

Abstract
Alzheimer’s Disease (AD) is the most prevalent form of dementia across the world. While its discovery and pathological manifestations are centered on protein aggregations of amyloid-beta (Aβ) and hyperphosphorylated tau protein, neuroinflammation has emerged in the last decade as a main component of the disease in both pathogenesis and progression. As the main innate immune cell type in central nervous system (CNS), microglia play a very important role in regulating neuroinflammation, which occurs commonly in neurodegenerative conditions including AD. Under inflammatory response, microglia undergo morphological changes and status transition from homeostatic to activated forms. Different microglia subtypes displaying distinct genetic profiles have been identified in AD, and these signatures often link to AD risk genes identified from the genome-wide association studies (GWAS), such as APOE and TREM2. Furthermore, many of AD risk genes are highly enriched in microglia and specifically influence the functions of microglia in pathogenesis, e.g. releasing inflammatory cytokines and clearing Aβ. Therefore, building up a landscape of these risk genes in microglia, based on current preclinical studies and in the context of their pathogenic or protective effects, would largely help us to understand the complexed etiology of AD and provide new insight for the unmet need of effective treatment.
Keywords: Alzheimer’s Disease, microglia, neuroinflammation, AD risk genes
Introduction
Alzheimer’s Disease (AD) is a chronic, devastating neurodegenerative disorder characterized by progressive impairment of behavioral and cognitive functions. It is the most common subtype of age-related dementia, affecting ~50 million individuals worldwide [1, 2]. Of note, the global prevalence of AD is still increasing and predicted to double every 20 years [2, 3]. As a major contributor to death and disability among the aging population, it places a formidable burden on the health-care systems [1, 4]. Despite enormous efforts in drug development, no effective cure currently exists [5]. Thus, there is an urgent need for an in-depth understanding of the etiology and pathogenesis of AD, which will facilitate the development of novel disease-modifying therapeutic strategies to prevent or slow down the progression of AD.
AD has cardinal pathological features including extracellular Aβ depositions often in the form of plaques, and formation of intracellular neurofibrillary tangles (NFTs) from hyperphosphorylated tau [5]. However, it is a heterogeneous disorder with complex etiology, particularly in the vast majority of sporadic cases. Accumulating evidence demonstrated that gliosis and neuroinflammation not only strongly correlate with AD pathological hallmarks, but also exert a causal role in disease pathogenesis [6, 7]. Instead of being a mere bystander activated by emerging senile plaques and NFTs, these processes may contribute as much to neuronal dysfunction and AD pathogenesis [6, 7]. Therefore, a comprehensive characterization of gliosis and neuroinflammation will provide critical insights into the underlying molecular mechanisms and identify potential effective targets for therapeutic intervention in AD.
Microglia, the brain-resident myeloid cells, play pleiotropic physiological roles in both developing and adult central nervous system (CNS) [8], by promoting phagocytic clearance and providing trophic support for brain homeostasis and tissue repair [9]. Aberrations in the cellular properties and functions of microglia contribute to a list of neurological conditions, ranging from neurodevelopmental disorders to neurodegenerative diseases, such as autism and AD, respectively [8, 10]. In particular, evidence from clinical and pre-clinical studies has clearly demonstrated the critical roles of microglia in the pathogenesis of AD [11]. Not only these data indicate a microglial diversity in AD, such as disease-associated microglia (DAM), plaque-associated microglia (PAM), and dark microglia [12–14], but also a variety of AD risk genes, such as TREM2, CD33, APOE4, MS4A, and PLCG2, which are preferentially or selectively expressed in microglia [11, 15]. Moreover, these risk genes are correlated with amyloid plaque burden and disease severity [16]. For example, mutations or genetic variants of specific genes encoding important immune receptors such as TREM2 (triggering receptor expressed on myeloid cells 2) and CD33 are differentially associated with risks for developing AD [17, 18]. These findings reveal the complexed interactions between risk genes and other signaling pathways in microglia, which might be a key to understand the neuroinflammation in AD.
In this review, we explored recent advances regarding microglial genetics and how phenotypes influence the pathogenesis of AD. Specifically, the risk genes highly expressed in microglia and their pathophysiological implications in AD pathology were emphasized. Further elucidation of the genetic mechanisms that regulate microglial activation and function is particularly important, which will lead to uncovering potential new targets for future therapeutic intervention.
Historical Perspectives
The name of microglia came from more than a century of research [19]. In 1856, “neuroglia” was defined by pathologist Rudolf Virchow for the first time based on their spacing around neurons and the ability to hold together like glue [20], while in fact, “neuroglia” refers more as a macrophage population comprising of astrocytes and oligodendrocytes. In the end of 1800’s, W. Ford Robertson introduced the term of “mesoglia” to describe mesoderm-derived phagocytic elements in the nervous system that has a distinct origin from “neuroglia” [21] and claimed that “mesoglia” appear differently from “neuroglia” because they seemed to be phagocytic based on platinum substitution modification of Golgi technique [22]. Later, Santiago Ramon y Cajal successfully used the gold chloride sublimate method to separate astrocytes from “apolar cells” and referred to “mesoglia” as “the third element of nervous system” [23]. In 1919, Pio del Rio-Hortega, a student of Cajal, found that the apolar cells consisted of two different cell types during histological examinations using the ammoniacal silver carbonate method [24], for which he named “microglia” and “interfascicular” oligodendrocytes to describe the non-neural, non-astrocytic “third element” [24].
Although the microglia have been studied for decades, their developmental origin is still debated. Evidence from initial studies, including those performed from Rio-Hortega, indicated that microglia arose from embryonic progenitors [24]. Based on similarities in morphology and phagocytic features, microglia were believed to be of myeloid origin like other macrophage populations, and originate from blood-circulating monocytes [19]. In the last 50 years, the Yolk Sac (YS) origin of microglia has been extensively discussed. By applying markers Mac-1 and F4/80 to label the cells dissociated from the forebrain, Alliot et al. found that embryonic brain parenchyma contained potential progenitors for microglia in C57BL/J6 mice [25]. Further investigations from the same group suggested that YS-derived microglia progenitors migrate into head mesenchyme and brain rudiment at embryonic day (ED) 8, where they further proliferate [26]. This theory of YS origin has been confirmed with a wide range of tracing techniques, including fate-mapping. For example, Ginhoux et al. used in vivo lineage tracing to demonstrate that Runx1-positive mouse myeloid progenitors in the YS seed the brain between ED8.5 and ED9.5 [27]. As Ncx−/− embryos, which have normal YS hematopoiesis but lack heartbeat and normally functional blood circulation [28], failed to develop any microglia at ED9.5 [27], which suggested that Runx1+ myeloid progenitors migrate to brain from YS through blood circulation before the establishment of the blood-brain barrier. Taken together, the endogenous microglial population is derived predominantly from primitive myeloid progenitors in the YS during early development, and maintained by local self-renewal, which is independent of hematopoiesis under pathophysiological conditions [29]. The potential contribution of peripherally derived microglial/macrophage-like cells in neurological diseases is more complicated and remains to be explored [30, 31].
