

Abstract
The incidence of low back pain caused by lumbar disc degeneration is high, and it can lead to loss of work ability and impose heavy social and economic burdens. The pathogenesis of low back pain is unclear, and there are no effective treatments. With age, the deposition of advanced glycation end products (AGEs) in intervertebral disc (IVD) gradually increases and is accelerated by diabetes and a high-AGEs diet, leading to destruction of the annulus fibrosus (AF), nucleus pulposus (NP), and cartilage endplate (CEP) and finally intervertebral disc degeneration (IDD). Reducing the accumulation of AGEs in IVD and blocking the transmission of downstream signals caused by AGEs have a significant effect on alleviating IDD. In this review, we summarize the mechanism by which AGEs induce IDD and potential treatment strategies.
1. Introduction
Lumbar intervertebral disc degeneration is considered to be the main cause of low back pain [1]. Low back pain refers to the pain in the lumbar spine. If the spinal cord or nerve roots are compressed, discomfort such as pain can also affect the lower limbs. In severe cases, the patient's mobility may be impaired. Low back pain is currently the most common musculoskeletal disease, with approximately 80% of the world's population experiencing low back pain at some point in their lives [2, 3], and it is the main reason why adults see physicians [4]. It is estimated that more than 600 million patients worldwide have low back pain, imposing considerable social and economic burdens [4, 5]. It is thus important to clarify the mechanism of intervertebral disc degeneration (IDD) and develop effective treatments.
The intervertebral disc (IVD) is the fibrocartilage tissue connecting the vertebral bodies of the spine and is comprised of the annulus fibrosus (AF), nucleus pulposus (NP), and cartilage endplate (CEP). The IVD plays an important role in bearing the impact of body movement, absorbing shocks, and distributing mechanical loads along the spine [6, 7]. The AF is composed of a ring rich in type I collagen, which is wound in a highly orderly manner around the NP. The collagen fibers are arranged at alternating angles to resist the circumferential stress on the NP during bending and twisting of the body and prevent its lateral displacement and collapse [8, 9]. The NP is located in the center of the IVD and is comprised of about 80% water, as a result of the permeability gradient of proteoglycan. The NP also contains an arrangement frame composed of type II collagen and elastin fibers, which combines with proteoglycans and transmits compressive stress to the AF and CEP [8, 10]. The CEP is the separation interface between the IVD and adjacent vertebral bodies and is mainly composed of a transparent hyaline cartilage matrix similar to joint cartilage [11]. IVD is nonvascular tissue. The transport of nutrients and metabolic wastes depends on the osmotic capacity of the CEP. The cell density of the NP is low (~3000/mm3) [12, 13]. The IVD's lack of perfusion and low cell density make it vulnerable to damage, and it has limited potential to repair such damage, causing accumulation of metabolic waste [14].
IDD is defined as the accumulation of degenerative factors leading to inappropriate cellular responses, which aggravates disease development and leads to loss of biological structural support and function [15]. Factors such as age, smoking, infection, biomechanical abnormalities, and malnutrition are implicated in IDD [16, 17]. In IDD, proinflammatory factors secreted by NP and AF cells, macrophages, T cells, and neutrophils [18–20] trigger a series of pathogenic reactions of IVD cells, promoting autophagy, senescence, and apoptosis [17, 21, 22]. IDD is characterized by changes in biochemical components and the resulting loss of biomechanical properties [23, 24], leading to a series of spinal diseases (such as intervertebral disc herniation and spinal stenosis).
Advanced glycation end products (AGEs) are metabolic derivatives of nonenzymatic reactions that occur between reducing sugars, free amines (mainly protein α-NH2 or ε-NH2 groups), and amino groups (from lipids and nucleic acids) [25]. AGEs modified by proteins undergo structural changes due to charge changes and cross-linking formation. This affects enzyme activity and function [26]. Therefore, the irreversible formation of AGEs results in their accumulation, especially in the presence of longevity proteins (e.g., collagen, serum albumin, and lens crystal) [27], leading to tissue damage and degeneration. AGEs are implicated in diabetes [28], cardiovascular diseases [29], kidney diseases [30], neurodegenerative diseases [27], and some cancers [31]. Here, we review the role of AGEs in IDD, focusing on the molecular mechanisms and therapeutic potential.
2. Advanced Glycation End Products (AGEs)
AGEs are a group of heterogeneous compounds formed by the nonenzymatic reactions. They are formed by the nonenzymatic glycation of free amino groups of proteins, lipids, and nucleic acids, mainly via reducing sugars and reactive aldehydes [32, 33]. The intermediate steps in AGE formation involve a series of rearrangement and cyclization reactions [34]. The first is the nonenzymatic reaction of a reducing sugar and an amino group to produce an unstable Schiff base. This reaction is reversible until the equilibrium is reached. The unstable Schiff base undergoes rearrangement to form a more stable Amadori product [35]. The Amadori products undergo a series of reactions, rearrangements, and dehydrations to produce highly reactive dicarbonyl compounds, such as methylglyoxal (MG), glyoxal (GO), or deoxyglucone (1-deoxyglucone [1-DG] and 3-deoxyglucone [3-DG]) [35–37]. Carbonyl stress is caused by continuous accumulation of dicarbonyl compounds [38]. These dicarbonyl compounds undergo oxidation, dehydration, and cyclization reactions to form AGEs [27, 39, 40] (Figure 1).
Figure 1.
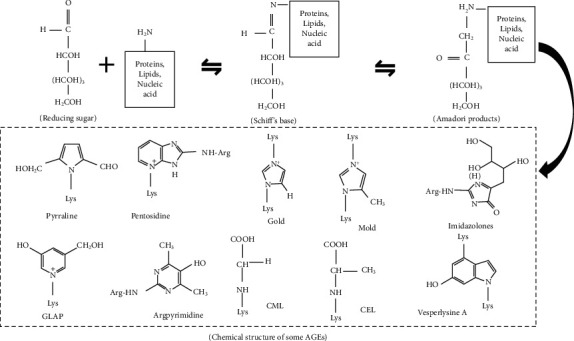
The general process of AGEs formation. CML: N-carboxymethyllysine; CEL: N-carboxyethyllysine; GOLD: glyoxal-lysine dimer; MOLD: methylglyoxal-lysine dimer; GLAP: glyceraldehyde-derived pyridinium compound.
AGEs may originate from exogenous sources, such as daily diet and smoking. A large number of AGEs are produced in food processing, particularly baking, frying, and barbecuing, and some are found in raw animal-derived food [41, 42]. Nɛ-Carboxymethyllysine (CML), pentosidine, methylglyoxal-lysine dimers (MOLD), and pyrrolidine are common AGEs in food [43]. AGEs are related to several age-related diseases [44]. Eating habits are an important variable, and only about 10% of ingested AGEs are absorbed and distributed in the tissues [45, 46]. More than 70% of AGEs escape absorption, because their cross-linking renders them resistant to enzymatic or acid hydrolysis [47]. Compared with nonsmokers, the serum AGEs levels of smokers are significantly higher [48], and AGEs formation from tobacco glycotoxins requires only a few hours [49].
AGEs are chemically modified proteins, lipids, or nucleic acids with stable chemical properties. AGEs may exert their effects as follows: (1) glycosylated proteins act as ligands to activate cell membrane receptors (such as RAGE), leading to oxidative stress and inflammation [50]; (2) glycosylated proteins form cross-links with other proteins, altering their activities and hardness [51]; and (3) saccharifying agents saccharify proteins, affecting their biological functions [32]. AGEs can also spread free radical reactions, thereby further damaging proteins, lipids, and/or nucleic acids [27].
More than 20 AGEs have been found in human blood, tissues, and food [52]. According to their chemical structure and fluorescence, they can be divided into four categories [52]: (1) fluorescent and cross-linked, (2) nonfluorescent and non-cross-linked, (3) nonfluorescent protein cross-linked, and (4) fluorescent non-cross-linked. The characteristics of several AGEs are listed in Table 1.
Table 1.
