

Abstract
Objective
The aim of this study was to investigate the pharmacokinetic, pharmacodynamic and safety profile of the glucagon‐like peptide‐1 receptor agonist, lixisenatide, for the treatment of type 2 diabetes (T2D) in pediatric individuals.
Materials and Methods
In this Phase 1, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group, ascending repeated dose study (NCT02803918), participants aged ≥10 and < 18 years were randomized 3:1 to receive once‐daily lixisenatide in 2‐week increments of 5, 10, and 20 μg (n = 18) or placebo (n = 5) for 6 weeks.
Results
Mean lixisenatide concentrations generally increased with increasing doses irrespective of anti‐drug antibody (ADA) status; however, mean lixisenatide concentrations and inter‐subject variability were higher for participants with positive ADA status. Improvements in fasting plasma glucose, post‐prandial glucose, AUC0–4.5, HbA1c, and body weight were observed with lixisenatide. Overall, the safety profile was consistent with the known profile in adults, with no unexpected side effects and no treatment‐emergent adverse events resulting in death or discontinuation. The most common events in the lixisenatide group were vomiting (11.1%) and nausea (11.1%). No symptomatic hypoglycemia was reported in either group. No clinically significant hematologic, biochemical or vital sign abnormalities were observed.
Conclusions
Mean lixisenatide concentrations generally increased with increasing dose, irrespective of ADA status. Lixisenatide was associated with improved glycemic control and a trend in body weight reduction compared with placebo. The safety and tolerability profile of repeated lixisenatide doses of up to 20 μg per day in children and adolescents with T2D was reflective of the established safety profile of lixisenatide in adults.
Keywords: antidiabetic drug, clinical trial, pharmacodynamics, pharmacokinetics, type 2 diabetes
1. INTRODUCTION
The incidence of type 2 diabetes (T2D) in children and adolescents worldwide has markedly increased over the last few decades, 1 for example, rising annually by 7% between 2002 and 2012 in youths aged 10–19 years in the United States. 2 There is a strong relationship between increasing incidence of obesity and increasing incidence of T2D among pediatric populations, with 18.5% of youth aged 2–19 years in the U.S. meeting the criteria for obesity in 2015–2016. 3 , 4 Evidence suggests that progression of T2D is faster in pediatric populations than in adult populations due to faster β‐cell function deterioration in young people. 5 This increases the likelihood of complications such as hypertension, dyslipidemia, fatty liver and microvascular complications in early adulthood. 3 , 6 , 7
Being a progressive condition, T2D treatment requires intensification to maintain good glycemic control. Current diabetes therapy guidelines recommend that people with T2D suboptimally controlled on oral antihyperglycemic drugs (OADs) should be intensified to a glucagon‐like peptide‐1 receptor agonist (GLP‐1 RA) as the first injectable therapy. 8 Lixisenatide is a GLP‐1 RA that lowers blood glucose in people with T2D by delaying gastric emptying, enhancing glucose‐dependent insulin secretion by β‐cells, and suppressing glucagon secretion by α‐cells. 9 , 10 This drug is approved in many countries, including the EU, where the addition of lixisenatide is indicated for adults with T2D suboptimally controlled on OADs and/or basal insulin together with diet and exercise, 11 and in the United States, where lixisenatide is indicated as an adjunct to diet and exercise to improve glycemic control in adults with T2D. 12
There are numerous challenges to achieving and maintaining good glycemic control in pediatric populations, including a complex interplay of family dynamics, mental health, ability to provide self‐care, supervision in the childcare and school environment, and affordability of medications as observed for adults with T2D. 13 , 14 This can reduce the impact of individual lifestyle changes such as increased exercise and improved nutrition for the treatment of T2D. 14 Despite the increase in childhood diabetes and the diverse range of oral and injectable agents available for the treatment of T2D in adults, most have not been studied in children. 15 The ability to perform long‐term interventional clinical studies in children and adolescents is limited due to insufficient pediatric trial infrastructure and inclusion/exclusion criteria that limit an already restricted participant pool. 16 Due to this paucity of clinical research in pediatric populations, pharmacological therapy options are limited to metformin, insulin (basal insulin or basal insulin plus premeal bolus insulin), liraglutide (since 2019) and extended‐release exenatide (since 2021). 14 , 17 , 18 Consequently, there is still limited evidence for the efficacy and safety of different GLP‐1 RAs in people <18 years of age, and thus controlled pediatric trials are required to expand the repertoire of available therapies. 19
The aim of this study was to evaluate the pharmacokinetics (PK), pharmacodynamics (PD) and safety of increasing doses of lixisenatide over 6 weeks of treatment in individuals aged ≥10 and < 18 years with T2D.
