

Abstract
Frontotemporal dementia (FTD) covers a spectrum of neurodegenerative disorders with different phenotypes, genetic backgrounds, and pathological states. Its clinicopathological diversity challenges the diagnostic process and the execution of clinical trials, calling for specific diagnostic biomarkers of pathologic FTD types. There is also a need for biomarkers that facilitate disease staging, quantification of severity, monitoring in clinics and observational studies, and for evaluation of target engagement and treatment response in clinical trials. This review discusses current FTD biofluid‐based biomarker knowledge taking into account the differing applications. The limitations, knowledge gaps, and challenges for the development and implementation of such markers are also examined. Strategies to overcome these hurdles are proposed, including the technologies available, patient cohorts, and collaborative research initiatives. Access to robust and reliable biomarkers that define the exact underlying pathophysiological FTD process will meet the needs for specific diagnosis, disease quantitation, clinical monitoring, and treatment development.
1. BACKGROUND
Frontotemporal dementia (FTD) is the second most prevalent neurodegenerative dementia in young‐onset patients (< 65 years old) and has a dramatic effect on life expectancy, with reported survival rates after onset varying from 3 to 14 years. 1 , 2 FTD causes a heavy financial burden with costs per patient estimated at two to three times higher for FTD than those for patients suffering from Alzheimer's disease (AD) dementia. 3 , 4 FTD is a heterogeneous and complex disorder comprising a wide range of subtypes from a clinical, genetic, and pathological perspective (Figure 1). Based on the clinical phenotype, patients may suffer primarily from behavioral changes and deficits in executive functions (behavioral FTD [bvFTD]) or language disturbances (primary progressive aphasia [PPA]). PPA encompasses semantic variant PPA (svPPA) and non‐fluent variant PPA (nfvPPA). 5 , 6 A logopenic variant of PPA (lvPPA) has also been described, but is usually associated with AD pathology. 5 , 6 The phenotypic spectrum also includes cases with concomitant amyotrophic lateral sclerosis (FTD‐ALS), 7 corticobasal syndrome (CBS), 8 and progressive supranuclear palsy (PSP; the classic PSP syndrome called Richardson syndrome). 9 Furthermore, the overlap of clinical features in bvFTD or svPPA with those observed in AD or psychiatric disorders represents additional challenges for clinical diagnosis. 10 , 11 , 12 , 13 These diverse clinical phenotypes can share different pathological and genetic backgrounds (Figure 1), underpinning the complexity of this disorder.
FIGURE 1.
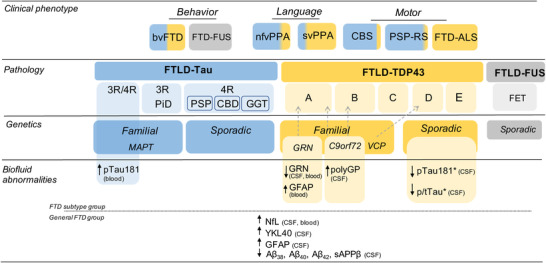
Clinicopathological and genetic classification of FTD spectrum. Diagram illustrates the clinicopathological correlations along the FTD spectrum and the corresponding genetic associations. Tau (blue), TDP‐43 (yellow), and FET (gray) pathologies, their corresponding immunohistochemical profiles and associated pathological diagnosis are annotated. Genetic forms of FTD are related to specific pathological aggregates (tau or TDP‐43). Gray arrows indicate specific associations between genetic mutation and immunohistochemical profiles. The biofluid‐based abnormalities indicate biomarkers that are dysregulated either between a specific FTLD subtype (upper panel) or undefined general FTD groups (bottom panel) and controls or between FTLD pathological subtypes (*) in either blood or CSF. Aβ, amyloid beta; bvFTD, behavioral variant frontotemporal dementia; CBD, corticobasal degeneration; CBS, corticobasal syndrome; CSF, cerebrospinal fluid; FET, family of RNA‐binding proteins including FUS, Ewing sarcoma and TAF15; FTD, frontotemporal dementia; FTD‐FUS, frontotemporal dementia fused in sarcoma variant; FTD‐ALS, frontotemporal dementia with concomitant amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration; GFAP, glial fibrillary acidic protein; GGT, globular glial tauopathy; NfL, neurofilament light; nfv, nonfluent variant; PiD, Pick's disease; PPA, primary progressive aphasia; PSP‐RS, progressive supranuclear palsy‐Richardson's syndrome; p‐tau, phosphorylated tau; sv, semantic variant; TDP‐43, TAR DNA‑binding protein 43 [Correction added on 17 March 2022, after first online publication: Figure 1 has been updated. Some text alignment as well as positioning has been corrected.]
1.1. FTD neuropathology
FTD is typically associated with focal degeneration of the frontal and temporal cortices, denoted by the term frontotemporal lobar degeneration (FTLD). Microscopically, ≈50% of FTLD patients are characterized by aggregates of TAR DNA‑binding protein 43 (TDP‑43 [FTLD‑TDP]) and 45% of FTLD patients develop aggregates of the microtubule‑associated protein tau (MAPT, i.e., FTLD‐tau). Less commonly (< 5% of the cases), FTLD features aggregates of RNA‑binding protein fused in sarcoma (FUS [FTLD‑FUS]) or ubiquitin‑positive inclusions (FTLD‑UPS). 14 , 15 , 16 Depending on the immunohistochemical profile, that is, density and distribution of the protein aggregates, FTLD‐TDP, FTLD‐tau, and FTLD‐FUS can be subclassified into different subtypes (Figure 1). 16 , 17 , 18 , 19
1.1.1. FTLD‐TDP
TDP‐43 is a nuclear DNA/RNA binding protein that regulates transcription and alternative splicing. 20 , 21 Mislocalization and aggregation of this protein in the cytoplasm leads to the pathological stress granules observed in FTLD. 22 TDP‐43 inclusions are also observed in > 95% of ALS, 22 supporting the notion that FTLD‐TDP and ALS are part of a clinicopathological continuum. 18 , 23 The heterogeneity within TDP‐43 histopathological patterns has prompted the definition of five different FTLD‐TDP subclassifications (FTLD type A, B, C, D, and E; Figure 1), depending on their shape, distribution, and cellular localization. 17 , 24 FTLD‐TDP can present with different clinical phenotypes including bvFTD, CBS, or PPA. 25 The presence of svPPA and FTD‐ALS show a strong correlation with TDP‐43 pathology. 25 , 26 Recent research suggests that psychiatric symptoms in FTD patients are associated with underlying TDP pathology. 27
1.1.2. FTLD‐tau
Misfolded or aggregated tau leads to the formation of pathological intraneuronal inclusion bodies and destabilization of microtubules. Alternative splicing leads to different tau isoforms with three or four microtubule‐binding domains (3‐repeat [3R] or 4‐repeat [4R] tauopathies). 28 Based on the predominant tau isoform present in such aggregates (tau 3R, 4R, or 3/4R) and the morphology of the inclusion bodies, FTLD‐tau can be classified and subdivided into several neuropathological diagnoses. Thus, FTLD‐tau includes Pick's disease (PiD), which features 3R tau pathology, and the 4R tauopathies PSP, corticobasal degeneration, and globular glial tauopathy (GGT; Figure 1). 18 , 19 FTLD‐tau can also present across the different clinical phenotypes. 18 Almost half of bvFTD have an underlying FTLD‐tau pathology, including PiD and to lesser extent CBD and PSP pathologies. FTLD‐tau has also been reported in patients with PPA and CBS phenotypes. The clinical syndrome of PSP highly correlates with PSP tau pathology. In addition, parkinsonism in association with bvFTD or PPA is a predictor of tau pathology. 26
1.1.3. FTLD‐FUS/UPS
FUS is also a nuclear DNA/RNA binding protein with diverse functions including transcription regulation, transport of RNA, and cell growth. 16 FUS inclusions are not very common in FTD; still, four different clinicopathological subclassifications have been proposed. 29 , 30 Patients with underlying FTLD‐FUS pathology often fulfill the diagnostic criteria of bvFTD. 30 Clinically, they are characterized by a disease onset before 40 years, a negative family history, and caudate atrophy on magnetic resonance imaging. 31
These data highlight that prediction of all these neuropathological subtypes on the basis of clinical phenotype is imperfect and varies across syndromes (Figure 1). Despite some syndromes being highly predictive of a specific underlying pathology (e.g., svPPA or FTD‐ALS with TDP or clinical PSP with tau), the most common sporadic bvFTD cases can have different neuropathologies that cannot be predicted by the clinical syndrome.
1.2. FTD genetics
FTD is highly heritable with approximately 10% to 20% of FTD cases caused by an autosomal dominant mutation. 32 , 33 Most of these variants have very poor genotype–phenotype correlations, but they are highly predictive of the underlying neuropathology (Figure 1). 18 , 34 The most common genes linked to familial FTD are MAPT, progranulin (GRN), and chromosome 9 open reading frame 72 (C9orf72). 35 , 36 , 37 , 38 We will only focus on these highly penetrant mutations, as exhaustive revisions outlining the heterogeneity of FTD genes have been previously published. 34 , 39 , 40
1.2.1. C9orf72
These carriers account for the largest amount of familial cases of FTD, in which abnormal GGGGCC expansions in the non‐coding region of the C9orf72 gene produce toxic RNA foci 41 , 42 and dipeptide repeat proteins (DPRs). 43 , 44 The number of expansions is directly correlated with pathogenicity, with most confirmed cases bearing hundreds of them. 40 These carriers mostly present with FTLD‐TDP A and B pathology, although type C has also been reported. 45 , 46 Clinically, FTD C9orf72 carriers can present with bvFTD, and in some cases also with PPA. 34
RESEARCH IN CONTEXT
Systematic Review: PubMed was used to search, identify, and evaluate the accumulated knowledge in relation to biofluid‐based biomarkers for frontotemporal dementia (FTD), together with data gathered through meetings, abstract, and presentations.
