

Abstract
Porcine deltacoronavirus is a newly emergent enteric pathogen affecting swine farms worldwide. It has been detected in several countries in Europe, Asia and North America; yet, it has not been reported in South America. In November 2019, an enteric disease outbreak in a pig farm located in San Martin, Peru, was reported along with submission of three intestinal samples from pigs who succumbed to the disease. Samples were processed for molecular detection by qRT‐PCR, viral isolation and further sequencing analysis. A taqman‐based RT‐PCR was performed to differentiate among the most relevant swine enteric coronaviruses described to date. All samples were positive to porcine deltacoronavirus with a cycle threshold (Ct) value between 9 and 14, revealing a high viral load, while testing negative to porcine epidemic diarrhea and transmissible gastroenteritis viruses. Following detection, viral isolation was performed using PK‐15 and Vero cell lines. After 5 days of inoculation, no cytopathic effect was observed. A second blind passage allowed the observation of cytopathic effect on PK‐15 cells, while it remained absent in Vero cells. A fluorescence test using an anti‐N monoclonal antibody confirmed viral replication. One sample was processed for whole genome sequencing (WGS). In short, raw reads were imported into CLC genomics and assembled de novo. Out of 479k reads generated from the sample, 436k assembled into a 25,501 bp contig which was 99.5% identical to a reference porcine deltacoronavirus strain from the USA within the North American phylogroup. Yet, there are relevant differences at the nucleotide and amino acid levels compared with previously described porcine deltacoronavirus strains. Altogether, our findings represent the first report of porcine deltacoronavirus in South America, which provides information of its evolutionary origin. Thus, this study offers new insights into the molecular epidemiology of porcine deltacoronavirus infections in the swine industry.
Keywords: diarrhea, emerging diseases, PDCoV isolation, Peru, porcine deltacoronavirus, veterinary epidemiology, whole genome sequencing
1. INTRODUCTION
Coronaviruses comprise a large group of single‐stranded, positive‐sense RNA viruses that infect a broad range of species such as avian and mammals, including humans. Coronaviruses belong to the order Nidovirales, family Coronaviridae, subfamily Coronavirinae. These enveloped viruses are the largest RNA viruses identified to date ranging from 24 to 32 kb. Members of the subfamily Coronavirinae have been recently grouped into four genus as Alphacoronaviruses, Betacoronaviruses, Gammacoronaviruses and Deltacoronaviruses by the International Committee for Taxonomy of Viruses (Lefkowitz et al., 2018; Woo et al., 2010). Interestingly, it appears that the first two groups have originated from bats, whereas the latter two emerged from wild birds (Woo et al., 2012).
Porcine deltacoronavirus (PDCoV) is an emergent virus that causes gastrointestinal disease such as diarrhea, vomiting, dehydration and death in young piglets representing a major threat to swine industry (Jung et al., 2015; Li et al., 2019; Zhang, 2016; Zhao et al., 2019). Although PDCoV by itself causes enteric disease, co‐infections with other coronaviruses such as porcine epidemic diarrhea virus (PEDV) and transmissible gastroenteritis virus (TGEV) or other viruses are commonly found (Ajayi et al., 2018; Mai et al., 2017; Marthaler et al., 2014a; Niederwerder, 2018; Song et al., 2015). In this context, PDCoV shows indistinguishable clinical signs from other forms of enteric disease such as PED, TGE and swine acute diarrhea syndrome. Thus, proper differential diagnostic relies on genetic detection‐based assays that offer a highly sensitive and specific method.
PDCoV has a unique genomic organization. Starting from 5′‐end, PDCoV has a 5′ untranslated region (UTR), replicase (ORF 1a/b), spike (S), envelope (E), membrane (M), non‐structural 6 (NS6), nucleocapsid (N), NS7 genes and 3′‐UTR (Woo et al., 2010; Zhang, 2016). Interestingly, PDCoV lacks ORF3 and NS1, present in other well‐known coronaviruses (Si et al., 2020). From these genes, S gene encodes a highly glycosylated protein responsible for binding, cell attachment and entry into target cells, and therefore highly immunogenic. Thus, S gene is commonly used for phylogenetic analysis and vaccine development.
The first identification of PDCoV dates back to 2012 in Hong Kong by Woo et al, followed by multiple outbreaks in the USA (Homwong et al., 2016; Marthaler et al., 2014b; Wang et al., 2014). Later, PDCoV was reported in Canada (Niederwerder, 2018), Korea (Jang et al., 2018; Lee & Lee, 2014; Lee et al., 2016), Japan (Suzuki et al., 2018), Thailand (Lorsirigool et al., 2017; Saeng‐Chuto et al., 2016) and Vietnam (Le et al., 2018; Saeng‐chuto et al., 2019). More recently, PDCoV was detected in Mexico (Pérez‐Rivera et al., 2019). In South America, coronavirus infections in porcine are commonly reported. For instance, multiple studies revealed that PEDV and TGEV are present in several countries such as Colombia (Piñeros & Mogollón Galvis, 2015), Ecuador (Barrera et al., 2017) and Peru (Castro‐Sanguinetti et al., 2017). However, PDCoV has never been reported in South America. Hence, we report the first isolation and whole genome sequencing of PDCoV in Peru, providing new insights into the molecular epidemiology of this emerging disease in swine.
2. MATERIALS AND METHODS
2.1. Samples and RNA extraction
In November 2019, a report of an enteric clinical disease outbreak in a farm located in San Martin, Peru (see Figure 1(c)) was made by a local veterinary professional to the National animal health authorities (SENASA). The report indicated high morbidity in young piglets. Intestinal samples were submitted by the farm professionals to the Laboratory of Virology in the Faculty of Veterinary Medicine at the Universidad Nacional Mayor de San Marcos in Lima, Peru, for testing and detection of PEDV, TGEV and PDCoV by qRT‐PCR. Intestinal content (1 ml) was processed accordingly. In short, samples were diluted in 5 ml of phosphate buffer solution and centrifuged at 250 g for 5 min. Following centrifugation, 1 ml of supernatant was processed for RNA extraction using the QIAmp viral RNA mini kit (Qiagen), following manufacturer's specifications.
FIGURE 1.
