

Abstract
Von Willebrand factor (VWF) is a glycoprotein that is secreted into the circulation and controls bleeding by promoting adhesion and aggregation of blood platelets at sites of vascular injury. Substantial inter‐individual variation in VWF plasma levels exists among the healthy population. Prior to secretion, VWF polymers are assembled and condensed into helical tubules, which are packaged into Weibel‐Palade bodies (WPBs), a highly specialized post‐Golgi storage compartment in vascular endothelial cells. In the inherited bleeding disorder Von Willebrand disease (VWD), mutations in the VWF gene can cause qualitative or quantitative defects, limiting protein function, secretion, or plasma survival. However, pathogenic VWF mutations cannot be found in all VWD cases. Although an increasing number of genetic modifiers have been identified, even more rare genetic variants that impact VWF plasma levels likely remain to be discovered. Here, we summarize recent evidence that modulation of the early secretory pathway has great impact on the biogenesis and release of WPBs. Based on these findings, we propose that rare, as yet unidentified quantitative trait loci influencing intracellular VWF transport contribute to highly variable VWF levels in the population. These may underlie the thrombotic complications linked to high VWF levels, as well as the bleeding tendency in individuals with low VWF levels.
Keywords: endoplasmic reticulum, endothelial cell, GBF1, Golgi, SEC22B, SNARE, STX5, Von Willebrand disease, Von Willebrand factor, Weibel‐Palade body
Endothelial cells secrete the haemostatic Von Willebrand factor (VWF) from secretory organelles, the Weibel‐Palade bodies. Quantitative deficiencies in VWF lead to the bleeding disorder Von Willebrand disease. Here, we discuss how a careful equilibrium between Arfs and SNAREs at the ER–Golgi interface controls the number, length and secretion competence of Weibel‐Palade bodies and how environmental cues act through this pathway to modify the secretory response of the endothelium.
Abbreviations
- ADP
adenosine diphosphate
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
5' adenosine monophosphate‐activated protein kinase
- AP‐1
adaptor protein 1
- Arf
ADP ribosylation factor
- COG
conserved oligomeric Golgi complex
- EC
endothelial cell
- ECFC
endothelial colony‐forming cell
- ER
endoplasmic reticulum
- GAP
GTPase activating protein
- GBF1
Golgi‐specific brefeldin A‐resistance guanine nucleotide exchange factor 1
- GEF
guanine nucleotide exchange factor
- GOSR1/2
Golgi SNAP receptor complex member 1/2
- GTPase
guanosine triphosphate hydrolase
- GWAS
genome‐wide association study
- KLF2
Krüppel‐like Factor 2
- MADD
MAP kinase‐activating death domain protein
- PI4K
phosphatidylinositol 4‐kinase
- SMAP1
stromal membrane‐associated protein 1
- SNARE
soluble N‐ethylmaleimide‐sensitive factor attachment proteins receptor
- STX2
syntaxin 2
- STX5
syntaxin 5
- STXBP5
syntaxin‐binding protein 5
- TGN
trans‐Golgi network
- VWD
Von Willebrand disease
- VWF
Von Willebrand factor
- WPB
Weibel‐Palade body
Introduction
Von Willebrand factor (VWF) is a large hemostatic glycoprotein that plays a central role in initiating blood coagulation and in minimizing bleeding. As a first response upon vascular injury, VWF is rapidly secreted from endothelial cells into the blood vessel lumen where it provides a mechanosensitive adhesive platform for platelets at sites of blood vessel damage. VWF in plasma also protects the coagulation factor VIII (FVIII) from premature clearance from the circulation. VWF is a pivotal player in hemostasis and thrombosis: low levels of VWF lead to an increased bleeding tendency, such as in Von Willebrand disease (VWD) [1]. Elevated levels of VWF are associated with a higher risk for cardiovascular morbidity resulting from arterial [2] and venous thrombosis [3].
VWD is the most common inherited bleeding disorder, affecting up to 1% of the world population [1]. VWD can be subdivided into qualitative (type 2; VWD2) or quantitative (type 1; VWD1 and type 3; VWD3) VWF deficiencies, which range in disease severity. In VWD2 and VWD3, pathogenic mutations in the VWF gene cause functional defects or complete absence of the protein, respectively. VWD1, the most prevalent subtype, is characterized by a partial VWF deficiency (< 30 IU·dL−1) and is often caused by heterozygous missense mutations in VWF that affect biosynthesis or plasma survival [4]. Individuals with levels between 30 and 50 IU·dL−1 are considered to have ‘low VWF’ and are also at higher risk of bleeding [5]. However, in ~ 30% of VWD1 patients and ~ 60% of individuals with ‘low VWF’ no pathogenic VWF mutation is identified [5, 6, 7], indicating other underlying causes for reduced VWF levels.
