

Abstract
Messenger ribonucleic acids (mRNAs) have been used in vaccines for various diseases and are attracting attention as a new pharmaceutical paradigm. The purification of mRNAs is necessary because various impurities, such as template DNAs and transcription enzymes, remain in the crude product after mRNA synthesis. Among the various purification methods, reversed-phase high-performance liquid chromatography (RP-HPLC) is currently attracting attention. Herein, we optimized the pore size of the packing materials, the mobile phase composition, and the temperature of the process; we also evaluated changes in the separation patterns of RNA strands of various lengths via RP-HPLC. Additionally, single-stranded (50–1000 nucleotides in length) and double-stranded (80–500 base pairs in length) RNAs were separated while their non-denatured states were maintained by performing the analysis at 60 °C using triethylammonium acetate as the mobile phase and octadecyl-based RNA-RP1 with super-wide pores (> 30 nm) as the column. Furthermore, impurities in a long-stranded RNA of several thousand nucleotides synthesized by in vitro transcription were successfully separated using an RNA-RP1 column. The columns used in this study are expected to separate various RNA strands and the impurities contained in them.
Graphical abstract

Keywords: Ribonucleic acid (RNA), High-performance liquid chromatography (HPLC), Octadecyl-based column, Super-wide pore, Separation and purification
Introduction
Messenger ribonucleic acids (mRNAs) have been used as vaccines for infectious diseases, such as coronavirus disease 2019 (COVID-19) and influenza, and various cancers; and they are attracting attention as novel pharmaceuticals [1–6]. mRNA used in COVID-19 vaccines is long-stranded, approximately 4000 nucleotide (4 knt) in length, and it is synthesized via in vitro transcription. By this approach, mRNAs are synthesized using a template DNA during transcription [4–6]. However, impurities, such as the template DNA itself and transcription enzymes, are left in the crude product after the transcription process [7]. Therefore, implementing a purification process is necessary for the medical use of mRNAs.
Various impurities are present in nucleic acid medicines, and the methods utilized to remove them include (1) cross-flow filtration (tangential flow filtration) for the removal of small molecules [8, 9], (2) cellulose coprecipitation with ethanol-containing solvents for double-stranded RNA (dsRNA) removal [10, 11], (3) preparative purification via polyacrylamide gel electrophoresis [12], and (4) lithium chloride precipitation for RNA purification [13]. However, conventional methods for the purification of nucleic acids suffer from some shortcomings, including (1) the fact that impurities, which because of their size, cannot pass through a cross-flow filter, cannot be removed; (2) the fact that impurities other than dsRNA cannot be removed; and (3) the fact that scaling up the purification process is difficult. In order to address these shortcomings, we focused on reversed-phase (RP) chromatography using ion pair reagents. In addition to RP chromatography, other methods for the separation and purification of RNA by high-performance liquid chromatography (HPLC) include ion-exchange chromatography [14] and size-exclusion chromatography [15]. Both of these techniques are characterized by few process parameters that can be changed. As ion-exchange chromatography uses the charge number of the molecules, and size-exclusion chromatography recognizes the apparent size of the molecules for separation, nucleotides having similar sizes but different sequences are difficult to separate using these two techniques. Preparation of similar sequences and chain lengths of nucleotides is difficult, because of the precise adjustment to sample elution that is required. By contrast, RP chromatography has many parameters related to analytical conditions, such as temperature, concentration and pH of the mobile phase, gradient, stationary phases, ion pair reagent, and organic solvent type, which can be changed to adjust the separation behavior.
For the reasons discussed above, we aimed to separate nucleic acids similar in structure via RP-HPLC. RP-HPLC making use of ion-pair reagents is a classic method for the HPLC-based separation of nucleic acid strands of several tens to hundreds of nucleotides (nt) in length; indeed, the inclusion of ion pair reagents in the mobile phase affords an increase in the retention time of nucleic acids compared to the separation process carried out in absence of such reagents. However, only a few studies have been published in which optimal separation conditions for the purification of long-stranded RNA (several thousand nt) have been determined. In this context, we believe that, by optimizing parameters like the mobile phase and purification temperature, various impurities present in long-length RNA reaction mixtures can be separated simultaneously, and the target nucleic acids can be purified.