2. METHODS
2.1. Study design
In this Phase 1, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group, ascending repeated dose study (NCT02803918), pediatric participants with T2D were randomized (3:1) using a central randomization method to receive once‐daily lixisenatide or placebo. Eligible individuals for inclusion in the study were male or female, aged ≥10 and < 18 years with: documented T2D suboptimally controlled with stable doses of metformin and/or basal insulin for 8 weeks prior to randomization with no use of other antihyperglycemic treatments within 1 month prior to screening; HbA1c at screening >6.5% and ≤ 11% (48 and 97 mmol/mol, respectively); Body Mass Index (BMI) >85th percentile for age and gender; BMI ≤50 kg/m2; fasting C‐peptide at screening >0.6 ng/mL (>0.20 nmol/L); a negative test at screening for anti‐insulinoma associated protein 2 and anti‐glutamic acid decarboxylase autoantibodies; no contraindication for GLP‐1 RAs. For females, eligibility required no ongoing pregnancy, or adequate contraception if sexually active.
Participants received lixisenatide 5 μg daily for the first 2 weeks, which was escalated to 10 μg and then 20 μg daily for 2 weeks each (Figure 1), or matching placebo. Treatment was blinded, but the dose was not. Lixisenatide 5 μg was administered subcutaneously using a pen‐type injector (Tactipen®), and lixisenatide 10 μg and 20 μg were administered subcutaneously using a disposable pre‐filled pen (Delta 14®). Subcutaneous injection alternated between the left and right anterolateral and the left and right posterolateral abdominal walls and, depending on the participant's maturity, they could either self‐inject or a parent could administer injections. If appropriate, a home nursing service was provided for the first three injections (or more if needed) to ensure good compliance. Compliance was defined as the actual number of days with at least one administration of lixisenatide compared to the planned number of days with lixisenatide administration during the treatment period up to treatment discontinuation. Lifestyle and diet therapies provided before screening were continued in a similar manner during the study. Doses and regimens of metformin and/or basal insulin at enrolment remained stable throughout the study, but adjustments of basal insulin were possible at the discretion of the investigator.
FIGURE 1.
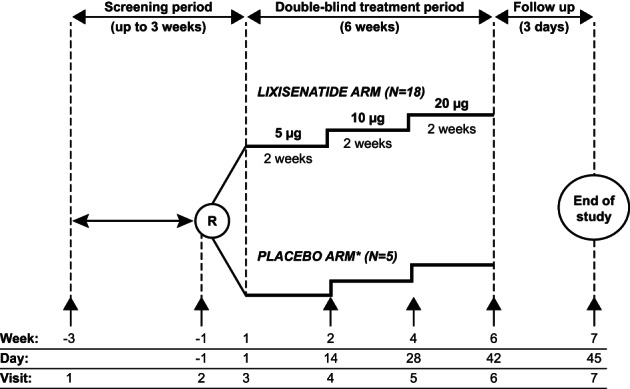
Study design. †Placebo solution and volume to be injected matching to lixisenatide solution. R, randomization; ↑, onsite visit
Participants were seen at seven visits and were required to have fasted for 8 h before breakfast on Visits 2, 4, 5, and 6 (Figure 1). Plasma glucose profiles were measured at baseline (Day −1) and Days 14, 28 and 42. At baseline and Day 42, plasma glucose profiles were measured from 30 min prior to a standardized liquid breakfast until up to 4.5 h later, while PK profiles were performed under the same conditions on Day 42 only. On Days 14 and 28, plasma glucose profiles and PK samples were taken from 30 min prior to a standardized liquid breakfast, until up to 2.5 h later. HbA1c was measured at screening and Day 42, while body weight was measured at all onsite study visits except Visit 3. Safety information was collected throughout the study duration.
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice, and the protocol was approved by institutional review boards or ethics committees at each study site. Each participant and their parent (or the participant's legal representative) provided written informed assent and consent, respectively.
2.2. Outcomes
The primary objective was to assess the safety of 14‐day repeated lixisenatide doses of 5, 10, and 20 μg compared with placebo, by occurrence of adverse events (AEs, coded according to the Medical Dictionary for Regulatory Activities [MedDRA v22.1]), treatment‐emergent AEs (TEAEs, defined as AEs that occurred, worsened or became serious during the on‐treatment phase), and clinical laboratory evaluations including vital signs, 12‐lead electrocardiogram (ECG) parameters, body temperature and physical examination. Other safety evaluations included the presence of anti‐lixisenatide antibodies in plasma samples taken at baseline (Day −1), and Days 14, 28, and 42. Anti‐lixisenatide antibody status and concentration were measured by a validated assay based on Surface Plasmon Resonance (SPR) using Biacore technology.
Secondary objectives evaluated PK and PD parameters after repeated doses of lixisenatide. PK assessments included plasma concentrations before and up to 2.5 h post final dose (for lixisenatide 5 and 10 μg doses), maximum plasma concentration (Cmax), time to reach Cmax (tmax), and area under the curve from 0 h to 4.5 h (AUC0–4.5) before and up to 4.5 h post dose (for lixisenatide 20 μg dose). PD assessments included change from baseline in plasma glucose AUC0–4.5 for lixisenatide 20 μg; change from baseline in fasting plasma glucose (FPG) and change from baseline in 2‐h post‐prandial glucose (PPG) excursion for lixisenatide 5, 10, and 20 μg. Other PD assessments included changes from baseline in HbA1c (Day 42), plasma glucose profiles (Days 14, 28, and 42), and changes in body weight and BMI (Days 14, 28, 42, and 45).
