, Durham, NC) were used. After PCR, 3 μl of amplicon was combined with 0.5 μl of λ-exonuclease (1 U/μl), 1.5 μl of 10 × λ-exonuclease buffer and 10 μl of nuclease-free water (final volume of 15 μl). This reaction was incubated for 10 min at room temperature to allow the λ-exonuclease to degrade the DNA strand containing the 5′ phosphate group. The resulting single-stranded DNA was hybridized to a fluorescein-labeled probe (Table 2, Karenia-1-PCR-R) by combining the following to form a 50 μl reaction: 1 μM probe, 10 μl of 5x hybridization buffer, 23 μl of nuclease-free H2O and 15 μl of the λ-exonuclease digested PCR product. The mixture was heated for 5 min at 80 °C and incubated for an additional 5 min at room temperature. A drop (40 μl) of hybridized PCR product was placed on a pre-washed sensor and analysis proceeded as described for the Rapid PCR-Detect assay except the final wash step was performed three times prior to placing the sensor into the AndCare 100 Sensor reader.
2.8. Multi-target electrochemical detection of bacteria, virus, and source tracking markers
A multi-target format was used for detection of bacterial and viral targets. Specifically, the assay targeted: (1) Enterococcus spp., which are used to indicate the presence of fecal pollution in environmental waters (Cabelli et al., 1979; Durfour, 1984; EPA, 2003); (2) the human-specific HF8 cluster of Bacteroides (Bernhard and Field, 2000); (3) the esp gene from E. faecium, which is used as a proxy for human fecal pollution (Scott et al., 2005); the waterborne bacterial pathogens; (4) E. coli 0157:H7, (5) Salmonella spp., (6) C. jejuni and (7) S. aureus (Percival et al., 2004); and (8) adenoviruses known to cause disease in humans via a waterborne route of transmission (Fong and Lipp, 2005).
Sample analysis was as described for the Rapid PCR-Detect assay except that 8-well carbon sensor strips and the AndCare 800 Sensor reader (Alderon Biosciences, Durham, NC) were used. That is, the method was identical to that described for Rapid PCR-Detect of K. brevis except that each surface of an eight-well sensor array (rather than a single well sensor) was exposed to a different microbial target.
2.9. Data analysis and controls
Student’s t-test and regression analysis were used to evaluate statistical significance and the electrochemical response versus DNA concentration (GraphPad Prism, San Diego, CA). Duplicate negative controls were run for each electrochemical assay and a positive response was calculated as an electrochemical signal greater than 2× the mean current signal of the negative controls. The negative controls contained PCR and electrochemical reagents except template DNA. Positive controls were included in each assay, consisting of all PCR and electrochemical reagents plus a known amount of target DNA. Triplicate PCR and triplicate electrodes were used to relate electrochemical signals to amount of template DNA. All environmental samples tested (Section 2.1) were previously shown not to contain PCR inhibitors (Goodwin et al., 2005; Sinigalliano et al., submitted for publication).
3. Results
The goal of this study was to demonstrate the concept that electrochemical biosensors can identify microbial contaminants in environmental samples. We hypothesized that the electrochemical system developed by Alderon Biosciences (Wojciechowski et al., 1999) primarily for medical markets could be adapted to detect microbes in natural water samples. To test this idea, we adapted two assays, Rapid and Hybrid PCR-Detect, to identify the toxic dinoflagellate K. brevis from environmental samples. In addition, we demonstrated the robustness, flexibility, and multi-target potential of these assays by: (1) modifying the Rapid and Hybrid PCR-Detect methods for detection of RNA (K. brevis), (2) developing protocols for electrochemical detection of bacteria or algae without purifying target DNA, and (3) showing the multiplexing potential of the methods by developing an 8-plexed Rapid PCR-Detect assay for simultaneous detection of fecal pollution indicator bacteria (Enterococcus spp.), two bacterial markers of human fecal pollution (the HF8 cluster of Bacteroides spp. and the esp gene of E. faecium), four human bacterial pathogens (E. coli 0157:H7, Salmonella spp., C. jejuni and S. aureus), and human adenovirus.
3.1. K. brevis assay sensitivity
Sensitivity of the Rapid and Hybrid PCR-Detect assays was determined by analyzing the electrochemical response versus the amount of DNA isolated from 10-fold serial dilutions of K. brevis DNA. The limit of detection for both assays was 10 K. brevis cells (genome equivalents) per PCR. The mean electrochemical signals obtained from 10 K. brevis cells were two times greater than the mean electrochemical signals collected from negative controls. Regression analysis of electrochemical signals obtained with the Rapid and Hybrid assays showed a positive correlation with DNA concentrations spanning five orders of magnitude (r ≥ 0.95; Fig. 1). The log-linear relationship between electrochemical signal and DNA concentration was consistent with results of (Litaker et al. (2001)) and (Aitichou et al. (2004)), which used the Hybrid PCR-Detect technique to detect algal and bacterial enterotoxin genes, respectively. The observed sensitivity of the assays is similar to results of Litaker et al. (Litaker et al., 2001), which showed detection of 10 genomic equivalents of cultured P. piscicida or Cryptoperidiniopsoid spp. using the Hybrid PCR-Detect method. The Rapid and Hybrid PCR-Detect assays are at least as sensitive as visualizing PCR products by the staining of agarose gels with ethidium bromide (e.g.,Fig. 4, 50 cells positive by gel electrophoresis and Rapid PCR-Detect). However, the electrochemical assays returned a digitized signal, making it straightforward to quickly and objectively determine whether or not a sample is positive for a target. In general, the electrochemical methods are faster than a typical gel run which takes about an hour, whereas the electrochemical methods take 30 min following completion of PCR. In addition, the electrochemical methods do not use human carcinogens for detecting PCR products, as is the case with ethidium bromide staining.
