

Abstract
In 2016–2017, France experienced a devastating epidemic of highly pathogenic avian influenza (HPAI) H5N8, with more than 400 outbreaks reported in poultry farms. We analyzed the spatiotemporal dynamics of the epidemic using a structured‐coalescent‐based phylodynamic approach that combined viral genomic data (n = 196; one viral genome per farm) and epidemiological data. In the process, we estimated viral migration rates between départements (French administrative regions) and the temporal dynamics of the effective viral population size (Ne) in each département. Viral migration rates quantify viral spread between départements and Ne is a population genetic measure of the epidemic size and, in turn, is indicative of the within‐département transmission intensity. We extended the phylodynamic analysis with a generalized linear model to assess the impact of multiple factors—including large‐scale preventive culling and live‐duck movement bans—on viral migration rates and Ne. We showed that the large‐scale culling of ducks that was initiated on 4 January 2017 significantly reduced the viral spread between départements. No relationship was found between the viral spread and duck movements between départements. The within‐département transmission intensity was found to be weakly associated with the intensity of duck movements within départements. Together, these results indicated that the virus spread in short distances, either between adjacent départements or within départements. Results also suggested that the restrictions on duck transport within départements might not have stopped the viral spread completely. Overall, we demonstrated the usefulness of phylodynamics in characterizing the dynamics of a HPAI epidemic and assessing control measures. This method can be adapted to investigate other epidemics of fast‐evolving livestock pathogens.
Keywords: emerging infectious diseases, modelling infectious disease dynamics, outbreak analytics, reconstruction of viral transmission, virus evolution, virus molecular epidemiology
1. INTRODUCTION
During the winter of 2016, an epidemic of the highly pathogenic avian influenza (HPAI) virus occurred in France. It resulted in 484 infected poultry farms and approximately 7 million culled birds (Guinat, Nicolas, et al., 2018). It remains critically important to learn as much as possible from this devastating epidemic, particularly as France once again experienced an HPAI outbreak starting in December 2020 (Adlhoch et al., 2021; Vergne et al., 2021). The 2016–17 epidemic was caused by an H5N8 virus of the lineage 2.3.4.4b (A/Gs/Gd/1/96 clade) of Asian origin that was likely introduced in France by migratory birds (Briand et al., 2021). The epidemic was largely concentrated in southwest France, encompassing the Occitanie and the Nouvelle‐Aquitaine régions (the largest French territorial administrative division; Bronner et al., 2017). The first suspicion in poultry was reported in a duck breeding farm in the Tarn département (administrative division equivalent to county or district) on 25 November 2016 and the infection was confirmed—based on lab diagnosis—on 28 November (Figure 1). Subsequently, the epidemic spread westwards (Guinat, Nicolas, et al., 2018), following the westerly distribution of poultry farms within the region. The last case of the epidemic was reported on 23 March 2017 (Guinat, Nicolas, et al., 2018).
FIGURE 1.
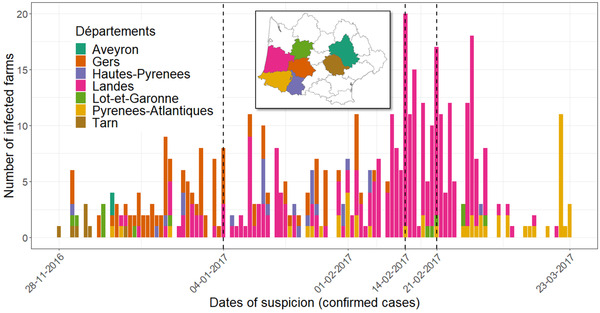
The spatiotemporal dynamics of the 2016‐17 H5N8 epidemic in southwest France. The bars represent the daily number of infected farms identified in each département, with each département being represented by a different colour. The number of infected farms represented those that were confirmed through PCR diagnostics during the epidemic period. The dates separating the three culling phases are marked by three vertical dashed lines. Inset: Map of southwest France with each study département marked with a specific colour corresponding to the source of samples.
Two major outbreak responses, namely preventive culling and stopping the transport of foie gras ducks between farms, were implemented during the epidemic. Culling was implemented locally in infected farms during the initial phase of the epidemic. However, these local measures proved to be insufficient in curbing the rapid spread of the virus. Consequently, there were three phases of large‐scale culling. On 4 January 2017, all duck farms located in 150 communes across four départements, namely Gers, Landes, Pyrénées‐Atlantiques and Hautes‐Pyrénées, were notified to be preventively culled (Figure 1). On 14 February, the number of communes under notification was doubled. On 21 February, the notified areas were increased for the last time to include more than 500 communes in total. Additionally, following Avian influenza H5 detection, ducks could not be moved from or to infected farms (Guinat et al., 2020).
Epidemiological studies based on incidence data have provided important insights into the HPAI epidemic patterns (Guinat, Nicolas, et al., 2018) and the control measures (Andronico et al., 2019; Guinat et al., 2020). Additionally, a recent phylogenetic study analyzed 196 viral sequences that were identified in poultry farms (Briand et al., 2021). The study reported that the genotype that caused the large‐scale epidemic in southwest France was associated with five geographic clusters (Briand et al., 2021). This association could be the consequence of a few long‐distance transmission events followed by local spread (Briand et al., 2021). However, the ecological and epidemiological drivers of these transmission patterns are still unknown. To fill this gap, we applied a phylodynamic framework to the 2016–2017 epidemic data. Viral phylodynamics is the study of viral phylogenies and how they are shaped by potential interactions between epidemiological, immunological and evolutionary processes (Holmes & Grenfell, 2009; Volz et al., 2013). These methods can detect spatiotemporal patterns of epidemics based on viral genomic data, which represent a powerful source of information and are complementary to epidemiological approaches (Guinat et al., 2021). Although phylodynamic methods are frequently used for reconstructing epidemic patterns, their application in assessing control measures is still rare (Dellicour et al., 2018). Here, we used a phylodynamic framework to quantify the 2016–2017 H5N8 epidemic in southwest France and tested the effect of large‐scale culling, along with other potential ecological and epidemiological drivers. Doing so, we underscore the versatility of phylodynamic methods in addressing both, specific control measure‐based questions, as well as more broad ones regarding viral spread history.
