

Abstract
Appropriately controlled gene expression is fundamental for normal growth and survival of all living organisms. In eukaryotes, the transcription of protein-coding mRNAs is dependent on RNA polymerase II (Pol II). The multi-subunit transcription cofactor Mediator complex is proposed to regulate most, if not all, of the Pol II-dependent transcription. Here we focus our discussion on two subunits of the Mediator complex, cyclin-dependent kinase 8 (CDK8) and its regulatory partner Cyclin C (CycC), because they are either mutated or amplified in a variety of human cancers. CDK8 functions as an oncoprotein in melanoma and colorectal cancers, thus there are considerable interests in developing drugs specifically targeting the CDK8 kinase activity. However, to evaluate the feasibility of targeting CDK8 for cancer therapy and to understand how their dysregulation contributes to tumorigenesis, it is essential to elucidate the in vivo function and regulation of CDK8-CycC, which are still poorly understood in multi-cellular organisms. We summarize the evidence linking their dysregulation to various cancers and present our bioinformatics and computational analyses on the structure and evolution of CDK8. We also discuss the implications of these observations in tumorigenesis. Because most of the Mediator subunits, including CDK8 and CycC, are highly conserved during eukaryotic evolution, we expect that investigations using model organisms such as Drosophila will provide important insights into the function and regulation of CDK8 and CycC in different cellular and developmental contexts.
Keywords: Mediator complex, CDK8, Transcription, Tumorigenesis, MD simulation
1. Introduction
The normal development and function of an organism require precise and coordinated control of gene expression. In general, cells respond to extracellular stimuli by activating and integrating distinct signal transduction pathways, which eventually process and relay the information from cell surface receptors to a variety of transcription factors, resulting in altered gene expressions. As a result, cells proliferate, differentiate, or commit apoptosis. Modulating gene expression is a critical step to ensure normal development and maintain tissue homeostasis.
Dysregulated transcription is at the root of many diseases, such as cancer—the major cause of mortality and morbidity in modern societies. The World Health Organization estimated that cancer alone accounts for approximately 13% of the total number of human deaths globally (7.9 million) in 2007 (McAloose and Newton, 2009). Genetic alterations, epigenetic perturbations, and environmental factors are often recognized as the multi-factorial causes of cancer. Genetic changes include gene mutations, deletions, and duplication as well as chromosomal abnormalities, which result in the loss of tumor-suppressor gene functions and/or the gain of oncogene functions (Kinzler and Vogelstein, 1997; Hanahan and Weinberg, 2000). Epigenetic changes include chromatin-mediated changes such as aberrant DNA methylation, histone modification, and loss of imprinting (Feinberg and Tycko, 2004; Sharma et al., 2010). Dysregulated gene transcription by these genetic and epigenetic changes is a universal feature of human cancers and many other diseases, which further highlights the importance of precisely regulated gene expression during development. Thus, the study of the transcriptional regulatory mechanisms is pivotal in understanding not only normal development and tissue homeostasis, but also tumorigenesis.
2. Function of the Mediator complex in transcription
Gene expression begins with the interactions between specific transcription factors and their DNA binding sites at the promoter region, and subsequent recruitment of the Mediator complex and the general transcription factors, such as TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH, and RNA polymerase II (Pol II), forming the pre-elongation (or preinitiation) complex (Hahn, 2004; Taatjes, 2010). To initiate transcription, these general transcription initiation factors are required for the recruitment of RNA Pol II to the promoters of target genes. Next, DNA surrounding the transcription start site is melted and allows the transcription initiation and elongation to occur. After the release of Pol II, several general transcription factors including TFIIA, TFIID, TFIIE, TFIIH, and the Mediator, remain behind at the promoter, forming the scaffold complex, which is thought to facilitate subsequent rounds of transcription or transcription re-initiation (Yudkovsky et al., 2000; Hahn, 2004).
