

Abstract
Background and Aims
We previously identified subsets of patients with NAFLD with different metabolic phenotypes. Here we align metabolomic signatures with cardiovascular disease (CVD) and genetic risk factors.
Approach and Results
We analyzed serum metabolome from 1154 individuals with biopsy‐proven NAFLD, and from four mouse models of NAFLD with impaired VLDL‐triglyceride (TG) secretion, and one with normal VLDL‐TG secretion. We identified three metabolic subtypes: A (47%), B (27%), and C (26%). Subtype A phenocopied the metabolome of mice with impaired VLDL‐TG secretion; subtype C phenocopied the metabolome of mice with normal VLDL‐TG; and subtype B showed an intermediate signature. The percent of patients with NASH and fibrosis was comparable among subtypes, although subtypes B and C exhibited higher liver enzymes. Serum VLDL‐TG levels and secretion rate were lower among subtype A compared with subtypes B and C. Subtype A VLDL‐TG and VLDL–apolipoprotein B concentrations were independent of steatosis, whereas subtypes B and C showed an association with these parameters. Serum TG, cholesterol, VLDL, small dense LDL5,6, and remnant lipoprotein cholesterol were lower among subtype A compared with subtypes B and C. The 10‐year high risk of CVD, measured with the Framingham risk score, and the frequency of patatin‐like phospholipase domain‐containing protein 3 NAFLD risk allele were lower in subtype A.
Conclusions
Metabolomic signatures identify three NAFLD subgroups, independent of histological disease severity. These signatures align with known CVD and genetic risk factors, with subtype A exhibiting a lower CVD risk profile. This may account for the variation in hepatic versus cardiovascular outcomes, offering clinically relevant risk stratification.

Abbreviations
- 0.1MCD
methionine (0.1%) and choline‐deficient diet
- ALT
alanine aminotransferase
- Apo
apolipoprotein
- AST
aspartate aminotransferase
- BMI
body mass index
- CL
cholesterol
- CoA
coenzyme A
- CVD
cardiovascular disease
- DG
diglycerides
- DNL
de novo lipogenesis
- FA
fatty acid
- FCR
fractional catabolic rate
- FRS
Framingham risk score
- GGT
gamma‐glutamyltransferase
- HbA1c
glycated hemoglobin
- HFD
high‐fat diet
- HOMA‐IR
homeostasis model assessment of insulin resistance
- IDL
intermediate‐density lipoprotein
- IHTG
intrahepatic triglyceride
- INR
international normalized ratio
- KO
knockout
- Ldlr
LDL receptor
- Mat1a
methionine adenosyltransferase 1a
- Mttp
microsomal triglyceride transfer protein
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PNPLA3
patatin‐like phospholipase domain‐containing protein 3
- RLP
remnant lipoprotein
- SAMe
S‐adenosylmethionine
- SM
sphingomyelin
- SR
secretion rate
- TG
triglycerides
- TM6SF2
transmembrane 6 superfamily 2
INTRODUCTION
NAFLD represents a spectrum of progressive liver disease characterized by increased intrahepatic triglyceride (IHTG) content in the absence of excessive alcohol consumption.[ 1 , 2 , 3 ] NAFLD ranges from simple steatosis to NASH, fibrosis, and ultimately cirrhosis and HCC. NAFLD is strongly associated with obesity, dyslipidemia, type 2 diabetes mellitus, and the metabolic syndrome. Importantly, cardiovascular disease (CVD) is the leading cause of mortality in patients with NAFLD.[ 4 ] Although NAFLD has been shown to be an independent risk factor for CVD, the mechanisms by which NAFLD contributes to CVD are unknown.
NAFLD represents a complex heterogeneous disease in which the histological features result from an imbalance in liver lipid metabolism caused by multiple nutritional and environmental exposures on a susceptible genetic background.[ 3 , 5 , 6 , 7 ] A better understanding of NAFLD heterogeneity and its biology will facilitate the development of personalized treatments. Overproduction of VLDL into circulation has been observed in patients with NAFLD and is thought to be, in part, a consequence of increased de novo lipogenesis (DNL) promoting hepatic triglyceride (TG) synthesis.[ 8 , 9 , 10 ] NAFLD is characterized by increased serum levels of TG and cholesterol (CL) in VLDL and intermediate‐density lipoproteins (IDL), increased concentration of smaller LDL subclasses, and decreased content of larger LDL subclasses, all of which are features associated with increased CVD risk.[ 11 , 12 ] Moreover, it has been suggested that increased levels of total lipids and CL in VLDL and LDL subclasses may be associated with NAFLD progression.[ 13 ] On the other hand, VLDL‐TG secretion is relatively impaired in a subset of patients with NAFLD and cannot compensate for increases in both fatty acid (FA) influx and DNL.[ 14 ] Genetic defects (e.g., microsomal triglyceride transfer protein [MTTP], transmembrane 6 superfamily member 2 [TM6SF2] and apolipoprotein B, [APO‐B]), which impair hepatic VLDL secretion, are also associated with reduced LDL‐CL levels and a favorable CVD risk profile, yet they still cause steatosis that may progress to NASH with fibrosis, even without obesity or insulin resistance.[ 15 , 16 , 17 , 18 , 19 , 20 ] In addition, the therapeutic benefits of LDL reduction in patients treated with antisense APO‐B or MTTP inhibitors are associated with hepatic steatosis, inflammation and fibrosis, which limits their utility.[ 21 , 22 , 23 ]
Our published data, using high‐throughput metabolomics, suggested that patients with NAFLD could be subclassified into two main metabolic subtypes.[ 24 ] One of these subtypes (which we here refer to as subtype A) corresponds to specific serum metabolic alterations that correlated with those observed in methionine adenosyltransferase 1A knockout (Mat1a‐KO) mice,[ 24 ] a model of NASH characterized by a reduction in hepatic S‐adenosylmethionine (SAMe),[ 25 ] impaired phosphatidylcholine (PC) synthesis, and VLDL secretion.[ 26 ] Importantly, patients with NAFLD often exhibit down‐regulation of MAT1A expression[ 27 , 28 ] and low SAMe.[ 29 ] The comparison of the serum lipidomic profile of LDL receptor (Ldlr)–KO mice fed a high‐fat diet[ 30 ] with the lipidomic profile of the cohort of patients with NAFLD mentioned previously revealed the existence of a second NAFLD subtype, referred to here as subtype C. The deletion of Ldlr in mice results in increased serum level of VLDL‐TG and LDL‐CL.[ 31 , 32 , 33 ] These findings raised the corollary question of whether patients with NAFLD with subtype A show impaired secretion rate (SR) of VLDL compared to patients with NAFLD with subtype C, which would show VLDL hypersecretion, and, consequently, both NAFLD subtypes could present different CVD risks. Based on these concepts, here we investigated the association between NAFLD subtypes with the main classes (VLDL, IDL, LDL, and HDL) and subclasses of serum lipoproteins, as well as with several common single‐nucleotide polymorphisms associated with hepatocyte TG accumulation and VLDL secretion.
MATERIALS AND METHODS
Study participants
NAFLD cases
Patients with biopsied NAFLD (n = 1154) were recruited at several clinical centers from Europe (Spain, UK, Italy, the Czech Republic), the USA (California and Florida), and Chile, as well as patients recruited by Galmed Pharmaceuticals (Israel) (Table 1). Patients fulfilled the following inclusion criteria: (1) age 18−75 years; (2) no known acute or chronic disease except for obesity, dyslipidemia, hypertension, or type 2 diabetes based on medical history, physical examination, and standard laboratory tests; and (3) alcohol consumption lower than 20 g/day for women and 30 g/day for men. Exclusion criteria included patients with NAFLD with cirrhosis and other causes of liver disease. The institutional review board of the participating hospitals approved the study, and written informed consent was obtained from all patients.[ 34 ]
TABLE 1.