2.3. Data analysis
The sample size for this study was based upon empirical considerations, with no formal sample size calculation performed. Safety analyses were performed in the safety population, defined as all randomized participants exposed to the study drug, regardless of the amount of treatment administered. Where statistical analysis was needed, safety data were evaluated using appropriate estimations and confidence intervals, otherwise evaluation was based upon the review of the individual values and descriptive statistics. PK analyses were performed for all participants with no major deviations related to administration of the study drug, who provided at least one blood sample for drug concentration measurement. Lixisenatide PK parameters were summarized using descriptive statistics for each dose group. All participants without any important deviation related to study drug administration for whom PD data were considered sufficient and interpretable were included in the PD population. Descriptive statistics for absolute values and changes from baseline in FPG, glucose AUC0–4.5 and 2‐h‐PPG excursions were calculated per treatment group and, for exploratory purposes, the changes from baseline in these parameters were analyzed using linear models. For HbA1c and body weight, descriptive analyses for absolute values and change from baseline were provided per treatment group.
3. RESULTS
3.1. Participant disposition and baseline characteristics
In total, 44 individuals were screened for study entry of whom 23 were eligible for inclusion and randomized to receive treatment: five with placebo and 18 with lixisenatide (Supplementary Figure 1). Study participants were recruited from 11 centers across the United States, Spain, Mexico, Mauritius, South Africa, and Turkey and were all between 13 and 17 years of age. The first participant was enrolled on May 17, 2017, and the last participant completed the study on January 27, 2020. Apart from one individual in the lixisenatide group who discontinued treatment due to poor compliance to the study protocol, all participants completed the study treatment period. Treatment compliance was >80–100% across both treatment groups.
Baseline demographics and disease characteristics are shown in Table 1. While some characteristics were similar between groups, there were some notable differences as expected with a small sample size, including differences in FPG, body weight, BMI, duration and age at onset of T2D, and creatinine clearance. Approximately, 30% of all participants received concomitant insulin at baseline (Table 1).
TABLE 1.
Baseline demographics and disease characteristics by treatment group
| Characteristic | Lixisenatide (N = 18) | Placebo (N = 5) |
|---|---|---|
| Age, years | ||
| Mean ± SD | 15.6 ± 1.0 | 15.4 ± 1.5 |
| Range | 14, 17 | 13, 17 |
| Duration of diabetes, years | ||
| Mean ± SD | 1.6 ± 1.2 | 3.5 ± 2.2 |
| Range | 0, 5 | 0, 6 |
| Age at diabetes onset, years | ||
| Mean ± SD | 14.3 ± 1.2 | 11.7 ± 3.1 |
| Range | 13, 17 | 8, 16 |
| Sex, female, n (%) | 13 (72.2) | 3 (60.0) |
| Race, n (%) | ||
| White | 12 (66.7) | 4 (80.0) |
| Black | 4 (22.2) | 0 |
| Asian | 0 | 0 |
| Other | 2 (11.1) | 1 (20.0) |
| Weight, kg | ||
| Mean ± SD | 91.3 ± 18.8 | 98.0 ± 14.7 |
| Range | 61, 123 | 88, 123 |
| BMI, kg/m2 | ||
| Mean ± SD | 33.2 ± 4.8 | 37.4 ± 3.6 |
| Range | 24, 42 | 34, 42 |
| HbA1c, % (mmol/mol) | ||
| Mean ± SD | 8.16 ± 0.93 | 8.14 ± 1.58 |
| (65.7 ± 10.2) | (65.4 ± 17.3) | |
| Range | 6.7, 10.3 | 6.8, 9.9 |
| (50, 89) | (51, 85) | |
| FPG, mmol/L | ||
| Mean ± SD | 9.6 ± 3.1 | 7.1 ± 2.2 |
| Range | 5.3, 16.5 | 4.7, 10.1 |
| Creatinine clearance, mL/min/1.73 m2 | ||
| Mean ± SD | 107.6 ± 17.0 | 129.8 ± 28.7 |
| Range | 81, 143 | 84, 157 |
| Background treatment at screening, n (%) | ||
| Metformin only | 13 (72.2) | 3 (60.0) |
| Basal insulin only | 1 (5.6) | 0 |
| Metformin + basal insulin | 4 (22.2) | 2 (40.0) |
Abbreviations: BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; SD, standard deviation.
3.2. PK outcomes
Mean lixisenatide concentrations generally increased with each increase in dose irrespective of anti‐drug antibody (ADA) status; however, mean lixisenatide concentrations and inter‐subject variability were generally higher for participants with positive ADA status (Table 2; Supplementary Figure 2).
TABLE 2.