From Homeostasis to activation
Microglia are the sentinels of the brain’s innate immune system, continuously monitoring the surrounding microenvironment [32]. Under normal physiological conditions, they exhibit a highly “ramified” morphology with a small soma; while in response to brain injury or in disease conditions, they can be activated rapidly and swiftly change to an “amoeboid” phenotype, which is characterized by shortened and extensively branched processes, as well as hypertrophy of the cell body [33]. There is a well-established classification system for various phenotypes microglia (Figure 1). M0 microglia, also known as the “resting” microglia, maintain the homeostasis of the CNS [33], which can further acquire either M1 or M2 phenotypes upon activation. The classical M1-like microglia exhibit cytotoxic and pro-inflammatory activities [6], and carry signature markers such as CD16, CD32, iNOS, IL-1β, IL-6 and TNF-α in mouse microglia and CD40, CD86 in human microglia [34–37]; in contrast, the alternatively activated M2-like microglia express signature markers such as CD206 (also known as macrophage mannose receptor 1), YM-1, FIZZ1, IL-10 as mouse marker and Arginase 1 (Arg-1), TREM2, and TGF-β for both human and murine. In addition, M2-like microglia often promote inflammation resolution and tissue regeneration [38–40]. According to distinct activation mechanisms and functions, M2-like microglia can be further divided in three subtypes, M2a, M2b, and M2c [41]. The M2a phenotype, typically stimulated by exposure to IL-4 and IL-13, is closely associated with anti-inflammatory and restorative processes [42]. By contrast, the M2b phenotype, often induced by the fusion of Fc gamma receptors (FcγRII) and immunoglobulin G (IgG) complexes, regulates selective phagocytosis and inflammatory responses [43]. Alternatively, the M2c phenotype, which occurs in response to IL-10 and TGF-β, plays crucial roles in immunosuppression, phagocytosis and tissue remodeling [44]. It is noteworthy that microglial phenotypes may be intermittent and dynamic in different pathophysiological conditions, and therefore further elucidation of these cellular and molecular changes will provide insights into the fundamental functions of microglia in CNS injuries and diseases.
Figure 1. Microglia subtypes and activation process.
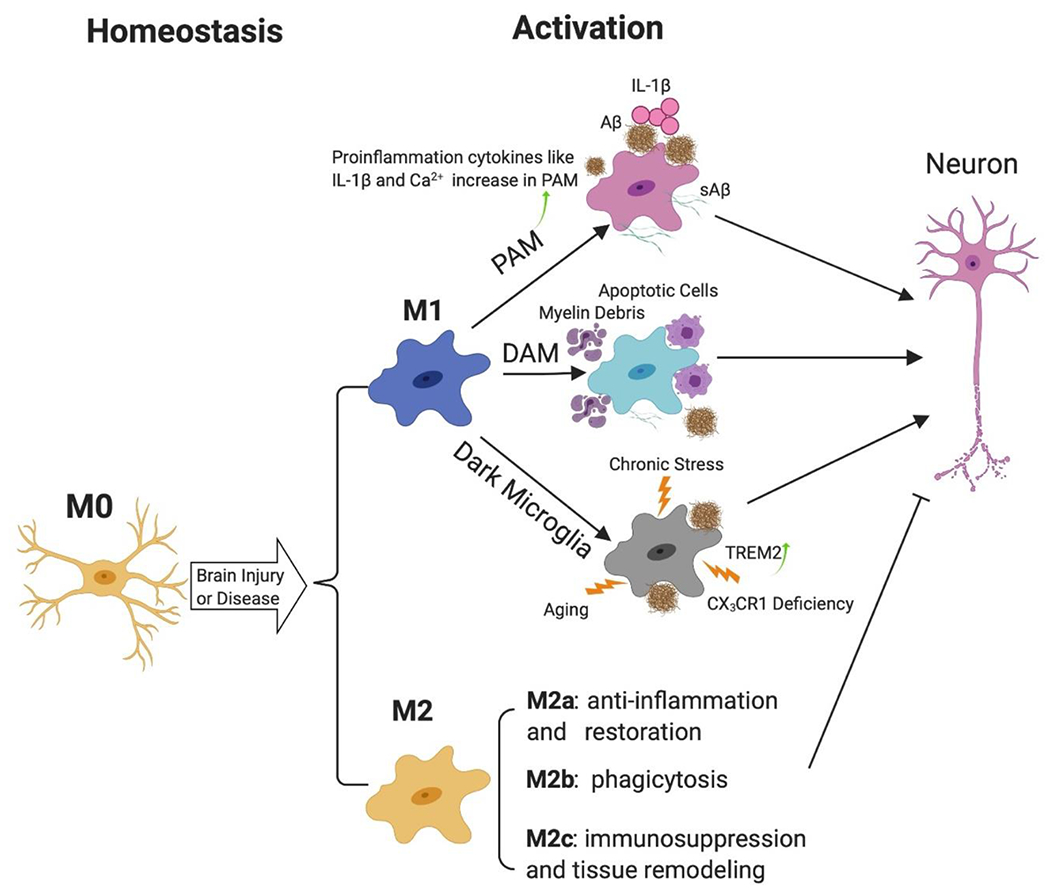
Microglia can be activated from homeostatic status (M0) to activate forms (M1 or M2), which can be further categorized into different subtypes. After activation, PAM expresses abundant of IL-1β and shows characteristic gene profile changes. Increased intracellular Ca2+ contributes to its hyperreactive immune response. DAM can be activated by various damaged-associated molecular patterns (DAMPs), which impair the phagocytosis and proteostasis functions. Dark Microglia can be activated by aging, chronic stress, AD pathologies or CX3CR1 deficiency. TREM2 is increasingly expressed in dark microglia near Aβ plaques. While the M2 subtypes, including M2a, M2b and M2c, in general are anti-inflammatory and promote proteostasis and repair. The inflammatory cytokines and neurotoxic factors produced by activated M1 microglia lead to neuro damage, which can stimulate more Aβ plaques made by neuron and go back to activate microglia further. Abbreviations: IL-1β: interleukin-1β; Aβ: β-amyloid; sAβ: soluble β-amyloid; PAM: Plaque-associated microglia; DAM: Disease-associated microglia.
Microglial transcriptomics in AD
Accumulating evidence has revealed microglia as an indispensable contributor to neuroinflammation and neurodegeneration [45], therefore understanding the diversity and subtype categorization of microglia is a key towards deciphering their fundamental roles in AD. Notably, microglial transcriptomes by bulk population sequencing have characterized the microglial diversity in age, neurodegenerative diseases and psychiatric disorders [46, 47]. However, the specific contributions from different microglia subtypes to brain pathologies remain unclear from bulk sequencing results, sometimes even controversial [33]. The advances in single-cell techniques, such as cytometry by time-of-flight mass spectrometry (CyTOF) as well as single-cell RNA sequencing, offer a good solution, as they detect cell types and intermediate cell states in an unbiased fashion. Such single-cell RNA technologies have revealed the spatio-temporal diversity of microglia under both homeostatic and pathological conditions in human and animal models [48]. For example, Hammond et al. discovered that there are at least 8 subtypes of microglia in the naïve mouse brain, and their transcriptomic changes are associated with age and gender [49], regional heterogenicity [50], and AD pathogenesis [51–53]. Therefore, in the following section, we will survey important microglial subtypes that are closely associated with AD at both pathological and transcriptomic levels.
Disease-associated microglia (DAM) is the most recently identified subtype of microglia [54], which has been detected in the 5xFAD transgenic mouse model by single-cell and single-nucleus RNA-seq analysis [52]. As reported, Keren-Shaul et al. sorted CD45+ immune cells from 5xFAD mouse brains and compared the gene profiling differences with control groups via massively parallel single-cell RNA-seq (MARS-Seq). In this study, five groups of immune cells including DAM were identified, which exhibited distinct cellular states in AD brains compared to that of wild-type controls. In addition, histological analysis confirmed that DAM is uniquely localized around intercellular Aβ plaque particles in both mouse and human AD samples, but are not present in healthy brain tissue [52]. More interestingly, the gene expression of TREM2, CTSD, TYROBP and APOE is significantly upregulated while the transcripts for P2RY12, P2RY13, CX3CR1, CD33 and TMEM119 are heavily downregulated in DAM. These findings indicate a potential crucial role of DAM in microglial phagocytic and/or endocytic functions, as well as inflammatory cascades [52].
Plaque-associated microglia (PAM) is known as a cell population of microglia which are stimulated by insoluble Aβ [54]. They can be detected in human AD brains by using immunohistochemical techniques [55]. Also, the transformed morphological features of PAM and their spatial distance to Aβ plaques was further revealed by 3D cell reconstruction in TgCRND8 mouse [56, 57]. Furthermore, Brawek et al. revealed substantial dysfunction of PAM in both APP23/PS45 [58] and APP/PS1 [59] mouse models, and demonstrated that aberrant intracellular calcium signaling may trigger release of toxic species from microglia in the plaque vicinity [60].