Fig. 1.

Detection limits of the (a) Rapid and (b) Hybrid PCR-Detect assays for the toxic dinoflagellate K. brevis. Points on the standard curves represent the mean of triplicate PCRs and electrodes. Isolated genomic DNA from approximately 101 K. brevis genome equivalents (analogous to 10 cells) was detected for both assays, with a signal-to-noise ratio of 2. Coefficients of correlation (r) are indicated.
Fig. 4.

Electrochemical detection (Rapid PCR-Detect) of K. brevis PCR products amplified directly from polycarbonate filters. The mean current from duplicate electrodes, filters, and PCRs are shown. The corresponding agarose gel (1.0%) is shown to the right with 10 μl of PCR product loaded per lane. bp = base pairs.
3.2. Assay reproducibility
Experiments were designed to compare the reproducibility of the Rapid and Hybrid PCR-Detect protocols. The variability of the electrochemical signal was assessed for separate PCR and sensors. Ten independent PCRs (five reactions for each assay type) were analyzed using DNA from 100 K. brevis cells (each using a separate Alderon sensor). Electrochemical detection of K. brevis was reproducible for both assays; however, the Rapid PCR-Detect assay was less variable (8% CV) than the Hybrid PCR-Detect assay (16% CV). The greater sample-to-sample variation observed for the Hybrid PCR-Detect assay could be a result of variability introduced during the enzymatic digestion and/or DNA probe hybridization steps of the assay.
3.3. K. brevis assay specificity
The probes and PCR primers used in both assay formats were specific for K. brevis when tested against DNA isolated from 12 different toxic dinoflagellates, including DNA from a close relative of K. brevis, Karenia mikimotoi (Table 1). K. mikimotoi differs from K. brevis by four nucleotides within the region targeted by the PCR primers and DNA probe. As predicted by our sequence analysis efforts, only PCRs containing K. brevis DNA gave rise to positive electrochemical signals (Table 1). A few recently described Karenia species differ from K. brevis by only one (K. asterichroma and K. bidigitata) or two (K. papilionacea) nucleotides within the region of the PCR primers/probes. Although published reports exist describing K. asterichroma, K. bidigitata and K. papilionacea (De Salas et al., 2004; Haywood et al., 2004), our attempts to obtain samples of these species were not successful.
3.4. Electrochemical detection of K. brevis in coastal water samples
The Rapid and Hybrid PCR-Detect assays developed for K. brevis successfully detected this toxic dinoflagellate in coastal water samples collected from the Rookery Bay NERR. For both assays, samples assigned by microscopy as containing ≤1000 K. brevis cells/l (“present” concentrations) yielded positive electrochemical signals in 6/6 samples (Fig. 2a). Five samples classified by microscopy as not containing K. brevis (“not present”) were tested and the electrochemical results were in 100% agreement with microscopic classification (Fig. 2b).
Fig. 2.

Electrochemical response for the Rapid and Hybrid PCR-Detect assays for 11 different environmental samples containing (a) ≤1000 K. brevis cells/l (“present”) or (b) samples in which K. brevis cells were “not present” as determined by microscopy. The mean current of duplicate PCRs and sensors is shown. “K. brevis gDNA” is a positive PCR containing genomic DNA from ∼1000 K. brevis cells and “Negative” is a PCR containing all reagents except DNA.
Electrochemical results for both assays differentiated between K. brevis that was “present” in the water versus K. brevis that was at “medium” concentrations (105 to <106 cells/l). A total of eight microscopically enumerated samples were used in this analysis. Four of the samples were classified as “present” and four were classified as having “medium” concentrations of K. brevis. Each sample was tested using replicate PCR amplifications. For both electrochemical assays, the “medium” group gave a higher mean electrochemical signal than the “present” group (Rapid PCR-Detect: 9.63 μA versus 3.14 μA; Hybrid PCR-Detect: 5.97 μA versus 1.86 μA; Fig. 3a and b). A Student’s t-test comparison of signals showed this difference to be statistically significant (p value <0.0001). These data show that the assays can differentiate between environmental samples containing K. brevis at “present” levels from those with “medium” levels.
Fig. 3.
