

Abstract
Despite intense elimination efforts, human malaria, caused by the infection of five Plasmodium species, remains the deadliest parasitic disease in the world. Even worse, with the emergence and spreading of the first‐line drug‐resistant Plasmodium parasites, therapeutic interventions based on novel plasmodial drug targets are more necessary than ever. Given that the blood‐stage parasites primarily rely on glycolysis for their energy supply, blocking glucose uptake, the rate‐limiting step of ATP generation, was considered a promising approach to kill these parasites. To achieve this goal, characterization of the plasmodial hexose transporter and development of selective inhibitors have been pursued for decades. Here, we review the identification and characterization of the Plasmodium falciparum hexose transporter (PfHT1) and summarize current advances in its inhibitor development.
Keywords: drug design, GLUT, hexose transporter, malaria, PfHT1
1. INTRODUCTION
Malaria, a life‐threatening parasite disease, is caused by the infection of mosquito‐borne Plasmodium parasites. Five species of Plasmodium parasites have been reported as the causal agents of human malaria, including P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi. 1 Of those parasites, P. falciparum represents the deadliest form and accounts for the majority of deaths in African. 2 Great efforts have been made to eliminate malaria since the 1990s, which resulted in an approximately 29% reduction of malaria cases over the past two decades. 3 Nonetheless, the situation is still alarming as malaria led to an estimated 241 million infections and 627 000 deaths worldwide in 2020. 4
Despite intense efforts to generate malarial vaccines, merely one vaccine, the RTS,S, is recommended by the World Health Organization (WHO) for the prevention of P. falciparum infection in children, 4 , 5 leaving antimalarial drugs as the current major therapeutic choice. 6 To date, more than 20 different antimalarial drugs have been approved for the prophylaxis and treatment of malaria. 7 Nonetheless, the rapid emergence and spreading of drug‐resistant parasites severely threaten the global malaria control. 8 , 9 , 10 , 11 Nowadays, all antimalarial drugs have encountered the issue of drug resistance, raising urgent demands for antimalarial agents based on novel therapeutic targets.
The sophisticated life cycle of Plasmodium parasites consists of the human liver‐stage, human blood‐stage, and mosquito‐stage. During the blood‐stage, Plasmodium parasites rapidly replicate asexually inside human erythrocytes, causing the clinical symptoms of malaria. 12 To meet the high energy demands for rapid proliferation, blood‐stage parasites rely on the fast but less efficient process of glycolysis for generating ATP. 13 Consequently, parasitized erythrocytes accelerate their glucose uptake up to 100‐fold to support the increased glucose consumption. 14 , 15 , 16 , 17 , 18 Moreover, due to the lack of fructose bisphosphatase, P. falciparum cannot synthesize energy stores like glycogen, making continuous glucose uptake an essential step for parasitic metabolism and survival. 19 Supporting this notion, deprivation of glucose from freed P. falciparum led to a reduction in glycolytic flux and a rapid decline in intracellular ATP levels, which severely perturbed its metabolic homeostasis. 20 , 21 , 22
Cutting off the energy supply of blood‐stage Plasmodium parasites through inhibition of their glucose uptake, therefore, represents an enticing strategy in impeding intraerythrocytic parasite proliferation. Accordingly, a major facilitator superfamily (MFS) transporter, PfHT1, was identified from the glucose permeable pathway of blood‐stage P. falciparum. 23 , 24 The PfHT1 is an essential hexose facilitator on parasite plasma membrane (PPM) for the glucose uptake of parasites. 25 Comparative investigation of P. falciparum hexokinase (PfHK) and PfHT1 revealed that glucose uptake through PfHT1 is the rate‐limiting step of intraerythrocytic parasite glycolysis 26 and inhibiting PfHT1 exhibited parasiticidal effects. 27 Therefore, PfHT1 is considered a novel antimalarial drug target for pharmacological intervention. Herein, we review the identification, characterization and inhibitor development of PfHT1 with emphasis on recent progresses on structural‐based rational inhibitor design.
2. THE DISCOVERY OF PFHT1
Morphological and physical changes of malaria‐infected erythrocytes have long been recognized, which result in the perturbation of erythrocytic substrate permeability of sodium, potassium, and amino acids. 28 , 29 , 30 , 31 The concept that glucose permeability of parasitized erythrocytes might also be altered can be dated back to 1966 when Herman and colleagues investigated the 14C‐d‐glucose utilization of P. gallinaceum‐infected chicken erythrocytes. 32 , 33 They found that the parasitized chicken erythrocytes produced more 14CO2 derived from 14C‐d‐glucose and speculated that the uptake of glucose was increased by parasite infection. 32
The first experimental evidence of accelerated glucose uptake in parasitized erythrocytes was reported in 1974. Through comparing 14C‐labeled d‐glucose and 3‐O‐methyl glucose (3‐OMG) uptake in P. lophurae infected or uninfected duck erythrocytes, Shereman and Tanigoshi proved an increase of glucose uptake in parasitized erythrocytes and reasoned that this phenotype is due to alterations of a simple diffusion component. 33 In the same year, Homewood and Neame published their observations on l‐glucose uptake by P. berghei‐infected mouse erythrocytes and concluded that the l‐glucose only entered the parasitized erythrocytes, but not the uninfected erythrocytes. 34 , 35
Contrary to the diffusion model, Izumo et al. proposed an active transport model in 1989. By comparing 3H‐labeled 2‐deoxy‐d‐glucose (2‐DOG) uptake in P. yoelii‐infected or P. yoelii‐uninfected mouse erythrocytes, Izumo et al. observed an 8‐fold 2‐DOG concentration increase inside the parasite compared to the parasitized erythrocyte cytosol. 36 Additionally, the transport of 2‐DOG was largely decreased by the supplement of a proton ionophore, carbonylcyanide m‐chlorophenylhydrazone (CCCP). They proposed that two components, a simple diffusion system on the erythrocyte plasma membrane (EPM) and a proton‐coupled active glucose transporter on the PPM, might be involved in the glucose uptake in parasitized erythrocytes. 36 , 37
Subsequent research from Kirk et al. confirmed the accumulation of14C‐2‐DOG in P. falciparum‐infected human erythrocytes and discovered that the phosphorylation of 2‐DOG, rather than active transport, accounted for the substrate accumulation. 36 , 38 Given that the intracellular/extracellular ratios of 3‐OMG and l‐glucose were kept below 1, Kirk et al. concluded that the glucose uptake of parasitized erythrocytes works through an equilibrium manner rather than via active transport. 38 Parallel to this report, Goodyer et al. also raised a similar concern regarding the possibility of 2‐DOG phosphorylation in the active transport model. 39 They employed 3H‐labeled 6‐deoxy‐d‐glucose (6‐DOG), a non‐phosphorylatable glucose analog, for an uptake and efflux assay. Based on kinetic analysis, Goodyer et al. proposed the existence of a saturable glucose transporter on the membrane of parasite compartment. 39 Due to the interference of a possible duct system on parasitophorous vacuole membrane (PVM), the specific location of this glucose transporter could not be distinguished in this research.
To eliminate the interference of the multi‐membrane system in malaria‐infected erythrocytes, Penny et al. used a Xenopus laevis oocytes heterologous expression system to validate plasmodial nutrient transporters. 23 By injecting mRNA isolated from asexual stages P. falciparum into X. laevis oocytes, Penny et al. observed a 7‐fold increase in 2‐DOG uptake, which therefore confirmed the existence of a P. falciparum‐encoded glucose transporter. 23 In the following milestone work, Woodrow et al. identified the sequence of Plasmodium falciparum hexose transporter, designated PfHT1, by searching the homologous sequence of human glucose transporter 1 (GLUT1). 24 Using the X. laevis oocytes expression system, the authors confirmed d‐glucose and 2‐DOG uptake by PfHT1. Additionally, PfHT1 was found to localize on the PPM rather than the EPM by immunofluorescence staining. 24 Henceforth, the approach to cut off glucose uptake for malarial therapies progressed toward the biochemical characterization of PfHT1 and its inhibitor development.