Compared to other general transcription factors, the Mediator is unique in that it is the largest complex, in terms of both the size (about 1.2 MDa) and the number of subunits (~30 polypeptides in different species) (Bjorklund and Gustafsson, 2005; Conaway et al., 2005; Kim and Lis, 2005; Malik and Roeder, 2005; Casamassimi and Napoli, 2007; Taatjes, 2010). The Mediator complex is proposed to regulate most, if not all, of the Pol II-mediated transcription (Kornberg, 2005). Despite extensive biochemical and molecular studies, together with elegant genetic studies in yeast, the function and regulation of most Mediator subunits in metazoans are still poorly understood. The shear size and enormous complexity of the Mediator complex provide many possible mechanisms to fine-tune gene expression in different biological contexts, but at the same time pose a daunting challenge for researchers to elucidate its function and regulation in vivo.
Biochemical studies support a general model for the role of the Mediator complex, which functions as a molecular bridge between specific transcription activators and the general transcription machinery (Fig. 1A). Most of the Mediator subunits are highly conserved in eukaryotes (Bourbon et al., 2004; Bourbon, 2008; references therein). Biochemical analyses have identified two distinct Mediator complexes: a “small” complex of over 20 polypeptides that activates the transcription of Pol II-dependent genes, and a “large” complex with four added subunits that generally represses transcription (Myers and Kornberg, 2000; Mittler et al., 2001; Näär et al., 2001; Bourbon et al., 2004; Malik and Roeder, 2005). The evolutionarily conserved, four-subunit module that defines the large Mediator complex comprises cyclin-dependent kinase 8 (CDK8), Cyclin C (CycC), MED12, and MED13 (Fig. 1B). The mammalian MED12 and CycC are required for the kinase activity of CDK8, while MED13 is necessary to recruit the CDK8 module to the small Mediator complex (Knuesel et al., 2009a, 2009b). Another difference between the small and the large Mediator complex is the subunit MED26 (ARC70 or CRSP70), which is only present in the small Mediator complex (Mittler et al., 2001; Taatjes et al., 2002; Sato et al., 2004; Ebmeier and Taatjes, 2010). Recently, human MED26 is proposed to regulate the transition from paused Pol II to productive elongation (Takahashi et al., 2011). It is still unclear how the conversion between the small and the large Mediator complexes is dynamically regulated during transcription.