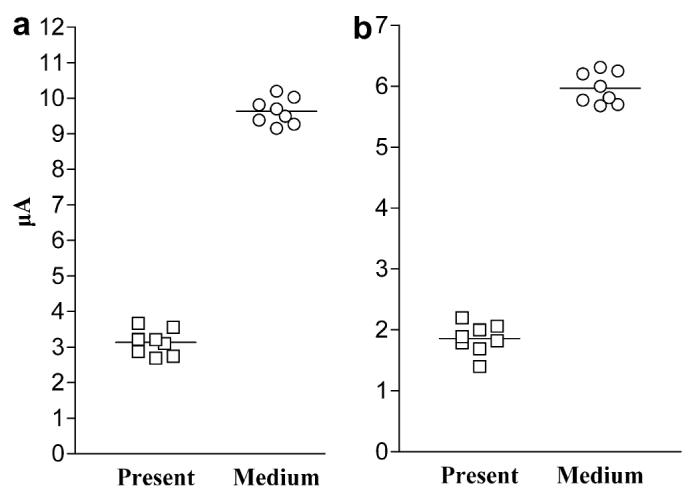
Scatter plots showing increasing electrochemical signal versus increasing concentration of K. brevis in environmental samples. A total of eight samples (four “present” and four “medium”) were tested by the (a) Rapid and (b) Hybrid PCR-Detect methods. Mean current values for replicate PCRs are shown, with each horizontal bar representing the mean of eight sensor readings.
3.5. Comparison of the K. brevis electrochemical assays to other molecular methods
The electrochemical results from both assays were consistent with two other molecular methods. Specifically, the environmental samples tested were also characterized by a colorimetric DNA hybridization assay and by DNA sequencing (Goodwin et al., 2005). All samples that yielded positive electrochemical results were positive via the color-imetric assay and the presence of K. brevis was verified by sequencing. Samples yielding negative electrochemical results were also negative via colorimetric assay and sequencing.
3.6. Detection of K. brevis RNA via the Rapid and Hybrid PCR-detect assays
The Rapid and Hybrid PCR-Detect methods were flexible in that RNA, as well as DNA, could be detected. Electrochemical detection of RNA by way of RT-PCR was achieved with the same primers and probes used to detect DNA (Table 2), and the electrochemical response was consistent with gel electrophoresis results. The mean (±STDEV) current signal response to the K. brevis RT-PCR sample was similar for the Rapid (7.5 ± 0.3 μA) and Hybrid (5.5 ± 0.5) PCR-Detect formats for duplicate electrodes. A negative response (Rapid PCR-Detect, 0.7 ± 0.2 μA; Hybrid PCR-Detect, 0.4 ± 0.2 μA) was returned for a no template reaction containing all RT-PCR reagents except RNA and for a reaction containing all RT-PCR reagents except Reverse Transcriptase (Rapid PCR-Detect, 0.9 ± 0.2 μA; Hybrid PCR-Detect, 0.5 ± 0.1 μA), which was run to test for residual genomic DNA.
3.7. Electrochemical detection without nucleic acid extraction
Extraction of nucleic acids poses a bottleneck that adds time and expense to molecular diagnostic tests. We were able to streamline the electrochemical assays presented here by using filter PCR (Kirchman et al., 2001) and crude lysate as a PCR template obtained through a bead-beat method (Haugland et al., 2005). Using filter PCR in conjunction with the Rapid PCR-Detect protocol, as few as 50 K. brevis cells per filter were detected (Fig. 4). At the lower K. brevis concentrations (50, 500 and 5000 K. brevis), the electrochemical signal increased with cell concentration (Fig. 4). However, the signal became saturated at the higher concentrations of K. brevis (≥50,000 cells per filter). This saturation effect was likely due to inhibition of the PCR due to an excess of nucleic acids in the PCR, as indicated by smearing on the agarose gel for the 50,000 and 500,000 lanes (Fig. 4). In addition, electrochemical detection of fecal-indicating bacteria (Enterococcus spp.) and a source tracking marker (HF8 cluster), was demonstrated using crude lysate obtained from a bead-beat method (Haugland et al., 2005) in conjunction with the Rapid PCR-Detect protocol (Fig. 5).
Fig. 5.

Electrochemical response from the Rapid PCR-Detect assay in multi-target format for fecal indicator bacteria, fecal source tracking markers, and human pathogens important to environmental water quality. Current measurements (using 2, 8-well sensor arrays) from individual PCRs are shown. “No template” = all PCR reagents except target DNA; “Target DNA” = PCRs with 1 ng target DNA; “Beach water” = PCRs with 5 μl of DNA extracted from 100 ml of natural beach water; “Contaminated beach water” = PCRs with 5 μl of DNA extracted from 100 mls of natural beach water spiked with 0.001% (w/v) human feces.
3.8. Comparison of two assay formats
Detection of K. brevis, with either the Rapid or Hybrid PCR-Detect assays yielded similar results in terms of performance. In an experiment comparing the mean current measurements (±STDEV) from five independent PCRs containing ∼102 K. brevis cells, the Rapid assay yielded a higher mean signal (4.87 ± 0.38 μA) with less sample-to-sample variability (8% CV) than did the Hybrid assay (2.26 ± 0.37 μA, 16% CV). However, the mean background was higher for the Rapid (1.33 μA; Fig. 1a) versus the Hybrid PCR-Detect assay (0.26 μA; Fig. 1b). Higher background signal was likely due to low levels of primer dimers, which we have observed can cause false positives (data not shown). This potential limitation of the Rapid assay can be overcome by empirically limiting the primer concentration during PCR. In the case of the Rapid PCR-Detect assay for K. brevis, decreasing the primer concentrations from 50 to 10 pmol 50 μl PCR eliminated false positives due to primer dimers. The Hybrid assay is not prone to false positives due to the formation of primer dimers and has the potential to be more selective than the Rapid assay. That is, in addition to PCR selectivity, the Hybrid assay has a second layer of selectivity by requiring a post-PCR, DNA probe-binding step to generate an electrochemical signal. The Hybrid PCR-Detect assay also is more amendable to multiplexing applications. For example, a pool of DNA containing several target species can be amplified in one PCR using genus specific primers, and individual target species identified by using a different species-specific DNA probe per sensor. However, the DNA probe-binding steps add time, complexity and cost to the assay in comparison to the Rapid Detect method.