Dark microglia were discovered as a new subtype of microglia in APP/PS1 mice with electron microscopy (EM) by Bisht et al. [12] Ultrastructural analysis demonstrated that they are strikingly different from the other phenotypes of microglia. Dark microglia show signs of oxidative stress including condensed cytoplasm and nucleoplasm with “dark” appearance under electron microscopy [12]. In addition, they are rarely observed in healthy brains, but often detected in individuals with chronic stress, aging and AD. Besides morphological changes, dark microglia have a specific gene profile with downregulated of homeostatic markers such as Iba1, P2RY12 and CX3XR1 [54], and upregulation in TREM2 expression, as well as increased immunoreactivity to the microglial-specific antibody 4D4 near amyloid plaques [12, 61]. Currently, the pathophysiological contribution of dark microglia to AD, however, remains unclear and warrants further investigations [54].
Human Alzheimer Microglia (HAM) refers to a special subtype of microglia found in AD patients which exhibits a unique gene expression patterns when compared to normal human microglia [54] or DAM from mouse AD models [62]. Srinivasan et al. applied a novel method for sorting myeloid cells from frozen post-mortem specimens of superior frontal gyrus (SFG) and fusiform gyrus in human AD tissue, the gene expression changes observed by RNA-Seq in these myeloid cells were not similar with DAM profile in mouse AD models. They named this new profile in human Alzheimer’s microglia/myeloid cells as HAM signature, in which half of these changes are consistent with healthy human aging profile while the other half only altered in AD-related genes like APOE, highlighting the significant differences of myeloid cells between human diseases and mouse models [62]. The potential reasons of such distinct differences between HAM and DAM may be driven by different immune system responses, timeline of AD progression or specific effects of microglia activation in human and mice [54].
AD-Associated Risk Genes in Microglia
While most AD cases are late-onset and sporadic, genetic inheritance is still a major determinant, and over 40 genes associated with the disease [63, 64] or loci have been identified over the last decade [15, 65]. The increased AD GWAS projects and samples size lead to a larger scale of AD discovery as well as improving statistics power of AD relative variants. In this part, we will focus on a subset of AD risk genes that are enriched in microglia and discuss their potential molecular mechanisms in AD pathology in the context of microglia phenotypes. We are fully aware that most of AD associated SNPs are unique to the human genome, and cannot be simply modeled in mice, and the biological difference between human and mice is substantial. They differ not only at the nucleotide sequences level, but also on epigenetic regulations. Although most of these are human SNPs, their relevant murine models have offered substantial insights to their biological functions in microglia and relationship to AD.
APOE
Apolipoprotein E (APOE) is a lipid-binding protein that transports cholesterol and other lipids between cells and organs. The human APOE gene has three major alleles, ε2, ε3 and ε4, which confer amino acid variations at position 112 and 158. APOE3 (Cys-112 and Arg-158) is the most common isoform in population and considered as the “neutral” isoform, while APOE4 (Arg-112 and Arg-158) is the most significant genetic risk factor for late onset of AD (LOAD). Two copies of ε4 increase the likelihood of developing AD by >12 folds, and lowers the age of onset by nearly 17 years [66]. On the other hand, APOE2 (Cys-112 and Cys-158) is considered to be neuroprotective and associated with a nearly 50% of reduction in AD risk. Clinical and preclinical evidence has demonstrated an isoform-dependent (APOE4 > APOE3 > APOE2) and dose-dependent association with AD risks and pathophysiology [67], however, both APOE4 and APOE2 are linked to cerebral amyloid angiopathy (CAA)-related hemorrhage [68]. The role of APOE4 in AD can be attributed to both loss of protective function and gain of toxic function [66, 69]. In contrast to APOE3, APOE4 harms multiple cellular functions in increasing APP expression and Aβ production from neuronal cells [70, 71], decreasing Aβ clearance [72, 73], promoting Aβ aggregation [74, 75], reducing cerebrovascular functions and BBB integrity [76, 77], as well as impairing synaptic function [78, 79] and neuronal activities [71, 80], and causing brain atrophy [81, 82]. However, the exact mechanisms of APOE isoform-dependent actions on different brain cell types are not fully understood yet.
APOE is mainly produced by astrocytes in normal brain [66]. However, it is highly upregulated in DAM, as revealed by recent single cell RNA sequencing studies in both humans and mice [51–53]. In addition, using single-molecule fluorescence in situ hybridization (smFISH) in APP knock-in mouse model (AppNL-G-F) [83], Frigerio et al. found the microglia were enriched in Apoe transcripts, and the intensity of signals increased in cells located closer to Aβ plaques [84], suggesting that DAM are potentially the major local contributor of APOE around the plaques. Besides amyloid pathology, APOE also influences tauopathy [66, 67]. By comparing P301S tau transgenic mouse with human APOE knock-in (ε2, ε3 or ε4) mouse model, or with murine Apoe knock-out (EKO) mice, Holtzman’s lab found P301S/ε4 mice had significantly higher level of tau pathology and neuroinflammation, when compared with P301S/ε2, P301S/ε3 or P301S/EKO [85], demonstrating a direct link between APOE4 and tauopathy in AD. This phenotype attributes to a pro-inflammatory effect which promotes activation of both microglia and astrocytes [85], as well as the upregulation of serpin superfamily of protease inhibitors, Serpina3 in particular [86].
TREM2
As a member of the triggering receptors expressed on myeloid cells (TREM) family, TREM2 is mainly expressed in microglia and infiltrating macrophages in brain. TREM2 protein belongs to the immunoglobin superfamily and plays a critical role in the innate immune system. The extracellular ligands for TREM2 include various lipoproteins like low density lipoprotein (LDL), malondialdehyde-modified LDL (MDA-LDL), high density lipoprotein (HDL), apolipoprotein E and clusterin (CLU, also known as apoJ), as well as phospholipids, glycolipids, apoptotic neurons and even bacteria [87] (Figure 2). The transmembrane domain of TREM2 interacts with the DNAX-activating protein 10 (DAP10) and 12 (DAP12), which are adaptor proteins that mediate intercellular signaling through Phosphoinositide 3-kinase (PI3K) and Spleen tyrosine kinase (SYK), respectively [87, 88]. TREM2 can be cleaved by disintegrin and metalloproteinase domain containing proteins ADAM10 and ADAM17 and shred its ectodomain sTREM2, which will deactivate the TREM2/DAP12 signaling [87]. sTREM2 levels in the cerebrospinal fluid (CSF) are correlated with tau and microglia changes in early stage of AD, and can be potentially used as a fluid biomarker [89, 90].
Figure 2. AD risk genes and signaling pathways in microglia.