Plasma VWF levels reflect the equilibrium between secretion by the endothelium and clearance from plasma by (predominantly) macrophages in the liver. The broad distribution of VWF plasma levels within the human population [8] is impacted by environmental (e.g. age, hormones, exercise, co‐morbidities, lifestyle) as well as genetic factors, with only a limited contribution of pathogenic mutations in the VWF locus itself (reviewed in [9]). Over the years, large genetic, transcriptomic and genome‐wide association studies (GWAS) have identified several genetic modifiers of VWF plasma levels, including the ABO locus, glycosyltransferases, clearance receptors, SNARE proteins and transcription factors, which explains some of the heritable inter‐individual VWF variations in VWD and low VWF patients as well as in the healthy population [10, 11, 12, 13, 14]. The increasing number of genetic modifiers of VWF plasma levels identified highlights that multiple pathways linked to plasma survival and clearance (reviewed in [15]) as well as secretion by endothelial cells (reviewed in [16]) contribute to the regulation of circulating VWF levels. But because it is unlikely that genetic association analyses alone will be capable of identifying all the molecular components of the regulatory networks involved [17], mechanistic studies will therefore remain vital for a full understanding of these mechanisms in health and disease.
Exocytosis of endothelial cell‐specific secretory organelles, Weibel‐Palade bodies (WPBs), has been the focus of intensive studies of endothelial VWF secretion, even before genetic associations between VWF plasma levels and SNAREs that take part in WPB exocytosis were uncovered. A complex regulatory network that engages with WPBs post‐Golgi enables the endothelium to carefully control the release of VWF from WPBs during steady state and upon cellular triggering [16]. But do other mechanisms exist in the endothelial secretory pathway to modulate VWF secretion? In this viewpoint, we will highlight recent insight into the impact of the early secretory pathway on biogenesis and release of WPBs and discuss the potential impact of this pathway on the regulation of VWF plasma levels in health and disease.
The long and short of VWF biosynthesis and secretion
VWF is expressed by both megakaryocytes and vascular endothelial cells (ECs) [18]. Since the vast majority of circulating VWF is derived from ECs [19] we will focus on the endothelial VWF secretory pathway. VWF is post‐translationally modified during its itinerary through the secretory pathway starting as a ProVWF monomer in the endoplasmic reticulum (ER). In the ER ProVWF is glycosylated on N‐linked sites and then dimerizes via disulfide bridges in the C‐terminus. ProVWF dimers are subsequently transported to the Golgi, where the VWF propeptide is proteolytically cleaved by furin, O‐linked and N‐linked sites are further modified by glycosyltransferases (including ABO glycosyltransferase), and N‐terminal head‐to‐head multimers are formed [20]. Aberrations at any of these processing steps underpin the pathogenesis of various subtypes of VWD [1]. VWF forms a spectrum of low to high molecular weight multimers that are sorted differentially at the level of the trans‐Golgi network (TGN): low molecular weight VWF multimers are released via constitutive secretion, while high molecular weight VWF multimers condense into tubules that are stored in WPBs [21]. Although WPBs also store many other bioactive compounds, their formation is entirely driven by VWF expression [22, 23, 24]. After emerging from the TGN, WPBs recruit transport and fusion machinery in a maturation‐dependent process that enables them to undergo exocytosis [16]. WPBs can be released via two pathways: unstimulated, basal secretion, which is responsible for the maintenance of VWF plasma levels, and regulated secretion following cellular activation such as during vascular injury. Upon exocytosis, soluble WPB content is released into the vessel lumen. Due to shear forces inside the blood vessel generated by blood flow, VWF multimers unfurl into long strings that can self‐associate and attach to collagen in the exposed subendothelial matrix at the site of vascular injury. Together this forms an adhesive substrate for platelets and circulating VWF, an essential first step in the formation of a platelet plug at sites of vascular damage.
WPBs have a distinct cigar‐shaped morphology driven by the tight packaging of VWF multimers into condensed tubules and they can range in length between 0.5 and 5 µm [25]. As their content gets released VWF strings form that can extend up to 1 mm. This is where WPBs size does matter: VWF string length, adhesion of platelets to VWF strings and association with plasma VWF are all proportional to WPB length [26], so some degree of control over the size of the storage organelles that are generated is pertinent to locally manage hemostatic function of VWF.