3.9. Multi-target detection of microbial contaminants
Electrochemical biosensors have potential as multi-target detection platforms as demonstrated by the assay developed here for simultaneous detection of microbes relevant to environmental water quality. The assay used the Rapid PCR-Detect format paired with a commercially available 8-well sensor array and sensor reader (Alderon Biosciences, Durham, NC) to simultaneously detect indicators of fecal pollution, source tracking markers, and human pathogens (bacterial and viral). Electrochemical detection of all eight targets (Table 3) was achieved with laboratory samples (Fig. 5) using previously published primers and monoplex PCR conditions with minimal modification. These data demonstrate that primers and amplification conditions optimized for detection by gel electrophoresis or real-time PCR can be readily adapted to an electrochemical format. The capacity of the multi-target assay to detect fecal contamination in coastal water samples was shown by analysis of natural beach water spiked with the feces of a healthy human volunteer. Rapid PCR-Detect analysis of the spiked sample was positive for enterococci. and human-specific Bacteroides spp. (Fig. 5). This spiking approach was pursued because beach water samples from our study site appeared to lack the targets (except enterococci) of the 8-plex assay, and thus were not appropriate for demonstrating multi-target detection from environmental samples.
Environmental samples (sediment and water) collected in New Orleans, Louisiana following Hurricanes Katrina and Rita were also tested with the multi-target assay. Limited DNA sample availability restricted testing to Enterococci and Bacteroides spp. The two water samples tested were collected from a canal before and after a water-pumping event. The sample taken before the pumping event (FDP-25) was reported to contain 115 enterococci/100 ml water, and the sample taken after the pumping event (FDP-26) was reported to contain 1481 enterococci/100 ml water (Sinigalliano et al., submitted for publication). By electrochemical analysis, FDP-25 was negative for enterococci and the human-marker of Bacteroides spp.; whereas, FDP-26 was positive for enterococci and the human-marker of Bacteroides spp. (Fig. 6). The sediment sample (YD3), reported to contain 1039 enterococci/g soil (Sinigalliano et al., submitted for publication), was positive for enterococci and negative for human-marker Bacteroides in this study (Fig. 6). These results indicate that the multi-target assay can be used to detect microbial contaminants from environmental sediment and water samples while returning information regarding the sources of fecal contamination.
Fig. 6.

Multi-target detection (Rapid PCR-Detect) of fecal indicator bacteria (Enterococcus spp.) and human-specific Bacteroides (HF8 cluster) amplified from sediment (YD3) and water (FDP-25 and FDP-26) samples collected from New Orleans, LA following Hurricane Katrina. Mean current measurements from duplicate sensors and PCRs are shown. “No template” = all PCR reagents except target DNA. Samples marked with an asterisk (*) are electrochemical positive, based on having a mean current >2x the mean current of no template PCR controls.
4. Discussion
The utility of electrochemical methods for water quality applications depends in part on availability, ease of use, and versatility. The reagents, biosensors, and sensor readers used in this study were all commercially available. The electrochemical formats, particularly the Rapid PCR-Detect, were relatively simple to use. For example, the multi-target assay design was based on previously published primers, and thus requires little modification to enable the 8-target sensor array to work “out of the box”. The methods described here were highly versatile as demonstrated by detection of both DNA and RNA of a toxic dinoflagellate and detection of a variety of bacterial and viral targets. The demonstrated ability to detect RNA could be useful for the differentiation of live versus dead microbes in the environment, as RNA is a better indicator of cellular viability (Keer and Birch, 2003). The successful detection of RNA also allows for the identification of microbes with an RNA genome. Furthermore, the developed assays were sensitive (Figs. 1, 2 and 4), rapid (can be completed in 3–5 h), used hand-held sensor readers that are relatively affordable (www.alderonbiosciences.com), returned a digitized signal which allowed rapid and objective determination of a positive result, and did not require the isolation of nucleic acids prior to detection (Figs. 4 and 5).
Although attention paid to electrochemical methods as microbial detection tools has grown (Drummond et al., 2003; Kerman et al., 2004; LaGier et al., 2005; Metfies et al., 2005), there is a lack of published investigations testing whether electrochemical methods can detect microbial contaminants in environmental samples. Without this key data, the usefulness of electrochemical sensors for environmental microbiology applications cannot be fully judged. In this study, electrochemical detection of the toxic dinoflagellate K. brevis was successfully demonstrated using coastal water samples. The assay was sensitive enough to detect K. brevis that was “present” (≤1000 cells) in samples without returning false positive results for samples in which K. brevis was “not present” (Fig. 2). Furthermore, the electrochemical response was significantly different for samples containing K. brevis at “present” versus “medium” concentrations (100,000 to <106 cells/l cells) (Fig. 3). Hence, the Rapid and Hybrid PCR-Detect assays could be useful research tools to screen for the presence or absence of K. brevis with the potential to differentiate between broad classes of abundance.