Pharmacokinetic parameters at Day 42, following administration of maximum lixisenatide dose (20 μg) for 14 days, presented by ADA‐status (PK population)
| ADA Negative (N = 4) | ADA Positive (N = 14) | |
|---|---|---|
| Cmax, pg/mL, mean ± SD (CV%) | 83.9 ± 25.2 (30) | 508 ± 453 a (89) |
| tmax, h, median (min, max) | 1.24 (0.98, 2.50) | 2.00 (0.50, 4.50) a |
| mean ± SD (CV%), AUC0–4.5, pg·h/mL | 267 ± 96.1 (36) | 2300 ± 1940 b (84) |
Abbreviations: ADA, anti‐drug antibody; AUC0–4.5, area under the curve from 0 h to 4.5 h; Cmax, maximum plasma concentration; CV%, inter‐subject variability; LLOQ, lower limit of quantification; PK, pharmacokinetic; SD, standard deviation; tmax, time to reach maximum concentration; tlast, time of last measurable concentration.
n = 11 as all results were < LLOQ for three participants.
n = 9; not calculable for two participants where tlast was 3.5 h or where there were <3 quantifiable samples, respectively.
At lixisenatide 20 μg, the median tmax was 1.24 h and 2.00 h for ADA‐negative and ADA‐positive participants, respectively, while mean Cmax and AUC0–4.5 were approximately 6‐ to 9‐fold higher, respectively, for ADA‐positive participants compared to those who were ADA‐negative.
3.3. PD outcomes
PD outcomes assessed in this study consisted of HbA1c, FPG, plasma glucose AUC0–4.5, 2‐h PPG excursion and change in body weight, BMI and BMI percentile. For every outcome assessed, participants treated with lixisenatide demonstrated an improvement compared with placebo by Day 42 (Table 3), including improved plasma glucose profiles (Supplementary Figure 3). Notably, at Day 42, 2‐h PPG excursion decreased by −4.0 ± 3.2 mmol/L from a baseline value of 4.0 ± 2.5 mmol/L in the lixisenatide group compared with a decrease of −0.1 ± 1.2 mmol/L from a baseline value of 4.2 ± 3.0 mmol/L in the placebo group (p = 0.0121).
TABLE 3.
Pharmacodynamic and other outcomes at Day 42, following administration of maximum lixisenatide dose (20 μg) for 14 days, presented by treatment group (PD population)
| Lixisenatide N = 18 | Placebo N = 5 | |
|---|---|---|
| HbA1c, % | ||
| Baseline | 8.2 ± 0.9 (6.7, 10.3) | 8.1 ± 1.6 (6.8, 9.9) |
| Day 42 | 7.9 ± 1.3 (5.7, 10.3) | 8.2 ± 1.1 (6.6, 9.5) |
| Change from baseline | −0.3 ± 1.2 (−3.3, 2.5) | 0.1 ± 1.1 (−1.4, 1.1) |
| HbA1c, mmol/mol | ||
| Baseline | 66 ± 10 (50, 89) | 65 ± 17 (51, 85) |
| Day 42 | 63 ± 14 (39, 89) | 66 ± 12 (49, 80) |
| Change from baseline | –3 ± 13 (−36, 27) | 1 ± 12 (−15, 12) |
| BMI, kg/m2 | ||
| Baseline | 33.2 ± 4.8 (24, 42) | 37.4 ± 3.6 (34, 42) |
| Day 42 | 33.4 ± 5.2 (24, 44) | 38.4 ± 4.0 (34, 42) |
| Change from baseline | 0.2 ± 0.6 (−1, 2) | 1.0 ± 0.9 (0, 3) |
| BMI percentile | ||
| Baseline | 98.28 ± 3.55 (86.30, 99.99) | 99.88 ± 0.14 (99.66, 99.98) |
| Day 42 | 98.07 ± 4.10 (84.19, 99.99) | 99.90 ± 0.12 (99.72, 99.99) |
| Change from baseline | −0.21 ± 0.56 (−2.11, 0.17) | 0.02 ± 0.03 (0.00, 0.06) |
| Body weight, kg | ||
| Baseline | 91.3 ± 18.8 (61.1, 123.0) | 98.0 ± 14.7 (88.4, 123.0) |
| Day 42 | 92.0 ± 19.7 (60.2, 123.2) | 100.8 ± 17.5 (90.0, 131.0) |
| Change from baseline | 0.7 ± 1.8 (−1.6, 5.0) | 2.8 ± 3.0 (0.9, 8.0) |
| FPG, mmol/L | ||
| Baseline | 9.6 ± 3.1 (5.3, 16.5) | 7.1 ± 2.2 (4.7, 10.1) |
| Day 42 | 8.4 ± 2.0 (4.9, 11.3) | 10.0 ± 3.6 (6.5, 16.0) |
| Change from baseline | −1.2 ± 2.1 (−6.2, 2.5) | 2.9 ± 3.7 (−0.3, 9.2) |
| Estimated treatment difference (95% CI); p‐value | −4.2 (−6.8, −1.6); p = 0.0030 | |
| Glucose AUC0–4.5, mmol·h/L | ||
| Baseline | 55.2 ± 16.9 (24.6, 85.2) | 44.0 ± 13.5 (32.1, 62.4) |
| Day 42 | 38.7 ± 10.7 (19.6, 62.6) | 57.8 ± 17.5 (40.4, 84.8) |
| Change from baseline | −17.3 ± 12.2 (−40.3, 9.0) | 13.8 ± 18.9 (0.5, 46.5) |
| Estimated treatment difference (95% CI); p‐value | −31.2 (−46.3, −16.1); p = 0.0004 | |
| 2‐hr PPG excursion, mmol/L | ||
| Baseline | 4.0 ± 2.5 (−0.2, 7.8) | 4.2 ± 3.0 (2.5, 9.5) |
| Day 42 | 0.1 ± 2.4 (−2.9, 6.3) | 4.1 ± 2.1 (2.6, 7.7) |
| Change from baseline | −4.0 ± 3.2 (−9.9, 1.1) | −0.1 ± 1.2 (−1.8, 1.4) |
| Estimated treatment difference (95% CI); p‐value | −3.9 (−6.8, −0.9); p = 0.0121 | |
Note: Data are presented as mean ± SD (range), unless otherwise stated.