WPB size control in the endothelial secretory pathway
The cellular mechanisms that control WPB shape are understood in broad strokes, but have long lacked molecular detail. Recently it has emerged that WPB length is determined by the number of laterally arranged, discrete VWF cargo units of ~ 0.5 µm at the TGN, so‐called VWF quanta, that are co‐packaged into budding WPBs. WPBs range in size in steps that are roughly equivalent to multiples of such VWF quanta [27]. Essential for the generation of long WPBs are (a) continuous, extended TGN cisternae that can accommodate a number of adjacent VWF quanta at the same time and (b) a sufficient flux of VWF through the early secretory pathway for several VWF quanta to arrive at the TGN adjacent to one another. In keeping with this model, WPBs in endothelial cells from patients with VWD1 caused by reduced or aberrant VWF biosynthesis are often small and/or lose their characteristic elongated morphology [28, 29].
A number of recent studies have now shown that ER and Golgi trafficking is critically important for the biogenesis, morphology and secretion competence of WPBs. Results from the Cutler group revealed that the ADP‐ribosylation factor (Arf) GTPases Arf1 and Arf4 and their guanine nucleotide exchange factor GBF1 (Golgi Brefeldin A Resistant Guanine Nucleotide Factor 1) are essential for the formation and shape of WPBs [30]. It is likely that this is partly due to activation‐dependent recruitment of Arf1 effectors at the TGN, such as the cargo adaptor AP‐1 [31] as well as the lipid‐modifying type II phosphatidylinositol 4‐kinases (PI4Ks) [32], which are both necessary for the formation of elongated WPBs (Fig. 1, step 5). Further evidence for the involvement of Arf GTPases in control of organelle size comes from Arf GTPase activating proteins (Arf GAPs). Depletion of Small Arf GAP 1 (SMAP1) also leads to shortening of WPBs [33], but if this depends on loss of GAP activity for Arf GTPases or its ability to act as an Arf effector that recruits clathrin remains unclear.
Fig. 1.
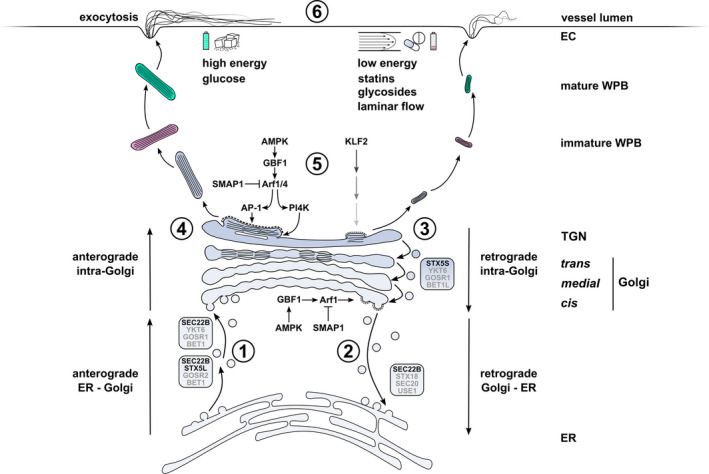
Regulatory mechanisms of WPB size and VWF secretion in the early secretory pathway of endothelial cells. Anterograde ER to Golgi trafficking of VWF via COPII vesicles is facilitated by SEC22B‐ and STX5‐ (STX5L: long isoform) containing SNARE complexes that mediate fusion at the Golgi. (2) Retrograde Golgi to ER membrane retrieval, which promotes cisternal maturation, anterograde transport of VWF through the Golgi and TGN exit is coordinated by active Arf1, which in turn recruits coatomer to COPI vesicles as they bud from the cis‐Golgi. Arf GAP SMAP1 deactivates Arf1 while Arf GEF GBF1 activates Arf1. GBF1 activity is modulated by the energy level‐dependent kinase AMPK. (3) Retrograde intra‐Golgi traffic via STX5‐containing SNARE complex (STX5S: short isoform). (4) Progression of VWF quanta through the Golgi stacks. The number of VWF quanta positioned directly next to one another at the TGN at the time of vesicle budding determines the length of the emerging WPB. (5) Arf1 and Arf4, regulated by SMAP1 and GBF1/AMPK, promote TGN exit by recruitment of the cargo adapter AP‐1 and clathrin to budding WPBs and via PI4K‐induced lipid modifications. Upregulation of the mechanosensitive transcription factor KLF2 promotes formation of smaller WPBs through a yet undefined mechanism that may involve Golgi fragmentation. (6) Dependent on type and strength of stimulus, mature WPBs are prioritized for exocytosis based on their size. Long WPBs release more VWF and produce longer VWF strings with increased platelet adhesive capacity, but require stronger stimulus. Environmental cues that are associated with long (left) and short (right) WPBs are indicated. Greyed out SNAREs (BET1, BET1L, GOSR1/2, SEC20, USE1 and YKT6) have been inferred from SNARE complexes in these pathways in other mammalian cells. Abbreviations: EC, endothelial cell; ER, endoplasmic reticulum; TGN, trans‐Golgi network; WPB, Weibel‐Palade body.