Fig. 1.
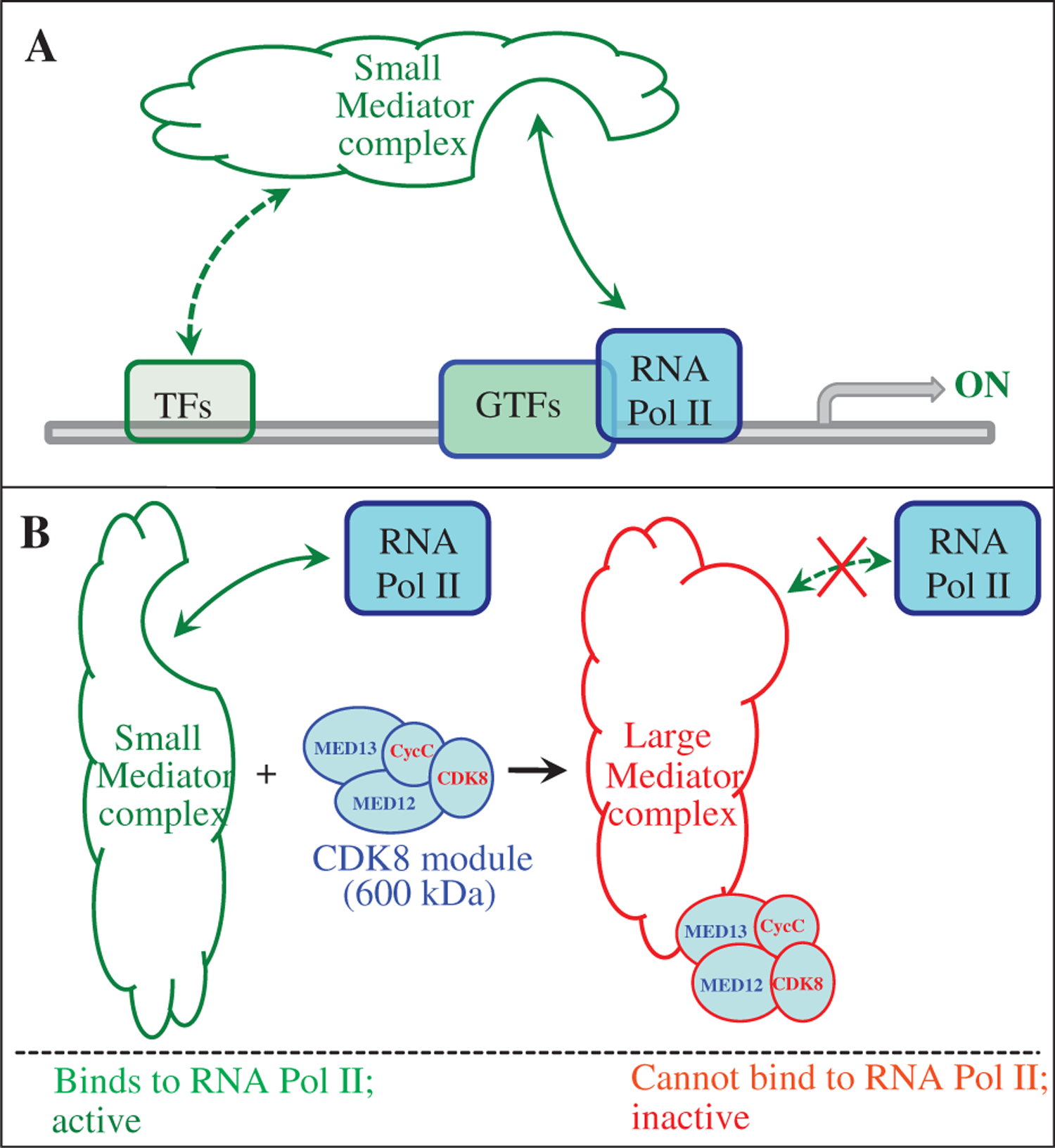
A model for Mediator complexes in regulating Pol II-dependent gene expression. A: Mediator complex serves as a molecular bridge between specific transcription factors (TFs) and the general transcription machinery, which is composed of the general transcription factors (GTFs) and RNA Polymerase II (RNA Pol II). B: CDK8 module represents a major difference between the small and the large Mediator complexes.
Several reviews were published in recent years, discussing how the CDK8 module and Mediator complexes modulate Pol II-dependent gene expression (Casamassimi and Napoli, 2007; Galbraith et al., 2010; Taatjes, 2010; Conaway and Conaway, 2011; Knuesel and Taatjes, 2011), and the role of CDK8 in regulating cell cycle progression (Ji and Dyson, 2010). Essentially, two major mechanisms have been proposed to explain how association of the CDK8 module with the small Mediator complex can repress transcription: first, the CDK8 submodule induces conformational change of the small Mediator complex that physically disrupts the interaction between RNA Pol II and the small Mediator complex, thereby blocks the subsequent rounds of transcription (i.e., transcription re-initiation) (Fig. 1B) (Näär et al., 2002; Taatjes et al., 2002; Samuelsen et al., 2003; Bjorklund and Gustafsson, 2005; Elmlund et al., 2006; Knuesel et al., 2009a; Ebmeier and Taatjes, 2010; Bernecky et al., 2011). Interestingly, this effect of the CDK8 module is independent of the kinase activity of CDK8. Second, CDK8 phosphorylates different substrates, which include several general transcription factors, such as RNA Pol II C-terminal domain, MED2, MED13, the Cyclin H subunit of the transcription initiation factor TFIIH, and certain transactivators, such as Notch intracellular domain and E2F1 in metazoans (Hengartner et al., 1998; Akoulitchev et al., 2000; Fryer et al., 2004; Hallberg et al., 2004; Liu et al., 2004; van de Peppel et al., 2005; Morris et al., 2008; Knuesel et al., 2009b). In the latter mechanism, the CDK8 kinase activity is required for the inhibitory effect of the CDK8 module, and this elegant mechanism explains why phosphorylation of transcription activators is often coupled with their degradation (Tansey, 2001). One common feature of both mechanisms is that the CDK8 module negatively regulates the rate of transcription re-initiation, thereby limiting the quantity of mRNA production. These mechanisms provide complex but efficient ways to fine-tune gene expression.