Abbreviations: AUC0–4.5, area under the curve from 0 h to 4.5 h; BMI, body mass index; CI, confidence interval; FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; PD, pharmacodynamic; PPG, post‐prandial glucose; SD, standard deviation.
3.4. Safety outcomes
All participants received their assigned treatment for at least 42 days, during which three participants (60%) in the placebo group reported four TEAEs and seven participants (39%) in the lixisenatide group reported 32 TEAEs (Table 4). Gastrointestinal (GI) disorders were reported by two participants for each lixisenatide dose level and by one participant in the placebo group (nausea), none of which were serious. Vomiting was the most commonly reported study drug‐related TEAE, with 11 events occurring in two participants at the 20 μg dose level of lixisenatide. Two vomiting events were reported as severe in one of the participants, who also reported seven nausea events and three dizziness events. There were no reports of symptomatic hypoglycemia in any treatment group. One event of injection site pain was reported by one participant in the lixisenatide group at the 20 μg dose level, which was considered as drug related by the investigator.
TABLE 4.
Adverse events by treatment group and dose (safety population)
| n (%) of participants with ≥1 TEAE in each category | Lixisenatide | Placebo (N = 5) | |||
|---|---|---|---|---|---|
| All (N = 18) | 5 μg (N = 18) | 10 μg (N = 18) | 20 μg (N = 18) | ||
| Any AESI (ALT increased) | 0 | 0 | 0 | 0 | 1 (20.0) |
| Any treatment‐emergent SAE (viral gastroenteritis) | 1 (5.6) | 0 | 0 | 1 (5.6) | 0 |
| Any TEAE | 7 (38.9) | 6 (33.3) | 3 (16.7) | 4 (22.2) | 3 (60.0) |
| Severe TEAE | 1 (5.6) | 0 | 0 | 1 (5.6) | 0 |
| Any TEAE leading to death | 0 | 0 | 0 | 0 | 0 |
| Any TEAE leading to permanent treatment discontinuation | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal disorders | 4 (22.2) | 2 (11.1) | 2 (11.1) | 2 (11.1) | 1(20.0) |
| Nausea | 2 (11.1) | 1 (5.6) | 2 (11.1) | 1 (5.6) | 1 (20.0) |
| Vomiting | 2 (11.1) | 0 | 0 | 2 (11.1) | 0 |
| Gastroesophageal reflux disease | 1 (5.6) | 1 (5.6) | 0 | 0 | 0 |
Abbreviations: AESI, adverse event of special interest; ALT, alanine aminotransferase; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
Few participants showed potentially clinically significant abnormalities (PCSAs) for hematology, clinical chemistry or vital signs, with no reported change in renal function, no clinically relevant change in amylase or lipase concentrations, and no obvious differences between the placebo and lixisenatide groups. In the lixisenatide group, one participant showed a PCSA for prolonged QRS interval on ECG, although the participant already had a borderline abnormal interval at baseline that only marginally increased by the end of study. Additionally, four participants showed PCSAs for prolonged QTc interval on ECG at end of study in the lixisenatide group, with two of these participants already having a prolonged QTc at baseline that increased marginally by 1 ms and 5 ms, respectively, by the end of study. Furthermore, one additional participant in the lixisenatide group showed an increase from baseline in the QTc interval of 30–60 ms from 358 ms to 396 ms. No participants had a QTc >500 ms or an increase from baseline >60 ms. No participants in the placebo group reported any ECG PCSAs.
All participants in the placebo group were negative for anti‐lixisenatide antibodies throughout the study, with one exception on Day 42 for one participant (thought to be a false positive). Of the 18 participants who received lixisenatide, 14 were positive for anti‐lixisenatide antibodies at Day 42.
One participant in the placebo group reported an AE of special interest (alanine aminotransferase increased), which was not considered study drug‐related, and one participant in the lixisenatide group experienced a serious event of viral gastroenteritis on Day 32, which was not considered as related to study drug and did not trigger treatment discontinuation. No participant reported any TEAE leading to death or permanent treatment discontinuation.
4. DISCUSSION
This study explored the PK, PD and safety of the GLP‐1 RA, lixisenatide, in comparison to placebo, for the treatment of T2D in pediatric populations aged ≥13 and < 18 years already on metformin and/or basal insulin.