The exact role of GBF1 in VWF trafficking is clearly more complex. GBF1 promotes anterograde traffic of VWF from ER to Golgi, judged by the reduced processing of ProVWF (which happens in the Golgi) and the accumulation of VWF in swollen ER cisternae when GBF1 is depleted [30]. Furthermore, GBF1 also (indirectly) controls anterograde transport within the Golgi by recruiting COPI to the cis‐Golgi via activation of Arf1 (Fig. 1, step 2). This facilitates retrograde Golgi‐to‐ER membrane retrieval during cisternal maturation, which is necessary for transport of secretory cargo to the TGN and exit from the TGN. Depletion of GBF1 led to the appearance of unusually long WPBs with a size up to 7–10 µm that very frequently were bended and that were secretagogue unresponsive. This would seem unexpected as delayed anterograde VWF transport from ER to the Golgi should lead to smaller WPBs according to the quanta model [27]. The current hypothesis is that the delay in ER‐to‐Golgi traffic is offset by the more severe delay in TGN exit, leading to a logjam of VWF quanta at the TGN that get incorporated in giant WPBs. Their loss of secretion competence also raises important questions about the recruitment of secretory machinery. Rab27A, one of the proximal components of the WPB release machinery, was not recruited to giant WPBs [30]. Rab27A normally gets recruited after WPBs separate from the TGN, catalyzed by the GEF MADD [34, 35]. Failure to recruit Rab27A may indicate that membrane proteins or lipids that normally signify a Golgi membrane have not been removed or have accumulated on giant WPBs due to defective retrograde membrane retrieval [30].
Another pathway to control WPB length and VWF secretion depends on regulation of membrane fusion steps during ER–Golgi trafficking, such as by the longin‐SNARE SEC22B [36]. Longin‐SNAREs are a subfamily of the extended SNARE protein family that control membrane fusion events in the secretory pathway [37]. SEC22B coordinates membrane fusion of COPII vesicles during anterograde ER‐to‐Golgi traffic and COPI vesicles during retrograde Golgi‐to‐ER transport (Fig. 1, steps 1‐2). Similar to GBF1, depletion of SEC22B led to reduced ProVWF processing and accumulation of VWF in aggregates within the ER, consistent with a role in anterograde ER‐to‐Golgi transport of VWF. But contrary to GBF1, silencing of SEC22B led to fragmentation of Golgi stacks, decreased WPB length and a significant reduction in histamine‐induced VWF secretion. A comparable phenotype was observed for the cognate SNARE syntaxin 5 (STX5) [38], which operates in retrograde intra‐Golgi traffic and (together with SEC22B) in anterograde ER‐to‐Golgi traffic (Fig. 1, steps 3 and 1) [39]. STX5 exists in a long (STX5L) and a short (STX5S) isoform as the result of an alternative start codon [40]. Both presumably act in distinct trafficking routes: STX5L is found at the ER, while STX5S, which lacks an N‐terminal ER‐retrieval motif, is primarily found at the Golgi. Upon silencing of STX5 in endothelial cells, reduced flux of VWF from ER to Golgi is compounded by disintegration of the Golgi, which explains why WPBs reduce in number and size. Silencing of STX5 completely abrogated histamine‐induced formation of VWF strings on the endothelial apical surface and also blunted VWF secretion after stimulation [38].
Together these studies firmly implicate the ER–Golgi SNAREs SEC22B and STX5 in VWF trafficking and WPB size control, but more research is needed to unravel the mechanisms involved. Key questions that should be addressed are at which secretory pathway subcompartment(s) SEC22B‐ and STX5‐containing SNARE complexes catalyse membrane fusion events and how this relates to their effects on ER‐to‐Golgi transport and Golgi architecture. Second, how do ER–Golgi SNARE interactions with ER and Golgi tethering complexes, such as the NRZ and COG complex, respectively, contribute to VWF trafficking [38]?