Classification of some AGEs based on chemical structure and ability to fluoresce.
| AGE compound | Cross-linking | Fluorescence |
|---|---|---|
| Pyrraline | No | No |
| N-Carboxymethyllysine (CML) | No | No |
| N-Carboxyethyllysine (CEL) | No | No |
| Imidazolones | No | No |
| Glyoxal-lysine dimer (GOLD) | Yes | No |
| Methylglyoxal-lysine dimer (MOLD) | Yes | No |
| Pentosidine | Yes | Yes |
| Argpyrimidine | Yes | Yes |
| Vesperlysine A | Yes | Yes |
3. Effect of AGEs on IVD
3.1. Nucleus Pulposus Cells (NP Cells)
NP cells are the main functional cells of the NP. The young, healthy human IVD contains notochord cells, which originate from the vacuolar cells of the embryonic notochord, and NP cells. The latter are small spherical cells with a unique phenotype similar to articular chondrocytes but express specific markers (e.g., ovos2, CA12, CD24, HIF-1α, and cytokeratin 8/18/19) [53–55]. The cell density of the NP is low (~3000/mm3) [12, 13]. NP cells synthesize and secrete extracellular matrix (ECM) rich in proteoglycan, type II collagen, and hyaluronic acid (HA), which maintains the osmotic pressure, and so also the biomechanical properties, of the spine [56].
AGEs accumulation in the IVD increases with age [57]. AGEs reduce the viability of NP cells by a variety of mechanisms, affect their proliferation, and promote their apoptosis [58]. The accumulation of AGEs in IVD tissue affects endoplasmic reticulum (ER) homeostasis [59]. ER-phagy is a type of selective autophagy. Some ER fragments are phagocytized by autophagosomes via specific receptors and transported to lysosomes for degradation, to restore the cellular energy level and ER homeostasis [60]. AGEs can trigger the accumulation of reactive oxygen species (ROS) in NP cells, activating ER-phagy mediated by FAM134B (a mammalian ER-phagy receptor). The overexpression of FAM134B alleviates ROS accumulation, apoptosis, and senescence in AGEs-treated NP cells [59]. The ER is responsible for protein synthesis, maturation, and quality control. Genetic and environmental pressures affect protein folding in the ER, resulting in the accumulation of unfolded/misfolded proteins, i.e., ER stress. Continuous ER stress can trigger cell self-destruction [61, 62]. AGEs induced a persistent increase of cytosolic Ca2+ ([Ca2+]c) and depletion of ER cavity Ca2+ ([Ca2+]er) in NP cells in a concentration- and time-dependent manner, resulting in ER stress [62]. AGEs alter the activity of ER Ca2+ channels in NP cells, including 1,4,5-triphosphate receptor channels (IP3R) and ryanodine receptor channels (RyR), and ER Ca2+-reuptake pumps, such as sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [63–65]. Pharmacological blockade of ER Ca2+ release by Ca2+ antagonists can improve the Ca2+ imbalance, ER stress, and apoptosis in NP cells and reduce the progression of IDD in vivo [63]. In addition, mesenchymal stem cell-derived exosomes (MSC-exos) can prevent the apoptosis of NP cells induced by ER stress by activating the AKT and ERK signaling pathways [63]. AGEs induce ER stress and activate the unfolded protein response (UPR) via key transmembrane proteins in the ER [66, 67]. After initiation of the UPR, the downstream C/EBP homologous protein (CHOP) is transcriptionally activated and controls the expression of apoptosis-related genes, thus inducing apoptosis under severe ER stress [68]. MSC-exos regulate ER stress by modulating the UPR and CHOP expression [69].
Because it constitutes the largest avascular tissue, anaerobic glycolysis is considered the main pathway of energy metabolism in the IVD [14, 70]. The NP is a hypoxic tissue due to a lack of vascularization [71], so NP cells stably express hypoxia-inducible factor-1α (HIF-1α) [72]. HIF-1α interacts with transcriptional coactivators, including p300/CBP, to upregulate genes such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH), glucose transporter (GLUT)-1, and GLUT-3, thus driving glycolytic metabolism [73–75]. HIF-1α expression is a biomarker of normal NP cells [72]. Treatment of NP cells with AGEs impaired the stability of HIF-1α. Following AGEs treatment, the receptor for activated C-kinase 1 (RACK1) competes with heat shock protein 90 (HSP90) for binding to HIF-1α, resulting in posttranslational HIF-1α degradation and RACK1-mediated proteasomal degradation, independently of the canonical iron-dependent prolyl-hydroxylase domain- (PHD-) mediated degradation pathway. Under normoxic conditions, PHD proteins promote HIF-1α degradation by the 26S proteasome [76, 77]. The degradation of HIF-1α disrupts the biological function of NP cells and promotes IDD.
The IVD generates energy by anaerobic sugar degradation, which has nothing to do with mitochondrial pathway, but mitochondria may be involved in the adaptive changes in the metabolic process of NP cells [78, 79]. The mitochondrial pathway regulates apoptosis via changes in mitochondrial membrane permeability and release of proapoptotic proteins [80]. AGEs increase the production of mitochondrial ROS, prolong the activation time of mitochondrial permeability transition pores, increase the level of mitochondrial Bax, decrease the Bcl-2 level, increase the intercellular ROS level, and promote the apoptosis of NP cells. This may involve functional impairment of SIRT3 (an NAD+-dependent deacetylase, which has deacetylase activity and maintains mitochondrial redox homeostasis and functional integrity) [58, 81, 82]. The above-mentioned changes in NP cells caused by AGEs can be rescued by nicotinamide mononucleotide (NMN) through the adenosine monophosphate-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) pathway, which restores SIRT3 function [58]. AGEs treatment of NP cells leads to mitochondrial dysfunction, which may involve mitochondrial quality control pathways [83]; this warrants further investigation.
In conclusion, ER stress/phagy and mitochondrial dysfunction are involved in AGEs-induced apoptosis of NP cells (Figure 2), and epigenetic modification may also be implicated [84, 85]. Therefore, several pathways are involved in AGEs-mediated damage to NP cells.
Figure 2.
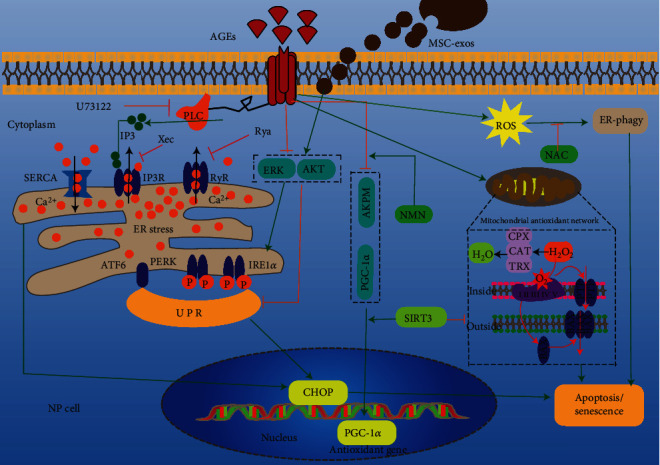
Mechanisms of AGEs-induced apoptosis (or senescence) of nucleus pulposus cells through endoplasmic reticulum and mitochondrial pathways. AGEs: advanced glycation end products; IP3: inositol 1,4,5-trisphosphate; PLC: phospholipase C; Rya: ryanodine; Xec: xestospongin C; RyR: ryanodine receptor; IP3R: inositol 1,4,5-triphosphate receptor (U73122, xec, and RYA are calcium antagonists of PLC, IP3R, and RyR, respectively); SERCA: sarco/endoplasmic reticulum Ca2+-ATPase; UPR: unfolded protein response; ROS: reactive oxygen species; CHOP: C/EBP homologous protein; ATF6: activating transcription factor 6; PERK: protein kinase-like endoplasmic reticulum kinase; IRE1α: inositol-requiring protein 1α; MSC-exos: mesenchymal stem cells-exosomes; AKT: protein kinase B; ERK: extracellular regulated protein kinases; AMPK: adenosine monophosphate-activated protein kinase; PGC-1α: peroxisome proliferator-activated receptor-γ coactivator 1α; NMN: nicotinamide mononucleotide; SIRT3: Sirtuin3; NAC: N-acetyl-L-cysteine; PTP: permeability transition pore; ER stress: endoplasmic reticulum stress; ER-phagy: endoplasmic reticulum-phagy.
