

Abstract
Since the release of the ICH E9(R1) (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Addendum on Estimands and Sensitivity Analysis in Clinical Trials to the Guideline on Statistical Principles for Clinical Trials) document in 2019, the estimand framework has become a fundamental part of clinical trial protocols. In parallel, complex innovative designs have gained increased popularity in drug development, in particular in early development phases or in difficult experimental situations. While the estimand framework is relevant to any study in which a treatment effect is estimated, experience is lacking as regards its application to these designs. In a basket trial for example, should a different estimand be specified for each subpopulation of interest, defined, for example, by cancer site? Or can a single estimand focusing on the general population (defined, for example, by the positivity to a certain biomarker) be used? In the case of platform trials, should a different estimand be proposed for each drug investigated? In this work we discuss possible ways of implementing the estimand framework for different types of complex innovative designs. We consider trials that allow adding or selecting experimental treatment arms, modifying the control arm or the standard of care, and selecting or pooling populations. We also address the potentially data‐driven, adaptive selection of estimands in an ongoing trial and disentangle certain statistical issues that pertain to estimation rather than to estimands, such as the borrowing of nonconcurrent information. We hope this discussion will facilitate the implementation of the estimand framework and its description in the study protocol when the objectives of the trial require complex innovative designs.
Since the release of the ICH E9(R1) (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Addendum on Estimands and Sensitivity Analysis in Clinical Trials to the Guideline on Statistical Principles for Clinical Trials) guideline, 1 estimands have become a fundamental part of clinical study protocols. By clearly describing the treatment, the end point, the population of interest in the trial, the intercurrent events (i.e., events occurring after treatment initiation that affect either the interpretation or the existence of the measurements associated with the clinical question of interest), and the summary statistic used to estimate the treatment effect, estimands aim at facilitating discussions between different stakeholders and at reaching agreement on the appropriate treatment effect to be estimated in the trial. 2 As the implementation of the estimand framework progresses, both trialists and regulators are gaining experience at respectively crafting and assessing estimands in many different therapeutic areas. 3 , 4 , 5
In parallel, complex innovative designs have gained increased popularity in drug development, in particular in early phase and/or in difficult experimental situations. They aim at accelerating drug development, for example by taking decisions earlier, leveraging external information, or reducing the sample size. Adaptive trials, which allow modifications of the design as data accrue, such as dropping an ineffective treatment arm during an interim analysis, are now current practice even in a confirmatory setting. 6 , 7 , 8 Master protocols (namely basket, umbrella, and platform trials), which study several treatments or disease subtypes within the same protocol, are gaining momentum, especially in oncology. 9 , 10 , 11 , 12 Large international platform trials were also set up to investigate the effect of several repurposed drugs to fight coronavirus disease 2019 (COVID‐19). 13 , 14 Another class of complex innovative designs includes trials using Bayesian models to borrow information from external or nonconcurrent sources 15 , 16 , 17 , 18 , 19 in order to potentially gain power or precision, or to reduce the sample size.
The ICH E9(R1) guideline covers in detail the estimand framework whenever a treatment effect is estimated, or a hypothesis related to a treatment effect is tested, whether related to efficacy or safety. This therefore applies in particular to complex innovative designs, but while describing the estimands of a classic randomized clinical trial (RCT) is becoming standard practice, experience is lacking in applying the estimand framework to these innovative studies. For basket trials, for example, should a different estimand be specified for each disease subtype? In a multi‐arm trial, does borrowing information between treatment arms or from historical studies modify the estimand?
Okwuokenye and Peace 20 have discussed the application of the estimand framework to trials with adaptations, such as sample size re‐estimation, group sequential designs, and enrichment designs. In this article this work is extended by reviewing the different types of complex innovative designs as defined by regulatory authorities 21 , 22 , 23 and by discussing ways of framing the estimands when implementing these designs. Although many aspects of estimands are admittedly trial‐specific and context‐specific, it is hoped this discussion will facilitate the choice and the description of estimands when planning complex innovative trials and writing their protocols, as well as contribute to a standardization of approaches.
The document starts by reminding the reader about the underlying principles of estimands and explains how they tie in with the objectives of clinical trials in general and among them, innovative complex designs. The following sections describe the use of estimands by three main types of adaptations and innovative characteristics: adding or selecting experimental treatment arms, modifying the control arm or the standard of care, and adding, selecting, or pooling populations. The last section disentangles certain statistical issues that pertain to estimation rather than to estimands, such as the borrowing of nonconcurrent information.
ESTIMANDS AND INNOVATIVE DESIGNS
Establishing the existence and estimating the magnitude of treatment effects are central questions in the development and approval of drugs: How does the outcome of the treatment compare with what would have happened to the same participants under an alternative treatment (i.e., if they had not received the treatment or had received another treatment)? Causal estimands are population quantities describing causal effects of treatments. They summarize at the population level what the results would look like in the same patients under different treatment conditions to be compared.