Environmental control of VWF secretion via the early secretory pathway
Endothelial cells can also adapt to their environment by tuning VWF expression and the size and secretability of their WPBs (Fig. 1, step 5). As blood flows through different types of blood vessels, the flow patterns and the shear forces to which endothelium is exposed can vary between vascular beds, as do the number of WPBs [41, 42]. The shear‐stress sensitive, anti‐thrombotic and anti‐atherogenic transcription factor KLF2, which is upregulated in endothelium exposed to laminar flow, reduces the size of WPBs through a yet undefined mechanism [43]. Lipid‐lowering 3‐hydroxy‐3‐methyl‐glutaryl‐CoA reductase inhibitors, also known as statins, improve endothelial function through upregulation of KLF2 expression [44, 45]. Statins also reduce the size of newly forming WPBs and the platelet‐adhesive capacity of VWF strings, partly through upregulation of KLF2 and through fragmentation of the Golgi [26]. GBF1 is a substrate for AMP kinase (AMPK). AMPK acts as a sensor for low energy levels, such as low glucose, and relays that signal by phosphorylation of its targets. Both activation of GBF1 by the AMPK agonist AICAR as well as low glucose levels led to a significant reduction of WPB length and agonist‐induced VWF release [30]. A recent screen of licensed drugs identified numerous additional compounds that reduced the size of WPBs in endothelial cells, such as microtubule depolymerizing agents (which unlink Golgi stacks), protein synthesis inhibitors (which lower a.o. VWF expression), statins and glycosides [46]. Apart from paving the way for the development of new anti‐thrombotic strategies, it also emphasizes that environmental cues linked to different metabolic pathways can regulate the output of the early secretory pathway. These findings also suggest that the “exposome” may modulate VWF levels, thereby contributing to the large inter‐individual variation in healthy individuals and VWD patients with the same genetic defect.
Conclusions and future perspectives
Collectively, these studies show that correct and continuous progression of sufficient VWF through the secretory pathway is the driving force that controls the number and length of WPBs. This is the result of a careful equilibrium between the action of Arf GTPase and SNARE networks that coordinate dispatch and delivery events during anterograde and retrograde trafficking. Apart from control over WPB pool size in terms of organelle numbers and dimensions, there are also implications on stimulus responsiveness (Fig. 1, step 6). Endothelial cells can selectively release WPBs through different fusion and expulsion modes based on their size [47]: short and intermediate WPBs are preferentially released after mild stimuli via full fusion or lingering kiss release [48, 49], while long WPBs require stronger stimuli and the formation of an actin ring [50]. Whether this is due to differences in recruitment of exocytotic machinery during WPB biogenesis or this reflects different requirements for cargo expulsion will need further study. By simultaneously affecting the size of the releasable pool, the hemostatic potential of its cargo and the sensitivity towards cellular triggering, subtle changes in trafficking of VWF can have significant implications for circulating levels of VWF in plasma and for the hemostatic response of the endothelium. A recent study of a cohort of patients with low VWF revealed that VWF levels and bleeding score were correlated with the number of rare nonsynonymous variants in VWF [14]. The biological mechanism needs further investigation, but we speculate that a possible mechanism could involve reduced synthesis due to suboptimal codon usage and/or alterations in the progression of VWF through the secretory pathway.
Current evidence from GWAS studies and functional follow‐up studies [51, 52] have now firmly implicated the SNARE proteins STX2 and STXBP5 in the regulation of VWF levels through their effect on WPB release. A significant proportion of the genetic regulation of VWF is still unaccounted for [9]. Based on the ability of the early secretory pathway to dynamically control biogenesis, release as well as the hemostatic function of VWF as outlined in this viewpoint, we anticipate that variants in genes encoding modulators of the ER–Golgi pathway also contribute to the inter‐individual variability of VWF levels in plasma. Next‐generation sequencing‐based studies are now underway to identify more genetic modulators of VWF levels [53]. Cell‐based approaches will be needed to address the functional significance of candidates using similar methods that have been utilized for addressing the functional consequences of GBF1 and SEC22B deficiencies. In addition, we anticipate that studies using patient‐derived endothelial colony‐forming cells (ECFCs) [28, 29, 54, 55, 56, 57] of individuals with unexplained quantitative defects of VWF or with (rare) sequence variants of unknown significance will be useful to establish their functional impact on biogenesis and release of WPBs from endothelial cells.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MK, CM, JV and RB conceptualized the manuscript. MK, JV and RB jointly wrote the manuscript.
Data availability statement
Data sharing is not applicable to this article as no new data were created in this study.