3.2. ECM Metabolism
Proteoglycan and type II collagen are the main components of the ECM of the IVD, and they maintain its osmotic pressure. Indeed, they are the material basis of the biomechanical properties of the IVD [56]. ECM catabolism and anabolism are in dynamic balance in the healthy IVD. However, in IDD, ECM catabolism is greater than anabolism [86]. Matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)—including MMP-2, MMP-3, MMP-9, MMP-13, ADAMTS-4, and ADAMTS-5—are the main catabolic enzymes in the NP [87, 88]. Decreased synthesis and increased catabolism of NP cells are important in ECM degradation. AGEs accumulation increases with age [57], triggering catabolism in IVD cells [89].
Immunohistochemistry confirmed the existence of AGEs and the receptor for advanced glycation end products (RAGE) in degenerating IVDs of human and oxtail. AGEs bind to RAGE and inhibit aggrecan secretion, which may be related to an inflammatory environment [89, 90]. The formation of endogenous AGEs occurs slowly during normal aging and is partly driven by sugar. In diabetes, the increase of blood sugar will accelerate the accumulation of AGEs [91]. The accumulation of AGEs in NP initiates the increase of MMP-2 expression related to ERK signaling pathway and promotes ECM decomposition [88]. Bromodomain-containing protein 4 (BRD4) passes MAPK and NF-κB signals and activates autophagy by upregulating MMP-13 in the diabetic IVD. Inhibition of BRD4 prevented ECM degradation in diabetic rats [88]. ADAMTS-5 and MMP-13 were upregulated in diabetic mice, and anti-inflammatory (pentosan polysulfate) and AGEs inhibitors (pyridoxamine) were effective [92]. The glycosaminoglycan (GAG) content in the IVD of diabetes rats is decreased significantly, which is related to endplate sclerosis and AGEs [93]. However, its relationship with the expression of catabolic enzymes in the ECM is unclear. In the IVD, AGEs accumulate mainly in long-lived proteins (e.g., aggrecan and collagen), which are chemically modified to prevent their repair and renewal [57]. The result is upregulation of matrix catabolic enzymes, promotion of ECM degradation, and acceleration of IDD (Figure 3).
Figure 3.
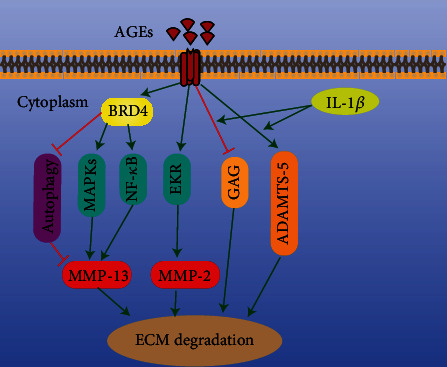
Mechanism of AGEs-induced ECM degradation. AGEs: advanced glycation end products; BRD4: bromodomain-containing protein 4; ERK: extracellular regulated protein kinase; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor kappa-B; GAG: glycosaminoglycan; ADAMTS-5: a disintegrin and metalloproteinase with thrombospondin motif-5.
3.3. Annulus Fibrosus(AF)
The AF is composed of 15–25 0.14–0.52 mm thick layers of fiber bundles arranged in a cross [94]. The AF comprises 20% proteoglycan and 60% collagen [95], mainly type I collagen arranged as concentric rings. The AF can be divided into the external AF, mainly composed of type I collagen fibers with high tensile strength, and the internal AF, which is the transitional area between the external AF and NP and has low density and little tissue [96]. AF cells have the characteristics of mesenchymal-derived long fibroblasts [97]. AGEs inhibit the proliferation and induce the apoptosis of AF cells [98]. AGEs not only significantly upregulated proapoptotic Bax and downregulated antiapoptotic Bcl-2 in AF cells but also promoted the release of cytochrome c (Cyto-c) from mitochondria to cytoplasm. This results in activation of caspase-9 and caspase-3, increases the level of reactive oxygen species (ROS), and reduces the mitochondrial membrane potential. These effects are reversed by the antioxidant, N-acetyl-L-cysteine (NAC) [98]. Therefore, the mitochondrial pathway is involved in age-induced apoptosis of AF cells (Figure 4).
Figure 4.
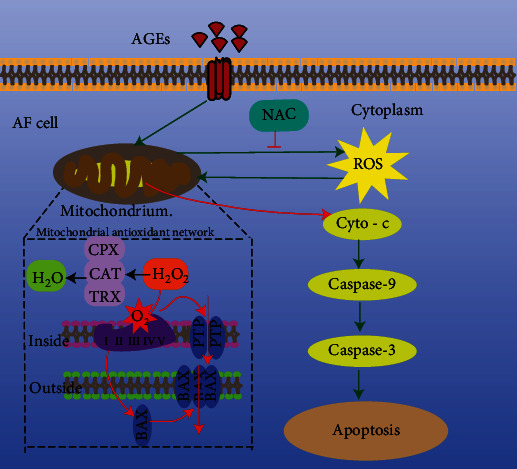
Mechanisms of AGEs-induced apoptosis of AF cells through mitochondrial pathways. AGEs: advanced glycation end products; NAC: N-acetyl-L-cysteine; ROS: reactive oxygen species; Cyto-c: cytochrome c.
Collagen fibers in the AF are arranged at alternating angles to resist the circumferential stress of the NP and prevent its lateral displacement and collapse [8, 9]. Animal experiments showed that a high-AGEs diet can lead to marked accumulation of AGEs in the IVD. Two-photon imaging indicated that collagen damage in the fiber ring was increased by a high-AGEs diet, and the damage was significantly greater in females than in males [99]. Another study using a similar method showed that dietary AGEs increase destruction of collagen fibers and decrease the total collagen level in a RAGE-dependent manner, which was caused by catabolic processes other than cross-linking [100]. However, it is puzzling and interesting that the latter study is not gender dependent under basically similar research conditions. The researchers think that the possible causes are as follows: AGEs may involve age receptor interacting with estrogen [99]; the local effects of AGEs on AF collagen may be gender independent, while other spinal tissues and characteristics have age-related and sex-dependent effects [100]. Whether the effect of AGEs on AF collagen is gender dependent is controversial, warranting further research. In summary, AGEs accumulation in the IVD damages AF collagen, promotes collagen degradation, and is thus implicated in IDD.
3.4. Cartilage Endplates (CEP)
The IVD is connected to the adjacent vertebral body via the cartilage endplates (CEPs) on the upper and lower sides. The IVD is the largest avascular tissue, and its transportation of nutrients and metabolic waste depends on infiltration of the CEP [101–103]. Any factor that affects infiltration of the CEP may trigger IVD and ultimately IDD. The increase of blood glucose will accelerate the accumulation of AGEs [91]. In diabetic nonobese mice, micro-X-ray computed tomography showed that CEP thickness increased by 21% and its porosity decreased by 41%. The change of CEP microarchitecture was significantly correlated with the GAG content, and both oxidative stress and RAGE expression increased [93]. Activation of the AGEs/RAGE axis may play a role in AGEs-mediated CEP sclerosis. The level of oxidative stress is increased by AGEs-induced pathological changes in target sites (e.g., the NP, AF, and CEP), implicating the mitochondrial pathway in the pathogenic effects of AGEs.