Clinical and biochemical characteristics of the patients with NAFLD included in the lipidomic study
| Total | Subtype A | Subtype B | Subtype C | p value | ||
|---|---|---|---|---|---|---|
| Demographics | ||||||
| n | 1154 | 541 | 305 | 308 | ||
| Age (years) | 50.0 ± 11.9 (1154) | 47.9 ± 12.2 (541) | 52.0 ± 11.3 (305) | 51.7 ± 11.3 (308) | 0.182 | |
| Female (%) | 576 (50%) | 266 (49%) | 151 (49%) | 159 (52%) | 0.778 | |
| BMI (kg/m2) | 34.6 ± 7.7 (1154) | 36.4 ± 9.0 (541) | 32.7 ± 6.0 (305) | 33.4 ± 5.9 (308) | 0.028 | |
| Biochemistry | ||||||
| TG (mg/dl) | 165.4 ± 106.1 (1109) | 140.6 ± 94.5 (525) | 177.5 ± 86.7 (292) | 197.6 ± 130.2 (292) | 2.84E‐17 | |
| Cholesterol (mg/dl) | 189.3 ± 44.4 (862) | 175.8 ± 41.1 (392) | 188.0 ± 37.3 (226) | 212.3 ± 46.4 (244) | 2.86E‐16 | |
| ALT (U/l) | 56.6 ± 44.9 (1112) | 53.1 ± 45.5 (509) | 56.3 ± 41.4 (297) | 62.6 ± 46.5 (306) | 6.20E‐04 | |
| AST (U/l) | 38.7 ± 28.2 (1113) | 35.2 ± 25.1 (509) | 40.3 ± 29.3 (297) | 43.1 ± 31.3 (307) | 8.00E‐06 | |
| GGT (U/l) | 66.5 ± 95.6 (687) | 60.3 ± 104.5 (328) | 70.5 ± 88.0 (190) | 73.9 ± 85.0 (169) | 2.09E‐04 | |
| HbA1c (%) | 6.5 ± 1.2 (722) | 6.5 ± 1.3 (259) | 6.4 ± 1.0 (220) | 6.5 ± 1.1 (243) | 0.854 | |
| HOMA‐IR | 7.0 ± 7.7 (597) | 6.7 ± 7.9 (204) | 7.1 ± 6.5 (186) | 7.3 ± 8.5 (207) | 0.412 | |
| Albumin (g/dl) | 4.4 ± 0.4 (897) | 4.4 ± 0.4 (379) | 4.5 ± 0.3 (261) | 4.4 ± 0.4 (257) | 3.49E‐04 | |
| INR | 1.0 ± 0.2 (419) | 1.1 ± 0.1 (138) | 1.1 ± 0.3 (146) | 1.0 ± 0.1 (135) | 5.00E‐06 | |
| Medication | ||||||
| Lipid‐lowering medication | N | 685 (79.4%) | 331 (76.3%) | 193 (82.5%) | 161 (82.6%) | 0.076 |
| Y | 178 (20.6%) | 103 (23.7%) | 41 (17.5%) | 34 (17.4%) | ||
| CVD risk (FRS) | ||||||
| Intermediate–high (>10%) | 46 (12.8%) | 28 (12.1%) | 8 (13.1%) | 10 (14.7%) | 0.851 | |
| High (>15%) | 21 (5.8%) | 10 (4.3%) | 6 (9.8%) | 5 (7.4%) | 0.221 | |
| Histology | ||||||
| NASH (%) | 735 (64%) | 359 (66%) | 187 (61%) | 189 (61%) | 0.208 | |
| Steatosis grade (%) | 1 | 48.31% | 44.92% | 52.13% | 46.75% | 0.118 |
| 2 | 31.81% | 34.01% | 30.82% | 29.22% | ||
| 3 | 19.88% | 21.07% | 17.05% | 24.03% | ||
| Inflammation grade (%) | 0 | 12.39% | 13.31% | 12.13% | 11.36% | 0.059 |
| 1 | 46.94% | 50.65% | 46.56% | 41.88% | ||
| 2 | 33.97% | 29.57% | 34.75% | 41.23% | ||
| 3 | 6.68% | 6.47% | 6.56% | 5.52% | ||
| Ballooning grade (%) | 0 | 29.54% | 25.88% | 35.08% | 35.71% | 0.013 |
| 1 | 45.73% | 49.17% | 42.30% | 43.83% | ||
| 2 | 24.71% | 24.95% | 22.62% | 20.45% | ||
| Fibrosis stage (%) | 0 | 25.92% | 26.80% | 29.51% | 28.25% | 0.447 |
| 1 | 35.66% | 38.26% | 37.70% | 37.34% | ||
| 2 | 19.32% | 22.74% | 18.36% | 18.18% | ||
| 3 | 12.88% | 12.20% | 14.43% | 16.23% |
Values for age, BMI, HbA1c, TG, ALT, AST, GGT, HbA1c, HOMA‐IR, albumin, and INR are given as means ± SD. Statistical significance was determined by Chi‐square test for categorical variables and Kruskal‐Wallis H‐test for continuous variables.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; CVD, cardiovascular disease; FRS, Framingham risk score; GGT, gamma‐glutamyl transferase; HbA1c, glycated hemoglobin; HOMA‐IR, homeostasis assessment model for insulin resistance; INR, international normalized ratio.
Diagnoses were established histologically in liver biopsy specimens. The histological diagnosis of NAFLD was established by a single liver pathologist in each hospital using the scoring system defined by Kleiner/Brunt.[ 35 , 36 ] Following assessment, patients were classified into two histological groups: (1) biopsies with only steatosis, steatosis with inflammation but without ballooning, or steatosis with ballooning but without inflammation were classified as nonalcoholic fatty liver, and (2) the histopathologic definition of NASH was determined by the joint presence of steatosis, lobular inflammation, and hepatocellular ballooning, independently of the total NAFLD activity score.
Blood was drawn under fasting conditions within 30 days of the diagnostic liver biopsy was performed. Serum was stored at −80°C until analysis as described.[ 34 ] Clinical data were collected retrospectively using patient records and laboratory values obtained at the time of biopsy. Serum samples from all 1154 patients with NAFLD were used to identify the main metabolic NAFLD subtypes by analyzing their serum metabolomic profiles. A subset of 197 patients with NAFLD from this cohort was used to study the association of the NAFLD metabolic subtype with the serum lipoprotein profile (Table S1). This cohort was determined by the availability of sufficient serum to perform the lipoprotein profiling (0.5 ml). For details see the Supporting Methods.
Controls
As a control for comparison of the lipoprotein profiling data, we included 350 serum samples from the general population[ 37 ] with similar gender and age to the cohort of patients with NAFLD used to determine the serum lipoprotein profile, and normal glucose, TG, CL, and alanine aminotransferase (ALT) (Table S2). For details, see the Supporting Methods.
Subjects with plasma VLDL‐TG secretion rate
Serum samples from women (n = 30) for which the hepatic VLDL‐TG metabolism and kinetics were previously studied[ 38 ] were used to determine the association of the NAFLD metabolic subtypes with the VLDL‐TG secretion rate (SR). Three groups were considered of the following characteristics: lean subjects with low VLDL‐TG SR (n = 10), obese subjects with NAFLD with low VLDL‐TG SR (n = 10), and obese subjects with NAFLD with high VLDL‐TG SR (n = 10).
Animals
Serum samples from five mouse models of NASH, all of them previously described, were used to compare their metabolomic profile with that of the patients with NAFLD mentioned previously: Mat1a‐KO mice,[ 24 ] methionine (0.1%) and choline‐deficient diet (0.1MCD)–fed mice,[ 39 ] Mttp‐LKO mice,[ 40 ] Tm6sf2‐LKO mice,[ 41 ] and LDLR‐KO mice fed a high‐fat diet (Ldlr−/−.Leiden/HFD).[ 30 ]
Metabolomics
Serum metabolomic profiles were semi‐quantified by liquid chromatography coupled with mass spectrometry.[ 24 ] For details see the Supporting Methods.
Lipoprotein analyses
Nuclear magnetic resonance lipoprotein analyses were performed in a subcohort of patients with biopsied NAFLD (n = 197; Supporting Table S1) and controls (n = 350; Supporting Table S2) as described.[ 37 ] For details, see the Supporting Methods.