Interpretation: The findings indicate that FTD clinicopathological diversity challenges the diagnostic process and the execution of clinical studies and trials, so there is a pressing need for reliable biofluid‐based biomarkers for different context of use (e.g., prognosis and diagnosis, monitoring, target engagement, drug efficacy). Among the main limitations and knowledge gaps encountered in the development and implementation of such markers are (a) the clinicopathological heterogeneity of FTD, (b) the lack of an established definition of the different disease stages of FTD (e.g., preclinical, prodromal, dementia), (c) a better understanding of the similarities and differences between sporadic and familial cases or pathological subclassifications.
Future Directions: We propose different strategies to overcome the identified challenges covering not only the study design but also the use of new innovative technologies as well as the importance of patients’ cohorts and collaborative research initiatives.
1.2.2. GRN
More than 70 pathogenic mutations of GRN have been described, mostly resulting in loss of function either due to aberrant transcription or to prevention of translation, which leads to GRN haploinsufficiency in FTD patients. 40 GRN mutations likely affect lysosomal function in a disease‐promoting manner. 47 GRN carriers often present with FTLD‐TDP A pathology and most patients have bvFTD clinical phenotype, though PPA has been also reported in these familial cases. 34
1.2.3. MAPT
Mutations in the MAPT gene (with ≥ 40 pathogenic mutations) lead to abnormal forms of the tau protein, promoting its aggregation and interfering with the polymerization and stabilization of microtubules. 48 These mutations are often associated with PSP and CBD pathology. 18 Genotype‐phenotype correlations showed that MAPT carriers are associated largely with bvFTD, PPA, and FTD with parkinsonism. 34 ,
The clinical, pathological, and genetic complexity of FTD requires the development of biomarkers for the molecular subtypes of FTD. Such biomarkers could be used to increase diagnostic accuracy; quantitate disease staging; and predict, monitor, and measure disease progression. 49 Depending on the context of use, general biomarkers of FTD or biomarkers for specific FTLD types and subtypes will be needed. Pathology‐specific biomarkers are especially relevant to define FTD biologically. Such biomarkers are urgently needed for recruitment for clinical trials, where homogeneous populations of patients with specific FTD pathological subtypes are crucial for testing candidate drugs that target these specific proteinopathies (e.g., tau or TDP). 50 In clinical trials, biomarkers are also needed to assess target engagement and evaluate drug response. Such markers are very important for FTD treatment development, as the heterogeneity of the clinical presentation prevents the use of clinical or imaging indices as outcome measures. 51 Biofluid‐based biomarkers (e.g., cerebrospinal fluid [CSF] and blood) can be especially helpful in clinical practice and clinical trials. 49 , 50 , 51 CSF has been the most widely used source for the development of fluid biomarkers for neurological disorders because it is viewed as reflecting ante mortem biochemical milieu and its changes during the neurodegenerative process, and concentrations of brain pathology‐specific proteins are expected to be higher than those measured in blood. 52 In AD, the implementation of CSF biomarkers in clinical practice has shown enormous benefits for early diagnosis and the testing of new compounds in AD clinical trials. 27 Lately, highly sensitive and specific immuno‐ and mass spectrometry‐based approaches have made blood‐based biomarkers feasible—a revolutionary development for AD research. 53 , 54 Compared to developments in the AD field, there has been significant progress in FTD biofluid biomarker research in the last 10 years, but this has also encountered difficulties that relate to the relative recency of the key neuropathology discoveries and to the clinical and pathologic heterogeneity of the syndromes. For instance, C9orf72 genetic expansions and the resulting pathological hallmarks were only revealed a decade ago 35 , 41 , 42 , 43 , 44 and pathological, genetic, and clinical associations have been expanded. 19 , 24 , 27 , 29 , 30 , 45 , 46 , 47 Leveraging these recent and upcoming discoveries warrants further developments within the FTD biomarker field.
Here, we review the key biofluid‐based biomarker developments in FTD (Figure 1 and Table 1) and identify the major opportunities and obstacles for the development and implementation in practice and research of biofluid‐based biomarkers for FTD. We discuss potential strategies to overcome the obstacles, taking into account the contexts of use, the different methods and technologies available, and the present‐day clinical/epidemiological cohorts and collaborative research initiatives.
TABLE 1.
Most widely studied biomarkers studies within the FTD field and its context‐of‐use [Correction added on 17 March 2022, after first online publication: The alignment of table column heads in Table 1 has been corrected.]
| Context‐of‐use | Sample matrix | Remarks | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Specific FTD diagnosis | |||||||||
| Biofluid based biomarker | vs. CON | vs. AD | vs. Psych.dis | Clinical/genetic/pathological subtypes | Staging and monitoring | Prognosis | Treatment Response | ||
| Tau, pTau (281/217/231), Aβ42, Aβ40, and their ratios | – | ++ | ns | + a | ns | ns | ns | CSF/plasma | AD biomarker; co‐pathology dependent |
| pTau181/tTau | – | – | + | + b | ns | ns | ns | CSF | AD biomarkers; limited accuracy |
| NfL | ++ | + | ++ | + | ++ | ++ | + | CSF/plasma/serum | General biomarker of neurodegeneration |
| sAPPβ | + | – | ns | ns | ns | ns | ns | CSF | Specifically decreased in FTD |
| Progranulin | ns | ns | ns | ++ c | ns | ns | + | CSF/plasma/serum | Specific genetic subtype |
| Poly(GP) | ns | ns | ns | ++ d | – | – | + | CSF | Specific genetic subtype |
Abbreviations; ++, validated biomarker (i.e., changes consistently observed across independent studies); +, promising biomarker; –, not useful; Aβ, amyloid beta; AD, Alzheimer's disease; CON, controls; CSF, cerebrospinal fluid; FTD, frontotemporal dementia; ns, not studied; Psych.dis., psychiatry disorders.
Note: Summary table of the most widely studied biomarkers within the FTD field and their corresponding context‐of‐use.
a Logopenic FTD variant.
b For TDP vs. tau.
c For GRN mutation carriers.
d For C9orf72 mutation carriers.
2. STATE OF THE ART OF BIOFLUID‐BASED BIOMARKERS FOR FTD
2.1. Biomarkers to differentiate FTD from other dementias and non‐degenerative disorders
An important challenge for both routine diagnosis and clinical trials is to identify cases within the FTD spectrum and differentiate these from those of related diseases, such as sporadic AD 55 or the non‐neurodegenerative primary psychiatric disorders. 56 In many AD studies of tau and amyloid beta (Aβ) fluid biomarkers, FTD has been included as a contrast group. It has been shown that elevated CSF concentrations of total tau (t‐tau) and tau phosphorylated at amino acids 181 and, more recently, 217 (p‐tau181 and p‐tau217, respectively) are surprisingly AD‐specific; tau‐associated FTD subtypes do not have elevated CSF t‐tau and p‐tau concentrations, 57 , 58 , 59 , 60 at least not to the levels present in AD. 61 Accordingly, an increased ratio of t‐tau or p‐tau to the 42 amino acid form of Aβ (Aβ42), i.e., the tau/Aβ42 ratio, is an AD‐specific finding that separates AD from FTD with high diagnostic accuracies (70% and 86% specificity when analyzing AD vs. bvFTD or svPPA, respectively). 62 Thus, these biomarkers can be used to identify patients who have frontal lobe dysfunction on the basis of AD pathology, rather than FTD, a condition sometimes called frontal variant AD. 63 Similarly, CSF biomarkers can be useful in the evaluation of the logopenic variant of PPA, which is typically associated with AD pathology, rather than FTD. 64 In this scenario, the association of elevated CSF tau levels and reduced Aβ42/40 ratio, supports AD pathology as the main etiology of the language syndrome. 64 It has also been repeatedly shown that Aβ species including Aβ38, Aβ40, Aβ42, and soluble amyloid precursor protein fragment (sAPPβ) are lower in CSF from FTD patients than in the compared controls. 65 , 66 The reason for this remains unclear, but in AD, cerebral Aβ pathology is associated with a relatively selective reduction in Aβ42 in CSF, while the reduction in FTD is seen for all measurable Aβ species and the decrease correlates with brain atrophy; 66 hence, the CSF Aβ42/Aβ40 ratio works well to differentiate amyloid pathology in AD (with reduced Aβ42/40 ratio) from the general reduction of APP‐derived peptides often seen in FTD (with Aβ42/40 ratio in the normal range, area under the curve [AUC]: 0.85). 65 Of note, many biomarker studies in FTD may be confounded by co‐occurring secondary AD pathology. 67 , 68 Therefore, when a patient with an FTD syndrome shows a positive AD CSF biomarker profile it is important to consider all clinical and radiological information to evaluate if the AD pathology is the primary pathology or a comorbid condition. Besides the classical AD CSF biomarkers, several studies have found much higher CSF neurofilament light (NfL, a general biomarker for neurodegeneration) levels in FTD compared to AD; 69 , 70 , 71 , 72 this finding, particularly in combination with negative AD biomarkers, strongly speaks for a non‐AD neurodegenerative disease. For instance, combination of AD CSF biomarkers (Aβ42 and p‐tau181) with CSF NfL could discriminate FTLD from AD with high performance (AUC > 0.90). 73 However, FTD should only be considered in the presence of a compatible clinical syndrome, as CSF NfL levels are increased in multiple related neurodegenerative disorders (e.g., ALS, PSP, multiple system). 74 For the differentiation between FTD and non‐neurodegenerative disorders, several CSF studies showed an excellent performance of NfL for discriminating FTD from psychiatric disorders (AUC > 0.90) 56 , 71 , 75 and consequent perceived added clinical use for the physician. 76
It is to be noted that the studies described above analyze mainly the performance of classical AD CSF biomarkers and thus are useful to exclude AD pathology rather than to diagnose FTD; or analyze general markers of neurodegeneration (e.g., NfL). These observations highlight the lack of well‐established FTD‐specific biomarkers. Promising biomarkers have been identified and are being investigated. 77 , 78 For example, a study combining CSF NfL, YKL40, and sAPPβ measurements from 159 cases with an FTLD‐related syndrome yielded high diagnostic accuracies (AUC > 0.90) in discriminating FTD from controls. 79 While YKL40 is also elevated in other neurodegenerative disorders including AD, 80 , 81 sAPPβ concentrations were specifically lower in FTLD syndromes. 79 Similar results were observed in an autopsy‐confirmed FTLD cohort. 82 Other pathways that are currently being examined in the quest for FTD‐specific biomarkers include neuroinflammation, lysosomal health, and synaptic health (for a recent review on the topic, please see Swift et al. 77 ). Combinations of CSF biomarkers (e.g., NfL, YKL40, p‐/t‐tau) do not seem to have added value over NfL alone, at least not for the discrimination of FTD from primary psychiatric disorders. 56
Regarding blood‐based biomarkers, recent AD studies showed that plasma concentrations of p‐tau181, p‐tau217 and p‐tau231 are elevated in AD, relative to controls, but not in FTD; 83 , 84 , 85 , 86 the two diseases can thus be separated with close to 100% accuracy using a simple blood test. These promising results were recently confirmed for plasma p‐tau181 and p‐tau231 in neuropathologically confirmed cohorts. 84 , 86 Similar to what was observed in CSF, serum and plasma NfL are elevated in FTD compared to AD, but with limited utility in discriminating FTD from AD. 87 , 88 , 89 At present, serum NfL has the highest diagnostic accuracy with AUC of up to 0.94 for differentiating FTD from primary psychiatric disorders. 71 , 90 , 91 However, normal NfL levels still do not rule out FTD. Because NfL levels are also associated with disease progression and survival, 85 , 92 normal NfL levels are probably associated with slowly progressive phenotypes of FTD.