Herein, we attempted to separate mixtures of RNA strands of different sizes via RP-HPLC. Additionally, the effects of the pore size of the packing material, the analysis temperature, and the mobile phase were investigated in detail by performing the separation of long RNA strands of various lengths. Furthermore, we analyzed the separation of in vitro-transcribed single-stranded RNA (ssRNA) of approximately 3.0 knt and 4.8 knt in length, which is longer than the RNA strand used in the COVID-19 vaccine.
Experimental
Chemicals
Triethylamine (TEA, guaranteed reagent grade), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, HPLC grade), acetic acid (guaranteed reagent grade), sodium dihydrogenphosphate (guaranteed reagent grade), disodium hydrogen phosphate (guaranteed reagent grade), methanol (HPLC grade), acetonitrile (HPLC grade), agarose for 50–800 base pair (bp) fragments (electrophoresis grade), loading dye brilliant color (6x) (electrophoresis grade), 10 x tris–acetate ethylenediaminetetraacetic acid (TAE) buffer (0.4 M tris(hydroxymethyl)aminomethane (Tris), 0.2 M acetic acid, and 10 mM ethylenediaminetetraacetic acid (EDTA), electrophoresis grade), and 0.44 mg/ml ethidium bromide solution (electrophoresis grade) were obtained from Nacalai Tesque, Inc. (Kyoto, Japan). Low-range ssRNA ladder, low-range dsRNA ladder, and wide-range ssRNA ladder were purchased from New England Biolabs, Inc. (Ipswich, MA, USA). The pBR322/MspI digest was purchased from NIPPON GENE CO., LTD. (Tokyo, Japan). The ΦX174/HaeIII digest and 50 bp DNA ladder (Dye Plus) were purchased from Takara Bio Inc. (Shiga, Japan).
Preparation of the mobile phase
The 400 mM HFIP-TEA buffer (pH 7.0) was prepared by titrating an HFIP solution prepared as described below with TEA [16, 17]. Specifically, a total of 67.23 g of HFIP was dissolved in 800 mL of ultrapure water. As the obtained mixture was stirred at room temperature, 360 µL of TEA was added to it slowly. After adjusting the obtained solution’s volume to 1 L using ultrapure water, the final concentration of HFIP was 400 mM.
Subsequently, 200 mM and 100 mM triethylammonium acetate (TEAA) buffer solutions (pH 7.0) were prepared by mixing appropriate volumes of TEA and acetic acid in ultrapure water [18, 19]. To obtain TEAA 200 mM, 11.44 mL of acetic acid was dissolved in 800 mL of ultrapure water; in the case of the TEAA 100 mM solution, 5.72 mL of acetic acid was dissolved in 800 mL of ultrapure water. While the acetic acid solutions were stirred at room temperature, 27.72 (200 mM) or 13.86 mL (100 mM) of TEA were added slowly to them. After adjusting the volumes of the two solutions to 1 L with ultrapure water, the final concentrations of TEA and acetic acid were 200 and 100 mM, respectively. A measuring pipette was used for volumes larger than 1 mL, and a micropipette was used for volumes smaller than 1 mL to add each reagent.
Phosphate buffer (PB; pH 6.8) at 100 mM concentration was prepared by mixing sodium dihydrogen phosphate and disodium hydrogen phosphate in ultrapure water. Specifically, a total of 47.5 mmol of sodium dihydrogenphosphate was dissolved in 800 mL of ultrapure water. Additionally, 52.5 mmol of disodium hydrogen phosphate was dissolved slowly into the mixture thus obtained. Subsequently, ultrapure water was added to a total volume of 1 L.
The 100 mM TEAA-20 mM PB (pH 7.0) solution (TEAA-PB) was prepared by mixing 500 mL of 200 mM TEAA (pH 7.0), 200 mL of 100 mM PB (pH 6.8), and 300 mL of ultrapure water.