Statistical analysis
Continuous variables were described by means ± SDs. Comparisons between pairs of groups were performed by two‐tailed independent Student t test or Kruskal‐Wallis H‐test. Chi‐square test was applied for categorical variables. Unadjusted p values were considered as statistically significant below 0.05.
Hierarchical clustering algorithms based on metabolites were used to visualize the differences in signatures between samples, considering the Ward’s minimum variance as the agglomeration method. The maximum of the average of the individual silhouette widths was calculated for the clusters.
A multinomial logistic regression classifier analysis was applied to generate a predictive metabolic signature capable of discriminating among NAFLD subtypes. A forward stepwise method was used as a variable selection criterion, in which the process started with a null model and optimum variables were added in each step until the best model was achieved. Logarithmic transformation of the data was applied. Validation process was performed through 5‐fold cross validation. References for libraries are included in the Supporting Methods.
RESULTS
The serum lipidome of patients with NAFLD with subtype A is similar to that of mouse models of NAFLD exhibiting defective VLDL secretion
Comparison of the serum metabolomic profile of the patients with NAFLD (n = 1154; Table 1) with that of different mouse models of NASH showing impaired (Mat1a‐KO, 0.1MCD, Mttp‐KO, and Tm6sf2‐KO) or increased (Ldlr−/−.Leiden/HFD) VLDL‐TG secretion using the experimental procedure previously described[ 24 ] resulted in the identification of three major metabolic NAFLD subtypes (A, B, and C) (Figure 1A, Figure S1). Patients with NAFLD (541, 47%) reflecting metabolomic features common to the four mouse models of NASH showing impaired VLDL secretion were classified as subtype A. Of the remaining subjects, 308 (27%) patients with NAFLD resembled the metabolic profile of the Ldlr−/−.Leiden/HFD NASH mouse model and were classified as subtype C, and the remaining 305 (26%) patients were classified as subtype B (Figure 1B, Table 1).
FIGURE 1.
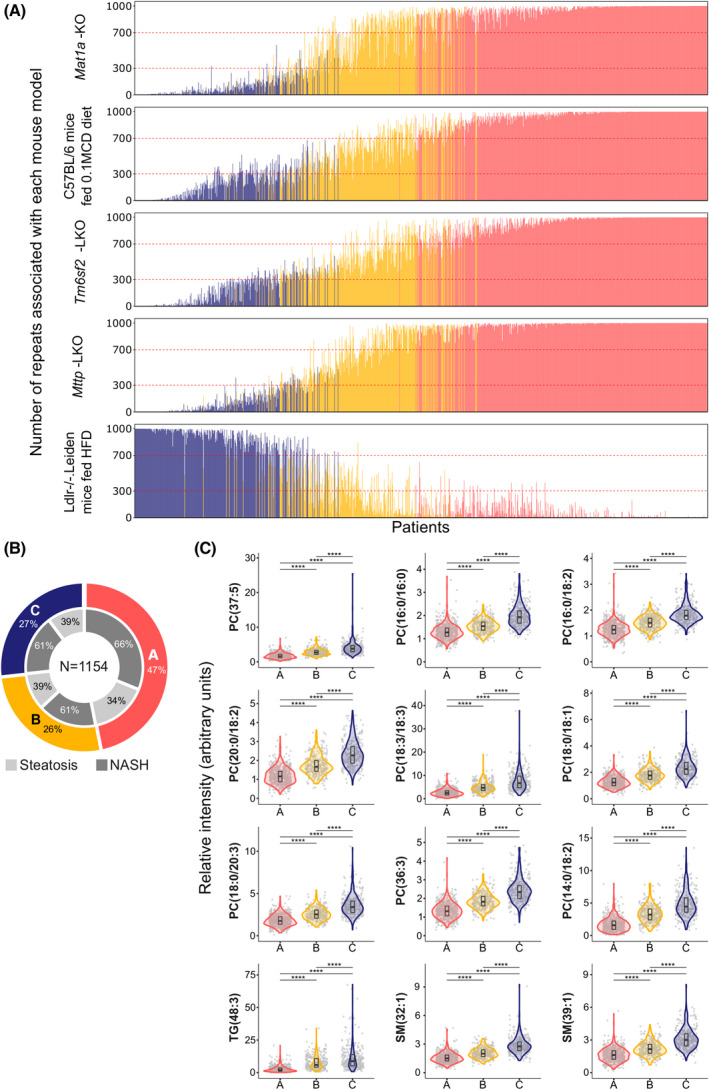
Classification of patients with NAFLD (n = 1242) into subtypes. (A) The frequency distribution of the patients with NAFLD (n = 1154) according to five mouse models of NAFLD. Frequencies were obtained after (1) random patient partition (50/50) into two cohorts with equal proportional representation of nonalcoholic fatty liver (NAFL)/NASH and males/females; (2) clustering analysis based on the 50 more significantly serum metabolites that differentiated more significantly between the mouse model of NAFLD and the respective control mice; (3) 1000‐fold repetition of the random partition with equal representation of NAFL/NASH and males/females. Representation of the number of repeats associated with methionine adenosyltransferase 1A knockout (Mat1a‐KO) mice, C57BL/6 mice fed 0.1% methionine and choline deficient diet (0.1MCD), liver‐specific microsomal triglyceride transfer protein KO (Mttp‐LKO), liver‐specific transmembrane 6 superfamily member 2 KO (Tm6sf2‐LKO), and germline Ldlr‐deficient (Ldlr−/−.Leiden) KO mice fed high‐fat diet (HFD). Patients with a frequency over 70% for Mat1a‐KO, 0.1MCD, Mttp‐LKO and Tm6sf2‐LKO, and lower than 70% for Ldlr−/−.Leiden/HFD, were classified as subtype A (colored in red). Patients with a frequency lower than 70% for Mat1a‐KO, 0.1MCD, Mttp‐LKO and Tms6f2‐LKO, and higher than 70% for the Ldlr−/−.Leiden/HFD, were classified as subtype C (colored in blue). Patients not classified as either subtype A or C were classified as subtype B (colored in yellow). (B) Percentage of patients classified as subtypes A, B, and C. Percentage of patients with NAFL and NASH per subtype is also indicated. (C) Serum levels of lipids that discriminate among NAFLD subtypes A, B, and C. Gray points in the background indicate the real values with a minimal random displacement to avoid overplotting. Densities are represented as violin plots, while main distributions (medians and first and third quartiles) are represented as internal box plots. Horizontal black lines are statistical comparisons (t test) between two groups, with the unadjusted p values as symbols above the lines (****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, ≥ 0.05). ns, not significant; PC, phosphatidylcholine; SM, sphingomyelin; TG, triglycerides
We then searched for lipid species that best differentiated among A, B, and C NAFLD subtypes (Figure 1C), and generated a statistical model in which these given lipids were assigned a relative weight depending on their importance for the classification of patients with NAFLD into subtypes (Table S3). This set of 12 NAFLD subtype biomarkers was gender‐independent. We also identified signatures that could differentiate steatosis from NASH for each subtype. These signatures included glycerolipids, glycerophospholipids, and sphingolipids (Figure S2).
Table 1 lists the main clinical data of patients with NAFLD classified by their metabolic subtype. No differences were found between the percentage of patients with NASH or stage of fibrosis and the NAFLD subtype. Moreover, no gender differences were indicated between subtypes. Patients with metabolic subtypes B and C were slightly older, with lower body mass index (BMI), and had more frequently higher TG, CL, ALT, aspartate aminotransferase (AST), and gamma‐glutamyltransferase (GGT), and less ballooning than subjects with subtype A. The homeostasis model assessment of insulin resistance (HOMA‐IR) index, glycated hemoglobin (HbA1c), albumin, and international normalized ratio (INR) were similar among the three subtypes. The Framingham CVD risk score (FRS) was calculated for 360 patients with NAFLD, who were chosen based on availability of clinical data. The percent of patients at intermediate CVD risk (FRS ≥ 10% at 10 years) was similar in subtypes A (12%), B (13%), and C (15%). When the percentage of patients at high CVD risk (FRS ≥ 15% at 10 years) was compared, patients with subtype A showed a lower score (4%) than patients with subtypes B (10%) or C (7%). We also found that the percent of patients with NAFLD treated with lipid‐lowering medication (statins and/or fibrates) was higher in subtype A (24%) than in subtypes B (17%) and C (17%), although it did not reach statistical significance (Table 1). These results indicate that lipid‐lowering medication has limited relevance to a particular NAFLD subtype.