Thus, in a number of contexts, the CDK8 module appears to inhibit the activities of the small Mediator complex (Näär et al., 2002; Taatjes et al., 2002; Malik et al., 2004; Malik and Roeder, 2005). However, accumulating evidence indicates that CDK8 can also positively regulate transcription, for examples, by directly phosphorylating p53, SMADs, and histone H3, or by facilitating assembly of Pol II elongation complex (Galbraith et al., 2010; Taatjes, 2010). These results suggest that the role of CDK8 on transcription is context-specific: it is dependent on the specific biological contexts and identity of transcription factors with which it interacts.
3. Distinguishing activities of CDK8 kinase versus the CDK8 module
To understand the context-specific functions of CDK8, it is important to distinguish the activities of CDK8 kinase with the activities of the CDK8 module. These concepts are different for the following two reasons. First, CDK8 may function outside of the Mediator complexes. CDK8, CycC, MED12, and MED13 form the CDK8 module in a 1:1:1:1 stoichiometry in vitro (Fig. 1B) (Knuesel et al., 2009b). Earlier work in yeast showed that all CDK8 module component deletions are viable, have similar phenotypes, and share common transcriptional defects, suggesting a uniform function for the four subunits (Carlson, 1997; van de Peppel et al., 2005). However, emerging evidence in yeast and metazoans is inconsistent with this picture. For examples, subunits of the CDK8 module have different roles in activating multidrug resistance gene expression in yeast: Med12 is uniquely required for the multidrug resistance transcription factor Pde3-induced PDR5 gene expression (Shahi et al., 2010). In addition, the mutants of Drosophila MED12 and MED13 behave differently from CDK8 and CycC mutants in genetic analyses, and MED12 and MED13 are particularly important in the differentiation of the eye and leg imaginal discs and the hematopoietic cells (Janody et al., 2003; Loncle et al., 2007; Gobert et al., 2010). In mammals, CDK8 knockout mice are lethal prior to compaction and implantation at embryonic days 2.5, suggesting a critical role of CDK8 for cell-fate determination in early embryos (Westerling et al., 2007). Conditional CDK8 knockout mice, or knockout mice for other subunits of the CDK8 module have not been reported. Nevertheless, MED12 is reported to recruit the histone H3 Lys9 methyltransferase G9a in a CDK8-independent manner, thereby inhibiting the expression of neuronal genes in non-neuronal cells (Ding et al., 2008; Ding et al., 2009). Furthermore, only a fraction of CDK8 is associated with MED12 and MED13 in mammalian cells, thus CDK8 may function outside of Mediator (Meyer et al., 2008; Knuesel et al., 2009a, 2009b). These observations suggest that the four subunits of the CDK8 module do not have identical roles and possibly that CDK8-CycC could have Mediator-independent functions that remain to be discovered.
Second, CDK8 module can inhibit transcription independent of the CDK8 kinase activity. The current models suggest that CDK8 is recruited to promoters through the small Mediator complex via MED13 subunit of the CDK8 module (Taatjes, 2010). CDK8 can inhibit Pol II-dependent transcription through the CDK8 module, which can induce the conformational change of the small Mediator complex, thereby disrupting the interaction between the large Mediator complex and RNA Pol II (Knuesel et al., 2009a; Taatjes, 2010; Bernecky et al., 2011). However, this effect is independent of the CDK8 kinase activity.