2.2. Biomarkers to define the underlying proteinopathies
Biomarkers for specific FTD‐related proteinopathies (TDP‐43, tau, or FUS 14 , 15 ) are a pressing need for drug development strategies that focus on specific pathological processes. Several independent studies have shown that CSF p‐tau/t‐tau ratio demarcate FTD‐TDP patients from FTD‐tau cases with sensitivity and specificity values around 82% and 62%, respectively. 93 , 94 , 95 , 96 , 97 Other studies have shown that the value of CSF tau measurements is limited to pure cases of FTLD (e.g., without copathologies). 61 , 67 Lower CSF p‐tau could discriminate FTD‐TDP from FTD‐tau in sporadic pure FTD cases after excluding AD. 67 As CSF t‐tau levels are not strongly associated with underlying FTLD‐tau, it would be worthwhile to study CSF pathological tau isoforms in more detail. 98
While it is possible to measure TDP‐43 in biofluids, current assays do not differentiate normal from pathological TDP‐43. 99 A recent study using real time quaking‐induced conversion assay, a test based on the amplification of misfolded proteins and used for detection of prion proteins, showed in vitro TDP‐43 aggregate formation in CSF. 100 In a recent proof‐of‐concept small clinical study this assay discriminated FTD and ALS patients from controls with 94% sensitivity and 85% specificity. 100 These studies await independent replication. However, the seed‐based testing paradigm may help to develop additional assays for inclusion‐specific forms of TDP‐43. Biomarkers that reflect metabolic changes downstream of TDP‐43 pathology might also be developed.
As far as we are aware, there is no biomarker of FUS pathology available. Ideally, a combined panel measuring markers of tau, TDP‐43, and FUS would allow for accurate pathologic diagnosis of FTD, but these have so far posed important challenges. Some studies have analyzed well‐established biomarkers that are not directly related to the specific proteinopathy. For example, higher levels of both CSF and plasma NfL have been observed in FTD‐TDP than in FTD‐tau cases, but with high within‐group variability. 87 , 101 , 102 Non‐hypothesis driven proteomics studies have identified multiple proteins that are differentially expressed in FTLD neuropathological or genetic subtypes. 95 , 103 , 104 These unbiased approaches have the potential to facilitate the identification of biomarkers specific for the main pathophysiological processes associated with the FTD subtypes (e.g., lysosomal degradation, autophagy). Replication and validation of these findings are of utmost importance to move the field forward.
2.3. Biomarkers for target engagement and treatment monitoring
Low CSF and blood progranulin levels have been found in GRN mutation carriers with an almost 100% diagnostic accuracy. 105 , 106 , 107 It is expected that disease‐modifying treatments aimed at restoring progranulin deficits can be evaluated by these serum GRN measurements. In C9orf72 expansion carriers, poly(GP), one of the DPR proteins produced by the C9orf72 expansions, is elevated as early as the presymptomatic phase. 108 , 109 , 110 Thus, DPR proteins may prove useful as pharmacodynamic biomarkers in gene‐silencing studies. While blood NfL has promise for measuring treatment effects in different neurodegenerative disorders, 51 , 92 , 111 , 112 its utility as an endpoint measure in GRN carriers appears to be limited by the observed longitudinal fluctuations. 92
2.4. Biomarkers for disease staging and prognosis
As observed in other dementias, accelerated rates of decline have been quantified during the symptomatic phase of both familial and sporadic FTD using clinical and imaging measures 113 and thus, markers reflecting the disease stage independent of the clinicopathological presentation would be desirable. Several studies have already indicated that baseline NfL concentrations in both CSF and blood could reflect the aggressiveness of the disease as it can predict rate of disease progression in the different clinicopathological phenotypes in both sporadic and familial FTD. 87 , 96 , 114 , 115 , 116 , 117 Future studies will clarify whether baseline CSF NfL levels can be used to stratify FTD severity (e.g., low, intermediate, high). Longitudinal analysis of CSF NfL shows that the levels of this marker change little during the symptomatic phase of FTD in individual patients. 114 However, a recent longitudinal multicenter study of familial FTD has shown in symptomatic GRN mutations carriers a rise in serum NfL levels as illness duration time and severity increased, notable from symptom onset on but with substantial fluctuations in the individual NfL trajectories. 92 Importantly, the study also showed that serum NfL levels increase in presymptomatic carriers 1 to 2 years before symptom onset. The study was limited by the number of FTD mutation carriers undergoing conversion to symptomatic phase (n = 9); nevertheless, it suggests that baseline serum NfL levels may discriminate those in the transition from normal to symptomatic status (i.e., converters) from those that are not in transition (i.e., non‐converters) with good accuracy, 92 indicating a potential utility of serum NfL as a prognostic marker and a selection criterion for clinical trials, at least in familial cases. Recent findings observed that higher levels of plasma glial fibrillary acidic protein (GFAP), an astrogliosis marker, was associated with a greater decrease in Mini‐Mental State Examination score and a poor cognitive outcome in both FTD and AD. Thus, in combination with FTD‐specific markers, plasma GFAP could also be potentially used to track disease severity or predict greater cognitive decline. 118
3. GAPS AND LIMITATIONS ON THE DEVELOPMENT OF BIOFLUID‐BASED BIOMARKERS FOR FTD
Taken together, except for NfL, the most robust FTD biofluid‐based biomarkers developed from the scientific efforts of the last decade are related to AD pathology rather than that of FTD. The high pathologic heterogeneity of FTD is a particular challenge; the current biomarkers are not specific for any pathologic subtype, and biomarkers may have different utilities depending on the FTD subtype.
3.1. Challenges in the development of biomarkers for specific diagnosis of FTD and its pathological subtypes
Given the high heterogeneity of FTD, it is not to be expected that a single FTD‐specific biofluid marker will be used to differentiate FTD from other neurodegenerative or non‐neurodegenerative disorders. Inclusion of non‐FTD dementias (e.g., AD) in biomarker studies will facilitate the distinction of FTD‐specific changes from those that are common to all neurodegenerative states. The fact that concomitant AD is not uncommon in FTD adds further complexity. 68 It will be important, therefore, to evaluate the effects of concomitant AD pathology in FTD biomarker studies, using standard AD CSF (and more recently also plasma) biomarkers. 67 , 68 , 83
The identification of biofluid‐based biomarkers that discriminate FTD pathologies (TDP‐43, tau, FUS), has often been hindered by the availability of pathologically characterized samples with known underlying pathology—these are usually selected based on autopsy confirmation or genetic mutation status (e.g., C9orf72 and GRN for TDP‐43 or MAPT for tau 18 , 22 , 35 , 37 ). The scarcity of these samples has prompted efforts to enrich cohorts with FTD syndromes that are highly correlated with a specific underlying neuropathology (e.g., FTD‐ALS for FTLD‐TDP 22 or PSP and CBS for FTLD‐tau 119 , 120 ) and/or to jointly analyze data from both combined cohorts of sporadic and familial cases with the same neuropathology background. It must be noted, however, that recent neuropathological studies have reported TDP‐43 copathology in a considerable amount of FTLD‐tau–related syndromes such as CBD, and to lesser extent PSP. 121 In addition, it is not clear to what extent the biochemical changes in those cases with FTD mutations resemble those of sporadic cases with the same proteinopathy, nor across the different immunohistochemical profiles within each pathological subtype (e.g., tau: 3R, 4R, 3R/4R; TDP: A, B, C, D, and E). Pathophysiological differences between hereditary and sporadic FTD have been reported. 122 , 123 Recent studies have shown that the performance of CSF p‐tau181 to discriminate between FTD pathological subtypes (i.e., FTLD‐tau vs. FTLD‐TDP) was considerably reduced when mutation carriers were analyzed separately (AUCs of 0.87 in sporadic pure cases vs. 0.58 in genetic cases). 61 , 67 Similarly, the levels of plasma p‐tau181 were approximately 2‐fold increased in MAPT mutation carriers with mixed 3/4R but not in those with 4R tau pathology. 83 Recent findings have shown that the levels of plasma GFAP are increased in symptomatic GRN carriers but not in those with C9orf72 expansions or MAPT mutations, and could distinguish these genetic forms with AUCs >0.70. 124 Furthermore, findings from molecular studies indicate that the mechanisms underlying TDP‐43 aggregation by C9orf72 expansions differ from those driven by GRN loss of function. 125 , 126 Taken together, these observations indicate a high heterogeneity also within each pathological and genetic subtype, in which different biological processes underlying familial and sporadic cases may ultimately result in similar proteinopathy profiles—but not necessarily in similar biomarker profiles. In other words, within a proteinopathy class different mechanisms may generate different biochemical profiles in body fluids, especially in the early stages of the disease. The biological complexity of FTLD subtypes needs to be considered in the design and interpretation of FTD biomarker studies.