While some differences in baseline characteristics between the two treatment groups were observed, these may be attributed to the overall small sample size of n = 23 participants. Similar to lixisenatide use in adults, 20 , 21 , 22 , 23 , 24 , 25 the observed TEAEs in the present study were primarily in the GI system class, although the small sample size and short study duration mean this study is not powered to evaluate less common TEAEs (occurring in <10% of participants) or comment on the overall safety profile of lixisenatide in pediatric populations. Only one treatment‐emergent serious AE was observed, which was not considered related to study drug, and no TEAEs led to treatment discontinuations or deaths. Few participants showed potentially clinically significant abnormalities (PCSAs) for hematology, clinical chemistry or vital signs. While there have been rare reports previously of pancreatitis following GLP‐1 RA treatment, 26 in the present study amylase and lipase were monitored and did not raise any concerns in the lixisenatide group, concurrent with multiple long‐term studies concluding no evidence for an increased risk of pancreatitis with GLP‐1 RA treatment. 27 , 28 No instances of symptomatic hypoglycemia were recorded in the trial, despite approximately 30% of all participants receiving concomitant insulin at baseline.
Vomiting was the most observed AE with 11 events in two participants in the highest‐dose group. In adults, GI AEs with lixisenatide occur primarily at the start of treatment (e.g. within the first 2 months) and then subside. 21 , 23 , 24 As this study examined lixisenatide treatment only for up to 42 days, further trials would be required to define the GI tolerability of lixisenatide over a longer period in pediatric populations.
Mean lixisenatide concentrations increased in a dose‐dependent manner and irrespective of ADA status; however, lixisenatide mean concentrations and inter‐subject variability were generally higher for participants with positive ADA status, as observed in previous studies with lixisenatide. 22 The increase in exposure with dose in pediatric participants with negative ADA status was comparable to that in adults with negative ADA status (Supplementary Figure 4), possibly owing to median body weights that broadly overlapped in these populations.
Approximately three‐quarters of the participants developed ADAs. This incidence is similar to the 71.2% observed in adults who received 20 μg lixisenatide twice daily. 29 Furthermore, no clinically relevant abnormalities in hematology, clinical chemistry or vital signs were observed in these pediatric individuals with ADA positive status. This is in line with results from studies in adults which have shown that antibody development does not appear to affect the efficacy or safety of lixisenatide. 23 Overall, exposure of lixisenatide and incidence of ADAs appear to be similar in children and adolescents compared with adults.
In adults, lixisenatide treatment is associated with statistically significant improvements in HbA1c versus placebo. 22 , 23 , 24 Although this study was not powered to demonstrate significant changes in HbA1c, a reduction in HbA1c of 0.3% (3 mmol/mol) was observed after only 6 weeks of treatment with lixisenatide, whereas HbA1c remained almost stable with placebo. The magnitude of this reduction most likely reflects the limited treatment duration and the fact that participants only received the maximum dose of lixisenatide for 2 weeks. Body weight increase was more pronounced with placebo versus lixisenatide, which reflected the trend for the positive effects of lixisenatide on body weight observed in the adult lixisenatide development program. 22 , 23 , 25 Since the short duration of this study may have limited the impact on body weight change, data generation in a larger study population would be required for confirmation of this observation. As with adult studies, lixisenatide treatment in children and adolescents was also associated with pronounced improvements in PPG and less substantial improvements in FPG when compared with placebo. 22 , 23 , 24 , 25
The strengths of this study include that it was a multicenter, randomized, double‐blind, placebo‐controlled, parallel group study of lixisenatide in a clinically‐relevant group of children and adolescents with T2D. However, this was a Phase 1 study of only 6 weeks duration, with a restricted number of participants included and without formal sample‐size calculation; therefore, assessment of the impact of lixisenatide on glycemic control is limited and would need to be confirmed in larger clinical trials. Additionally, the paucity of clinical trials in pediatric populations with T2D limits the ability to compare these results to other antihyperglycemic drugs. However, HbA1c lowering effects of liraglutide were also observed in pediatric populations after 5 weeks in one study, 30 and after 6 months in a longer‐term study. 31
In conclusion, repeated doses of lixisenatide of up to 20 μg per day were associated with a safety and tolerability profile in children and adolescents with T2D that is reflective of the established safety profile of lixisenatide in adults. Further, larger, long‐term investigations are required to confirm if the safety profiles are consistent between pediatric and adult populations. Lixisenatide concentrations increased in a dose‐dependent manner and lixisenatide treatment was also associated with improvements in glycemic control, while a trend towards a lower increase in body weight was seen compared with placebo, although this was not the primary focus of the study. The positive findings from this Phase 1 trial support further research in this population.