These concepts have recently been popularized within the clinical trial community with the publication of the ICH E9(R1) guideline on estimands and sensitivity analyses. 1 This guideline is driven by efforts to describe an aligned framework for planning, conducting, analyzing, and interpreting clinical trials. This new framework emphasizes the importance of a precise description of the treatment effect of interest by defining the population, the variable, and the treatment. This description needs to explicitly account for events which occur after randomization (“intercurrent events”), e.g., treatment discontinuation due to an adverse event, the use of rescue medications, death, etc. Finally, a population‐level summary for the variable should be specified, providing a basis for comparison between treatment conditions. Regulatory interest in the application of the principles outlined will be greater for confirmatory clinical trials and, where used to generate confirmatory conclusions, for data integrated across trials.
The principles and the underlying estimand thinking process outlined in the ICH E9(R1) guideline are relevant whenever a treatment effect is estimated, or a hypothesis related to a treatment effect is tested, and are therefore applicable to complex innovative designs. For example, assume that the main objective of the trial is to investigate the effect of a marketed drug in three related niche indications. The scientific objective(s) is (are) clear and because it is a marketed drug with a presumably known safety profile, a phase III program can be considered. It could also be run as a series of parallel randomized trials, each with the objective to investigate the treatment effect in a particular disease, together with a dedicated estimand discussion. One could argue that these objectives do not change when running a basket trial, so that a dedicated estimand discussion for each trial objective should take place. For example, when developing tumor‐independent cancer drugs, a basket trial might enroll patients in nonoverlapping subpopulations (e.g., three different types of cancer with the same genetic alteration). Because there are three primary trial objectives (one for each indication), a dedicated discussion for each of the three related estimands should take place, following the thinking process shown in Figure 1 . These estimands will be different because three different diseases and therefore three different patient populations are targeted (even if they are related to each other). There could be other differences as well; see Table 2. Once these three estimands have been established, one can then think about a suitable analysis approach. For example, if it is clinically plausible to assume enough similarity across the diseases, one may consider using a hierarchical model, 24 as long as it is aligned with the target estimands. Sensitivity analyses will be important to investigate the assumptions underlying the analysis approach (e.g., the hierarchical model). A different situation occurs when the original primary scientific question concerns the efficacy in the overall population, possibly followed by subsequent discussions on estimands and analysis approaches to assess the treatment effect in the individual subpopulations. 9 Focus on the overall population might be reasonable when the three related niche indications can be considered as one overall disease.
Figure 1.

Estimand thinking process to be implemented at the design stage (adapted from ref. 54).
ESTIMANDS BY TYPE OF ADAPTATION AND INNOVATIVE CHARACTERISTICS
In this section the estimand framework is discussed for common types of adaptation and element of innovation, as listed in European Medicines Agency (EMA) and US Food and Drug Administration (FDA) regulatory guidance. 21 , 22 , 23
Adding or selecting experimental treatment arms
Consider an RCT in which two experimental treatments are compared with a common control arm in a given target population. It seems reasonable to expect the protocol to specify a specific estimand for each comparison of an experimental treatment to control. As described in Table 1 , some attributes of these estimands would share a certain degree of similarity, as the population, while others could theoretically be completely different, such as certain intercurrent events. Indeed, some intercurrent event might only occur with one of the experimental treatments but neither with the control nor with the other experimental treatment. Such an intercurrent event should therefore be reflected in each estimand associated with this experimental treatment. For example, assume that one of the experimental treatments is a biologic (such as a targeted anticancer agent or an immunotherapy). These biological drugs may trigger immune responses that lead to the formation of antidrug antibodies (ADAs). 25 , 26 ADAs may be directed against immunogenic parts of the drug and may affect its efficacy or safety, or they may bind to regions of the protein which do not affect safety or efficacy, with little to no clinical effect. ADA positivity is triggered by treatment, appears post randomization, and has the potential to affect the interpretation of the outcome. It can thus be considered an intercurrent event in the language of the ICH E9(R1) guideline, 27 , 28 and it is then specific to the biological treatment in the three‐arm trial (assuming the other treatments are not biologic).
Table 1.
Differences and similarities between the estimands of several treatments for the same disease studied within the same trial or master protocol
| Estimand attribute | Guidance |
|---|---|
| A. Treatment | “Different for each treatment investigated but common comparator” |
| B. Population | “Similar for each treatment investigated” |
| C. Variable | Could vary with the drug as different treatments could target different aspects of the disease (e.g., remission, disease severity, pain, etc.) |
| D. Intercurrent events | Population‐specific IEs would be similar (e.g., change in background medication), whereas treatment‐specific IEs would vary |
| E. Population‐level summary | Would change with the variable |
IEs, intercurrent events.