3.2. Challenges in the development of biomarkers for disease stage, prognosis, and drug efficacy
Biofluid‐based biomarkers for the different stages of FTD, including the preclinical and prodromal phases, are still needed. This is especially challenging in FTD as, unlike in AD, there is still not an established definition of the preclinical/prodromal stages (e.g., mild cognitive impairment due to FTD). Diagnosis is typically delayed for up to 6 years. 100 Moreover, the first and core symptom of FTD is a deficit in social cognition, which is not readily recognized as such until the illness is well established. The construct of social cognition, including emotion recognition, theory of mind, empathy, and moral reasoning, has been proven difficult to measure in clinical practice. 71 Use of newly developed psychometric measures for characterizing all stages of symptoms (e.g., FTLD‐Clinical Dementia Rating, Multidomain Impairment Rating) in familial FTD research will facilitate the discovery and validation of early FTD biomarkers. 51 , 127 As highlighted above, the identification of early diagnostic markers will likely be more effective when accounting for the pathological heterogeneity of the disease.
A major challenge in the field of neurodegenerative dementias is to predict whether and when an individual will develop the specific disease, an important question as early application is expected to increase the value and efficacy of treatments aiming to prevent or delay disease progression. Because disease progression in most neurodegenerative diseases is non‐linear, longitudinal studies characterizing the behavior of a biomarker over time should include at least three time points. 128 In this respect, blood‐based biomarkers are more suitable than CSF biomarkers. Considering the lack of preclinical/prodromal markers for sporadic FTD, studies of familial FTD are crucial for such purposes. The utility of serum NfL as prognostic marker should be replicated in larger cohorts, 92 analyzing also the potential influence of the different FTD genetic backgrounds. It is also essential to understand whether findings from familial FTD research can be generalized to sporadic FTD. These data also indicate that biochemical changes can be detected in blood before the appearance of clinical symptoms, and it thus boosts the search for additional biomarker candidates. Despite the need for more studies, it should be noted that the presymptomatic phase of FTD, at least in familial cases, might be shorter than that observed in familial forms of AD; as in the latter, NfL changes were observed already 10 years before symptom onset. 129 Considering that serum NfL is also a general marker of neurodegeneration with similar NfL changes in presymptomatic familial AD cases, 129 the quest for predictive biomarkers specific for FTD especially in sporadic cases is warranted.
To date, there are no established biomarkers to measure target engagement or drug efficacy in FTD clinical trials, especially for sporadic cases. The increasing number of trials in the industry pipeline accentuates the need to develop markers for different contexts of use that account for the clinical and pathologic diversity of FTD. 51
4. FILLING THE GAPS ON THE DEVELOPMENT OF BIOFLUID‐BASED BIOMARKERS FOR FTD
4.1. Emerging technologies and approaches
The identification of novel biomarkers will be facilitated by leveraging a variety of technologies. Mass spectrometry (MS) has the advantage of an unbiased screening, which has facilitated the identification of novel biomarkers for FTLD subtypes. 103 , 104 Novel proteins identified through MS approaches can then be measured as single analytes using immunoassays or incorporated into panels of protein arrays for further validation studies. 95 , 103 , 104 , 130 , 131 Conventional unbiased MS methods are, unfortunately, still not optimal for blood proteomic analysis. This limitation is partially overcome by novel and highly sensitive high‐throughput protein arrays, such as aptamer‐based technologies, immunobased proximity extension assays, or antibody suspension beads arrays, which can readily measure large sets of protein libraries in CSF and blood samples. 132 , 133 , 134 , 135 , 136 , 137 , 138 An additional advantage of such protein arrays is that the reagents used for biomarker discovery with specific protein binders (e.g., antibodies) can in principle be also used for validation, and for translation to the clinical setting in which immunoassays are a cornerstone of clinical chemistry analyses. 52 , 139 The different proteome profiling technologies currently available can be useful to cross‐validate findings, an essential step toward the development of optimal biomarkers. It is, however, expected that these technologies will also complement each other as protein libraries partially differ, and the distinct platforms can detect the proteins in different conformation/states (denatured in MS vs. native in protein arrays).
It is also conceivable that the search should be continued in alternative matrices or alternative targets, such as RNA in platelets or extracellular vesicles. Blood platelet mRNA analysis has shown potential for multiple sclerosis. 140 Panels (proteins, nRNA, metabolites) may be needed to capture the full complexity of the pathological differences among the FTD subtypes, and within them, and thus multiplex technologies and computer assisted algorithms need to be used. These could be extended or incorporated into multimodal approaches that include not only biofluid‐based biomarkers (e.g., CSF and blood), but also other types of measures (e.g., imaging results, genetic variation, neuropsychological findings, etc.), which can be especially helpful for both clinical practice and trials. 49 , 51
4.2. Collaborative initiatives
The identification of biofluid‐based biomarkers for FTD pathologies will require the analysis of data and samples from large and well‐characterized cohorts with longitudinal and autopsy‐confirmed cases. Study design and analyses should account for the potential influence of specific mutations or pathological subclassifications, especially within the earliest stages of the disease. This would also involve more systematic post mortem research in FTD, together with deep phenotyping. Considering the limited availability of highly characterized samples, such studies will only be successful when multiple centers are involved. The extension of existing cohorts and development of new ones will be extremely valuable and, therefore, there is a need to reinforce the interest and engagement of the advocacy community, patients and families, and the public in FTD research.
Several initiatives have facilitated collaborative studies worldwide, including the Genetic Frontotemporal Dementia Initiative (GENFI), the ARTFL‐LEFFTDS Longitudinal Frontotemporal Lobar Degeneration (ALLFTD), and their coming together in the worldwide FTD Prevention Initiative (FPI). These consortia are continuously gathering invaluable information from multiple FTD cohorts worldwide and are the reference for many of the FTD biomarker studies performed to date, especially in familial FTD. The FTD professional interest area (FTD‐PIA) has been recently created within the Alzheimer's Association International Society to Advance Alzheimer's Research and Treatment (ISTAART), which is collaborating with the biofluid‐based biomarkers PIA (BBB‐PIA) to facilitate the development and optimization of biomarkers for FTD.
For validation and implementation of the identified markers, the field can leverage the developments from more advanced fields, such as the AD field. This especially applies to preanalytical aspects and the development of standard operating procedures for sample handling and analysis, such as for AD biomarkers in CSF and blood, 141 — 144 and to quality control programs, 145 which are a requirement for in vitro diagnostic tests. These activities are promoted through different organizations such as the Society for CSF Analysis and Clinical Neurochemistry or the Global Biomarkers Standardization Consortium (GBSC). Moreover, there is a potential for synergism from initiatives to obtain regulatory approval and reimbursement for, for example, NfL for other diseases.
4.3. Understanding the cautions and caveats in the development of FTD biofluid‐based biomarkers
It is valuable to specify some of the potential pitfalls and shortcomings when biofluid‐based biomarkers are translated from the level of discovery and research to that of clinical dogma. Many of these points will be well known to the experts but perhaps less so to others not familiar with biomarker‐based diagnosis and staging of neurodegenerative diseases. Reflection on the state‐of‐the‐art reveals both “known unknowns” and “unknown unknowns.” Examples of some of the known unknowns are the potential contributions of vascular and immune mechanisms as we contemplate the full picture from disease initiation to disease progression. Vascular and immunological mechanisms have been proposed for many neurodegenerative diseases and, for some, have been demonstrated to play key roles in the development and progression of clinical syndromes. Even when, for example, vascular and/or immune factors have been implicated, there is insufficient knowledge about which biomarkers can be combined to give a more complete picture. For vascular and immunological abnormalities, reliable biomarkers are infrequent, imprecise, or lacking altogether. Besides the imprecision that this brings to nosology, the complexity of multiple concurrent pathologies will certainly hamper the ability to test interventions that are aimed at only one component of the concurrent pathologies. In other words, the more complex the concurrent pathologies, the greater the challenge in designing trials that are sufficiently sensitive to generate interpretable clinical or biomarker endpoints. It is also worth noting that age‐, sex‐, race‐, and ethnicity‐based variations; the frequency of comorbidities; and the effects of genetic diversity must always be kept in mind, as observed already in AD and ALS, 146 , 147 , 148 , 149 , 150 , 151 , 152 , 153 , 154 , 155 , 156 , 157 but also within the FTD field. 158 , 159 , 160 Indeed, it is far more prudent to assume that these parameters are more likely to be important than to dismiss them.
The unknown category is illustrated by the proliferation of newly recognized clinicopathological entities. For example, primary aging‐related tauopathy (PART), limbic age‐related TDP‐43 encephalopathy (LATE), and hippocampal sclerosis may be associated with amnestic syndromes that are only slowly progressive and may be associated clinically with a “chronic mild cognitive impairment.” Slow progression syndromes have the potential to confound the interpretation of biomarker characterizations and clinical trial outcomes when they are misclassified in studies as examples of typical cases. It is therefore important to acquire the genetic, biofluid, and neuroimaging panels that will facilitate the characterization and classification of atypical proteinopathies and the syndromes with which they are associated. While biomarker‐based diagnosis has made a quantum leap forward in the past decade, there remain important caveats as they are translated from the research setting into more general application in clinics worldwide.
5. CONCLUSIONS AND FUTURE DIRECTIONS
In current clinical practice, the place of CSF analysis is to exclude AD pathology and verify neurodegeneration—it does not have utility for affirmative diagnosis of FTD. 5 The blood‐based biomarker NfL is now being incorporated in clinical practice for FTD, especially to differentiate bvFTD from primary psychiatric disorders. 71 , 76 Several novel biomarker candidates reflecting different pathological processes associated with FTD beyond the classical proteinopathies have been identified (lysosomal markers, inflammatory markers, circulating nucleic acids, synaptic markers), 77 and are in need of thorough validation in well characterized independent cohorts of familial and sporadic FTD. There is a need to identify and validate biomarkers for different purposes (differential diagnosis, pathology typing, staging, prognostication, treatment monitoring) using robust approaches that allow their incorporation into clinical diagnostic criteria. It is expected that the technical developments of the last years (e.g., ultrasensitive technologies, large high‐throughput protein panels) will facilitate such developments. CSF biomarker research is still essential to support the association of the different markers with pathophysiological processes, but translation into blood‐based biomarkers would be ideal for repeated measurement in longitudinal studies and broad use, eventually, in practice. The high complexity and heterogeneity of FTD discourages the use of small sample sizes in biomarker analyses and calls for collaborative large multicohort studies with thoroughly characterized samples (i.e., with carefully ascertained and measured clinical, genetic, and pathological indices). A fuller understanding of the molecular factors and biomarkers that underlie FTD pathophysiology at different stages of the disease will optimize clinical and molecular diagnosis and provide an invaluable resource for the development and testing of novel therapies.