Serum VLDL‐TG concentration is lower among patients with NAFLD with subtype A compared with the subtype C
Because the lipidomic profile of patients with NAFLD with subtype A resembled that of mouse models of NAFLD deficient in VLDL‐TG secretion, we hypothesized that the serum concentration of VLDL‐TG and its subclasses would also be lower in patients with NAFLD with subtype A as compared to patients with NAFLD with subtype C, and that patients with NAFLD with subtype B would show an intermediate content of total VLDL‐TG and subclasses.
Comparison of the serum lipoprotein profile of a subset of 197 samples from the NAFLD cohort (Table S4) confirmed this hypothesis. Table S1 lists the main clinical data of this subset of patients with NAFLD classified by their metabolic subtype. As expected, our results indicate that, independently of their subtype, patients with NAFLD have higher serum TG levels (p < 0.0001) than controls (Figure 2, Table S4). Our data also show that the concentration of serum TG was higher (p < 0.0001) among patients with NAFLD with subtype C than among subjects with NAFLD with subtype A, with intermediate levels of TG in patients with NAFLD with subtype B (Figure 2, Table S4). In accordance with former studies in patients with NAFLD,[ 10 , 13 ] we observed that, independently of the subtype, the concentration of TG in VLDL was higher in the patients with NAFLD (p < 0.0001) compared with controls (Figure 2, Table S4). Furthermore, our results show that the concentration of TG in VLDL, IDL, and LDL was lower (p < 0.0001) in patients with subtype A than among patients with subtype C, with intermediate levels of VLDL‐TG, IDL‐TG, and LDL‐TG in patients with NAFLD with subtype B (Figure 2). The concentration of serum VLDL‐Apo‐B (a surrogate for VLDL particle number) was higher among patients with NAFLD with subtype C than among subjects with subtype A (p < 0.0001), with intermediate levels of VLDL‐Apo‐B in patients with subtype B (Figure 2). The difference in serum IDL‐Apo‐B concentration between the A and C subtypes, although statistically significant (p < 0.0001), was less pronounced than for VLDL‐Apo‐B (Figure 2). For LDL‐Apo‐B, patients with subtype C showed higher levels than patients with subtype A (p < 0.0001), with intermediate levels in patients with subtype B (Figure 2). The concentration of LDL‐Apo‐B was higher among the control group than among patients with subtype A (p < 0.0001), and lower than in the cases with subtype C (Figure 2). This observation is reflected when the total concentration of Apo‐B was determined, which follows the order: subtype A < control group < subtype C (Figure 2). Given that a reduction in VLDL‐TG in patients with NAFLD with subtype A might reflect partitioning of acetyl–coenzyme A (CoA) toward ketone body synthesis, we determined the serum concentration of acetoacetate, β‐hydroxybutyrate, and acetone in the control group and the NAFLD subtypes. Our results showed that there are no differences in the concentration of ketone bodies among subtypes (Figure S3).
FIGURE 2.
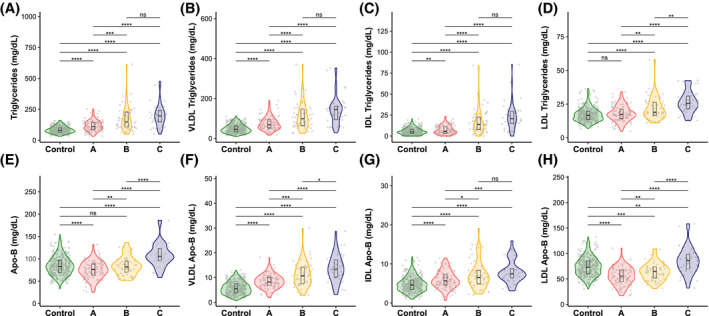
Blood lipoprotein TG (A–D) and apolipoprotein B (Apo‐B) (E–H) concentrations for NAFLD subtypes and control subjects. Gray points in the background indicate the real values with a minimal random displacement to avoid overplotting. Densities are represented as violin plots, while main distributions (medians and first and third quartiles) are represented as internal box plots. Horizontal black lines are statistical comparisons (t test) between two groups, with the unadjusted p values as symbols above the lines (****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, ≥ 0.05). IDL, intermediate‐density lipoprotein
In patients with NAFLD with subtype A, the serum concentration of VLDL‐TG and VLDL‐Apo‐B was independent of the degree of steatosis
We next examined the relationship between serum VLDL‐TG and VLDL‐Apo‐B concentrations and the grade of steatosis for each NAFLD subtype. Liver steatosis was graded histologically based on the percentage of hepatocytes showing fat accumulation[ 36 ]: S0 (normal, <5%), S1 (mild, 5%–33%), S2 (moderate, 34%–66%), and S3 (severe, >66%). When the serum concentrations of VLDL‐TG and VLDL‐Apo‐B from patients with NAFLD (n = 197) and the control group (n = 350) were plotted against the grade of steatosis for each NAFLD subtype, we found that patients with subtype C showed a curvilinear relationship between these parameters with a peak at S2, whereas in patients with subtype B increased linearly from S1 to S3 (Figure 3). In contrast, patients with subtype A exhibited lower concentrations of serum VLDL‐TG and VLDL‐Apo‐B, regardless of the steatosis grade (Figure 3). Serum albumin and INR were comparable among subtypes (Table 1), indicating that altered hepatic VLDL production alone does not account for the differences in hepatic steatosis in subtype A.
FIGURE 3.
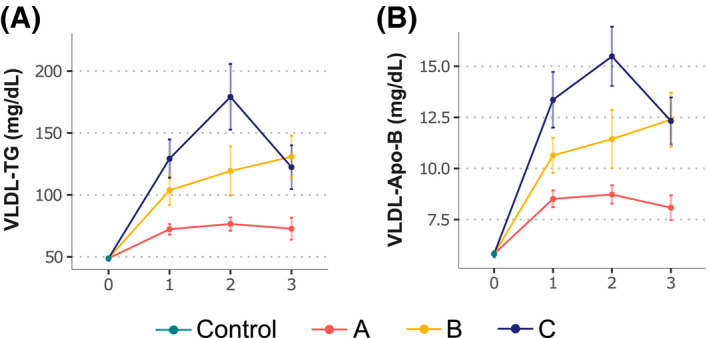
VLDL‐TG and VLDL‐Apo‐B concentration by grade of steatosis for each NAFLD subtype and control cohort. (A,B) Total VLDL‐TG (A) and VLDL‐Apo‐B (B). Values are represented as means (points) and standard errors (vertical lines)
Cardiovascular risk factors are lower among patients with NAFLD with subtype A compared with subtype C
Compared to the control group, patients with NAFLD with subtype C exhibited an increase in the serum concentration of VLDL and small dense LDL (LDL5 and LDL6) particles, lower total HDL‐CL, higher LDL‐CL/HDL‐CL and Apo‐B/Apo‐A1 ratios, and increased concentration of remnant lipoprotein cholesterol (RLP‐CL, the sum of the CL associated with VLDL and IDL) (Figure 4, Table S4), which are all markers of CVD risk and have previously been associated with NAFLD.[ 4 ] Our data also show that the concentration of VLDL and small dense LDL (LDL5 and LDL6) particles, the HDL‐CL content, and the RLP‐CL concentration were lower among patients with subtype A than among subjects with subtype C, whereas the LDL‐CL/HDL‐CL ratio showed similar values in both subtypes (Figure 4). Patients with B subtype exhibited intermediate values for these parameters. These results indicate that, compared to the C subtype, patients with NAFLD with subtype A exhibited a favorable CVD risk profile.
FIGURE 4.