Chronic (18 months of postweaning) nondiabetic mice on a high-AGEs diet developed ectopic calcification of the CEP and hypertrophy of NP cells (increased expression of COL-X) [104]. In cadaveric specimens, ectopic calcification of the CEP was found in IVDs at different stages of degeneration and was colocalized with methylglyoxalhydroimidazolone-1- (MG-H1-) positive cells. MG-H1 also colocalized with collagen 10 (COL10) and osteopontin (OPN) [105]. This implicates AGEs in CEP calcification and the osteogenic differentiation of NP cells. Similar results were found in bovine tail and cadaveric NP cells, and the AGEs/RAGE axis may be involved in AGEs-induced hypertrophy and osteogenic differentiation of NP cells [105]. Therefore, RAGE has potential as a therapeutic target.
CEP is the main channel for material exchange between the IVD and the body. CEP calcification results in cell loss and lacunar occlusion, restricting the diffusion of nutrients and metabolic waste [14]. This leads to a decrease in glucose concentration and an increase in metabolic waste in the IVD [14], inducing degenerative changes. CEP sclerosis restricts the entry of drugs and biological agents into the IVD from the blood, influencing IVD-related diseases. AGEs promote CEP thickening and calcification [93]. Reducing AGEs deposition in the IVD and blocking the related signal pathway could ameliorate CEP calcification and reduce the level of metabolic waste in the IVD. In an in vitro experiment, MMP-8 treatment of the cadaveric lumbar CEP reduced the sulfated GAG and local collagen levels and altered collagen structure, thereby improving the diffusion of a small solute (376 Da). Also, the effect of MMP-8 was negatively correlated with AGEs content [106]. There is a marked difference between the in vitro cadaver experiment and the in vivo environment. Cellular electrical activities and the osmotic pressure between tissues need to be considered.
3.5. Inflammation
A painful IDD is in a chronic inflammatory state, and proinflammatory cytokines are upregulated in symptomatic IDD [107–109]. Age, smoking, infection, biomechanical abnormalities, and malnutrition can result in abnormal IVD cells and the production of cytokines and catabolic factors [17, 109–112]. Although the importance of these factors is unclear, they decrease the water signal of the IVD on T2-weighted MRI, known as black disc, as well as inflammation and NP herniation [109]. However, AGEs accumulation is also associated with increased inflammation [113]. AGEs accumulation in the IVD leads to increased expression of the proinflammatory factor TNF-α [92], which is associated with disc herniation and nerve stimulation and ingrowth [114, 115]. Intradiscal injection of AGEs in mice increased IL-23 expression and decreased the level of the anti-inflammatory cytokine IL-10 [84]. This implicates AGEs in the development of intradiscal inflammation. High mobility group box 1 (HMGB1) and IL-1β regulate the release of inflammatory factors from the degenerated IVD via RAGE. Because it is a multiligand receptor [116], it is uncertain whether RAGE is associated with AGEs. Cytokines are important players in IDD [109], and further work should focus on their regulation by AGEs in IDD.
3.6. Discogenic and Radicular Pain
Discogenic low back pain and radicular pain caused by IDD are common musculoskeletal diseases of unknown pathogenesis with no effective treatment. The serum methylglyoxal (MG) level in patients with lumbar disc herniation (LDH) with pain is higher than in painless or normal volunteers and is correlated with the visual analog scale score [117]. In an NP implantation animal model, simulated lumbar disc herniation increased the MG level in serum and the dorsal root ganglion (DRG), leading to mechanical pain and increased DRG neuron activity. This is accompanied by a decrease in the activity of glyoxalase 1 (catalyzes MG hydrolysis). The MG scavenger aminoguanidine can reduce MG accumulation in the DRG and ameliorate the mechanical pain caused by NP implantation and enhanced DRG neuron activity [117]. Also, activation of the RAGE/STAT3 pathway is key in LDH-induced persistent pain and so may be a therapeutic target [118]. Diabetic mice showed prolonged radicular pain-related behaviors, likely to be associated with prolonged inflammation and nerve regeneration under diabetic conditions [118].
AGEs accumulation destroys the normal structure of the IVD [92], leading to diseases such as intervertebral disc herniation. In disc herniation, CD68+ macrophages, neutrophils, and T cells (CD4+ and CD8+) migrate into the disc due to the ingrowth of blood vessels [119, 120]; nerve fibers from the DRG also invade IVD tissue [121, 122]. The stimulation of nerve fibers by components of the IVD may cause discogenic and/or radicular pain. Although AGEs destroy the integrity of the IVD structure, it may not only be the initiating factor in discogenic and radicular pain but also may be a potential therapeutic target in terms of its impact on the excitability of the DRG.
3.7. Intervertebral Disc Biomechanics
AGEs accumulation in the IVD gradually increases with age. A high-AGEs diet led to AGEs accumulation in the IVD, increasing IVD compressive stiffness, torque range, and torque to failure; these effects were more pronounced in females and were attributed to a marked increase in AGEs cross-linking in the AF [98]. AGEs accumulation also leads to collagen damage but does not affect its biomechanical properties and induces disc degeneration [99]. A high-fat diet can cause structural damage to the spine, and the degree of damage differs by gender [123]. Gender dependence is thus characteristic of the effects of several factors on spinal injury, but the mechanism is unclear.
Diabetes accelerates AGEs accumulation due to hyperglycemia [91]. In diabetic mice, AGEs accumulation resulted in a 97% increase in disc hardness [93]. In cadaver specimens, AGEs increased IVD hardness and AF mechanical stiffness, altering the biomechanical properties of the IVD [124]. AGEs reduce the water content of the AF and NP, which are related to the mechanical properties of the IVD, in a dose-dependent manner [125]. Although hyperglycemia in diabetes can lead to AGEs accumulation and destruction the of IVD, in the early stage of diabetes, especially in young patients, hyperglycemia may precede AGEs and cause changes in the structure of IVD. These changes may explain the development of IDD in patients with late-stage diabetes [126]. In general, with the increase of age, AGEs accumulate in the IVD, promoting collagen cross-linking and structural changes, decreasing metabolism, increasing catabolism, destroying the normal structure, and degrading the biomechanical properties of the IVD.
4. Conclusions and Perspectives
The integrity of the IVD is the necessary basis for its biomechanical properties. Any factor that causes structural changes will interfere with the normal function of the IVD. The accumulation of AGEs in IVD has caused extensive damage to various structures of IVD, including NP, AF, and CEP, resulting in the occurrence of IDD and becoming the basis of spinal degenerative diseases such as disc herniation and spinal stenosis. AGEs derived from endogenous and exogenous pathways (such as daily diet and smoking) can be deposited in IVD through the CEP and play a pathogenic role through the mitochondrial pathway, ER pathway, AGEs-RAGE axis, etc. The effect of high-AGEs diet on IVD may be gender dependent, and the effect on women is more obvious, but it is controversial. AGEs inhibitor treatment has a relatively clear effect on improving lesions. However, the route of administration must be considered. The operability of in vitro and in vivo tests is different, and oral administration may be more acceptable. However, CEP sclerosis may be unfavorable to the absorption of drugs and biological agents. There are few studies on improving permeability in patients with endplate sclerosis, and there is no effective treatment at present. There are many factors leading to IDD, and it is not clear which is relatively important at present. Existing studies show that AGEs have a wide impact on IVD and may be one of the important factors. Although we have a certain understanding of its pathogenic mechanism, there are still few studies on the whole, and some of them are controversial. There may be interference between different mechanisms. The underlying mechanism still needs to be further revealed to find potential molecular targets for effective treatment.
Acknowledgments
This work is supported by the Gansu Province Health Industry Scientific Research Program (No. GSWSKY-2019-48) and Gansu Youth Science and Technology Fund Program, China (No. 21JR11RA198).
Contributor Information
Xuewen Kang, Email: ery_kangxw@lzu.edu.cn.
Bing Ma, Email: mabing-2005@163.com.
Data Availability
Contact the corresponding author to obtain relevant data.
Conflicts of Interest
The authors declare no competing interests.
Authors' Contributions
Fengguang Yang, Daxue Zhu, Zhaoheng Wang, Yingping Ma, and Liangzeng Huang contributed equally to this work, and all authors contributed to the revision and approved the submitted version.