CONFLICTS OF INTEREST
Support received for the submitted work: None of the authors received support for the present manuscript. MC is supported by the attraction talent fellowship from Comunidad de Madrid and the ‘Europe Research’ 2020 dynamization actions from Research and innovation Spanish government, payment made to institution. SG has a joint appointment at Icahn School of Medicine (1 FTE) and James J. Peters VA Medical Center (5/8th) which constitutes 100% of his total professional responsibilities (TPE). The combined TPE at both institutions does not exceed 12 person months (PM). Dr. Gandy's joint appointment is documented under an MOU between Mount Sinai and the VA. There is no dual compensation for the same work, nor is there an actual or apparent conflict of interest regarding such work. Institution: Icahn School of Medicine at Mount Sinai Type of Appointment: Academic (Professor) Full Time: 1.00 FTE Active support R01AG067312 (MPI: Huffman (contact), Tu) NIH/NIA/Albert Einstein College of Medicine; Co‐I P30AG066514 (PI: Sano) NIH/NIA TC Mount Sinai Alzheimer's Disease Research Center; Associate Director RF1AG062661 (PI: Salton, Ehrlich) NIH/NIA; Co‐I RF1AG058469 (MPI: Ehrlich (contact), Gandy) NIH/NIA; MPI U01AG046170 (MPI: Zhang (contact), Gandy, Ehrlich, Haroutunian NIH/NIA; MPI R01AG061894 (MPI: Gandy, Noggle (contact), Fossati) NIH/NIA/New York; MPI RF1AG059319 (MPI: Gandy (contact), Ehrlich) NIH‐NIA; MPI R03AG070506 (PI: Wang) NIH/NIA; Co‐I R01AG057907 (MPI: Zhang, Haroutunian, Ehrlich) NIH/NIA; Co‐I R21 AG063068 (PI: Readhead) NIH/NIA/Arizona State University; Role: Subaward Co‐I R6AG066431 (MPI: Huffman, Gandy) NIH/NIA/Albert Einstein College of Medicine; MPI (PI: Gandy) Alzheimer Drug Discovery Foundation; PI Pending (JIT Award under consideration) R03AG070710 (PI: Haure‐Mirande) NIH NIA; Co‐I R21 (PI: Vaska) NIH/Stony Brook; Subaward PI Lab Personnel Paid Directly by Third Party Entities: None Overlap: There is no scientific or budgetary overlap Institution: James J. Peters VA Medical Center Type of Appointment: 5/8th appt Active I01RX002333 (Gandy Role PI) Department of Veterans Affairs. Pending: None Lab Personnel Paid Directly by Third Party Entities: None Overlap: There is no scientific or budgetary overlap. CO received research grants from Biogen and Alector; payments were made to the institution. FO received support from FAPESP ‐ The State of São Paulo Research Foundation, grant #2015/10109‐5, payment made to FO. CU has nothing to disclose. AL reports payment to the institution from Health Department of the Government of Catalonia (grant PERIS SLT002/16/00408), Fondo de Investigaciones Sanitario (FIS), Instituto de Salud Carlos III (PI17/01896; AC/0013), CIBERNED (COEN), BBVA foundation. CET's research is supported by the European Commission (Marie Curie International Training Network, grant agreement No 860197 (MIRIADE), and for bPRIDE (JPND), Health Holland, the Dutch Research Council (ZonMW), Alzheimer Drug Discovery Foundation, The Selfridges Group Foundation, Alzheimer Netherlands, Alzheimer Association. CT is recipient of ABOARD, which is a public‐private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). More than 30 partners participate in ABOARD. ABOARD also receives funding from Edwin Bouw Fonds and Gieskes‐Strijbisfonds. IV is appointed on a research grant by Alzheimer Nederland (NL‐17004). CET has a collaboration contract with ADx Neurosciences, Quanterix, and Eli Lilly; performed contract research or received grants from AC‐Immune, Axon Neurosciences, Biogen, Brainstorm Therapeutics, Celgene, EIP Pharma, Eisai, PeopleBio, Roche, Toyama, Vivoryon. YP has nothing to disclose. All authors are members of the Biofluid Based Biomarkers or Frontotemporal Dementia and Related Disorders Professional Interest Areas (ISTAART). MC has given a lecture at the annual meeting of Spanish Neurology Society, payment made to MC. HZ has served on scientific advisory boards for Alector, Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies, and CogRx. HZ has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, and Biogen. HZ is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. All payments made to HZ. HZ is a chair of the Alzheimer's Association Global Biomarker Standardization Consortium and the AA Biofluid Based Biomarkers Professional Interest Area. No payments made. SG has provided expert testimony to Vernick & Associates, Annapolis, MD, and Post & Schell, Philadelphia, PA. SG has a patent issued method for iPSC derived basal forebrain cholinergic neurons (issued). SG has served as advisor for Ritrova Therapeutics. CO provides technical or medicolegal advice to Alector, Goodell Devries Leech & Dann. Payment made to CO. CO is part of the medical Advisory Council of the Association for Frontotemporal Degeneration and the Scientific Advisory Board of the FTD Disorders Registry. No payments involved. FO has received consulting fees for acting as a healthcare council member for Gerson Lehrman Group, Atheneum Partners, Guidepoint, Health Advances, and Lionbridge. FO received support from the International Parkinson and Movement Disorder Society to attend the 22nd International Congress of Parkinson's Disease and Movement Disorders in Hong Kong, October 2018. From 2018 to 2021, FO was a member of the ISTAART Advisory Council (unpaid). Starting in 2021, FO is a current member of the International Subcommittee of the American Academy of Neurology (unpaid) and of the Awards Committee of the International Parkinson and Movement Disorder Society (unpaid). FO has also been a member of the Community of Experts of the European Science Foundation since 2019 (and receives payments for each review of a grant). CU is an executive Committee member of Biofluid Based Biomarkers Professional Interest Area (ISTAART) (unpaid) and board of Trustees member of the British Society for neuroendocrinology (unpaid). AL has a patent licensed of synaptic markers in neurodegenerative diseases, payments to the institution and to AL. AL received personal fees for serving on scientific advisory boards from Fujirebio‐Europe, Nutricia, Biogen, and Roche Diagnostics. AL received personal fees for lectures from Nutricia and Zambon. CET has given lectures for Roche and serves on editorial boards of Medidact Neurologie/Springer, Alzheimer Research and Therapy, Neurology: Neuroimmunology & Neuroinflammation, and is editor of a Neuromethods book from Springer.
ACKNOWLEDGMENTS
This manuscript was facilitated by the Alzheimer's Association International Society to Advance Alzheimer's Research and Treatment (ISTAART), through the Biofluid Based Biomarkers and Frontotemporal Dementia and Related Disorders professional interest areas (PIAs). The views and opinions expressed by the authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer's Association. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C and #ADSF‐21‐831377‐C), the Olav Thon Foundation, the Erling‐Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019‐0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. CUO is supported by The Robert and Nancy Hall Brain Research Fund.
del Campo M, Zetterberg H, Gandy S, et al. New developments of biofluid‐based biomarkers for routine diagnosis and disease trajectories in frontotemporal dementia. Alzheimer's Dement. 2022;18:2292–2307. 10.1002/alz.12643
[Correction added on 17 March 2022, after first online publication: The first author's name has been corrected from “Martha del Campo” to “Marta del Campo”.]