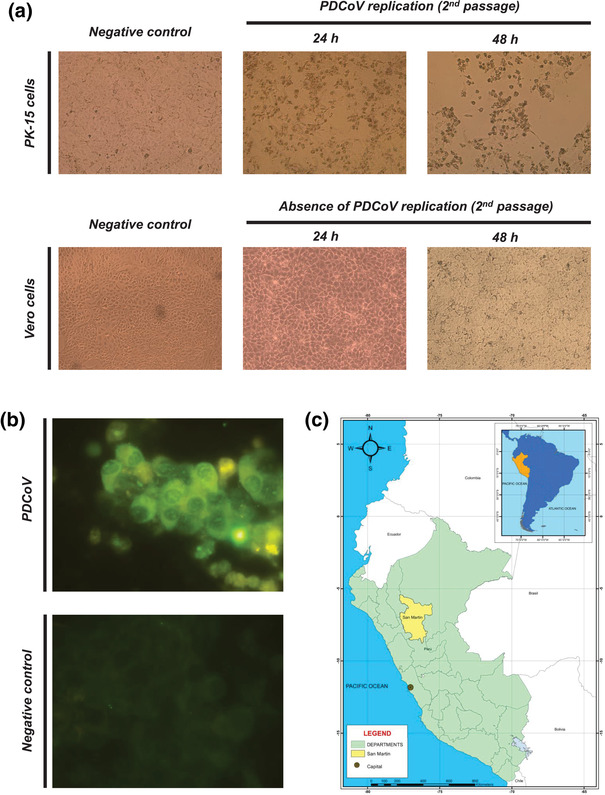
PDCoV isolation in PK‐15 cells and geographical map indicating locations where the outbreak was reported. Following PDCoV detection, intestinal content was filtered, and TPCK‐trypsin treated for isolation in cell lines known to be permissible for PDCoV replication. Following 5 days of inoculation, no cytopathic effect (cpe) was evidenced under light microscopy in both cell lines. A second passage evidenced cpe after 24 h, with initial cell rounding and monolayer disruption in PK‐15 cells but absent in Vero cells. Following 48 h of second passage, cpe was evidenced in almost 70% of cell monolayer, characterized by cell rounding, cell detaching, pyknosis in PK‐15 cell lines but no cytopathic effect was detected in Vero cell lines (a). Moreover, an immunofluorescence test with a PDCoV nucleoprotein monoclonal antibody confirmed viral replication in PK‐15 infected cells (b). A representative map of South America (blue) highlighting Peru (orange) is shown in the upper right side. Additionally, a geographical map of San Martin department (yellow) located in the north of Peru (light green) is represented below (c)
2.2. PCR differentiation and detection
Samples were assayed by qRT‐PCR to differentiate three main enteric coronaviruses affecting pigs as PEDV, TGEV and PDCoV. Thus, we used the EZ‐PED/TGE/PDCoV MPX 1.1 kit (Tetracore) following manufacturer's specifications.
2.3. Viral isolation
For viral isolation, we tested two different cell lines known to support coronavirus replication. We used PK‐15 and Vero cells that were plated in 24‐well plates at 90% of confluence. PK‐15 cells were kindly provided by Dr. Jhon Pasick from the Canadian Food Inspection Agency (Ontario, Canada) and Vero cells by the Naval Medical Research Unit Six (NAMRU‐6). Briefly, 100 μl of intestinal content was filtered and treated with TPCK‐trypsin (10 μg/ml) to allow ligand cleavage for further virus attachment to cell receptors. Treated samples were inoculated and incubated for 1 h, followed by replacement of maintenance media with trypsin. Cells were incubated for 5 days at 37°C with 5% CO2, with a second blind passage. Cytopathic effect was evaluated daily, and pictures recorded under light microscopy (Leica Microsystems) using a Leica MC170 HD camera (Leica Microsystems). Following isolation, we performed an immunofluorescence test using a PDCoV anti‐nucleoprotein monoclonal antibody (SD55‐197), kindly provided by Drs. Eric Nelson and Steven Lawson from the South Dakota State University (South Dakota, USA), to confirm the presence of the PDCoV.
2.4. Whole genome sequencing
Viral RNA was purified from one sample and processed for next‐generation sequencing (Illumina, Inc), following manufacturer's specifications. Reads were imported into CLC genomics and assembled de novo. Whole genome (n = 44) and ORF 1a/b (n = 23) sequences of PDCoV were obtained from GenBank for phylogenetic analysis. For analysis based on S gene, we also included those from Mexico (n = 61). General information of nucleotide sequences used is listed in Table 1.
TABLE 1.
Nucleotide sequences for phylogenetic analysis
| Full name | Short name | Country | Length (nt) | Accession number | |
|---|---|---|---|---|---|
| 1 | Porcine deltacoronavirus strain USA/NorthCarolina452/2014, complete genome | PDCoV/USA/NorthCarolina452/2014 | USA | 25394 | KR265858.1 |
| 2 | Porcine deltacoronavirus strain USA/Minnesota/2013, complete genome | PDCoV/USA/Minnesota/2013 | USA | 25394 | KR265853.1 |
| 3 | Porcine deltacoronavirus strain USA/Illinois449/2014, complete genome | PDCoV/USA/Illinois449/2014 | USA | 25394 | KR265852.1 |
| 4 | Porcine deltacoronavirus strain USA/Minnesota159/2014, complete genome | PDCoV/USA/Minnesota159/2014 | USA | 25401 | KR265859.1 |
| 5 | Deltacoronavirus PDCoV/USA/Illinois134/2014 from USA, complete genome | PDCoV/USA/Illinois134/2014 | USA | 25404 | KJ601778.1 |
| 6 | Porcine deltacoronavirus strain USA/Nebraska210/2014, complete genome | PDCoV/USA/Nebraska210/2019 | USA | 25404 | KR265861.1 |
| 7 | Porcine deltacoronavirus genomic RNA, complete genome, strain: YMG/JPN/2014 | PDCoV/JPN/YMG/2014 | Japan | 25362 | LC260044.1 |
| 8 | Porcine deltacoronavirus strain USA/Michigan448/2014, complete genome | PDCoV/USA/Michigan448/2014 | USA | 25394 | KR265850.1 |
| 9 | Porcine deltacoronavirus strain USA/Michigan447/2014, complete genome | PDCoV/USA/Michigan447/2014 | USA | 25393 | KR265849.1 |
| 10 | Deltacoronavirus PDCoV/USA/Ohio137/2014 from USA, complete genome | PDCoV/USA/Ohio137/2014 | USA | 25404 | KJ601780.1 |
| 11 | Porcine deltacoronavirus strain USA/Indiana453/2014, complete genome | PDCoV/USA/Indiana453/2014 | USA | 25394 | KR265851.1 |
| 12 | Porcine deltacoronavirus strain USA/Minnesota455/2014, complete genome | PDCoV/USA/Minnesota455/2014 | USA | 25394 | KR265855.1 |
| 13 | Porcine deltacoronavirus strain USA/Minnesota454/2014, complete genome | PDCoV/USA/Minnesota454/2014 | USA | 25394 | KR265854.1 |
| 14 | Porcine deltacoronavirus strain USA/Arkansas61/2015, complete genome | PDCoV/USA/Arkansas61/2015 | USA | 25398 | KR150443.1 |
| 15 | Deltacoronavirus PDCoV/USA/Illinois121/2014 from USA complete genome | PDCoV/USA/Illinois121/2014 | USA | 25406 | KJ481931.1 |
| 16 | Porcine deltacoronavirus 8734/USA‐IA/2014 complete genome | PDCoV/USA‐IA/8734 | USA | 25422 | KJ567050.1 |
| 17 | Porcine deltacoronavirus strain OH‐FD22 P7 complete genome | PDCoV/USA/OH‐FD22 | USA | 25438 | MZ291567.1 |