The multi-target assay was designed to return an indication of fecal contamination along with information on whether contamination was from a human source and if selected pathogens were present. Such an tool would be useful for managers of recreational water quality (Bower et al., 2005) because even though high levels of fecal-indicating bacteria appear to pose a public health risk (Cabelli et al., 1979; Dufour, 1984; EPA, 2003; Wade et al., 2003), the presence of indicators is not always correlated with the presence of pathogens (Lipp et al., 2001; Noble and Furhman, 2001), and standard methods (EPA, 2003) do not identify the source of the fecal contamination (Scott et al., 2005).
The multi-target assay was tested against laboratory samples and beach water spiked with human feces. Rapid PCR-Detect analysis successfully detected all eight targets in laboratory samples, and the beach water sample contaminated with human feces was positive for enterococci and human-specific Bacteroides (Fig. 5). Both Enterococcus spp. and Bacteroides spp. are commonly found in human feces (Tortora et al., 2001), and the human-specific Bacteroides marker (HF8) is estimated to be present at 5 × 1010 copies/g of feces (Bernhard and Field, 2000). In addition, the reaction for detecting the human-specific Bacteroides marker has been shown to be sensitive, with a positive visualization of PCR products from sewage samples diluted 1:150,000 (Bower et al., 2005). Lack of detection for the other targets may have been due to lack of presence in the human fecal sample, but was not due to PCR inhibition (data not shown). The sample was from a healthy volunteer, thus the pathogens Salmonella spp., C. jejuni, E. coli 0157:H7, and human adenovirus were not expected in the sample. The esp gene is not found in all humans (Scott et al., 2005), and only 30–50% of humans carry S. aureus (Youmans et al., 1985). The lack of an esp signal also might be due the relatively low sensitivity of the PCR used to amplify the esp gene, which in our hands was 10×less sensitive then the PCR used to amplify Bacteroides belonging the HF8 cluster (10 plasmid copies/PCR for esp versus 1 plasmid/PCR for HF8).
In addition, the multi-target assay was tested against water and sediment samples collected from flooded sites within New Orleans following Hurricane Katrina. The water samples were collected before (FDP-25) and after (FDP-26) a pumping event. Visual observation confirmed the infusion of fecal waste after pumping (Sinigalliano et al., submitted for publication). The electrochemical data showed that the enterococci detected after pumping were associated with feces of human origin (Fig. 6) and suggest the usefulness of the electrochemical assay as a source tracking tool. The lack of an enterococci signal in sample FDP-25 is likely due to the constraint of DNA availability. The filter for water sample FDP-25 should have contained 115 enterococci prior to DNA extraction. Even assuming no loss of target DNA during the extraction procedure, each PCR (containing 1 μl of genomic DNA) run on FDP-25 would have contained ∼2 genome equivalents of enterococci, which is less than the reported sensitivity of the enterococci primers (five genome equivalents per PCR) (He and Jiang, 2005).
The sediment sample (YD3) was positive for enterococci and negative for human-marker Bacteroides (Fig. 6), indicating that the enterococci signal was not the result of recent human contamination. Nonetheless, this sample was positive by PCR for Bfidobacteria adolescentis (King et al., 2007; Sinigalliano et al., submitted for publication), a species with a human-dominated host range (Bonjoch et al., 2004). The lack of a HF8 signal might be a result of PCR-detectable Bacteroides not persisting in aerobic environments for more than 1–2 days, based on results for Bacteroides distasonis (Kreader, 1998). Alternatively, perhaps B. adolescentis is more prevalent in human feces in comparison to the HF8 marker, and thus more likely to be detected.
Although the multi-target assay requires more field-testing, the data (Figs. 5 and 6) reinforce single-plex results for K. brevis (Figs. 2 and 3) demonstrating the capacity of electrochemical methods to detect microbial contaminants from environmental samples. The multi-target data also suggest that electrochemical methods could be used to simultaneously return information on fecal indicators, source tracking markers, and perhaps pathogen presence, particularly if adequate sample concentration and efficient nucleic acid recovery can be achieved. A limitation of using the multi-target assay for analyzing large numbers of field samples is the use of monoplex PCR prior to electrochemical detection (i.e. simultaneous electrochemical detection using sensor arrays). Multiplex PCR coupled with the Hybrid PCR-Detect method may be utilized to more efficiently analyze large sets of field samples. In addition, the integration of the multi-target electrochemical assays into automated devices should minimize limitations associated with monoplex PCR (Farmer et al., 2006).
Efficient upstream processing of environmental samples is critical to the success of downstream molecular detection methods, but sample concentration and nucleic acid extraction pose major obstacles to the development of rapid and practical methods for water quality monitoring (Noble and Weisberg, 2005). The sensitivity achieved with isolated DNA (Fig. 1) typically is not observed with filtered environmental water samples (Figs. 2 and 6, sample FDP-25) in part because of poor DNA recovery during the extraction and isolation of nucleic acids. For instance, average extraction efficiencies associated with commercial kits can range from 2–30% for environmental soil samples (Mumy and Findlay, 2004). Thus, in addition to streamlining the upstream processing of environmental samples for molecular analysis, eliminating the nucleic acid isolation step (Figs. 4 and 5) may minimize losses in sensitivity due to poor extraction efficiencies.