Assume now that the trial is a platform trial in which recruitment is open‐ended and that new treatments can be added dynamically to the design, as, e.g., the I‐SPY2 (Investigation of Serial studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis) trial (https://www.ispytrials.org/i‐spy‐platform/i‐spy2). In practice, adding a new treatment arm to this platform trial would simply mean adding another objective (of comparing the new treatment vs. control) and therefore another estimand to the protocol. Importantly, the introduction of the new estimand would not necessarily need to be prespecified at the start of the platform trial but rather before the addition of the new treatment arm. Important considerations about adding an arm to a platform trial can be found in the literature. 13 , 29 , 30 , 31
Assume now the trial includes an interim analysis aiming at dropping ineffective treatment arms based on accrued data. Examples of such trials are the GATSBY (Trastuzumab emtansine versus taxane use for previously treated HER2‐positive locally advanced or metastatic gastric or gastro‐oesophageal junction adenocarcinoma) trial 32 , 33 in which a dose could be dropped at interim, or the RECOVERY (Randomized Evaluation of COVID‐19 Therapy) trial in COVID‐19 14 (www.recoverytrial.net) in which hydroxychloroquine was dropped at interim. 34 In practice, this means that prespecified estimands are selected at interim, and that the trial continues with the remaining estimands. Methods for these designs are well described in the literature relative to adaptive designs 7 and some supplementary considerations are given in Points to consider on estimation.
Modifying the control arm
Consider again a three‐arm RCT comparing two experimental treatments, E 1 and E 2 against a control arm C. As described in the previous section, two specific estimands Est1 and Est2 would need to be specified. Now assume that while running this trial, new evidence arises which leads to a modification of the treatment given in the control arm. Such a change could, e.g., be the approval of a new therapy replacing the original standard of care. This would thus generate two stages in the trial: one with control treatment 1 and one with control treatment 2 (Figure 2 ). This would in turn potentially induce four estimands in total: Est ij where i = 1,2 refers to the comparison of experimental treatment i to the control and j = 1,2 refers to the stage, i.e., prior to or after the control arm changes.
Figure 2.
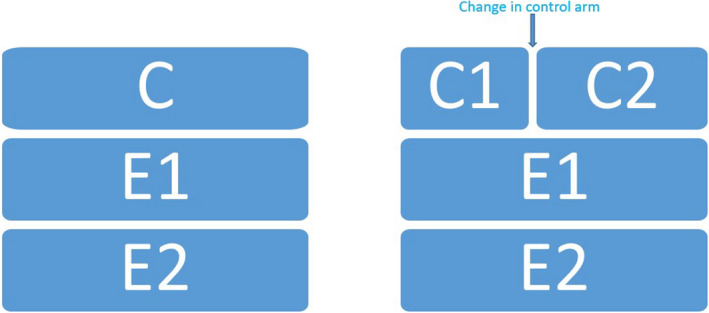
Control and two experimental arms without (left) and with (right) change of control arm. C, control; E, experimental.
Deriving the estimands Est i 2 at Stage 2 from the estimands Est i 1 at Stage 1 potentially affects three estimand attributes:
Treatment: The change to the control arm obviously needs to be reflected in the treatment attribute of the estimand.
A modification of the control arm might induce a change to the population given patients might not be eligible anymore for this new treatment. It might also have an impact regarding the generalizability of the results of the updated trial on the initially intended target population.
Intercurrent event: Both a new control treatment and a population change may lead to the occurrence of intercurrent events for Est i 2 that were not initially considered for Est i 1.
As regards estimation, the situation is very similar to a platform trial where a new treatment is added as data accrue. Participants recruited to the control arm prior to the addition of the new treatment are nonconcurrent, and whether or not including them in the estimation of the effect of the new treatment, particularly in phase III, has been extensively discussed in the literature. 9 , 29 , 30
Alternatively, if the main interest is to compare both experimental treatments with a control arm regardless of any changes to the standard of care throughout the trial, then the treatment attribute of Est i would remain aspecific and would then simply mention that the experimental treatment is compared with a state‐of‐the‐art control therapy. In this case, no update to Est i would be needed and the treatment effect of both experimental arms would be compared with C using the full data sample.
Instead of adapting the estimand in order to reflect the change in the control treatment, an alternative option would be to keep the initial clinical trial objective and estimand and assess if the latter can still be estimated given the collected data are now different. Estimation and operating characteristics of an estimator based on this data might however be inferior to what was originally intended, even when adding external data sources or using network meta‐analysis, and the trialist will therefore have to decide, potentially following a discussion with regulatory agencies, which solution is more appropriate.
Adding, selecting, or pooling populations
In this section the discussion is illustrated by examples from oncology, e.g., with subpopulations defined by a biomarker or tumor types, but the reasoning would hold for any type of subpopulations, e.g., mild vs. severe.
Biomarker‐defined subpopulations
It is increasingly common that a covariate biomarker is expected to be predictive for the relative efficacy of an experimental treatment, E, as compared with standard‐of‐care control, C. From a regulatory 35 and public health perspective, it is often desirable that data are generated both in biomarker‐positive (BM+) and biomarker‐negative (BM−) patients.