| 18 | Porcine deltacoronavirus strain SHJS/SL/2016, complete genome | PDCoV/SHJS/SL/2016 | China | 25414 | MF041982.1 |
| 19 | Porcine deltacoronavirus strain CH/SXD1/2015, complete genome | PDCoV/CH/SXD1/2015 | China | 25419 | KT021234.1 |
| 20 | Porcine deltacoronavirus strain CHN‐LYG‐2014, complete genome | PDCoV/CHN‐LYG/2014 | China | 25370 | KU665558.1 |
| 21 | Porcine deltacoronavirus isolate PDCoV/CHJXNI2/2015, complete genome | PDCoV/CHJXNI2/2015 | China | 25438 | KR131621.1 |
| 22 | Porcine deltacoronavirus isolate CHN‐AH‐2004, complete genome | PDCoV/CHN‐AH/2004 | China | 25420 | KP757890.1 |
| 23 | Porcine deltacoronavirus strain CH/Hunan/2014 complete genome | PDCoV/CH/Hunan/2014 | China | 25413 | KY513724.1 |
| 24 | Porcine deltacoronavirus PDCoV/CH/SXD1/2015 | PDCoV/CH/SXD1/2015 | China | 25419 | KT021234.1 |
| 25 | Porcine deltacoronavirus isolate 104–553 complete genome | PDCoV/104‐553 | China | 25418 | MW854634.1 |
| 26 | Porcine deltacoronavirus strain CHN/GS/2017/1 complete genome | PDCoV/CHN/GS/2017 | China | 25420 | MF642324.1 |
| 27 | Porcine deltacoronavirus strain CHN/QH/2017/1 complete genome | PDCoV/CHN/QH/2017 | China | 25420 | MF642325.1 |
| 28 | Porcine deltacoronavirus isolate CHN‐HN‐1601 complete genome | PDCoV/CHN/HN1601/2017 | China | 25419 | MG832584.1 |
| 29 | Porcine deltacoronavirus isolate CHN/Sichuan/2019 complete genome | PDCoV/ CHN/Sichuan/2019 | China | 25380 | MK993519.1 |
| 30 | Porcine deltacoronavirus isolate CH/JXJGS01/2016 complete genome | PDCoV/CH/JXJGS01/2016 | China | 25446 | MK625641.1 |
| 31 | Porcine deltacoronavirus isolate P1 16 BTL 0115/PDCoV/2016/Lao complete genome | PDCoV/Lao/2016 | Lao | 25405 | KX118627.1 |
| 32 | Porcine deltacoronavirus strain PDCoV/Swine/Vietnam/HaNoi6/2015, complete genome | PDCoV/Vietnam/HaNoi6/2015 | Vietnam | 25406 | KX834351.1 |
| 33 | Porcine deltacoronavirus strain PDCoV/Swine/Vietnam/Binh21/2015, complete genome | PDCoV/Vietnam/Binh21/2015 | Vietnam | 25406 | KX834352.1 |
| 34 | Porcine deltacoronavirus strain PDCoV/Swine/Thailand/S5015L/2015, complete genome | PDCoV/Thailand/S5015L/2015 | Thailand | 25405 | KU051649.1 |
| 35 | Porcine deltacoronavirus strain PDCoV/Swine/Thailand/S5011/2015, complete genome | PDCoV/Thailand/S5011/2015 | Thailand | 25405 | KU051641.1 |
| 36 | Porcine deltacoronavirus isolate KNU16‐07 complete genome | PDCoV/SouthKorea/KNU16‐07 | South Korea | 25422 | KY364365.1 |
| 37 | Porcine deltacoronavirus isolate KNU16‐11 complete genome | PDCoV/SouthKorea/KNU16‐11 | South Korea | 25419 | KY926512.1 |
| 38 | Porcine deltacoronavirus genomic RNA complete genome strain: IWT/JPN/2014 | PDCoV/JPN/IWT/2014 | Japan | 25362 | LC260041.1 |
| 39 | Porcine deltacoronavirus genomic RNA complete genome strain: MYZ/JPN/2014 | PDCoV/JPN/MYZ/2014 | Japan | 25362 | LC260042.1 |
| 40 | Porcine deltacoronavirus genomic RNA complete genome strain: AKT/JPN/2014 | PDCoV/JPN/AKT/2014 | Japan | 25362 | LC260038.1 |
| 41 | Porcine deltacoronavirus genomic RNA complete genome strain: OKN/JPN/2014 | PDCoV/JPN/OKN/2014 | Japan | 25362 | LC260043.1 |
| 42 | Porcine deltacoronavirus isolate PDCoV/Haiti/Human/0256‐1/2015 complete genome | PDCoV/Haiti/Human/0256‐1/2015 | Haiti | 25447 | MW685623.1 |
| 43 | Porcine deltacoronavirus isolate PDCoV/Haiti/Human/0329‐4/2015 complete genome | PDCoV/Haiti/Human/0329‐4/2015 | Haiti | 25444 | MW685624.1 |
| 44 | Porcine deltacoronavirus isolate PDCoV/Haiti/Human/0081‐4/2014 complete genome | PDCoV/Haiti/Human/0081‐4/2014 | Haiti | 25444 | MW685622.1 |
| 45† | Porcine deltacoronavirus isolate CD‐HNZK‐02 S protein gene complete cds | PDCoV/CD‐HZNK‐02 | China | 3480 | MZ326690.1 |
| 46† | Porcine deltacoronavirus strain CC‐HNZK‐02 S protein gene complete cds | PDCoV/CC‐HNZK‐02 | China | 3480 | MK248485.1 |
| 47† | Porcine deltacoronavirus isolate SD‐01‐2018 spike glycoprotein gene complete cds | PDCoV/SD/01/2018 | China | 3480 | MN173803.1 |
| 48† | Porcine deltacoronavirus strain CH‐XS‐2018 spike protein gene complete cds | PDCoV/CH/XS/2018 | China | 3480 | MK040452.1 |
| 49† | Porcine deltacoronavirus strain CH‐HX‐2018 spike protein gene complete cds | PDCoV/CH/HX/2018 | China | 3480 | MK040448.1 |
| 50† | Porcine deltacoronavirus strain CH‐WH‐2017 spike protein gene complete cds | PDCoV/CH/WH/2017 | China | 3480 | MK040451.1 |
| 51† | Pocine deltacoronavirus PDCoV/CH HN 2016 S protein (S) gene complete cds | PDCoV/CH HN 2016 | China | 3480 | KY496312.1 |
| 52† | Porcine deltacoronavirus isolate CHN‐GD16‐13 spike protein gene complete cds | PDCoV/CHN‐GC16‐13 | China | 3480 | KY078916.1 |
| 53† | Porcine deltacoronavirus isolate CHN‐GC16‐12 spike protein gene complete cds | PDCoV/CHN‐GC16‐12 | China | 3480 | KY078915.1 |
| 54† | Porcine deltacoronavirus isolate CH/XJYN/2016 spike protein (S) gene complete cds | PDCoV/CH/XJYN/2016 | China | 3483 | MN064712.1 |
| 55† | Porcine deltacoronavirus isolate GX05‐2017 spike glycoprotein gene complete cds | PDCoV/GX05‐2017 | China | 3480 | MT505449.1 |
| 56† | Porcine deltacoronavirus strain CH‐DH2‐2017 spike protein gene complete cds | PDCoV/CH‐DH2‐2017 | China | 3480 | MK040450.1 |
| 57† | Porcine deltacoronavirus isolate SD‐12‐2018 spike glycoprotein gene complete cds | PDCoV/SD‐12‐2018 | China | 3480 | MN173812.1 |
| 58† | Porcine deltacoronavirus isolate SD‐10‐2018 spike glycoprotein gene complete cds | PDCoV/SD‐10‐2018 | China | 3480 | MN173810.1 |
| 59† | Porcine deltacoronavirus isolate GX03‐2017 spike glycoprotein gene complete cds | PDCoV/GX03‐2017 | China | 3474 | MT505447.1 |