It is noteworthy that the nutrient uptake of intraerythrocytic parasites is much more complicated due to the existence of a PVM and the adaptation changes of parasitic and host membrane systems. For instance, blood‐stage parasites are encompassed by PVM, making the passage through PVM as a precondition for glucose entry to PPM. 40 A non‐selective channel on PVM is responsible for the permeation of soluble macromolecules up to 1400 Da. 41 , 42 Despite no direct evidence for glucose permeation, the PVM channel has been considered a permeable path for glucose uptake given its permeability to a hexose‐like polyol glucoronate. 41 Moreover, a duct system on PVM has also been reported to offer direct access to extracellular macromolecules. 39 , 43 , 44 Additionally, a tubovesicular membrane (TVM) extended from the PVM is relevant to the delivery of nucleosides and amino acids to parasites. 23 , 45 Both the duct system and the TVM are undercharacterized and their roles in glucose uptake need further investigations. As for the host erythrocyte membrane, except for GLUT1, a parasite‐induced new permeability pathway (NPP) is formed during trophozoite maturation. 46 , 47 It has been reported that NPP could conduct glucose. 48 Nonetheless, glucose penetration of parasitized erythrocytes is dominated by GLUT1‐mediated facilitated diffusion. 49
Taken together, the major glucose permeable pathway for intraerythrocytic parasites can be described by a classic three membrane model including EPM, PVM, and PPM (Figure 1). In this model, extracellular glucose is first transported across the EPM by GLUT1 in an equilibrium manner. 50 Intracellular glucose then penetrates through PVM via a non‐selective channel. Finally, PfHT1 mediates the facilitated diffusion of glucose across the PPM. The subsequent phosphorylation of glucose in glycolysis can maintain the concentration gradient toward parasitic cytosol and drives the glucose influx from extracellular serum.
FIGURE 1.
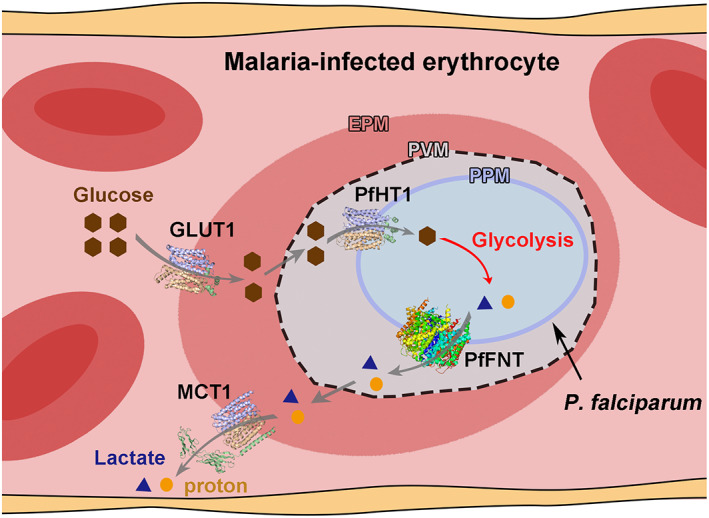
Schematic diagram for the glucose uptake and metabolism of the asexual blood‐stage P. falciparum. Three membrane model of the malaria‐infected erythrocytes. The NPP on EPM and the duct and TVM on PVM are omitted. Glucose and lactate are represented by brown hexagons and blue triangles, respectively. Protons are presented as orange circles. The PDB codes for the GLUT1, PfHT1, PfFNT, and MCT1 structures are 4PYP, 6M20, 7E26, and 6LZ0, respectively. All structure figures were prepared with PyMOL
3. FUNCTIONAL CHARACTERIZATION OF PFHT1
PfHT1 was first identified as a putative transporter gene on P. falciparum chromosome 2. As the hybridization of the P. falciparum genome with low stringency PfHT1 sequence cannot yield close sequence, PfHT1 was defined as a single‐copy gene without closed paralogues. 24 A 504‐amino‐acid transmembrane protein, located on the parasite plasma membrane, is encoded by PfHT1. PfHT1 was originally assumed as a glucose transporter for its ability to increase the d‐glucose and 2‐DOG uptake in X. laevis oocytes. 24 Further competition binding assays revealed d‐fructose as a competitor of d‐glucose or 2‐DOG uptake, indicating that d‐fructose could be a potential substrate for PfHT1. Subsequently, this hypothesis was verified by the PfHT1‐mediated 14C‐d‐fructose uptake in X. laevis oocytes. 24 , 51 Therefore, PfHT1 is responsible for both the d‐glucose and d‐fructose uptake of parasites.
Using the X. laevis oocytes heterologous expression system, the transport property and substrate recognition of PfHT1 were analyzed through an in vitro transport and competition binding assay. The glucose and fructose uptake assay revealed PfHT1 as a saturable transporter, whose transport activity is independent of the sodium and proton gradient. 24 , 52 The transport activity of PfHT1 can be inhibited by conventional glucose transporter inhibitors like Phloretin, Phloridizin, and Cytochalasin B (CCB). l‐Glucose cannot inhibit the PfHT1‐mediated uptake of d‐glucose and 2‐DOG, classifying PfHT1 as a stereospecific transporter. 24 Together with the evidence that glucose uptake of intraerythrocytic parasites is mediated by a saturable transporter in an equilibrium manner, PfHT1 was defined as a stereospecific facilitator to mediate the diffusion of d‐glucose and d‐fructose across parasite plasma membrane. Kinetic analysis revealed that the affinities (K m) of PfHT1 for d‐glucose and d‐fructose were 0.97 and 11.6 mM, respectively. 52 In contrast, its human counterpart GLUT1 facilitates glucose uptake with a K m of 3 mM, and another counterpart GLUT5 solely contributes to fructose uptake with a K m of 6 mM. 50 , 53 The higher glucose affinity of PfHT1 might help with glucose influx into intraerythrocytic parasites. Notably, despite PfHT1‐mediated fructose uptake supporting the parasite survival in vitro, fructose is unlikely the main energy source for blood‐stage parasites under physiological conditions, as the physiological fructose concentration is relatively low. 51 , 54
Stage‐dependent expression of PfHT1 was first characterized by measuring its mRNA levels during the blood‐stage. 24 PfHT1 is constitutively expressed throughout the blood‐stage with the highest, intermediate, and lowest levels occurring in the early ring stage, trophozoites and meronts, and gametocytes, respectively. 24 Consistently, knock‐out of PfHT1 led to a lethal phenotype in intraerythrocytic P. falciparum, whereas complementation of episomal PfHT1 rescued the transgenic parasite. 55 These results suggested that PfHT1 is an essential component for the blood‐stage P. falciparum. To extend the investigation to the whole plasmodial life cycle, P. berghei, a rodent malarial species amenable to in vivo life cycle studies, was further characterized to detect the expression of P. berghei hexose transporter (PbHT1). By monitoring fluorescence signal from a GFP‐tagged PbHT1 or immunofluorescence signal from a hemagglutinin‐tagged PbHT1 in transgenic P. berghei, PbHT1 was found to be constitutively expressed in both mosquito‐stage and liver‐stage parasites. 55 , 56 , 57 Therefore, Plasmodium hexose transporters, like PfHT1 and PbHT1, are not only essential for the blood‐stage parasites but may also play important roles in the rest of the plasmodial life cycle.
4. MOLECULAR MECHANISM OF PFHT1 AND ITS SUBSTRATE RECOGNITION
4.1. Competition analysis and site‐directed mutagenesis
Before experimental structures were available, analysis of PfHT1 substrate recognition relied on transport and competition binding assays using an X. laevis oocytes heterologous expression system. A variety of hexose analogs were applied to inhibit PfHT1‐mediated d‐glucose uptake. 24 , 51 The calculated inhibition constant (Ki) of the competitors enabled evaluation of the relative binding strengths of different hexose analogs, revealing the structural requirements for PfHT1 substrate recognition. 24
In aqueous solvents, d‐glucose mainly exists in pyranose form as approximately 33% α‐anomer and 66% β‐anomer (Figure 2A). To characterize the functionalities of hydroxyl groups on the pyranose form of d‐glucose, analogs with hydroxyl group substitutions were first interrogated by competition binding assays. The K i of 2‐DOG, 5‐thio‐d‐glucose, and 6‐DOG are similar to the K m of d‐glucose. On the other hand, K i of 1‐DOG and 3‐DOG increased substantially, indicating that 1‐ and 3‐hydroxyl groups are involved in stronger interactions with PfHT1 compared to other hydroxyl groups. 51 Interestingly, 3‐fluoro‐3‐DOG and 3‐OMG restored the inhibition to PfHT1, suggesting that the 3‐hydroxyl group of d‐glucose might contribute to substrate binding as a hydrogen bond recipient. 24 , 51 Consistently, epimer analysis demonstrated that the affinity of C2 epimer (d‐mannose) is similar to d‐glucose but C3 epimer (d‐allose) has a much lower affinity. d‐Galactose (C4 epimer) has an intermediate affinity to PfHT1, suggesting that both orientations of 4‐hydroxyl group can contribute to the interaction. 51 To access the anomer preference, 1‐fluoro‐1‐deoxy‐α‐ and 1‐fluoro‐1‐deoxy‐β‐d‐glucose were analyzed in the competition binding assay, both of which possess comparable affinities as 1‐DOG. Therefore, similar to human glucose transporters, PfHT1 can accommodate both α‐ and β‐anomer of d‐glucose. 51 The two major forms of fructose in water solution include approximately 25% β‐fructofuranose and 65% β‐fructopyranose (Figure 2B). 2‐deoxy‐β‐fructofuranose, a β‐furanose analog, maintained a high affinity to PfHT1, whereas l‐sorbopyranose, the β‐pyranose analog and C5 epimer of d‐fructose, failed to inhibit fructose uptake of PfHT1. These results suggest that PfHT1 transports d‐fructose in the β‐furanose form. 51
FIGURE 2.