REFERENCES
- 1. Onyike CU, Diehl‐Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25:130‐137. 10.3109/09540261.2013.776523. (Abingdon, England). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kansal K, Mareddy M, Sloane KL, et al. Survival in frontotemporal dementia phenotypes: a meta‐analysis. Dement Geriatr Cogn Disord. 2016;41:109‐122. 10.1159/000443205 [DOI] [PubMed] [Google Scholar]
- 3. Kandiah N, Wang V, Lin X, et al. Cost related to dementia in the young and the impact of etiological subtype on cost. J Alzheimer's Dis. 2015;49:277‐285. 10.3233/JAD-150471 [DOI] [PubMed] [Google Scholar]
- 4. Galvin JE, Howard DH, Denny SS, Dickinson S, Tatton N. The social and economic burden of frontotemporal degeneration. Neurology. 2017;89:2049‐2056. 10.1212/WNL.0000000000004614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456‐2477. 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006‐1014. 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131‐146. 10.1080/17482960802654364 [DOI] [PubMed] [Google Scholar]
- 8. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496‐503. 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP International Workshop. Neurology. 1996;47:1‐9. 10.1212/WNL.47.1.1 [DOI] [PubMed] [Google Scholar]
- 10. Harris JM, Gall C, Thompson JC, et al. Sensitivity and specificity of FTDC criteria for behavioral variant frontotemporal dementia. Neurology. 2013;80:1881‐1887. 10.1212/WNL.0b013e318292a342 [DOI] [PubMed] [Google Scholar]
- 11. Balasa M, Gelpi E, Martín I, et al. Diagnostic accuracy of behavioral variant frontotemporal dementia consortium criteria (FTDC) in a clinicopathological cohort. Neuropathol Appl Neurobiol. 2015;41:882‐892. 10.1111/nan.12194 [DOI] [PubMed] [Google Scholar]
- 12. Lanata SC, Miller BL. The behavioural variant frontotemporal dementia (bvFTD) syndrome in psychiatry. J Neurol Neurosurg Psychiatry. 2016;87:501‐511. 10.1136/jnnp-2015-310697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woolley JD, Khan BK, Murthy NK, Miller BL, Rankin KP. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease. J Clin Psychiatry. 2011;72:126‐133. 10.4088/JCP.10m06382oli [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mackenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1‐4. 10.1007/s00401-009-0612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 2007;114:5‐22. 10.1007/s00401-007-0237-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, MacKenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922‐2931. 10.1093/BRAIN/AWP214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD‐TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134:65‐78. 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Irwin DJ, Cairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2014. 10.1007/s00401-014-1380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forrest SL, Kril JJ, Stevens CH, et al. Retiring the term FTDP‐17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2018;141(2):521‐534. 10.1093/brain/awx367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position‐dependent splicing regulation by TDP‐43. Nat Neurosci. 2011;14:452‐458. 10.1038/NN.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buratti E, Baralle FE. Multiple roles of TDP‐43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867‐878. 10.2741/2727 [DOI] [PubMed] [Google Scholar]
- 22. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130‐133. 10.1126/SCIENCE.1134108. (New York, NY). [DOI] [PubMed] [Google Scholar]
- 23. Geser F, Martinez‐Lage M, Robinson J, et al. Clinical and pathological continuum of multisystem TDP‐43 proteinopathies. Arch Neurol. 2009;66:180‐189. 10.1001/ARCHNEUROL.2008.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mackenzie IRA, Neumann M, Baborie A, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;122:111‐113. 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59:952‐962. 10.1002/ANA.20873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boeve BF. Links between frontotemporal lobar degeneration, corticobasal degeneration, progressive supranuclear palsy, and amyotrophic lateral sclerosis. Alzheimer Dis Assoc Disord. 2007;21. 10.1097/WAD.0b013e31815bf454. Alzheimer Dis Assoc Disord. [DOI] [PubMed] [Google Scholar]
- 27. Scarioni M, Gami‐Patel P, Timar Y, et al. Frontotemporal dementia: correlations between psychiatric symptoms and pathology. Ann Neurol. 2020;87:950‐961. 10.1002/ana.25739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mesulam MM. Primary progressive aphasia and the language network: the 2013 H. Houston Merritt Lecture. Neurology. 2013;81:456‐462. 10.1212/WNL.0B013E31829D87DF [DOI] [PubMed] [Google Scholar]
- 29. MacKenzie IRA, Munoz DG, Kusaka H, et al. Distinct pathological subtypes of FTLD‐FUS. Acta Neuropathol. 2011;121:207‐218. 10.1007/S00401-010-0764-0 [DOI] [PubMed] [Google Scholar]
- 30. Chornenka K, Hirsch‐Reinshagen V, Perez‐Rosendahl M, et al. Expanding the phenotype of frontotemporal lobar degeneration with FUS‐positive pathology (FTLD‐FUS). J Neuropathol Exp Neurol. 2020;79:809‐812. 10.1093/JNEN/NLAA045 [DOI] [PubMed] [Google Scholar]
- 31. Seelaar H, Klijnsma KY, De Koning I, et al. Frequency of ubiquitin and FUS‐positive, TDP‐43‐negative frontotemporal lobar degeneration. J Neurol. 2010;257:747‐753. 10.1007/s00415-009-5404-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451‐1456. 10.1212/WNL.0b013e3181bf997a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266:2075‐2086. 10.1007/s00415-019-09363-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gossye H, Van Broeckhoven C, Engelborghs S. The use of biomarkers and genetic screening to diagnose frontotemporal dementia: evidence and clinical implications. Front Neurosci. 2019;13:757. 10.3389/fnins.2019.00757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron. 2011;72:257‐268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker M, Kwok JB, Kucera S, et al. Localization of frontotemporal dementia with parkinsonism in an Australian kindred to chromosome 17q21‐22. Ann Neurol. 1997;42:794‐798. 10.1002/ana.410420516 [DOI] [PubMed] [Google Scholar]
- 37. Baker M, Mackenzie IR, Pickering‐Brown SM, et al. Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916‐919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- 38. Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin‐positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988‐3001. 10.1093/hmg/ddl241 [DOI] [PubMed] [Google Scholar]
- 39. Raffaele F, Claudia M, John H. Genetics and molecular mechanisms of frontotemporal lobar degeneration: an update and future avenues. Neurobiol Aging. 2019;78:98‐110. 10.1016/J.NEUROBIOLAGING.2019.02.006 [DOI] [PubMed] [Google Scholar]
- 40. Van Mossevelde S, Engelborghs S, Van Der Zee J, Van Broeckhoven C. Genotype‐phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol. 2018;14:363‐378. 10.1038/S41582-018-0009-8 [DOI] [PubMed] [Google Scholar]
- 41. Mizielinska S, Lashley T, Norona FE, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845‐857. 10.1007/S00401-013-1200-Z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gendron TF, Bieniek KF, Zhang YJ, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829‐844. 10.1007/S00401-013-1192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mizielinska S, Grönke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science. 2014;345:1192‐1194. 10.1126/science.1256800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kwon I, Xiang S, Kato M, et al. Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139‐1145. 10.1126/science.1254917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mackenzie IR, Neumann M. Subcortical TDP‐43 pathology patterns validate cortical FTLD‐TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020;139:83‐98. 10.1007/S00401-019-02070-4/TABLES/5 [DOI] [PubMed] [Google Scholar]
- 46. Nishihira Y, Gefen T, Mao Q, et al. Revisiting the utility of TDP‐43 immunoreactive (TDP‐43‐ir) pathology to classify FTLD‐TDP subtypes. Acta Neuropathol. 2019;138:167‐169. 10.1007/S00401-019-02024-W/FIGURES/1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ward ME, Chen R, Huang HY, et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 2017;9. 10.1126/scitranslmed.aah5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5’‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature. 1998;393:702‐704. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 49. Rosen HJ, Boeve BF, Boxer AL. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: recent findings from ARTFL and LEFFTDS. Alzheimers Dement. 2020;16(1):71‐78. 10.1002/alz.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Desmarais P, Rohrer JD, Nguyen QD, et al. Therapeutic trial design for frontotemporal dementia and related disorders Neurodegeneration. J Neurol Neurosurg Psychiatry. 2019;90:412‐423. 10.1136/jnnp-2018-318603 [DOI] [PubMed] [Google Scholar]
- 51. Boxer AL, Gold M, Feldman H, et al. New directions in clinical trials for frontotemporal lobar degeneration: methods and outcome measures. Alzheimers Dement. 2020;16:131‐143. 10.1016/j.jalz.2019.06.4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Teunissen CE, Otto M, Engelborghs S, Herukka S‐K, Lehmann S. White paper by the society for CSF analysis and clinical neurochemistry: overcoming barriers in biomarker development and clinical translation. Alzheimer's Res Ther . 2018;12:23. 10.1186/s13195-018-0359-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jack CR. The transformative potential of plasma phosphorylated tau. Lancet Neurol. 2020;19:373‐374. 10.1016/S1474-4422(20)30112-5 [DOI] [PubMed] [Google Scholar]
- 54. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood‐based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2021. 10.1016/S1474-4422(21)00361-6 [DOI] [PubMed] [Google Scholar]
- 55. Irwin DJ, Trojanowski JQ, Grossman M. Cerebrospinal fluid biomarkers for differentiation of frontotemporal lobar degeneration from Alzheimer's disease. Front Aging Neurosci. 2013;5:6. 10.3389/fnagi.2013.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vijverberg EGB, Dols A, Krudop WA, et al. Cerebrospinal fluid biomarker examination as a tool to discriminate behavioral variant frontotemporal dementia from primary psychiatric disorders. Alzheimers Dement. 2017;7:99‐106. 10.1016/j.dadm.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Diaz‐Lucena D, Escaramis G, Villar‐Piqué A, et al. A new tetra‐plex fluorimetric assay for the quantification of cerebrospinal fluid β‐amyloid42, total‐tau, phospho‐tau and α‐synuclein in the differential diagnosis of neurodegenerative dementia. J Neurol. 2020;267:2567‐2581. 10.1007/s00415-020-09870-9 [DOI] [PubMed] [Google Scholar]
- 58. Portelius E, Zetterberg H, Skillbäck T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain. 2015;138:3373‐3385. 10.1093/brain/awv267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Itoh N, Arai H, Urakami K, et al. Large‐scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at serine 199 for the antemortem diagnosis of Alzheimer's disease. Ann Neurol. 2001;50:150‐156. 10.1002/ana.1054 [DOI] [PubMed] [Google Scholar]