| 60† | Porcine deltacoronavirus isolate GX05‐2019 spike glycoprotein gene complete cds | PDCoV/GX05‐2019 | China | 3483 | MT505460.1 |
| 61† | Porcine deltacoronavirus isolate GX03‐2018 spike glycoprotein gene complete cds | PDCoV/GX03‐2018 | China | 3483 | MT505452.1 |
| 62† | Porcine deltacoronavirus isolate GX03‐2019 spike glycoprotein gene complete cds | PDCoV/GX03‐2019 | China | 3483 | MT505458.1 |
| 63† | Porcine deltacoronavirus isolate GX05‐2018 spike glycoprotein gene complete cds | PDCoV/GX05‐2018 | China | 3483 | MT505454.1 |
| 64† | Porcine deltacoronavirus isolate GX04‐2019 spike glycoprotein gene complete cds | PDCoV/GX04‐2019 | China | 3483 | MT505459.1 |
| 65† | Porcine deltacoronavirus CH‐HA2‐2017 spike protein gene complete cds | PDCoV/CH‐HA2‐2017 | China | 3480 | MK040454.1 |
| 66† | Porcine deltacoronavirus strain CH‐HA3‐2017 spike protein gene complete cds | PDCoV/CH‐HA3‐2017 | China | 3483 | MK040455.1 |
| 67† | Porcine deltacoronavirus isolate YUC/UICMPR/2015 spike glycoprotein (S) gene, complete cds | PDCoV/Mexico/YUC/UICMPR/2015 | Mexico | 3483 | MK478381.1 |
| 68† | Porcine deltacoronavirus isolate MEXICO/OAX/UI1253CMPR/2017 spike glycoprotein (S) gene, complete cds | PDCoV/Mexico/OAX/UI1253CMPR/2017 | Mexico | 3483 | MK478383.1 |
| 69† | Porcine deltacoronavirus isolate EDOMEX/UI202CMPR/2017 spike glycoprotein (S) gene, complete cds | PDCoV/Mexico/UI202CMPR/2017 | Mexico | 3482 | MK478382.1 |
| 70† | Porcine deltacoronavirus isolate QRO/UI689CMPR/2017 spike glycoprotein (S) gene, complete cds | PDCoV/Mexico/QRO/UI689CMPR/2017 | Mexico | 3482 | MK478380.1 |
| 71† | Porcine deltacoronavirus isolate KNU16‐08 spike protein (S) gene complete cds | PDCoV/SouthKorea/KNU16‐08 | South Korea | 3480 | KY926511.1 |
| 72† | Porcine deltacoronavirus isolate SL5 spike protein gene complete cds | PDCoV/SouthKorea/SL5 | South Korea | 3483 | KR060083.1 |
| 73† | Porcine deltacoronavirus isolate SL2 spike protein gene complete cds | PDCoV/SouthKorea/SL2 | South Korea | 3483 | KR060082.1 |
Nucleotide sequences included for phylogenetic analysis of S gene.
2.5. Phylogenetic analysis
The PDCoV whole genome, ORF 1a/b and S nucleotide sequences in this study were aligned using Clustal W from MEGA X software (Kumar et. Et al., 2018). We used PDCoV genome sequences obtained from GenBank isolated in the USA (16), China (13), Japan (5), Haiti (3), South Korea (2), Vietnam (2), Thailand (2) and Laos (1). For the spike analysis, we included sequences from the USA (13), China (28), Japan (5), Mexico (n = 4), South Korea (3), Haiti (3), Vietnam (2), Thailand (2) and Laos (1). A general time reversible nucleotide substitution model with gamma distribution among site rate variation was used, with a maximum likelihood estimation model for phylogenetic reconstruction. Bootstrap analysis was carried out on 1000 data sets. The percentage of nucleotide sequence identity was also calculated.
3. RESULTS
3.1. Molecular detection confirms the presence of PDCoV genome in intestinal samples of pigs suffering from an enteric disease outbreak in Peru
We took advantage of a widely used qRT‐PCR assay that allows accurate differentiation of most common enteric coronaviruses in swine such as PEDV, TGEV and PDCoV. qRT‐PCR results confirmed the presence of PDCoV RNA in all (n = 3) samples analyzed, which tested negative to PEDV and TGEV. In addition to its qualitative feature, this assay allows relative quantification. Thus, the viral load was quantified through the amount of genetic target of PDCoV amplified during the process. Samples had low Ct values to PDCoV (Ct = 9–14) which indicated a high PDCoV viral load. From these results, we inferred that PDCoV was involved in the enteric disease with high viral titres.
3.2. PDCoV replicated in PK‐15 while Vero cells did not allow PDCoV propagation
Following PDCoV detection, our objective was to isolate in vitro using PK‐15 and Vero cell lines. Despite the high viral load detected by PCR, we did not see evidence of cytopathic effect during the first 5 days following inoculation in any of the cell line tested. Thus, we performed a second blind passage to evidence viral replication. Interestingly, 48 h post inoculation, cytopathic effect was observed in PK‐15 cell line, but morphological changes in Vero cell line were absent. These findings were also confirmed by qRT‐PCR. Within the major cellular changes observed are: pyknosis, cell rounding, monolayer disruption, cell detachment, which correspond to typical coronavirus cytopathic effect previously described (see Figure 1(a)). Furthermore, the immunofluorescence test in PK‐15‐infected cells confirmed the presence of PDCoV (see Figure 1(b)). Following isolation, a sample was selected and prepared for genome sequencing.
3.3. Whole genome and ORF 1 a/b sequencing analysis indicates that Peruvian PDCoV isolate originated from a PDCoV strain from the USA
The nucleotide sequence of Peruvian PDCoV isolate, identified as PDCoV/Peru/isolate/2019, was submitted to GenBank under the accession number MT227371. Our Peruvian PDCoV genome follows similar patterns with other PDCoV genome sequences deposited in GenBank. Thus, this isolate is 25,501 nt in length and consists of, excluding the polyA tail: 5′‐UTR (1–480 nt), ORF1a/b (481–11,368 nt, 11,368–19,283 nt), S (19,265–22,747 nt), E (22,741–22,992 nt), M (22,985–23,638 nt), NS6 (23,638–23,922 nt), N (23,943–24,971 nt), NS7 (24,037–24,639 nt) and 3′‐UTR (24,972–25,501 nt). A graphical representation of the characterized PDCoV isolate is shown in Figure 2.
FIGURE 2.