HPLC measurements
HPLC experiments were performed using a Prominence HPLC system (Shimadzu Corporation, Kyoto, Japan) equipped with an LC-20 AD intelligent pump. Separations were conducted on a COSMOSIL RNA-RP1 (2.0 mm I. D. x 100 mm, particle size; 5 µm, Nacalai Tesque) column whose temperature was kept in the 40 °C–70 °C range by using a CTO-20AC column oven (Shimadzu Corporation). The pore sizes of the RNA-RP1-based columns used for pore size optimization are approximately 12 nm for normal pore and > 30 nm for super-wide pore. The flow rate was 0.2 mL/min. Ultraviolet (UV) detection was carried out at 260 nm. HFIP-TEA, TEAA, and TEAA-PB were used as solvent A. Methanol, acetonitrile, and methanol-acetonitrile mixture (50/50, vol/vol) were used as solvent B. All samples were separated using the gradient elution mode.
Agarose gel electrophoresis
Agarose gel electrophoresis experiments were conducted using 3% agarose gel. Notably, the 3% agarose gels were prepared by suspending 3 g of agarose powder in 100 mL of 1 x TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA), dissolving the obtained suspension in a microwave oven while heating, cooling it to approximately 60 °C and pouring the melted mixture into a gel plate. Subsequently, 2 µL of the analyte sample, 1 µL of loading dye brilliant color (6x), and 3 µL of ultrapure water were mixed together. A 6 µL aliquot of each sample was loaded onto the gel, and an electrophoresis experiment was carried out at 100 V for 30–35 min at room temperature. The gels were stained with ethidium bromide for 40 min and imaged using a ChemiDoc Touch MP Imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
In vitro transcription of RNA
RNA strands that were 3.0 knt and 4.8 knt in length were generated from the relevant DNA templates via in vitro transcriptions conducted utilizing T7 RNA polymerase (TOYOBO Co., LTD., Osaka, Japan) followed by a purification process using lithium chloride precipitation. The DNA templates were constructed via the ligation of blunt-ended HindIII and BamHI fragments from pQBI T7-GFP (Qbiogene, Inc., USA) performed to eliminate the T7 terminator, followed by a linearization process conducted either in the presence of EagI and BglII or of BglII alone; as a result, DNA fragments of 3.0 kbp and 4.8 kbp lengths, respectively, were prepared, which included a T7 promoter at each upstream edge.
Results and discussions
Effect of the packing material’s porosity and pore size on the separation of long-stranded RNAs
Several researchers have reported that porous packing materials yield better oligonucleotide separation than their non-porous counterparts [20–22]. Therefore, we first confirmed whether pore size and the presence or absence of pores affected the separation of long-stranded RNAs. The separation patterns of low-range ssRNAs and wide-range ssRNAs were compared using non-porous packing materials and packing materials characterized by pores of common sizes (about 12 nm; normal pores) and pores of sizes larger than 30 nm (super-wide pores). Clear peaks were absent for either the low- or wide-range ssRNAs separated using the non-porous packing material (Fig. 1). Although the use of the normal pore packing material afforded the observation of clear peaks, poor separation was obtained for the low- and wide-range ssRNAs. On the other hand, the use of the super-wide pore packing materials afforded high sensitivity, and low- and wide-range ssRNAs could be separated using this material (Fig. 1). These results indicate that packing materials with super-wide pores are suitable for the separation of long-stranded RNAs.
Fig. 1.

HPLC chromatograms of a low-range and b wide-range ssRNAs obtained using COSMOSIL RNA-RP1-type columns (2.0 mm I. D. x 100 mm; pore sizes are reported in the figure). HPLC analysis was performed with 100 mM TEAA (pH 7.0) using a linear gradient from a 0% to 25% and b 7.5% to 17.5% with acetonitrile over 20 min at a flow rate of 0.2 mL/min and a temperature of 65 °C. UV detection was performed at 260 nm
Effects of the mobile phases on RNA separation
Next, we optimized the mobile phase conditions for the ssRNAs separation. The separation patterns of low-range ssRNAs were compared using an octadecyl (C18)-based COSMOSIL RNA-RP1 column with super-wide pores using three different mobile phases consisting of mixtures of HFIP-TEA [23, 24] and TEAA [25, 26], which are commonly used for oligonucleotide separation. Since acetonitrile did not dissolve in the HFIP-containing mixture, when such a mixture was used as solvent A, only methanol was used as solvent B. Nucleic acids are easily adsorbed on various surfaces, so in HPLC analysis, when the peak of the target nucleic acid is not detected or the analysis data are not reproducible, the nucleic acid may have been adsorbed on the column or piping. The adsorption of nucleic acids can be prevented by adding a buffer solution, such as PB, to the mobile phase. Indeed, the TEAA buffer containing PB is used to inhibit RNA adsorption.