References
- 1.Frapin L., Clouet J., Delplace V., Fusellier M., Guicheux J., Le Visage C. Lessons learned from intervertebral disc pathophysiology to guide rational design of sequential delivery systems for therapeutic biological factors. Advanced Drug Delivery Reviews . 2019;149-150:49–71. doi: 10.1016/j.addr.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Borenstein D. Mechanical low back pain--a rheumatologist's view. Nature Reviews Rheumatology . 2013;9(11):643–653. doi: 10.1038/nrrheum.2013.133. [DOI] [PubMed] [Google Scholar]
- 3.Kalkidan Hassen Abate C. A., Abbas K. M., HALE Collaborators Global, Regional, and National Disability-Adjusted Life-Years (DALYs) for 333 Diseases and Injuries and Healthy Life Expectancy (HALE) for 195 Countries and Territories, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. The Lancet . 2017;390(10100):1260–1344. doi: 10.1016/S0140-6736(17)32130-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katz J. N. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. The Journal of Bone and Joint Surgery American Volume . 2006;88(Supplementray 2):21–24. doi: 10.2106/JBJS.E.01273. [DOI] [PubMed] [Google Scholar]
- 5.March L., Smith E. U., Hoy D. G., et al. Burden of disability due to musculoskeletal (MSK) disorders. Best Practice & Research Clinical Rheumatology . 2014;28(3):353–366. doi: 10.1016/j.berh.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Z., Gao M., Wei F., et al. Shock absorbing function study on denucleated intervertebral disc with or without hydrogel injection through static and dynamic biomechanical tests in vitro. BioMed Research International . 2014;2014:7. doi: 10.1155/2014/461724.461724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Setton L. A., Chen J. Cell mechanics and mechanobiology in the intervertebral disc. Spine . 2004;29(23):2710–2723. doi: 10.1097/01.brs.0000146050.57722.2a. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro I. M., Vresilovic E. J., Risbud M. V. Is the spinal motion segment a diarthrodial polyaxial joint: what a nice nucleus like you doing in a joint like this? Bone . 2012;50(3):771–776. doi: 10.1016/j.bone.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambard D., Cherblanc F. Mechanical behavior of annulus fibrosus: a microstructural model of fibers reorientation. Annals of Biomedical Engineering . 2009;37(11):2256–2265. doi: 10.1007/s10439-009-9761-7. [DOI] [PubMed] [Google Scholar]
- 10.Jahnke M. R., McDevitt C. A. Proteoglycans of the human intervertebral disc. Electrophoretic heterogeneity of the aggregating proteoglycans of the nucleus pulposus. The Biochemical Journal . 1988;251(2):347–356. doi: 10.1042/bj2510347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nachemson A., Elfström G. Intravital dynamic pressure measurements in lumbar discs. A study of common movements, maneuvers and exercises. Scandinavian Journal of Rehabilitation Medicine Supplement . 1970;1:1–40. [PubMed] [Google Scholar]
- 12.Fontana G., See E., Pandit A. Current trends in biologics delivery to restore intervertebral disc anabolism. Advanced Drug Delivery Reviews . 2015;84:146–158. doi: 10.1016/j.addr.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Maroudas A., Stockwell R. A., Nachemson A., Urban J. Factors involved in the nutrition of the human lumbar intervertebral disc: cellularity and diffusion of glucose in vitro. Journal of Anatomy . 1975;120(1):113–130. [PMC free article] [PubMed] [Google Scholar]
- 14.Urban J. P., Smith S., Fairbank J. C. Nutrition of the intervertebral disc. Spine . 2004;29(23):2700–2709. doi: 10.1097/01.brs.0000146499.97948.52. [DOI] [PubMed] [Google Scholar]
- 15.Vo N. V., Hartman R. A., Patil P. R., et al. Molecular mechanisms of biological aging in intervertebral discs. Journal of Orthopaedic Research . 2016;34(8):1289–1306. doi: 10.1002/jor.23195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urban J. P., Roberts S. Degeneration of the intervertebral disc. Arthritis Research & Therapy . 2003;5(3):120–130. doi: 10.1186/ar629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts S., Evans H., Trivedi J., Menage J. Histology and pathology of the human intervertebral disc. The Journal of Bone and Joint Surgery American Volume . 2006;88(Supplementary 2):10–14. doi: 10.2106/JBJS.F.00019. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto J., Maeno K., Takada T., et al. Fas ligand plays an important role for the production of pro-inflammatory cytokines in intervertebral disc nucleus pulposus cells. Journal of Orthopaedic Research . 2013;31(4):608–615. doi: 10.1002/jor.22274. [DOI] [PubMed] [Google Scholar]
- 19.Rand N., Reichert F., Floman Y., Rotshenker S. Murine nucleus pulposus-derived cells secrete interleukins-1-beta, -6, and -10 and granulocyte-macrophage colony-stimulating factor in cell culture. Spine . 1997;22(22):2598–2601. doi: 10.1097/00007632-199711150-00002. discussion 2602. [DOI] [PubMed] [Google Scholar]
- 20.Kepler C. K., Markova D. Z., Hilibrand A. S., et al. Substance P stimulates production of inflammatory cytokines in human disc cells. Spine . 2013;38(21):E1291–E1299. doi: 10.1097/BRS.0b013e3182a42bc2. [DOI] [PubMed] [Google Scholar]
- 21.Purmessur D., Walter B. A., Roughley P. J., Laudier D. M., Hecht A. C., Iatridis J. A role for TNFα in intervertebral disc degeneration: a non-recoverable catabolic shift. Biochemical and Biophysical Research Communications . 2013;433(1):151–156. doi: 10.1016/j.bbrc.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen C., Yan J., Jiang L. S., Dai L. Y. Autophagy in rat annulus fibrosus cells: evidence and possible implications. Arthritis Research & Therapy . 2011;13(4):p. R132. doi: 10.1186/ar3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter B. A., Torre O. M., Laudier D., Naidich T. P., Hecht A. C., Iatridis J. C. Form and function of the intervertebral disc in health and disease: a morphological and stain comparison study. Journal of Anatomy . 2015;227(6):707–716. doi: 10.1111/joa.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scharf B., Clement C. C., Yodmuang S., et al. Age-related carbonylation of fibrocartilage structural proteins drives tissue degenerative modification. Chemistry & Biology . 2013;20(7):922–934. doi: 10.1016/j.chembiol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vlassara H., Striker G. E. AGE restriction in diabetes mellitus: a paradigm shift. Nature Reviews Endocrinology . 2011;7(9):526–539. doi: 10.1038/nrendo.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki A., Yabu A., Nakamura H. Advanced glycation end products in musculoskeletal system and disorders. Methods (San Diego, Calif) . 2022;203:179–186. doi: 10.1016/j.ymeth.2020.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Grillo M. A., Colombatto S. Advanced glycation end-products (AGEs): involvement in aging and in neurodegenerative diseases. Amino Acids . 2008;35(1):29–36. doi: 10.1007/s00726-007-0606-0. [DOI] [PubMed] [Google Scholar]
- 28.Vlassara H., Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Current Diabetes Reports . 2014;14(1):p. 453. doi: 10.1007/s11892-013-0453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharifi-Zahabi E., Sharafabad F. H., Abdollahzad H., Malekahmadi M., Rad N. B. Circulating advanced glycation end products and their soluble receptors in relation to all-cause and cardiovascular mortality: a systematic review and meta-analysis of prospective observational studies. Advances in Nutrition (Bethesda, Md) . 2021;12(6):2157–2171. doi: 10.1093/advances/nmab072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rabbani N., Thornalley P. J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney International . 2018;93(4):803–813. doi: 10.1016/j.kint.2017.11.034. [DOI] [PubMed] [Google Scholar]
- 31.Takino J., Yamagishi S., Takeuchi M. Cancer malignancy is enhanced by glyceraldehyde-derived advanced glycation end-products. Journal of Oncology . 2010;2010:8. doi: 10.1155/2010/739852.739852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulrich P., Cerami A. Protein glycation, diabetes, and aging. Recent Progress in Hormone Research . 2001;56(1):1–22. doi: 10.1210/rp.56.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Cho S. J., Roman G., Yeboah F., Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Current Medicinal Chemistry . 2007;14(15):1653–1671. doi: 10.2174/092986707780830989. [DOI] [PubMed] [Google Scholar]