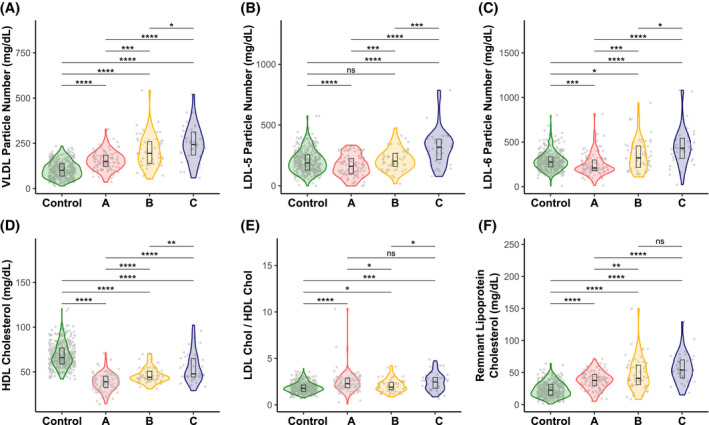
Blood lipoprotein particle numbers and cholesterol concentrations for NAFLD subtypes (A, B, and C) and control subjects. (A) VLDL particle number. (B) LDL‐5 particle number. (C) LDL‐6 particle number. (D) HDL cholesterol. (E) LDL cholesterol/HDL cholesterol ratio. (F) Remnant lipoprotein cholesterol. Gray points in the background indicate the real values with a minimal random displacement to avoid overplotting. Densities are represented as violin plots, while main distributions (medians and first and third quartiles) are represented as internal box plots. Horizontal black lines are statistical comparisons (t test) between two groups, with the unadjusted p values as symbols above the lines (****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, ≥ 0.05)
VLDL‐TG SR is lower in subjects with NAFLD with subtype A compared to the B and C subtypes
To address the question of whether patients with NAFLD with subtype A have lower VLDL‐TG SR than patients with non‐A subtype, we examined the relationship between the NAFLD subtype, the plasma VLDL‐TG concentration, and the VLDL‐TG SR in a group of obese women (n = 20) with IHTG content assessed by MR spectroscopy. A group of lean women (n = 10) with low VLDL‐TG SR and normal IHTG content was included for comparison (Table 2). These subjects are a subgroup of a larger cohort previously used to study VLDL‐TG SR kinetics.[ 38 ] Subtyping of the obese subjects with NAFLD was performed using the classification algorithm described in Table S2. Of the 20 obese subjects with NAFLD studied, 12 fell into subtype A, 5 into subtype B, and 3 into subtype C (Table 2). Subjects with subtypes B and C were grouped together for comparative reasons. Obese subjects with NAFLD with subtype A had similar age, BMI, and IHTG content than patients with subtypes B and C. As hypothesized, VLDL‐TG SR was higher (2.7‐fold) among obese subjects with NAFLD with subtypes B and C than in obese subjects with NAFLD with subtype A (Table 2). The fractional catabolic rate (FCR, the fraction of the intravascular pool of VLDL catabolized per hour) was similar among obese subjects with NAFLD with subtype A that in the patients with subtypes B and C (Table 2). Obese patients with NAFLD with subtype A had similar plasma TG, VLDL‐TG, and VLDL‐TG SR than lean subjects (Table 2).
TABLE 2.
Relationship between NAFLD subtypes and plasma VLDL‐TG secretion rate in obese women with NAFLD (n = 20) and lean women (n = 10)
| Lean | Obese A | Obese B/C | p value | |||
|---|---|---|---|---|---|---|
| n = 10 | n = 12 | n = 8 | Lean vs. obese A | Lean vs. obese B/C | Obese A vs. obese B/C | |
| Age (years) | 42.50 ± 20.10 | 39.50 ± 10.28 | 38.75 ± 11.46 | 0.6759 | 0.626161 | 0.88344 |
| BMI (kg/m²) | 21.71 ± 1.89 | 36.14 ± 4.33 | 36.48 ± 4.74 | 2.02e‐08 | 1.9e‐05 | 0.87137 |
| IHTG (%) | 1.02 ± 0.37 | 12.09 ± 13.90 | 13.76 ± 8.90 | 0.0329 | 0.004851 | 0.76145 |
| Plasma TG (mg/dl) | 80.90 ± 21.43 | 92.08 ± 49.21 | 161.25 ± 79.62 | 0.4879 | 0.024660 | 0.05158 |
| VLDL‐TG (mmol/l) | 0.21 ± 0.12 | 0.37 ± 0.43 | 0.83 ± 0.47 | 0.2592 | 0.007241 | 0.04268 |
| VLDL‐TG SR (µmol/l/h) | 113.18 ± 43.91 | 169.70 ± 134.47 | 467.72 ± 189.10 | 0.1924 | 0.000974 | 0.00241 |
| FCR (pools/h) | 0.63 ± 0.30 | 0.76 ± 0.44 | 0.74 ± 0.37 | 0.4017 | 0.506677 | 0.89100 |
Obese women classified as NAFLD subtypes B and C were group together for comparative reasons. For each group, the variables are summarized as means and SDs. p values between pairs of groups were calculated from two‐tailed independent Student’s t test.
Abbreviations: FCR, fractional catabolic rate; IHTG, intrahepatic triglycerides; SR, secretion rate.
Relevance of the genotype to a particular NAFLD subtype
To study the relevance of the genotype to a particular NAFLD metabolic phenotype, we tested the association with the three NAFLD subtypes for four of the major known genetic risk factors, namely, rs9992651 (17β‐hydroxysteroid dehydrogenase type 13), rs641738 (membrane‐bound O‐acyltransferase domain‐containing protein 7), rs738409 (patatin‐like phospholipase domain‐containing protein 3 [PNPLA3]), and rs58542926 (TM6SF2).[ 5 ] Only one variant (PNPLA3) associated with metabolic subtype distributions (p = 0.0003) (Table 3), being that this allele is overrepresented in individuals with NAFLD subtypes B and C. The I148M protein variant of PNPLA3 accounts for the largest proportion of genetic predisposition to NAFLD in all populations.[ 42 , 43 , 44 ] This variant has been associated with increased hepatic fat and elevated liver enzymes, which goes in accordance with the biological inferences observed in patients with NAFLD with subtypes B and C.
TABLE 3.
Percent of risk alleles by single‐nucleotide polymorphism (gene) and subtype
| SNP (gene) risk alleles | Subtype A | Subtype B | Subtype C | p value | n |
|---|---|---|---|---|---|
| rs9992651‐G or rs72613567‐T (HSD17B13) | 0.6358 | 391 | |||
| 0 | 16 (6.18%) | 3 (4.35%) | 2 (3.17%) | ||
| 1 | 74 (28.57%) | 22 (31.88%) | 14 (22.22%) | ||
| 2 | 169 (65.25%) | 44 (63.77%) | 47 (74.60%) | ||
| rs641738‐T (MBOAT7) | 0.7730 | 335 | |||
| 0 | 62 (28.05%) | 16 (30.77%) | 21 (33.87%) | ||
| 1 | 111 (50.23%) | 28 (53.85%) | 30 (48.39%) | ||
| 2 | 48 (21.72%) | 8 (15.38%) | 11 (17.74%) | ||
| rs738409‐G (PNPLA3) | 0.0003 | 433 | |||
| 0 | 131 (46.13%) | 30 (39.47%) | 17 (23.29%) | ||
| 1 | 96 (33.80%) | 20 (26.32%) | 26 (35.62%) | ||
| 2 | 57 (20.07%) | 26 (34.21%) | 30 (41.10%) | ||
| rs58542926‐T (TM6SF2) | 0.2939 | 410 | |||
| 0 | 234 (85.71%) | 66 (94.29%) | 61 (91.04%) | ||
| 1 | 35 (12.82%) | 4 (5.71%) | 5 (7.46%) | ||
| 2 | 4 (1.47%) | 0 (0.00%) | 1 (1.49%) |
Risk alleles are indicated next to SNP code. p values were calculated with Fisher’s exact test.
Abbreviations: HSD17B13, 17β‐hydroxysteroid dehydrogenase type 13; MBOAT7, membrane‐bound O‐acyltransferase domain‐containing protein 7; PNPLA3, patatin‐like phospholipase domain‐containing protein 3; SNP, single‐nucleotide polymorphism.