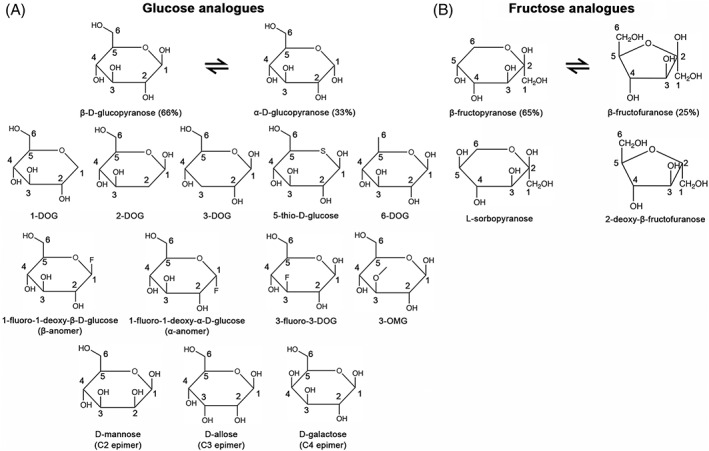
Representative analogs used in the substrate recognition studies of PfHT1. (A) Chemical structures of glucose analogs. (B) Chemical structures of fructose analogs
Site‐directed mutagenesis of PfHT1 was employed to analyze Gln169 on the transmembrane segment (TM) 5 and 302SGL triplet motif on TM7; the latter triplet motif was shown to be irrelevant to substrate binding in experimental structures. 51 , 58 , 59 , 60 Q169N variant reserved the ability of d‐glucose uptake, but failed to transport d‐fructose. Consistently, the K is of d‐glucose analogs, 1‐fluoro‐1‐deoxy‐α‐ and 1‐fluoro‐1‐deoxy‐β‐d‐glucose, are comparable between Q169N variant and wild‐type PfHT1, whereas the affinity of d‐fructose analog, 2‐deoxy‐β‐fructofuranose, to Q169N variant decreased significantly compared to wild‐type PfHT1. 51 These results indicated Gln169 is involved in d‐fructose binding and transport. Furthermore, a 4‐fold decrease in Ki for 5‐thio‐d‐glucose and 4‐fold increase in Ki for 6‐DOG were observed in the Q169N variant, suggesting that Gln169 interacts with 5‐ and 6‐hydroxyl group of d‐glucose. 58 To uncover substrate recognition and transport details, a putative three‐dimensional model of PfHT1 (PDB code 1LV1) was further established based on a predicted GLUT1 model (PDB code 1JA5). 58 Limited by the inaccuracy of the initial model and piecemeal strategy for helical assignment and prediction, structural information and mechanistic explanations based on the predicted PfHT1 model are unreliable, 61 and will not be further discussed here.
4.2. Crystal structure of PfHT1 bound with d‐glucose
Most recently, we and another group independently reported two crystal structures of d‐glucose‐bound PfHT1 at 2.6 and 3.65 Å, respectively. 59 , 60 The discrepancy of resolution might be due to the different detergents used for protein purification. Nonetheless, PfHT1 molecules in both structures were largely identical with a root‐mean‐square deviation (RMSD) of 0.444 Å over 364 Cα atoms. Henceforth, I will use our high‐resolution structure to elaborate the substrate recognition and transport mechanism of PfHT1.
Similar to other sugar porter (SP) family members like GLUT1 and GLUT3, the overall structure of PfHT1 exhibits a canonical MFS fold with 12 TMs forming two 6‐helical bundles. The intervening sequence between the N and C domains, together with the C‐terminal segment, forms five short helices, which constitute an intracellular helical (ICH) domain (Figure 3A). d‐Glucose resides in the central pocket located approximately in the middle of lipid bilayer. Detailed analysis of the central pocket revealed a highly conserved glucose‐binding pattern; the glucose‐binding residues and their configurations in PfHT1 were almost identical to those of GLUT1 and GLUT3 (Figure 3A). Four residues on the C domain and one residue on the N domain form polar contacts with d‐glucose. Specifically, the 1‐hydroxyl group of d‐glucose is stabilized by Gln169 on TM5, Gln305 on TM7a, and Trp412 on TM10. The 2‐hydroxyl group interacts with Gln305 on TM7a. The 3‐ and 4‐hydroxyl groups form two hydrogen bonds with Asn311 on TM7b. The 6‐hydroxyl group interacts with Gln169 on TM5 and Asn341 on TM8. An additional hydrogen bond is formed between Gln169 and the hemiacetal oxygen of glucose.
FIGURE 3.
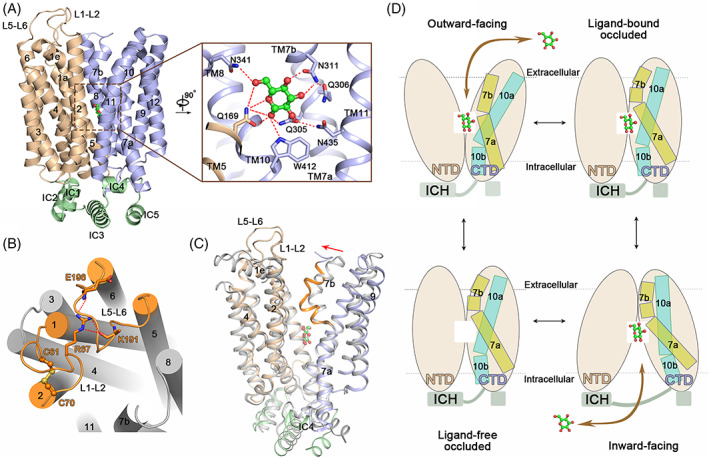
Substrate recognition and transport mechanisms of PfHT1. (A) Overall structure of d‐glucose‐bound PfHT1. Inset: d‐glucose‐binding pocket. The N‐, C‐domains, and intracellular helices are colored light brown, blue, and green, respectively. d‐glucose is shown as ball‐and‐stick and colored green. (B) The unique extracellular loop regions and disulfide bond of PfHT1. The L1–2 and L5–6 loops are colored orange. The disulfide bond is presented as the ball‐and‐stick. Residues involved in the extracellular loop interaction are shown as sticks. (C) Conformational changes between the outward‐occluded GLUT3 and occluded PfHT1. The glucose‐bound PfHT1 structure is colored domain‐wise. The glucose‐bound GLUT3 structure is colored gray. The TM7b of PfHT1‐glucose complex is highlighted in orange. The red arrow indicates the movement of TM7b. TM8 and TM11 are omitted for clarity. The PDB codes of outward‐occluded GLUT3 and occluded PfHT1 structures are 4ZW9 and 6M20, respectively. (D) Alternating access cycle for d‐glucose transport of PfHT1. The local shifts of TM7 and TM10 are highlighted through the cartoon representation
The residues adjacent to the substrate binding site are less conserved between PfHT1 and GLUTs, resulting in a larger central cavity for substrate accommodation in PfHT1. This might be one of the reasons why PfHT1 can transport variety of hexoses. Notably, a series of PfHT1 mutants were generated by the Drew group to rationalize substrate preferences. 60 Contrary to the previous report, Q169N variant abolished both the d‐glucose and d‐fructose transport of PfHT1. The authors identified another two variants, W412A and N435A, that impaired d‐fructose transport rather than d‐glucose transport. Nonetheless, the molecular mechanism for PfHT1 substrate promiscuity remains unclear. 60
Besides the common features like GLUTs, PfHT1 also possesses unique regions. Similar to the GLUT3 structure, the extracellular half of TM1 forms a membrane‐parallel helix, designated TM1e in PfHT1; however, the TM1e of PfHT1 is much shorter than that of GLUT3. The intervening sequence between TM1e and TM2 forms a long extracellular loop named L1–2, which tightly coordinates with another long extracellular loop L5–6 through hydrogen bonds between Arg67 on L1–2 and Glu196 and Lys191 on L5–6 (Figure 3B). A disulfide bond between Cys61 and Cys70 connects TM1 and TM2 together, which is important for the d‐glucose transport of PfHT1 (Figure 3B). Additionally, TM7b bends to a larger degree toward the substrate‐binding site of PfHT1, which seals the central cavity from the extracellular side. Together with the closed intracellular tunnel, the glucose‐bound PfHT1 structure presents as an occluded state, which is a missing piece in the alternating access cycle of GLUTs homologue structures (Figure 3C). Combined with other conformational states obtained from GLUTs structures, hexose transport by PfHT1 can be explained by the same alternating access model (Figure 3D). 62
5. INHIBITOR DEVELOPMENT TARGETING PFHT1
Given its important roles for parasite survival and development in all life cycle stages, pharmacological interventions targeting PfHT1 have long been pursued for novel malarial therapies. Developing selective inhibitors to discriminate PfHT1 and GLUTs is the first step in achieving this goal.