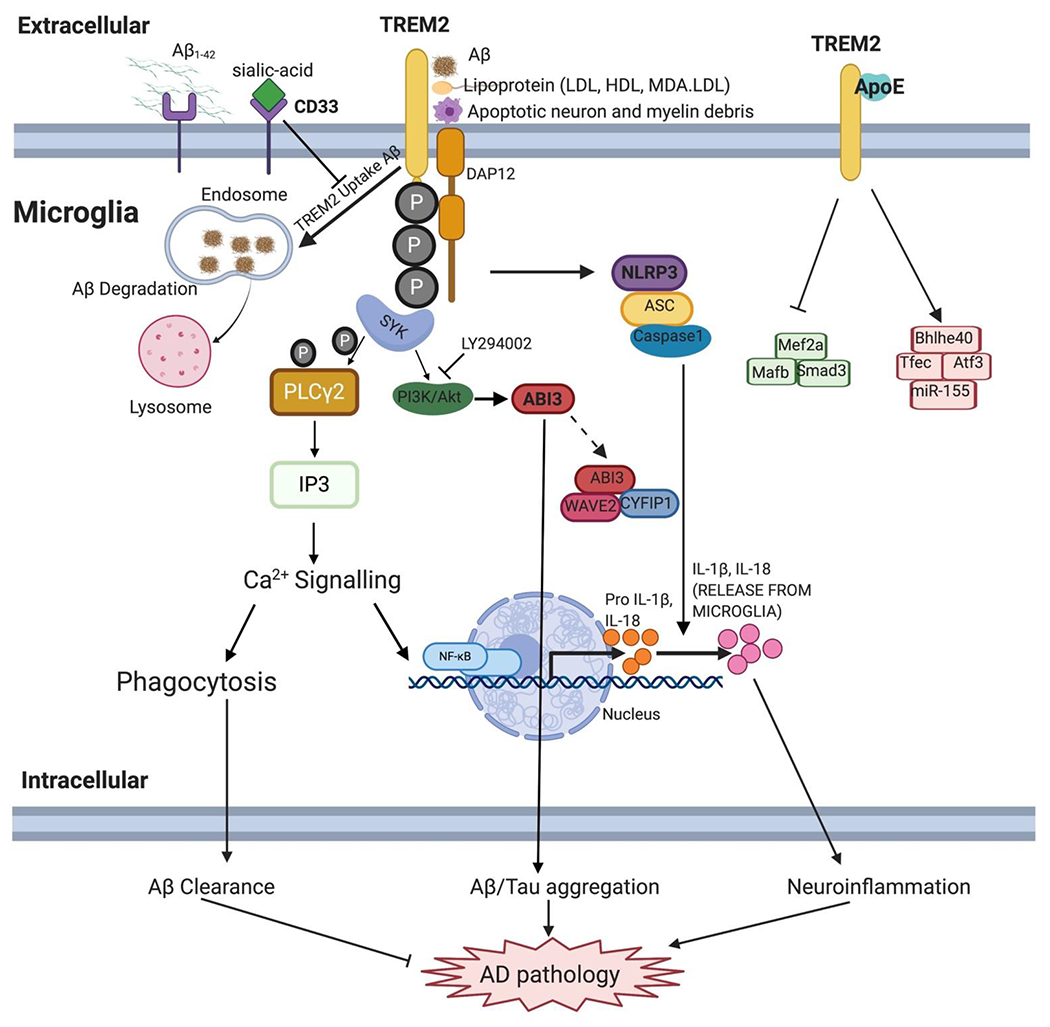
As a triggering receptor expressed on myeloid cells, TREM2 have various ligands from extracellular, including Aβ, LDL, HDL, apoptotic cells and ApoE. The combination of TREM2 and its ligand can phosphorylate ITAM of DAP12, then triggering SYK, PI3K/Akt and GSK3β downstream signaling. The interaction between APOE and TREM2 triggers the transcription regulation and microglia dysfunction. Microglia speck-like protein containing a CARD (ASC) and Capase-1 upregulate proinflammation cytokines IL-1β and IL-18. In the presence of sialic-acid, CD33 inhibits the TREM-dependent uptake of Aβ. LY294002, an inhibitor of PI3K/Akt pathway; Abi3 variants is associated with Aβ or tau aggregation in microglia and ABI3 may combine with CYFIP1 and WAVE2 to form wave regulator complex (WRC) and regulate microglia activation in AD. PLCγ2 can be phosphorylated by SYK and stimulate IP3, which then triggering Ca2+ signaling, modulating phagocytosis of microglia or activating NF-κB pathway. Abbreviations: Aβ: β-amyloid; LDL: low density lipoprotein; HDL: high density lipoprotein; MDA-LDL: malondialdehyde-modified LDL; DAP12: DNAX-activating protein; IL-1β: interleukin-1β; IL-18: interleukin-18.
Nonsense mutations in TREM2 and DAP12 are linked to Nasu-Hakola disease, a rare autosomal recessive condition of early onset frontotemporal dementia with leukoencephalopathy and basal ganglia calcification [91]. In 2013, a rare R47H variant of TREM2 (rs75932628) was identified as a risk allele for AD [18, 92], and the R47H mutation was predicted to be partial loss-of-function [18]. Data from NIMH AD Genetics Initiative Study and AD Sequencing Project indicated additional risk rare coding variants including R62H, D87N and H157Y, and a protective mutation R62C [93]. Yeh et al. used bio-layer interferometry (BLI) to test the effects of missense mutations on apolipoprotein and lipoprotein binding, and found R47H, D87N and R62H on hTREM2 all exhibited significant loss of binding abilities with LDL, CLU and APOE [94]. In addition, Atagi et al. confirmed R47H mutation significantly reduced the binding ability of TREM2 on all three isoforms of APOE [95].
During AD pathogenesis, microglia cluster around plaques forms a physical barrier limiting amyloid pathology and neurotoxicity [96, 97]. Microglia can efficiently uptake Aβ-LDL complex; however, this ability is dramatically inhibited in Trem2-KO microglia in vitro [94]. Although the amyloid phenotypes in Trem2-KO mice when crossed with different AD models were reported to be model-specific, age- and region-dependent [98], impaired microglia responses to amyloid are commonly observed regardless of model and disease stage [96–100]. The TREM2-dependent microglial endophenotype in vivo is consistent with transcriptomic changes at the single-cell level [51–53]. For example, Keren-Shaul et al. reported that the activation of DAM in AD requires a TREM2-dependent pathway in Trem2−/−; 5xFAD mice [52], for example, the regulation of APOE to DAM requires TREM2-dependent pathway [101]. In addition, the TREM2-APOE signaling associated with neurodegeneration can be characterized by inhibition of homeostatic transcription factors such as PU.1, MEF2A, MAFB, SALL1 and SMAD3, and activation of pro-inflammatory transcription factors such as BHLHE40, TFEC and ATF3, and MIR-155 [101]. Therefore, targeting TREM-APOE axis may represents a novel approach in restoring microglial homeostasis in AD.
CD33
CD33, a member of the sialic-acid binding immunoglobulin-like lectins (SIGLETs), anchors on the cytomembrane of microglia, macrophages, myeloid progenitor cells and monocytes. The extracellular part of CD33 consists of a N-terminal immunoglobulin domain is responsible for sialic acid recognition, as well as a C2-type immunoglobulin repeat. Intracellularly, human CD33 has two conserved cytoplasmic tyrosine-based motifs: a membrane-proximal ITIM and a membrane-distal ITIM-like motif; while the murine form lacks the ITIM motif [102]. CD33 plays important roles in immune cells, including cell adhesion, endocytosis, and inhibition of cytokines release [103].
CD33 was found modulating microglia activation and inhibiting Aβ by regulating its expression and alternatively splicing [104]. GWAS has identified that rs3865444 and rs12459419 are the two main CD33 variants that confer the risk for AD [105–107]. The major allele rs3865444C is linked to an increased risk of AD in Chinese, European, and North American populations [108] and is associated with cognitive decline in AD [109]. In contrast, the minor allele rs3865444A exerts a protective effect against AD which is also associated with increased CD33 expression level [110, 111].
CD33 is enriched on the surface of microglia, and inhibits microglia uptake of Aβ in the presence of sialic-acid [112, 113]. The amount of CD33-immunoreactive microglia positively correlates with Aβ burden in the same brain region [113]. Interestingly, TREM2 and CD33 exert opposite effects on Aβ pathology and microglia activation in 5xFAD mice. Compared to the exacerbated pathologies in Trem2−/−;5xFAD mice, CD33 deletion in Cd33−/−;5xFAD mice promoted microglia phagocytosis and neuroprotection [17]. This protection is abolished in the double knockout Cd33−/−;Trem2−/−;5xFAD mice, suggesting that TREM2 acts downstream of CD33 in modulating microglial pathology in AD [17].