The Rapid and Hybrid PCR-Detect assays use hand-held instruments, compact sensors, and produce digitized results and thus are amendable to field detection. However, the PCR and electrochemical steps of the assays should be integrated into a single device to be more practical for field applications. Ongoing efforts include building electrochemical instruments for multi-target microbial detection (Farmer et al., 2006).
Acknowledgements
We thank Dr. Marek Wojciechowski and the Alderon Biosciences staff for providing AndCare Sensor readers during the course of this study. We thank G. Scorzetti, L. Brand, and S. Morton for dinoflagellate samples; J. Rose, T. Scott, S. Jiang, and N. Cirino for bacterial and viral samples; and the Rookery Bay NERR for environmental samples. We are grateful to H. Solo-Gabriele and C. Sinigialliano of the University of Miami Center of Excellence for Oceans and Human Health (NSF #0CE0432368/ NIEHS #P50 ES12736) for post-Katrina samples. Financial support is gratefully acknowledged from the National Science Foundation (OCE-332918), the NIEHS Marine and Freshwater Biomedical Sciences Center Grant (ES 05705) to the University of Miami, the Cooperative Institute of Estuarine and Environmental Technology (CICEET), and the NOAA Oceans and Human Health Initiative Capacity Building Grant. This research was carried out in part under the auspices of the Cooperative Institute for Marine and Atmospheric Studies (CIMAS), a joint institute of the University of Miami and the National Oceanic and Atmospheric Administration, cooperative agreement #NA17RJ1226.
References
- Aitichou M, Henkens R, Sultana AM, Ulrich RG, Ibrahim M. Sofi. Detection of Staphylococcus aureus enterotoxin a and b genes with PCR-EIA and a hand-held electrochemical sensor. Molecular and Cellular Probes. 2004;18:373–377. doi: 10.1016/j.mcp.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Andersen RA, Morton SL, Sexton JP. Provasoli-Guillard national center for culture of marine phytoplankton 1997 list of strains. Journal of Phycology. 1997;33(suppl.):1–75. [Google Scholar]
- Attatippaholkun WH, Attatippaholkum MK, Nisalak A, Vaughn DW, Innis BL. A novel method for the preparation of large cDNA fragments from dengue-3 RNA genome by long RT-PCR amplification. Southeast Asian Journal of Tropical Medicine and Public Health. 2003;31:126–133. [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short Protocols in Molecular Biology. 4th John Wiley and Sons; New York: 1999. [Google Scholar]
- Bernhard AE, Field KG. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Applied and Environmental Microbiology. 2000;66:4571–4574. doi: 10.1128/aem.66.10.4571-4574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonjoch X, Balleste E, Blanch AR. Multiplex PCR with 16S rRNA Gene-Targeted Primers of Bifidobacterium spp. To Identify Sources of Fecal Pollution. Applied and Environmental Microbiology. 2004;70:3171–3175. doi: 10.1128/AEM.70.5.3171-3175.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower PA, Scopel CO, Jensen ET, Depas MM, McLellan SL. Detection of genetic markers of fecal indicator bacteria in Lake Michigan and determination of their relationship to Escherichia coli densities using standard microbiological methods. Applied and Environmental Microbiology. 2005;71:8305–8313. doi: 10.1128/AEM.71.12.8305-8313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand LE. The isolation and culture of microalgae for biotechnological applications. In: Labeda DP, editor. Isolation of Biotechnological Organisms from Nature. McGraw-Hill; New York: 1990. pp. 81–115. [Google Scholar]
- Cabelli VJ, Dufour AP, Levin MA, McCabe J, Haberman PW. Relationship of microbial indicators to health effects at bathing beaches. American Journal of Public Health. 1979;69:690–696. doi: 10.2105/ajph.69.7.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culverhouse PF, Williams R, Reguera B, Herry V, González-Gil S. Do experts make mistakes? A comparison of human and machine identification of dinoflagellates. Marine Ecology Progress Series. 2003;247:17–25. [Google Scholar]
- De Salas MF, Bolch CJS, Hallegraeff GM. Karenia asterichroma spp. nov. (Gymnodiniales, Dinophyceae), a new dinoflagellate species associated with finfish aquaculture mortality in Tasmania, Australia. Phycologia. 2004;43:624–631. [Google Scholar]
- Drummond TG, Hill MG, Barton JK. Electrochemical DNA sensors. Nature Biotechnology. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- Dufour AP.Health effects criteria for fresh recreational waters 1984. EPA-600/1-84/004. Office of Research and Development, USEPA.
- Durfour AP. Bacterial indicators of recreational water quality. Can. J. Public Health. 1984;75:49–56. [PubMed] [Google Scholar]
- Dwight RH, Fernandez LM, Baker DB, Semenza JC, Olson BH. Estimating the economic burden from illnesses associated with recreational coastal water pollution—a case study in Orange County, California. Journal of Environmental Management. 2005;76:95–103. doi: 10.1016/j.jenvman.2004.11.017. [DOI] [PubMed] [Google Scholar]
- EPA Guidelines establishing test procedures for the analysis of pollutants; Analytical methods for biological pollutants in ambient water; Final Rule 2003. Federal Register V68, No. 139 40 CFR Part 136,43272–43283.