Indeed, many trials have the dual objectives of assessing efficacy both in BM+ and in the total population, sometimes referred to as all comers (AC) (see ref. 36 for an example). In this case, two separate estimands, for AC and BM+, should be prespecified. In general, these estimands would differ only in the definition of the population, while other attributes, the end point, for example, would be the same.
While the trial sponsor may be interested in showing efficacy in AC (or BM+), regulators and payers may ask whether a proven efficacy in AC should lead to approval and reimbursement in the full population or, say, only in BM+. To inform this decision, the efficacy in BM− is crucial. 37 Whether or not defined in the study protocol, the estimand for BM− may therefore be important. Its definition will typically follow directly from those of the other estimands, i.e., will mainly differ by the ”population” attribute.
As the relative treatment efficacy is a priori expected to be lower in BM− than in BM+, it is often considered to discontinue the BM− subpopulation at an interim if it is deemed futile to continue. The trial would therefore proceed with a single objective and consequently a single estimand, focused on the BM+ subpopulation.
Although the population is often divided into BM+ and BM−, the underlying biomarker is often continuous. One example is PD/L1 protein expression, for which different trials 38 have been using very different cutoffs to define BM+. A trial could be designed to adaptively choose an optimal cutoff, at an interim or the final analysis. In this case, a continuum of subpopulations can be prespecified and defined as PD/L1 expression > x%, for a range of values x. Consequently, there will be a continuum of predefined estimands, of which one will finally be chosen.
Basket trials
The same two treatments, E and C, may be compared in the same trial in several different settings. A basket trial may, e.g., include multiple tumor types. If the objective of the trial is to assess treatment efficacy in each tumor type separately, then each type needs its own prespecified estimand and these estimands may potentially differ in several different ways (Table 2 ), as discussed in Section Estimands and innovative designs.
Table 2.
Differences and similarities between the estimands of a given treatment tested in different diseases/subtypes within the same master protocol
| Estimand attribute | Guidance |
|---|---|
| A. Treatment | Same for each disease / subtype |
| B. Population | Different for each disease / subtype (but could have a common denominator, e.g., a positive biomarker) |
| C. Variable |
Could vary as the same drug could target different aspects in different populations (e.g., in oncology: OS, PFS, ORR) |
| D. Intercurrent events | Population‐specific IEs would be different, whereas treatment‐specific IEs would be the same |
| E. Population‐level summary | Would change with the variable and the population (e.g., a same binary variable could lead to percent difference in one population and odds ratio in the other if adjustment is needed) |
IEs, intercurrent events; OS, overall survival; ORR, observed response rate; PFS, progression‐free survival.
An adaptive design could also select a more informative subset of these tumor types for further investigation in the latter part of the trial, which would correspond to a selection of corresponding estimands, e.g., at interim.
New tumor types could be added during the trial (e.g., after in vitro/in vivo models indicating treatment potential). This would add new objectives to the trial and therefore each new tumor type would need a dedicated estimand.
In basket trials, it is not always feasible to adequately power separate treatment comparisons for each tumor type. Provided there is a clinical and biological rationale to do so, one objective of the study could therefore be to estimate the treatment effect in the pooled set of populations. Such pooling is facilitated if the end point and intercurrent events are similar across tumor types. To this objective would therefore correspond a specific single estimand whose “population” attribute focuses on the pooled set of populations. When pooling is applied, it is likely that some interest will still be placed on efficacy by tumor type. Thus, the tumor‐specific estimands would still be of relevance. An alternative to pooling tumor types is to borrow information between them as discussed in the next section.
Other types of adaptations
Other types of trial design modifications are frequently encountered in drug development and cannot be grouped in the previous classification:
Sample size reassessment 39 consists in potentially increasing the sample size based on an interim readout in order to boost the power of the trial. As explained in ref. 20, such an adaptation is linked to the estimation process rather than the objectives of the study and this would therefore not be reflected in the estimand of the study.
The same would logically apply to a change to the randomization ratio.
For a similar reason, early stop for efficacy or futility in group sequential designs 7 would not trigger a modification of the objective or of the estimand of the study. 20
An unplanned change of primary end point would likely reflect a change to the primary objective of the trial. Furthermore, a modification of the definition of the end point or the way it is measured would be associated with a modification of the corresponding estimand attribute. An example in a diabetes and chronic kidney disease trial 40 was the modification of the components of a primary, composite end point based on various cardiovascular events. This decision was enforced in order to increase the number of events after the trial fell short of funding and would therefore not provide the power needed for the primary end point as initially defined. In practice, the end point attribute of the primary estimand of the trial should have been updated accordingly.