The shapes of the peaks of the low-range ssRNAs were broad in the HFIP-TEA mixture (Fig. 2a). In contrast, in the TEAA and TEAA-PB mixtures of methanol and acetonitrile, although the buffer conditions were different, somewhat similar separation patterns were obtained; moreover, the peak intensities were considerably larger when these two mobile phases were utilized than when HFIP-TEA was used (Fig. 2b, c). These results indicate that TEAA and TEAA-PB mixtures are suitable for use as mobile phases to realize the separation of the oligomeric constituent of the low-range ssRNAs. Additionally, the chromatograms obtained employing TEAA and TEAA-PB exhibited similar separation patterns, indicating that no RNA adsorption on the column had occurred in either case. Furthermore, the effect of the solvent used as solvent B was evaluated in the presence of the TEAA and TEAA-PB mixtures; evidence indicated that the use of methanol as solvent B tended to afford better separation of short-stranded RNAs between 50 and 300 nt, while the use of acetonitrile afforded better separation of long-stranded RNAs between 300 and 1000 nt. As for the elution order, the nucleic acids were eluted in order of short chain length. In general, hydrophobicity increases as nucleic acid chain length increases. Therefore, it is thought that the sample was eluted from short- to long-stranded RNAs.
Fig. 2.

HPLC chromatograms obtained for a-c low-range and d wide-range ssRNAs using a COSMOSIL RNA-RP1 column (2.0 mm I. D. x 100 mm; particle size, 5 μm). HPLC analysis was performed with a 400 mM HFIP-TEA buffer, b 100 mM TEAA, and c-d 100 mM TEAA-20 mM PB (pH 7.0) using a linear gradient from a 15% to 25%, b 10% to 25% (solvent B; methanol) and 5% to 20% (solvent B; acetonitrile), c 8.5% to 21% (solvent B; methanol) and 3.5% to 16% (solvent B; acetonitrile), and d 14% to 17% (solvent B; methanol) and 7% to 10% (solvent B; acetonitrile) with methanol or acetonitrile over 20 min at a flow rate 0.2 mL/min and a temperature of 65 °C. UV detection was performed at 260 nm
We then compared the separation patterns of low-range and wide-range ssRNAs using TEAA-PB as solvent A and methanol and acetonitrile as solvent B. When methanol was utilized as solvent B, chromatographic peaks due to 5000, 7000, and 9000 nt RNA strands completely overlapped with each other, and all peaks were broad (Fig. 2d). In contrast, when acetonitrile was used as solvent B, although chromatographic peaks of 7000 and 9000 nt RNAs overlapped completely with each other, the 5000 nt RNA peak was separated from the rest (Fig. 2d). Additionally, the peak shapes were also better with acetonitrile than with methanol. The detailed analysis of each peak revealed that methanol shows broad peak shapes compared to acetonitrile, but with a large Δtr (difference in retention time of each peak) values for various RNA lengths (Table 1 and Table 2). From the above, methanol-acetonitrile mixtures were evaluated for separation of various RNA lengths without collapse of peak shape. When a methanol-acetonitrile mixture was used as solvent B, the Δtr values on low-range and wide-range RNAs were lower than methanol but higher than acetonitrile, and the peak shape was similar to that of acetonitrile (Fig. 3a, b and Table 1, 2). Based on this evidence, in the experiments described after this section, TEAA or TEAA-PB were used as solvent A and acetonitrile or methanol-acetonitrile mixture were used as solvent B in the separation of long-stranded RNAs.
Table 1.