- 34.Mustafa I., Ahmad S., Dixit K., Moinuddin A. J., Ali A. Glycated human DNA is a preferred antigen for anti-DNA antibodies in diabetic patients. Diabetes Research and Clinical Practice . 2012;95(1):98–104. doi: 10.1016/j.diabres.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Thornalley P. J., Langborg A., Minhas H. S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. The Biochemical Journal . 1999;344:109–116. [PMC free article] [PubMed] [Google Scholar]
- 36.Eggen M. D., Glomb M. A. Analysis of glyoxal- and methylglyoxal-derived Advanced glycation end products during grilling of porcine meat. Journal of Agricultural and Food Chemistry . 2021;69(50):15374–15383. doi: 10.1021/acs.jafc.1c06835. [DOI] [PubMed] [Google Scholar]
- 37.Poulsen M. W., Hedegaard R. V., Andersen J. M., et al. Advanced glycation endproducts in food and their effects on health. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association . 2013;60:10–37. doi: 10.1016/j.fct.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 38.Negre-Salvayre A., Salvayre R., Augé N., Pamplona R., Portero-Otín M. Hyperglycemia and glycation in diabetic complications. Antioxidants & redox signaling . 2009;11(12):3071–3109. doi: 10.1089/ars.2009.2484. [DOI] [PubMed] [Google Scholar]
- 39.Lapolla A., Traldi P., Fedele D. Importance of measuring products of non-enzymatic glycation of proteins. Clinical Biochemistry . 2005;38(2):103–115. doi: 10.1016/j.clinbiochem.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Nowotny K., Jung T., Höhn A., Weber D., Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules . 2015;5(1):194–222. doi: 10.3390/biom5010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldberg T., Cai W., Peppa M., et al. Advanced glycoxidation end products in commonly consumed foods. Journal of the American Dietetic Association . 2004;104(8):1287–1291. doi: 10.1016/j.jada.2004.05.214. [DOI] [PubMed] [Google Scholar]
- 42.Uribarri J., del Castillo M. D., de la Maza M. P., et al. Dietary advanced glycation end products and their role in health and disease. Advances in Nutrition (Bethesda, Md) . 2015;6(4):461–473. doi: 10.3945/an.115.008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C. H., Huang S. M., Lin J. A., Yen G. C. Inhibition of advanced glycation endproduct formation by foodstuffs. Food & Function . 2011;2(5):224–234. doi: 10.1039/c1fo10026b. [DOI] [PubMed] [Google Scholar]
- 44.Singh R., Barden A., Mori T., Beilin L. Advanced glycation end-products: a review. Diabetologia . 2001;44(2):129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 45.Koschinsky T., He C. J., Mitsuhashi T., et al. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proceedings of the National Academy of Sciences of the United States of America . 1997;94(12):6474–6479. doi: 10.1073/pnas.94.12.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He C., Sabol J., Mitsuhashi T., Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes . 1999;48(6):1308–1315. doi: 10.2337/diabetes.48.6.1308. [DOI] [PubMed] [Google Scholar]
- 47.O'Brien J., Morrissey P. A., Ames J. M. Nutritional and toxicological aspects of the Maillard browning reaction in foods. Critical Reviews in Food Science and Nutrition . 1989;28(3):211–248. doi: 10.1080/10408398909527499. [DOI] [PubMed] [Google Scholar]
- 48.Peppa M., Uribarri J., Vlassara H., Advanced glycoxidation Advanced Glycoxidation: A new risk factor for cardiovascular disease. Cardiovascular Toxicology . 2002;2(4):275–288. doi: 10.1385/CT:2:4:275. [DOI] [PubMed] [Google Scholar]
- 49.Nicholl I. D., Bucala R. Cellular and Molecular Biology . 7. Vol. 44. France: Noisy-le-Grand; 1998. Advanced Glycation Endproducts and Cigarette Smoking; pp. 1025–1033. [PubMed] [Google Scholar]
- 50.Sparvero L. J., Asafu-Adjei D., Kang R., et al. RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. Journal of Translational Medicine . 2009;7(1):p. 17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willett T. L., Kandel R., De Croos J. N., Avery N. C., Grynpas M. D. Enhanced levels of non-enzymatic glycation and pentosidine crosslinking in spontaneous osteoarthritis progression. Osteoarthritis and Cartilage . 2012;20(7):736–744. doi: 10.1016/j.joca.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 52.Perrone A., Giovino A., Benny J., Martinelli F. Advanced glycation end products (AGEs): biochemistry, signaling, analytical methods, and epigenetic effects. Oxidative Medicine and Cellular Longevity . 2020;2020:18. doi: 10.1155/2020/3818196.3818196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trout J. J., Buckwalter J. A., Moore K. C. Ultrastructure of the human intervertebral disc: II. Cells of the nucleus pulposus. The Anatomical Record . 1982;204(4):307–314. doi: 10.1002/ar.1092040403. [DOI] [PubMed] [Google Scholar]
- 54.Chen J., Yan W., Setton L. A. Molecular phenotypes of notochordal cells purified from immature nucleus pulposus. European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research. Society . 2006;15:S303–S311. doi: 10.1007/s00586-006-0088-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Colombier P., Camus A., Lescaudron L., Clouet J., Guicheux J. Intervertebral disc regeneration: a great challenge for tissue engineers. Trends in Biotechnology . 2014;32(9):433–435. doi: 10.1016/j.tibtech.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 56.Yang X., Li X. Nucleus pulposus tissue engineering: a brief review. European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society . 2009;18(11):1564–1572. doi: 10.1007/s00586-009-1092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sivan S. S., Tsitron E., Wachtel E., et al. Age-related accumulation of pentosidine in aggrecan and collagen from normal and degenerate human intervertebral discs. The Biochemical Journal . 2006;399(1):29–35. doi: 10.1042/BJ20060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song Y., Li S., Geng W., et al. Sirtuin 3-dependent mitochondrial redox homeostasis protects against AGEs- induced intervertebral disc degeneration. Redox Biology . 2018;19:339–353. doi: 10.1016/j.redox.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luo R., Li S., Li G., et al. FAM134B-mediated ER-phagy upregulation attenuates AGEs-induced apoptosis and senescence in human nucleus pulposus cells. Oxidative Medicine and Cellular Longevity . 2021;2021:19. doi: 10.1155/2021/3843145.3843145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delorme-Axford E., Popelka H., Klionsky D. J. TEX264 is a major receptor for mammalian reticulophagy. Autophagy . 2019;15(10):1677–1681. doi: 10.1080/15548627.2019.1646540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oakes S. A., Papa F. R. The role of endoplasmic reticulum stress in human pathology. Annual review of pathology . 2015;10(1):173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grootjans J., Kaser A., Kaufman R. J., Blumberg R. S. The unfolded protein response in immunity and inflammation. Nature reviews Immunology . 2016;16(8):469–484. doi: 10.1038/nri.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo R., Song Y., Liao Z., et al. Impaired calcium homeostasis via advanced glycation end products promotes apoptosis through endoplasmic reticulum stress in human nucleus pulposus cells and exacerbates intervertebral disc degeneration in rats. The FEBS Journal . 2019;286(21):4356–4373. doi: 10.1111/febs.14972. [DOI] [PubMed] [Google Scholar]