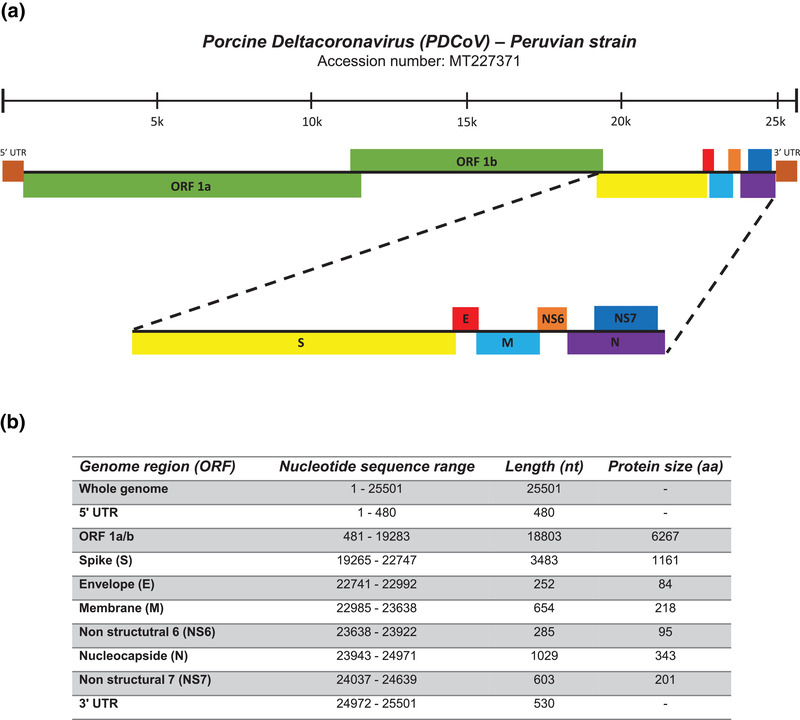
Genomic organization of the Peruvian PDCoV isolate. Whole genome sequence of the Peruvian PDCoV isolate was performed by next‐generation sequencing (NGS). PDCoV whole genome sequence is 25501 nt. Starting from 5′ end, the genome is structured as follows: 5′‐UTR, ORF 1a/b, S, E, M, NS6, N, NS7 and 3′‐UTR (a). Genomic regions, nucleotide sequence range and nucleotide and protein lengths are depicted at the bottom (b)
Phylogenetic analysis has typically been performed using key major genes of any organism of interest. However, this analysis tends to limit the analysis to a certain gene or group genes. Conversely, whole genome sequencing offers a more complete and deeper genetic characterization compared with partial approaches. In our study, we took advantage of next generation sequencing of our PDCoV isolate to track its evolutionary origin. Our results indicated that our Peruvian strain belongs to the North American phylogroup and is closely related to a PDCoV strain from the USA isolated in 2015 (99.5% of nucleotide identity). Genetic distance of the Peruvian PDCoV strain with other PDCoV analyzed reveals high similarity between 97.1 and 99.5%. Compared with the strains from the USA, the Peruvian PDCoV has a nucleotide identity between 99.45 and 99.51%. Percentages range from 98.6 to 98.74% when compared with the Chinese strains. Finally, nucleotide identity is 97% and 97.5% for Thai and Vietnamese strains, respectively. A summary of nucleotide identity is shown in Table 2. Further analysis based on ORF 1 a/b showed identical topology to the whole genome sequence phylogenetic tree. Altogether, these results indicate that the virus detected in Peru has emerged from a North American ancestor (see Figures 3(a) and (b)). Similarly, PDCoV protein sequence analysis resembled the topology of the nucleotide analysis.
TABLE 2.
Genetic distances of PDCoV nucleotide sequences (percentages) with the Peruvian PDCoV strain
| Whole genome | ORF 1a/b | S | E | M | NS6 | N | NS7 | |
|---|---|---|---|---|---|---|---|---|
| USA | 99.45–99.51 | 99.56–99.63 | 99.15–99.30 | 99.6 | 99.38–99.85 | 99.29–99.65 | 99.21–99.61 | 98.99–99.67 |
| China | 98.60–98.74 | 98.72–98.86 | 97.65–98.38 | 98.36–98.78 | 98.26–99.07 | 98.93–99.29 | 98.10–98.91 | 98.12–98.99 |
| Japan | 99.5 | 99.61 | 99.24 | 99.6 | 99.85 | 99.29 | 99.51 | 99.67 |
| Thailand | 97.10–97.11 | 97.27–97.28 | 95.51–95.54 | 99.19 | 98.26 | 98.57 | 96.83 | 97.04 |
| Vietnam | 97.46–97.5 | 97.50–97.53 | 95.95–96.05 | 99.6 | 98.91 | 98.93 | 98.61 | 98.12 |
| Mexico | – | – | 98.65–99.21 | – | – | – | – | – |
FIGURE 3.
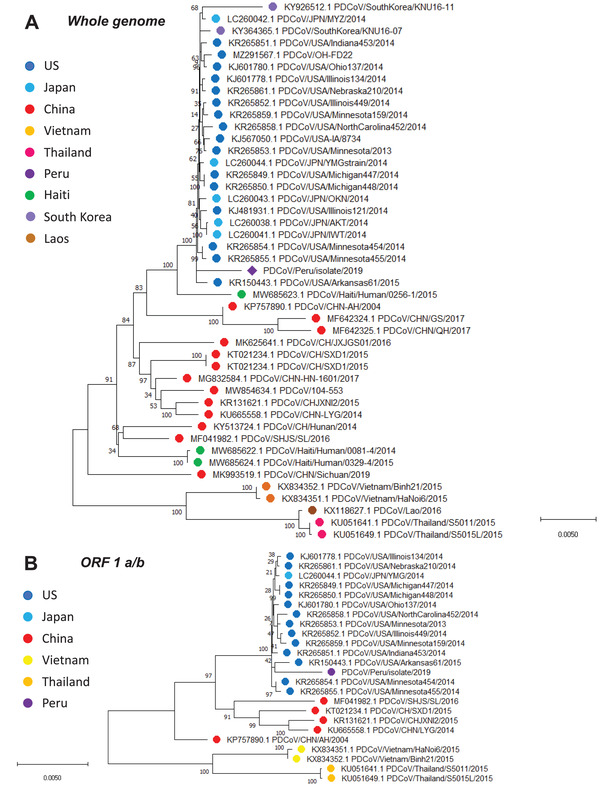
Whole genome and ORF 1a/b phylogenetic analysis of Peruvian PDCoV strain reveals its evolutionary origin from a North American PDCoV strain. For both whole genome and ORF 1a/b, the evolutionary history was inferred by using the maximum likelihood method and general time reversible model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete gamma distribution was used to model evolutionary rate differences among sites. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The whole genome analysis involved 45 nucleotide sequences, whereas the ORF 1 a/b involved 24 nucleotide sequences. Evolutionary analyses were conducted in MEGA X
3.4. Phylogenetic analysis of Peruvian PDCoV S gene shows close relationship to the Mexican PDCoV within the North American phylogroup
S gene is one of the most variable genes among coronaviruses. This is due to its function in cell attachment and viral replication. This high polymorphism makes the S gene a powerful tool to estimate the evolutionary relationship among virus strains belonging to the same genetic group. Hence, we performed a phylogenetic analysis using the PDCoV S nucleotide sequences publicly available with our Peruvian S nucleotide sequence to evaluate their evolutionary distance (see Figure 4(a)). Our results indicate that our PDCoV isolate has close relationship to the Mexican PDCoV strains (98.65–99.21% of nucleotide identity) within the North American phylogroup. Similarly, S protein phylogenetic analysis reveal similar evolutionary relationship among strains analyzed. Yet, S phylogenetic analysis evidence that Peruvian PDCoV has a certain degree of divergency from the North American phylogroup.
