

Summary
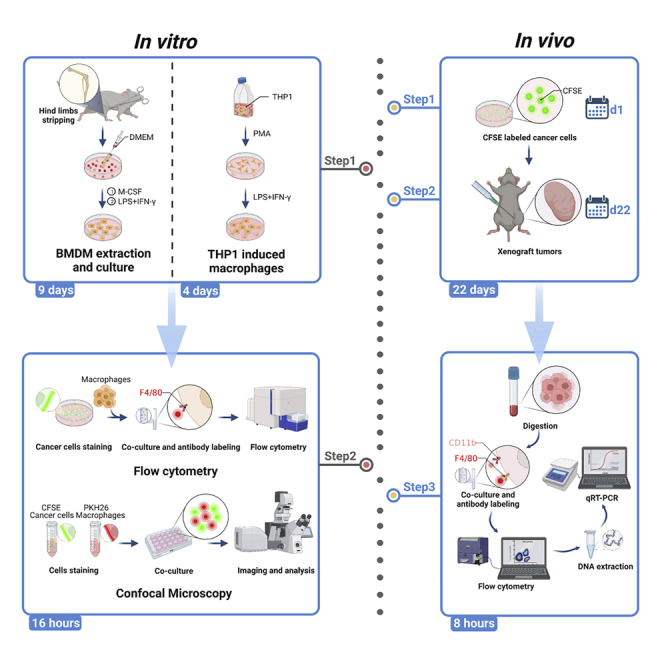
Here, we present optimized approaches to identify the efficiency of cancer cell phagocytosis by macrophages in vitro and in vivo. We describe the preparation and co-culture of macrophages and cancer cells, followed by in vitro phagocytosis assay using flow cytometry and confocal microscopy, respectively. We then detail the establishment of xenograft tumor mouse model and the in vivo detecting of phagocytosis efficiency by flow cytometry and qRT-PCR. This protocol provides a convenient way to assess macrophage-mediated phagocytosis of cancer cells.
For complete details on the use and execution of this protocol, please refer to Xu et al.1
Subject areas: Cell Biology, Cell culture, Flow Cytometry/Mass Cytometry, Cancer, Immunology, Microscopy, Model Organisms, Molecular Biology
Graphical abstract
Highlights
-
•
Co-culture of macrophages and cancer cells for assessing phagocytosis in vitro
-
•
Detection of cancer cell phagocytosis in vitro by flow cytometry or confocal microscopy
-
•
Establishment of a xenograft tumor mouse model for assessing in vivo phagocytosis
-
•
Flow cytometry and qRT-PCR detecting of cancer cells phagocytosed by macrophages in vivo
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present optimized approaches to identify the efficiency of cancer cell phagocytosis by macrophages in vitro and in vivo. We describe the preparation and co-culture of macrophages and cancer cells, followed by in vitro phagocytosis assay using flow cytometry and confocal microscopy, respectively. We then detail the establishment of xenograft tumor mouse model and the in vivo detecting of phagocytosis efficiency by flow cytometry and qRT-PCR. This protocol provides a convenient way to assess macrophage-mediated phagocytosis of cancer cells.
Before you begin
Macrophages are immune cells present in nearly all tissues that actively remove dead and damaged cells, bacteria, cancerous cells, and cellular debris from the body.2 The process by which macrophages engulf and digest cells is called phagocytosis.3 CD47 is a ubiquitously expressed trans-membrane protein that interacts with SIRPα to trigger a “don’t eat me” signal to inhibit the phagocytosis of macrophages. This protocol describes methods for detecting the efficiency of phagocytosis of cancer cells by macrophages in vitro and in vivo. The first part is in vitro testing. The procedure can be divided into 3 main steps: bone marrow-derived macrophage (BMDM) or THP1-induced macrophage preparation, the co-culture of macrophages and tumor cells, and analysis of phagocytosis index by flow cytometry or confocal imaging method. The second part is the detection of phagocytosis efficiency in vivo. The procedure includes the establishment of xenograft tumors, sequential tumor digestion, and analysis of phagocytosis by flow cytometry and qRT-PCR.
Institutional permissions
The Institutional Animal Care and Use Committee of the Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China approved all the experiments. These experiments conform to the relevant regulatory standards. Scientists should acquire permission from the relevant institutions before undertaking this protocol.
Buffers and materials preparation
Timing: 3 h
-
1.
See materials and equipment for information on preparing needed materials.
-
2.
Autoclave surgical tools, including ophthalmic scissors and forceps.
-
3.
CFSE 5 mM is stored at −20°C; M-CSF 50 μg/mL, PMA 100 μg/mL, IFN-γ 20 μg/mL, and LPS 200 μg/mL are stored at −80°C.