- 64.Raffaello A., Mammucari C., Gherardi G., Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends in Biochemical Sciences . 2016;41(12):1035–1049. doi: 10.1016/j.tibs.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mackrill J. J. Ryanodine receptor calcium channels and their partners as drug targets. Biochemical Pharmacology . 2010;79(11):1535–1543. doi: 10.1016/j.bcp.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 66.Chan C. M., Huang D. Y., Huang Y. P., et al. Methylglyoxal induces cell death through endoplasmic reticulum stress- associated ROS production and mitochondrial dysfunction. Journal of Cellular and Molecular Medicine . 2016;20(9):1749–1760. doi: 10.1111/jcmm.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo R., Liu W., Liu B., Zhang B., Li W., Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: an insight into endoplasmic reticulum stress response mechanism. International Journal of Cardiology . 2015;191:36–45. doi: 10.1016/j.ijcard.2015.04.245. [DOI] [PubMed] [Google Scholar]
- 68.Kim I., Xu W., Reed J. C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature Reviews Drug Discovery . 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 69.Liao Z., Luo R., Li G., et al. Exosomes from mesenchymal stem cells modulate endoplasmic reticulum stress to protect against nucleus pulposus cell death and ameliorate intervertebral disc degeneration in vivo. Theranostics . 2019;9(14):4084–4100. doi: 10.7150/thno.33638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grunhagen T., Wilde G., Soukane D. M., Shirazi-Adl S. A., Urban J. P. Nutrient supply and intervertebral disc metabolism. The Journal of Bone and Joint Surgery American Volume . 2006;88(Suppl 2):30–35. doi: 10.2106/JBJS.E.01290. [DOI] [PubMed] [Google Scholar]
- 71.Risbud M. V., Schipani E., Shapiro I. M. Hypoxic regulation of nucleus pulposus cell survival: from niche to notch. The American Journal of Pathology . 2010;176(4):1577–1583. doi: 10.2353/ajpath.2010.090734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Risbud M. V., Schoepflin Z. R., Mwale F., et al. Defining the phenotype of young healthy nucleus pulposus cells: recommendations of the spine research interest group at the 2014 annual ORS meeting. Journal of Orthopaedic Research . 2015;33(3):283–293. doi: 10.1002/jor.22789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajpurohit R., Risbud M. V., Ducheyne P., Vresilovic E. J., Shapiro I. M. Phenotypic characteristics of the nucleus pulposus: expression of hypoxia inducing factor-1, glucose transporter-1 and MMP-2. Cell and Tissue Research . 2002;308(3):401–407. doi: 10.1007/s00441-002-0563-6. [DOI] [PubMed] [Google Scholar]
- 74.Zeng Y., Danielson K. G., Albert T. J., Shapiro I. M., Risbud M. V. HIF-1 alpha is a regulator of galectin-3 expression in the intervertebral disc. Journal of Bone and Mineral Research : The Official Journal of the American Society for Bone and Mineral Research . 2007;22(12):1851–1861. doi: 10.1359/jbmr.070620. [DOI] [PubMed] [Google Scholar]
- 75.Gogate S. S., Nasser R., Shapiro I. M., Risbud M. V. Hypoxic regulation of β-1,3-glucuronyltransferase 1 expression in nucleus pulposus cells of the rat intervertebral disc: role of hypoxia-inducible factor proteins. Arthritis and Rheumatism . 2011;63(7):1950–1960. doi: 10.1002/art.30342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y. C., Gu Y., Yang J. Y., et al. RACK1 mediates the advanced glycation end product-induced degradation of HIF-1α in nucleus pulposus cells via competing with HSP90 for HIF-1α binding. Cell Biology International . 2021;45(6):1316–1326. doi: 10.1002/cbin.11574. [DOI] [PubMed] [Google Scholar]
- 77.Semenza G. L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annual Review of Pathology . 2014;9(1):47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 78.Martin J. A., Martini A., Molinari A., et al. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthritis and Cartilage . 2012;20(4):323–329. doi: 10.1016/j.joca.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McElroy G. S., Chandel N. S. Mitochondria control acute and chronic responses to hypoxia. Experimental Cell Research . 2017;356(2):217–222. doi: 10.1016/j.yexcr.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kroemer G., Galluzzi L., Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews . 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 81.Morris B. J. Seven sirtuins for seven deadly diseases of aging. Free Radical Biology & Medicine . 2013;56:133–171. doi: 10.1016/j.freeradbiomed.2012.10.525. [DOI] [PubMed] [Google Scholar]
- 82.Kumar S., Lombard D. B. Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxidants & Redox Signaling . 2015;22(12):1060–1077. doi: 10.1089/ars.2014.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pickles S., Vigié P., Youle R. J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Current Biology : CB . 2018;28(4):R170–r185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang X., Chen D. LncRNA FAM83H-AS1 maintains intervertebral disc tissue homeostasis and attenuates inflammation-related pain via promoting nucleus pulposus cell growth through miR-22-3p inhibition. Annals of Translational Medicine . 2020;8(22):p. 1518. doi: 10.21037/atm-20-7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niu C. C., Lin S. S., Yuan L. J., et al. Upregulation of miR-107 expression following hyperbaric oxygen treatment suppresses HMGB1/RAGE signaling in degenerated human nucleus pulposus cells. Arthritis Research & Therapy . 2019;21(1):p. 42. doi: 10.1186/s13075-019-1830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le Maitre C. L., Pockert A., Buttle D. J., Freemont A. J., Hoyland J. A. Matrix synthesis and degradation in human intervertebral disc degeneration. Biochemical Society Transactions . 2007;35(4):652–655. doi: 10.1042/BST0350652. [DOI] [PubMed] [Google Scholar]
- 87.Tsai T. T., Ho N. Y., Lin Y. T., et al. Advanced glycation end products in degenerative nucleus pulposus with diabetes. Journal of Orthopaedic Research . 2014;32(2):238–244. doi: 10.1002/jor.22508. [DOI] [PubMed] [Google Scholar]
- 88.Yokosuka K., Park J. S., Jimbo K., et al. Advanced glycation end-products downregulating intervertebral disc cell production of proteoglycans in vitro. Journal of Neurosurgery Spine . 2006;5(4):324–329. doi: 10.3171/spi.2006.5.4.324. [DOI] [PubMed] [Google Scholar]
- 89.Yoshida T., Park J. S., Yokosuka K., et al. Up-regulation in receptor for advanced glycation end-products in inflammatory circumstances in bovine coccygeal intervertebral disc specimens in vitro. Spine . 2009;34(15):1544–1548. doi: 10.1097/BRS.0b013e3181a98390. [DOI] [PubMed] [Google Scholar]
- 90.Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Research and Clinical Practice . 2005;67(1):3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 91.Illien-Junger S., Grosjean F., Laudier D. M., Vlassara H., Striker G. E., Iatridis J. C. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLoS One . 2013;8(5, article e64302) doi: 10.1371/journal.pone.0064302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fields A. J., Berg-Johansen B., Metz L. N., et al. Alterations in intervertebral disc composition, matrix homeostasis and biomechanical behavior in the UCD-T2DM rat model of type 2 diabetes. Journal of Orthopaedic Research . 2015;33(5):738–746. doi: 10.1002/jor.22807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Marchand F., Ahmed A. M. Investigation of the laminate structure of lumbar disc anulus fibrosus. Spine . 1990;15(5):402–410. doi: 10.1097/00007632-199005000-00011. [DOI] [PubMed] [Google Scholar]
- 94.Fujita K., Nakagawa T., Hirabayashi K., Nagai Y. Neutral proteinases in human intervertebral disc. Role in degeneration and probable origin. Spine . 1993;18(13):1766–1773. doi: 10.1097/00007632-199310000-00009. [DOI] [PubMed] [Google Scholar]
- 95.Raj P. P. Intervertebral disc: anatomy-physiology-pathophysiology-treatment. Pain . 2008;8(1):18–44. doi: 10.1111/j.1533-2500.2007.00171.x. [DOI] [PubMed] [Google Scholar]