- 60. Hanes J, Kovac A, Kvartsberg H, et al. Evaluation of a novel immunoassay to detect p‐tau Thr217 in the CSF to distinguish Alzheimer disease from other dementias. Neurology. 2020;95:e3026‐e3035. 10.1212/wnl.0000000000010814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Irwin DJ, Lleó A, Xie SX, et al. Ante mortem cerebrospinal fluid tau levels correlate with postmortem tau pathology in frontotemporal lobar degeneration. Ann Neurol. 2017;82:247‐258. 10.1002/ana.24996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Paterson RW, Slattery CF, Poole T, et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer's disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther. 2018;10. 10.1186/s13195-018-0361-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Townley RA, Graff‐Radford J, Mantyh WG, et al. Progressive dysexecutive syndrome due to Alzheimer's disease: a description of 55 cases and comparison to other phenotypes. Brain Commun. 2020;2. 10.1093/braincomms/fcaa068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Norise C, Ungrady M, Halpin A, et al. Clinical correlates of Alzheimer's disease cerebrospinal fluid analytes in primary progressive aphasia. Front Neurol. 2019;10. 10.3389/fneur.2019.00485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gabelle A, Roche S, Gény C, et al. Decreased sAβPPβ, Aβ38, and Aβ40 cerebrospinal fluid levels in frontotemporal dementia. J Alzheimers Dis. 2011;26:553‐563. 10.3233/JAD-2011-110515 [DOI] [PubMed] [Google Scholar]
- 66. Illán‐Gala I, Pegueroles J, Montal V, et al. APP‐derived peptides reflect neurodegeneration in frontotemporal dementia. Anna Clin Transl Neurol. 2019;6:2518‐2530. 10.1002/acn3.50948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lleó A, Irwin DJ, Illán‐Gala I, et al. A 2‐step cerebrospinal algorithm for the selection of frontotemporal lobar degeneration subtypes. JAMA Neurol. 2018. 10.1001/jamaneurol.2018.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Toledo JB, Brettschneider J, Grossman M, et al. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol. 2012;124:23‐35. 10.1007/s00401-012-0983-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Goossens J, Bjerke M, Van Mossevelde S, et al. Diagnostic value of cerebrospinal fluid tau, neurofilament, and progranulin in definite frontotemporal lobar degeneration. Alzheimers Res Ther. 2018;10. 10.1186/s13195-018-0364-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bridel C, Van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76:1035‐1048. 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ducharme S, Dols A, Laforce R, Devenney E, Kumfor F, Van Den Stock J, et al. Recommendations to distinguish behavioural variant frontotemporal dementia from psychiatric disorders. Brain. 2020;143:1632‐1650. 10.1093/brain/awaa018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sjögren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology. 2000;54:1960‐1964. 10.1212/WNL.54.10.1960 [DOI] [PubMed] [Google Scholar]
- 73. de Jong D, Jansen MMRW, Pijnenburg YAL, et al. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry. 2007;78:936‐938. 10.1136/jnnp.2006.107326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bridel C, Van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019:1‐14. 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eratne D, Loi SM, Li QX, et al. Cerebrospinal fluid neurofilament light chain is elevated in Niemann–Pick type C compared to psychiatric disorders and healthy controls and may be a marker of treatment response. Aust N Z J Psychiatry. 2020;54:648‐649. 10.1177/0004867419893431 [DOI] [PubMed] [Google Scholar]
- 76. Willemse EAJ, Scheltens P, Teunissen CE, Vijverberg EGB. A neurologist's perspective on serum neurofilament light in the memory clinic: a prospective implementation study. Alzheimer's Res Ther. 2021;13. 10.1186/S13195-021-00841-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Swift IJ, Sogorb‐Esteve A, Heller C, et al. Fluid biomarkers in frontotemporal dementia: past, present and future. J Neurol Neurosurg Psychiatry. 2020. 10.1136/jnnp-2020-323520 [DOI] [PubMed] [Google Scholar]
- 78. Woolley JD, Khan BK, Natesan A, et al. Satiety‐related hormonal dysregulation in behavioral variant frontotemporal dementia. Neurology. 2014;82:512‐520. 10.1212/WNL.0000000000000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Alcolea D, Vilaplana E, Suárez‐Calvet M, et al. CSF sAPPβ, YKL‐40, and neurofilament light in frontotemporal lobar degeneration. Neurology. 2017;89:1‐11. 10.1212/WNL.0000000000004088 [DOI] [PubMed] [Google Scholar]
- 80. Craig‐Schapiro R, Perrin RJ, Roe CM, et al. YKL‐40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry. 2010;68:903‐912. 10.1016/j.biopsych.2010.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91:e867‐e877. 10.1212/WNL.0000000000006082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Alcolea D, Irwin DJ, Illán‐Gala I, et al. Elevated YKL‐40 and low sAPPβ:yKL‐40 ratio in antemortem cerebrospinal fluid of patients with pathologically confirmed FTLD. J Neurol Neurosurg Psychiatry. 2019;90:180‐186. 10.1136/jnnp-2018-318993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thijssen EH, La JoieR, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration Adam L. Boxer 1 ✉ and Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) investigators*. Nat Med. 2020. 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422‐433. 10.1016/S1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 85. Benussi A, Karikari TK, Ashton N, et al. Diagnostic and prognostic value of serum NfL and p‐Tau 181 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2020;91:960‐967. 10.1136/jnnp-2020-323487 [DOI] [PubMed] [Google Scholar]
- 86. Ashton NJ, Pascoal TA, Karikari TK, et al. Plasma p‐tau231: a new biomarker for incipient Alzheimer's disease pathology. Acta Neuropathol. 2021;141:709‐724. 10.1007/s00401-021-02275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Illán‐Gala I, Lleo A, Karydas A, et al. Plasma tau and neurofilament light in frontotemporal lobar degeneration and Alzheimer's disease. Neurology. 2021;96(5). 10.1212/wnl.0000000000011226. 10.1212/WNL.0000000000011226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Steinacker P, Anderl‐Straub S, Diehl‐Schmid J, et al. Serum neurofilament light chain in behavioral variant frontotemporal dementia. Neurology. 2018;91:e1390‐e1401. 10.1212/WNL.0000000000006318 [DOI] [PubMed] [Google Scholar]
- 89. Forgrave LM, Ma M, Best JR, DeMarco ML. The diagnostic performance of neurofilament light chain in CSF and blood for Alzheimer's disease, frontotemporal dementia, and amyotrophic lateral sclerosis: a systematic review and meta‐analysis. Alzheimers Dement. 2019;11:730‐743. 10.1016/j.dadm.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Al Shweiki MR, Steinacker P, Oeckl P, Hengerer B, Danek A, Fassbender K, et al. Neurofilament light chain as a blood biomarker to differentiate psychiatric disorders from behavioural variant frontotemporal dementia. J Psychiatr Res. 2019;113:137‐140. 10.1016/j.jpsychires.2019.03.019 [DOI] [PubMed] [Google Scholar]
- 91. Katisko K, Cajanus A, Jääskeläinen O, et al. Serum neurofilament light chain is a discriminative biomarker between frontotemporal lobar degeneration and primary psychiatric disorders. J Neurol. 2020;267:162‐167. 10.1007/s00415-019-09567-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van der Ende EL, Meeter LH, Poos JM, et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019;18:1103‐1111. 10.1016/S1474-4422(19)30354-0 [DOI] [PubMed] [Google Scholar]
- 93. Hu WT, Watts K, Grossman M, et al. Reduced CSF p‐Tau 181 to Tau ratio is a biomarker for FTLD‐TDP study sponsorship : author disclosure. Neurology. 2013;81:1945‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Borroni B, Benussi A, Archetti S, et al. Csf p‐tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener. 2014;16:86‐91. 10.3109/21678421.2014.971812 [DOI] [PubMed] [Google Scholar]
- 95. del Campo M, Galimberti D, Elias N, et al. Novel CSF biomarkers to discriminate FTLD and its pathological subtypes. Ann Clin Trans Neurol. 2018;5(10):1163‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Meeter LHH, Vijverberg EG, Del CampoM, et al. Clinical value of neurofilament and phospho‐tau/tau ratio in the frontotemporal dementia spectrum class of evidence criteria for rating therapeutic and diagnostic studies. Neurology. 2018;90(14). 10.1212/WNL.0000000000005261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pijnenburg YAL, Verwey NA, van der Flier WM, Scheltens P, Teunissen CE. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement. 2015;1:505‐c512. 10.1016/j.dadm.2015.11.001. (Amsterdam, Netherlands). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Luk C, Compta Y, Magdalinou N, et al. Development and assessment of sensitive immuno‐PCR assays for the quantification of cerebrospinal fluid three‐ and four‐repeat tau isoforms in tauopathies. J Neurochem. 2012;123:396‐405. 10.1111/j.1471-4159.2012.07911.x [DOI] [PubMed] [Google Scholar]
- 99. Steinacker P, Barschke P, Otto M. Biomarkers for diseases with TDP‐43 pathology. Mol Cell Neurosci. 2019;97:43‐59. 10.1016/j.mcn.2018.10.003 [DOI] [PubMed] [Google Scholar]
- 100. Scialò C, Tran TH, Salzano G, et al. TDP‐43 real‐time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020;2. 10.1093/braincomms/fcaa142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pijnenburg YAL, Verwey NA, Van Der Flier WM, Scheltens P, Teunissen CE. CSF biomarkers discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement. 2015;1:505‐512. 10.1016/j.dadm.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Landqvist Waldö M, Santillo AF, Passant U, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol. 2013;54. 10.1186/1471-2377-13-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Teunissen CE, Elias N, Koel‐Simmelink MJA, et al. Novel diagnostic cerebrospinal fluid protein biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement. 2016;2:86‐94. 10.1016/j.dadm.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. van der Ende EL, Meeter LH, Stingl C, et al. Novel CSF biomarkers in genetic frontotemporal dementia identified by proteomics. Ann Clin Transl Neurol. 2019;6:698‐707. 10.1002/acn3.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Huin V, Barbier M, Bottani A, et al. Homozygous GRN mutations: new phenotypes and new insights into pathological and molecular mechanisms. Brain. 2020;143:303‐319. 10.1093/brain/awz377 [DOI] [PubMed] [Google Scholar]
- 106. Meeter LHH, Patzke H, Loewen G, et al. Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra. 2016;6:330‐340. 10.1159/000447738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Goossens J, Bjerke M, Van Mossevelde S, et al. Diagnostic value of cerebrospinal fluid tau, neurofilament, and progranulin in definite frontotemporal lobar degeneration. Alzheimers Res Ther. 2018;10:31. 10.1186/s13195-018-0364-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Meeter LHH, Gendron TF, Sias AC, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. 2018;5:583‐597. 10.1002/acn3.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lehmer C, Oeckl P, Weishaupt JH, et al. Poly‐ GP in cerebrospinal fluid links C9orf72 ‐associated dipeptide repeat expression to the asymptomatic phase of ALS /FTD. EMBO Mol Med. 2017;9:859‐868. 10.15252/emmm.201607486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9. 10.1126/scitranslmed.aai7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289‐300. 10.2307/2346101 [DOI] [Google Scholar]