Δtr (difference in retention time of each peak) values of low-range ssRNAs
| Δtr(80–50nt) | Δtr(150–80nt) | Δtr(300–150nt) | Δtr(500–300)nt | Δtr(1000–500nt) | |
|---|---|---|---|---|---|
| 100% Methanol | 2.31 | 1.08 | 1.44 | 0.47 | 0.27 |
| Methanol/Acetonitrile (50%/50%) | 1.87 | 1.05 | 1.26 | 0.48 | 0.39 |
| 100% Acetonitrile | 1.39 | 0.94 | 1.05 | 0.45 | 0.40 |
Table 2.
Δtr (difference in retention time of each peak) values of wide-range ssRNAs
| Δtr(1000–500nt) | Δtr(2000-1000nt) | Δtr(3000–2000nt) | Δtr(5000–3000nt) | Δtr(7000–5000nt) | |
|---|---|---|---|---|---|
| 100% Methanol | 1.99 | 4.46 | 1.89 | 2.27 | 0 |
| Methanol/Acetonitrile (50%/50%) | 2.24 | 3.17 | 1.03 | 1.36 | 0.78 |
| 100% Acetonitrile | 1.28 | 2.19 | 0.72 | 0.96 | 0.67 |
Fig. 3.
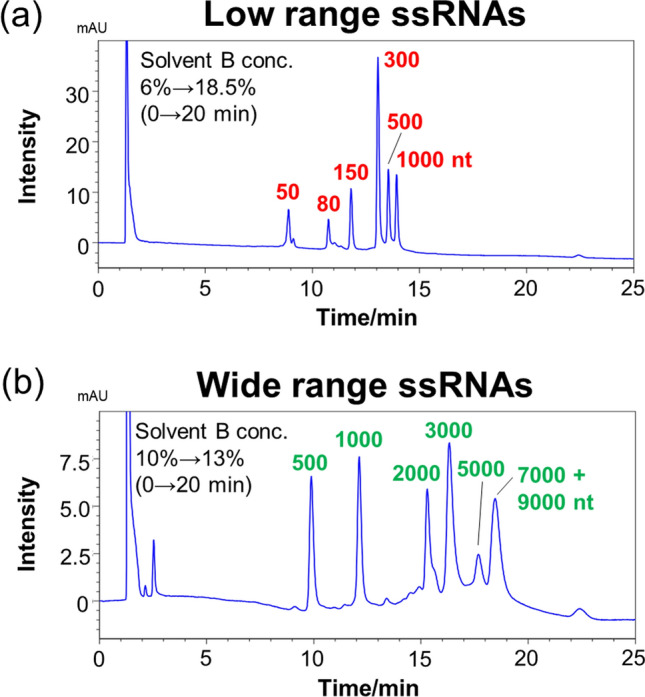
HPLC chromatograms obtained for a low-range and b wide-range ssRNAs using a COSMOSIL RNA-RP1 column (2.0 mm I. D. x 100 mm; particle size, 5 μm). HPLC analysis was performed with 100 mM TEAA-20 mM PB (pH 7.0) using a linear gradient from a 6% to 18.5% and b 10% to 13% with methanol/acetonitrile (50/50, vol/vol) over 20 min at a flow rate of 0.2 mL/min and a temperature of 65 °C. UV detection was performed at 260 nm
Effect of temperature on RNA separation
Subsequently, the separation patterns of low-range ssRNAs and low-range dsRNAs were determined at different temperatures in the 40 °C–70 °C range, based on the consideration that the structure of nucleic acids is affected by temperature. In general, dsDNAs and dsRNAs are more strongly retained than single-stranded oligonucleotides, so single-stranded oligonucleotides elute early in the chromatogram and are well separated from the peaks of dsDNAs and dsRNAs [27]. The column utilized in these experiments was the COSMOSIL RNA-RP1 column, and TEAA was used as the mobile phase. In the case of low-range ssRNAs, the separation pattern did not vary significantly with the temperature (Fig. 4a). However, the peak intensity decreased as the temperature decreased. Nucleic acids longer than 100 nt tended to form structures and under low-temperature conditions, resulting in decreased UV absorption. In the case of low-range dsRNAs, the separation pattern was not significantly different between the situations whereby the separation procedures were conducted at 40 °C and at 60 °C (Fig. 4b). In contrast, when the separation was conducted at 70 °C, the dsRNA-derived peaks that had been observed at 40 °C and 60 °C disappeared, while a new peak due to ssRNAs was observed in the 9.0–12.5 min retention time range (Fig. 4b). These results indicate that at 70 °C, dsRNA underwent denaturation to produce ssRNAs, so that separating these RNA segments while maintaining their double-stranded structure proved difficult.