2. MATERIALS AND METHODS
2.1. Genomic data, bioinformatics and phylogenetic methods
Note that 196 HPAI H5N8 viruses were isolated amongst the 484 farms that experienced an outbreak between November 2016 and March 2017 (study period). The viral genomes were sequenced (one sequence per farm) at the French national reference laboratory for avian influenza (ANSES‐Ploufragan, France; Briand et al., 2021). Sequence metadata, including Accession numbers, are provided in Supporting Information Appendix Tables S1 and S2. Each sequence was geocoded and associated with the département where the outbreak was located along with the collection date (Supporting information S3). The départements we studied are as follows: Aveyron (n = 2), Gers (n = 57), Hautes‐Pyrénées (n = 14), Landes (n = 96), Lot‐et‐Garonne (n = 5), Pyrénées‐Atlantiques (n = 15) and Tarn (n = 7). The whole concatenated virus genomic sequences were aligned using the MUSCLE multiple sequence alignment algorithm (Edgar, 2004) implemented in MegaX software with default parameters (Kumar et al., 2018). Influenza viral genome is particularly prone to frequent genomic reassortments (Lycett et al., 2019), which, if undetected, may provide spurious genomic signals. We, therefore, checked for the absence of recombination and reassortments in our aligned sequences using five different algorithms, namely BootScan, CHIMERA, MaxCHI, RDP and SisScan, implemented in RDP v4.1 (Martin et al., 2015). None of the algorithms detected any recombination events. Therefore, we used all available sequences (n = 196) across the seven départements for downstream analyses.
We conducted an exploratory phylogenetic analysis, based on a maximum likelihood method (Minh et al., 2020), to ensure the geographic fidelity of the samples and that the samples collected earlier were closer to the root of the phylogenetic tree. Following this analysis, we conducted a structured and more computationally intensive phylodynamic analysis.
2.2. Phylodynamic reconstruction of viral phylogeny and spread history
In our study, we used the GTR+Gamma4 substitution model which was selected as the best model by the SMS algorithm in the Programme PhyML ver. 3 (Guindon et al., 2010). We also used a strict molecular clock as is common for avian influenza viruses (Fourment & Holmes, 2015; Si et al., 2017) and also because the root‐to‐tip genetic distances from the maximum likelihood tree and sampling dates of the samples were strongly positively correlated. The phylogenetic temporal structure was assessed using Tempest ver. 1.5.3 (Rambaut et al., 2016).
2.3. Phylodynamic and generalized linear models
To infer the spread of H5N8, we used a recently developed phylodynamic model called the marginal approximation of the structured coalescent (MASCOT; Müller et al., 2018). This model was adapted in order to describe the coalescence of lineages within and the migration of lineages between departments and has been applied several times to study the spread of pathogens (J. Yang et al., 2019; Müller et al., 2019). To identify putative predictors of viral spread, we used the MASCOT model where the migrations rates and effective population sizes through time are defined as a generalized linear model (GLM; Müller et al., 2019).
2.4. Migration rate predictors
To investigate the effects of large‐scale culling of ducks, we compared viral spread before and after preventive culling was notified by the French government. The assumption was that if culling was effective then viral spread should be curtailed after the date of notification. To test this scenario, we created a predictor based on 4 January 2017. This was the date of the first notification when preventive culling was implemented in 150 communes located at the border between Gers, Landes, Pyrénées‐Atlantiques and Hautes‐Pyrénées départements. We also created another predictor based on 14 February, when a second notification was issued to extend the culling to other communes. The last predictor of culling was based on 21 February, when the last notification was issued to include yet more communes under culling control measure. In Figure 1, we showed the daily distribution of the number of outbreaks in each of the seven départements and marked the three culling notification dates.
The movement of ducks between different farms is a major feature of the foie‐gras production in southwest France. Ducks, in large flocks of several thousands, are first raised in breeding farms for up to 12 weeks. Then, they are divided into small groups of hundreds and transported in batches to force‐feeding farms across the region. Small batches are necessary because the process of force‐feeding is labour intensive and can optimally be done only in small batches. At the force‐feeding farms, the ducks are fattened for 12 days and then are sent off to the slaughterhouse for harvest. As a result of this process, farms are continuously receiving and sending out shipments of ducks potentially facilitating the spread of the virus as well. To test any link between duck movement and viral spread between départements, we considered the total number of duck shipments exchanged between départements. These data were obtained from the professional organization of fattening duck producers (CIFOG) for the period between October 2016 and February 2017 (Guinat et al., 2019). This predictor was considered time independent.
The density of livestock is often linked to pathogen spread (Cantrell et al., 2020; Craft, 2015; Tildesley et al., 2019) and the density of poultry farms in southwest France was found to be associated the occurrence of outbreaks in the case of the French HPAI 2016–2017 epidemic (Guinat et al., 2019). To test the role of poultry density in driving viral spread, we considered the densities of duck and chicken farms per département as predictors. During epidemics in livestock, humans can spread pathogens between farms, either as fomites directly or through sharing of equipment (Mansley, 2004; van Andel et al., 2021). Hence, we also considered the human population per département as a separate predictor. These time‐independent variables were computed from the French Directorate‐General for Food (DGAl) and CIFOG databases (Guinat et al., 2019).
We also considered the total number of infected farms in a département to be a potential predictor of viral spread as départements with high number of cases could represent a major source of virus for other departments. This variable is strongly positively correlated with sample sizes, that is, the numbers of sequences obtained in each département (S4), which can be a potential predictor as well (Müller et al., 2019; Yang et al., 2019). This correlation suggests that the proportion of outbreaks that were sequenced was of similar magnitude across départements, reflecting accurately the processes underlying differences in outbreak sizes. This is important because spatial sampling design is known to impact phylodynamics patterns and may lead to biased conclusions in case sampling design does not represent the epidemic dynamics (Guindon & Maio, 2021). In a preliminary analysis, we considered sample size as a potential predictor along with the total number of infected farms. Both predictors showed similar associations with viral spread (data not shown). It is a common practice to drop one of the collinear predictors to reduce the redundancy of collinear predictors. We decided to keep the total number of infected farms predictor in the later analyses as we thought it is of more interest over sample size. As the first case was detected in Tarn, we considered a potential predictor that represented Tarn to be the most likely source of viral spread for all départements.