POINTS TO CONSIDER ON ESTIMATION
The estimand framework distinguishes between the estimand, defined as the target of estimation, and the estimator, the method of estimation. 1
Master protocols and platform trials typically have multiple objectives and therefore multiple estimands. These may correspond, for example, to different subgroups or treatments. Furthermore, in platform trials not all estimands are defined at the start of the trial but new estimands will be added when new substudies are added to the platform. In addition, estimands may be dropped or selected in interim analyses, e.g., if treatment arms are stopped for futility, efficacy, or safety reasons or are based on external information. The data‐dependent selection of estimands poses additional challenges to define reliable estimators, confidence intervals, and hypothesis tests. Especially if the selection of the estimand depends on information internal to the trial, estimators that do not account for the selection may be biased. For example, if experimental arms are selected based on interim outcomes (see Section Adding or selecting experimental treatment arms), selecting arms with the most promising interim results will lead to positively biased estimates and invalid confidence intervals and hypothesis tests if not appropriately accounted for. 41 , 42 , 43 Similarly, if subpopulations are selected based on interim data (as discussed in Section Adding, selecting or pooling populations) the resulting estimates will be biased and appropriately adjusted confidence intervals and hypothesis tests have to be applied to control the error probabilities for the selected estimand. 7 , 44 Bias associated with the selection of estimands is closely related to the multiplicity problem that arises when testing multiple hypotheses. To which extent an adjustment for multiplicity (and thus a correction for the related selection bias) is required will depend on the trial objective and is also under discussion for confirmatory platform trials. 9 In addition, besides the bias, the utility of estimators is also determined by their variability. Especially in settings with large variability, as in small trials in rare disease settings, reducing the variability at the cost of a moderate bias can lead to more accurate estimators in terms of the mean squared error. The trade‐off between variance and bias has to be accounted for when assessing estimators.
Estimation becomes even more challenging if the estimand changes in an ongoing trial. For example, if the control arm changes (Section Modifying the control arm), then the trial objective and the estimands are also likely to be changed during the trial. If the estimand changes, also the corresponding estimator needs to be adapted accordingly. In addition, if the change of estimand is dependent on interim data, estimators that do not account for the data‐dependent change may have poor operating characteristics.
For trials in difficult experimental situations, where adequately powered clinical trials are not feasible (as, e.g., in small subgroups, rare diseases, or the pediatric population), it has been proposed to use information borrowing as a tool to reduce the sample size or to improve the power of statistical tests and increase the precision of treatment effect estimates. 45 This tool can also be used to construct more precise estimators for estimands in platform trials. If the estimand of the trial from which information is borrowed (the source trial) differs from the estimand of the trial for which inference should be made (the target trial), the estimator from the source trial will typically not be appropriate, and corresponding adjustments (e.g., for inclusion and exclusion criteria, covariates, or if populations differ) will be required. If such an adjustment is not possible, as, for example, in settings where information from a disjoint population is borrowed (e.g., from adults to children, or different tumor types), information borrowing relies on prior assumptions on the similarity of the estimands in the two populations. Information borrowing can also be applied within a platform trial or a basket trial where information is borrowed across respective treatments or populations. 46 Furthermore, for platform trials where treatment arms are added during the trial, 30 it has been proposed to borrow information from nonconcurrent controls. Borrowing information within a trial is less prone to biases than borrowing from external trials, as many aspects of trial conduct are standardized and are less likely to cause bias. 47 However, risk of bias remains, if, e.g., time trends are not appropriately adjusted for when using nonconcurrent controls in treatment‐to‐control comparisons. 48 , 49 , 50
The choice of estimands in master protocols has to be paired with an assessment of feasibility, i.e., if valid estimators are available, in spite of the selection and adaptive modifications of the estimand. If no valid estimator for an estimand is available, several iterations between formulating the target questions and the assessment of feasibility of estimation may be required. Besides the definition of the main estimator, sensitivity analysis to assess the robustness of the estimators with respect to the underlying assumptions should be specified. Here simulation studies and tipping point analyses can play an important role. They can support the assessment of the properties of estimators under a range of scenarios, corresponding to different assumptions on the distribution of outcomes, intercurrent events, missing data mechanisms, and other trial characteristics. For platform trials, special emphasis must be put on the potential impact of time trends caused by adaptations, such as the addition/dropping of treatment arms or changes in the allocation ratios. In addition, external time trends, as changes in the standard of care or seasonal variation, need to be considered. 48
It is important to realize that multiple key scientific questions and related estimands may be needed in a trial. A stratified comparison vs. different standard‐of care controls 13 or a pooled analysis, over different tumor types or subpopulations, may prove overall efficacy. Still, the corresponding estimands differ from those of the more targeted comparisons vs. the most recent standard of care in a certain subpopulation.
CONCLUSIONS
Trials can have multiple estimands, and it is essential to clearly define those in advance. When planning an adaptive design, it is particularly important to anticipate changes in estimands that would occur by design. Regulators can be faced with the task to make several decisions at the same time; for this it is essential to have transparently documented the estimand discussion and conveyed the characteristics of each of them. In particular, the consequences of planned adaptations, such as patient selection (e.g., all comers vs. BM+), can and should be described in terms of objectives and estimands at the start of the trial.