FIGURE 4.
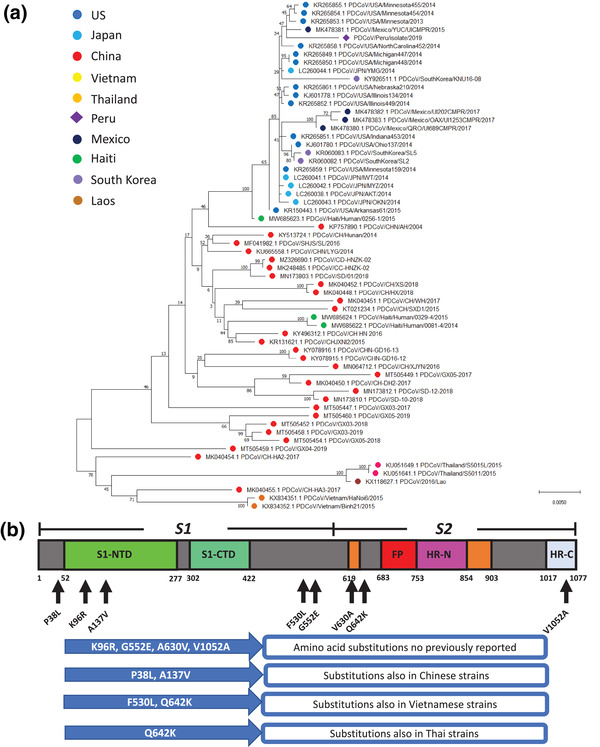
Phylogenetic analysis of S gene reveals that Peruvian PDCoV strain has a close relationship with a Mexican strain within the North American phylogroup. The evolutionary history was inferred by using the maximum likelihood method and general time reversible model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete gamma distribution was used to model evolutionary rate differences among. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 62 nucleotide sequences. Evolutionary analyses were conducted in MEGA X (a). Schematic representation of the porcine deltacoronavirus S protein highlighting the amino acid substitutions found in the present study. Black arrows represent the site of these substitutions. A multiple amino acid sequence alignment of S protein was performed using Clustal W in MEGA X. Eight amino acid modifications were detected within the Peruvian sequence compared to others (b)
3.5. Peruvian PDCoV Spike amino acid sequence reveals unique substitutions compared to other PDCoV strains
As we observed multiple changes at the nucleotide level in the S gene, we were interested in evaluating whether these changes represent modifications at the protein level. Thus, we found multiple changes in the S protein sequence (see Figure 4(b)). Most relevant changes are K96R, G552E, A630V and V1052A that represent unique variations compared with other PDCoV strains. Other amino acid changes have been also found in Chinese strains such as P38L and A137V. We also detected F530L, like the one found in the Vietnamese strains. Furthermore, the Peruvian strains have a Q642K, like those in Vietnamese and Thai strains. These results provide evidence that PDCoV has undergone to unique changes that indicate a degree of genetic diversity in the Peruvian PDCoV strain. Based on previous studies of PDCoV Spike protein characterization (Shang et al., 2018), amino acid substitutions observed here are located randomly across both S protein subunits (S1 and S2) but none of them is located in the RBD region (S1‐CTD).
4. DISCUSSION
PDCoV is one of the most recent and relevant coronaviruses of swine industry. It represents a major threat for swine productivity, and it is responsible for large economic losses worldwide. Yet, PDCoV remains poorly studied despite major efforts made recently. In Peru, multiple cases of enteric disease have occurred; however, these cases are not properly addressed and frequently misdiagnosed. Hence, this study represents the first report of isolation and phylogenetic characterization of a Peruvian PDCoV isolate using the whole genome sequence and its major S protein, revealing unique aspects compared to other PDCoV strains.
Although multiple studies have proven that ST, LLC‐PK (Hu et al., 2015) and IPI‐2I (Zhang et al., 2019) are the primary option for PDCoV isolation, we showed that PK‐15 might be a valid alternative when is needed. This is also demonstrated by Jiang et al. (2019), in the assessment of innate immune activation following PDCoV infection. Nevertheless, it is possible that efficacy in viral replication might vary among different cell lines and PK‐15 might not be as permissive as others. This should be considered when viral infectivity is the main objective. In addition, since we detected viral replication in the presence of trypsin, we speculate that PK‐15 cells might offer a compared PDCoV permissibility in LLC‐PK while differing from that observed in ST. Furthermore, our viral isolation findings contrasted with those detected by qRT‐PCR. This provided evidence that a large proportion of viral particles were unable to replicate into a cell line support. This has also been reported by others indicating that low viral isolation rates might be attributed to sample degradation and viral viability (Hu et al., 2015). On the other hand, multiple authors have shown that successful viral isolation are due to other factors such as cell line permissibility and enzyme treatment (Jung et al., 2016; Yang et al., 2020; Zhao et al., 2019). In our study, it remains unclear whether trypsin concentration (10 μg/ml) might have played a role in the lack of viral replication in Vero cells. Additional studies, at different trypsin concentration, will elucidate Vero cell line permissibility to PDCoV. Interestingly, we did not observe cell toxicity in our assays, as that was absent following the first 5 days of inoculation and PDCoV cytopathic effect was evidenced in the second passage. These results contrast with other claims that cell toxicity is common during PDCoV isolation using intestinal content or foecal samples (Hu et al., 2015). Nevertheless, further studies are required to clarify the implications of cell permeability to PDCoV in viral replication and its effects on clinical presentation.
Whole genome analysis revealed that our isolate was closely related to North American strains. The close relationship within the Peruvian and the North American strains indicate they share a common phylogenetic ancestor and revealed that the Peruvian isolate emerged from a strain from the USA. In 2019, Perez‐Rivera et al. reported the first phylogenetic analysis of PDCoV in Mexico, focusing the analysis on the S gene nucleotide sequence, and no report of PDCoV whole genome sequence was made (Pérez‐Rivera et al., 2019). Thus, we were unable to evaluate the phylogenetic relationship using the whole genome sequence of PDCoV from Peru and Mexico. We believe that this would add deeper understanding about its appearance in Peru and contribute to its epidemiology in South America.
Due to its high variability, S gene nucleotide sequences have been used to estimate the genetic relationship of PDCoV strains worldwide. Perez‐Rivera et al. (2019) demonstrated that Mexican PDCoV formed two clades: the group including strains isolated in 2015 and those PDCoV isolated in 2017. In our study, phylogenetic analysis using S gene nucleotide sequences reveals that Peruvian PDCoV grouped closely to the Mexican strain isolated in 2015 within the North American phylogroup. This indicates a close relationship among PDCoV strains from Mexico, the USA and Peru, sharing a common ancestor and evidencing a dissemination route of PDCOV from North America to South America. Interestingly, multiple non‐silent mutations were found in the Peruvian PDCoV strain compared with other genomes. Even though some of these mutations have been described in other PDCoV strains, some are unique to the Peruvian isolate, revealing that this isolate has undergone phenotypical changes after its emergence in North America. Although these substitutions were neither located in critical regions of glycosylation sites nor in the RBD region (S1‐CTD), they might have influence in ligand/receptor interaction. Further studies are required to clarify whether these modifications have implications in the pathobiology and development of the clinical disease.