5.1. Semi‐rational basis design
The semi‐rational basis approach described the identification of the first synthetic PfHT1 specific inhibitor C3361, whose activity was confirmed through large‐scale screening with X. laevis oocytes‐based competition binding assay. 25 , 63 This pioneering work was initiated as the substrate requirements of PfHT1 were elucidated. Knowing that O‐3 hexose derivatives possess higher potency against PfHT1 than GLUT1, the Krishna group systematically examined O‐1 to O‐6 glucose derivatives, exemplified by 1‐, 2‐, 3‐, 4‐, 5‐, and 6‐OMG, for their inhibition of PfHT1‐ or GLUT1‐mediated d‐glucose uptake. 27 , 51 The 3‐OMG exhibited the highest selectivity, while 2‐ and 4‐OMG also presented intermediate selectivity. Longer O‐3 substitutions based on 3‐OMG were generated for further analysis, which yielded a selective PfHT1 inhibitor 3‐O‐(undec‐10‐en)‐1‐yl‐d‐glucose (C3361) 27 (Figure 4A). The C3361 selectively inhibited PfHT1 rather than GLUT1 with a selectivity index SIglucose (KiGLUT1 /KiPfHT1 ) of 62. The selectivity between PfHT1 and GLUT5 is higher as inhibition against GLUT5 was not observed. 27 Further characterization revealed that C3361 also inhibited hexose transporters from P. vivax (PvHT1), P. knowlesi (PkHT1), and P. yoelii (PyHT1) with comparable potency to PfHT1, suggesting C3361 as a broad‐spectrum hexose transporter inhibitor for different Plasmodium spp. 27 , 64
FIGURE 4.
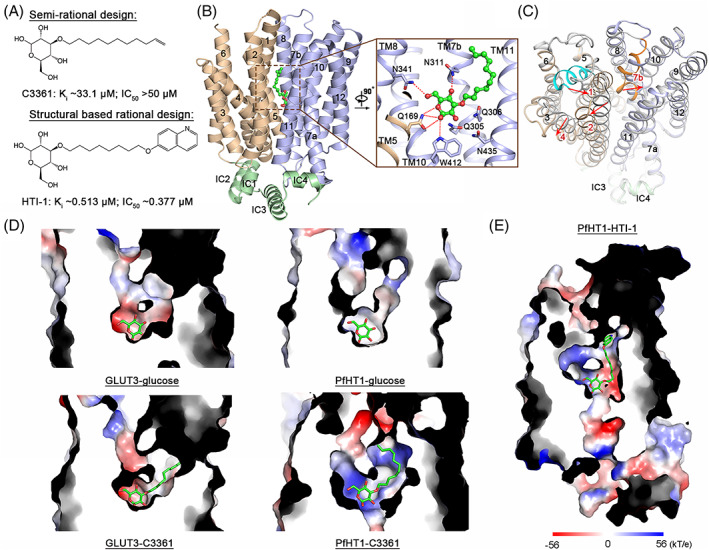
Inhibition mechanism of C3361 and structural‐guided lead optimization. (A) Chemical structures of C3361 and HTI‐1. The Ki represents the inhibition constant of PfHT1‐mediated 3 H‐d‐glucose uptake. The IC50 represents the inhibition constant of P. falciparum 3D7 growth. (B) Structure of C3361‐bound PfHT1. Inset: binding pose of C3361. The N‐, C‐domains, and intracellular helices are colored light brown, blue, and green, respectively. C3361 is presented as the ball‐and‐stick and colored green. The PDB code of C3361‐bound PfHT1 is 6M2L. (C) C3361‐induced conformational change. C3361‐bound PfHT1 is colored domain‐wise. Glucose‐bound PfHT1 is colored gray. The TM1e and TM7b of PfHT1‐glucose complex are highlighted with cyan and orange, respectively. The red arrows indicate the shifts of TM1, TM2, TM4, and TM7b. The PDB codes of PfHT1‐glucose and PfHT1‐C3361 complexes are 6 M20 and 6M2L, respectively. (D) Substrate binding pockets of GLUT3‐glucose, GLUT3‐C3361, PfHT1‐glucose, and PfHT1‐C3361. The PDB codes of GLUT3‐glucose, GLUT3‐C3361, PfHT1‐glucose, and PfHT1‐C3361 complexes are 4ZW9, 7CRZ, 6 M20, and 6M2L, respectively. (E) Cut‐open side view of the PfHT1‐HTI‐1 complex. The model was generated from the molecular dynamic simulation. All surface electrostatic potential maps were calculated by PyMOL
The parasiticidal activity of C3361 were assessed through drug treatments on in vitro cultured blood‐stage P. falciparum 3D7 (chloroquine sensitive strain) and K1 (multidrug resistant strain); the growth of both strains was inhibited by C3361. 27 A further study revealed that C3361 also inhibited schizont maturation of patient‐isolated P. vivax. 64 Life‐cycle studies using P. berghei demonstrated that C3361 could inhibit both hepatic‐stage and vector‐parasite‐stage parasites, and therefore exhibited transmission blocking activity. 56 , 57 , 65 Moreover, the in vivo potency of C3361 was evaluated by a P. berghei‐infected mouse model where significant suppression of parasitaemia was observed in C3361 treated mouse. 27 Therefore, C3361 served as a proof of principle for antimalarial drug development targeting of PfHT1.
Following the establishment of C3361, chemical modifications were employed to explore its structure–affinity relationships. By replacing the aliphatic tail of C3361 with other 3‐O‐substituents of different length, size and hydrophilicity, the optimum structures that maintained high affinity to PfHT1 were found to be those of 3‐O‐substituents with C8–C13 lipophilic chains. Meanwhile, none of their derivatives exhibited significant inhibition to GLUT1. 66 Subsequently, the optimum position for undec‐en‐10‐yl substituent on d‐glucose was investigated. Compared to other O‐substitution derivatives, 2‐O‐(undec‐10‐en)‐yl‐d‐glucose stood out for its approximately 25‐fold higher affinity to PfHT1 than C3361, and negligible inhibition of GLUT1. 67
5.2. Structural‐based rational design
Despite the identification and characterization of C3361 shedding light on novel antimalarial drug development, the optimization of further leads has been hampered by insufficient mechanistic insight about its inhibition. A “lollipop” model was proposed by the Krishna group after their discovery of C3361; that is to say, the glucose moiety of C3361 (the ball of the lollipop) occupies the sugar‐binding site while the aliphatic chain (the stick) associates with the lipid bilayer. 27 This hypothesis was raised based on the following observations: (1) the Q169N mutant of PfHT1 was less susceptible to C3361, indicating that its glucose moiety might interact with amino acids in the middle of TM5. 27 (2) the affinities of C3361 derivatives decreased with the increment of hydrophilicity of their aliphatic tail, which suggested that the aliphatic tail of C3361 is accommodated in a hydrophobic environment. 66
To explore the binding pose of C3361, structural models of PfHT1 were built with homology modeling, followed by molecular docking and dynamic simulations of C3361. 61 The best hit model was generated based on homology modeling of the crystal structure of FucP (PDB code 3O7Q). Given that FucP is an Escherichia coli l‐fructose/proton symporter, its substrate transport path and central cavity are very different from those of eukaryotic hexose facilitator, PfHT1, which led to inaccurate model building and C3361 docking. 61 , 68 Therefore, the molecular mechanism of C3361 inhibition remained enigmatic until the PfHT1‐C3361 structure was determined.
The experimental model of C3361‐bound PfHT1 was elucidated by a 3.7 Å resolution crystal structure. 59 The overall structure of C3361‐bound PfHT1 retains the occluded conformational state that seals the substrate transport tunnel from both extracellular and intracellular sides (Figure 4B). The binding pose of the glucose moiety of C3361 is almost identical to d‐glucose in the PfHT1‐glucose complex structure (Figure 4B). Remarkably, conformational changes of PfHT1 were observed in the neighborhood of the C3361 aliphatic chain; TM1e and TM7b straighten up with C3361 association, while TM2 and TM4 slightly swap away from the center pocket 59 (Figure 4C). The conformational shifts form a hydrophobic tunnel to accommodate the alkenyl tail of C3361. More importantly, an unprecedented allosteric pocket, encompassed by residues on TM1, 2, 7b, and 11, was induced, encaging the last five carbons of the aliphatic chain with an unoccupied extra space 59 (Figure 4D). Consistently, the density of the last three carbons on the aliphatic chain cannot be clearly resolved, indicating high mobility at the end of the tail. Notably, no conformational shift was observed between the C3361‐bound GLUT3 structure and the d‐glucose‐bound GLUT3 structure 69 , 70 (Figure 4D). Therefore, it is instinctive to pursue a high potent and selective PfHT1 inhibitor by simultaneously occupying both the substrate binding pocket and the novel allosteric pocket.