- 112. Sormani MP, Haering DA, Kropshofer H, et al. Blood neurofilament light as a potential endpoint in Phase 2 studies in MS. Ann Clin Transl Neurol. 2019;6:1081‐1089. 10.1002/acn3.795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rosen HJ, Boeve BF, Boxer AL. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: recent findings from ARTFL and LEFFTDS. Alzheimers Dement. 2020;16:71‐78. 10.1002/alz.12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ljubenkov PA, Staffaroni AM, Rojas JC, et al. Cerebrospinal fluid biomarkers predict frontotemporal dementia trajectory. Ann Clin Transl Neurol. 2018;5:1250‐1263. 10.1002/acn3.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3:623‐636. 10.1002/acn3.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75:116‐126. 10.1002/ana.24052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies molecular networks in alzheimer's disease progression. Neuron. 2020;105:975‐991.e7. 10.1016/j.neuron.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhu N, Santos‐Santos M, Illán‐Gala I, et al. Plasma glial fibrillary acidic protein and neurofilament light chain for the diagnostic and prognostic evaluation of frontotemporal dementia. Transl Neurodegener. 2021;10:50. 10.1186/S40035-021-00275-W [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ingelsson M, Ramasamy K, Russ C, et al. Increase in the relative expression of tau with four microtubule binding repeat regions in frontotemporal lobar degeneration and progressive supranuclear palsy brains. Acta Neuropathol. 2007;114:471‐479. 10.1007/s00401-007-0280-z [DOI] [PubMed] [Google Scholar]
- 120. Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935‐946. [DOI] [PubMed] [Google Scholar]
- 121. Koga S, Kouri N, Walton RL, et al. Corticobasal degeneration with TDP‐43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype HHS Public Access. Acta Neuropathol. 2018;136:389‐404. 10.1007/s00401-018-1878-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135:794‐806. 10.1093/brain/aws001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. MacKenzie IR, Arzberger T, Kremmer E, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico‐pathological correlations. Acta Neuropathol. 2013;126:859‐879. 10.1007/s00401-013-1181-y [DOI] [PubMed] [Google Scholar]
- 124. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020:263‐270. 10.1136/jnnp-2019-321954 [DOI] [PubMed] [Google Scholar]
- 125. Zhang J, Velmeshev D, Hashimoto K, et al. Neurotoxic microglia promote TDP‐43 proteinopathy in progranulin deficiency. Nature. 2020:1‐5. 10.1038/s41586-020-2709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cook CN, Wu Y, Odeh HM, Gendron TF, et al. C9orf72 poly(GR) aggregation induces TDP‐43 proteinopathy. Sci Transl Med. 2020;12. 10.1126/scitranslmed.abb3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Miyagawa T, Brushaber D, Syrjanen J, et al. Use of the CDR® plus NACC FTLD in mild FTLD: data from the ARTFL/LEFFTDS consortium. Alzheimers Dement. 2020;16:79‐90. 10.1016/j.jalz.2019.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. McGhee DJM, Ritchie CW, Thompson PA, Wright DE, Zajicek JP, Counsell CE. A systematic review of biomarkers for disease progression in alzheimer's disease. PLoS One. 2014;9. 10.1371/journal.pone.0088854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25:277‐283. 10.1038/s41591-018-0304-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. del Campo M, Pijnenburg YAL, Chen‐Plotkin A, et al. Sex hormone‐binding globulin (Shbg) in cerebrospinal fluid does not discriminate between the main ftld pathological subtypes but correlates with cognitive decline in ftld tauopathies. Biomolecules. 2021;11. 10.3390/biom11101484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Van Der Ende EL, Xiao M, Xu D, et al. Original research: neuronal pentraxin 2: a synapse‐derived CSF biomarker in genetic frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91:612. 10.1136/JNNP-2019-322493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sattlecker M, Kiddle SJ, Newhouse S, et al. Alzheimer's disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimers Dement. 2014;10:724‐734. 10.1016/j.jalz.2013.09.016 [DOI] [PubMed] [Google Scholar]
- 133. Whelan CD, Mattsson N, Nagle MW, et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer's disease. Acta Neuropathol Commun. 2019. 10.1186/s40478-019-0795-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Drobin K, Nilsson P, Schwenk JM. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods Mol Biol. 2013;1023:137‐145. 10.1007/978-1-4614-7209-4_8. (Clifton, NJ). [DOI] [PubMed] [Google Scholar]
- 135. Bergström S, Öijerstedt L, Remnestål J, et al. A panel of CSF proteins separates genetic frontotemporal dementia from presymptomatic mutation carriers: a GENFI study. Mol Neurodegener. 2021;16. 10.1186/S13024-021-00499-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Remnestål J, Öijerstedt L, Ullgren A, et al. Altered levels of CSF proteins in patients with FTD, presymptomatic mutation carriers and non‐carriers. Transl Neurodegener. 2020;9:1‐13. 10.1186/S40035-020-00198-Y/FIGURES/5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhong W, Edfors F, Gummesson A, Bergström G, Fagerberg L, Uhlén M. Next generation plasma proteome profiling to monitor health and disease. Nat Commun. 2021;12:1‐12. 10.1038/s41467-021-22767-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jiang Y, Zhou X, Ip FC, et al. Large‐scale plasma proteomic profiling identifies a high‐performance biomarker panel for Alzheimer's disease screening and staging. Alzheimers Dement. 2021. 10.1002/ALZ.12369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. del Campo M, Jongbloed W, Twaalfhoven H. Facilitating the validation of novel protein biomarkers for dementia: an optimal workflow for the development of sandwich immunoassays. Front Neurol. 2015;6:1‐10. 10.3389/fneur.2015.00202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sol N, Leurs CE, ’t VeldSGIn, Strijbis EM, Vancura A, Schweiger MW, et al. Blood platelet RNA enables the detection of multiple sclerosis. Mult Scler J. 2020;6. 10.1177/2055217320946784. 2055217320946784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Del Campo M, Mollenhauer B, Bertolotto A, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer's and Parkinson's disease cerebrospinal fluid biomarkers: an update. Biomark Med. 2012;6:419‐430. 10.2217/bmm.12.46 [DOI] [PubMed] [Google Scholar]
- 142. Hok‐A‐Hin YS, Willemse EAJ, Teunissen CE, Del Campo M. Guidelines for CSF processing and biobanking: impact on the identification and development of optimal CSF protein biomarkers. Methods Mol Biol. 2019;2044:27‐50. 10.1007/978-1-4939-9706-0_2. Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- 143. Rózga M, Bittner T, Batrla R, Karl J. Preanalytical sample handling recommendations for Alzheimer's disease plasma biomarkers. Alzheimer's and Dementia: diagnosis. Alzheimers Dement (Amst). 2019;11:291‐300. 10.1016/j.dadm.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Verberk IMW, Misdorp EO, Koelewijn J, et al. Characterization of pre‐analytical sample handling effects on a panel of Alzheimer's disease‐related blood‐based biomarkers: results from the Standardization of Alzheimer's Blood Biomarkers (SABB) working group. Alzheimers Dement. 2021. 10.1002/ALZ.12510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Mattsson N, Andreasson U, Persson S, et al. The Alzheimer's association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement. 2011;7. 10.1016/j.jalz.2011.05.2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in Alzheimer disease—the gateway to precision medicine. Nat Rev Neurol. 2018;14:457‐469. 10.1038/S41582-018-0032-9 [DOI] [PubMed] [Google Scholar]
- 147. Lleó A, Suárez‐Calvet M. Race and Alzheimer disease biomarkers. Neurology Genetics. 2021;7:e574. 10.1212/NXG.0000000000000574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. García‐Redondo A, Dols‐Icardo O, Rojas‐García RR, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. 2013;34:79‐82. 10.1002/HUMU.22211 [DOI] [PubMed] [Google Scholar]
- 149. Meeker KL, Wisch JK, Hudson D, et al. Socioeconomic status mediates racial differences seen using the AT(N) framework. Ann Neurol. 2021;89:254‐265. 10.1002/ANA.25948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schindler SE, Cruchaga C, Joseph A, et al. African Americans have differences in CSF soluble TREM2 and associated genetic variants. Neurology Genetics. 2021;7:e571. 10.1212/NXG.0000000000000571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Brickman AM, Manly JJ, Honig LS, et al. Plasma p‐tau181, p‐tau217, and other blood‐based Alzheimer's disease biomarkers in a multi‐ethnic, community study. Alzheimers Dement. 2021;17(8):1353‐1364. 10.1002/alz.12301. alz.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. BM R, Tijms B, Scheltens P, et al. Sex differences in CSF biomarkers vary by Alzheimer disease stage and APOE ε4 genotype. Neurology. 2020;95:e2378‐e2388. 10.1212/WNL.0000000000010629 [DOI] [PubMed] [Google Scholar]
- 153. Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology. JAMA Neurol. 2019:1‐14. 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Nel M, Mahungu AC, Monnakgotla N, et al. Revealing the mutational spectrum in Southern Africans with amyotrophic lateral sclerosis. Neurol Genet. 2022;8:e654. 10.1212/NXG.0000000000000654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nel M, Mavundla T, Gultig K, et al. Repeats expansions in ATXN2, NOP56, NIPA1 and ATXN1 are not associated with ALS in Africans. IBRO Neurosci Rep. 2021;10:130‐135. 10.1016/J.IBNEUR.2021.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Brand D, Polak M, Glass JD, Fournier CN. Comparison of phenotypic characteristics and prognosis between black and white patients in a tertiary ALS clinic. Neurology. 2021;96:e840‐e844. 10.1212/WNL.0000000000011396 [DOI] [PubMed] [Google Scholar]
- 157. Nel M, Agenbag GM, Henning F, Cross HM, Esterhuizen A, Heckmann JM. C9orf72 repeat expansions in South Africans with amyotrophic lateral sclerosis. J Neurol Sci. 2019;401:51‐54. 10.1016/J.JNS.2019.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Illán‐Gala I, Casaletto KB, Borrego‐Écija S, et al. Sex differences in the behavioral variant of frontotemporal dementia: a new window to executive and behavioral reserve. Alzheimers Dement. 2021;17:1329‐1341. 10.1002/ALZ.12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kohli MA, John‐Williams K, Rajbhandary R, et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer's disease in Caucasians. Neurobiol Aging. 2013;34:1519.e5‐1519.e12. 10.1016/J.NEUROBIOLAGING.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Wallon D, Rovelet‐Lecrux A, Deramecourt V, et al. Definite behavioral variant of frontotemporal dementia with C9ORF72 expansions despite positive Alzheimer's disease cerebrospinal fluid biomarkers. J Alzheimers Dis: JAD. 2012;32:19‐22. 10.3233/JAD-2012-120877 [DOI] [PubMed] [Google Scholar]