Acknowledgements
Work in our laboratory was funded by grants from the Landsteiner Stichting voor Bloedtransfusie Research (LSBR‐1707 and LSBR‐2005) and the Dutch Thrombosis Foundation (TSN 2017‐01).
References
- 1. Leebeek FWG, Eikenboom JCJ. Von Willebrand’s Disease. N Engl J Med. 2016;375:2067–80. [DOI] [PubMed] [Google Scholar]
- 2. Wieberdink RG, van Schie MC, Koudstaal PJ, Hofman A, Witteman JCM, de Maat MPM, et al. High von Willebrand Factor Levels Increase the Risk of Stroke: The Rotterdam Study. Stroke. 2010;41:2151–6. [DOI] [PubMed] [Google Scholar]
- 3. Koster T, Vandenbroucke JP, Rosendaal FR, Briët E, Rosendaal FR, Blann AD. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep‐vein thrombosis. Lancet. 1995;345:152–5. [DOI] [PubMed] [Google Scholar]
- 4. de Jong A, Eikenboom J. Von Willebrand disease mutation spectrum and associated mutation mechanisms. Thromb Res. 2017;159:65–75. [DOI] [PubMed] [Google Scholar]
- 5. Lavin M, Aguila S, Schneppenheim S, Dalton N, Jones KL, O’Sullivan JM, et al. Novel insights into the clinical phenotype and pathophysiology underlying low VWF levels. Blood. 2017;130:2344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM‐1VWD). Blood. 2007;109:112–21. [DOI] [PubMed] [Google Scholar]
- 7. James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–54. [DOI] [PubMed] [Google Scholar]
- 8. Sadler JE. von Willebrand factor: two sides of a coin. J Thromb Haemost. 2005;3:1702–9. [DOI] [PubMed] [Google Scholar]
- 9. Swystun LL, Lillicrap D. Genetic regulation of plasma von Willebrand factor levels in health and disease. J Thromb Haemost. 2018;16:2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith NL, Chen M‐H, Dehghan A, Strachan DP, Basu S, Soranzo N, et al. Novel associations of multiple genetic loci with plasma levels of Factor VII, Factor VIII, and von Willebrand Factor. Circulation. 2010;121:1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desch KC, Ozel AB, Siemieniak D, Kalish Y, Shavit J, Thornburg CD, et al. Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome‐wide association. Proc Natl Acad Sci. 2013;110:588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Loon J, Dehghan A, Weihong T, Trompet S, McArdle WL, Asselbergs FW, et al. Genome‐wide association studies identify genetic loci for low von Willebrand factor levels. Eur J Hum Genet. 2016;24:1035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sabater‐Lleal M, Huffman JE, de Vries PS , Marten J, Mastrangelo MA, Song C, et al. Genome‐wide association transethnic meta‐analyses identifies novel associations Regulating Coagulation Factor VIII and von Willebrand Factor Plasma Levels. Circulation. 2019;139:620–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sadler B, Christopherson PA, Haller G, Montgomery RR, Di Paola J. von Willebrand factor antigen levels are associated with burden of rare nonsynonymous variants in the VWF gene. Blood. 2021;137:3277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O’Sullivan JM, Ward S, Lavin M, O’Donnell JS. von Willebrand factor clearance ‐ biological mechanisms and clinical significance. Br J Haematol. 2018;183:185–95. [DOI] [PubMed] [Google Scholar]
- 16. Schillemans M, Karampini E, Kat M, Bierings R. Exocytosis of Weibel‐Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome‐wide association studies. Nat Rev Genet. 2019;20:467–84. [DOI] [PubMed] [Google Scholar]
- 18. Karampini E, Bierings R, Voorberg J. Orchestration of primary hemostasis by platelet and endothelial lysosome‐related organelles. Arterioscler Thromb Vasc Biol. 2020;40:1441–53. [DOI] [PubMed] [Google Scholar]
- 19. Nichols TC, Samama CM, Bellinger DA, Roussi J, Reddick RL, Bonneau M, et al. Function of von Willebrand factor after crossed bone marrow transplantation between normal and von Willebrand disease pigs: effect on arterial thrombosis in chimeras. Proc Natl Acad Sci. 1995;92:2455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124:1412–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCormack JJ, Lopes da Silva M, Ferraro F, Patella F, Cutler DF. Weibel‐Palade bodies at a glance. J Cell Sci. 2017;130:3611–7. [DOI] [PubMed] [Google Scholar]
- 22. Wagner DD, Saffaripour S, Bonfanti R, Sadler JE, Cramer EM, Chapman B, et al. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell. 1991;64:403–13. [DOI] [PubMed] [Google Scholar]