- Farmer AS, LaGier MJ, Goodwin KD, Ivanov S, Steimle G, Fries D. Portable sensor development towards PCR-based electrochemical detection. Florida Marine Biotechnology Summit. 2006;V:18. [Google Scholar]
- Fong TT, Lipp EK. Enteric viruses of humans and animals in aquatic environments: health risks, detection, and potential water quality assessment tools. Microbiol. Mol. Biol. Rev. 2005;69:357–371. doi: 10.1128/MMBR.69.2.357-371.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin KD, Cotton SA, Scorzetti G, Fell JW. A DNA hybridization assay to identify toxic dinoflagellates in coastal waters: detection of Karenia brevis in the Rookery Bay National Estuarine Research Reserve. Harmful Algae. 2005;4:411–422. [Google Scholar]
- Griffin DW, Lipp EK, McLaughlin MR, Rose JB. Marine recreation and public health microbiology: quest for the ideal indicator. BioScience. 2001;51:817–825. [Google Scholar]
- Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP. Comparison of Enterococcus measurements in freshwater at two recreational beaches by quantitative polymerase chain reaction and membrane filter culture analysis. Water Research. 2005;39:559–568. doi: 10.1016/j.watres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Haywood AJ, Steidinger KA, Truby EW, Bergquist PR, Bergquist PL, Adamson J, Mackenzie L. Comparative morphology and molecular phylogenetic analysis of three new species of the genus Karenia (Dinophyceae) from New Zealand. Journal of Phycology. 2004;40:165–179. [Google Scholar]
- He JW, Jiang S. Quantification of Enterococci and Human adenoviruses in environmental samples by real-Time PCR. Applied and Environmental Microbiology. 2005;71:2250–2255. doi: 10.1128/AEM.71.5.2250-2255.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keer JT, Birch L. Molecular methods for the assessment of bacterial viability. Journal of Microbiological Methods. 2003;53:175–183. doi: 10.1016/s0167-7012(03)00025-3. [DOI] [PubMed] [Google Scholar]
- Kerman K, Kobayashi M, Tamiya E. Recent trends in electrochemical DNA biosensing technology. Measurement Science Technology. 2004;15:R1–R11. [Google Scholar]
- Kiesling TL, Wilkinson E, Rabalais J, Ortner PB, McCabe MM, Fell JW. Rapid identification of adult and macular stages of copepods using DNA hybridization methodology. Marine Biotechnology. 2002;4:30–39. doi: 10.1007/pl00021689. [DOI] [PubMed] [Google Scholar]
- King EL, Bachoon DS, Gates KW. Rapid detection of human fecal contamination in estuarine environments by PCR targeting of Bifidobacteria adolescentis. Journal of Microbiological Methods. 2007;68:76–81. doi: 10.1016/j.mimet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Kirchman DL, Yu L, Fuchs BM, Amaan R. Structure of communities in aquatic systems as revealed by filter PCR. Aquatic Microbial Ecology. 2001;26:13–22. [Google Scholar]
- Kirkpatrick B, Fleming LE, Squicciarini D, Backer LC, Clark R, Abraham W, Benson J, Cheng YS, Johnson D, Pierce R. Literature review of Florida red tide: implications for human health effects. Harmful Algae. 2004;3:99–115. doi: 10.1016/j.hal.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong RYC, Lee SKY, Lee TFW, Law SHW, Wu RSS. Rapid detection of six types of bacterial pathogens in marine waters by multiplex PCR. Water Research. 2002;36:2802–2812. doi: 10.1016/s0043-1354(01)00503-6. [DOI] [PubMed] [Google Scholar]
- Kreader CA. Persistence of PCR-detectable bacteroides distasonis from human feces in river water. Applied and Environment Microbiology. 1998;64:4103–4105. doi: 10.1128/aem.64.10.4103-4105.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGier MJ, Scholin CA, Fell JW, Wang J, Goodwin KD. An electrochemical RNA hybridization assay for detection of the fecal indicator bacterium Escherichia coli. Marine Pollution Bulletin. 2005;50:1251–1261. doi: 10.1016/j.marpolbul.2005.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGier MJ, Joseph LA, Passaretti TV, Musser KA, Cirino NM. A real-time multiplexed PCR assay for rapid detection and differentiation of Campylobacter jejuni and Campylobacter coli. Molecular and Cellular Probes. 2004;18:275–282. doi: 10.1016/j.mcp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Landsberg JH. The effects of harmful algal blooms on aquatic organisms. Reviews in Fisheries Science. 2002;10:113–390. [Google Scholar]
- Leclerc H, Schwarzbrod L, Dei-Cas E. Microbial agents associated with waterborne diseases. Critical Reviews in Microbiology. 2002;28:371–409. doi: 10.1080/1040-840291046768. [DOI] [PubMed] [Google Scholar]
- Lidie KB, Ryan JC, Barbier M, Van Dolah FM. Gene expression in Florida red tide dinoflagellate Karenia brevis: analysis of an expressed sequence tag library and development of DNA microarray. Marine Biotechnology. 2005;7:481–493. doi: 10.1007/s10126-004-4110-6. [DOI] [PubMed] [Google Scholar]
- Lipp EK, Farrah SA, Rose JB. Assessment and impact of microbial fecal pollution and human enteric pathogens in a coastal community. Marine Pollution Bulletin. 2001;42:286–293. doi: 10.1016/s0025-326x(00)00152-1. [DOI] [PubMed] [Google Scholar]