A different situation is the need to adapt a trial protocol to unplanned events, COVID‐19 being the most prominent example at the moment. Developers must rediscuss the original estimand to determine whether it has been impacted by any measures taken outside their own development that could impact the attributes of the originally intended estimand. In particular changes to the population and new emerging intercurrent events that could not have been anticipated at the planning stage must be considered. 51 , 52 Another aspect requiring consideration is the rapidly changing treatment landscape that has an impact on the population as a whole, the control arm, and the treatment itself. In general, the study team will have to decide, potentially following a discussion with regulatory agencies, whether the original estimand can still be estimated or whether the objectives of study have been impacted by those changes. In fact, the estimand framework is a perfect tool to explore such impacts by providing a comprehensive and agreed‐upon language to developers and regulators.
Finally, it is important to emphasize that the estimand framework applies to any study in which a treatment effect is estimated, including complex innovative designs. This was illustrated in this article for several examples of adaptions and innovative elements. In general, more experience with the estimand framework is needed on both sides of the regulatory divide. In particular more examples of implementation of the estimand framework to areas such as noninferiority trials, early‐phase trials, pharmacokinetic/pharmacodynamic studies, observational studies, and studies using real‐world data 53 , 54 would be useful.
FUNDING
EU‐PEARL has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No. 853966.
CONFLICT OF INTEREST
C.‐F.B. is an employee of and shareholder of AstraZeneca. O.C. is an employee of and shareholder of GSK. F.B. is an employee of and shareholder of Novartis. K.R. is an employee of and shareholder of Roche. All other authors declared no competing interests for this work.
ACKNOWLEDGMENTS
Frank Bretz, Carl‐Fredrik Burman and Martin Posch are members of the EU Patient Centric Clinical Trial Platforms (EU‐PEARL). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA and Children’s Tumor Foundation, Global Alliance for TB Drug Development nonprofit organization, Springworks Therapeutics Inc. This publication reflects the authors‘ views. Neither IMI nor the European Union, EFPIA, nor any Associated Partners are responsible for any use that may be made of the information contained herein.
- 1. International Council For Harmonisation of Technical Requirements For Pharmaceuticals For Human Use (ICH) . ICH E9(R1) addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials <https://database.ich.org/sites/default/files/E9‐R1_Step4_Guideline_2019_1203.pdf> (2019).
- 2. Akacha, M. , Bretz, F. , Ohlssen, D.I. , Rosenkranz, G.K. & Schimdli, H. Estimands and their role in clinical trials. Stat. Biopharm. Res. 9, 268–271 (2017). [Google Scholar]
- 3. Akacha, M. , Bretz, F. & Ruberg, S. Estimands in clinical trials–broadening the perspective. Stat. Med. 36, 5–19 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Mallinckrodt, C. et al. Aligning estimators with estimands in clinical trials: putting the ICH E9 (R1) guidelines into practice. Ther. Innov. Regul. Sci. 54, 353–364 (2020). [DOI] [PubMed] [Google Scholar]
- 5. Ratitch, B. et al. Choosing estimands in clinical trials: putting the ICH E9 (R1) into practice. Ther. Innov. Regul. Sci. 54, 324–341 (2020). [DOI] [PubMed] [Google Scholar]
- 6. Bauer, P. , Bretz, F. , Dragalin, V. , König, F. & Wassmer, G. Twenty‐five years of confirmatory adaptive designs: opportunities and pitfalls. Stat. Med. 35, 325–347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bretz, F. , Koenig, F. , Brannath, W. , Glimm, E. & Posch, M. Adaptive designs for confirmatory clinical trials. Stat. Med. 28, 1181–1217 (2009). [DOI] [PubMed] [Google Scholar]
- 8. Collignon, O. et al. Adaptive designs in clinical trials: from scientific advice to marketing authorisation to the European Medicine Agency. Trials 19, 642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Collignon, O. et al. Current statistical considerations and regulatory perspectives on the planning of confirmatory basket, umbrella, and platform trials. Clin. Pharmacol. Ther. 107, 1059–1067 (2020). [DOI] [PubMed] [Google Scholar]
- 10. Meyer, E.L. et al. The evolution of master protocol clinical trial designs: a systematic literature review. Clin. Ther. 42, 1330–1360 (2020). [DOI] [PubMed] [Google Scholar]
- 11. Verweij, J. , Hendriks, H.R. , Zwierzina, H. , Cancer Drug Development Forum . Innovation in oncology clinical trial design. Cancer Treat. Rev. 74, 15–20 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Collignon, O. , Posch, M. & Schiel, A. Assessment of tumour‐agnostic therapies in basket trials. Lancet Oncol. 23, e8 (2022). [DOI] [PubMed] [Google Scholar]