- 23. Voorberg J, Fontijn R, Calafat J, Janssen H, van Mourik JA, Pannekoek H. Biogenesis of von Willebrand factor‐containing organelles in heterologous transfected CV‐1 cells. EMBO J. 1993;12:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schillemans M, Kat M, Westeneng J, Gangaev A, Hofman M, Nota B, et al. Alternative trafficking of Weibel‐Palade body proteins in CRISPR/Cas9‐engineered von Willebrand factor‐deficient blood outgrowth endothelial cells. Res Pract Thromb Haemost. 2019;3:718–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel‐Palade bodies. Blood. 2011;117:5033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferraro F, Lopes‐da‐Silva M, Grimes W, Lee HK, Ketteler R, Kriston‐Vizi J, et al. Weibel‐Palade body size modulates the adhesive activity of its von Willebrand Factor cargo in cultured endothelial cells. Sci Rep. 2016;6:32473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferraro F, Kriston‐Vizi J, Metcalf DJ, Martin‐Martin B, Freeman J, Burden JJ, et al. A two‐tier Golgi‐based control of organelle size underpins the functional plasticity of endothelial cells. Dev Cell. 2014;29:292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J‐W, Bouwens EAM, Pintao MC, Voorberg J, Safdar H, Valentijn KM, et al. Analysis of the storage and secretion of von Willebrand factor in blood outgrowth endothelial cells derived from patients with von Willebrand disease. Blood. 2013;121:2762–72. [DOI] [PubMed] [Google Scholar]
- 29. Starke RD, Paschalaki KE, Dyer CEF, Harrison‐Lavoie KJ, Cutler JA, McKinnon TAJ, et al. Cellular and molecular basis of von Willebrand disease: studies on blood outgrowth endothelial cells. Blood. 2013;121:2773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lopes‐da‐Silva M, McCormack JJ, Burden JJ, Harrison‐Lavoie KJ, Ferraro F, Cutler DF. A GBF1‐dependent mechanism for environmentally responsive regulation of ER‐Golgi transport. Dev Cell. 2019;49:786–801.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lui‐Roberts WWY, Collinson LM, Hewlett LJ, Michaux G, Cutler DF. An AP‐1/clathrin coat plays a novel and essential role in forming the Weibel‐Palade bodies of endothelial cells. J Cell Biol. 2005;170:627–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. da Silva ML, O’Connor MN, Kriston‐Vizi J, White IJ, Al‐Shawi R, Simons JP, et al. Type II PI4‐kinases control Weibel‐Palade body biogenesis and von Willebrand factor structure in Human endothelial cells. J Cell Sci. 2016;129:2096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watanabe A, Hataida H, Inoue N, Kamon K, Baba K, Sasaki K, et al. Arf GTPase‐activating proteins SMAP1 and AGFG2 regulate the size of Weibel‐Palade bodies and exocytosis of von Willebrand factor. Biol Open. 2021;10:bio058789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bierings R, Hellen N, Kiskin N, Knipe L, Fonseca A‐V, Patel B, et al. The interplay between the Rab27A effectors Slp4‐a and MyRIP controls hormone‐evoked Weibel‐Palade body exocytosis. Blood. 2012;120:2757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kat M, Bürgisser PE, Janssen H, De Cuyper IM, Conte IL, Hume AN, et al. GDP/GTP exchange factor MADD drives activation and recruitment of secretory Rab GTPases to Weibel‐Palade bodies. Blood Adv. 2021;5:5116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karampini E, Bürgisser PE, Olins J, Mulder AA, Jost CR, Geerts D, et al. Sec22b determines Weibel‐Palade body length by controlling anterograde ER‐Golgi transport. Haematologica. 2021;106:1138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daste F, Galli T, Tareste D. Structure and function of longin SNAREs. J Cell Sci. 2015;128:4263–72. [DOI] [PubMed] [Google Scholar]