DISCUSSION
FAs in the liver largely originate from the blood derived from lipolysis of TG in adipose tissue, and from glucose derived from DNL. FAs can be re‐esterified into lipids or be transported to the mitochondria to be oxidized into acetyl‐CoA, which in turn can be condensed with oxaloacetate to form citrate entering the tricarboxylic acid (TCA) cycle for complete oxidation, or enter the ketogenic pathway, where it is converted to acetoacetate and β‐hydroxybutyrate. Alternatively, citrate may be redirected to DNL when the amount of acetyl‐CoA exceeds the capacity of the TCA cycle to oxidize it. Some of the TG synthesized by the liver combine with Apo‐B to form VLDL, and are exported into the blood stream. IDL and LDL are formed after lipases hydrolyze VLDL‐TG for delivery of FAs to peripheral tissues. TGs that are not secreted into the blood form lipid droplets in hepatocytes, whose accumulation is a defining feature of NAFLD.[ 45 ] Understanding to which extent alterations in these metabolic pathways contribute to NAFLD development and its progression to NASH is crucial not only in providing granularity to understand its pathogenesis, but also to understand to which extent different mechanisms leading to disease may contribute to the clinical manifestations of other pathologies associated with NAFLD, such as CVD.[ 2 , 3 , 5 , 6 ]
The current study confirms and extends prior observations on NAFLD subtypes[ 24 ] with extensive metabolomic profiling of a large international cohort of 1154 biopsy‐confirmed patients with NAFLD. Here we demonstrate the existence of three distinctive, clinically potentially relevant NAFLD subtypes characterized by serum lipidomic signatures. Among these, subtype A, which accounts for 47% of patients with NAFLD, shows a lipidomic signature like that observed in four different mouse models with defective VLDL‐TG secretion. Alterations in these four models ranged from the deletion of Mttp (the gene encoding MTTP, a protein playing a central role in VLDL assembly)[ 40 ] and Tm6sf2 (the gene encoding TM6SF2, a protein involved in the lipidation of nascent VLDL particles)[ 41 ] to the deletion of Mat1a (a protein‐encoding gene that catalyzes the synthesis of SAMe, which plays a necessary role in the liver synthesis of PC,[ 46 ] the major phospholipid component of VLDL) and the induction of NAFLD by feeding mice a MCD diet, which also results in a lower PC/phosphatidylethanolamines (PE) ratio.[ 47 ] The geographical distribution of NAFLD cases among subtypes A, B, and C varied significantly between Europe (higher proportion of patients with subtype A), the United States (higher proportion of patients with subtype C), and Israel and Chile (with similar proportions among subtypes) (Table S5). Based on these results, we propose that subtype A stems from multiple nutritional and environmental factors that, acting on a susceptible genetic background, converge in reduced synthesis and export of VLDL‐TG, resulting in lower circulating levels of both VLDL‐Apo‐B and VLDL‐TG.
Consistent with this hypothesis, we found that the concentration of TG, VLDL‐TG, IDL‐TG, and LDL‐TG was lower among patients with subtype A compared to patients with subtypes B or C, and similar to that observed in controls. The same pattern was also observed when the content of phospholipids, total CL, and CL esters in VLDL, IDL, and LDL was analyzed (Table S4). Moreover, the content of VLDL‐Apo‐B, IDL‐Apo‐B, and LDL‐Apo‐B was also lower among patients with subtype A compared to patients with subtypes B and C, and similar to controls. The ratio of VLDL‐TG (TG content in the particle) to VLDL Apo‐B (number of particles) was similar among the three subtypes, suggesting that secreted VLDL particles are similar in size and lipid composition. It is tempting to speculate that PC availability for VLDL assembly is lower in patients with subtype A, as is the case in Mat1a‐KO and 0.1MCD mice,[ 47 , 48 ] and VLDL synthesis is therefore saturated at a lower IHTG content than in the subtypes B and C. That speculation, however, will require formal evaluation.
The finding that the concentration of VLDL‐TG and VLDL‐Apo‐B in the serum of patients with subtype A was independent of the grade of steatosis, whereas in subjects with subtypes B or C the concentration of serum VLDL‐TG and VLDL‐Apo‐B increased with the grade of steatosis, also supports the hypothesis that VLDL‐TG SR is saturated in patients with subtype A at a lower IHTG concentration.
Evidence that patients with NAFLD with subtype A exhibit reduced VLDL‐TG SR was obtained by subtyping a cohort of 20 obese women with elevated IHTG and different VLDL‐TG SR. VLDL‐TG SR was 2.7‐fold lower in obese individuals with NAFLD classified as subtype A compared to the obese subjects with NAFLD with subtypes B and C. The FCR was similar among subjects with subtype A compared to subtypes B and C, which indicates that reduced catabolism of VLDL‐TG does not contribute to the higher concentration of VLDL‐TG in subjects with subtypes B and C. Finally, subjects with NAFLD with subtype A showed similar VLDL‐TG SR, VLDL‐TG, and TG compared with lean subjects, despite having a 12‐fold increase in IHTG, which further supports the hypothesis that the VLDL‐TG SR in these subjects likely becomes saturated at low levels of IHTG.
NAFLD is associated with increased CVD risk in association with alterations in circulating lipid levels and lipoprotein metabolism.[ 11 , 13 ] We observed that patients with NAFLD with subtype C display hypertriglyceridemia, elevated VLDL‐TG, and VLDL‐Apo‐B, increased small dense LDL particles (LDL5 and LDL6), and RLP‐CL, compared with control subjects. All of these features were significantly lower among patients with NAFLD with subtype A as compared to subtype C, indicating that patients with subtype A exhibited a favorable CVD risk profile, likely the result of reduced VLDL secretion. The percentage of patients with NAFLD at high CVD risk (FRS ≥ 15% at 10 years) was lower in subtype A than in subtypes B and C. This reduction in FRS did not reach statistical significance, probably due to the small number of high‐risk CVD cases. The percent of patients receiving lipid‐lowering medication was not statistically different between subtypes. Liver enzymes (ALT, AST, and GGT), which are also associated with increased risk of CVD,[ 49 ] were higher in patients with subtype C than in subjects with subtype A. The HOMA‐IR index and HbA1c, two independent CVD risk factors,[ 50 , 51 ] were similar among the three subtypes. NASH lipidomic signatures were characterized by changes in TG, PC, and sphingomyelins (SM). Up to 56% of TG (49 of 84), 24% of PC (23 of 92), and 19% of SM (6 of 31) analyzed changed significantly in patients with NASH with subtype C. Diglycerides (DG; 9 of 13), PE (4 of 24), phosphatidylinositols (PI; 2 of 3), and ceramides (Cer; 1 of 12) also changed significantly in patients with NASH with subtype C. Compared with subtype C, NASH lipidomic signatures in patients with subtype A displayed fewer changes. These included TG (23 of 84), DG (6 of 13), PC (6 of 92), PE (4 of 24), and SM (1 of 31). NASH lipidomic signature in patients with subtype B showed changes in TG (23 of 84), DG (5 of 13), PC (15 of 92), PE (4 of 24), PI (1 of 3), Cer (1 of 12), and SM (18 of 31). These data provide evidence of the existence of distinct metabolic mechanisms associated with NAFLD progression varying between subtypes.
We also acknowledge some limitations in the interpretation of our findings. The small number of genotyped individuals limits our ability to characterize in depth the role of genetics in NAFLD metabolic subtypes. Despite this limitation, the association of PNPLA3 risk allele with NAFLD subtypes B and C is robust. The high frequency of these subtypes in the Chilean cohort, nonetheless, introduces a note of caution regarding the actual strength of this association. This cohort likely harbors genetic differences with the other European counterparts; hence, larger studies that are focused on homogeneous genetic ancestry settings will be useful to validate our observation. The main limitation of our study is the lack of CVD outcomes and the relatively small number of cases (360 individuals) for which the FRS was available. The lower content of serum TG, VLDL, LDL, and Apo‐B, and the lower FRS ≥ 15% at 10 years, suggest an improved CVD risk profile in patients with NAFLD with subtype A compared to subjects with subtypes B and C, which can provide clinically useful information in the management of patients with NAFLD.[ 52 ] Further work should be carried out to confirm this hypothesis. We further emphasize a strength of this study, which is the inclusion of large international cohorts of patients with biopsy‐proven NAFLD/NASH. This study shows NAFLD/NASH lipidomic phenotyping according to CVD risks profile, the latter representing the leading cause of death in patients with NAFLD/NASH.