- 13. Collignon, O. , Burman, C.F. , Posch, M. & Schiel, A. Collaborative platform trials to fight COVID‐19: methodological and regulatory considerations for a better societal outcome. Clin. Pharmacol. Ther. 110, 311–320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horby, P.W. et al. Convalescent plasma in patients admitted to hospital with COVID‐19 (RECOVERY): a randomised, controlled, open‐label, platform trial. Lancet 397, 2049–2059 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collignon, O. , Schritz, A. , Senn, S.J. & Spezia, R. Clustered allocation as a way of understanding historical controls: components of variation and regulatory considerations. Stat. Methods Med. Res. 29, 1960–1971 (2020). [DOI] [PubMed] [Google Scholar]
- 16. Collignon, O. , Schritz, A. , Spezia, R. & Senn, S.J. Implementing historical controls in oncology trials. Oncologist 26, e859–e862 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghadessi, M. et al. A roadmap to using historical controls in clinical trials—by Drug Information Association Adaptive Design Scientific Working Group (DIA‐ADSWG). Orphanet J. Rare Dis. 15, 69 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lim, J. et al. Reducing patient burden in clinical trials through the use of historical controls: appropriate selection of historical data to minimize risk of bias. Ther. Innov. Regul. Sci. 54, 850–860 (2020). [DOI] [PubMed] [Google Scholar]
- 19. Schmidli, H. , Häring, D.A. , Thomas, M. , Cassidy, A. , Weber, S. & Bretz, F. Beyond randomized clinical trials: Use of external controls. Clin. Pharmacol. Ther. 107, 806–816 (2020). [DOI] [PubMed] [Google Scholar]
- 20. Okwuokenye, M. & Peace, K.E. Adaptive design and the estimand framework. Ann. Biostat. Biomet. Appl. 1 (2019). 10.33552/ABBA.2019.01.000524 [DOI] [Google Scholar]
- 21. European Medicines Agency (EMA) . Reflection paper on methodological issues in confirmatory clinical trials planned with an adaptive design <https://www.ema.europa.eu/en/documents/scientific‐guideline/reflection‐paper‐methodological‐issues‐confirmatory‐clinical‐trials‐planned‐adaptive‐design_en.pdf> (2007).
- 22. US Food and Drug Administration . Guidance document: adaptive design clinical trials for drugs and biologics guidance for industry <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/adaptive‐design‐clinical‐trials‐drugs‐and‐biologics‐guidance‐industry> (2019).
- 23. US Food and Drug Administration . Interacting with the FDA on complex innovative trial designs for drugs and biological products <https://www.fda.gov/media/130897/download> (2020).
- 24. Neuenschwander, B. , Wandel, S. , Roychoudhury, S. & Bailey, S. Robust exchangeability designs for early phase clinical trials with multiple strata. Pharm. Stat. 15, 123–134 (2016). [DOI] [PubMed] [Google Scholar]
- 25. van Brummelen, E.M.J. , Ros, W. , Wolbink, G. , Beijnen, J.H. & Schellens, J.H.M. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist 21, 1260–1268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moussa, E.M. et al. Immunogenicity of therapeutic protein aggregates. J. Pharm. Sci. 105, 417–430 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Bornkamp, B. et al. Principal stratum strategy: potential role in drug development. Pharm. Stat. 20, 737–751 (2021). [DOI] [PubMed] [Google Scholar]
- 28. Kong, S. , Heinzmann, D. , Lauer, S. & Tian, L. Weighted approach for estimating effects in principal strata with missing data for a categorical post‐baseline variable in randomized controlled trials. Stat. Biopharm. Res. 1–11 (2021). 10.1080/19466315.2021.2009020 [DOI] [Google Scholar]
- 29. Cohen, D.R. , Todd, S. , Gregory, W.M. & Brown, J.M. Adding a treatment arm to an ongoing clinical trial: a review of methodology and practice. Trials 16, 179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee, K.M. , Brown, L.C. , Jaki, T. , Stallard, N. & Wason, J. Statistical consideration when adding new arms to ongoing clinical trials: the potentials and the caveats. Trials 22, 203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee, K.M. , Wason, J. & Stallard, N. To add or not to add a new treatment arm to a multiarm study: a decision‐theoretic framework. Stat. Med. 38, 3305–3321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carreras, M. , Gutjahr, G. & Brannath, W. Adaptive seamless designs with interim treatment selection: a case study in oncology. Stat. Med. 34, 1317–1333 (2015). [DOI] [PubMed] [Google Scholar]
- 33. Thuss‐Patience, P.C. et al. Trastuzumab emtansine versus taxane use for previously treated HER2‐positive locally advanced or metastatic gastric or gastro‐oesophageal junction adenocarcinoma (GATSBY): an international randomised, open‐label, adaptive, phase 2/3 study. Lancet Oncol. 18, 640–653 (2017). [DOI] [PubMed] [Google Scholar]
- 34. RECOVERY Collaborative Group . Effect of hydroxychloroquine in hospitalized patients with Covid‐19. N. Engl. J. Med. 383, 2030–2040 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. EMA . Guideline on the clinical evaluation of anticancer medicinal products <https://www.ema.europa.eu/en/documents/scientific‐guideline/draft‐guideline‐evaluation‐anticancer‐medicinal‐products‐man‐revision‐6_en.pdf> (2020).