-
4.
L-929 cell culture supernatant: Incubate L-929 cells with DMEM complete medium at 37°C, 5% CO2 incubator for 5–7 days, collect the cell supernatant, filter it by a 0.45 μm sterile syringe filter, and store it at −80°C.
Macrophages preparation
Bone marrow-derived macrophage (BMDM) preparation
-
5.
Sacrifice a mouse by CO2 inhalation and soak it in 75% Ethyl Alcohol for 5 min (Figure 1Ai).
-
6.
Make an incision at the base of the thigh.
-
7.
Expose the hind limbs by clipping outward with ophthalmic scissors (Figure 1Aii).
-
8.
Separate the entire thigh, remove the skin and cut the bones at the hip joint (Figure 1Aiii).
Note: Try to maintain the integrity of joints at the ends of the thigh.
-
9.
Wash the separated hind limbs with PBS containing 5% Penicillin-Streptomycin (P/S).
-
10.
Wash the separated hind limbs with PBS containing 1% P/S.
-
11.
Place the hind limbs in the biosafety cabinet.
Note: All procedures and reagents/equipment need to be sterile after this point.
-
12.
Remove the Achilles tendon, knee, and hip joint to obtain femurs and tibias (Figures 1Bi–1Biii).
Note: Each mouse provides two femurs and two tibias from the hind limbs.
-
13.
Use scissors and forceps to remove the muscles on the femurs and tibias as much as possible.
-
14.
Remove the upper and lower ends of the femur and tibia with ophthalmic scissors to expose the bone marrow cavities.
-
15.
Absorb PBS with a 1 mL syringe, insert the needle (26G) into the upper bone foramen, and slowly flush out the bone marrow with PBS.
-
16.
Repeat rinse three times or until the PBS buffer is clear (Figure 1Ci) (troubleshooting 1).
-
17.
Filter the flushed cells through a 70 μm cell strainer and centrifuge at 4°C, 300 × g for 5 min (Figure 1Cii).
-
18.
Discard the supernatant and resuspend cells with 2 mL red blood cell lysis buffer to lyse the erythrocytes and incubate on ice for 10–15 min.
Note: Red blood cell lysis buffer from different manufacturers are used under different temperature. Please refer to the manufacturer's instructions.
-
19.
Add 10 mL PBS to the lysed cells to balance the osmotic pressure.
-
20.
Centrifuge at 300 × g for 5 min at 4°C and discard the supernatant.
-
21.
Repeat wash steps from 19–20 and resuspend the cells in a complete DMEM medium.
-
22.
Count the cells and seed 5 × 106 cells in each 100 mm petri dish with 10 mL complete DMEM medium.
-
23.
Thaw 50 μg/mL M-CSF on ice.
-
24.
Add 10 μL M-CSF to the dish to achieve the final 50 ng/mL concentration.
Alternatives: It is possible to add 20% L-929 cell culture supernatant in the complete DMEM medium to replace M-CSF.
Note: The status of BMDM changes from suspension growth to adherent growth. After 3 days of primary culture, a few protrusions begin to appear and the morphology became irregular. After 7 days of primary culture, the cells completely adhere to the bottom of the petri dish completely. Most cells begin to stretch into elongated spindle shape, with multiple long strip protrusions, and some cells show star shape. After the addition of LPS and IFN-γ, the protrusion part of the cells gradually decreases. 2 days later, most of the cells become round with loose cytoplasm, large nucleus, and obvious organelles (Figure 2A).
Figure 1.

Isolation and extraction of bone marrow-derived macrophages
(A) Separate the entire thigh. i. Mice are sacrificed by CO2 inhalation and soaked in 75% Ethyl Alcohol for 5 min. ii. Make an incision at the base of the thigh and expose the hind limbs by clipping outward with ophthalmic scissors. iii. Separate the entire thigh and remove the skin.
(B) Collect femurs and tibias. i. The separated thigh is washed with PBS containing P/S and placed in the biosafety cabinet. ii. Remove all muscle from the hind limbs and separate the joints to obtain femurs and tibias (achilles tendon, hip joint, and knee joint). iii. The ends of the femur and tibia are cut off to expose the bone marrow cavities. Scale bar: 1 cm or 4 cm.
(C) Flush out the bone marrow. i. The 1 mL of syringe needle (26G) is inserted into the bone marrow cavity, and the bone marrow cells are gently flushed from top to bottom into a 50 mL centrifuge tube. ii. The flushed cells are filtered through a 70 μm cell strainer.
Figure 2.