Fig. 4.

HPLC chromatograms of a low-range ssRNAs and b low-range dsRNAs obtained using a COSMOSIL RNA-RP1 column (2.0 mm I. D. x 100 mm, particle size; 5 μm) at a flow rate of 0.2 mL/min in the 40 °C–70 °C temperature range. HPLC chromatogram of low-range ssRNA and dsRNA mixtures obtained using a COSMOSIL RNA-RP1 column (2.0 mm I.D. x 100 mm, particle size; 5 μm) at a flow rate of 0.2 mL/min and at a temperature of c 60 °C and d 65 °C. HPLC analysis was performed with 100 mM TEAA (pH 7.0) using a linear gradient from 5% to 20% with acetonitrile over 20 min. UV detection was performed at 260 nm
We subsequently attempted to simultaneously separate ssRNAs and dsRNAs at 60 °C; in this case, a good separation was achieved for ssRNAs and dsRNAs. The chromatogram reported in Fig. 4c comprises peaks that appeared at the same retention times as those observed in the chromatograms of individual ssRNAs and dsRNAs mixtures subjected to separation at the same temperature of 60 °C (see Fig. 4a, b, respectively). When the separation was conducted at 65 °C, 80 bp dsRNA was denatured and completely overlapped with the peak of 80 nt ssRNA (Fig. 4d). These results indicate that ssRNAs and dsRNAs can be separated in a non-denatured state by carefully controlling the mobile phase and temperature.
Precise separation of DNA strands of various lengths
Subsequently, the simultaneous separation of nucleic acids of various chain lengths was attempted. We also compared the separation capability of HPLC and gel electrophoresis. A pBR 322 sample digested with MspI (to produce 16 dsDNA segments ranging in size from 67 to 622 bp) and a ΦX174 sample digested with HaeIII (to produce 11 dsDNA segments ranging in size from 72 to 1353 bp) were used as the nucleic acid strands to be separated. For the electrophoresis experiments, agarose gels were selected to separate dsDNAs from tens to thousands of bp in length. All dsDNA strands ranging in length from short to long, except for the 238 and 242 bp-long strands obtained from the MspI-driven digestion of pBR 322, were separated by HPLC (Fig. 5b, d). In contrast, agarose gel electrophoresis did not afford the separation of some dsDNAs with similar strand lengths (Fig. 5a, c). Additionally, the detection sensitivity of short-length dsDNAs (67, 76, and 90 bp) was very low in the case of the electrophoresis-based separation process (Fig. 5a). These results indicate that the separation of nucleic acid strands of various lengths is difficult to realize by agarose gel electrophoresis. Notably, the detection sensitivity of short-length dsDNAs achieved by HPLC-based separation was higher than that achieved by agarose gel electrophoresis-based separation. These results indicate that HPLC can be used to separate samples that are difficult to separate simultaneously by electrophoresis. Moreover, impurities in the synthesized long-length RNAs that cannot be detected by electrophoresis may be detected by HPLC.
Fig. 5.