Based on the structural information, dozens of C3361 derivatives were rationally designed with modifications of their sugar moieties, aliphatic linkers and substituents at the end of the aliphatic tail. By introducing aromatic or heteroaromatic groups of various sizes to the end of the aliphatic tail, the quinolin‐6‐yloxyl group was identified as the best substituent to occupy the novel allosteric pocket. 59 Further structure–activity relationship (SAR) studies of derivatives with different sugar moieties and aliphatic linkers confirmed the following facts: (1) replacement of the d‐glucose moiety by d‐fructose or l‐glucose moiety decreased the potency of the inhibitor; 59 (2) the optimum length for aliphatic linker is between 8 and 10 carbons 59 , 69 ; (3) adding a hydrophilic group to the linker negates the inhibition activity of the inhibitor 69 ; (4) O‐2 and O‐3 sites of the d‐glucose moiety are the optimum positions for substitution. 69 Finally, 3‐O‐(8‐[quinolin‐6‐yloxy]octyl)‐d‐glucose, designated HTI‐1, was validated as the best hit for its 64‐fold higher affinity to PfHT1 compared to C3361 59 (Figure 4A,E). Proteoliposome‐based inhibition assays confirmed that HTI‐1 selectively inhibits PfHT1 over GLUT1. Also, HTI‐1 exhibits good parasiticidal activity to blood‐stage P. falciparum 3D7 and Dd2 (multidrug resistant strain) while exhibiting low cytotoxicity in both HEK293T/17 and HepG2 cell lines. 59 Together, the structural characterization of C3361‐bound PfHT1 led to the serendipitous discovery of an unexploited allosteric pocket. The following structural‐guided rational inhibitor design exploited both the sugar‐binding pocket and novel allosteric pocket and succeeded in generating the highly potent and selective inhibitor, HTI‐1, paving the way for the development of next‐generation orthosteric–allosteric dual inhibitor targeting PfHT1.
5.3. HTS for PfHT1 inhibitors
Except for the semi‐rational and structural‐based rational drug design, high‐throughput screens (HTS) provide another powerful path to identifying pre‐leads targeting PfHT1. To date, three different systems have been established for HTS, including a PfHT1‐complemented Saccharomyces cerevisiae system, 56 a glucose transporter null mutant (Δlmxgt) of Leishmania mexicana system, 71 and a fluorescence resonance energy transfer (FRET)‐based HTS system. 72 The PfHT1‐complemented yeast system was only validated by its susceptibility to C3361‐based growth inhibition, whereas the latter two HTS systems were implemented for PfHT1 inhibitor identification. 56 , 72 , 73 , 74
The L. mexicana HTS system was established in a glucose transporter knock out cell line Δlmxgt. 71 Through complementary expression of PfHT1 or GLUT1, the 3H‐d‐glucose uptake of Δlmxgt was restored, and defined as the complementary null mutant lines, Δlmxgt[pPfHT1] and Δlmxgt[pGLUT1]. Treatment of C3361 led to specific inhibition of 3H‐d‐glucose uptake of Δlmxgt[pPfHT1]. To increase throughput, the 3H‐d‐glucose was replaced by a live cell permeable dye, alamarBlue, to monitor the viability of L. Mexicana in the beginning. Further characterization confirmed that C3361 could specifically inhibit the growth of Δlmxgt[pPfHT1] and not Δlmxgt[pGLUT1], which served as a proof of principle for this HTS system. 71 In practical screening, the indicator was changed to the DNA‐binding dye, SYBR green, to monitor cell proliferation. 73
Using the Δlmxgt[pPfHT1/pGLUT1] L. mexicana HTS system, the Landfear group screened two “focused” libraries, whose parasiticidal effects against P. falciparum have been validated in vitro. 73 The first library, Tres Cantos antimalarial compound set (TCAMS), contained 13 533 compounds. The second, Malaria Box from Medicines for Malaria Venture (MMV), contained 400 compounds. Combined with two rounds of screening of 3H‐d‐glucose uptake assays and one round of 3H‐l‐proline uptake assays to rule out nonspecific binding, 6 hits from the TCAMS library and 3 hits from Malaria Box were identified. 73 Further dose–response characterization revealed only two hits, including TCMDC‐125163 and MMV009085 (also called GNF‐Pf‐3184), which exhibited high potency to Δlmxgt[pPfHT1] and low potency to Δlmxgt[pGLUT1] (Figure 5A). The parasiticidal effects of TCMDC‐125163 and its analogs were confirmed by both P. falciparum 3D7 and K1. The cytotoxicities of these compounds were insignificant to human foreskin fibroblasts cells (EC50 > 25 μM). 73 However, further optimization of TCMDC‐125163 is needed due to its poor pharmacokinetic property.
FIGURE 5.
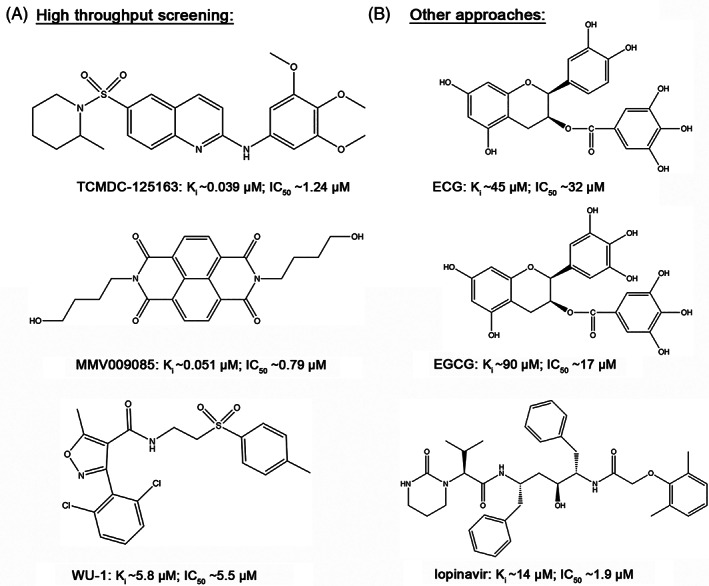
Representative compounds obtained from HTS and other approaches. Chemical structures of the representative compounds obtained from high‐throughput screening (A) and other approaches (B). The Ki represents the inhibition constant of PfHT1‐mediated d‐glucose/2‐DOG uptake. The IC50 represents the inhibition constant of P. falciparum 3D7 growth. Notably, the measurements of Ki and IC50 may vary in different research studies
The FRET‐based HTS system was designed to circumvent the weaknesses of the isotope‐labeled substrate transport assay and cell death assay, which involve (1) the manipulation of radiolabeled substrate limiting the throughput of the transport assay and (2) both approaches being difficult to distinguish PfHT1 inhibition from the inhibition of other targets. 72 Therefore, a direct glucose indicator is needed to monitor glucose uptake and its inhibition. FLII 12 Pglu‐700μδ6 (FLIP) is a chimeric protein with an architecture of CFP‐MgIB‐YFP. 75 The intermediate MgIB is a glucose‐/galactose‐binding domain flanked terminally by fluorescence proteins CFP and YFP. Upon glucose binding, conformational change of MgIB pulls CFP and YFP together, resulting in a resonance energy transfer from CFP to YFP. By exciting CFP and scanning the emission from YFP, glucose uptake can be directly monitored by the FRET signal of FLIP. HEK293 cells were transfected with plasmids of PfHT1 and FLIP to generate a stable PfHT1‐FLIP cell line. Endogenous GLUT1 expression was further knocked down by shRNA to eliminate background glucose absorption. 72
Using the FRET‐based PfHT1‐FLIP cell HTS system, 400 compounds of Malaria Box from MMV were first screened, yielding 5 primary hits. Combined with the results from 14C‐2‐DOG uptake inhibition and the parasite growth inhibition assay, the MMV009085 was identified as a PfHT1 inhibitor. 72 Inhibition of GLUT1‐4 mediated 3 H‐2‐DOG uptake demonstrated that MMV009085 has little effect on GLUTs‐mediated glucose transport. 72 Therefore, MMV009085 is a PfHT1 specific inhibitor, consistent with the preliminary finding from the screening using the L. mexicana HTS system. 72 , 73 In further research, the Hruz group screened 14 399 compounds from the Maybridge HitFinder collection, yielding 407 hits. 74 After removing 212 compounds with intrinsic fluorescence, the remaining 195 hits were narrowed down to 6, named WU‐1 to WU‐6, by their inhibition of a PfHT1‐mediated 3 H‐2‐DOG uptake assay. Furthermore, the four hits, WU‐1, WU‐2, WU‐3, and WU‐5, were characterized for their inhibition of GLUT1‐4, revealing WU‐1 as a PfHT1 selective inhibitor and WU‐2 as a less selective PfHT1 inhibitor. Subsequent parasite growth assays revealed that WU‐1 can inhibit P. falciparum 3D7 growth with a lower IC50 than it of WU‐2. Given that the IC50s for the glucose uptake inhibition (5.8 ± 0.6 μM) and the parasitic growth inhibition (5.5 ± 0.6 μM) are similar, it was proposed that WU‐1 kills P. falciparum by inhibiting PfHT1. Together, WU‐1 was validated as a lead that kills P. falciparum by selectively inhibiting PfHT1 74 (Figure 5A).