Representative images of macrophages after induction
(A) The images show the morphology of BMDM on day 1, 3, 7, and 9, respectively. Scale bar: 100 μm.
(B) The images show the morphology of THP1 cells at 0, 24, 48, and 96 h, respectively. Scale bar: 100 μm.
-
25.
Add 3 mL fresh complete DMEM medium with M-CSF (50 ng/mL) on the 3rd day and 5th day, and finally obtain mature BMDM on the 7th day.
-
26.
Replace the medium with 10 mL fresh complete DMEM medium.
-
27.
Add 20 μL IFN-γ and 10 μL LPS to stimulate the cells for 48 h.
THP1 derived macrophages preparation
-
28.
Culture THP1 cells with complete RPMI-1640 medium.
-
29.
Harvest the THP1 cells into a 15 mL centrifuge tube.
-
30.
Centrifuge at 300 × g for 5 min at 20°C–25°C and discard culture supernatant.
-
31.
Resuspend the cell pellet with 1 mL complete RPMI-1640 medium and count the cells.
-
32.
Add RPMI-1640 medium to adjust the THP1 cell density to 5 × 105 cells/mL.
-
33.
Seed 2 mL cells/well in a 6-well plate.
-
34.
Add 2 μL PMA to each well to achieve a final 100 ng/mL concentration for 48 h.
-
35.
Replace the medium with 2 mL fresh complete RPMI-1640 medium.
Note: The status of THP1 changes from suspension growth to adherent growth, from circular to irregular morphology. The cell volume increases, the cytoplasm is loose, the nucleus is enlarged significantly, a large number of apparent organelles are visible, and a small number of protrusions can be seen around the cell membrane (Figure 2B).
-
36.
Add 2 μL IFN-γ and 2.4 μL LPS to each well to achieve a final concentration of IFN-γ 20 ng/mL and LPS 240 ng/mL, respectively.
-
37.
Replace the conditioned medium 48 h later with a fresh complete RPMI-1640 medium.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Rat Anti-Mouse CD11b (1:100) | SouthernBiotech | 1561-11 |
| PE-Cy7 Rat Anti-Mouse F4/80 (1:100) | BD Bioscience | #565410 |
| SIRP-γ antibody (LSB2.20) | Santa Cruz | sc-53604 |
| Ig κ chain antibody (L1C1) | Santa Cruz | sc-59265 |
| Chemicals, peptides, and recombinant proteins | ||
| PMA | Absin | abs9107 |
| M-CSF | Peprotech | AF-300-25 |
| LPS | Sigma-Aldrich | 297-473-0 |
| IFN-γ | Peprotech | AF-315-05 |
| Collagenase Type I | STEM CELL | #07415 |
| Collagenase Type IV | STEM CELL | #07426 |
| 75% Ethyl Alcohol | CHRON CHEMICALS | 64-17-5 |
| Critical commercial assays | ||
| PKH26 Red Fluorescent Cell Linker MINI Kit | Sigma-Aldrich | MINI26 |
| Genomic DNA Extraction Kit | TIANGEN | DP304 |
| TB Green® Premix Ex Taq™ II | TaKaRa | RR820Q |
| BioTracker 488 CFSE | Sigma-Aldrich | SCT110 |
| RPMI Medium 1640 basic (1×) | Gibco | C22400500BT |
| Fetal Bovine Serum | NEWZERUM | FBS-E500 |
| Trypsin-EDTA (0.25%), phenol red | Gibco | 25200072 |
| Penicillin-streptomycin (10,000 U/mL) (P/S) | Gibco | #15140122 |
| Red blood cell lysis buffer | Solarbio | R1010 |
| Dulbecco’s modified Eagle’s medium (DMEM medium) | Gibco | C11995500BT |
| PBS, 1× (pH7.2–7.4, 0.01 M, cell culture) | Solarbio | P1020 |
| Experimental models: Cell lines | ||
| A549 passage numbers: 5–20 | ATCC | CCL-185™ |
| NCI-H1975 passage numbers: 5–20 | ATCC | CRL-5908™ |
| THP1 passage numbers: 5–20 | ATCC | TIB-202™ |
| L-929 passage numbers: 5–20 | ATCC | CCL-1™ |
| NCI-H1975 shControl passage numbers: 5–20 | Xu et al.1 | N/A |
| NCI-H1975 shCD47 passage numbers: 5–20 | Xu et al.1 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 mice, age 4–6 weeks, either gender | Beijing Huafukang Biological Technology Co., Ltd. | N/A |