CONFLICT OF INTEREST
Dr. Alonso is employed by OWL Metabolomics. Dr. Anstee is the coordinator of the EU IMI‐2 LITMUS consortium. He received grants from, consults for, and is on the speaker’s bureau for Allergan. He received grants from and consults for AstraZeneca, Boehringer Ingelheim, Intercept, Novartis, and Pfizer. He consults for and is on the speakers’ bureau for BMS, Genfit SA, and Gilead. He consults for 89 Bio, Abbvie, Akero, Altimentiv, Alitimmune, Axcella, Blade, BNN Cardio, Cirius, CymaBay, EcoR1, E3Bio, Eli Lilly & Company Ltd, Galmed, Genentech, Grunthal, HistoIndex, Indalo, Inventiva, IQVIA, Janssen, Johnson & Johnson, Madrigal, MedImmune, Medpace, Merck, Metacrine, NGMBio, North Sea Therapeutics, Novo Nordisk, PathAI, Poxel, ProSciento, Raptor Pharma, Roche, Servier, Shionogi, Terns, The Medicines Company, and Viking Therapeutics. He is on the speakers’ bureau for Abbott Laboratories, Clinical Care Options, Falk, Fishawack, Integritas Communications, Kenes, and Medscape. He received grants from GlaxoSmithKline and Glympse Bio. He receives royalties from Elsevier. Dr. Arretxe is employed by OWL Metabolomics. Dr. Banales advises OWL Metabolomics. Dr. Bugianesi consults for and received grants from Gilead. She consults for NovoNordisk, Lilly, and Merck. Dr. Crespo is on the speakers’ bureau for Intercept and Shionogui. He received grants from Gilead and AbbVie. Dr. Cusi consults for Arrowhead, AstraZeneca, 89 Bio, Lilly, Madrigal, and Quest. He advises Sagimet, Sonic Incytes, and Terns. He received grants from Echosens, Inventiva, Novo, Poxel, and Labcorp. Dr. Iruarrizaga‐Lejarreta is employed by OWL Metabolomics. Dr. Hayardeny owns stock in and is employed by Galmed. Dr. Mato consults and advises Abbott. He owns stock in, consults for, and advises OWL Metabolomics. He consults for Galmed. Dr. mayo is employed by OWL Metabolomics. Mr. Martinez‐Arranz is employed by OWL Metabolomics. Mrs. Mincholé is employed by OWL Metabolomics. Dr. Noureddin owns stock in and received grants from Viking. He advises and received grants from Gilead, Madrigal, and Pfizer. He consults for Perspectum. He advises 89 Bio, Altimmune, CohBar, Cytodyn, Intercept, Novo Nordisk, Blade, EchoSens, Fractyl, NorthSea, Terns, Siemens, and Roche. He received grants from Allergan, BMS, Galmed, Galectin, Genfit, Conatus, Enanta, Novartis, Shire, and Zydus. He owns stock in Anaetos and Rivus Pharma.
AUTHOR CONTRIBUTIONS
Study concept and design: Mazen Noureddin, Shelly C. Lu, Nicholas O. Davidson, Cristina Alonso and José M. Mato. Funding obtainment and study supervision: José M. Mato. Data acquisition: Chiara Bruzzone, Maider Bizkarguenaga, David Fernández‐Ramos, Fernando Lopitz‐Otsoa, Rebeca Mayo, Nieves Embade, Elizabeth Newberry, Bettina Mittendorf, Laura Izquierdo‐Sánchez, Vaclav Smid, Jorge Arnold, Paula Iruzubieta, Ylenia Pérez Castaño, Marcin Krawczyk, Martine C. Morrison, Robert Kleemann, and Cristina Alonso. Data analysis: Ibon Martínez‐Arranz, Ruben Gil‐Redondo, Enara Arretxe, Marta Iruarrizaga‐Lejarreta, Urko M. Marigorta, Oscar Millet, Cristina Alonso, and José M. Mato. Data interpretation: Ibon Martínez‐Arranz, Mazen Noureddin, Ruben Gil‐Redondo, Enara Arretxe, Marta Iruarrizaga‐Lejarreta, Urko M. Marigorta, Oscar Millet, Cristina Alonso, and José M. Mato. Statistical and informatics analysis: Itziar Mincholé. Animal experimentation: David Fernández‐Ramos, Fernando Lopitz‐Otsoa, Elizabeth Newberry, Martine C. Morrison, and Robert Kleemann. Acquisition of human samples and data: Antonio Martín‐Duce, Liat Hayardeny, Libor Vitek, Radan Bruha, Rocío Aller de la Fuente, Javier Crespo, Manuel Romero‐Gomez, Jesus M. Banales, Marco Arrese, Kenneth Cusi, Elisabetta Bugianesi, Samuel Klein, and Quentin M. Anstee. Manuscript draft: Cristina Alonso and José M. Mato. Critical revision of the manuscript for important intellectual content: Ibon Martínez‐Arranz, Mazen Noureddin, Ruben Gil‐Redondo, Urko M. Marigorta, Libor Vitek, Radan Bruha, Jesus M. Banales, Shelly C. Lu, Quentin M. Anstee, Oscar Millet, and Nicholas O. Davidson. Final approval of the version to be published: Mazen Noureddin, Shelly C. Lu, Quentin M. Anstee, Nicholas O. Davidson, Cristina Alonso, and José M. Mato.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Table S1‐S5
Supplementary Material
Martínez‐Arranz I, Bruzzone C, Noureddin M, Gil‐Redondo R, Mincholé I, Bizkarguenaga M, et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology. 2022;76:1121–1134. 10.1002/hep.32427
Ibon Martínez‐Arranz, Chiara Bruzzone, and Mazen Noureddin contributed equally to the study.
Funding information
National Institutes of Health (R01DK123763, R01DK119437, HL151328, P30DK52574, P30DK56341, and UL1TR002345); Ministerio de Economía y Competitividad de España (SAF2017‐88041‐R); Ministerio de Economía y Competitividad de España for the Severo Ochoa Excellence Accreditation (SEV‐2016‐0644); CIBERehd (Biomedical Research Center in Hepatic and Digestive Diseases) and Netherlands Organization for Applied Scientific Research Program (PMC13 and PMC15); Spanish Carlos III Health Institute (PI15/01132 and PI18/01075); Miguel Servet Program (CON14/00129 and CPII19/00008); Fondo Europeo de Desarrollo Regional, CIBERehd, Department of Industry of the Basque Country (Elkartek: KK‐2020/00008); La Caixa Scientific Foundation (HR17‐00601); Liver Investigation: Testing Marker Utility in Steatohepatitis consortium funded by the Innovative Medicines Initiative Program of the European Union (777377), which receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA; Newcastle NIHR Biomedical Research Center; Czech Ministry of Health (RVO‐VFN64165/2020); Fondo Nacional De Ciencia y Tecnología de Chile (1191145); and the Comisión Nacional de Investigación, Ciencia y Tecnología (AFB170005, CARE Chile UC); Agencia Nacional de Investigación y Desarrollo (ANID ACE 210009); European Union's Horizon 2020 Research and Innovation Program (825510).
Contributor Information
Cristina Alonso, Email: calonso@owlmetabolomics.com.
José M. Mato, Email: director@cicbiogune.es.