To date, it is unclear how PDCoV was introduced in Peru. However, there is a long history of commerce between Peru and North American countries that has expanded in recent years. The National Service of Animal Health in Peru (SENASA) reported the import of a large number of purebred animals (∼150 tons) during the 2014 and 2018 period. Furthermore, Peru imports feed ingredients for swine farms mainly from the USA (MINAGRI, 2020). Altogether, this might explain the possible routes for PDCoV entrance into the country, similar to that described for other viruses of importance for swine industry (Dee et al., 2018; Ramírez et al., 2019). Interestingly, there is no report of PDCoV in other South American countries so the introduction from those is unlikely. Nevertheless, further studies are needed to understand the epidemiology of this disease in Peru and its relationship to other countries.
In conclusion, Peruvian PDCoV strain was successfully sequenced, isolated and phylogenetically analyzed demonstrating that this isolate has been derived from a strain identified in the USA. To our knowledge, this is the first report of a PDCoV strain detected in South America and offers new insights about the epidemiology of PDCoV worldwide.
CONFLICT OF INTEREST
All authors have declared no conflict of interest.
ETHICAL STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to. No ethical approval was required as research samples were obtained in accordance with guidelines from the Peruvian National authorities in animal health.
ACKNOWLEDGEMENTS
The authors would like to express their gratitude to Drs. Mariluz Arainga‐Ramirez and Steven Lawson for their contribution, and the Universidad Nacional Mayor de San Marcos for its financial support. We also would like to acknowledge the farm professionals and the national health authorities (SENASA‐Peru) for the information provided.
More‐Bayona, J. A. , Ramirez‐Velasquez, M. , Hause, B. , Nelson, E. , & Rivera‐Geronimo, H. (2022). First isolation and whole genome characterization of Porcine Deltacoronavirus from pigs in Peru. Transboundary and Emerging Diseases, 69, e1561–e1573. 10.1111/tbed.14489
Contributor Information
Juan A. More‐Bayona, Email: jmoreb@unmsm.edu.pe.
Mercy Ramirez‐Velasquez, Email: mramirezv@unmsm.edu.pe.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study were submitted to the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) with accession number MT227371 for the Peruvian strain of PDCoV obtained.
REFERENCES
- Ajayi, T. , Dara, R. , Misener, M. , Pasma, T. , Moser, L. , & Poljak, Z. (2018). Herd‐level prevalence and incidence of porcine epidemic diarrhoea virus (PEDV) and porcine deltacoronavirus (PDCoV ) in swine herds in Ontario, Canada. Transboundary and Emerging Diseases, 65, 1197–1207. 10.1111/tbed.12858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera, M. , Garrido‐Haro, A. , Vaca, M. S. , Granda, D. , Acosta‐Batallas, A. , & Pérez, L. J. (2017). Tracking the origin and deciphering the phylogenetic relationship of porcine epidemic diarrhea virus in Ecuador. BioMed Research International, 2017, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Sanguinetti, G. , Ramírez, V. M. , More ,B. J. , Manchego, S. A. , & Rivera, G. H. (2017). Isolation and molecular detection of emerging porcine epidemic diarrhea virus strains in Lima, Peru. Revista de Investigaciones Veterinarias Del Peru, 28(4), 1010‐1019. 10.15381/rivep.v28i4.13885 [DOI] [Google Scholar]
- Dee, S. A. , Bauermann, F. V. , Niederwerder, M. C. , Clement, A. T. , De Lima, M. , Long, C. , Patterson, G. , Sheahan, M. A. , Stoian, A. M. M. , Petrovan, V. , Jones, C. K. , De Jong, J. , Ji, J.u , Spronk, G. D. , Minion, L. , Christopher‐Hennings, J. , Zimmerman, J. J. , Rowland, R. R. R. , Nelson, E. , … Diel, D. G. (2018). Survival of viral pathogens in animal feed ingredients under transboundary shipping models. PLoS One, 14(3), 1–18. 10.1371/journal.pone.0214529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homwong, N. , Jarvis, M. C. , Lam, H. C. , Diaz, A. , Rovira, A. , Nelson, M. , & Marthaler, D. (2016). Characterization and evolution of porcine deltacoronavirus in the United States. Preventive Veterinary Medicine, 123(2014), 168–174. 10.1016/j.prevetmed.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , Jung, K. , Vlasova, A. N. , Chepngeno, J. , Lu, Z. , Wang, Q. , & Saif, L. J. (2015). Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. Journal of Clinical Microbiology, 53(5), 1537–1548. 10.1128/JCM.00031-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, G. , Kim, S. ‐. H. , Lee, Y. J. , Kim, S. , Lee, D.u S. , Lee, K.‐K.i , & Lee, C. (2018). Isolation and characterization of Korean porcine deltacoronavirus strain KNU16‐07. Journal of Veterinary Science, 19(4), 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, S. , Li, F. , Li, X. , Wang, L. , Zhang, L.i , Lu, C. , Zheng, L.i , & Yan, M. (2019). Transcriptome analysis of PK‐15 cells in innate immune response to porcine deltacoronavirus infection. Plos One, 14(10), 1–20. 10.1371/journal.pone.0223177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , Eyerly, B. , Lu, Z. , Chepngeno, J. , & Saif, L. J. (2015). Pathogenicity of 2 porcine deltacoronavirus strains in gnotobiotic pigs. Emerging Infectious Diseases, 21(4), 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , & Saif, L. J. (2016). Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. In Virus Research (Vol., 226, pp. 50–59). Elsevier B.V. 10.1016/j.virusres.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, V. P. , Song, S. , An, B. ‐. H. , Park, G. ‐. N. , Pham, N. T. , Le, D. Q. , Nguyen, V. T. , Vu, T. T. H. , Kim, K.i‐S. , Choe, S. , & An, D. ‐. J. (2018). A novel strain of porcine deltacoronavirus in Vietnam. Archives of Virology, 163(1), 203–207. 10.1007/s00705-017-3594-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. H. , Chung, H. C. , Nguyen, V. G. , Moon, H. J. , Kim, H. K. , Park, S. J. , Lee, C. H. , Lee, G. E. , & Park, B. K. (2016). Detection and phylogenetic analysis of porcine deltacoronavirus in Korean Swine Farms , 2015. Transboundary and Emerging Diseases, 63(2013), 248–252. 10.1111/tbed.12490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2014). Complete genome characterization of Korean porcine deltacoronavirus strain KOR/KNU14‐04/2014. Genome Announcements, 2(6), 4–5. 10.1128/genomeA.01191-14.Copyright [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz, E. J. , Dempsey, D. M. , Hendrickson, R. C. , Orton, R. J. , Siddell, S. G. , & Smith, D. B. (2018). Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Research, 46(2017), D708‐D717. 10.1093/nar/gkx932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Zheng, L. , Li, H. , Ding, Q. , Wang, Y. , & Wei, Z. (2019). Porcine deltacoronavirus causes diarrhea in various ages of field‐infected pigs in China. Bioscience Reports, 39, 1–10. 10.1042/BSR20190676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsirigool, A. , Saeng‐Chuto, K. , Madapong, A. , Temeeyasen, G. , Tripipat, T. , Kaewprommal, P. , Tantituvanont, A. , Piriyapongsa, J. , & Nilubol, D. (2017). The genetic diversity and complete genome analysis of two novel porcine deltacoronavirus isolates in Thailand in 2015. Virus Genes, 53(2), 240–248. 10.1007/s11262-016-1413-z [DOI] [PubMed] [Google Scholar]
- Mai, K. , Feng, J. , Chen, G. , Li, D. , Zhou, L. , Bai, Y. , Wu, Q. , & Ma, J. (2017). The detection and phylogenetic analysis of porcine deltacoronavirus from Guangdong Province in Southern China. Transboundary and Emerging Diseases, 65, 166‐173. 10.1111/tbed.12644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Jiang, Y. , Collins, J. , & Rossow, K. (2014). Complete genome sequence of strain SDCV/USA/Illinois121/2014, a porcine deltacoronavirus from the United States. Genome A, 2(2), 1–2. 10.1128/genomeA.00218-14.Copyright [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Raymond, L. , Jiang, Y. , Collins, J. , Rossow, K. , & Rovira, A. (2014). Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerging Infectious Diseases, 20(8), 1347–1350. 10.3201/eid2008.14-0526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINAGRI . (2020). Anuario estadistico de Comercio Exterior Agrario 2019. 77.