CONFLICT OF INTEREST
M Barrientos‐Pérez—has served as an investigator for trials in pediatrics for Sanofi, Novo Nordisk, Eli Lilly, Janssen, OKPO and Boehringer Ingelheim; and has received honoraria for lectures from Pfizer, Abbott, Novo Nordisk and Eli Lilly. D Hsia—has no conflicts to disclose, funds related to the content of this study were paid directly to his institution. O Mungur—has no conflicts to disclose. H Nell—has no conflicts to disclose. L Sloan—has served as an investigator for Amgen, Boehringer Ingelheim, GSK, Janssen, Lilly, Merck and Sanofi. He serves on advisory boards for and has received honoraria from AstraZeneca, Bayer, Boehringer Ingelheim, Janssen and Lilly. L Hovsepian, R Spranger and N Yang—are employees of Sanofi. E Niemoeller and W Schmider—are employees of Sanofi and hold shares in Sanofi.
AUTHOR CONTRIBUTIONS
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. All authors participated in the interpretation of the data, the writing, reviewing and editing of the manuscript, and had final responsibility for approving the published version.
ETHICS APPROVAL
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice, and the protocol was approved by institutional review boards or ethics committees at each study site.
PATIENT CONSENT
Each participant and their parent (or the participant's legal representative) provided written informed assent and consent, respectively.
Supporting information
Supplementary Figure 1Participant disposition
Supplementary Figure 2.Mean ± SD lixisenatide plasma concentration‐time profiles following repeat administration of 5, 10, or 20 μg lixisenatide for (A) ADA‐negative or (B) ADA‐positive participants
Supplementary Figure 3.Mean plasma glucose concentration‐time profiles by treatment group on Days 14, 28 and 42
Supplementary Figure 4.Comparison between AUC in pediatric and adult individuals with T2D with negative ADA status by lixisenatide dose: Box and whisker plots represent the adult and blue dots the pediatric population (data for adult populations taken from Sanofi data on file).
ACKNOWLEDGMENTS
We thank Dr Jens Stechel for his contributions to this study who sadly passed away before the study was completed. We also thank Ana Merino‐Trigo, PhD, (Sanofi) for coordinating the development, facilitating author discussions, and review of this manuscript, and Thomas Frank, PhD, (Sanofi) for the statistical analysis in Supplementary Figure 2. Editorial assistance was provided by Jo Bentley, PhD, of Fishawack Communications Ltd, part of Fishawack Health.
Barrientos‐Pérez M, Hsia DS, Sloan L, et al. A study on pharmacokinetics, pharmacodynamics and safety of lixisenatide in children and adolescents with type 2 diabetes. Pediatr Diabetes. 2022;23(6):641‐648. doi: 10.1111/pedi.13343
Funding InformationSponsorship for this study was funded by Sanofi, Paris, France. Editorial assistance provided by Fishawack Communications Ltd, part of Fishawack Health, was funded by Sanofi.
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to patient‐level data and related documents. Patient‐level data will be anonymised, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi;s data sharing criteria, eligible studies, and process for requesting access can be found at http://www.clinicalstudydatarequest.com.
REFERENCES
- 1. Nadeau KJ, Anderson BJ, Berg EG, et al. Youth‐onset type 2 diabetes consensus report: current status, challenges, and priorities. Diabetes Care. 2016;39:1635‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mayer‐Davis EJ, Dabelea D, Lawrence JM. Incidence trends of type 1 and type 2 diabetes among youths, 2002–2012. N Engl J Med. 2017;377:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: epidemiology and treatment. Curr Diab Rep. 2014;14:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief. 2017;288:1‐8. [PubMed] [Google Scholar]
- 5. Hannon TS, Arslanian SA. The changing face of diabetes in youth: lessons learned from studies of type 2 diabetes. Ann N Y Acad Sci. 2015;1353:113‐137. [DOI] [PubMed] [Google Scholar]
- 6. International Diabetes Federation . IDF Diabetes Atlas. 9th ed. Belgium; 2019. [Google Scholar]
- 7. Temneanu OR, Trandafir LM, Purcarea MR. Type 2 diabetes mellitus in children and adolescents: a relatively new clinical problem within pediatric practice. J Med Life. 2016;9:235‐239. [PMC free article] [PubMed] [Google Scholar]
- 8. American Diabetes Association . 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes ‐ 2022. Diabetes Care. 2022;45:S125‐S143. [DOI] [PubMed] [Google Scholar]
- 9. Shaefer CF Jr. Lixisenatide: a new member of the glucagon‐like peptide 1 receptor agonist class of incretin therapies. Clin Diabetes. 2016;34:81‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker RH, Stechl J, Steinstraesser A, Golor G, Pellissier F. Lixisenatide reduces postprandial hyperglycaemia via gastrostatic and insulinotropic effects. Diabetes Metab Res Rev. 2015;31:610‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanofi‐aventis group. Lyxumia (lixisenatide) . Summary of product characteristics; 2021.
- 12. Adlyxin (lixisenatide) . Prescribing information; 2019.