REFERENCES
- 1. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. 2020;158:1851–64. [DOI] [PubMed] [Google Scholar]
- 2. Stefan N, Häring HU, Cusi K. Non‐alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019;7:313–24. [DOI] [PubMed] [Google Scholar]
- 3. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Francque SM, van der Graaff D, Kwanten WJ. Non‐alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol. 2016;65:425–43. [DOI] [PubMed] [Google Scholar]
- 5. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome‐wide association study of non‐alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol. 2020;73:505–15. [DOI] [PubMed] [Google Scholar]
- 6. Masoodi M, Gastaldelli A, Hyötyläinen T, Arretxe E, Alonso C, Gaggini M, et al. Metabolomics and lipidomics in NAFLD: biomarkers and noninvasive diagnostic tests. Nat Rev Gastro Hep. 2021;18:835–56. [DOI] [PubMed] [Google Scholar]
- 7. Arrese M, Arab JP, Barrera F, Kaufmann B, Valenti L, Feldstein AE. Insights into Nonalcoholic Fatty‐Liver Disease heterogeneity. Semin Liver Dis. 2021;41:421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130:1453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adiels M, Taskinen M‐R, Packard C, Caslake MJ, Soro‐Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–65. [DOI] [PubMed] [Google Scholar]
- 11. Siddiqui MS, Fuchs M, Idowu MO, Luketic VA, Boyett S, Sargeant C, et al. Severity of nonalcoholic fatty liver disease and progression to cirrhosis are associated with atherogenic lipoprotein profile. Clin Gastroenterol Hepatol. 2015;13:1000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corey KE, Misdraji J, Gelrud L, Zheng H, Chung RT, Krauss RM. Nonalcoholic steatohepatitis is associated with an atherogenic lipoprotein subfraction profile. Lipids Health Dis. 2014;13:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Männistö VT, Simonen M, Soininen P, Tiainen M, Kangas AJ, Kaminska D, et al. Lipoprotein subclass metabolism in nonalcoholic steatohepatitis. J Lipid Res. 2014;55:2676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Filippo M, Moulin P, Roy P, Samson‐Bouma ME, Collardeau‐Frachon S, Chebel‐Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol. 2014;61:891–902. [DOI] [PubMed] [Google Scholar]
- 16. Bonnefont‐Rousselot D, Condat B, Sassolas A, Chebel S, Bittar R, Federspiel M‐C, et al. Cryptogenic cirrhosis in a patient with familial hypocholesterolemia due to a new truncated form of apolipoprotein B. Eur J Gastroenterol Hepatol. 2009;21:104–8. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y‐L, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non‐alcoholic fatty liver disease. Nat Commun. 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506–14. [DOI] [PubMed] [Google Scholar]
- 19. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin W, Sundaram M, Wang Y, Zhou HU, Zhong S, Chang C‐C, et al. Missense mutation in APOC3 within the C‐terminal lipid binding domain of human ApoC‐III results in impaired assembly and secretion of triacylglycerol‐rich very low‐density lipoproteins: evidence that ApoC‐III plays a major role in the formation of lipid precursors within the microsomal lumen. J Biol Chem. 2011;286:27769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56. [DOI] [PubMed] [Google Scholar]
- 22. Cuchel M, Blom DJ, Averna MR. Clinical experience of lomitapide therapy in patients with homozygous familial hypercholesterolaemia. Atherosclerosis Supplements. 2014;15:33–45. [DOI] [PubMed] [Google Scholar]
- 23. Hashemi N, Odze RD, McGowan MP, Santos RD, Stroes ES, Cohen DE. Liver histology during Mipomersen therapy for severe hypercholesterolemia. J Clinical Lipidol. 2014;8:606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alonso C, Fernández‐Ramos D, Varela‐Rey M, Martínez‐Arranz I, Navasa N, Van Liempd SM, et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology. 2017;152:1449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu SC, Alvarez L, Huang Z‐Z, Chen L, An W, Corrales FJ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci. 2001;98:5560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mato JM, Alonso C, Noureddin M, Lu SC. Biomarkers and subtypes of deranged lipid metabolism in non‐alcoholic fatty liver disease. World J Gastroenterol. 2019;25:3009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology. 2014;59:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease‐specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18:296–302. [DOI] [PubMed] [Google Scholar]
- 29. Guo T, Dai Z, You KE, Battaglia‐Hsu S‐F, Feng J, Wang F, et al. S‐adenosylmethionine upregulates the angiotensin receptor‐binding protein ATRAP via the methylation of HuR in NAFLD. Cell Death Dis. 2021;12:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morrison MC, Verschuren L, Salic K, Verheij J, Menke A, Wielinga PY, et al. Obeticholic acid modulates serum metabolites and gene signatures characteristic of human NASH and attenuates inflammation and fibrosis progression in Ldlr‐/‐.leiden mice. Hepatol Commun. 2018;2:1513–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knowles JW, Maeda N. Genetic modifiers of atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2000;20:2336–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21:434–40. [DOI] [PubMed] [Google Scholar]
- 33. Khan A, Waqar K, Shafique A, Irfan R, Gul A. In vitro and in vivo animal models: the engineering towards understanding human diseases and therapeutic interventions. Omics Technologies and Bio‐Engineering Towards Improving Quality of Life. 2018;1:431–48. [Google Scholar]
- 34. Hardy T, Wonders K, Younes R, Aithal GP, Aller R, Allison M, et al. The European NAFLD Registry: a real‐world longitudinal cohort study of nonalcoholic fatty liver disease. Contemp Clin Trials. 2020;98:106175. [DOI] [PubMed] [Google Scholar]
- 35. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. [DOI] [PubMed] [Google Scholar]
- 36. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bruzzone C, Bizkarguenaga M, Gil‐Redondo R, Diercks T, Arana E, García de Vicuña A, et al. SARS‐CoV‐2 infection dysregulates the metabolomic and lipidomic profiles of serum. iScience. 2020;23:101645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mittendorfer B, Yoshino M, Patterson BW, Klein S. VLDL triglyceride kinetics in lean, overweight, and obese men and women. J Clin Endocrinol Metab. 2016;101:4151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iruarrizaga‐Lejarreta M, Varela‐Rey M, Fernández‐Ramos D, Martínez‐Arranz I, Delgado TC, Simon J, et al. Role of aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun. 2017;1:911–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newberry EP, Xie Y, Kennedy SM, Graham MJ, Crooke RM, Jiang H, et al. Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology. 2017;65:836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Newberry EP, Hall Z, Xie Y, Molitor EA, Bayguinov PO, Strout GW, et al. Liver specific deletion of mouse Tm6sf2 promotes steatosis, fibrosis and hepatocellular cancer. Hepatology. 2021;74:1203–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sookoian S, Pirola CJ, Valenti L, Davidson NO. Genetic pathways in nonalcoholic fatty liver disease: insights from systems biology. Hepatology. 2020;72:330–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jonas W, Schürmann A. Genetic and epigenetic factors determining NAFLD risk. Mol Metab. 2021;50:101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu SC, Mato JM. S‐adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res. 2008;49:1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bernardo‐Seisdedos G, Bilbao J, Fernández‐Ramos D, Lopitz‐Otsoa F, Gutierrez de Juan V, Bizkarguenaga M, et al. Metabolic landscape of the mouse liver by quantitative 31 P‐Nuclear Magnetic Resonance analysis of the phosphorome. Hepatology. 2021;74:148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monami M, Bardini G, Lamanna C, Pala L, Cresci B, Francesconi P, et al. Liver enzymes and risk of diabetes and cardiovascular disease: results of the Firenze Bagno a Ripoli (FIBAR) study. Metabolism. 2008;57:387–92. [DOI] [PubMed] [Google Scholar]
- 50. Bonora E, Formentini G, Calcaterra F, Lombardi S, Marini F, Zenari L, et al. HOMA‐estimated insulin resistance is an independent predictor of cardiovascular disease in type 2 diabetic subjects: prospective data from the Verona Diabetes Complications Study. Diabetes Care. 2002;25:1135–41. [DOI] [PubMed] [Google Scholar]
- 51. Cavero‐Redondo I, Peleteiro B, Álvarez‐Bueno C, Rodriguez‐Artalejo F, Martínez‐Vizcaíno V. Glycated haemoglobin A1c as a risk factor of cardiovascular outcomes and all‐cause mortality in diabetic and nondiabetic populations: a systematic review and meta‐analysis. BMJ Open. 2017;7:e015949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non‐alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo‐controlled phase 3 trial. Lancet. 2019;394:2184–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material
Table S1‐S5
Supplementary Material