- 36. Jenkins, M. , Stone, A. & Jennison, C. An adaptive seamless phase II/III design for oncology trials with subpopulation selection using correlated survival endpoints. Pharm. Stat. 10, 347–356 (2011). [DOI] [PubMed] [Google Scholar]
- 37. Edgar, K. , Jackson, D. , Rhodes, K. , Duffy, T. , Burman, C.F. & Sharples, L.D. Frequentist rules for regulatory approval of subgroups in phase III trials: a fresh look at an old problem. Stat. Methods Med. Res. 30, 1725–1743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brody, R. et al. PD‐L1 expression in advanced NSCLC: insights into risk stratification and treatment selection from a systematic literature review. Lung Cancer 112, 200–215 (2017). [DOI] [PubMed] [Google Scholar]
- 39. Gould, A.L. Planning and revising the sample size for a trial. Stat. Med. 14, 1039–1051 (1995). [DOI] [PubMed] [Google Scholar]
- 40. Bhatt, D.L. et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N. Engl. J. Med. 384, 129–139 (2021). [DOI] [PubMed] [Google Scholar]
- 41. Bauer, P. , Koenig, F. , Brannath, W. & Posch, M. Selection and bias—two hostile brothers. Stat. Med. 29, 1–13 (2010). [DOI] [PubMed] [Google Scholar]
- 42. Posch, M. , Maurer, W. & Bretz, F. Type I error rate control in adaptive designs for confirmatory clinical trials with treatment selection at interim. Pharm. Stat. 10, 96–104 (2011). [DOI] [PubMed] [Google Scholar]
- 43. Posch, M. , Koenig, F. , Branson, M. , Brannath, W. , Dunger‐Baldauf, C. & Bauer, P. Testing and estimation in flexible group sequential designs with adaptive treatment selection. Stat. Med. 24, 3697–3714 (2005). [DOI] [PubMed] [Google Scholar]
- 44. Magirr, D. , Jaki, T. , Posch, M. & Klinglmueller, F. Simultaneous confidence intervals that are compatible with closed testing in adaptive designs. Biometrika 100, 985–996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schmidli, H. , Gsteiger, S. , Roychoudhury, S. , O’Hagan, A. , Spiegelhalter, D. & Neuenschwander, B. Robust meta‐analytic‐predictive priors in clinical trials with historical control information. Biometrics 70, 1023–1032 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Chen, N. & Lee, J.J. Bayesian cluster hierarchical model for subgroup borrowing in the design and analysis of basket trials with binary endpoints. Stat. Methods Med. Res. 29, 2717–2732 (2020). [DOI] [PubMed] [Google Scholar]
- 47. Burger, H.U. et al. The use of external controls: to what extent can it currently be recommended? Pharm. Stat. 20, 1002–1016 (2021). [DOI] [PubMed] [Google Scholar]
- 48. Dodd, L.E. , Freidlin, B. & Korn, E.L. Platform trials – beware the noncomparable control group. N. Engl. J. Med. 384, 1572–1573 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee, K.M. & Wason, J. Including non‐concurrent control patients in the analysis of platform trials: is it worth it? BMC Med. Res. Methodol. 20, 165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sridhara, R. et al. Use of nonconcurrent common control in master protocols in oncology trials: report of an American statistical association biopharmaceutical section open forum discussion. Stat. Biopharm. Res. 1–5 (2021). 10.1080/19466315.2021.1938204 [DOI] [Google Scholar]
- 51. Degtyarev, E. et al. Assessing the impact of COVID‐19 on the clinical trial objective and analysis of oncology clinical trials – application of the estimand framework. Stat. Biopharm. Res. 12, 427–437 (2020): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyer, R.D. et al. Statistical issues and recommendations for clinical trials conducted during the COVID‐19 pandemic. Stat. Biopharm. Res. 12, 399–411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Levenson, M. et al. Biostatistical considerations when using RWD and RWE in clinical studies for regulatory purposes: a landscape assessment. Stat. Biopharm. Res. 1–11 (2021). [Google Scholar]
- 54. International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) . ICH E9(R1) Step 2 training material <https://database.ich.org/sites/default/files/E9‐R1_EWG_Step2_TrainingMaterial.pdf> (2018).
- 55. Akacha, M. et al. Estimands‐What they are and why they are important for pharmacometricians. CPT Pharmacometrics Syst Pharmacol. 10, 279–282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]