- 96.Hu Y., Shao Z., Cai X., et al. Mitochondrial pathway is involved in advanced glycation end products-induced apoptosis of rabbit annulus fibrosus cells. Spine . 2019;44(10):E585–e595. doi: 10.1097/BRS.0000000000002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Krishnamoorthy D., Hoy R. C., Natelson D. M., et al. Dietary advanced glycation end-product consumption leads to mechanical stiffening of murine intervertebral discs. Disease Models & Mechanisms . 2018;11(12) doi: 10.1242/dmm.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hoy R. C., D'Erminio D. N., Krishnamoorthy D., et al. Advanced glycation end products cause RAGE-dependent annulus fibrosus collagen disruption and loss identified using in situ second harmonic generation imaging in mice intervertebral disk in vivo and in organ culture models. JOR Spine . 2020;3(4, article e1126) doi: 10.1002/jsp2.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Whatley B. R., Wen X. J. M. S. Intervertebral disc (IVD): Structure, degeneration, repair and regeneration. Materials Science and Engineering . 2012;32(2):61–77. doi: 10.1016/j.msec.2011.10.011. [DOI] [Google Scholar]
- 100.Lotz J. C., Fields A. J., Liebenberg E. C. The role of the vertebral end plate in low back pain. Global Spine Journal . 2013;3(3):153–164. doi: 10.1055/s-0033-1347298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bartels E. M., Fairbank J. C., Winlove C. P., Urban J. P. Oxygen and lactate concentrations measured in vivo in the intervertebral discs of patients with scoliosis and back pain. Spine . 1998;23(1):1–7. doi: 10.1097/00007632-199801010-00001. discussion 8. [DOI] [PubMed] [Google Scholar]
- 102.Illien-Jünger S., Lu Y., Qureshi S. A., et al. Chronic ingestion of advanced glycation end products induces degenerative spinal changes and hypertrophy in aging pre-diabetic mice. PLoS One . 2015;10(2, article e0116625) doi: 10.1371/journal.pone.0116625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Illien-Jünger S., Torre O. M., Kindschuh W. F., Chen X., Laudier D. M., Iatridis J. C. AGEs induce ectopic endochondral ossification in intervertebral discs. European Cells & Materials . 2016;32:257–270. doi: 10.22203/eCM.v032a17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dolor A., Sampson S. L., Lazar A. A., Lotz J. C., Szoka F. C., Fields A. J. Matrix modification for enhancing the transport properties of the human cartilage endplate to improve disc nutrition. PLoS One . 2019;14(4, article e0215218) doi: 10.1371/journal.pone.0215218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weiler C., Nerlich A. G., Bachmeier B. E., Boos N. Expression and distribution of tumor necrosis factor alpha in human lumbar intervertebral discs: a study in surgical specimen and autopsy controls. Spine . 2005;30(1):44–53. doi: 10.1097/01.brs.0000149186.63457.20. discussion 54. [DOI] [PubMed] [Google Scholar]
- 106.Le Maitre C. L., Hoyland J. A., Freemont A. J. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Research & Therapy . 2007;9(4):p. R77. doi: 10.1186/ar2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Risbud M. V., Shapiro I. M. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nature Reviews Rheumatology . 2014;10(1):44–56. doi: 10.1038/nrrheum.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Battié M. C., Videman T., Kaprio J., et al. The Twin Spine Study: contributions to a changing view of disc degeneration†. The Spine Journal : Official Journal of the North American Spine Society . 2009;9(1):47–59. doi: 10.1016/j.spinee.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 109.Adams M. A., Freeman B. J., Morrison H. P., Nelson I. W., Dolan P. Mechanical initiation of intervertebral disc degeneration. Spine . 2000;25(13):1625–1636. doi: 10.1097/00007632-200007010-00005. [DOI] [PubMed] [Google Scholar]
- 110.Wang D., Nasto L. A., Roughley P., et al. Spine degeneration in a murine model of chronic human tobacco smokers. Osteoarthritis and Cartilage . 2012;20(8):896–905. doi: 10.1016/j.joca.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cai W., Ramdas M., Zhu L., Chen X., Striker G. E., Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proceedings of the National Academy of Sciences of the United States of America . 2012;109(39):15888–15893. doi: 10.1073/pnas.1205847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hayashi S., Taira A., Inoue G., et al. TNF-alpha in nucleus pulposus induces sensory nerve growth: a study of the mechanism of discogenic low back pain using TNF-alpha-deficient mice. Spine . 2008;33(14):1542–1546. doi: 10.1097/BRS.0b013e318178e5ea. [DOI] [PubMed] [Google Scholar]
- 113.Murata Y., Onda A., Rydevik B., Takahashi I., Takahashi K., Olmarker K. Changes in pain behavior and histologic changes caused by application of tumor necrosis factor-alpha to the dorsal root ganglion in rats. Spine . 2006;31(5):530–535. doi: 10.1097/01.brs.0000201260.10082.23. [DOI] [PubMed] [Google Scholar]
- 114.Stern D., Yan S. D., Yan S. F., Schmidt A. M. Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Advanced Drug Delivery Reviews . 2002;54(12):1615–1625. doi: 10.1016/S0169-409X(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 115.Liu C. C., Zhang X. S., Ruan Y. T., et al. Accumulation of methylglyoxal increases the advanced glycation end-product levels in DRG and contributes to lumbar disk herniation-induced persistent pain. Journal of Neurophysiology . 2017;118(2):1321–1328. doi: 10.1152/jn.00745.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang X. S., Li X., Luo H. J., et al. Activation of the RAGE/STAT3 pathway in the dorsal root ganglion contributes to the persistent pain hypersensitivity induced by lumbar disc herniation. Pain Physician . 2017;20(5):419–427. [PubMed] [Google Scholar]
- 117.Shamji M. F., Setton L. A., Jarvis W., et al. Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis and Rheumatism . 2010;62(7):1974–1982. doi: 10.1002/art.27444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kokubo Y., Uchida K., Kobayashi S., et al. Herniated and spondylotic intervertebral discs of the human cervical spine: histological and immunohistological findings in 500 en bloc surgical samples. Laboratory Investigation. Journal of Neurosurgery Spine . 2008;9(3):285–295. doi: 10.3171/SPI/2008/9/9/285. [DOI] [PubMed] [Google Scholar]
- 119.Melrose J., Roberts S., Smith S., Menage J., Ghosh P. Increased nerve and blood vessel ingrowth associated with proteoglycan depletion in an ovine anular lesion model of experimental disc degeneration. Spine . 2002;27(12):1278–1285. doi: 10.1097/00007632-200206150-00007. [DOI] [PubMed] [Google Scholar]
- 120.D'Erminio D. N., Krishnamoorthy D., Lai A., et al. High fat diet causes inferior vertebral structure and function without disc degeneration in RAGE-KO mice. Journal of Orthopaedic Research . 2022;40(7):1672–1686. doi: 10.1002/jor.25191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wagner D. R., Reiser K. M., Lotz J. C. Glycation increases human annulus fibrosus stiffness in both experimental measurements and theoretical predictions. Journal of Biomechanics . 2006;39(6):1021–1029. doi: 10.1016/j.jbiomech.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 122.Jazini E., Sharan A. D., Morse L. J., et al. Alterations in T2 relaxation magnetic resonance imaging of the ovine intervertebral disc due to nonenzymatic glycation. Spine . 2012;37(4):E209–E215. doi: 10.1097/BRS.0b013e31822ce81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lintz M., Walk R. E., Tang S. Y., Bonassar L. J. The degenerative impact of hyperglycemia on the structure and mechanics of developing murine intervertebral discs. JOR Spine . 2022;5(1, article e1191) doi: 10.1002/jsp2.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang G. Z., Liu M. Q., Chen H. W., et al. NF-κB signalling pathways in nucleus pulposus cell function and intervertebral disc degeneration. Cell Proliferation . 2021;54(7, article e13057) doi: 10.1111/cpr.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hastreiter D., Ozuna R. M., Spector M. Regional variations in certain cellular characteristics in human lumbar intervertebral discs, including the presence of alpha-smooth muscle actin. Journal of Orthopaedic Research . 2001;19(4):597–604. doi: 10.1016/S0736-0266(00)00069-3. [DOI] [PubMed] [Google Scholar]
- 126.Freemont A. J., Watkins A., Le Maitre C., et al. Nerve growth factor expression and innervation of the painful intervertebral disc. The Journal of Pathology . 2002;197(3):286–292. doi: 10.1002/path.1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Contact the corresponding author to obtain relevant data.