| Mouse: Balb/c nude mice, age 4–6 weeks, either gender | Beijing Huafukang Biological Technology Co., Ltd. | N/A |
| Oligonucleotides | ||
| Primer: HUMAN PTGER2 Forward | Tsingke Biotechnology | GCTGCTTCTCATTGTCTCGG |
| Primer: HUMAN PTGER2 Reverse | Tsingke Biotechnology | CCAGGAGAATGAGGTGGTC |
| Primer: MOUSE PTGER2 Forward | Tsingke Biotechnology | CCTGCTGCTTATCGTGGCTG |
| Primer: MOUSE PTGER2 Reverse | Tsingke Biotechnology | GCCAGGAGAATGAGGTGGTC |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| Microsoft Excel | Microsoft | https://www.microsoft.com/zh-cn/microsoft-365/excel |
| Flowjo v10 | BD Bioscience | https://www.flowjo.com |
| Other | ||
| 70 μm cell strainers | Corning | #352350 |
| 0.45 μm sterile syringe filter | Sartorius | #16533 |
| 1 mL syringe with 26G needle | xjshifeng | SBI-080 |
| 15 mL centrifuge tubes | BIOFIL | CFT411150 |
| 50 mL centrifuge tubes | BIOFIL | CFT011500 |
| 100 mm Petri dish | NEST | 704202 |
| 6-well plate | BIOFIL | TCP011006 |
| 24-well plate | BIOFIL | TCP011024 |
| FACS tube | FALCON | 352063 |
| 15 mm glass bottom cell culture dish | NEST | 801002 |
| Fuchs-Rosenthal Counting Chamber | MARIENFELD | AP0650030 |
| Incubator | ESCO | CLM240B8CN |
| Sorvall Legend Micro 21R | Thermo Fisher | 75002445 |
| Soorvall ST 16R Centrifuge | Thermo Fisher | ST16R |
| CFX Connect Real-Time PCR Detection System | Bio-Rad | CFX96 |
| FACS AriaII Flow cytometer | BD | N/A |
| Nikon A1R Confocal | Nikon | N/A |
Materials and equipment
5% P/S PBS buffer
| Final concentration | Amount | |
|---|---|---|
| PBS | 1× | 95 mL |
| P/S | 500 U/mL | 5 mL |
| Total | N/A | 100 mL |
Note: Store at 4°C for up to 1 month.
1% P/S PBS buffer
| Final concentration | Amount | |
|---|---|---|
| PBS | 1× | 99 mL |
| P/S | 100 U/mL | 1 mL |
| Total | N/A | 100 mL |
Note: Store at 4°C for up to 1 month.
Culture medium (complete 1640 medium)
| Final concentration | Amount | |
|---|---|---|
| RPMI 1640 | N/A | 450 mL |
| P/S | 100 U/mL | 5 mL |
| Fetal bovine serum | 10% | 50 mL |
| Total | N/A | 505 mL |
Note: Store at 4°C for up to 1 month.
Culture medium (complete DMEM medium)
| Final concentration | Amount | |
|---|---|---|
| DMEM | N/A | 450 mL |
| P/S | 100 U/mL | 5 mL |
| Fetal bovine serum | 10% | 50 mL |
| Total | N/A | 505 mL |
Note: Store at 4°C for up to 1 month.
FACS buffer
| Final concentration | Amount | |
|---|---|---|
| PBS | 1× | 98 mL |
| FBS | 2% | 2 mL |
| Total | N/A | 100 mL |
Note: Store at 4°C for up to 1 month.
Starving medium
| Final concentration | Amount | |
|---|---|---|
| RPMI 1640/DMEM | N/A | 100 mL |
| Fetal bovine serum | 0.1% | 100 μL |
| Total | N/A | 100 mL |
Note: Store at 4°C for up to 1 month.
Digestion medium
| Final concentration | Amount | |
|---|---|---|
| DMEM | N/A | 50 mL |
| Collagenase types I | 1.5 mg/mL | 75 mg |
| Collagenase types IV | 1.5 mg/mL | 75 mg |
| Total | N/A | 50 mL |
Note: Store at −20°C for up to 1 month.
Step-by-step method details
Part one in vitro phagocytosis assay
Preparation of CFSE-labeled cancer cells
This section describes the process of collecting cancer cells and staining with CFSE.
-
1.
Count and harvest 2 × 106 shControl and shCD47 H1975 cancer cells into two 15 mL centrifuge tubes, respectively.
-
2.
Wash the cells with 5 mL PBS buffer and resuspend with 1 mL complete DMEM medium.
-
3.
Add 1 μL CFSE to the cell suspension to make the final concentration 5 μM and incubate at 37°C for 10 min in darkness.
CRITICAL: Invert several times to mix thoroughly.
-
4.
Add 10 mL of complete DMEM medium and invert to mix.
-
5.
Centrifuge at 400 × g for 5 min at 20°C–25°C.
-
6.
Wash the cells twice with 5 mL complete DMEM medium.
-
7.
Resuspend the cancer cell pellet with 1 mL complete DMEM medium.
In vitro phagocytosis assay based on flow cytometry
This section completes the co-culture of tumor cells and macrophages, and phagocytosis efficiency detection by flow cytometry.
-
8.
Culture macrophages (BMDM from step 27 above or THP1 induced macrophages from step 37 above) in a 100 mm dish with 10 mL starved media or 6 well plate with 3 mL starved medium per well for 12 h.
-
9.
Trypsinize the macrophages by 0.25% Trypsin-EDTA for 20–30 min at 37°C and terminate with equal volume of complete medium, and harvest in 15 mL centrifuge tube.
-
10.
Count and add 2 × 104 macrophages and an equal number of GFP-labeled (CFSE staining) cancer cells into the FACS tube in a total volume of 200 μL.
-
11.
Incubate the co-culture tubes at 37°C for 2 h.
-
12.
Add 1 μL PE Cy7-conjugated anti-mouse F4/80 antibody in the FACS tube and incubate at 4°C for 30 min in darkness.
Note: It is critical to stain the cells immediately after co-incubation and acquired detection immediately after staining.
-
13.
Wash the cells three times with FACS buffer (troubleshooting 2).
-
14.
Assess the phagocytosis efficiency by flow cytometry (BD FACS) (Figure 3).
Figure 3.

Gating strategy for in vitro phagocytosis assay is performed by co-culture of cancer cells (H1975-shcontrol and H1975-shCD47 cells) and BMDMs
FitC: GFP positive cancer cells; PE-Cy7: F4/80 positive macrophages.
In vitro phagocytosis assay based on confocal microscopy
This section describes how to stain and co-culture the macrophages and cancer cells and phagocytosis efficiency detection for a microscopy-based phagocytosis assay. The staining of macrophages is based on the PKH26 Red Fluorescent Cell Linker MINI Kit from Sigma-Aldrich, designed for the staining of membranes of live macrophages (see manufacturer protocol at https://www.sigmaaldrich.cn/CN/zh/product/sigma/mini26). The staining of macrophages and cancer cells enables us to perform in vitro confocal microscopy-based phagocytosis assay.
-
15.
Place a suspension containing 2 × 106 single macrophages in a conical bottom polypropylene tube and wash once using serum-free DMEM medium.
-
16.
Centrifuge the cells 400 × g for 5 min into a loose pellet.
-
17.
Aspirate the supernatant carefully.
-
18.
Prepare a 2×Cell Suspension by adding 100 μL of Diluent C (Catalog Number CGLDIL) to the cell pellet and resuspend with gentle pipetting to ensure complete dispersion.
-
19.
Prepare a 2×Dye Solution (4 μm) in Diluent C by adding 0.4 μL of the PKH26 ethanolic dye solution (Catalog Number P9691) to 1 mL of Diluent C in a polypropylene centrifuge tube and mix well to disperse.
-
20.
Add 100 μL of 2×Cell Suspension (step 18) to 100 μL of 2×Dye Solution (step 19) rapidly and mix the sample by pipetting immediately.
-
21.
Incubate the cell/dye suspension from step 20 for 3 min with periodic mixing in darkness.
-
22.
Stop the staining by adding 200 μL FBS and incubate for 1 min to allow for binding of excess dye.
-
23.
Centrifuge the cells at 400 × g for 10 min at 20°C–25°C and carefully remove the supernatant without removing cells.
-
24.
Resuspend cell pellet in 10 mL complete DMEM medium.
-
25.
Transfer the suspension to a fresh sterile conical polypropylene tube and centrifuge at 400 × g for 5 min at 20°C–25°C.
-
26.
Wash the cell pellet twice with 10 mL of complete DMEM medium to ensure the removal of unbounded dye.
-
27.
Resuspend the cell pellet in 10 mL of complete medium.
-
28.
Count and seed 2 × 104 macrophages in each well of the 24-well tissue culture plate overnight (troubleshooting 3).
-
29.
Incubate macrophages in the starved medium for 12 h.
-
30.
Add 2 × 104 CFSE-labelled cancer cells and incubate at 37°C for 2 h (troubleshooting 4).
-
31.
Wash the non-phagocytic cancer cells with PBS more than three times and image the cells live under a confocal microscope (Figure 4)1 (troubleshooting 5).
Figure 4.

Phagocytosis of CFSE-labeled A549 cells by PKH26-labeled BMDMs is assessed by confocal microscopy
Red, macrophages; green, tumor cells. Scale bar: 50 μm.
Part two phagocytosis assay in vivo
This section describes the construction of subcutaneously implanted tumor model in Balb/c nude mice, the sorting of macrophages in the subcutaneous tumor and the detecting of phagocytosis efficiency by flow cytometry.
-
32.
Purchase 4–6 weeks aged Balb/c nude mice from qualified companies and maintain in SPF rated conditions for one week.6,7
-
33.
Count CFSE labeled A549 lung cancer cells and adjust the cell density to 5 × 106/100 μL with DMEM (troubleshooting 6).
-
34.
Inoculate 5 × 106 CFSE labeled A549 cells subcutaneously in the right flank of the prepared Balb/c nude mice.
-
35.
Give the mice intraperitoneal injection of monoclonal LSB2.20 or IgG every 2 days for a total of 4 injections (IgG 50 μg per injection; LSB2.20 at a dose of 25 μg, 50 μg or 100 μg per injection).
-
36.
Sacrifice the mice by CO2 inhalation 21 days later.
Note: Since CFSE labeling detection relies on the cell proliferation rate, the range of days that phagocytosis detection in vivo can vary in different mouse tumor models.
-
37.
Mince the tumor tissue in a 50 mL centrifuge tube, add 5 mL digestion buffer, digest at 37°C, shaken at 120 × rpm for 1.5 h, and prepare a single-cell suspension.
-
38.
Filter the suspension through a 70 μm cell strainer.
-
39.
Wash the cells three times with DMEM and resuspend in cold FACS buffer.
-
40.
Adjust the cell density to 1 × 106/100 μL with FACS buffer.
-
41.
Add 1 μL APC labeled rat anti-mouse CD11b and PE-Cy7-labeled rat anti-mouse F4/80 antibody per 100 μL cell suspension and incubate at 4°C for 30 min in darkness.
-
42.
Centrifuge the cell suspension at 300 × g for 5 min and resuspend in cold FACS buffer.
-
43.
Flow cytometric data are obtained using BD FACS AriaII and analyze with FlowJo software (Figure 5).
Figure 5.

Gating strategy for in vivo phagocytosis assay
A549 cells are used to construct a mouse subcutaneous xenograft model. The phagocytosis efficiency is represented by the percentage of GFP+F4/80+CD11b+ cells in total F4/80+CD11b+ cells.
In vivo phagocytosis efficiency of macrophages by qRT-PCR
This step uses the lg (DNA amount) value and the Ct value (qRT-PCR) of DNA mixture to obtain the standard curve and calculate the ratio of human to mouse DNA in sorted macrophages.1
-
44.
To sort the intra-tumoral macrophage, adjust the tumor sample concentration prepared in step 42 to make the target cells passing through the sample chamber by 100–300 cells/s, preferably around 200 cells/s.
-
45.
Centrifuge the sorted macrophages at 300 × g for 5 min and discard the culture supernatant.
-
46.
Extract total DNA from sorted macrophages, M-CSF induced macrophages and A549 tumor cells using TIANamp Genomic DNA Kit, respectively.
Note: TIANamp Genomic DNA Kit or other total genomic DNA extraction methods from other companies are permitted.
-
47.
Add 100 ng, 10 ng, 1 ng, 0.1 ng and 0.01 ng DNA from A549 cells (human DNA) to mouse macrophages DNA (mouse DNA) to form a total of 200 ng DNA mixture, respectively.
-
48.
Perform qRT-PCR using the DNA mixture prepared in step 47 and equal amount of DNA sample from sorted macrophages, respectively.
-
49.
Obtain the standard curve by using the lg (DNA amount) value and the DNA mixtures’ Ct value (qRT-PCR).
-
50.
Calculate the ratio of human to mouse DNA in sorted macrophages according to standard curve (Figure 6).
-
51.
The primers below are used to test the ratio of human to mouse DNA in sorted mouse xenograft macrophages.
| Gene | Primer sequence (5′→3′) | Amplification size (bp) |
|---|---|---|
| HUMAN PTGER2 |
F: GCTGCTTCTCATTGTCTCGG R: CCAGGAGAATGAGGTGGTC |
189 |
| MOUSE PTGER2 |
F: CCTGCTGCTTATCGTGGCTG R: GCCAGGAGAATGAGGTGGTC |
186 |
The specific qRT-PCR reaction system and reaction procedure are as follows.
PCR reaction system
| Reagent | Usage amount |
|---|---|
| TB Green Premix Ex Taq II (2×) | 25 μL |
| PCR Forward Primer (10 μm) | 2.0 μL |
| PCR Reverse Primer (10 μm) | 2.0 μL |
| DNA | 200 ng (4 μL) |
| ddH2O | 17 μL |
| Total | 50 μL |
Reaction process
| Process | Temperature | Time | Cycle |
|---|---|---|---|
| Initial denaturation | 95°C | 30 s | 1 cycle |
| PCR reaction | 95°C | 3 s | 40 cycles |
| 60°C | 30 s | ||
| Hold | 4°C | forever | |
Figure 6.
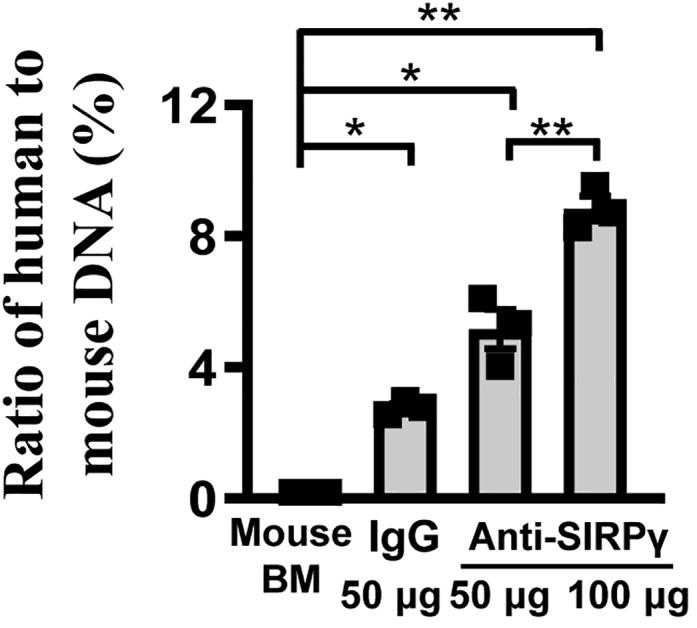
The ratio of human DNA to mouse DNA in sorted tumor macrophages
Expected outcomes
The phagocytosis assay determines the efficiency of target cancer cells engulfed by macrophages. Suppose that the target cancer cells have broken the “don’t eat me” signal or enhanced the “eat me” signal through various signaling pathways. In that case, macrophages can recognize and phagocytose the cancer cells efficiently.5 Here, the results presented in Figures 3, 4, 5 demonstrate that, as expected, knockdown CD47 or SIRPγ or blocking SIRPγ signaling by antibody in A549 or H1975 cells can enhance the phagocytosis efficiency of macrophages compared to the control group (see also).1 The methods allow quantification of phagocytosis efficiency in vitro and in vivo on cancer cells.
M1 macrophages have better phagocytosis ability compared to M0 macrophages. Thus, we encourage researchers to carefully choose what type of macrophages suitable for their investigation.
Limitations
We strongly recommended researchers using a combination of flow cytometry, confocal microscopy imaging, and qRT-PCR to assess the phagocytosis efficiency for both in vivo and in vitro assay.
Troubleshooting
Problem 1
BMDMs are in low quantity or poor quality (step 16 in macrophages preparation).
Potential solution
The quantity and quality of BMDMs could be affected by the age of mice. Ensure that the mice used for collecting BMDMs are between 4–6 weeks. Be careful to collect most of the bone marrow from the hind limbs of the mice. This will ensure to have enough cells for conducting the following experiment.
Problem 2
After the staining, co-culture, and centrifugation procedures, the cells left are insufficient for flow cytometry detection (step 14 in PART one in vitro phagocytosis assay).
Potential solution
The number of cancer cells and macrophages for co-culture may require optimization for different types of cancer cells. We recommend that the number of cells for co-culture are between 2 × 104 cells to 1 × 105 cells. The researchers may adjust the speed and time of centrifugation according to the varied size of cancer cells. After centrifugation, a cell pellet should be able to be seen at the bottom of the tube.
Problem 3
In the confocal microscopy-based phagocytosis assay, macrophages do not attach, or only a few do, and they do not look flat and even (step 28 in PART one in vitro phagocytosis assay).
Potential solution
The status of macrophages could affect the adhesion to the bottom of the culture plate. Researchers should conduct pre-experiment to determine the optimal digestion time, staining time, and centrifugal speed of cells to ensure the best status of macrophages. Researchers should try to mix the sample as well as possible and avoid shaking the 24-well plate violently to make the cells look flat and even.
Problem 4
The phagocytosis efficiency is too low in flow cytometry-based and microscopy-based phagocytosis assays (step 30 in PART one in vitro phagocytosis assay).
Potential solution
The incubation time may require optimization for different cancer cells. The incubation time is usually 2–24 h. A time-response phagocytosis efficiency should be detected initially to determine a suitable incubation time. In detail, 2 × 104 cancer cells and an equal number of macrophages are co-cultured at 37°C for 2, 4, 8, 16, and 24 h, respectively. Choose the best co-culture time at which the phagocytosis efficiency is best for the next step. Under natural conditions, macrophages phagocytose cancer cells in a lower proportion. Therefore, positive controls, such as CD47 blocking antibody treatment, can be employed to ensure the normal operation of the phagocytosis system.
Problem 5
The non-phagocytic cancer cells after co-culture could not be washed away thoroughly (step 31 in PART one in vitro phagocytosis assay).
Potential solution
In vitro microscopy-based co-culture system, we recommend using the cancer cells with a long adhesion time to make them easier to wash away after co-culture. The number of cancer cells and macrophages should be adjusted according to the size of cells and the bottom area of the co-culture plate, so that the proportion of phagocytosis can be conveniently counted even if the non-phagocytic cells cannot be washed away thoroughly.
Problem 6
The CFSE-labeled cancer cells used for in vivo phagocytosis assay cannot be detected well by flow cytometry (step 33 in PART two phagocytosis assay in vivo).
Potential solution
The uneven and impertinent labeling of cancer cells may affect the detection and analysis of phagocytosis assay based on flow cytometry. Therefore, it is essential to stain the cells just before use and choose dyes that decay slowly. Another option is to use lysosomal stable fluorescent proteins (i.e., ZsGreen) that can be engineered into tumor cells lines for phagocytosis detecting, especially in vivo study.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hui-Kuan Lin (hulin@wakehealth.edu).
Materials availability
This protocol does not generate unique reagents.
Acknowledgments
This work is supported by funding from the National Natural Science Foundation of China (81873048) to C.X. and Endowment funds for Anderson Discovery Professor for Cancer Research and Start-up Funds and Wake Innovation Catalyst Funds from Wake Forest School of Medicine to H.-K.L.
Author contributions
Conception and design, data analysis and interpretation, manuscript writing, and funding acquisition, H.-K.L., C.X.; Collection and assembly of data (performance of experiments), FACS sorting, confocal microscopy, and manuscript writing, H.W.; Collection and assembly of data, and manuscript writing, C.X., H.W., Y.L.; Collection and assembly of data, F.L. Scientific inputs, R.K.M.
Declaration of interests
H.-K.L. is a consultant for Stablix, Inc. All other authors have declared that no competing interests exist.
Contributor Information
Chuan Xu, Email: xuchuan100@163.com.
Hui-Kuan Lin, Email: hulin@wakehealth.edu.
Data and code availability
This study does not generate unique datasets or code.
References
- 1.Xu C., Jin G., Wu H., Cui W., Wang Y.H., Manne R.K., Wang G., Zhang W., Zhang X., Han F., et al. SIRPgamma-expressing cancer stem-like cells promote immune escape of lung cancer via Hippo signaling. J. Clin. Invest. 2022;132:e141797. doi: 10.1172/JCI141797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies L.C., Jenkins S.J., Allen J.E., Taylor P.R. Tissue-resident macrophages. Nat. Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon S. Phagocytosis: an immunobiologic process. Immunity. 2016;44:463–475. doi: 10.1016/j.immuni.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Tseng D., Volkmer J.P., Willingham S.B., Contreras-Trujillo H., Fathman J.W., Fernhoff N.B., Seita J., Inlay M.A., Weiskopf K., Miyanishi M., Weissman I.L. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA. 2013;110:11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramachandran P., Pellicoro A., Vernon M.A., Boulter L., Aucott R.L., Ali A., Hartland S.N., Snowdon V.K., Cappon A., Gordon-Walker T.T., et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA. 2012;109:E3186–E3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X., Fan J., Wang S., Li Y., Wang Y., Li S., Luan J., Wang Z., Song P., Chen Q., et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol. Res. 2017;5:363–375. doi: 10.1158/2326-6066.CIR-16-0398. [DOI] [PubMed] [Google Scholar]
- 7.Schürch C.M., Roelli M.A., Forster S., Wasmer M.H., Brühl F., Maire R.S., Di Pancrazio S., Ruepp M.D., Giger R., Perren A., et al. Targeting CD47 in anaplastic thyroid carcinoma enhances tumor phagocytosis by macrophages and is a promising therapeutic strategy. Thyroid. 2019;29:979–992. doi: 10.1089/thy.2018.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study does not generate unique datasets or code.