- Litaker W, Sundseth R, Wojciechowski M, Bonaventura C, Henkens R, Tester P. Harmful Algal Blooms 2000. Paris: 2001. Electrochemical detection of DNA or RNA from harmful algal bloom species; pp. 242–245. [Google Scholar]
- Mason WJ, Blevins JS, Beenken K, Wibowo N, Ojha N, Smeltzer MS. Multiplex PCR protocol for the diagnosis of Staphylococcal infection. Journal of Clinical Microbiology. 2001;39:3332–3338. doi: 10.1128/JCM.39.9.3332-3338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer JJ, Schmidt D, Petrosko P, Sanchez S, Bolton L, Lee MD. Development of primers to O-antigen biosynthesis genes for specific detection of Escherichia coli O157 by PCR. Applied and Environmental Microbiology. 1999;65:2954–2960. doi: 10.1128/aem.65.7.2954-2960.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metfies K, Huljic S, Lange M, Medlin LK. Electrochemical detection of the toxic dinoflagellate Alexandrium ostenfeldi with a DNA-biosensor. Biosensors and Bioelectronics. 2005;20:1349–1357. doi: 10.1016/j.bios.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Millie DF, Schofield OM, Vinyard BT. Detection of harmful algal blooms using photopigments and absorption signatures: a case study of the Florida red tide dinoflagellate, Gymnodinium breve. Limnology and oceanography. 1997;42:1240–1251. [Google Scholar]
- Mumy KL, Findlay RH. Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. Journal of Microbiological Methods. 2004;57:259–268. doi: 10.1016/j.mimet.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Noble RT, Furhman JA. Enteroviruses detected by reverse transcriptase polymerase chain reaction from the coastal waters of Santa Monica Bay, California: low correlation to bacterial indicator levels. Hydrobiologia. 2001;460:175–184. [Google Scholar]
- Noble RT, Weisberg SB. A review of technologies being developed for rapid detection of bacteria in recreational waters. Journal of Water and Health. 2005;3:381–392. doi: 10.2166/wh.2005.051. [DOI] [PubMed] [Google Scholar]
- Percival SL, Chalmers RM, Embrey M, Hunter PR, Sellwood J, Wyn-Jones P. Microbiology of Waterborne Diseases. Elsevier Academic Press; 2004. pp. 21–209. [Google Scholar]
- Rizzo PJ, Jones M, Ray SM. Isolation and properties of isolated nuclei from the Florida red tide dinoflagellate Gymnodinium breve. Journal of Phycology. 1982;29:217–222. doi: 10.1111/j.1550-7408.1982.tb04014.x. [DOI] [PubMed] [Google Scholar]
- Scott TM, Jenkins TM, Lukasik J, Rose JB. Potential use of a host associated molecular marker in Enterococcus faecium as an index of human fecal pollution. Environmental Science and Technology. 2005;39:283–287. [PubMed] [Google Scholar]
- Shibata T, Solo-Gabriele HM, Fleming LE, Elmir S. Monitoring marine recreational water quality using multiple microbial indicators in an urban tropical environment. Water Research. 2004;38:3119–3131. doi: 10.1016/j.watres.2004.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinigalliano CD, Gidley ML, Shibata T, Dixon TH, Whitman D, Bachoon D, Brand L, Amaral-Zettler L, Gast R, Nigro OD, Steward GF, Hou A, Mathews J, Laws E, Fujioka R, Solo-Gabriele HM, Fleming LE.submitted for publication.Microbial measurements in water and sediments of the New Orleans area in the aftermath of hurricanes Katrina and Rita [DOI] [PMC free article] [PubMed]
- Steidinger KA, Landsberg JH, Truby EW, Roberts BS. First report of Gymnodinium pulchellum (Dinophyceae) in North America and associated fish kills in the Indian River, Florida. Journal of Phycology. 1998;34:431–437. [Google Scholar]
- Tester PA, Steidinger KA. Gymnodinium breve red tide blooms: initiation, transport, and consequences of surface circulation. Limnology and oceanography. 1997;42:1039. [Google Scholar]
- Tortora GJ, Funke BR, Case CL. Microbiology, An Introduction. 7th Addison Wesley Longman, Inc.; San Francisco: 2001. [Google Scholar]
- Wade TJ, Pai N, Eisenberg JNS, Colford JM., Jr. Do U.S. Environmental Protection Agency water quality guidelines for recreational waters prevent gastrointestinal illness? A systematic review and meta-analysis. Environmental Health Perspectives. 2003;111:1102–1109. doi: 10.1289/ehp.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Analytical Electrochemistry. 2nd John Wiley and Sons Press; New York: 2000. [Google Scholar]
- Wojciechowski M, Sundseth R, Moreno M, Henkens R. Multichannel electrochemical detection system for quantitative monitoring of PCR amplification. Clinical Chemistry. 1999;45:1690–1693. [PubMed] [Google Scholar]
- Youmans GP, Paterson PY, Sommers HM. The Biological and Clinical Basis of Infectious Diseases. 3rd The W.B. Saunders Company; Philadelphia: 1985. [Google Scholar]
- Zhang Y, Kim HH, Heller A. Enzyme-amplified amperometric detection of 3000 copies of DNA in a 10-μl droplet at 0.5 fM concentration. Analytical Chemistry. 2003;85:3267–3269. doi: 10.1021/ac034445s. [DOI] [PubMed] [Google Scholar]