- 38. Kat M, Karampini E, Hoogendijk AJ, Bürgisser PE, Mulder AA, Van Alphen FPJ, et al. Syntaxin 5 determines Weibel‐Palade body size and Von Willebrand factor secretion by controlling Golgi architecture. Haematologica. 2022. 10.3324/haematol.2021.280121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Linders PTA, van der Horst C, Ter Beest M, van den Bogaart G. Stx5‐Mediated ER‐Golgi transport in mammals and yeast. Cells. 2019;8:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Linders PTA, Gerretsen ECF, Ashikov A, Vals M‐A, de Boer R, Revelo NH, et al. Congenital disorder of glycosylation caused by starting site‐specific variant in syntaxin‐5. Nat Commun. 2021;12:6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krüger‐Genge A, Blocki A, Franke R‐P, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. 2019;20:4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gebrane‐Younès J, Drouet L, Caen JP, Orcel L. Heterogeneous distribution of Weibel‐Palade bodies and von Willebrand factor along the porcine vascular tree. Am J Pathol. 1991;139:1471–84. [PMC free article] [PubMed] [Google Scholar]
- 43. Van Agtmaal EL, Bierings R, Dragt BS, Leyen TA, Fernandez‐Borja M, Horrevoets AJG, et al. The Shear Stress‐Induced Transcription Factor KLF2 Affects Dynamics and Angiopoietin‐2 Content of Weibel‐Palade Bodies. PLoS One. 2012;7:e38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, García‐Cardeña G. Statins Exert Endothelial Atheroprotective Effects via the KLF2 Transcription Factor. J Biol Chem. 2005;280:26714–9. [DOI] [PubMed] [Google Scholar]
- 45. Sen‐Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, et al. Kruppel‐Like Factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–6. [DOI] [PubMed] [Google Scholar]
- 46. Ferraro F, Patella F, Costa JR, Ketteler R, Kriston‐Vizi J, Cutler DF. Modulation of endothelial organelle size as an antithrombotic strategy. J Thromb Haemost. 2020;18:3296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCormack JJ, Harrison‐Lavoie KJ, Cutler DF. Human endothelial cells size‐select their secretory granules for exocytosis to modulate their functional output. J Thromb Haemost. 2020;18:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conte IL, Cookson E, Hellen N, Bierings R, Mashanov G, Carter T. Is there more than one way to unpack a Weibel‐Palade body? Blood. 2015;126:2165–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Babich V, Meli A, Knipe L, Dempster JE, Skehel P, Hannah MJ, et al. Selective release of molecules from Weibel‐Palade bodies during a lingering kiss. Blood. 2008;111:5282–90. [DOI] [PubMed] [Google Scholar]
- 50. Nightingale TD, White IJ, Doyle EL, Turmaine M, Harrison‐Lavoie KJ, Webb KF, et al. Actomyosin II contractility expels von Willebrand factor from Weibel‐Palade bodies during exocytosis. J Cell Biol. 2011;194:613–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu Q, Yamakuchi M, Ture S, de la Luz G‐H, Ko KA, Modjeski KL, et al. Syntaxin‐binding protein STXBP5 inhibits endothelial exocytosis and promotes platelet secretion. J Clin Invest. 2014;124:4503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu QM, Ko KA, Ture S, Mastrangelo MA, Chen M‐H, Johnson AD, et al. Novel Thrombotic Function of a Human SNP in STXBP5 Revealed by CRISPR/Cas9 Gene Editing in Mice. Arterioscler Thromb Vasc Biol. 2017;37:264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sadler B, Minard CG, Haller G, Gurnett CA, O’Brien SH, Wheeler A, et al. Whole‐exome analysis of adolescents with low VWF and heavy menstrual bleeding identifies novel genetic associations. Blood Adv. 2022;6:420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Van Breevoort D, Snijders AP, Hellen N, Weckhuysen S, Van Hooren KWEM, Eikenboom J, et al. STXBP1 promotes Weibel‐Palade body exocytosis through its interaction with the Rab27A effector Slp4‐a. Blood. 2014;123:3185–94. [DOI] [PubMed] [Google Scholar]
- 55. Schillemans M, Karampini E, van den Eshof BL, Gangaev A, Hofman M, van Breevoort D, et al. Weibel‐palade body localized syntaxin‐3 modulates von willebrand factor secretion from endothelial cells. Arterioscler Thromb Vasc Biol. 2018;38:1549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karampini E, Schillemans M, Hofman M, van Alphen F, de Boer M, Kuijpers TW, et al. Defective AP‐3‐dependent VAMP8 trafficking impairs Weibel‐Palade body exocytosis in Hermansky‐Pudlak Syndrome type 2 blood outgrowth endothelial cells. Haematologica. 2019;104:2091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bowman ML, Pluthero FG, Tuttle A, Casey L, Li L, Christensen H, et al. Discrepant platelet and plasma von Willebrand factor in von Willebrand disease patients with p.Pro2808Leufs*24. J Thromb Haemost. 2017;15:1403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created in this study.