5.4. Other PfHT1 inhibitors
Except for the aforementioned approaches, catechins and lopinavir are another two compound classes that have been identified as PfHT1 inhibitors through similar routes. 76 That is to say, preliminary clues from other research revealed antimalarial activity and GLUTs‐binding properties for both compounds. It is thus of interest to determine whether catechins and lopinavir can kill the parasites through inhibition of PfHT1.
In the case of catechins, previous research found that catechins could inhibit the asexual blood‐stage P. falciparum growth. 77 Also, it was reported that gallated catechins are inhibitors of GLUT1. 78 To explore their antimalarial mechanism, four catechins extracted from green tea [(−)‐epicatechin (EC), (−)‐epicatechin‐gallate (ECG), (−)‐epigallocatechin (EGC) and (−)‐epigallocatechin‐gallate (EGCG)] were assessed with respect to their abilities to inhibit d‐glucose uptake via PfHT1 and GLUT1. 76 The gallated catechins (ECG and EGCG) were identified as PfHT1 and GLUT1 inhibitors with submillimolar Kis (Figure 5B). It was also found that gallated catechins could moderately inhibit GLUT5‐mediated d‐fructose transport. Further P. falciparum 3D7 growth inhibition assays revealed that IC50s of gallated catechins were comparable to their affinities of PfHT1. However, the parasite growth inhibition was not altered by different external d‐glucose concentrations. Therefore, while this research identified gallated catechins as nonselective inhibitors of PfHT1, GLUT1, and GLUT5, their antimalarial mechanisms remain elusive. 76
As for lopinavir, it was originally developed as a human immunodeficiency virus (HIV) protease inhibitor (PI). Further characterization revealed its antimalarial activity through drug susceptibility assays against P. falciparum. 79 The research of PI‐induced insulin resistance revealed that PIs directly interact with GLUT4. 80 These hints raised the possibility that lopinavir might achieve antimalarial activity through inhibiting PfHT1. 81 By competing with the 14 C‐2‐DOG uptake of parasites, lopinavir was found to block P. falciparum glucose uptake at a submillimolar IC50. Using a PfHT1‐overexpression HEK293 cell line, the inhibition of PfHT1‐mediated 3 H‐2‐DOG uptake by lopinavir was confirmed. The IC50 of PfHT1 inhibition (~14 μM) is like that of its inhibition of parasite glucose uptake (~16 μM), suggesting that lopinavir suppresses P. falciparum growth and glucose uptake by inhibiting PfHT1 81 (Figure 5B).
6. DISCUSSION
Despite the huge progress that has been made for malaria control, the global malaria burden remains heavy due to the malaria prevalence in Africa and Southeast Asia. New biological threats, exemplified by multidrug‐resistant parasites, are compromising the campaign for malaria elimilation. 82 Development of novel antimalarial therapies based on new parasitic targets are urgently needed to conquer drug‐resistant variants. Playing an essential role in the hexose uptake of asexual blood‐stage Plasmodium parasites, PfHT1 represents such a promising and unexploited drug target. Characterization of PfHT1 has uncovered its expression, functionality, enzymatic properties, and structural models, providing opportunities for further antimalarial agent development. Combining the semi‐rational design, structural‐based rational design, HTS, and other approaches, a series of leads with various chemical scaffolds have been identified or generated.
The first synthetic PfHT1 specific inhibitor, C3361, is the prototype for selective inhibition of PfHT1 compared to GLUTs. Characterization of the in vitro and in vivo parasiticidal activities of C3361 serves as a proof of principle for therapeutic intervention against malaria by inhibiting PfHT1. However, C3361 is not considered a drug‐like molecule. 73 , 83 Structural characterization of C3361‐bound PfHT1 identified unprecedented conformational changes, which lead to the formation of a novel allosteric pocket. By generating C3361 derivatives to occupy the unexploited pocket, an orthosteric–allosteric dual inhibitor, HTI‐1, which specifically and potently inhibits PfHT1 with high potency and efficiently suppresses the growth of the asexual blood‐stage P. falciparum, was rationally designed and generated. Nonetheless, it is unclear whether HTI‐1 can kill parasites in vivo. Further optimization is expected to increase the affinity of HTI‐1 while retaining its selectivity to PfHT1. Besides, the pharmacokinetics of HTI‐1 are not well characterized and require further investigations.
Given the progress from the structural guided rational design of HTI‐1, it is clear that structural models are of great importance for the understanding of the inhibition mechanism and optimization of lead compounds. It is therefore intuitive to investigate whether other PfHT1 inhibitors share a similar inhibition mechanism to C3361 and HTI‐1. If so, optimization of PfHT1 inhibitors with other chemical scaffolds can be achieved following a similar principle to HTI‐1. Otherwise, identifying novel inhibition mechanisms will urgently be needed. Despite the limited insight, hints for the existence of other inhibition mechanisms have been provided via the investigations of lopinavir and WU‐1, where both are suggested to bind to an inward‐facing state PfHT1. 74 , 81 It is therefore worth elucidating the molecular mechanism of PfHT1 binding to new‐scaffold inhibitors in future research.
Due to the limitations of current methods, life‐cycle studies of PfHT1 rely on the P. berghei infected mouse model and proteomic characterization. 55 The expression and functionality of PfHT1 were mainly characterized in blood‐stage parasites. Given that PbHT1 can express in all stages of the plasmodial life cycle, the parasiticidal activities of C3361 against liver‐stage and mosquito‐stage parasites have been obtained from the P. berghei model. Therefore, further characterizations of PfHT1 in the whole life cycle are needed, which could extend the functionality of PfHT1 inhibitors from symptomatic phase treatments to prophylactic and transmission‐blocking agents.
In an extended view, blocking the lactate export might be as efficient as inhibiting PfHT1‐mediated glucose uptake. 84 , 85 Given that glycolysis dominates the ATP generation of asexual blood‐stage parasites, the end product, lactate, must be continuously exported through P. falciparum lactate transporter (PfFNT) and human monocarboxylate transporter (MCT1) (Figure 1). PfFNT has been intensively interrogated for inhibitor development, which identified another Malarial Box compound, MMV007839, as a PfFNT‐specific inhibitor. 86 , 87 , 88 , 89 MMV007839 and its derivatives can efficiently kill parasites in vitro. 90 Therefore, therapeutic interventions targeting PfFNT may supplement antimalarial therapies by restricting plasmodial energy supply.
In summary, combined multiple drug design and screening methods, further therapeutic development targeting PfHT1 are promising to generate novel antimalarial agents that may circumvent multidrug‐resistant Plasmodium parasites and support the global fight against malaria.
CONFLICT OF INTEREST
A patent application was filed for HTI‐1. The author is one of the inventors for this patent application. Applicant institution: Tsinghua University. Application number: PCT/CN2020/074258. Status of application: not yet published.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26351.
ACKNOWLEDGMENT
Thanks to Dr Armella Zadoorian and Dr Ivan Lukmantara from the University of New South Wales for the critical reading of the manuscript. Open access publishing facilitated by University of New South Wales, as part of the Wiley ‐ University of New South Wales agreement via the Council of Australian University Librarians.