Agarose gel electrophoresis images of a pBR322/MspI and c ΦX174/HaeIII digests. HPLC chromatograms of b pBR322/MspI and d ΦX174/HaeIII digests obtained using a COSMOSIL RNA-RP1 column (4.6 mm I.D. x 100 mm, particle size; 5 μm). HPLC analysis was performed with 100 mM TEAA (pH 7.0) using a linear gradient from b 10% to 17% and d 12% to 17% with acetonitrile over 20 min at a flow rate of 1.0 mL/min and a temperature of 40 °C. UV detection was performed at 260 nm
Separation of impurities in synthesized long-length RNA
Finally, an RNA-RP1 column was used to confirm the separation of impurities in the synthesized long-length RNAs. Generally, RNA-based vaccines use mRNA strands that are 3000–4000 nt in length. Therefore, we synthesized 3.0 knt and 4.8 knt pQBI T7-GFP RNA by in vitro transcription and went on to separate the impurities in that RNA (Fig. 6a). Electrophoresis was employed to confirm that the pQBI T7-GFP RNA produced by in vitro transcription had been synthesized at high purity (Fig. 6a). When magnifying the HPLC chromatogram of the mixture containing 3.0 knt RNA, some peaks other the main peak, which resulted from impurities, were identified (Fig. 6b). The HPLC chromatogram of the mixture containing 4.8 knt RNA included several peaks from impurities in addition to the main peak, but not as much as 3.0 knt RNA (Fig. 6c). These results indicate that, although gel electrophoresis could not achieve the separation of impurities, HPLC did separate the peaks hypothesized to be due to impurities from the main peak.
Fig. 6.

a Scheme and electrophoresis image of the synthesized 3.0 knt and 4.8 knt pQBI T7-GFP RNA. HPLC chromatograms of b 3.0 knt and c 4.8 knt RNA obtained using a COSMOSIL RNA-RP1 (2.0 mm I.D. x 100 mm; particle size, 5 µm) column. HPLC analysis was performed with 100 mM TEAA-20 mM PB (pH 7.0) using a linear gradient from 11% to 13% with methanol/acetonitrile (50/50, vol/vol) over 20 min at a flow rate of 0.2 mL/min and a temperature of 60 °C. UV detection was performed at 260 nm
Conclusions
Packing materials with super-wide pores are better than non-porous packing materials and porous packing materials containing normal-sized pores for the separation of RNA strands of several hundred nucleotides. TEAA and TEAA-PB were more effective as mobile phases than HFIP-TEA for the separation of RNA strands by several hundred nucleotides in length. Additionally, short RNAs were better separated when methanol was used as solvent B in the HPLC-based separation, while longer RNAs were better separated when acetonitrile or a methanol-acetonitrile mixture was used as solvent B. A temperature of 60 °C for the HPLC purification process was suitable for the separation of ssRNA and dsRNA using TEAA as the mobile phase; moreover, at that temperature, ssRNA and dsRNA could be separated simultaneously without any denaturation of dsRNA being observed. Digested DNA strands characterized by similar lengths, which are difficult to separate by agarose gel electrophoresis, can be separated using the C18-based RNA-RP1 column. Furthermore, impurities present in the 3.0 knt and 4.8 knt pQBI T7-GFP RNA strands synthesized by in vitro transcription, which cannot be separated by electrophoresis, were separated using the RNA-RP1 column. Notably, the RNA strands synthesized in this study were large, which caused difficulties in the identification of impurities using mass spectrometry. Therefore, we are considering the separation and identification of impurities, which should be described in future reports by our group.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declarations
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
The original online version of this article was revised: Ref. 23 and 26 were incorrect.
Tomomi Kuwayama and Makoto Ozaki are equally contributed to this work.
Change history
3/2/2023
A Correction to this paper has been published: 10.1007/s44211-023-00310-y
References
- 1.Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Marc GP, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Jr, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC. the C4591001 Clinical Trial Group. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T. the COVE Study Group. N. Engl. J. Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freyn AW, Silva JR, Rosado VC, Bliss CM, Pine M, Mui BL, Tam YK, Madden TD, Ferreira LCS, Weissman D, Krammer F, Coughlan L, Palese P, Pardi N, Nachbagauer R. Mol. Ther. 2020;28:1569–1584. doi: 10.1016/j.ymthe.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen Y, Kruse V, Corthals J, Schats K, Dam P-J, Seremet T, Heirman C, Brochez L, Kockx M, Thielemans K, Neyns B. Cancer Immunol. Immunother. 2020;69:2589–2598. doi: 10.1007/s00262-020-02618-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim M, Xia Y, Bettegowda C, Weller M. Nat. Rev. Clin. Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 6.Qin S, Tang X, Chen Y, Chen K, Fan N, Xiao W, Zheng Q, Li G, Teng Y, Wu M, Song X. Signal Transduct. Target. Ther. 2022;7:166. doi: 10.1038/s41392-022-01007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Granados-Riveron JT, Aquino-Jarquin G. Biomed. Pharmacother. 2021;142:111953. doi: 10.1016/j.biopha.2021.111953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.M. Heartlein, F. Derosa, A. Dias, S. Karve, WO2014152966A1, 2014.