- Niederwerder, M. C. , & Hesse, R. A. (2018). Swine enteric coronavirus disease : A review of 4 years with porcine epidemic diarrhoea virus and porcine deltacoronavirus in the United States and Canada. Transboundary and Emerging Diseases, 65(2017), 660–675. 10.1111/tbed.12823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Rivera, C. , Ramírez‐Mendoza, H. , Mendoza‐Elvira, S. , Segura‐Velázquez, R. , & Sánchez‐Betancourt, J. I. (2019). First report and phylogenetic analysis of porcine deltacoronavirus in Mexico. Transboundary and Emerging Diseases, 66(4), 1436–1441. 10.1111/tbed.13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñeros, R. , & Mogollón Galvis, J. D. (2015). Coronavirus in pigs: Significance and presentation of swine epidemic diarrhea virus (PEDV) in Colombia. Revista de Medicina Veterinaria, 29, 73–89. [Google Scholar]
- Ramírez, M. , Bauermann, F. V. , Navarro, D. , Rojas, M. , Manchego, A. , Nelson, E. A. , Diel, D. G. , & Rivera, H. (2019). Detection of porcine reproductive and respiratory syndrome virus (PRRSV) 1‐7‐4‐type strains in Peru. Transboundary and Emerging Diseases, 66(3), 1107–1113. 10.1111/tbed.13134 [DOI] [PubMed] [Google Scholar]
- Saeng‐Chuto, K. , Jermsutjarit, P. , Stott, C. J. , Vui, D. T. , Tantituvanont, A. , & Nilubol, D. (2019). Retrospective study, full‐length genome characterization and evaluation of viral infectivity and pathogenicity of chimeric porcine deltacoronavirus detected in Vietnam. Transboundary and Emerging Diseases, 67, 183‐198. 10.1111/tbed.13339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeng‐Chuto, K. , Lorsirigool, A. , Temeeyasen, G. , Vui, D. T. , Stott, C. J. , Madapong, A. , Tripipat, T. , Wegner, M. , Intrakamhaeng, M. , Chongcharoen, W. , Tantituvanont, A. , Kaewprommal, P. , Piriyapongsa, J. , & Nilubol, D. (2016). Different lineage of porcine deltacoronavirus in Thailand, Vietnam and Lao PDR in 2015. Transboundary and Emerging Diseases, 64, 3‐10. 10.1111/tbed.12585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Zheng, Y. , Yang, Y. , Liu, C. , Geng, Q. , Tai, W. , Du, L. , Zhou, Y. , Zhang, W. , & Li, F. (2018). Cryo‐electron microscopy structure of porcine deltacoronavirus spike protein in the prefusion state. Journal of Virology, 92(4), 1–14. 10.1128/JVI.01628-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si, F. , Hu, X. , Wang, C. , Chen, B. , Wang, R. , Dong, S. , Yu, R. , & Li, Z. (2020). Enhances viral proliferation by inhibiting apoptosis of infected cells. Viruses, 12(214), 1–18. 10.3390/v12020214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , Zhou, X. , Peng, Q. , Chen, Y. , Zhang, F. , Huang, T. , Zhang, T. , Li, A. , Huang, D. , Wu, Q. , He, H. , & Tang, Y. (2015). Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: Identification, prevalence and full‐length genome sequence analysis. Transboundary and Emerging Diseases, 62, 575–580. 10.1111/tbed.12399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Shibahara, T. , Imai, N. , Yamamoto, T. , & Ohashi, S. (2018). Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infection, Genetics and Evolution, 176–182. 10.1016/j.meegid.2018.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerging Infectious Diseases, 20(7), 1227–1230. 10.3201/eid2007.140296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Huang, Y.i , Lau, S. K. P. , & Yuen, K. ‐. Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses, 2, 1804–1820. 10.3390/v2081803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Lau, S. K. P. , Lam, C. S. F. , Lau, C. C. Y. , Tsang, A. K. L. , Lau, J. H. N. , Bai, R. , Teng, J. L. L. , Tsang, C. C. C. , Wang, M. , Zheng, B. ‐ J. , Chan, K. ‐ H. , & Yuen, K. ‐ Y. (2012). Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavi. Journal of Virology, 86(7), 3995–4008. 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. ‐. L. , Meng, F. , Qin, P. , Herrler, G. , Huang, Y. ‐. W. , & Tang, Y. ‐. D. (2020). Trypsin promotes porcine deltacoronavirus mediating cell‐to‐cell fusion in a cell type‐ dependent manner. Emerging Microbes and Infections, 9, 457–468. 10.1080/22221751.2020.1730245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. (2016). Porcine deltacoronavirus : Overview of infection dynamics , diagnostic methods , prevalence and genetic evolution. Virus Research, 226, 71–84. 10.1016/j.virusres.2016.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Chen, J. , Shi, D.a , Shi, H. , Zhang, X. , Liu, J. , Cao, L. , Zhu, X. , Liu, Y.e , Wang, X. , Ji, Z. , & Feng, L.i (2019). Porcine deltacoronavirus enters cells via two pathways: A protease‐mediated one at the cell surface and another facilitated by cathepsins in the endosome. Journal of Biological Chemistry, 294(25), 9830–9843. 10.1074/jbc.RA119.007779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Qu, H. , Hu, J. , Fu, J. , Chen, R. , Li, C. , Cao, S. , Wen, Y. , Wu, R. , Zhao, Q. , Yan, Q. , Wen, X. , & Huang, X. (2019). Characterization and pathogenicity of the porcine deltacoronavirus isolated in Southwest China. Viruses, 11(1074), 1–22. 10.3390/v11111074 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study were submitted to the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) with accession number MT227371 for the Peruvian strain of PDCoV obtained.