- 13. Alzaid A, Ladrón de Guevara P, Beillat M, Lehner Martin V, Atanasov P. Burden of disease and costs associated with type 2 diabetes in emerging and established markets: systematic review analyses. Expert Rev Pharmacoecon Outcome Res. 2020;21:1‐14. [DOI] [PubMed] [Google Scholar]
- 14. American Diabetes Association . Children and adolescents: standards of medical care in diabetes – 2022. Diabetes Care. 2022;45:S208‐S231. [DOI] [PubMed] [Google Scholar]
- 15. Gao YQ, Gao M, Xue Y. Treatment of diabetes in children. Exp Ther Med. 2016;11:1168‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huff RA, Maca JD, Puri M, Seltzer EW. Enhancing pediatric clinical trial feasibility through the use of Bayesian statistics. Pediatr Res. 2017;82:814‐821. [DOI] [PubMed] [Google Scholar]
- 17. Arslanian S, Bacha F, Grey M, Marcus MD, White NH, Zeitler P. Evaluation and management of youth‐onset type 2 diabetes: a position statement by the American Diabetes Association. Diabetes Care. 2018;41:2648‐2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LP AP . Bydureon BCise (exenatide extended‐release). Prescribing Information; 2021.
- 19. Oberle MM, Kelly AS. It is time to consider glucagon‐like peptide‐1 receptor agonists for the treatment of type 2 diabetes in youth. Front Endocrinol (Lausanne). 2019;10:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei ZG, Wang MC, Zhang HH, et al. PRISMA‐efficacy and safety of lixisenatide for type 2 diabetes mellitus: a meta‐analysis of randomized controlled trials. Medicine (Baltimore). 2018;97:e13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenstock J, Raccah D, Koranyi L, et al. Efficacy and safety of lixisenatide once daily versus exenatide twice daily in type 2 diabetes inadequately controlled on metformin: a 24‐week, randomized, open‐label, active‐controlled study (GetGoal‐X). Diabetes Care. 2013;36:2945‐2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Riddle MC, Forst T, Aronson R, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled with newly initiated and continuously titrated basal insulin glargine: a 24‐week, randomized, placebo‐controlled study (GetGoal‐duo 1). Diabetes Care. 2013;36:2497‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riddle MC, Aronson R, Home P, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled by established basal insulin: a 24‐week, randomized, placebo‐controlled comparison (GetGoal‐L). Diabetes Care. 2013;36:2489‐2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pinget M, Goldenberg R, Niemoeller E, Muehlen‐Bartmer I, Guo H, Aronson R. Efficacy and safety of lixisenatide once daily versus placebo in type 2 diabetes insufficiently controlled on pioglitazone (GetGoal‐P). Diabetes Obes Metab. 2013;15:1000‐1007. [DOI] [PubMed] [Google Scholar]
- 25. Ahrén B, Leguizamo Dimas A, Miossec P, Saubadu S, Aronson R. Efficacy and safety of lixisenatide once‐daily morning or evening injections in type 2 diabetes inadequately controlled on metformin (GetGoal‐M). Diabetes Care. 2013;36:2543‐2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lyxumia (lixisenatide) . EPAR ‐ Public assessment report. https://www.ema.europa.eu/en/documents/assessment-report/lyxumia-epar-public-assessment-report_en.pdf. Accessed August 2021.
- 27. Monami M, Nreu B, Scatena A, et al. Safety issues with glucagon‐like peptide‐1 receptor agonists (pancreatitis, pancreatic cancer and cholelithiasis): data from randomized controlled trials. Diabetes Obes Metab. 2017;19:1233‐1241. [DOI] [PubMed] [Google Scholar]
- 28. Pinto LC, Falcetta MR, Rados DV, Leitão CB, Gross JL. Glucagon‐like peptide‐1 receptor agonists and pancreatic cancer: a meta‐analysis with trial sequential analysis. Sci Rep. 2019;9:2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ratner RE, Rosenstock J, Boka G, DRI6012 Study Investigators . Dose‐dependent effects of the once‐daily GLP‐1 receptor agonist lixisenatide in patients with type 2 diabetes inadequately controlled with metformin: a randomized, double‐blind, placebo‐controlled trial. Diabet Med. 2010;27:1024‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klein DJ, Battelino T, Chatterjee DJ, Jacobsen LV, Hale PM, Arslanian S. Liraglutide's safety, tolerability, pharmacokinetics, and pharmacodynamics in pediatric type 2 diabetes: a randomized, double‐blind, placebo‐controlled trial. Diabetes Technol Ther. 2014;16:679‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamborlane WV, Barrientos‐Pérez M, Fainberg U, et al. Liraglutide in children and adolescents with type 2 diabetes. N Engl J Med. 2019;381:637‐646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1Participant disposition
Supplementary Figure 2.Mean ± SD lixisenatide plasma concentration‐time profiles following repeat administration of 5, 10, or 20 μg lixisenatide for (A) ADA‐negative or (B) ADA‐positive participants
Supplementary Figure 3.Mean plasma glucose concentration‐time profiles by treatment group on Days 14, 28 and 42
Supplementary Figure 4.Comparison between AUC in pediatric and adult individuals with T2D with negative ADA status by lixisenatide dose: Box and whisker plots represent the adult and blue dots the pediatric population (data for adult populations taken from Sanofi data on file).
Data Availability Statement
Qualified researchers may request access to patient‐level data and related documents. Patient‐level data will be anonymised, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi;s data sharing criteria, eligible studies, and process for requesting access can be found at http://www.clinicalstudydatarequest.com.