- 9.Kis Z, Kontoravdi C, Dey AK, Shattock R. J. Adv. Manuf. Process. 2020;2:1–10. doi: 10.1002/amp2.10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franklin RM. Proc. Nat. Acad. Sci. 1966;55:1504–1511. doi: 10.1073/pnas.55.6.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baiersdörfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U, Karikó K. Mol. Ther. Nucleic. Acids. 2019;15:26–35. doi: 10.1016/j.omtn.2019.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petrov A, Wu T, Puglisi EV, Puglisi JD. Meth. Enzymol. 2013;530:315–330. doi: 10.1016/B978-0-12-420037-1.00017-8. [DOI] [PubMed] [Google Scholar]
- 13.Walker SE, Lorsch J. Meth. Enzymol. 2013;530:337–343. doi: 10.1016/B978-0-12-420037-1.00019-1. [DOI] [PubMed] [Google Scholar]
- 14.Easton LE, Shibata Y, Lukavsky PJ. RNA. 2010;16:647–653. doi: 10.1261/rna.1862210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukavsky PJ, Puglisi JD. RNA. 2004;10:889–893. doi: 10.1261/rna.5264804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. J. Chromatogr. A. 1997;777:3–21. doi: 10.1016/S0021-9673(97)00256-2. [DOI] [PubMed] [Google Scholar]
- 17.Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. Anal. Chem. 1997;69:1320–1325. doi: 10.1021/ac960916h. [DOI] [PubMed] [Google Scholar]
- 18.Fritz H-J, Belagaje R, Brown EL, Fritz RH, Jones RA, Lees RG, Khorana HG. Biochemistry. 1978;17:1257–1267. doi: 10.1021/bi00600a020. [DOI] [PubMed] [Google Scholar]
- 19.Fountain KJ, Gilar M, Gebler JC. Rapid Commun. Mass Spectrom. 2003;17:646–653. doi: 10.1002/rcm.959. [DOI] [PubMed] [Google Scholar]
- 20.Bagge J, Enmark M, Leśko M, Limé F, Fornstedt T. J. Chromatogr. A. 2020;1634:461653. doi: 10.1016/j.chroma.2020.461653. [DOI] [PubMed] [Google Scholar]
- 21.Close ED, Nwokeoji AO, Milton D, Cook K, Hindocha DM, Hook EC, Wood H, Dickman MJ. J. Chromatogr. A. 2016;1440:135–144. doi: 10.1016/j.chroma.2016.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner BM, Schuster SA, Boyes BE, Shields TJ, Miles WL, Haynes MJ, Moran RE, Kirkland JJ, Schre MR. J. Chromatogr. A. 2017;1489:75–85. doi: 10.1016/j.chroma.2017.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Z, Jayaseelan S, Hebert J, Seo H, Niu L. Anal. Biochem. 2013;435:35–43. doi: 10.1016/j.ab.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wetzel C, Limbach PA. Analyst. 2016;141:16–23. doi: 10.1039/C5AN01797A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nwokeoji AO, Kumar S, Kilby PM, Portwood DE, Hobbs JK, Dickman MJ. Analyst. 2019;144:4985–4994. doi: 10.1039/C9AN00954J. [DOI] [PubMed] [Google Scholar]
- 26.Yamauchi Y, Taoka M, Nobe Y, Izumikawa K, Takahashi N, Nakayama H, Isobe T. J. Chromatogr. A. 2013;1312:87–92. doi: 10.1016/j.chroma.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy SM, Gilar M, Gebler J. Anal. Biochem. 2009;390:181–188. doi: 10.1016/j.ab.2009.03.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.