Jiang X. An overview of the Plasmodium falciparum hexose transporter and its therapeutic interventions. Proteins. 2022;90(10):1766‐1778. doi: 10.1002/prot.26351
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed
REFERENCES
- 1. Loy DE, Liu W, Li Y, et al. Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax . Int J Parasitol. 2017;47(2–3):87‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perkins DJ, Were T, Davenport GC, Kempaiah P, Hittner JB, Ong'echa JM. Severe malarial anemia: innate immunity and pathogenesis. Int J Biol Sci. 2011;7(9):1427‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization . World Malaria Report. World Health Organization; 2019. [Google Scholar]
- 4. World Health Organization . World Malaria Report. World Health Organization; 2021. [Google Scholar]
- 5. Rts SCTP. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015;386(9988):31‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wicht KJ, Mok S, Fidock DA. Molecular mechanisms of drug resistance in Plasmodium falciparum malaria. Annu Rev Microbiol. 2020;74:431‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haldar K, Bhattacharjee S, Safeukui I. Drug resistance in plasmodium. Nat Rev Microbiol. 2018;16(3):156‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sinha S, Medhi B, Sehgal R. Challenges of drug‐resistant malaria. Parasite. 2014;21:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trape JF. The public health impact of chloroquine resistance in Africa. Am J Trop Med Hyg. 2001;64(1–2 Suppl):12‐17. [DOI] [PubMed] [Google Scholar]
- 10. Sibley CH, Hyde JE, Sims PF, et al. Pyrimethamine‐sulfadoxine resistance in Plasmodium falciparum: what next? Trends Parasitol. 2001;17(12):582‐588. [DOI] [PubMed] [Google Scholar]
- 11. Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin‐resistant Plasmodium falciparum malaria. Nature. 2014;505(7481):50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Primers. 2017;3:17050. [DOI] [PubMed] [Google Scholar]
- 13. Sherman IW. Biochemistry of plasmodium (malarial parasites). Microbiol Rev. 1979;43(4):453‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khabir PA, Manwell RD. Glucose consumption of Plasmodium hexamerium . J Parasitol. 1955;41(6):595‐603. [PubMed] [Google Scholar]
- 15. Manwell RD, Feigelson P. Glycolysis in Plasmodium gallinaceum . Proc Soc Exp Biol Med. 1949;70(4):578‐582. [DOI] [PubMed] [Google Scholar]
- 16. McKee RW, Ormsbee RA, Anfinsen CB, Geiman QM, Ball EG. Studies on malarial parasites: vi. The chemistry and metabolism of normal and parasitized (P. knowlesi) monkey blood. J Exp Med. 1946;84(6):569‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warren L, Manwell RD. Rate of glucose consumption by malarial blood. Exp Parasitol. 1954;3(1):16‐24. [DOI] [PubMed] [Google Scholar]
- 18. Roth EF Jr. Malarial parasite hexokinase and hexokinase‐dependent glutathione reduction in the Plasmodium falciparum‐infected human erythrocyte. J Biol Chem. 1987;262(32):15678‐15682. [PubMed] [Google Scholar]
- 19. Krishna S, Webb R, Woodrow C. Transport proteins of Plasmodium falciparum: defining the limits of metabolism. Int J Parasitol. 2001;31(12):1331‐1342. [DOI] [PubMed] [Google Scholar]
- 20. Saliba KJ, Kirk K. pH regulation in the intracellular malaria parasite, Plasmodium falciparum. H(+) extrusion via a V‐type H(+)‐ATPase. J Biol Chem. 1999;274(47):33213‐33219. [DOI] [PubMed] [Google Scholar]
- 21. van Niekerk DD, Penkler GP, du Toit F, Snoep JL. Targeting glycolysis in the malaria parasite Plasmodium falciparum . FEBS J. 2016;283(4):634‐646. [DOI] [PubMed] [Google Scholar]
- 22. Saliba KJ, Krishna S, Kirk K. Inhibition of hexose transport and abrogation of pH homeostasis in the intraerythrocytic malaria parasite by an O‐3‐hexose derivative. FEBS Lett. 2004;570(1–3):93‐96. [DOI] [PubMed] [Google Scholar]
- 23. Penny JI, Hall ST, Woodrow CJ, Cowan GM, Gero AM, Krishna S. Expression of substrate‐specific transporters encoded by Plasmodium falciparum in Xenopus laevis oocytes. Mol Biochem Parasitol. 1998;93(1):81‐89. [DOI] [PubMed] [Google Scholar]
- 24. Woodrow CJ, Penny JI, Krishna S. Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem. 1999;274(11):7272‐7277. [DOI] [PubMed] [Google Scholar]
- 25. Joet T, Krishna S. The hexose transporter of Plasmodium falciparum is a worthy drug target. Acta Trop. 2004;89(3):371‐374. [DOI] [PubMed] [Google Scholar]
- 26. Tjhin ET, Staines HM, van Schalkwyk DA, Krishna S, Saliba KJ. Studies with the Plasmodium falciparum hexokinase reveal that PfHT limits the rate of glucose entry into glycolysis. FEBS Lett. 2013;587(19):3182‐3187. [DOI] [PubMed] [Google Scholar]
- 27. Joet T, Eckstein‐Ludwig U, Morin C, Krishna S. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci USA. 2003;100(13):7476‐7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weidekamm E, Wallach DF, Lin PS, Hendricks J. Erythrocyte membrane alterations due to infection with Plasmodium berghei . Biochim Biophys Acta. 1973;323(4):539‐546. [DOI] [PubMed] [Google Scholar]
- 29. Overman RR. Reversible cellular permeability alterations in disease; in vivo studies on sodium, potassium and chloride concentrations in erythrocytes of the malarious monkey. Am J Physiol. 1948;152(1):113‐121. [DOI] [PubMed] [Google Scholar]
- 30. Sherman IW, Virkar RA, Ruble JA. The accumulation of amino acids by Plasmodium lophurae (avian malaria). Comp Biochem Physiol. 1967;23(1):43‐57. [DOI] [PubMed] [Google Scholar]
- 31. Dunn MJ. Alterations of red blood cell sodium transport during malarial infection. J Clin Invest. 1969;48(4):674‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herman YF, Ward RA, Herman RH. Stimulation of the utilization of 1‐14C‐glucose in chicken red blood cells infected with Plasmodium gallinaceum . Am J Trop Med Hyg. 1966;15(3):276‐280. [DOI] [PubMed] [Google Scholar]
- 33. Sherman IW, Tanigoshi L. Glucose transport in the malarial (Plasmodium lophurae) infected erythrocyte. J Protozool. 1974;21(4):603‐607. [DOI] [PubMed] [Google Scholar]
- 34. Homewood CA, Neame KD. Malaria and the permeability of the host erythrocyte. Nature. 1974;252(5485):718‐719. [DOI] [PubMed] [Google Scholar]
- 35. Neame KD, Homewood CA. Alterations in the permeability of mouse erythrocytes infected with the malaria parasite, Plasmodium berghei . Int J Parasitol. 1975;5(5):537‐540. [DOI] [PubMed] [Google Scholar]
- 36. Izumo A, Tanabe K, Kato M, Doi S, Maekawa K, Takada S. Transport processes of 2‐deoxy‐D‐glucose in erythrocytes infected with Plasmodium yoelii, a rodent malaria parasite. Parasitology. 1989;98:371‐379. [DOI] [PubMed] [Google Scholar]
- 37. Tanabe K. Glucose transport in malaria infected erythrocytes. Parasitol Today. 1990;6(7):225‐229. [DOI] [PubMed] [Google Scholar]
- 38. Kirk K, Horner HA, Kirk J. Glucose uptake in Plasmodium falciparum‐infected erythrocytes is an equilibrative not an active process. Mol Biochem Parasitol. 1996;82(2):195‐205. [DOI] [PubMed] [Google Scholar]
- 39. Goodyer ID, Hayes DJ, Eisenthal R. Efflux of 6‐deoxy‐D‐glucose from Plasmodium falciparum‐infected erythrocytes via two saturable carriers. Mol Biochem Parasitol. 1997;84(2):229‐239. [DOI] [PubMed] [Google Scholar]
- 40. Kirk K, Lehane AM. Membrane transport in the malaria parasite and its host erythrocyte. Biochem J. 2014;457(1):1‐18. [DOI] [PubMed] [Google Scholar]
- 41. Desai SA, Krogstad DJ, McCleskey EW. A nutrient‐permeable channel on the intraerythrocytic malaria parasite. Nature. 1993;362(6421):643‐646. [DOI] [PubMed] [Google Scholar]
- 42. Desai SA, Rosenberg RL. Pore size of the malaria parasite's nutrient channel. Proc Natl Acad Sci USA. 1997;94(5):2045‐2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pouvelle B, Spiegel R, Hsiao L, et al. Direct access to serum macromolecules by intraerythrocytic malaria parasites. Nature. 1991;353(6339):73‐75. [DOI] [PubMed] [Google Scholar]
- 44. Wilairat P, Auparakkitanon S. Plug for the parasitophorous duct: a solution of two conundra. Malar J. 2020;19(1):370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lauer SA, Rathod PK, Ghori N, Haldar K. A membrane network for nutrient import in red cells infected with the malaria parasite. Science. 1997;276(5315):1122‐1125. [DOI] [PubMed] [Google Scholar]
- 46. Ginsburg H, Krugliak M, Eidelman O, Cabantchik ZI. New permeability pathways induced in membranes of Plasmodium falciparum infected erythrocytes. Mol Biochem Parasitol. 1983;8(2):177‐190. [DOI] [PubMed] [Google Scholar]
- 47. Kutner S, Breuer WV, Ginsburg H, Aley SB, Cabantchik ZI. Characterization of permeation pathways in the plasma membrane of human erythrocytes infected with early stages of Plasmodium falciparum: association with parasite development. J Cell Physiol. 1985;125(3):521‐527. [DOI] [PubMed] [Google Scholar]
- 48. Ginsburg H, Kutner S, Krugliak M, Cabantchik ZI. Characterization of permeation pathways appearing in the host membrane of Plasmodium falciparum infected red blood cells. Mol Biochem Parasitol. 1985;14(3):313‐322. [DOI] [PubMed] [Google Scholar]