Finally, two time‐independent spatial variables were considered as previous studies identified spatial patterns in the epidemic spread (Guinat et al., 2019; Guinat, Nicolas, et al., 2018). We tested the effect of the Euclidean distance between départements centroids as well as the border sharing between départements on the viral spread between departments.
2.5. Effective population size (Ne) predictors
In the case of Ebola virus epidemics, the number of detected cases was found to be highly predictive of the viral effective population size (Ne; Dellicour et al., 2018; Dudas et al., 2017; Müller et al., 2019). It is derived from the Wright–Fisher population genetic model and is assumed to be proportional to viral population size (Frost & Volz, 2010). In our analysis, we, therefore, used the number of infected farms that were officially reported in each département per day, which was smoothed using a 7‐day moving average. However, if virus lineages are erroneously time‐stamped due to unknown sampling delay, a GLM analysis may result in spurious association between case number and Ne (Dellicour et al., 2018). So, for our second case number‐based predictor, we artificially incorporated a 1‐week delay in the case number distribution to check for any delay (Müller et al., 2019).
To test any link between duck movement and Ne within a département, we considered the total number of shipments exchanged between farms within a département as another potential predictor. Livestock density plays an important role in viral transmission and hence could be linked to Ne (Meadows et al., 2018). We used the densities of duck and chicken farms per département as potential predictors. Humans, as fomites, can also influence Ne and hence human population density was also used as a predictor. Both these predictors were time independent. Also, all non‐binary variables were log transformed and standardized so that their means and standard deviations equalled 0 and 1, respectively. It must be noted that while Ne is a useful and popular population genetic measure of epidemic size, it may not always track epidemic size faithfully during the initial phase of an acute outbreak (Frost & Volz, 2010). Therefore, its interpretation should be cautious and context specific. We also tested multi‐collinearity between linear predictors of the viral effective population size GLM (Supporting Information S1).
The modelling exercises and the Markov chain Monte Carlo (MCMC) simulations were conducted in BEAST v2.6.3 (Bouckaert et al., 2014). For posterior sampling, we used the coupled MCMC package (Müller & Bouckaert, 2020) with 100 million iterations. The convergence and mixing of the MCMC chains were checked visually in Tracer v1.7.1 (Rambaut et al., 2018). An analysis was considered successful if the effective sample size of each of the parameters was at least 200. The time‐scaled phylogenetic tree with the most likely département of each lineage was created in FigTree v1.4.3 (Rambaut, 2016).
2.6. Selection of generalized linear model
Model selection requires extensive computation, particularly for models with many parameters. Hence, MASCOT provides an efficient model averaging approach, the Bayesian stochastic search variable selection (BSSVS), that is used to calculate a binary indicator variable (Lemey et al., 2009; Müller et al., 2019). This variable indicates if its predictor has contributed (1 = yes; 0 = No) to the GLM. BSSVS results in an estimate of the posterior inclusion probability or support for each predictor. Each predictor is also associated with a coefficient (Müller et al., 2019). The purpose of the coefficient is to express the predictor's effect size and the value can range between ‐∞ to ∞. We also measured the confidence in the result based on Bayes factors (K), which is a Bayesian alternative to classical Null‐Hypothesis Significance Testing (Benjamin et al., 2018; Keysers et al., 2020). Based on convention (Kass & Raftery, 1995), we decided the confidence to be not substantial (1 > K < 3.2), substantial (3.2 > K < 10), strong (10 > K < 100) or decisive (K > 100). In the final model, we included only the predictors with at least substantially confident result. These statistical analyses were conducted in R v4.0.3 (R Core Team, 2020).
3. RESULTS
3.1. Viral phylogeny and spread history
Figure 2a shows the evolutionary relationship between viral lineages from different départements in the form of a time‐scaled summarized phylogenetic tree. It also shows the most likely viral spread history of the lineages between départements, each of which is represented by a different colour. A change in colour across tree branches represents spread events between départements. We inferred Tarn to be the most likely source of all the viral lineages in southwest France (median posterior probability = 0.75, 95% credible interval; CI = 0.16–1). The median day of the emergence was estimated to be the middle of November 2016 (95% CI = 27 Oct.–22 Nov.). The clustering of basal sequences indicated a single introduction event in southwest France, followed by epidemic transmission (short terminal branches) mainly toward the west.
FIGURE 2.
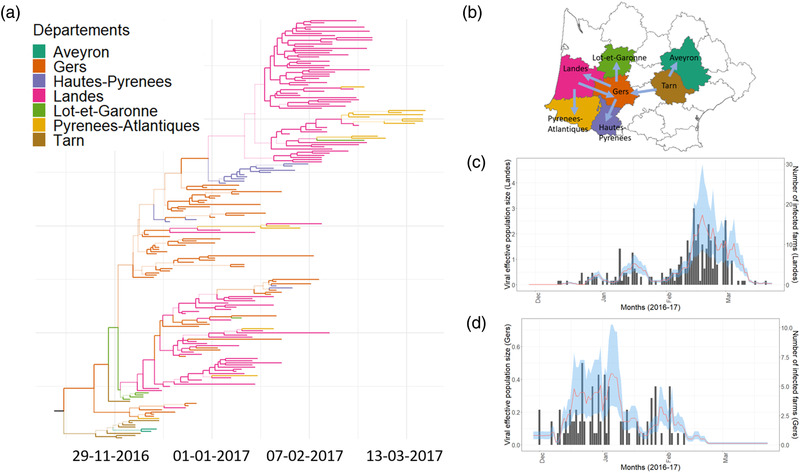
Inferred epidemics dynamics based on structured coalescent population model. (a) A time‐scaled maximum clade credibility (MCC) phylogenetic tree representing the evolutionary relationship between viral lineages. The colour of a branch indicates the inferred location (see legend) with the highest posterior support. A colour change on a branch indicates a virus spread event. Numbers of all the nodes are shown in the supplementary figure; (b) A schematic of the prominent (maximum probability of inferred location > 0.7) viral spread events between départements. (c) Estimates of viral effective population size (line in red; 95% highest posterior density; HPD in blue) and the time series of the infected number of farms in Landes (black bar chart), the most affected département. (d) Estimates of viral population size (line in red; 95% HPD in Blue) and the time series of the infected number of farms in Gers (black bar chart), the second most affected département
According to the inferred migration history (Figure 2b), the virus mostly spread between neighbouring départements. The only exception to this pattern was migration from Tarn to Gers. In most cases, the virus migrated in one direction, from one département to another and did not come back to the source. However, in a few instances—between Gers and Landes and occasionally between Landes and Pyrénées‐Atlantiques, we observed bidirectional spread of the virus. Gers was the most frequent source of spread and was responsible for spread into three other départements, namely Lot‐et‐Garonne (once), Landes (on multiple occasions) and Haute‐Pyrenees (once). On the other hand, both Gers and Landes were the départements where viruses were introduced from more than one département—namely Tarn (once) and Landes (in multiple occasions) into Gers and Gers (in multiple occasions) and Pyrénées‐Atlantiques (at least twice) and Landes (in multiple occasions). On the other end of the spectrum, Hautes‐Pyrénées and Aveyron were the départements that received viruses in single events and never passed it forward to any other départements. Towards the end of the outbreak, lineages reached Pyrénées‐Atlantiques on multiple occasions, all exclusively from Landes despite sharing its border with two other départements (Gers and Hautes‐Pyrenees) as well.
The estimated viral effective population size Ne distribution for Landes—most affected in terms of case numbers—and Gers—second most affected—départements were proportional to the corresponding time series of case numbers (Figure 2c,d).
3.2. Identifying the predictors of viral spread between départements
In Figure 3a, we show the viral spread predictors and their level of support in the form of the Bayes factor (K). Predictors with at least substantial support (K > 3.2) were included in the final GLM (Kass & Raftery, 1995). In Figure 3b, we show the estimated coefficients, which represent the strength and direction (positive or negative) of each predictor's effect on the between‐département viral spread. The analysis demonstrated that sharing of borders (K > 3.2) as well as the period of culling of the epidemic (before and after 4 January) (K > 10) were important predictors of the observed spread patterns associated with viral spread. We inferred both predictors to positively predict spread, meaning that spread was greater between départements with shared borders and that spread was reduced after the 4 January culling initiation date.
FIGURE 3.
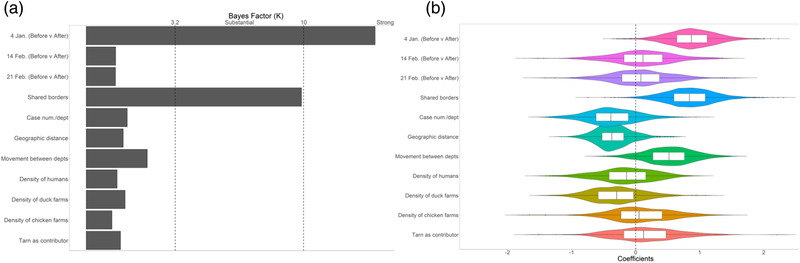
The generalized linear models (GLMs) of the viral spread between départements. (a) The viral spread predictors and their level of support are based on indicator probability, which was estimated based on Bayesian Stochastic Search Variable Selection (BSSVS). Different strengths of support are marked by vertical lines. Predictors whose indicators had at least substantial support (Bayes factor, BF = 3.2) were included in the final GLM. (b) The boxplots depict the strength and direction of each predictor's effect
3.3. Identifying the predictors of viral effective population size Ne
Figure 4 shows the results of a phylodynamic GLM analysis where we investigated the relationship between viral effective population size Ne and a number of potential predictors. In Figure 4a, we show the Ne predictors and their level of support based on indicator probability. The predictors whose indicators had at least substantial support (Bayes factor, K = 3.2) were included in the final GLM (Figure 4a). In Figure 4b, we showed the estimated coefficients, which represented the strength and direction of each predictor's effect. Based on our inclusion criterion, the final GLM again included only two predictors, namely the case numbers (K > 100, decisive) and the duck movement within département (K > 3.2).
FIGURE 4.
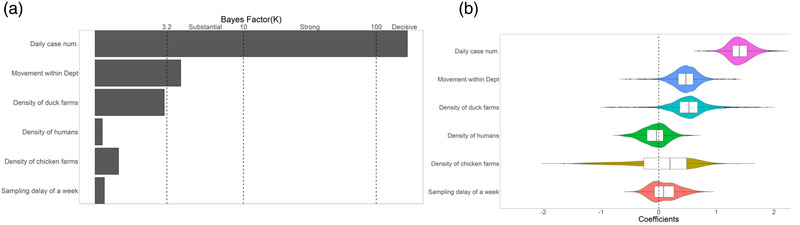
The generalized linear models (GLMs) of viral effective population size Ne within départements. (A) The Ne predictors and their level of support are based on indicator probability, which was estimated based on Bayesian Stochastic Search Variable Selection (BSSVS). Different strengths of support are marked by vertical lines. Predictors whose indicators had at least substantial support (Bayes factor, BF = 3.2) were included in the final GLM. (b) The boxplots depict the strength and direction of each predictor's effect.
4. DISCUSSION
In 2016–2017, France experienced a devastating epidemic of highly pathogenic avian influenza (HPAI) epidemic caused by an H5N8 subtype of the lineage 2.3.4.4b (A/Gs/Gd/1/96 clade). This outbreak caused major economic losses to the industry and contributed to the implementation of stricter farm biosecurity measures (Andronico et al., 2019). Unfortunately, the virus returned to France in the winter of 2020 causing yet another epidemic in the southwestern part of the country, highlighting the importance of understanding its transmission dynamics and the effectiveness of the control strategies that were implemented. Employing phylodynamic methods that combine genomic and epidemiological data, we showed that the large‐scale preventive culling measure initiated on 4 January 2017 was successful in reducing viral spread between départements. At the regional scale, we showed that viral spread between départements occurred more frequently between départements that shared borders than between départements that were far apart, which is consistent with the geographical clusters identified in Briand et al. (2021). Our results could not find links between the viral spread between départements and duck transport. Within départements, we found that duck movements were positively but weakly associated with the viral effective population size. However, the number of infected farms was a powerful predictor of the effective population size. Additionally, our phylodynamic analysis estimated the date of viral emergence close to the date of the first detection, traced the origin of the epidemic to Tarn and exhibited detailed viral spread history between départements.
To curb the epidemic, preventive culling was implemented by pre‐emptively depopulating yet uninfected (or undetected due to lack of clinical signs) farms. This measure created a spatial gap in the path of the epidemic. This gap is supposed to act as a barrier—similar to a Firebreak—to slow or stop the progress of epidemic between infected and yet uninfected zones. Preventive culling has been shown to be an effective control measure for fast‐spreading pathogens (Ferguson et al., 2001; Keeling et al., 2001; Le Menach et al., 2006). In the case of the French HPAI 2016–17 epidemic, a mathematical modelling study showed that preventive culling has halved the epidemic size, as compared to a scenario without preventive culling (Andronico et al., 2019). Our results also support the efficacy of preventive culling in reducing the regional, that is, long‐distance spread of the virus, which may not be controlled by culling of infected farms only. Taken together with earlier studies (Andronico et al., 2019; Guinat et al., 2020), our study can inform decision‐making on preventive culling, even though it would require a more detailed understanding of the optimal conditions for this strategy, particularly with regard to the timeliness and location of its implementation.
In our study, we also investigated the role of the two extensions of preventive culling, which were successively implemented on 14 and 21 February 2017. Despite including more communes for culling, our results could not find evidence that these two culling phases reduced the transmission of H5N8 beyond the effect that the first culling phase on 4 January 2017 had.
Preventive culling is often seen as a controversial strategy due to the killing of healthy livestock and high operational costs (Lederman, 2016; te Beest et al., 2011). In the case of southwest France, which experienced yet another devastating epidemic of HPAI during 2020–2021, it is likely that alternatives to preventive culling are required, including structural changes of the poultry production systems. A hypothesis that would be worth investigating is how a decrease in the duck farm density in the southwest region of France could improve the resilience of the poultry sector to the risk of HPAI in the long term because palmiped farms have been shown to be both more susceptible and more infectious than galliform farms during the 2016–2017 epidemic (Andronico et al., 2019). Decreasing duck farm density—in combination with stronger biosecurity practices—may at least slow down the spread of the virus, even if it may not prevent an epidemic. This is still useful because the slowing down may provide additional time to veterinary services to implement outbreak interventions, which, in the end, would help reducing the number of preventively culled flocks.
Our results did not find evidence that the amount of duck movements between départements impacted the spread of the virus between départements. This finding—along with the evidence of between‐département viral spread only between adjacent départements—suggests that most live‐duck movements probably did not play any role in the viral spread. This is consistent with observations that the duck movements that could have been responsible for the spread of the virus were quite limited and mostly occurred at the very early phases of the epidemic before stringent movement bans were implemented (Guinat et al., 2020). This could also be explained by the implementation of a PCR test for all duck flocks that were about to be transported. One other explanation of this pattern could be that the ban on movements successfully disrupted the association between viral spread and long‐distance duck movements. However, we did not consider the duck movements as a time‐varying predictor, which could have impacted our ability to detect duck movements as a predictor of viral transmission between départements. This hypothesis could be tested in the future with duck movement data collected over time (time‐dependent variable). Alternatively, our results could also mean that the virus did not spread in the long‐distance through duck movements, but by some other mechanisms. Two major contenders for alternative mechanisms of viral spread are humans and farm equipment as fomites and wild birds as carriers. We did not find any significant association between human population density—which acted as a proxy for human activity—and viral spread. This suggested that human populations, which were not specifically involved in the duck‐producing industry, did not play a major role in spreading the virus. An interesting development of this analysis would be to include equipment sharing data, although collection of these data are challenging as such records are probably not maintained methodically. Regarding the wild‐bird related mechanisms, a study on Chinese poultry industry indicated that wild birds did not play a major role in spreading avian influenza viruses along the value chain across the country, in contrast to between‐country spread for which wild birds play a prominent role (Yang et al., 2020). This is likely to be the case in southwest France as well. Another potential mechanism could be wind‐borne transmission, which is suspected in the case of HPAI (Scoizec et al., 2018; Ypma et al., 2013; Zhao et al., 2019). But, a previous study did not find any link between the direction of wind during the epidemic and the direction of viral spread, suggesting that the transmission was unlikely to be wind‐borne (Guinat, Rouchy, et al., 2018). However, the study was limited to a specific period during the epidemic, hence the role of wind in spreading the virus will still need to be investigated.
Our results indicated that the virus entered southwest France through Tarn and that this event represented a single source of the epidemic. This observation is likely to be robust because (i) all the samples from Tarn are monophyletic and connect to the root, (ii) terminal branches are short, and (iii) it is consistent with a recent study based on phylogenetic approaches (Briand et al., 2021). Although Tarn itself is not considered to have a high exposure risk from migratory birds, the single introduction pattern is likely to be explained by the European routes of migratory waterfowls because the estimated emergence interval coincided with the winter migration season (October–November) of many wild waterfowl species. These species are now known to carry and spread multiple avian influenza A viruses in poultry around the world, Still, we cannot completely reject the possibility of multiple introductions of a virus on the basis of the phylogeny alone (da Silva Filipe et al., 2021; Hill et al., 2015). Indeed, if the genomic sequences from distinct introductions are similar and in turn form monophyletic clusters, then the number of introductions could be underestimated. However, given avian influenza viruses are RNA viruses with high mutations rates, this alternative hypothesis is unlikely to hold. Our finding of a single introduction that acted as the source of the whole epidemic supports a previous phylogenetic study (Briand et al., 2021) and is consistent with outbreak investigations and epidemiological analyses (Andronico et al., 2019; Guinat, Nicolas, et al., 2018). Indeed, the index case in poultry was suspected and confirmed in a duck‐breeding farm in Tarn, the day after that farm sent live‐ducks to force‐feeding farms in Gers and Lot‐et‐Garonne. This event is likely to have triggered the viral spread (Guinat et al., 2020).
Overall, by successfully analyzing the impact of control measures, we showed the versatility of viral phylodynamic methods in providing both a broad understanding of epidemics and the means to test hypotheses related to specific control measures. Finally, the methodology used in this study can be efficiently adapted to study the impact of control measures on many other viral epidemics.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to. No ethical approval was required because the data was already published.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
This work was financially supported by the FEDER/Région Occitanie Recherche et Sociétés 2018—AI‐TRACK and the PREDYT project (Fonds pour la Recherche sur l'Influenza Aviaire). This study was also supported by the “Chair for Avian Biosecurity”, hosted by the National Veterinary College of Toulouse and funded by the Direction Generale de l'Alimentation, Ministère de l'Agriculture et de l'Alimentation, France. NFM is funded by the Swiss National Science Foundation (P2EZP3_191891). CG is funded by the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No. 842621. DC gratefully acknowledges the contribution of the INRAE MaIAGE unit in the form of free access to their MIGALE computer cluster. We also thank all the members of the EPIDESA team for their support and help.
Chakraborty, D. , Guinat, C. , Müller, N. F. , Briand, F.‐X. , Andraud, M. , Scoizec, A. , Lebouquin, S. , Niqueux, E. , Schmitz, A. , Grasland, B. , Guerin, J.‐L. , Paul, M. C. , & Vergne, T. (2022). Phylodynamic analysis of the highly pathogenic avian influenza H5N8 epidemic in France, 2016–2017. Transboundary and Emerging Diseases, 69, e1574–e1583. 10.1111/tbed.14490
DATA AVAILABILITY STATEMENT
All data, including genetic data accession numbers, XML files and R scripts, are available in the following repository, https://github.com/dc27708/H5N8_MASCOT. We have also added the viral sequence accession numbers from Briand et al., 2021 as Supporting Information S2 and other metadata information as Supporting Information S3.
REFERENCES
- Adlhoch, C. , Fusaro, A. , Gonzales, J. L. , Kuiken, T. , Marangon, S. , Niqueux, É. , Staubach, C. , Terregino, C. , Guajardo, I. M. , Lima, E. , & Baldinelli, F. (2021). Avian influenza overview December 2020 – February 2021. EFSA Journal, 19(3), e06497. 10.2903/j.efsa.2021.6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronico, A. , Courcoul, A. , Bronner, A. , Scoizec, A. , Lebouquin‐Leneveu, S. , Guinat, C. , Paul, M. C. , Durand, B. , & Cauchemez, S. (2019). Highly pathogenic avian influenza H5N8 in south‐west France 2016–2017: A modeling study of control strategies. Epidemics, 28, 100340. 10.1016/j.epidem.2019.03.006 [DOI] [PubMed] [Google Scholar]
- Benjamin, D. J. , Berger, J. O , Johannesson, M. , Nosek, B. A. , Wagenmakers, E.‐J. , Berk, R. , Bollen, K. A. , Brembs, B. , Brown, L. , Camerer, C. , Cesarini, D. , Chambers, C. D. , Clyde, M. , Cook, T. D. , De Boeck, P. , Dienes, Z. , Dreber, A. , Easwaran, K. , Efferson, C. , … Johnson, V. E. , (2018). Redefine statistical significance. Nature Human Behaviour, 2(1), 6–10. 10.1038/s41562-017-0189-z [DOI] [PubMed] [Google Scholar]
- Bouckaert, R. , Heled, J. , Kühnert, D. , Vaughan, T. , Wu, C.‐H. , Xie, D. , Suchard, M. A. , Rambaut, A. , & Drummond, A. J. (2014). BEAST 2: A software platform for Bayesian evolutionary analysis. Plos Computational Biology, 10(4), e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand, F.‐X. , Niqueux, E. , Schmitz, A. , Martenot, C. , Cherbonnel, M. , Massin, P. , Kerbrat, F. , Chatel, M. , Guillemoto, C. , & Guillou‐Cloarec, C. (2021). Highly pathogenic avian influenza A (H5N8) virus spread by short‐and long‐range transmission, France, 2016–17. Emerging Infectious Diseases, 27(2), 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner, A. , Niqueux, E. , Schmitz, A. , Le Bouquin, S. , Huneau‐Salaün, A. , Guinat, C. , Paul, M. C. , Courcoul, A. , & Durand, B. (2017). Description de l’épisode d'influenza aviaire hautement pathogène en France en 2016–2017. Bulletin épidémiologique, santé animale etalimentation, 79, 13–17. [Google Scholar]
- Cantrell, D. L. , Groner, M. L. , Ben‐Horin, T. , Grant, J. , & Revie, C. W. (2020). Modeling pathogen dispersal in marine fish and shellfish. Trends in Parasitology, 36(3), 239–249. 10.1016/j.pt.2019.12.013 [DOI] [PubMed] [Google Scholar]
- Craft, M. E. (2015). Infectious disease transmission and contact networks in wildlife and livestock. Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1669), 20140107. 10.1098/rstb.2014.0107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Filipe, A. , Shepherd, J. G. , Williams, T. , Hughes, J. , Aranday‐Cortes, E. , Asamaphan, P. , Ashraf, S. , Balcazar, C. , Brunker, K. , Campbell, A. , Carmichael, S. , Davis, C. , Dewar, R. , Gallagher, M. D. , Gunson, R. , Hill, V. , Ho, A. , Jackson, B. , James, E. , … Thomson, E. C. (2021). Genomic epidemiology reveals multiple introductions of SARS‐CoV‐2 from mainland Europe into Scotland. Nature Microbiology, 6(1), 112–122. 10.1038/s41564-020-00838-z [DOI] [PubMed] [Google Scholar]
- Dellicour, S. , Baele, G. , Dudas, G. , Faria, N. R. , Pybus, O. G. , Suchard, M. A. , Rambaut, A. , & Lemey, P. (2018). Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. Nature Communications, 9(1), 2222. 10.1038/s41467-018-03763-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas, G. , Carvalho, L. M. , Bedford, T. , Tatem, A. J. , Baele, G. , Faria, N. R. , Park, D. J. , Ladner, J. T. , Arias, A. , Asogun, D. , Bielejec, F. , Caddy, S. L. , Cotten, M. , D'Ambrozio, J. , Dellicour, S. , Di Caro, A. , Diclaro, J. W. , Duraffour, S. , Elmore, M. J. , … Rambaut, A. (2017). Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature, 544(7650), 309–315. 10.1038/nature22040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, N. M. , Donnelly, C. A. , & Anderson, R. M. (2001). The foot‐and‐mouth epidemic in Great Britain: Pattern of spread and impact of interventions. Science, 292(5519), 1155–1160. 10.1126/science.1061020 [DOI] [PubMed] [Google Scholar]
- Fourment, M. , & Holmes, E. C. (2015). Avian influenza virus exhibits distinct evolutionary dynamics in wild birds and poultry. BMC Evolutionary Biology, 15(1), 120. 10.1186/s12862-015-0410-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, S. D. W. , & Volz, E. M. (2010). Viral phylodynamics and the search for an ‘effective number of infections’. Philosophical Transactions of the Royal Society B: Biological Sciences, 365(1548), 1879–1890. 10.1098/rstb.2010.0060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinat, C. , Artois, J. , Bronner, A. , Guérin, J. L. , Gilbert, M. , & Paul, M. C. (2019). Duck production systems and highly pathogenic avian influenza H5N8 in France, 2016–2017. Scientific Reports, 9(1), 6177. 10.1038/s41598-019-42607-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinat, C. , Durand, B. , Vergne, T. , Corre, T. , Rautureau, S. , Scoizec, A. , Lebouquin‐Leneveu, S. , Guérin, J.‐L. , & Paul, M. C. (2020). Role of live‐duck movement networks in transmission of avian influenza, France, 2016–2017. Emerging Infectious Diseases Journal, 26(3). 10.3201/eid2603.190412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinat, C. , Nicolas, G. , Vergne, T. , Bronner, A. , Durand, B. , Courcoul, A. , Gilbert, M. , Guérin, J.‐L. , & Paul, M. C. (2018). Spatio‐temporal patterns of highly pathogenic avian influenza virus subtype H5N8 spread, France, 2016 to 2017. Eurosurveillance, 23(26), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinat, C. , Rouchy, N. , Camy, F. , Guérin, J. L. , & Paul, M. C. (2018). Exploring the wind‐borne spread of highly pathogenic avian influenza H5N8 during the 2016–2017 epizootic in France. Avian Diseases, 63(1s), 246–248. 10.1637/11881-042718-ResNote.1 [DOI] [PubMed] [Google Scholar]
- Guinat, C. , Vergne, T. , Kocher, A. , Chakraborty, D. , Paul, M. C. , Ducatez, M. , & Stadler, T. (2021). What can phylodynamics bring to animal health research? Trends in Ecology and Evolution, 36(9), 837–847. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.‐F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Guindon, S. , & Maio, N. D. (2021). Accounting for spatial sampling patterns in Bayesian phylogeography. Proceedings of the National Academy of Sciences, 118(52). 10.1073/pnas.2105273118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, S. C. , Lee, Y.‐J. , Song, B.‐M. , Kang, H.‐M. , Lee, E.‐K. , Hanna, A. , Gilbert, M. , Brown, I. H. , & Pybus, O. G. (2015). Wild waterfowl migration and domestic duck density shape the epidemiology of highly pathogenic H5N8 influenza in the Republic of Korea. Infection, Genetics and Evolution, 34, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, E. C. , & Grenfell, B. T. (2009). Discovering the phylodynamics of RNA viruses. PLOS Computational Biology, 5(10), e1000505. 10.1371/journal.pcbi.1000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, R. E. , & Raftery, A. E. (1995). Bayes factors. Journal of the American Statistical Association, 90(430), 773–795. [Google Scholar]
- Keeling, M. J. , Woolhouse, M. E. J. , Shaw, D. J. , Matthews, L. , Chase‐Topping, M. , Haydon, D. T. , Cornell, S. J. , Kappey, J. , Wilesmith, J. , & Grenfell, B. T. (2001). Dynamics of the 2001 UK foot and mouth epidemic: Stochastic dispersal in a heterogeneous landscape. Science, 294(5543), 813–817. 10.1126/science.1065973 [DOI] [PubMed] [Google Scholar]
- Keysers C., Gazzola V., & Wagenmakers E.‐J. (2020). Using Bayes factor hypothesis testing in neuroscience to establish evidence of absence. Nature Neuroscience, 23(7), 788–799. 10.1038/s41593-020-0660-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman, Z. (2016). One Health and culling as a public health measure. Public Health Ethics, 9(1), 5–23. 10.1093/phe/phw002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Menach, A. , Vergu, E. , Grais, R. F. , Smith, D. L. , & Flahault, A. (2006). Key strategies for reducing spread of avian influenza among commercial poultry holdings: Lessons for transmission to humans. Proceedings of the Royal Society B: Biological Sciences, 273(1600), 2467–2475. 10.1098/rspb.2006.3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey, P. , Suchard, M. , & Rambaut, A. (2009). Reconstructing the initial global spread of a human influenza pandemic. PLoS Currents, 1. 10.1371/currents.RRN1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycett, S. J. , Duchatel, F. , & Digard, P. (2019). A brief history of bird flu. Philosophical Transactions of the Royal Society B, 374(1775), 20180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansley, L. M. (2004). The challenge of FMD control in the 2001 UK FMD epidemic. Animal production in Europe: The way forward in a changing world “in‐between” Congress of the International Society for Animal Hygiene. Citeseer. [Google Scholar]
- Martin, D. P. , Murrell, B. , Golden, M. , Khoosal, A. , & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows, A. J. , Mundt, C. C. , Keeling, M. J. , & Tildesley, M. J. (2018). Disentangling the influence of livestock vs. farm density on livestock disease epidemics. Ecosphere, 9(7), e02294. 10.1002/ecs2.2294 [DOI] [Google Scholar]
- Minh, B. Q. , Schmidt, H. A. , Chernomor, O. , Schrempf, D. , Woodhams, M. D. , Von Haeseler, A. , & Lanfear, R. (2020). IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37(5), 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, N. F. , & Bouckaert, R. R. (2020). Adaptive parallel tempering for BEAST 2. BioRxiv. 10.1101/603514 [DOI] [Google Scholar]
- Müller, N. F. , Dudas, G. , & Stadler, T. (2019). Inferring time‐dependent migration and coalescence patterns from genetic sequence and predictor data in structured populations. Virus Evolution, 5(2), vez030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, N. F. , Rasmussen, D. , & Stadler, T. (2018). MASCOT: Parameter and state inference under the marginal structured coalescent approximation. Bioinformatics, 34(22), 3843–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2020). R: A language and environment for statistical computing. . (4.0.3) [Computer software]. R Foundation for Statistical Computing, Vienna, Austria. https://www.R‐project.org/ [Google Scholar]
- Rambaut, A. (2016). FigTree (1.4.3) [Computer software]. http://tree.bio.ed.ac.uk/software/figtree/
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Lam, T. T. , Max Carvalho, L. , & Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evolution, 2(1), vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoizec, A. , Niqueux, E. , Thomas, R. , Daniel, P. , Schmitz, A. , & Le Bouquin, S. (2018). Airborne detection of H5N8 highly pathogenic avian influenza virus genome in poultry farms, France. Frontiers in Veterinary Science, 5. 10.3389/fvets.2018.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si, Y.‐J. , Lee, I. W. , Kim, E.‐H. , Kim, Y.‐I. , Kwon, H.‐I. , Park, S.‐J. , Nguyen, H. D. , Kim, S. M. , Kwon, J.‐J. , Choi, W.‐S. , Beak, Y. H. , Song, M.‐S. , Kim, C.‐J. , Webby, R. J. , & Choi, Y.‐K. (2017). Genetic characterisation of novel, highly pathogenic avian influenza (HPAI) H5N6 viruses isolated in birds, South Korea, November 2016. Eurosurveillance, 22(1), 30434. 10.2807/1560-7917.ES.2017.22.1.30434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Beest, D. E. , Hagenaars, T. J. , Stegeman, J. A. , Koopmans, M. P. , & van Boven, M. (2011). Risk based culling for highly infectious diseases of livestock. Veterinary Research, 42(1), 81. 10.1186/1297-9716-42-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tildesley, M. J. , Brand, S. , Brooks Pollock, E. , Bradbury, N. V. , Werkman, M. , & Keeling, M. J. (2019). The role of movement restrictions in limiting the economic impact of livestock infections. Nature Sustainability, 2(9), 834–840. 10.1038/s41893-019-0356-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Andel, M. , Tildesley, M. J. , & Gates, M. C. (2021). Challenges and opportunities for using national animal datasets to support foot‐and‐mouth disease control. Transboundary and Emerging Diseases, 68(4), 1800–1813. [DOI] [PubMed] [Google Scholar]
- Vergne, T. , Gubbins, S. , Guinat, C. , Bauzile, B. , Delpont, M. , Chakraborty, D. , Gruson, H. , Roche, B. , Andraud, M. , Paul, M. , & Guérin, J.‐L. (2021). Inferring within‐flock transmission dynamics of highly pathogenic avian influenza H5N8 virus in France, 2020. Transboundary and Emerging Diseases, 68(6), 3151–3155. 10.1111/tbed.14202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz, E. M. , Koelle, K. , & Bedford, T. (2013). Viral phylodynamics. PLOS Computational Biology, 9(3), e1002947. 10.1371/journal.pcbi.1002947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Müller, N. F. , Bouckaert, R. , Xu, B. , & Drummond, A. J. (2019). Bayesian phylodynamics of avian influenza A virus H9N2 in Asia with time‐dependent predictors of migration. PLOS Computational Biology, 15(8), e1007189. 10.1371/journal.pcbi.1007189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Zhao, X. , Lemey, P. , Suchard, M. A. , Bi, Y. , Shi, W. , Liu, D. , Qi, W. , Zhang, G. , Stenseth, N. C. , Pybus, O. G. , & Tian, H. (2020). Assessing the role of live poultry trade in community‐structured transmission of avian influenza in China. Proceedings of the National Academy of Sciences, 117(11), 5949–5954. 10.1073/pnas.1906954117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ypma, R. J. F. , Jonges, M. , Bataille, A. , Stegeman, A. , Koch, G. , van Boven, M. , Koopmans, M. , van Ballegooijen, W. M. , & Wallinga, J. (2013). Genetic data provide evidence for wind‐mediated transmission of highly pathogenic avian influenza. The Journal of Infectious Diseases, 207(5), 730–735. 10.1093/infdis/jis757 [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Richardson, B. , Takle, E. , Chai, L. , Schmitt, D. , & Xin, H. (2019). Airborne transmission may have played a role in the spread of 2015 highly pathogenic avian influenza outbreaks in the United States. Scientific Reports, 9(1), 11755. 10.1038/s41598-019-47788-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
All data, including genetic data accession numbers, XML files and R scripts, are available in the following repository, https://github.com/dc27708/H5N8_MASCOT. We have also added the viral sequence accession numbers from Briand et al., 2021 as Supporting Information S2 and other metadata information as Supporting Information S3.