PICALM
Phosphatidylinositol binding clathrin assembly protein (PICALM) regulates the formation of the clathrin lattice during endocytosis. Multiple single nucleotide polymorphisms (SNPs) within or close to the PICALM gene, such as rs3851179, have been associated with LOAD [114, 115]. It was first functionally linked to AD for its key function in mediating Aβ clearance across the blood-brain barrier [116]. Specifically, PICALM regulates endothelial internalization of Aβ that is bound to the low-density lipoprotein receptor-related protein-1 (LRP1), guides Aβ trafficking to Rab5-positive early endosomes and Rab11-positive sorting endosomes, and eventually leads to Aβ transcytosis from brain to circulation. Therefore, animal model with Picalm deficiency develops accelerated AD pathology. PICALM is also known to be highly expressed in microglia, and further upregulated in LOAD patients [117]. Although its exact function in AD-related microglia phenotypes is yet to be investigated, single cell sequencing has identified two gene-trait correlation modules in AD microglia, carrying a common molecular signature specific to AD risk genes including APOE, TREM2 and PICALM [53], suggesting a potential interaction between these genes in microglia.
CR1
Complement receptor 1, also known as c3b/c4b receptor or CD35, is a member of receptors of complement activation (RCA) family. The human CR1 gene is located in the complement related genes cluster on chromosome 1. It encodes type 1 membrane glycoprotein enriched in innate immune cells including microglia. CR1 modulates complement activation in multiple pathways contributing to inflammation and AD pathogenesis, for example, CR1 expressed in microglia is participated in immune clearance of Aβ in AD brain, it is upregulated when exposed to Aβ, and its blockage with neutralizing antibody attenuated microglia phagocytosis of Aβ [118]. In 2009, studies in nearly 4,000 AD cases from Belgium, Finland, Italy and Spain found CR1 rs6656401 polymorphism is associated with AD risk, with odds ratio (OR) of 1.21 [119]. Similar association between multiple CR1 polymorphisms and AD risks were later confirmed in different populations [120, 121], and remain highly significant in GWAS meta-analysis [114, 115]. Yet, the exact mechanism how CR1 variants are linked to AD pathogenesis remains unknown.
MS4A
Membrane-spanning 4-domains subfamily A (MS4A) is a cluster of genes that encode CD-20 like proteins with four transmembrane-spanning regions. Members of MS4A such as MS4A1, MA4A2, and MS4A4B participate in regulation of intracellular Ca2+ signaling. Two SNPs of the MS4A gene cluster, rs610932 and rs670139, were first reported as susceptibility loci associating with AD in 2011 with OR 0.91 and 1.08, respectively [106]. One of them, rs610932, is significantly associated with AD in Chinese Han population [122]. In addition, rs1562990 was found in Spain general population with an OR of 0.88[123]; and rs1582763 was the top SNP reached by GWAS using sTREM2 concentration in patient CSF samples (p=1.15×10−15) [124]. Furthermore, human macrophages treated with MS4A4A antibody showed decreased sTREM2 production, suggesting a functional interaction between MS4A and TREM2 in microglia.
ABCA7
ATP-binding cassette subfamily A member 7 (ABCA7) is a multi-pass transmembrane protein that harnesses energy from ATP hydrolysis and actively transfers molecules across cell membranes. It is expressed in microglia, macrophages, and subtypes of neurons in both humans and mice. ABCA7 variants with potential loss-of-functions are associated with AD risk, which nearly doubled in African-Americans [125]. Abca7 haplodeficiency in microglia impairs the immune response by disrupting the expression of CD14 after acute lipopolysaccharide stimulation in mouse models, as well as leading excessive microglial Aβ accumulation with increased endosomal compartments [126], suggesting Abca7 loss-of-functions in microglia may contribute to AD pathogenesis. ABCA7 also directly regulates phagocytic pathways in mouse microglia [126], and Abca7 deficiency in Abca7−/−;APP/PS1 mice accelerated amyloid pathology and neurodegeneration [127, 128].
ABI3
ABI3 is a member of Abelson (Abl) interactor (Abi) family of adaptor proteins. The ABI family regulates the actin cytoskeleton mammalian cells and commonly has three functional domains: a homeobox homology domain, a proline rich region and a Src-homology 3 (SH3) domain, besides that, ABI3 also contains a coiled-coil domain for dimerization. In invasive cancer cell lines, ABI3’s SH3 domain interacts with p21-activated kinase (PAK) to modulate ABI3 expression and cell motility [129]. ABI3 is also a key component of the ABI3/WAVE2 complex (AWC), which regulates actin polymerization during T cell activation [130]. The ABI3 common variant rs55978930 is association with decreased t-tau level in CSF in progressive mild cognitive impairment (pMCI) patients, and rs16947151 is associated with cognition decline in MCI patients [131]. More importantly, a missense change p.S209F in ABI3 at rs616338 was found by Sims et al. as a rare-coding risk variant using GWS association signals with LOAD. p.S209F is consistently associated with increased LOAD risk cross all the stages of AD [132]. ABI3 is enriched in microglia, however its role in microglia-related AD pathology remained underexplored.
PLCG2
Phospholipase C gamma 2 (PLCG2) encodes the 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-2 (PLCγ2), which is a transmembrane signaling enzyme producing inositol 1,4,5-trisphosphare (IP3) and diacylglycerol (DAG) as second messengers in immune cells (Figure 2). PLCγ2 has multiple conserved domains including PH, PU-PLC X-box, SH2 1, SH2 2, SH3, PI-PLC Y-box and C2. In microglia, PLCγ2 can promote many cellular processes like survival, proliferation and phagocytosis, as well as production of cytokines and chemokines[133]. A missense variant of PLCG2 p.P552R at rs72824905 was identified as a protective variant in AD by Sims et al [132]. Rare coding variants of ABI3 and PLCG2 are differentially association with AD risks in different cohorts across the world [134, 135], which indicates that ABI3 p.S209F functions as pathogenic variant while PLCG2 p.P522R is protective. This is confirmed in a recent report [136] demonstrating that PLCG2 p.P522R carriers had a much slower progression in cognition decline and lower pTau181 levels in CSF compared to non-carriers. In addition, there is a functional protein interplaying network among PLCG2, APOE and TREM2 in microglia, as the gene co-expressed with PLCG2 significantly overlap with the ones that co-expressed with TREM2 (P=1.37X10−33) and APOE (p=7.49×10−34) [136]. However, how these variants influence the protein normal functions in microglia and contribute to AD pathogenesis needs further understanding.
NLRP3 inflammasome
Increasing evidence suggested that innate immunity and inflammasome response strongly contribute to AD pathogenesis in brain. NLRP3 inflammasome is highly important for microglia activation, and its indispensable roles in Aβ plaque formation and tauopathy have been demonstrated [137]. The nucleotide-binding oligomerization domain-, leucine-rich repeat- and pyrin domain-containing 3 (NLRP3) inflammasome is a subcellular multiprotein complex, which can sense a wide range of exogenous and endogenous stimuli such microbes, aggregated and misfolded proteins, and adenosine triphosphate, which result in activation of caspase-1 [138]. NLRP3 inflammasome is comprised of a sensor NLRP3, an adaptor protein apoptosis associated speck-like protein containing a CARD (ASC) and an effector caspase-1[139]. Activated caspase-1 subsequently leads to the processing of pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18) and mediates rapid cell death [140]. Both IL-1β and IL-18 drive inflammatory responses through diverse downstream signaling pathways leading to neuronal damage and other neurological pathogenesis [139, 141–143].
Based on single cell RNA sequencing in humans, NLRP3 is mainly expressed in microglia [144]. It plays a critical role for initiating and maintaining neuroinflammation, as well as in the development of neurological disorders including AD [143]. Specifically, Nlrp3 is activated in AD and contributes to pathology in APP/PS1 mice because NLRP3 or caspase-1 deficiency leads to the alteration of microglia phenotypes and the decreased Aβ deposition [145]. Recent evidence showed that loss of NLRP3 inflammasome function in microglia reduced tau hyperphosphorylation and aggregation in Tau/Nlrp3−/− mouse model [146], identifying an important role of microglia and NLRP3 inflammasome activation in the pathogenesis of tauopathies. These studies clearly demonstrate that the NLRP3 inflammasome activation increases the risk for driving AD pathology and suggest that the manipulation of NLRP3 inflammasome activity is potentially promising way to treat AD. Indeed, targeting NLRP3 inflammasome signaling with small molecular inhibitors has been proven to ameliorate proinflammatory response in microglia and alleviate AD pathogenesis in preclinical studies [142, 147].
Developing AD therapeutic strategies on AD risk genes
There is currently no effective treatment for AD. Most AD therapeutic strategies mainly focus on preventing Aβ aggregation by modulating its enzymatic processing or clearance [148]. The genetic studies on TREM2 and its variants in microglia have enabled new advances in AD therapeutics. Monoclonal antibody AL002, which activates the TREM2 receptor and microglial immune response, has shown promising efficacy in preclinical studies for Aβ clearance and cognitive improvement [149]. Its human variant is now in phase 2 clinical trials. On the other hand, anti-APOE antibodies, such as HJ6.3 that binds to APOE associated with Aβ plaques [150] and HAE-4 that binds to non-lipidated APOE [151], have shown success in preclinical animal models by recruiting microglia to plaques and significantly reducing Aβ deposition. In addition, increasing APOE’s lipidation, designing APOE mimetics or interfering APOE and Aβ interaction all performed effective in mouse models [152]. As NLRP3 activation in microglia has been proved as a contributor for neuroinflammation and pathogenesis in AD [145, 146], therapeutics antagonizing the NLRP3 inflammasome are also under development right now.
Conclusion and Future Directions
In this review, we briefly introduced historical perspectives on microglia including the initial discovery and definition, demonstrated the classic activation of microglia from homeostatic (M0) to activated (M1/M2) status and summarized the diverse subtypes of microglia after being activated. Most importantly, we discussed the heterogeneity of the AD risk gene profile in microglia, like TREM2 and APOE which are well-established genetic risk factors, or ABI3 and PLCG2 which are novel contributors for AD progression. Among these risk genes closely associated with AD or other neurodegenerative disorders, some own their specific pathway to trigger pathologies of AD; some directly interacte with Aβ and modulate the phagocytosis of microglia; in addition, some risk genes closely collaborate within complex network stimulating microglia changing from homeostatic status to DAM. However, there are still many of them remaining unknown about their mechanisms for being an AD risk gene. The post-GWAS analysis is important for assessing the potential role of AD risk genes, the various hypotheses will lead more comprehensive biological mechanism uncovered and better characterize functional variants within AD [63]. Understanding microglia’s dynamics on morphology, activation, spatial distribution and gene expression profile enable the discovery of neurodegenerative diseases much easier and widely. With the advanced and continually improved technologies and analysis based on large clinical data from different populations of world-wide range, countless risk genes will show up for people to study, even though nowadays many of them keep remain unclear and unknown for us, identification and characterization of them can provide promising targets and benefits for developing treatments in clinical therapy for AD and other neurodegenerative diseases.
Table 1.
Alzheimer’s Disease Risk genes
| Gene | Activities in AD | Cellular Functions | |
|---|---|---|---|
| Microglia | Other cell types | ||
| TREM2 | TREM 2 variants R47H, D87N, R62C and H157Y cause partial loss of functions on microglia’s phagocytosis and clearance of Aβ, and increase AD risks. | Interacting with and phosphorylating DAP12 adaptor, mediating SYK, PI3K/Akt, GSK3β pathways and rising inflammasome NLRP3. | Cleaved by ADAM10 in dendritic cells; a negative regulator for TLR-4 expression in astrocytes; attenuate macrophage activation. |
|
| |||
| CD33 | Increased CD33 expression on microglia reduces the uptake of Aβ. rs3885444A allele is protective for AD. | A receptor on microglia activated by sialic acid-containing glycoproteins and glycolipids, recruiting proteins to inhibit phagocytosis. | Phosphorylates ITIM, docks SH2 domain-containing protein and inhibits phagocytosis in myeloid cells. |
|
| |||
| APOE | APOE4 is the strongest genetic risk for LOAD, it initiates and accelerates Aβ accumulation, aggregation and deposition in brain | ApoE is a main lipid ligand for TREM2 on microglia. | ApoE4 decreases uptake of palmitate and increases fatty acid oxidation and accumulates in astrocytes; stimulates expression of ADP-ARF6 and traps ABCA1 in endosome. |
|
| |||
| PICALM | Clearance of Aβ, playing a protective role in recognizing and shipping APP-CTF in autophagic degradation process. | Mediating trafficking endocytic protein during endocytosis. | PLCALM regulates internalization of Aβ, and guides Aβ trafficking and clearance in brain endothelial cells. |
|
| |||
| ABI3 | ABI3 variants may cause neurodegeneration through adjusting total level of tau protein in CSF or modulating cognitive function. | ABI3 participates in regulation of actin cytoskeleton and inhibits ectopic metastasis of tumor cells, as well as cell migration. | ABI3, CYFIP1 and WAVE constitute WRC, and regulates cytoskeleton dynamics in follicular thyroid cells, which is related to cancer development and metastasis. ABI3 also regulate cell migration in endothelial cells. |
|
| |||
| PlCG2 | Variant rs72824905G is a protective variant in AD and other neurodegenerative diseases. | PLCγ2 catalyzes the hydrolysis of phospholipids and produces diacylglycerols and water-soluble phosphorylated derivatives of the lipid head groups. | PLCγ2 involved in cell proliferation, transformation and tumor growth. |
|
| |||
| CR1 | The activity of microglial phagocytosis on Aβ is positively related with activation of CR1. | CR1 is a glycoprotein expressed in microglia and a main receptor for C3b which participates in immune activation. | CR1 regulates the binding between immune complex in erythrocytes. In B cells, CR1 forms a complex with CR2 to facilitate C3b or iC3b coated protein antigen uptake. |
|
| |||
| ABCA7 | The premature termination codon mutation in ABCA7 can cause increased risk on AD. ABCA7 haplodeficiency leads to excessive microglial Aβ accumulation. | ABCA7 has an important function to regulate homeostasis of phospholipids and cholesterol in CNS and peripheral tissues and involves in phagocytosis process. | N/A |
|
| |||
| MS4A | MS4A involves in AD pathology through controlling intercellular Ca2+, MS4A family overexpression activates T cell, in which Th1 and Th17 can increase microglial production of inflammatory cytokines. | MS4A family is adaptor protein or ion transport modulator in regulating cell activation, growth and development. | MS4A4 express at the plasma membrane in monocytes, dendritic cell and macrophages. MS4A4 is important for macrophage differentiation and polarization, and involves in dectin-1-dependent activation of NK cell-mediated resistance to metastasis. |
Abbreviations: ADP-ARF6, ribosylation factor 6; ABCA1, ATP binding cassette A1; ALS: Amyotrophic lateral sclerosis; APP-CTF, APP C-terminal fragment; CYFIP1, Cytoplasmic FMR1 Interacting Protein 1; WAVE, WASP-family verprolin-homologous protein.
REFERENCE
- [1].GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18(1): 88–106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 7(3): 137–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 16(3): 391–460 (2020). [DOI] [PubMed] [Google Scholar]
- [4].Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 80(19): 1778–1783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 179(2): 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14(4): 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 153(3): 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 23(9): 1018–1027 (2017). [DOI] [PubMed] [Google Scholar]
- [9].Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain. 138(Pt 5): 1138–1159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mosser C-A, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol. 149–150: 1–20 (2017). [DOI] [PubMed] [Google Scholar]
- [11].Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 217(2): 459–472 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bisht K, Sharma KP, Lecours C, Sánchez MG, El Hajj H, Milior G, et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia. 64(5): 826–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A. 114(41): E8788–E8797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol Aging. 55: 115–122 (2017). [DOI] [PubMed] [Google Scholar]
- [15].Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wes PD, Sayed FA, Bard F, Gan L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia. 64(10): 1710–1732 (2016). [DOI] [PubMed] [Google Scholar]
- [17].Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron. 103(5): 820–835.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 368(2): 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. 7(8): a020537 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chvátal A, Verkhratsky A. An Early History of Neuroglial Research: Personalities. Neuroglia. 1(1): 245–257 (2018). [Google Scholar]
- [21].Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 7: 45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rezaie P, Male D. Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. J Hist Neurosci. 11(4): 325–374 (2002). [DOI] [PubMed] [Google Scholar]
- [23].y Cajal SR. Contribucion al conocimiento de la neuroglia del cerebro humano. (1913). [Google Scholar]
- [24].Rio-Hortega P. THE MICROGLIA. The Lancet. 233(6036): 1023–1026 (1939). [Google Scholar]
- [25].Alliot F, Lecain E, Grima B, Pessac B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci USA. 88(4): 1541–1545 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 117(2): 145–152 (1999). [DOI] [PubMed] [Google Scholar]
- [27].Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science. 330(6005): 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koushik SV, Wang J, Rogers R, Moskophidis D, Lambert NA, Creazzo TL, et al. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 15(7): 1209–1211 (2001). [DOI] [PubMed] [Google Scholar]
- [29].Ginhoux F, Garel S. The mysterious origins of microglia. Nat Neurosci. 21(7): 897–899 (2018). [DOI] [PubMed] [Google Scholar]
- [30].Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 14(9): 1142–1149 (2011). [DOI] [PubMed] [Google Scholar]
- [31].Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U-K, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 10(12): 1544–1553 (2007). [DOI] [PubMed] [Google Scholar]
- [32].Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 6(4): 193–201 (2010). [DOI] [PubMed] [Google Scholar]
- [33].Stratoulias V, Venero JL, Tremblay M, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 38(17) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cameron B, Tse W, Lamb R, Li X, Lamb BT, Landreth GE. Loss of interleukin receptor-associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer’s disease. J Neurosci. 32(43): 15112–15123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hjorth E, Zhu M, Toro VC, Vedin I, Palmblad J, Cederholm T, et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-β42 by human microglia and decrease inflammatory markers. J Alzheimers Dis. 35(4): 697–713 (2013). [DOI] [PubMed] [Google Scholar]
- [36].Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 11(1): 56–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lan X, Han X, Li Q, Yang Q-W, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 13(7): 420–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 11: 98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cherry JD, Olschowka JA, O’Banion MK. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J Neuroinflammation. 12: 203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Weekman EM, Sudduth TL, Abner EL, Popa GJ, Mendenhall MD, Brothers HM, et al. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J Neuroinflammation. 11: 127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Joers V, Tansey MG, Mulas G, Carta AR. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol. 155: 57–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ghosh M, Xu Y, Pearse DD. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J Neuroinflammation. 13: 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chhor V, Le Charpentier T, Lebon S, Oré M-V, Celador IL, Josserand J, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 32: 70–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mecha M, Yanguas-Casás N, Feliú A, Mestre L, Carrillo-Salinas FJ, Riecken K, et al. Involvement of Wnt7a in the role of M2c microglia in neural stem cell oligodendrogenesis. J Neuroinflammation. 17(1): 88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 21(10): 1359–1369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 356(6344) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. 20(8): 1162–1171 (2017). [DOI] [PubMed] [Google Scholar]
- [48].Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 30(5): 1271–1281 (2020). [DOI] [PubMed] [Google Scholar]
- [49].Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 50(1): 253–271.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tan Y-L, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. 25(2): 351–367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Grubman A, Gabriel Chew, John F. O, Guizhi S, Xinyi C, Catriona M. A single cell brain atlas in human Alzheimer’s disease. Biorxiv. (2019). [Google Scholar]
- [52].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 169(7): 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- [53].Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 570(7761): 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hashemiaghdam A, Mroczek M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J Neuroimmunol. 341: 577185 (2020). [DOI] [PubMed] [Google Scholar]
- [55].Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 24(3): 173–182 (1989). [DOI] [PubMed] [Google Scholar]
- [56].Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 6(1): 6176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in Microglia Regulates Brain Amyloid Deposition through Selective Protofibrillar Amyloid- Phagocytosis. Journal of Neuroscience. 30(50): 17091–17101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ly PTT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 123(1): 224–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, et al. Accelerated Amyloid Deposition in the Brains of Transgenic Mice Coexpressing Mutant Presenilin 1 and Amyloid Precursor Proteins. Neuron. 19(4): 939–945 (1997). [DOI] [PubMed] [Google Scholar]
- [60].Brawek B, Schwendele B, Riester K, Kohsaka S, Lerdkrai C, Liang Y, et al. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 127(4): 495–505 (2014). [DOI] [PubMed] [Google Scholar]
- [61].Bisht K, Sharma K, Tremblay M-È. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiology of Stress. 9: 9–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, Brug MP van der, Foreman O, et al. Alzheimer’s patient brain myeloid cells exhibit enhanced aging and unique transcriptional activation. bioRxiv. 610345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bellenguez C, Grenier-Boley B, Lambert J-C. Genetics of Alzheimer’s disease: where we are, and where we are going. Current Opinion in Neurobiology. 61: 40–48 (2020). [DOI] [PubMed] [Google Scholar]
- [64].Takatori S, Wang W, Iguchi A, Tomita T. Genetic Risk Factors for Alzheimer Disease: Emerging Roles of Microglia in Disease Pathomechanisms. Adv Exp Med Biol. 1118: 83–116 (2019). [DOI] [PubMed] [Google Scholar]
- [65].Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2(10) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu C-C, Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 9(2): 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 15(9): 501–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Biffi A, Sonni A, Anderson CD, Kissela B, Jagiella JM, Schmidt H, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol. 68(6): 934–943 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proceedings of the National Academy of Sciences. 103(15): 5644–5651 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, et al. Apolipoprotein (apo) E4 enhances amyloid peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proceedings of the National Academy of Sciences. 102(51): 18700–18705 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Huang Y-WA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell. 168(3): 427–441.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci USA. 110(19): E1807–1816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sagare AP, Bell RD, Srivastava A, Sengillo JD, Singh I, Nishida Y, et al. A Lipoprotein Receptor Cluster IV Mutant Preferentially Binds Amyloid-β and Regulates Its Clearance from the Mouse Brain. J Biol Chem. 288(21): 15154–15166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Liu C-C, Zhao N, Fu Y, Wang N, Linares C, Tsai C-W, et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron. 96(5): 1024–1032.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s Disease: Accidental Encounters or Partners? Neuron. 81(4): 740–754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 485(7399): 512–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 581(7806): 71–76 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proceedings of the National Academy of Sciences. 107(26): 12011–12016 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiology of Disease. 18(2): 390–398 (2005). [DOI] [PubMed] [Google Scholar]
- [80].Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Agosta F, Vossel KA, Miller BL, Migliaccio R, Bonasera SJ, Filippi M, et al. Apolipoprotein E ε4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. PNAS. 106(6): 2018–2022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rusinek H, De Santi S, Frid D, Tsui W-H, Tarshish CY, Convit A, et al. Regional Brain Atrophy Rate Predicts Future Cognitive Decline: 6-year Longitudinal MR Imaging Study of Normal Aging. Radiology. 229(3): 691–696 (2003). [DOI] [PubMed] [Google Scholar]
- [83].Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 17(5): 661–663 (2014). [DOI] [PubMed] [Google Scholar]
- [84].Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 27(4): 1293–1306.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 549(7673): 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhao N, Ren Y, Yamazaki Y, Qiao W, Li F, Felton LM, et al. Alzheimer’s Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 14(11): 667–675 (2018). [DOI] [PubMed] [Google Scholar]
- [88].Hammond TR, Marsh SE, Stevens B. Immune Signaling in Neurodegeneration. Immunity. 50(4): 955–974 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Schlepckow K, Araque Caballero MÁ, Franzmeier N, et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener. 14(1): 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Suárez-Calvet M, Kleinberger G, Araque Caballero MÁ, Brendel M, Rominger A, Alcolea D, et al. sTREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 8(5): 466–476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Humphrey MB, Xing J, Titus A. The TREM2-DAP12 signaling pathway in Nasu–Hakola disease: a molecular genetics perspective. RRBC. 89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 368(2): 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimer’s & Dementia. 13(4): 381–387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 91(2): 328–340 (2016). [DOI] [PubMed] [Google Scholar]
- [95].Atagi Y, Liu C-C, Painter MM, Chen X-F, Verbeeck C, Zheng H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem. 290(43): 26043–26050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 90(4): 724–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 213(5): 667–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee S-H, Srinivasan K, et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J Neurosci. 40(9): 1956–1974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 212(3): 287–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 22(2): 191–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 47(3): 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Estus S, Shaw BC, Devanney N, Katsumata Y, Press EE, Fardo DW. Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol. 138(2): 187–199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 7(4): 255–266 (2007). [DOI] [PubMed] [Google Scholar]
- [104].Zhao L. CD33 in Alzheimer’s Disease - Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology. 65(4): 323–331 (2019). [DOI] [PubMed] [Google Scholar]
- [105].Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 83(5): 623–632 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 43(5): 429–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 43(5): 436–441 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Li X, Shen N, Zhang S, Liu J, Jiang Q, Liao M, et al. CD33 rs3865444 Polymorphism Contributes to Alzheimer’s Disease Susceptibility in Chinese, European, and North American Populations. Mol Neurobiol. 52(1): 414–421 (2015). [DOI] [PubMed] [Google Scholar]
- [109].Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE. 7(11): e50976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 33(33): 13320–13325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Raj T, Ryan KJ, Replogle JM, Chibnik LB, Rosenkrantz L, Tang A, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. 23(10): 2729–2736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Salminen A, Kaarniranta K. Siglec receptors and hiding plaques in Alzheimer’s disease. J Mol Med. 87(7): 697–701 (2009). [DOI] [PubMed] [Google Scholar]
- [113].Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 78(4): 631–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Alzheimer Disease Genetics Consortium (ADGC), The European Alzheimer’s Disease Initiative (EADI), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES), Kunkle BW, Grenier-Boley B, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 51(3): 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 45(12): 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. 18(7): 978–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ando K, Brion J-P, Stygelbout V, Suain V, Authelet M, Dedecker R, et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 125(6): 861–878 (2013). [DOI] [PubMed] [Google Scholar]
- [118].Zhu X-C, Yu J-T, Jiang T, Wang P, Cao L, Tan L. CR1 in Alzheimer’s disease. Mol Neurobiol. 51(2): 753–765 (2015). [DOI] [PubMed] [Google Scholar]
- [119].Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 41(10): 1094–1099 (2009). [DOI] [PubMed] [Google Scholar]
- [120].Antúnez C, Boada M, López-Arrieta J, Moreno-Rey C, Hernández I, Marín J, et al. Genetic association of complement receptor 1 polymorphism rs3818361 in Alzheimer’s disease. Alzheimers Dement. 7(4): e124–129 (2011). [DOI] [PubMed] [Google Scholar]
- [121].Kucukkilic E, Brookes K, Barber I, Guetta-Baranes T, ARUK Consortium, Morgan K, et al. Complement receptor 1 gene (CR1) intragenic duplication and risk of Alzheimer’s disease. Hum Genet. 137(4): 305–314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Deng Y-L, Liu L-H, Wang Y, Tang H-D, Ren R-J, Xu W, et al. The prevalence of CD33 and MS4A6A variant in Chinese Han population with Alzheimer’s disease. Hum Genet. 131(7): 1245–1249 (2012). [DOI] [PubMed] [Google Scholar]
- [123].Antúnez C, Boada M, González-Pérez A, Gayán J, Ramírez-Lorca R, Marín J, et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med. 3(5): 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Deming Y, Filipello F, Cignarella F, Cantoni C, Hsu S, Mikesell R, et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med. 11(505) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Berg CN, Sinha N, Gluck MA. The Effects of APOE and ABCA7 on Cognitive Function and Alzheimer’s Disease Risk in African Americans: A Focused Mini Review. Front Hum Neurosci. 13: 387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Aikawa T, Ren Y, Yamazaki Y, Tachibana M, Johnson MR, Anderson CT, et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci USA. 116(47): 23790–23796 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fu Y, Hsiao J-HT, Paxinos G, Halliday GM, Kim WS. ABCA7 Mediates Phagocytic Clearance of Amyloid-β in the Brain. JAD. 54(2): 569–584 (2016). [DOI] [PubMed] [Google Scholar]
- [128].Sakae N, Liu C-C, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, et al. ABCA7 Deficiency Accelerates Amyloid-β Generation and Alzheimer’s Neuronal Pathology. J Neurosci. 36(13): 3848–3859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Ichigotani Y, Yokozaki S, Fukuda Y, Hamaguchi M, Matsuda S. Forced expression of NESH suppresses motility and metastatic dissemination of malignant cells. Cancer Res. 62(8): 2215–2219 (2002). [PubMed] [Google Scholar]
- [130].Sekino S, Kashiwagi Y, Kanazawa H, Takada K, Baba T, Sato S, et al. The NESH/Abi-3-based WAVE2 complex is functionally distinct from the Abi-1-based WAVE2 complex. Cell Commun Signal. 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Niu L-D, Yin R, Li J-Q, Cao X-P, Yu J-T, Tan L, et al. Common Variants in ABI3 Influence Cerebrospinal Fluid Total Tau Levels and Cognitive Decline in Progressive Mild Cognitive Impairment Patients. J Alzheimers Dis. 70(1): 17–23 (2019). [DOI] [PubMed] [Google Scholar]
- [132].Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 49(9): 1373–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y, et al. Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 11(1): 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Dalmasso MC, Brusco LI, Olivar N, Muchnik C, Hanses C, Milz E, et al. Transethnic meta-analysis of rare coding variants in PLCG2, ABI3, and TREM2 supports their general contribution to Alzheimer’s disease. Transl Psychiatry. 9(1): 55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Conway OJ, Carrasquillo MM, Wang X, Bredenberg JM, Reddy JS, Strickland SL, et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener. 13(1): 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kleineidam L, Chouraki V, Próchnicki T, van der Lee SJ, Madrid-Márquez L, Wagner-Thelen H, et al. PLCG2 protective variant p.P522R modulates tau pathology and disease progression in patients with mild cognitive impairment. Acta Neuropathol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hanslik KL, Ulland TK. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front Neurol. 11: 570711 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends in biochemical sciences. 41(12): 1012–1021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 19(8): 477–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 20(13) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Mamik MK., Power C. Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain. 140(9): 2273–2285 (2017). [DOI] [PubMed] [Google Scholar]
- [142].Shao B-Z, Cao Q, Liu C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Front Mol Neurosci. 11: 320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Song L, Pei L, Yao S, Wu Y, Shang Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front Cell Neurosci. 11: 63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, et al. Molecular Architecture of the Mouse Nervous System. Cell. 174(4): 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 493(7434): 674–678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 575(7784): 669–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Zahid A., Li B., Kombe JK., Jin T., Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Frontiers in immunology. 10: 2538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 6(1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Price BR, Sudduth TL, Weekman EM, Johnson S, Hawthorne D, Woolums A, et al. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J Neuroinflammation. 17(1): 238 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Kim J, Eltorai AEM, Jiang H, Liao F, Verghese PB, Kim J, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. Journal of Experimental Medicine. 209(12): 2149–2156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. Journal of Clinical Investigation. 128(5): 2144–2155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Yamazaki Y, Painter MM, Bu G, Kanekiyo T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs. 30(9): 773–789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]