- 49. Krishna S, Woodrow CJ, Burchmore RJ, Saliba KJ, Kirk K. Hexose transport in asexual stages of Plasmodium falciparum and kinetoplastidae. Parasitol Today. 2000;16(12):516‐521. [DOI] [PubMed] [Google Scholar]
- 50. Augustin R. The protein family of glucose transport facilitators: It's not only about glucose after all. IUBMB Life. 2010;62(5):315‐333. [DOI] [PubMed] [Google Scholar]
- 51. Woodrow CJ, Burchmore RJ, Krishna S. Hexose permeation pathways in Plasmodium falciparum‐infected erythrocytes. Proc Natl Acad Sci USA. 2000;97(18):9931‐9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Joet T, Holterman L, Stedman TT, et al. Comparative characterization of hexose transporters of Plasmodium knowlesi, Plasmodium yoelii and Toxoplasma gondii highlights functional differences within the apicomplexan family. Biochem J. 2002;368(Pt 3):923‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Slavic K, Krishna S, Derbyshire ET, Staines HM. Plasmodial sugar transporters as anti‐malarial drug targets and comparisons with other protozoa. Malar J. 2011;10:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Slavic K, Straschil U, Reininger L, et al. Life cycle studies of the hexose transporter of plasmodium species and genetic validation of their essentiality. Mol Microbiol. 2010;75(6):1402‐1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blume M, Hliscs M, Rodriguez‐Contreras D, et al. A constitutive pan‐hexose permease for the Plasmodium life cycle and transgenic models for screening of antimalarial sugar analogs. FASEB J. 2011;25(4):1218‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Slavic K, Delves MJ, Prudencio M, et al. Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite's life cycle. Antimicrob Agents Chemother. 2011;55(6):2824‐2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manning SK, Woodrow C, Zuniga FA, et al. Mutational analysis of the hexose transporter of Plasmodium falciparum and development of a three‐dimensional model. J Biol Chem. 2002;277(34):30942‐30949. [DOI] [PubMed] [Google Scholar]
- 59. Jiang X, Yuan Y, Huang J, et al. Structural basis for blocking sugar uptake into the malaria parasite Plasmodium falciparum . Cell. 2020;183:258‐268.e12. [DOI] [PubMed] [Google Scholar]
- 60. Qureshi AA, Suades A, Matsuoka R, et al. The molecular basis for sugar import in malaria parasites. Nature. 2020;578(7794):321‐325. [DOI] [PubMed] [Google Scholar]
- 61. Fonseca AL, Nunes RR, Braga VM, et al. Docking, QM/MM, and molecular dynamics simulations of the hexose transporter from Plasmodium falciparum (PfHT). J Mol Graph Model. 2016;66:174‐186. [DOI] [PubMed] [Google Scholar]
- 62. Yan N. A glimpse of membrane transport through structures‐advances in the structural biology of the GLUT glucose transporters. J Mol Biol. 2017;429(17):2710‐2725. [DOI] [PubMed] [Google Scholar]
- 63. Joet T, Morin C, Fischbarg J, et al. Why is the Plasmodium falciparum hexose transporter a promising new drug target? Expert Opin Ther Targets. 2003;7(5):593‐602. [DOI] [PubMed] [Google Scholar]
- 64. Joet T, Chotivanich K, Silamut K, Patel AP, Morin C, Krishna S. Analysis of Plasmodium vivax hexose transporters and effects of a parasitocidal inhibitor. Biochem J. 2004;381(Pt 3):905‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Talman AM, Prieto JH, Marques S, et al. Proteomic analysis of the plasmodium male gamete reveals the key role for glycolysis in flagellar motility. Malar J. 2014;13:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fayolle M, Ionita M, Krishna S, Morin C, Patel AP. Probing structure/affinity relationships for the Plasmodium falciparum hexose transporter with glucose derivatives. Bioorg Med Chem Lett. 2006;16(5):1267‐1271. [DOI] [PubMed] [Google Scholar]
- 67. Ionita M, Krishna S, Leo PM, Morin C, Patel AP. Interaction of O‐(undec‐10‐en)‐yl‐D‐glucose derivatives with the Plasmodium falciparum hexose transporter (PfHT). Bioorg Med Chem Lett. 2007;17(17):4934‐4937. [DOI] [PubMed] [Google Scholar]
- 68. Dang S, Sun L, Huang Y, et al. Structure of a fucose transporter in an outward‐open conformation. Nature. 2010;467(7316):734‐738. [DOI] [PubMed] [Google Scholar]
- 69. Huang J, Yuan Y, Zhao N, et al. Orthosteric‐allosteric dual inhibitors of PfHT1 as selective antimalarial agents. Proc Natl Acad Sci USA. 2021;118(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Deng D, Sun P, Yan C, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526(7573):391‐396. [DOI] [PubMed] [Google Scholar]
- 71. Feistel T, Hodson CA, Peyton DH, Landfear SM. An expression system to screen for inhibitors of parasite glucose transporters. Mol Biochem Parasitol. 2008;162(1):71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kraft TE, Heitmeier MR, Putanko M, et al. A novel fluorescence resonance energy transfer‐based screen in high‐throughput format to identify inhibitors of malarial and human glucose transporters. Antimicrob Agents Chemother. 2016;60(12):7407‐7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ortiz D, Guiguemde WA, Johnson A, et al. Identification of selective inhibitors of the plasmodium falciparum hexose transporter PfHT by screening focused libraries of anti‐malarial compounds. PLoS One. 2015;10(4):e0123598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heitmeier MR, Hresko RC, Edwards RL, et al. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG‐mediated protein engineering. PLoS One. 2019;14(5):e0216457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hou BH, Takanaga H, Grossmann G, et al. Optical sensors for monitoring dynamic changes of intracellular metabolite levels in mammalian cells. Nat Protoc. 2011;6(11):1818‐1833. [DOI] [PubMed] [Google Scholar]
- 76. Slavic K, Derbyshire ET, Naftalin RJ, Krishna S, Staines HM. Comparison of effects of green tea catechins on apicomplexan hexose transporters and mammalian orthologues. Mol Biochem Parasitol. 2009;168(1):113‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sannella AR, Messori L, Casini A, et al. Antimalarial properties of green tea. Biochem Biophys Res Commun. 2007;353(1):177‐181. [DOI] [PubMed] [Google Scholar]
- 78. Naftalin RJ, Afzal I, Cunningham P, et al. Interactions of androgens, green tea catechins and the antiandrogen flutamide with the external glucose‐binding site of the human erythrocyte glucose transporter GLUT1. Br J Pharmacol. 2003;140(3):487‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nsanzabana C, Rosenthal PJ. In vitro activity of antiretroviral drugs against Plasmodium falciparum . Antimicrob Agents Chemother. 2011;55(11):5073‐5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hresko RC, Hruz PW. HIV protease inhibitors act as competitive inhibitors of the cytoplasmic glucose binding site of GLUTs with differing affinities for GLUT1 and GLUT4. PLoS One. 2011;6(9):e25237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kraft TE, Armstrong C, Heitmeier MR, Odom AR, Hruz PW. The glucose transporter PfHT1 is an antimalarial target of the HIV protease inhibitor Lopinavir. Antimicrob Agents Chemother. 2015;59(10):6203‐6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. World Health Organization . World Malaria Report. World Health Organization; 2020. [Google Scholar]
- 83. Staines HM, Derbyshire ET, Slavic K, Tattersall A, Vial H, Krishna S. Exploiting the therapeutic potential of Plasmodium falciparum solute transporters. Trends Parasitol. 2010;26(6):284‐296. [DOI] [PubMed] [Google Scholar]
- 84. Marchetti RV, Lehane AM, Shafik SH, Winterberg M, Martin RE, Kirk K. A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum . Nat Commun. 2015;6:6721. [DOI] [PubMed] [Google Scholar]
- 85. Wu B, Rambow J, Bock S, et al. Identity of a plasmodium lactate/H(+) symporter structurally unrelated to human transporters. Nat Commun. 2015;6:6284. [DOI] [PubMed] [Google Scholar]
- 86. Golldack A, Henke B, Bergmann B, et al. Substrate‐analogous inhibitors exert antimalarial action by targeting the plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog. 2017;13(2):e1006172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hapuarachchi SV, Cobbold SA, Shafik SH, et al. The malaria Parasite's lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog. 2017;13(2):e1006180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lyu M, Su CC, Kazura JW, Yu EW. Structural basis of transport and inhibition of the Plasmodium falciparum transporter PfFNT. EMBO Rep. 2021;22:e51628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Peng X, Wang N, Zhu A, et al. Structural characterization of the Plasmodium falciparum lactate transporter PfFNT alone and in complex with antimalarial compound MMV007839 reveals its inhibition mechanism. PLoS Biol. 2021;19(9):e3001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Walloch P, Hansen C, Priegann T, Schade D, Beitz E. Pentafluoro‐3‐hydroxy‐pent‐2‐en‐1‐ones potently inhibit FNT‐type lactate transporters from all five human‐pathogenic plasmodium species. ChemMedChem. 2020;16:1283‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed