

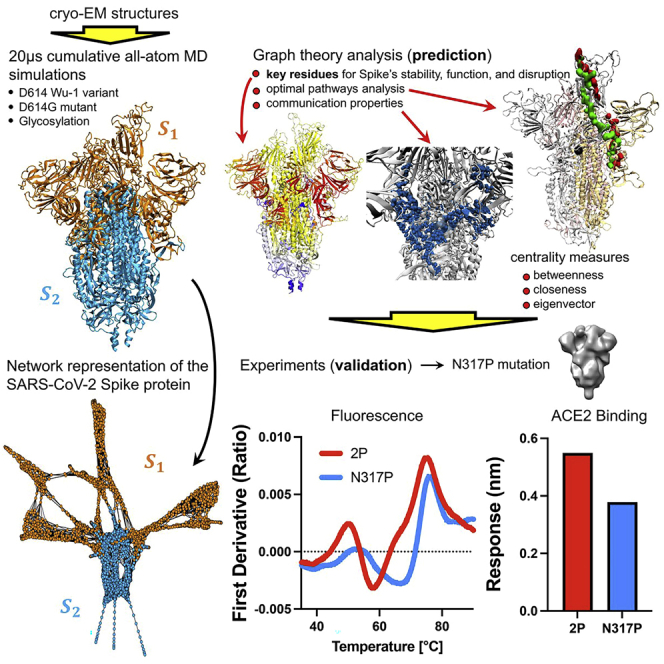
Summary
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has triggered myriad efforts to understand the structure and dynamics of this complex pathogen. The spike glycoprotein of SARS-CoV-2 is a significant target for immunogens as it is the means by which the virus enters human cells, while simultaneously sporting mutations responsible for immune escape. These functional and escape processes are regulated by complex molecular-level interactions. Our study presents quantitative insights on domain and residue contributions to allosteric communication, immune evasion, and local- and global-level control of functions through the derivation of a weighted graph representation from all-atom MD simulations. Focusing on the ancestral form and the D614G-variant, we provide evidence of the utility of our approach by guiding the selection of a mutation that alters the spike’s stability. Taken together, the network approach serves as a valuable tool to evaluate communication “hot-spots” in proteins to guide design of stable immunogens.
Subject areas: Biological sciences, Immunology, Virology, Structural biology
Graphical abstract
Highlights
-
•
The main hubs of communication of the protein are located in the NTD and CT0 regions
-
•
The D614G variant is more resilient to disruption than the ancestral form
-
•
Empirical model validation shows that a N317P substitution could impact ACE2 binding
-
•
A network analysis approach could guide the design of effective immunogens
Biological sciences; Immunology; Virology; Structural biology
Introduction
The emergence and subsequent worldwide spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing COVID-191,2 led to an ongoing global health emergency that, according to the World Health Organization, has taken more than 6 million lives as of August 2022.3 COVID-19 is a highly contagious respiratory illness initiated by viral entry into host cells leading to infection and symptoms. Viral entry is contingent upon the binding of the spike glycoprotein, located at the surface of the SARS-CoV-2 virion, with the human host receptor angiotensin-converting enzyme 2 (ACE2).4,5,6
The viral spike protein is a homotrimeric class I viral fusion protein able to adopt different conformations according to the state of its receptor binding site region (the receptor-binding domain or RBD) of each of its protomers. Cryo-electron microscopy at the atomic level7 has shown that the spike adopts two predominant conformations: the all-down conformation, in which all three protomers are in a closed state, or the one-up conformation, in which a single protomer is in an open state while the remaining two are in a closed state. The one-up conformation promotes host-receptor binding due to heightened exposure of the RBD. Each protomer in the spike protein can be grouped into two main functional subunits, S1 and S2, delimited by the furin cleavage site (FCS) that is located within a loop at residues 675–690. The S1 subunit (residues 14–685) includes the RBD,8,9,10 while the S2 subunit (residues 686–1273) mediates virus-host cell membrane fusion and subsequent viral entry. Here, for ease of analysis, as we have done previously,11 we divide each protomer into 12 domains (Figure 1).
Figure 1.

Structure and graph representations of the SARS-CoV-2 spike protein
Top and bottom left panels illustrate the graph and structure representations, respectively, of the spike protein divided into pre-assigned protomer domains (colored regions/nodes). In the bottom right panel, we provide the full domain names, the domain abbreviations, and the residues that comprise them. Each amino acid residue of the protein forms a node in the graph representation. All molecular images were created using VMD (https://doi.org/10.1016/0263-7855(96)00018-5). The graph was drawn using the interface IGraph/M for Wolfram mathematica (https://doi.org/10.5281/zenodo.1134932).
While COVID-19 has shown some immunologically relevant variant accumulation over time, a notable early evolutionary event that has stably persisted through the subsequent evolution of new variants was selection of the G clade, a lineage defined by 4 mutations, including one in the spike protein that was located outside of neutralizing antibody-targeting sites: an amino acid substitution in residue D614G where an aspartic acid (D) was replaced by a glycine (G).12 This variant (the D614G form) began to emerge at a significant level in early March of 2020 and by the summer of 2020 had become the dominant form of the virus globally. It showed an enhanced infection capability over the original (D form),12,13,14,15,16 by virtue of its greater tendency to take on the ACE2 binding-capable RBD-open conformation.11,17 Other studies have implicated alterations in stability18 and fitness.19 Another potential factor impacting the infectivity of the G form is the higher efficiency of furin cleavage.13 The furin cleavage site (FCS) loop is an advantage acquired by the CoV-2 spike over other known sarbecoviruses, wherein a furin-mediated S1-S2 proteolytic cleavage site was acquired, resulting in more efficient cleavage and higher infectivity.
Significantly more infectious new variants have emerged over the course of the pandemic, such as Delta and Omicron, each bringing with them additional challenges for vaccines and immunotherapies.20,21 (A summary of past variants, including a fasta file with representative forms, can be found in the Los Alamos National Laboratory download section of GISAID, https://www.gisaid.org/). The D614G mutation persists in almost all of these variants, although the ancestral D614 form was retained in several minor variants that surfaced in 2021 (e.g. Pango lineages A23.1 and A27). Most known N-terminal domain (NTD)-specific neutralizing monoclonal antibodies (mAbs) target a specific region of the NTD (known as the NTD supersite) that coincides with the occurrence of these high-frequency mutations. This supersite is composed of residues 14–20 (loop), 140–158 (-hairpin), and 245–264 (loop).22,23 Among the variants, recurrent insertions and deletions and accelerated substitution rates in the NTD drive adaptive antigenic evolution and can confer resistance to neutralizing antibodies.24,25,26 It is still not mechanistically understood why the NTD is an effective target for these neutralizing antibodies, and its relevance within the protein at large is poorly understood. Furthermore, mutations near the furin cleavage sites may be contributing to enhanced infectivity of emerging variants.27 On the other hand, the functioning of the spike protein is largely driven by allostery, with coupled dynamics between critical moving parts such as the NTD, RBD, FCS, and the D614G-loop in CT2, modulating its overall behavior.28,29 The manner by which these domains effectively communicate over long distances, however, remains unanswered. Here, we implement a mathematical framework that sheds light on these questions and complements the current body of knowledge on the virus.
In this work, we construct a weighted graph representation of the protein in which the nodes are residues and the edges are weighted by the strength of the correlations between the nodes.30 We extract contact and correlation matrices from extensive all-atom molecular dynamics simulations of the D and G forms in their dominant conformation states (all-RBD down or all-down, and 1-RBD up or one-up).11 Within the derived graph theoretic framework, we quantitatively describe the dynamical relationships between different residues and how they are impacted by different sequences and simulation conditions, as well as the inter-chain communication between either the three symmetrically down protomers or the up protomer with the left and right chains on either side (U, L, R-protomers, respectively).
In this article, using graph theoretic techniques, we identify the critical residues (i.e., nodes of the graph) and domains, and assess their roles in communication and control of protein allostery. From this analysis, we predict that modification of these critical residues could dramatically alter essential long-range molecular interactions and stability. Our analysis shows that these key residues tend not to overlap with those where most of the mutation/deletions occur within RBD and NTD domains. It is likely that the high mutation rate in these domains occur as a consequence of fitness pressures toward immune escape of receptor interactions. In contrast, graph theory analysis finds critical residues for communications in these domains and other regions of the spike that are indicative of their potential importance for protein structure and function. We provide experimental support for this hypothesis by showing that a single point mutation in a network hub identified in our calculations is capable of altering the spike protein conformation.
Results
We address the communication differences between four spike protein-based networks: the closed and open conformations in the D614 and the D614G forms. We illustrate the differences by exploring several aspects of the communication properties of each network, and how these differences impact infectivity and overall network stability. Then, we carry out additional calculations that assess the importance of the poorly understood NTD domain and the furin cleavage site. Next, we evaluate emerging mutations and identify the critical sites that can be used for immunogen design. Finally, we provide experimental evidence that the residues identified by our network calculations indeed impact stability. Additional network properties of the spike protein, as well as further details of the results presented in this section are provided in the supplemental information. For calculations involving the RBD, we focus on the subset of residues that bind to the ACE2 receptor—the receptor-binding motif (RBM, residues 438–508)—which are the most relevant for infectivity. In the main text, we generally report results from simulations without glycans, pointing out where these networks are unaffected by glycosylation. The SARS-CoV-2 spike protein typically has 22 glycans per protomer.11 We extract communication features that are independent of glycan modifications to the protein, although glycan occupancy and glycoforms of carbohydrates at a given site can vary depending on cell lines and enzyme repertoires, and the nature of the glycoforms at particular sites can in turn affect the dynamics of the proteins.31 Some network properties do change with the presence of glycans, which we note and discuss further in the supplemental information.
Graph measure uncovers the communication center of the spike protein
The ability of a particular node or group of nodes to form a bridge between distant regions of a network is quantified by its betweenness centrality. Mathematically, the betweenness centrality of a node is the number of optimal pathways (geodesics) that run through it as part of connecting any other pair of nodes in the network. In the context of our graph-based analysis of the spike, residues having higher betweenness centrality take part in a greater number of “shortest pathways”—i.e., the most highly correlated series of contacts—connecting any other two residues of the protein. Residues with high betweenness centrality act as critical intra-protein “hubs” that influence many optimal pathways of protein allosteric communication. We compute the betweenness centrality for each node (individual residue) of our four networks and assess its implications at the level of the entire protein, at the level of the domains, and at the level of the individual residues.
At the entire protein level, we find that the regions with the highest values of betweenness centrality form a closed ring around the protein, which represents the structural communication core of the whole system. Differences in the communication core between networks highlight the impact of the D614G shift in the all-down and one-up states (Figure 2A). The ring straddles the equatorial plane of the spike at the base of the NTD, extending via the CT0 to the base of the RBD. This high-centrality band of residues extends to the beta-strands at the nadir of the furin cleavage hairpin, and comprises residues of the NTD, CT0, CT1, and CT2 domains from all three protomers (Table S2). This holds for both forms (D and G) and both conformations (all-down and one-up). However, we find that the communication ring in the G form is comprised of a greater fraction of residues of CT0 and a lower fraction of residues of CT2. This core ring directly connects and is crucial for communication between the three peripheral domains of the spike, the NTD, RBD, and FCS, and the central region of the protein. Interestingly, this high centrality core avoids the NTD supersite (highlighted in green in Figure 2A), where many antibodies have shown particular binding preference and effectiveness.22,24,32 The specific residues comprising the core ring for each protein and both conformations are listed in Table S3 in the supplemental information. The dynamics of all three domains involved in the core ring have been established to play significant roles in spike protein function.14,33,34 Our analysis elucidates the allosteric linkage pathways of these important moving parts of the spike and demonstrates that there is a lack of overlap between sites of preferential antibody binding and sites that impact protein communication.
Figure 2.

Communication center of the spike protein
(A) SARS-CoV-2 spike protein in the structure representation highlighting the NTD supersite (green), alongside the top-50 residues with highest betweenness centrality for the D form only (orange), the G form only (blue), and both forms (black), for the all-down (left panel) and one-up (right panel) conformations.
(B) Betweenness centrality broken down by domain for the D and G form and the one-up and all-down conformations.
(C) Ranked betweenness centrality for individual residues of the D form (left) and the G form (right) and the all-down (red) and one-up (green) conformations.
D614G variant is more resilient to communication disruption compared to the ancestral form
At the domain level, we find that CT0, despite being a small domain comprised of only 25 residues in each protomer (Figure 1), is associated with the highest overall betweenness centrality. Indeed, it is the highest ranking domain for both conformations of the G-form and the all-down conformation of the D-form (CT1 ranks slightly higher for the one-up D-form conformation) (Figure 2B). This domain, located at the heart of the spike, appears to be crucial for communication between distant regions of the protein and hence could become a strategic target for intervention (i.e., drug target and antibody therapy). Our findings corroborate a speculation based on a previous experimental cryo-EM study of the spike ectodomain35 that suggested that this NTD-RBD linker acted as a modulator of conformational changes connecting distal domains, and we further quantify the relative significance of each residue in this role.
At the individual residue level, plotting the betweenness versus the node rank reveals that the all-down configurations contain nodes with higher values of betweenness, while the difference in betweenness for the two configurations is larger for the D-form than for the G-form (Figure 2C). In other words, the structural variation the spike protein undergoes when transitioning from the all-down to the one-up configuration has a greater impact on its communication properties in the D-form than in the G-form. The G-form possesses more stable patterns of communication in which key structural and functional aspects of the protein should be less affected by the transition to the one-up state. This result agrees with the previously observed greater symmetrization of the inter-domain correlations and inter-protomer contacts in the G-form between the all-down and one-up states.11 The high betweenness in the all-down conformation of the D-form indicates that the optimal paths connecting any two residues in the protein go through a reduced number of residues when comparing with the all-down conformation in the G-form. This occurs when the correlated movement across regions in the protein is more localized, which restricts the optimal paths to go through these correlated regions producing a high betweenness. Thus, the reduced localized interactions and the accompanying heightened long-distance correlations in G614 allow for this variant to undergo RBD down-up allosteric transitions more frequently compared to the D-form, as has been observed in previous experiments and simulations.11,14 Structural rearrangements required for the infection-capable RBD-up conformation is more easily attained in the G614 protein, while the original D-form down conformation remains relatively stable through the elevated localized correlations. Moreover, from a purely network perspective, the large values of betweenness in the all-down configuration of the D-form compared to those in the G-form uncover an increased vulnerability of the D-form, in which a small group of nodes is involved in a greater portion of the communication network. Our analysis points to three residues in the NTD, S297, L296, and Y279 that hold significantly higher values of betweenness in the all-down conformation of the D-form compared to that in the G-form. These residues are holding combined values that are four times greater in the D-form compared to those in the G-form. These residues are located at the base of the NTD where the domain meets the trunk of the spike through CT0. Interactions in this region should influence the relative motion between NTD and the rest of the spike, which in turn has been implicated to influence RBD conformational changes.36 Furthermore, these positions have been indicated among others as possible distant ACE2 allosteric drug targets,37,38 demonstrating that they are involved in RBD modulation. Reduced betweenness of these residues from D614 to G614 forms should thus contribute to enhanced RBD transitions. Perturbation of this group of nodes (e.g., through mutation, or through binding to different partners) more heavily influences the conformation of the spike in the D-form than a perturbation intervention to the G-form, since a network with a more uniform distribution of betweenness centrality among its nodes tends to be more resilient to external interventions causing network disruption. This finding is in agreement with experimental and structural studies that demonstrated that the occurrence of S1/S2 dissociation is more likely in G-form than in D.15,18 On the other hand, the structurally dominant residues according to their betweenness values that are common in these two spike proteins in the one-up conformation are V595, S596, and V597 of the CT2 subregion, and R44, Y279, D290, L296, and S297 from the NTD. This patch of residues belongs to the same NTD-CT0 interface discussed above, at the kink where NTD interacts with the rest of the Spike. The occurrence of high-centrality residues in this region along with the highest D versus G betweenness differences here evince this region as the critical bridge that relays NTD conformational effects to the RBD through the CT0 strands. Finally, from a statistical perspective, we compare the distributions of betweenness scores of both proteins in the same conformational state and find that their respective p values (computed via a KS test) are greater than 0.05. This is not surprising given the extremely similar conformational topology of the spike proteins from D and G forms. However, when restricting the test to the top-50 residues with high betweenness scores, the comparison yields the p value which is well below the threshold (, D-statistic 0.48). Taken together, it indicates that while the overall networks between the all-down conformations of D and G forms are very similar, a few critical residues differentially alter the communication within the all-down conformations in a statistically significant manner. It is imperative, therefore, to perform detailed mutational studies of these residues to obtain detailed understanding of spike allostery.
NTD and residues 306–330 (CT0) are structurally efficient signal sources in the spike protein
As its name suggests, closeness centrality measures the proximity of a particular node to the rest of the nodes in the network. Mathematically, it is defined as the inverse of the average path length between a particular node and all of the remaining nodes in the network. Higher values of this measure are associated with greater network-scale communication efficiency. This quantity answers the question of which nodes are most efficient at transmitting a signal (e.g., warning, cue, and infection) to the rest of the network. In the context of the spike protein, residues with a moderate-to-high closeness centrality are initiators of effective allosteric communication able to efficiently reach every other residue of the protein. Unlike betweenness centrality, which measures the number of shortest paths going through a particular node, closeness centrality measures the length of shortest paths between this particular node and the rest of the sites in the network. Therefore, these quantities provide complementary information about the communication properties individual nodes possess in the network.
Computing this centrality measure for each of our four graphs unveiled some similarities and some contrasts at the protein, domain, and residue levels. At the protein level, in all four networks (D- and G-form, one-up and down), the S1 dominates S2 in terms of high centrality (Figure 3A). Analyzing the residues that are within the top 33% in closeness, we find that a minimum of 98.76% and a maximum of 99.64% of them belong to S1, depending on the network. Though this is not necessarily surprising given the key role of the S1 subunit in regulating the receptor recognition/binding process, the finding provides a good check that supports the approach and provides additional backing for our other findings. At the domain level, we find that the NTD, CT0, CT1, and CT2 regions score the highest in closeness centrality. This indicates that these domains are capable of drastically and efficiently impacting the entire protein. In the context of RBD antibody neutralization, antibody binding can directly interfere with ACE2 interactions and directly inhibit infection.25 If an antibody binds to the NTD, even if it is not exactly binding to the highest centrality residues, it will affect the NTD dynamics, which are highly correlated with the RBD dynamics, and therefore NTD antibodies are targeting areas of the protein that are directly relevant for protein function—possibly through the modulation of the up/down-RBD transition.
Figure 3.

Closeness centrality highlights the NTD, CT0, CT1, and CT2 domains
(A–C) Structural representation of the SARS-CoV-2 spike protein in the one-up conformation of the G forms colored by the closeness centrality. This measure is also presented broken down by protein region (B) and by individual residues ranked from highest to lowest (C), for the D and the G form, and for the all-down and the one-up conformations.
Comparing the closeness centrality of the different networks, a general trend is found where the closeness for the all-down conformation is higher than that of the one-up, and the closeness of the G-form is higher than that of the D-form (Figure 3B). This statement also holds at the level of the individual residues (Figure 3C). A ranking of the nodes shows a smooth behavior in which, as opposed to the betweenness centrality of the top residues, the difference in closeness for consecutive ranked residues does not change drastically, and the overall trend of the closeness values of ranked residues agrees with the domain-level trend. This tendency is explained by looking into the pair correlations of linked residues (i.e., edge weights) for the different protein systems. We find that the average pair correlation between contact residues for the all-down conformation in both, the D- and the G-form, is always higher than that of the one-up conformation, which in turn, makes the average path length shorter in the all-down conformation (Table S1). Moreover, we find that the G-form has shorter path lengths, on average, than the D-form, which makes the spike of the D614G variant more efficient at establishing effective allosteric communication pathways than the ancestral D614 form. As an important note, our conclusions regarding the communication centrality measures (i.e., betweenness and closeness) are not affected by the influence of glycosylations in the spike protein (see supplemental information).
NTD is a center of influence with a far-reaching role in protein functionality
While centralities of communication, such as betweenness and closeness, focus on shortest paths, centralities of influence, such as degree and eigenvector, take into account additional network features providing further information about the relevance of specific nodes at the local level as well as the network at large. Degree centrality quantifies the strength of the connections of a given node. For unweighted networks, this is simply the number of edges a particular node has. For weighted networks, each edge is scaled by its respective weight, and the sum of these weights is known as weighted degree or vertex strength.39 Since it includes the nearest neighbors only, vertex strength is considered a measure of influence at the local level. An extension of this measure is eigenvector centrality, which computes the influence of a particular node based on the influence of its neighbors. Thus, the centrality score of a given node takes into account the score of its neighbors, which in turn, take into account the scores of their neighbors, which are the second degree neighbors of node , and so on. Therefore, this centrality intrinsically includes interactions with higher degree of neighbors and hence quantifies the global influence of a given node. Mathematically, the eigenvector centrality of the node is the -th entry of the adjacency matrix eigenvector associated with the largest eigenvalue. In contrast to optimal paths analysis, in which edge weights indicate path lengths and therefore the smaller the weight the higher the importance of the edge, in influence centrality measures, the edge weights directly indicate importance. Therefore, we employ an edge weighting for influence centrality that monotonically increases with the correlation between two residues (see STAR Methods) and which is the inverse of the weight used for optimal communication paths calculations.40
Computing the vertex strength and the eigenvector centrality of the graph representation of the spike protein reveals that the NTD dominates in terms of both. At the domain level, a high vertex strength indicates that strong correlations between nearest neighbors in the NTD are common across the domain (Figure S1). This result is surprising: since the NTD is the largest domain of the spike, containing 292 residues, one might expect a high number of pairs with lower correlations would drive down the average node strength. Other domains associated with strong intra-domain interactions are CT1, RBD, and CT0. In terms of eigenvector centrality, the NTD far outstrips any of the other domains in the protein, which highlights its hierarchical importance for the network at large (Figures 4A–4C). This finding demonstrates in a quantitative manner that the critical residues in this domain, highlighted by the eigenvector centrality, exert wide-reaching influence to the protein at large, and that alteration in these nodes will affect the entire network. Interestingly, the residues with the highest eigenvector centrality in the G form are in close contact with the up-RBD but are not located within the NTD supersite (Figures 4A and 4B). Identical calculations for the glycosylated spike in the all-down conformation show equivalent results. However, for the one-up conformation, we report a shift in the high centrality region to the up-RBD due to the interaction with the glycans (see supplemental information). It is likely that the experimentally observed comparatively rapid mutation in the supersite occurs as a consequence of fitness pressures toward immune escape, while the lack of mutation in the nearby high eigenvector centrality residues further underscores their importance for protein function.
Figure 4.

Eigenvector centrality highlights dominant role of NTD
(A) Graph representation of the SARS-CoV-2 spike protein in the one-up conformation of the G form colored by individual node eigenvector centrality.
(B) Structure close-up for the NTD and neighboring RBD region with eigenvector centrality highlighted by residue.
(C) Domain-level eigenvector centrality for the D and the G forms, and for the all-down and the one-up conformations.
The allosteric communication pathway between RBD and furin cleavage site
We explore the effects of the D614G variant at altering specific pathways relevant for viral infectivity through a path length analysis. Up to date, variants of concern are characterized by a combination of increased transmissibility and/or immune response escape.20,21,25 Several studies have shown an impact of furin cleavage on the RBD up/down dynamics.17,35,36 This suggests, variations in the communication pathways between distally coupled regulatory sites such as D614G, FCS, and RBD become a key question to address in order to establish the impact of mutations in the transmissibility.
To this end, we analyze the optimal paths between the furin cleavage region and the RBM (Figure 5), and between residue 614 and the RBM (Figure S4). We employ the Floyd-Warshall algorithm to determine the most efficient route connecting any two nodes in a given network from the contact matrix and the node pair correlations. The path length between these sites is defined as the sum of the edge weights of edges lying between the end point sites. As previously mentioned, high-magnitude correlations correspond to small edge weights and hence shorter paths, whereas low-magnitude correlations correspond to large edge weights and hence longer paths. Chains of highly correlated residues thus form shorter (i.e., more efficient) pathways than chains with the same number of links comprised by uncorrelated residues. A computation of the optimal pathways reveals that residue 614 lies near the shortest path from the FCS to the RBM region. This highlights its relationship to the up/down protomer states. Moreover, we find that the frequent mutation sites near FCS41 do not generally lie on the optimal pathway to the RBM. We hypothesize that these sites do not overlap with the shortest-path sites due to the fact that changing the shortest-path sites would also change the communication pathway from the RBM to the cleavage site. Further evaluation of optimal pathways between the different source/target regions suggests G-form enjoys viral infectivity advantages over the D-form at the cost of a higher RBD vulnerability to antibodies and glycans tend to alleviate this vulnerability cost (see supplemental information).
Figure 5.

Optimal pathways to RBM from residue 614 and furin cleavage sites
SARS-CoV-2 spike protein in the structure representation highlighting the optimal intra-chain (left panel) and inter-chain (right panel) pathways from the furin cleavage (residue 685) to the RBM region (residue 501) for the all-down only (red), one-up only (green), and both conformations (yellow) in the G form. The protomers are labeled R, L, and U. Protomers R and L are set in the down state, while the up or down state of the U protomer defines the conformation of the spike as one-up or all-down, respectively.
NTD mutational sites in delta are efficient communication initiators
The Delta variant possesses, in addition the D614G amino acid substitution, three modifications and two deletions in the NTD supersite (T19R, G142D, R158G, and deletions at E156- and E157-, respectively), two modifications in the RBM (L452R and T478K), one modification near the FCS (P681R), and two additional modifications: one in the NTD (T95I) and another in the heptad repeat 1 (HR1) (D950N). The T95I mutation is found in roughly half of Delta variants. The location of these sites in the spike protein is shown in Figures 6A and 6B.
Figure 6.

Relevance of sites modified/deleted by the Delta variant
(A) Structural representation of the SARS-CoV-2 spike protein in the one-up conformation of the G-form highlighting the residues altered in the Delta variant. Deletions are marked in bold font and the symbol.
(B) Analogous to (A) but in the graph representation of the spike protein.
(C) Left panel shows the closeness centrality of individual residues ranked from highest to lowest in the one-up conformation of the spike protein. The horizontal grid lines mark the values of closeness associated to the residues modified/deleted by the Delta variant. The colors match the table in the right panel, and shows detailed information concerning the individual ranking position, the identity of the residue (bold font indicates deletions), and the value of closeness in units of .
(D) Average closeness centrality and fluctuations (error bars) of the NTD mutation sites of the Delta variant compared to that of the NTD supersite for the all-down and the one-up conformation.
We characterize the sites modified by the Delta variant with our network analysis, particularly the centrality measures of betweenness, closeness, and eigenvector. In terms of betweenness and eigenvector, we find that these residues rank very low for both conformations of the RBD, pointing to a scarce ability to bridge together or exert control over distant residues. Among the sites studied, the most relevant residue for communication (i.e., betweenness) is L452, through which runs a small fraction (5%) of the optimal communication pathways, in comparison to the highest ranked residue of the protein (V320 for all-down and V597 for one-up). As a background, residue 452 was extensively characterized as part of the California variant, and it impacted infectivity and resistance to neutralizing antibodies.42 Deletions in this variant account for nearly 0.5% of the number of pathways carried by these highest ranked residues. Similar values are found when we examine the betweenness centrality of the modification at the FCS, and when we look at the eigenvector centrality of all the mutations (Figure S3). Therefore, modification/deletion of these residues is not expected to severely affect the protein in connectivity or large-scale control.
By contrast, examining the closeness centrality of these critical residues modified by the Delta variant, we find that those of the NTD rank high in their centrality measures (see Figures 6C and S7) compared to the rest of the residues in the spike. This means that these residues are good at initiating efficient pathways capable of reaching every region of the protein. They possess little capacity to serve as communication bridges (i.e., low betweenness), but they possess outstanding capacity to serve as pathway initiators (i.e., high closeness). These NTD residues effectively reach every other residue in the protein, with efficiencies ranging between 80% and 95% with respect to the highest ranked residue in closeness centrality (V597 for all-down, and K310 for one-up). Figure 6D compares the average closeness centrality for these sites with that of the residues comprising the NTD supersite (residues 14–20, 140–158, and 242–264). We find that the average closeness of the NTD residues modified/deleted in the Delta variant is higher than the average closeness of the NTD supersite overall.
Our analysis provides a quantitative argument for why these critical sites are being modified/deleted in the Delta variant: changing residues with low betweenness does little to hinder the overall allosteric dynamics of the protein, but changing residues with high closeness could impact antibody binding. Modifications in the sites G142 and R158 in specific have already been shown to be directly associated to mAbs neutralization escape.22 Thus, our results suggest that the closeness centrality may be used as a quantitative measure to determine critical residues in the spike protein with the potential of serving as effective neutralizing epitopes via allosteric mechanisms.
Experimental validation of a network-predicted residue that is high in closeness and betweenness centralities
We next tested experimentally whether a central line of communication in the network connecting S2 to S1 would alter the spike conformation. Our network analysis has identified the CT0 region (residues 306 to 330) which is a structural bridge connecting NTD to RBD to act as a hub of communication with both high betweenness and closeness measures, as detailed in the previous sections. The capacity of this region to serve as a communication hub remains unchanged upon glycosylation of the protein, with consistently high betweenness and closeness centralities. Moreover, a recent cryo-EM analysis has also indicated the linking peptide between the NTD and RBD can refold depending on the arrangement of the S1 domains.43 Consequently, we chose this beta-stranded region to test the effects of perturbing the spike communication hub. We hypothesized that introduction of a backbone dihedral restrictive proline (P) substitution in the linking region would modify the spike conformation by limiting communication between nodes in the network.
The N317P mutation was specifically selected for experimental studies based on its position in the SD2 paired beta-sheet and prominent score in both betweenness and closeness centrality. This residue position, belonging to the CT0 region, scores high in betweenness in closeness centrality (top 5% of scores among all residues), and is invariant to glycosylation effects. The mutation was prepared in the original D614 Wuhan SARS-CoV-2 sequence backbone with the S2 stabilizing 2P mutations. ACE2 binding is enabled when at least one of the RBD is in the UP conformation. Our binding measurements are the average over all conformations populated in solution, including contributions from 2-UP and 3-UP conformations. One-third of the population exists with at least 1-UP RBD in the original D614 form. In the mutant form, the 1-UP population is slightly reduced and a significant population of RBD is half way UP, likely hindering RBD interface to ACE2.
The N317P mutant displayed a reduction in ACE2 binding by biolayer interferometry (Figure 7A) and differential scanning fluorimetry showed a marked shift in the N317P thermal denaturation profile toward higher temperatures (Figure 7B). Consistent with these observations, negative stain electron microscopy 3D classification indicated a reduction in down state particles with the appearance of a prominent state in which the RBD is stalled in an intermediate state between the up and down states (Figures 7C–7F, S15, and S16). These results together indicate that disruption of the NTD to RBD linker modifies the spike conformation, consistent with an important role for communication between the S1 domains observed in the network analysis.
Figure 7.

The N317P mutation alters the SARS-CoV-2 spike RBD disposition
(A) Binding of ACE2 to the 2P and N317P 2P spike ectodomains of the ancestral form of the virus. Error bars are standard deviation.
(B) Differential scanning fluorimetry profiles for triplicate measures of the 2P and N317P 2P spikes.
(C) Negative stain electron microscopy 3D reconstructions of the 1-up and 3-down states of the 2P spike indicating the relative proportion of particles assigned to each state.
(D) Negative stain electron microscopy 3D reconstructions of the 1-up, 1/2-up, and 3-down states of the 2P-N317P spike indicating the relative proportion of particles assigned to each state.
(E) Refined negative stain electron microscopy 3D reconstruction of the 2P 1-up state with fit coordinates (PDB ID).
(F) Refined negative stain electron microscopy 3D reconstruction of the 2P-N317P 1/2-up state with fit coordinates (PDB ID). Red circles indicate position of coordinates outside density.
Discussion
A graph-based analysis from MD simulations has been a valuable tool for deciphering complex molecular interactions and their roles in regulating cellular processes.30,44,45,46 Many of these efforts have focused on the identification of the critical residues that facilitate effective allosteric interactions, how they are impacted by different conformational states, and how their modification could affect communication pathways.30 Thus it is an approach capable of guiding efforts aimed at the disruption of coordinated motion between distant and important functional regions in specific proteins.
In this article, we have implemented an approach of this nature to identify and analyze the critical residues pertaining to communication efficiency and wide-reaching control in the SARS-CoV-2 spike protein. We studied a total of 30 s of all-atom MD simulations of the full protein trimer in the two widely observed dominant conformations of the original D614 Wu-1 type, as well as the dominant D614G variant. For the latter, we also carried out equivalent MD simulations of the glycosylated protein (detailed in the supplemental information) allowing us to assess which properties of the protein could these external interactions affect or otherwise. Our results are consistent with experimental and computational findings pertaining to the enhanced infectivity of the the D614G virus,12,15,47 its greater vulnerability to RBD-binding antibodies,16,48 and also suggests that the D614G mutation increases the stability of the spike protein.15,18 Experimental results here showed that introduction of a dynamically restrictive proline substitution in a hub of communication identified in the network resulted in marked changes in the spike protein conformation and stability. Thus, the network analysis can be used to guide structure-based immunogen design, highlighting regions with potential to modify epitope presentation.
Long-distance interactions and linkages in proteins are challenging to capture experimentally. For example, the mutational perturbation from D to G at position 614 is known to increase the occurrence of open spike conformation49 and enhance protease cleavage at FCS,35 but the exact mechanism of such information transfer has not been determined. Our computational graph approach has elucidated optimal pathways by which such distant sites of the spike can communicate with each other and ultimately lead to concerted dynamics and allosteric modulations. Moreover, previous studies have highlighted the importance of hydrogen bonds and structural reorganization of the RBD linkers acting as hinges,35,50 and in this study, we have established how critical communication through these hinges is more balanced in the D614G variant, elucidating a mechanism for its phenotypic behavior (Figure S5).
While previous studies have implemented similar network-based approaches using relatively shorter simulations with the aim of identifying overall subdomains and hubs in the spike,51,52,53 our analysis provides detailed and new insights into the communication structure and the control cores of the protein at large and the role of glycosylation. We identify a critical communication ring connecting peripheral regions of the protein, such as the NTD supersite, the up-RBD, and the furin cleavage sites, to each other as well as to the core of the protein, where crucial processes take place upon host receptor binding (e.g. membrane fusion). The structural ring (Figure 2A) is comprised of residues of the NTD (14–305), CT0 (306–330), CT1 (528–590), and CT2 (591–685) domains (Figure 1). These regions also showed a remarkable ability to impact the entire network, where pathways initiated in these regions are able to reach every corner of the protein more efficiently. This provides a quantitative interpretation as to why there is a preference of some mAbs to bind to regions such as the NTD, and highlights the CT0, CT1, and CT2 domains as potential targets for functionality disruption and protein inhibition. Equally important regions of the NTD were also highlighted by the eigenvector centrality, which is a control measure. The functional region found by our analysis does not overlap with the NTD supersite, or more generally, with high-frequency mutable residues (Table S6), suggesting that substitution/deletion events in this domain tend to avoid sites that are relevant for protein functionality. The dominant region is adjacent and in contact with the up-RBD pointing to a potential role of the NTD at facilitating RBD binding or initial virus attachment to the host cell surface via recognition of specific sugar molecules, or possibly aiding the prefusion-to-postfusion transition, as found in other coronaviruses.54,55,56
A specific examination of the NTD residues modified/deleted in the Delta variant (section 2.5) reveals that these mutated residues are associated with an on-average greater closeness centrality than those of the NTD supersite, providing a quantitative explanation as to why these particular residues have been selected for immune escape. Extending this analysis to the Alpha, Beta, and Gamma variants, a similar trend as Figure 7D is found (Figure S6). This indicates that sites for immune escape in NTD could be characterized by their centrality values. With this in mind, we further examined the average centrality scores of all the residues in the spike from Alpha, Beta, Gamma, and Delta variants reveal that the NTD supersite has low betweenness- and eigen centralities with mid-level closeness values (Figure S6). Indeed, the generalizability of possible association between specific centrality measures and immune escape sites will require further studies. Finally, we inspected residues 50 and 417 from the NTD and RBD, respectively, that were implied in a potential recombination event from pangolin RBD to the RBD found in SARS-CoV-2 leading to a significantly higher open conformation in CoV-2 as compared to earlier pangolin spike.57 Our network analysis indicates that the combination of relatively high closeness score and low betweenness score is maintained at these sites.
Post-translational glycan modifications are characteristic of spike proteins and so far their extent of influence beyond shielding has not been well understood. Our network analysis indicates the intrinsic properties of protein communications that are unchanged by protein glycosylation or glycan interactions. Measures such as betweenness and closeness centralities that are unaltered by glycosylation should therefore be resilient to different experimental conditions that can affect glycoforms. So far, our knowledge is extremely limited in terms of proper control over glycoforms, which can complicate rational therapeutic design.58 These glycan-invariant properties therefore give us individual residue-level measures of the spike that can be tapped for improving immunogen candidates irrespective of glycan presence.
In summary, the outcomes of our study can be briefly stated as follows: (1) The communication structure in the D614G variant is more resilient to disruption than that of the ancestral form. (2) The main hubs of communication are located in the NTD and CT0 regions of the protein. (3) Certain residues in the NTD are critically positioned and hierarchically connected to exert wide-reaching control of the protein at large. (4) The D614G-form promotes efficient allosteric pathways to the receptor-binding motif (RBM) from distant regions such as the furin cleavage site, at the cost of heightened vulnerability to RBD-binding antibodies. (5) The NTD residues altered in the Delta variant are equally efficient at impacting the full protein as the ancestral residues. (6) Finally, we provide validation of our model by experimentally demonstrating the predicted importance of a key residue, showing that a N317P substitution could impact ACE2 binding, protein stability, and thermal denaturation, and could shift the protein toward a partially open state.
Limitations of the study
Current technological limitations restrict the length of our computational MD simulations of the spike protein, which excludes timescales and hence processes occurring at the millisecond and higher timescales. For example, we are unable to simulate dynamical transitions between the RBD up and down conformational states. Moreover, we cannot rule out inaccuracies in the force fields used in the simulations, which in turn, would have implication (e.g., biases) in the network representation of the protein. In reference to the critical residues identified, we are unable to tell the exact effect of a given mutation on them and on the full protein. Our analysis highlights these residues as relevant for communication/control, but does not tell us the precise consequence of altering such a residue (e.g., through mutation). For example, if the residue is found relevant for communication (i.e., high betweenness), mutating it could either improve or disrupt communications in the region of the protein. We cannot precisely predict which way it will go since it depends on the specifics of the mutation. Networks are complex objects which makes any comparison analysis challenging. Our results are limited by the scope of the theoretical measures of centralities and shortest path analysis, and they do not fully capture the complexity of these objects. Finally, network representations are also limited to pairwise interactions, which neglect collective behavior (e.g., coordination) involving three or more units. The effect of these higher order interactions is yet to be determined.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human Fc-ACE2 | This paper | N/A |
| Critical commercial assays | ||
| Octet Biolayer Interferometry Instrument | FortѐBio, Sartorius | https://www.sartorius.com/en/products/protein-analysis/octet-bli-detection/octet-label-free-detection-systems |
| Anti-Human Fc (AHC) Octet Biosensors | Sartorius | Cat#18–5064 |
| Deposited data | ||
| Plasmid for the N317P SARS-CoV-2 Spike ectodomain | https://www.addgene.org/196378/ | [Addgene]: [pαH-S-GSAS-2P-N317P] and accession number 196378 |
| Initial Structure Network Models |
https://www.rcsb.org/structure/6VXX https://www.rcsb.org/structure/6VYB Ref. 5 |
PBD: 6VXX & 6VYB |
| ACE2-bound RBD substructure | https://www.rcsb.org/structure/6M0J Ref.10 | PDB: 6M0J |
| Experimental models: Cell lines | ||
| FreeStyle293F cells | ThermoFisher Scientific | Cat#R79007 |
| Turbo293 | Speed Biosystems | Cat#PXX1002 |
| StrepTactin resin | IBA | Cat#2-1208-002 |
| Recombinant DNA | ||
| SARS-CoV-2 Spike construct plasmids (Parent 2P and 2P containing N317P ectodomain) | GenBank | GenBank: MN_908947 |
| Software and algorithms | ||
| GROMACS 5.1.4 | University of Groningen, Royal Institute of Technology, Uppsala University | https://www.gromacs.org/ |
| Wolfram Mathematica 12.0 | Wolfram Research | https://www.wolfram.com/mathematica/ |
| IGraph/M 0.5.1 (October 12, 2020) | The igraph core team | https://igraph.org/ |
| AMBER 2016 | Amber Software Group, University of California – San Francisco | https://ambermd.org/index.php |
| Visual Molecular Dynamics (VMD) 1.9.3 | University of Illinois Urbana-Champaign | https://www.ks.uiuc.edu/Research/vmd/vmd-1.9.3/ |
| CHARMM36 and CHARMM36m forcefield | Martin Karplus – Harvard University; Alexander MacKerell – Maryland University | https://www.charmm.org/archive/charmm/resources/charmm-force-fields/ |
Resource availability
Lead contact
Requests for resources should be directed to and will be fulfilled by the lead contact, S. Gnanakaran (gnana@lanl.gov)
Materials availability
The plasmid for the N317P SARS-CoV-2 Spike ectodomain construct generated in this study have been deposited to Addgene with name pαH-S-GSAS-2P-N317P and accession number 196378.
Experimental model and subject details
Protein production
The parent 2P and 2P containing N317P SARS-CoV-2 Spike ectodomain (residues 1 to 1208; Gen-Bank: MN908947) constructs were produced via transient transfection in FreeStyle293F cells using Turbo293 (SpeedBiosystems). Proteins were purified from the filtered supernatant six days post-transfection using a Strep-Tactin XT Sepharose column (Cytiva). Purity and structural morphology were assessed using reduced and non-reduced SDS-PAGE and negative stain electron microscopy, respectively. Mutagenesis was conducted by GeneImmune Biotechnology (Rockville, MD). Each construct contained a furin cleavage site substitution ( GSAS), a T4 fibritin trimerization motif at the C-terminus, an HRV3C cleavage site, and an 8XHisTag. Transient transfection of the p H vector expression plasmid was performed in FreeStyle293F cells using Turbo293 (SpeedBiosystems) with purification performed using StrepTactin resin (IBA) and size-exclusion chromatography. The human Fc-ACE2 fusion protein was prepared similarly with purification performed using a Protein A affinity column. Samples were prepared in buffer containing 2 mM Tris, 200 mM NaCl, and 0.02% NaN at pH 8.0.
Thermal shift analysis
Thermal shift analysis was conducted using the NanoTemper Technologies Tycho NT.6 instrument. Triplicate measures of were performed in individual capillaries measuring intrinsic fluorescence at 330 nm and 350 nm while heating from 35°C to 95°C at a rate of 3°C per minute. Plots indicate the first derivative of the ratio of fluorescence at 330 nm and 350 nm.
Biolayer interferometry
Binding analysis was performed using the Octet biolayer interferometry instrument (FortèBio). Antihuman Fc (AHC) Octet biosensors were dipped into 10 nM Fc-ACE2 containing wells for a total of 60 s to capture Fc-ACE2 on the senor tip followed by immersion of the tip into solution containing 200 nM spike for 300 s. A blank using Fc-ACE2 capture tips dipped into buffer was collected for signal subtraction. After blank subtraction, the signal at the end of the associate phase was recorded. Measurements were performed in triplicate for both the 2P and N317P constructs.
Negative stain electron microscopy
An aliquot provided freshly at RT and sat at room temperature overnight. Sample was then diluted to 100 g/mL with 5 g/dL Glycerol in HBS (20mM HEPES, 150mM NaCl pH 7.4) buffer containing 8 mM glutaraldehyde. After 5 min incubation, glutaraldehyde was quenched by adding sufficient 1 M Tris stock, pH 7.4, to give 75 mM final Tris concentration and incubated for 5 min. Quenched sample was applied to a glow-discharged carbon-coated EM grid for 10–12 second, then blotted, and stained with 2 g/dL uranyl formate for 1 min, blotted and air-dried. Grids were examined on a Philips EM420 electron microscope operating at 120 kV and nominal magnification of 49,000x, and 137 images were collected on a 76 Mpix CCD camera at 2.4 Å/pixel. Images were analyzed by 2D and 3D class averages using standard protocols with Relion 3.0.59
Method details
Molecular modeling and simulation
For this work, we constructed a network model from the residue-wise correlation matrix of a series of extensive all-atom simulations we previously reported.11 In brief, we constructed the initial structures based on experimentally-resolved Cryo-EM structures of the one-up and all-down states (PDB ID 6VXX and 6VYB5). Regions–largely disordered loops– that were unresolved in these structures were built with a data-driven homology-modeling approach. Missing residues in the RBD specifically were built from an ACE2-bound RBD substructure (PDB ID 6M0J10). The G-form was created from the D-form through manual mutation of residue 614, but otherwise the initial structures were identical.
Atomistic molecular dynamics simulations were performed using the Gromacs software suite with the CHARMM36m force field. We ran five 1.2- s simulations for each of the four systems. The final 1000ns of each trajectory was considered as the production set, after equilibration, for a total of 20 s of simulation in neutral solution, using the Berendsen barostat, the particle mesh Ewald method for electrostatics and temperature coupling at 310K with a Langevin thermostat.
To compare the performance of glycosylated simulations, five glycosylated structures each for the G-form all-down and 1-up conformations were randomly selected from our previously reported ensembles.11 In short, the most probable glycan types at each glycosylation site were selected based on mass spectrometry results60 and added by ab initio modeling using ALLOSMOD61 with CHARMM36 forcefield. Five trajectories of 1.2- s MD simulations were run for both all-down and 1-up conformations of the G-form starting from these structures. The AMBER16 software package62 with CHARMM36m forcefield and TIP3P water model63 was used for the simulations. A cubic box with a minimum of 15 angstrom padding was used to allow for possible glycan extensions during the simulations. The systems were neutralized at an ionic concentration of 150 mM of KCl. Steepest decent energy minimization was performed with gradual release of constraints on the glycan heavy atoms and protein backbone. This was followed by a 2 ns equilibration in an NVT ensemble and a 10 ns equilibration in an NPT ensemble. Constant temperature of 310 K was maintained using Langevin thermostat64 and constant isotropic pressure of 1 bar was maintained by a Berendsen barostat.65 Unrestrained production simulations were performed in the NPT ensemble. Coulombic interactions were computed with the Particle Mesh Ewald (PME) method,66 and covalent bond lengths were constrained with the SHAKE algorithm.67
Graph construction
We use the contact matrix in combination with the cross-correlation matrix to define the edges and weights of our networks, respectively. An edge between two residues is defined when heavy atoms of each residue were at a Euclidean distance of 6Å or less for at least 75 of the analyzed simulation frames. The cross-correlation matrix is defined as:
| (Equation 1) |
where are the fluctuations of atom with respect to its average coordinates. The weights for the communication graphs edges are defined as .30 We use this definition of weights for calculations based on shortest paths analysis (e.g., betweenness and closeness centralities), while its inverse for control/influence analysis40 (e.g. degree and eigenvector centralities). The average coordinates and the fluctuations were calculated after aligning all the frames of the trajectories by residues 856–1067 which constitute the S2 domain helical cores.
Definitions of network measures
In graph theory, centrality measures quantify the importance of a particular node within the network at large.68 Since the importance can be interpreted in several different ways, there are different types of centrality measures according to specific criteria. In undirected networks, the most employed measures are degree, eigenvector, closeness, and betweenness centrality.
Degree centrality quantifies the number of edges a particular node has. It is interpreted as a measure of popularity and influence a node has at the local level. In weighted networks, each edge is leaden by a specific weight and the degree of a node is the sum of the weights of the associated edges and it is hence also known as vertex strength. Mathematically, the degree centrality of node in a weighted graph can be computed as:
| (Equation 2) |
where is the adjacency matrix defining the connections of the graph (, if is in contact distance with and , otherwise), and is the weight matrix defining the strength in the connection between any two nodes. Hence, if the graph is unweighted for all and . The weighted clustering coefficient for residue is computed using Vespignani et. al.39 expression for weighted networks:
| (Equation 3) |
where is the unweighted degree.
Eigenvector centrality measures the influence a node has at a wider scale than degree. It quantifies the influence of a node given that of its neighbors. For example, a given node has many edges and hence it would have a high degree centrality. But if its neighboring nodes have few or no connections, the influence of them is rather small and it will lead to a small eigenvector centrality for the node . On the other hand, if the node is connected with a few other nodes, it would hold a small value of degree centrality, but if these neighboring nodes are robustly connected, node would hold an indirect influence over the additional nodes, which makes the eigenvector centrality of the node higher. Thus, eigenvector centrality is a measure of influence on a larger scale. Mathematically, the eigenvector centrality of the node is given by the -th entry of the eigenvector of the adjacency matrix associated to its largest eigenvalue . In closed form, the full eigenvector satisfies the following equation:
| (Equation 4) |
Closeness centrality measures how far a particular node is from all the other nodes in the network according to their associated pathlength, which quantifies the distance between any two nodes in a network. For weighted networks, the pathlength between node and node , is the sum of the weights associated to the edges that comprise the network path between node and node . Therefore, nodes holding higher values of closeness centrality are located, on average, at a shorter distance from all the rest of the nodes and hence are able spread information more efficiently throughout the graph. For a network with number of nodes, the closeness centrality of the -th node is defined as the inverse of the average distance between a particular node and the rest of the nodes.
| (Equation 5) |
Finally, betweenness centrality quantifies the number of shortest pathways (also called geodesics) a particular node takes part on that connects any two other nodes. Hence, nodes with high betweenness are critical at establishing efficient bridges between distant regions of the network. The removal of these high-betweenness nodes can ultimately lead to the disruption of the network, and therefore their role is key for the stability and resiliency of it. Formally, betweenness centrality of node in an arbitrary network can be expressed as:
| (Equation 6) |
where is the total number of geodesics connecting nodes and , while is the number of geodesics connecting node to node that pass through node .
Quantification and statistical analysis
The KS goodness-of-fit analysis that we used to obtain the -values and D-statistic of the distributions of betweenness scores was carried out through the standard statistical package available in Wolfram Mathematica version 12.0. The threshold of significance used for the computed -values is 0.05.
Acknowledgments
P.D.M. and R.M. were supported by Los Alamos National Laboratory (LANL) Director’s Fellowship. S.C. was supported by the Center of Nonlinear Studies Postdoctoral program. K.N. and S.G. were partially supported by the DOE national laboratories focused on response to COVID-19, with funding providing by the Coronavirus CARES Act. B.K. and S.G. were partially supported by LANL LDRD project 20200706ER. This research used computational resources from LANL Institutional Computing Program. R.H. and P.A. were supported by NIAID project number R01AI165947, and by the Translating Duke Health Initiative.
Author contributions
Conceptualization, P.D.M. and S.G.; Methodology, P.D.M., S.C., R.H., and S.G.; Investigation, P.D.M., S.C., R.H., R.J.E., R.M., K.N., V.S., C.S., and K.M.; Writing - Original Draft, P.D.M., S.C., R.M., and R.H.; Writing – Review & Editing, P.D.M., S.C., and S.G.; Funding Acquisition, S.G., B.K., and P.A.; Resources, S.G., B.K., and P.A.; Supervision, S.G., B.K., and P.A.
Declaration of interests
The authors declare no competing interests.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105855.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. https://europepmc.org/articles/PMC7095418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265—269. doi: 10.1038/s41586-020-2008-3. https://europepmc.org/articles/PMC7094943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Who coronavirus (covid-19) dashboard (latest seen on 11/23/2022 at 12:23 pm et) 2022. https://covid19.who.int/ [Google Scholar]
- 4.Kirchdoerfer R.N., Cottrell C.A., Wang N., Pallesen J., Yassine H.M., Turner H.L., Corbett K.S., Graham B.S., McLellan J.S., Ward A.B. Pre-fusion structure of a human coronavirus spike protein. Nature. 2016;531:118–121. doi: 10.1038/nature17200. https://europepmc.org/articles/PMC4860016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell. 2020;183:1735. doi: 10.1016/j.cell.2020.11.032. https://europepmc.org/articles/PMC7833104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Y., Yang C., Xu X.-F., Xu W., Liu S.-W. Structural and functional properties of sars-cov-2 spike protein: potential antivirus drug development for covid-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. https://europepmc.org/articles/PMC7396720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. https://www.science.org/doi/abs/10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The architecture of sars-cov-2 transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. https://europepmc.org/articles/PMC7179501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y., et al. Structural and functional basis of sars-cov-2 entry by using human ace2. Cell. 2020;181:894–904.e9. doi: 10.1016/j.cell.2020.03.045. https://europepmc.org/articles/PMC7144619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the sars-cov-2 spike receptor-binding domain bound to the ace2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 11.Mansbach R.A., Chakraborty S., Nguyen K., Montefiori D.C., Korber B., Gnanakaran S. The sars-cov-2 spike variant d614g favors an open conformational state. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abf3671. https://europepmc.org/articles/PMC8051874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., Hastie K.M., Parker M.D., Partridge D.G., Evans C.M., Freeman T.M., de Silva T.I., Sheffield COVID-19 Genomics Group. McDanal C., Perez L.G., Tang H., Moon-Walker A., Whelan S.P., LaBranche C.C., Saphire E.O., Montefiori D.C. Tracking changes in sars-cov-2 spike: evidence that d614g increases infectivity of the covid-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. https://europepmc.org/articles/PMC7332439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peacock T.P., Goldhill D.H., Zhou J., Baillon L., Frise R., Swann O.C., Kugathasan R., Penn R., Brown J.C., Sanchez-David R.Y., et al. The furin cleavage site in the sars-cov-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021;6:899–909. doi: 10.1038/s41564-021-00908-w. [DOI] [PubMed] [Google Scholar]
- 14.Benton D.J., Wrobel A.G., Roustan C., Borg A., Xu P., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. The effect of the d614g substitution on the structure of the spike glycoprotein of sars-cov-2. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2022586118. https://europepmc.org/articles/PMC7936381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Jackson C.B., Mou H., Ojha A., Peng H., Quinlan B.D., Rangarajan E.S., Pan A., Vanderheiden A., Suthar M.S., et al. Sars-cov-2 spike-protein d614g mutation increases virion spike density and infectivity. Nat. Commun. 2020;11:6013. doi: 10.1038/s41467-020-19808-4. https://europepmc.org/articles/PMC7693302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weissman D., Alameh M.-G., de Silva T., Collini P., Hornsby H., Brown R., LaBranche C.C., Edwards R.J., Sutherland L., Santra S., et al. D614g spike mutation increases sars cov-2 susceptibility to neutralization. Cell Host Microbe. 2021;29:23–31.e4. doi: 10.1016/j.chom.2020.11.012. https://europepmc.org/articles/PMC7707640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Sars-cov-2 and bat ratg13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. https://europepmc.org/articles/PMC7610980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Cai Y., Xiao T., Lu J., Peng H., Sterling S.M., Walsh R.M., Rits-Volloch S., Zhu H., Woosley A.N., et al. Structural impact on sars-cov-2 spike protein by d614g substitution. Science (New York, N.Y.) 2021;372:525–530. doi: 10.1126/science.abf2303. https://europepmc.org/articles/PMC8139424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plante J.A., Liu Y., Liu J., Xia H., Johnson B.A., Lokugamage K.G., Zhang X., Muruato A.E., Zou J., Fontes-Garfias C.R., et al. Spike mutation d614g alters sars-cov-2 fitness. Nature. 2021;592:116–121. doi: 10.1038/s41586-020-2895-3. https://europepmc.org/articles/PMC8158177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauring A.S., Hodcroft E.B. Genetic variants of sars-cov-2-what do they mean? JAMA. 2021;325:529–531. doi: 10.1001/jama.2020.27124. [DOI] [PubMed] [Google Scholar]
- 21.McCormick K.D., Jacobs J.L., Mellors J.W. The emerging plasticity of sars-cov-2. Science (New York, N.Y.) 2021;371:1306–1308. doi: 10.1126/science.abg4493. [DOI] [PubMed] [Google Scholar]
- 22.McCallum M., De Marco A., Lempp F.A., Tortorici M.A., Pinto D., Walls A.C., Beltramello M., Chen A., Liu Z., Zatta F., et al. N-terminal domain antigenic mapping reveals a site of vulnerability for sars-cov-2. Cell. 2021;184:2332–2347.e16. doi: 10.1016/j.cell.2021.03.028. https://europepmc.org/articles/PMC7962585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerutti G., Guo Y., Zhou T., Gorman J., Lee M., Rapp M., Reddem E.R., Yu J., Bahna F., Bimela J., et al. Potent sars-cov-2 neutralizing antibodies directed against spike n-terminal domain target a single supersite. Cell Host Microbe. 2021;29:819–833.e7. doi: 10.1016/j.chom.2021.03.005. https://europepmc.org/articles/PMC7953435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., et al. A neutralizing human antibody binds to the n-terminal domain of the spike protein of sars-cov-2. Science (New York, N.Y.) 2020;369:650–655. doi: 10.1126/science.abc6952. https://europepmc.org/articles/PMC7319273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy K.R., Rennick L.J., Nambulli S., Robinson-McCarthy L.R., Bain W.G., Haidar G., Duprex W.P. Recurrent deletions in the sars-cov-2 spike glycoprotein drive antibody escape. Science (New York, N.Y.) 2021;371:1139–1142. doi: 10.1126/science.abf6950. https://europepmc.org/articles/PMC7971772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer W., Giorgi E.E., Chakraborty S., Nguyen K., Bhattacharya T., Theiler J., Goloboff P.A., Yoon H., Abfalterer W., Foley B.T., Tegally H., San J.E., de Oliveira T., Network for Genomic Surveillance in South Africa NGS-SA. Gnanakaran S., Korber B. Network for Genomic Surveillance in South Africa (NGS-SA), S. Gnanakaran, B. Korber, Hiv-1 and sars-cov-2: patterns in the evolution of two pandemic pathogens. Cell Host Microbe. 2021;29:1093–1110. doi: 10.1016/j.chom.2021.05.012. https://europepmc.org/articles/PMC8173590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saito A., Irie T., Suzuki R., Maemura T., Nasser H., Uriu K., Kosugi Y., Shirakawa K., Sadamasu K., Kimura I., et al. Genotype to Phenotype Japan G2P-Japan Consortium Enhanced fusogenicity and pathogenicity of sars-cov-2 delta p681r mutation. Nature. 2022;602:300–306. doi: 10.1038/s41586-021-04266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray D., Le L., Andricioaei I. Distant residues modulate conformational opening in sars-cov-2 spike protein. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100943118. https://europepmc.org/articles/PMC8639331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spinello A., Saltalamacchia A., Borišek J., Magistrato A. Allosteric cross-talk among spike’s receptor-binding domain mutations of the sars-cov-2 south african variant triggers an effective hijacking of human cell receptor. J. Phys. Chem. Lett. 2021;12:5987–5993. doi: 10.1021/acs.jpclett.1c01415. https://europepmc.org/articles/PMC8247780 [DOI] [PubMed] [Google Scholar]
- 30.Sethi A., Eargle J., Black A.A., Luthey-Schulten Z. Dynamical networks in trna:protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. https://europepmc.org/articles/PMC2672494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen K., Chakraborty S., Mansbach R.A., Korber B., Gnanakaran S. Exploring the role of glycans in the interaction of sars-cov-2 rbd and human receptor ace2. Viruses. 2021;13:927. doi: 10.3390/v13050927. https://europepmc.org/articles/PMC8156887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suryadevara N., Shrihari S., Gilchuk P., VanBlargan L.A., Binshtein E., Zost S.J., Nargi R.S., Sutton R.E., Winkler E.S., Chen E.C., et al. Neutralizing and protective human monoclonal antibodies recognizing the n-terminal domain of the sars-cov-2 spike protein. Cell. 2021;184:2316–2331.e15. doi: 10.1016/j.cell.2021.03.029. https://europepmc.org/articles/PMC7962591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu C., Wang Y., Liu C., Zhang C., Han W., Hong X., Wang Y., Hong Q., Wang S., Zhao Q., et al. Conformational dynamics of sars-cov-2 trimeric spike glycoprotein in complex with receptor ace2 revealed by cryo-em. Sci. Adv. 2021;7:eabe5575. doi: 10.1126/sciadv.abe5575. https://europepmc.org/articles/PMC7775788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmin T., Kalbermatter D., Harder D., Plattet P., Fotiadis D. Structures and dynamics of the novel s1/s2 protease cleavage site loop of the sars-cov-2 spike glycoprotein. J. Struct. Biol. X. 2020;4 doi: 10.1016/j.yjsbx.2020.100038. https://europepmc.org/articles/PMC7534663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gobeil S.M.-C., Janowska K., McDowell S., Mansouri K., Parks R., Manne K., Stalls V., Kopp M.F., Henderson R., Edwards R.J., et al. D614g mutation alters sars-cov-2 spike conformation and enhances protease cleavage at the s1/s2 junction. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2020.108630. https://europepmc.org/articles/PMC7762703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benton D.J., Wrobel A.G., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Receptor binding and priming of the spike protein of sars-cov-2 for membrane fusion. Nature. 2020;588:327–330. doi: 10.1038/s41586-020-2772-0. https://europepmc.org/articles/PMC7116727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra A., Rathore A.S. Pharmacophore screening to identify natural origin compounds to target rna-dependent rna polymerase (rdrp) of sars-cov2. Mol. Divers. 2022;26:2613–2629. doi: 10.1007/s11030-021-10358-5. https://europepmc.org/articles/PMC8742708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dutta K. Allosteric site of ace-2 as a drug target for covid-19. ACS Pharmacol. Transl. Sci. 2022;5:179–182. doi: 10.1021/acsptsci.2c00003. https://europepmc.org/articles/PMC8864771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrat A., Barthélemy M., Pastor-Satorras R., Vespignani A. The architecture of complex weighted networks. Proc. Natl. Acad. Sci. USA. 2004;101:3747–3752. doi: 10.1073/pnas.0400087101. https://www.pnas.org/doi/abs/10.1073/pnas.0400087101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antoniou I.E., Tsompa E.T. Statistical analysis of weighted networks. Discrete Dynam Nat. Soc. 2008;2008:1–16. [Google Scholar]
- 41.Xing Y., Li X., Gao X., Dong Q. Natural polymorphisms are present in the furin cleavage site of the sars-cov-2 spike glycoprotein. Front. Genet. 2020;11:783. doi: 10.3389/fgene.2020.00783. https://europepmc.org/articles/PMC7379507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCallum M., Bassi J., De Marco A., Chen A., Walls A.C., Di Iulio J., Tortorici M.A., Navarro M.-J., Silacci-Fregni C., Saliba C., et al. Sars-cov-2 immune evasion by the b.1.427/b.1.429 variant of concern. Science (New York, N.Y.) 2021;373:648–654. doi: 10.1126/science.abi7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gobeil S.M.-C., Henderson R., Stalls V., Janowska K., Huang X., May A., Speakman M., Beaudoin E., Manne K., Li D., et al. Structural diversity of the sars-cov-2 omicron spike. Mol. Cell. 2022;82:2050–2068.e6. doi: 10.1016/j.molcel.2022.03.028. https://europepmc.org/articles/PMC8947964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sethi A., Tian J., Derdeyn C.A., Korber B., Gnanakaran S. A mechanistic understanding of allosteric immune escape pathways in the hiv-1 envelope glycoprotein. PLoS Comput. Biol. 2013;9 doi: 10.1371/journal.pcbi.1003046. https://europepmc.org/articles/PMC3656115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fokas A.S., Cole D.J., Ahnert S.E., Chin A.W. Residue geometry networks: a rigidity-based approach to the amino acid network and evolutionary rate analysis. Sci. Rep. 2016;6 doi: 10.1038/srep33213. https://europepmc.org/articles/PMC5021933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melo M.C.R., Bernardi R.C., de la Fuente-Nunez C., Luthey-Schulten Z. Generalized correlation-based dynamical network analysis: a new high-performance approach for identifying allosteric communications in molecular dynamics trajectories. J. Chem. Phys. 2020;153 doi: 10.1063/5.0018980. [DOI] [PubMed] [Google Scholar]
- 47.Groves D.C., Rowland-Jones S.L., Angyal A. The d614g mutations in the sars-cov-2 spike protein: implications for viral infectivity, disease severity and vaccine design. Biochem. Biophys. Res. Commun. 2021;538:104–107. doi: 10.1016/j.bbrc.2020.10.109. https://europepmc.org/articles/PMC7643658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinto D., Park Y.-J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of sars-cov-2 by a human monoclonal sars-cov antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 49.Yurkovetskiy L., Wang X., Pascal K.E., Tomkins-Tinch C., Nyalile T.P., Wang Y., Baum A., Diehl W.E., Dauphin A., Carbone C., et al. Structural and functional analysis of the d614g sars-cov-2 spike protein variant. Cell. 2020;183:739–751.e8. doi: 10.1016/j.cell.2020.09.032. https://europepmc.org/articles/PMC7492024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meirson T., Bomze D., Markel G. Structural basis of sars-cov-2 spike protein induced by ace2. Bioinformatics. 2021;37:929–936. doi: 10.1093/bioinformatics/btaa744. https://europepmc.org/articles/PMC7558967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halder A., Anto A., Subramanyan V., Bhattacharyya M., Vishveshwara S., Vishveshwara S. Surveying the side-chain network approach to protein structure and dynamics: the sars-cov-2 spike protein as an illustrative case. Front. Mol. Biosci. 2020;7 doi: 10.3389/fmolb.2020.596945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verkhivker G.M., Di Paola L. Dynamic network modeling of allosteric interactions and communication pathways in the sars-cov-2 spike trimer mutants: differential modulation of conformational landscapes and signal transmission via cascades of regulatory switches. J. Phys. Chem. B. 2021;125:850–873. doi: 10.1021/acs.jpcb.0c10637. https://europepmc.org/articles/PMC7839160 [DOI] [PubMed] [Google Scholar]
- 53.Ghorbani M., Brooks B.R., Klauda J.B. Exploring dynamics and network analysis of spike glycoprotein of sars-cov-2. Biophys. J. 2021;120:2902–2913. doi: 10.1016/j.bpj.2021.02.047. https://europepmc.org/articles/PMC7939993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krempl C., Schultze B., Laude H., Herrler G. Point mutations in the s protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 1997;71:3285–3287. doi: 10.1128/jvi.71.4.3285-3287.1997. https://europepmc.org/articles/PMC191465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Künkel F., Herrler G. Structural and functional analysis of the surface protein of human coronavirus oc43. Virology. 1993;195:195–202. doi: 10.1006/viro.1993.1360. https://europepmc.org/articles/PMC7130786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu G., Wang Q., Gao G.F. Bat-to-human: spike features determining ’host jump’ of coronaviruses sars-cov, mers-cov, and beyond. Trends Microbiol. 2015;23:468–478. doi: 10.1016/j.tim.2015.06.003. https://europepmc.org/articles/PMC7125587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wrobel A.G., Benton D.J., Xu P., Calder L.J., Borg A., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Structure and binding properties of pangolin-cov spike glycoprotein inform the evolution of sars-cov-2. Nat. Commun. 2021;12:837. doi: 10.1038/s41467-021-21006-9. https://europepmc.org/articles/PMC7864994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buettner M.J., Shah S.R., Saeui C.T., Ariss R., Yarema K.J. Improving immunotherapy through glycodesign. Front. Immunol. 2018;9:2485. doi: 10.3389/fimmu.2018.02485. https://europepmc.org/articles/PMC6224361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-em structure determination in relion-3. Elife. 2018;7 doi: 10.7554/elife.42166. https://europepmc.org/articles/PMC6250425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the sars-cov-2 spike. Science (New York, N.Y.) 2020;369:330–333. doi: 10.1126/science.abb9983. https://europepmc.org/articles/PMC7199903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guttman M., Weinkam P., Sali A., Lee K.K. All-atom ensemble modeling to analyze small-angle x-ray scattering of glycosylated proteins. Structure. 1993;21:321–331. doi: 10.1016/j.str.2013.02.004. https://europepmc.org/articles/PMC3840220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salomon-Ferrer R., Götz A.W., Poole D., Le Grand S., Walker R.C. Routine microsecond molecular dynamics simulations with amber on gpus. 2. explicit solvent particle mesh ewald. J. Chem. Theor. Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 64.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 65.Van Gunsteren W.F., Berendsen H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simulat. 1988;1:173–185. doi: 10.1080/08927028808080941. [DOI] [Google Scholar]
- 66.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 67.Ryckaert J.-P., Ciccotti G., Berendsen H.J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. https://www.sciencedirect.com/science/article/pii/0021999177900985 [DOI] [Google Scholar]
- 68.Newman M. Oxford University Press; 2010. Networks: An Introduction. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.