Taken together, the activities of CDK8 kinase and transcription function of CDK8 module are not the same. The universal requirement of the CDK8 kinase function in various cellular and developmental contexts and the specific requirements for other conserved module members are unknown. Distinguishing these two concepts may have important implications in developing therapeutic approaches (see Section 6).
For the following two reasons we focus our discussion and analyses on two subunits of the Mediator complex: CDK8 and CycC with an emphasis on CDK8. First, both CDK8 and CycC are dysregulated in a variety of human cancers (see below). Second, the function and regulation of CDK8 and CycC in vivo are still poorly understood. Because CDK8 and CycC are highly conserved in evolution (Bourbon, 2008), we expect that studies using model organisms such as Drosophila may provide important insights into the function and regulation of CDK8 during development.
4. Dysregulation of CDK8 and CycC in human cancers
As summarized in this section, genes encoding CDK8 (CDK8) and CycC (CCNC) are found to be frequently dysregulated in various human cancers. There are only a few cases where the role and potential mechanisms of CDK8 dysregulation in tumorigenesis have been studied to some extent. These include colorectal cancers and melanoma in humans, and skin cancer in walleye, a freshwater fish native to North America. The causes and consequences of CDK8 and CycC dysregulation in other cancers have not been explored.
The dysregulation of CDK8 is best studied in colorectal cancers. CDK8 was identified as an oncoprotein that promotes the proliferation of colorectal cancer cells (Firestein et al., 2008). In this study, two RNAi screens were carried out to search for kinases and phosphatases required for both colon cancer cell proliferation and β-catenin-dependent transcription. Nine kinases were identified, but CDK8 was the only one exhibiting frequent copy number gain in human colorectal cancers: CDK8 gene is amplified in ~47% of colorectal adenocarcinoma patient samples (N = 123). This observation is consistent with other reports showing that the chromosomal region that harbors CDK8 in chromosome 13 (13q12.13) is gained in ~60% of colorectal cancers (Tsafrir et al., 2006; Martin et al., 2007; Sheffer et al., 2009). Ectopic expression of wild-type CDK8, but not the kinase dead mutant (CDK8-D173A), in immortal murine fibroblasts (untransformed NIH 3T3 cells) led to anchorage-independent colony growth and tumor formation in immunodeficient mice, while the expression of the CDK8-D173A inhibited β-catenin-induced 3T3 cell transformation (Firestein et al., 2008). These results suggest that the gain of CDK8 activity is sufficient to transform 3T3 cells, and CDK8 activity is necessary for β-catenin-driven transformation of the 3T3 cells. Finally, knocking down CDK8, but not neighboring genes, significantly slowed tumor cell growth. An important implication of this observation is that CDK8 may serve as a promising drug target. These seminal observations established that CDK8 is a bona fide oncoprotein in colorectal cancers and that CDK8 is required for the expression of β-catenin target genes (Firestein et al., 2008; Firestein and Hahn, 2009).
Following up work with large cohorts of clinical samples further confirmed the importance of CDK8 overexpression in colorectal cancers. For example, CDK8 expression is increased in 70% of colorectal cancer samples (N = 470), which is significantly correlated with increased colon cancer-specific mortality (Firestein et al., 2010). However, no correlation between the expression of CDK8 and clinical outcome among rectal cancers was observed, suggesting that CDK8 is a particularly valuable molecular biomarker for the prognosis of a subset of colon cancer patients (Firestein et al., 2010). The expression of CDK8 is significantly higher in samples from women than from men but is not correlated with tumor stage, body-mass index, or age of patients at diagnosis (Firestein et al., 2010). At the molecular level, CDK8 expression is correlated with high β-catenin activation, expression of tumor suppressor p53, and overexpression of FASN (fatty acid synthase), suggesting that CDK8 activity may regulate multiple pathways involved in colorectal tumorigenesis (Firestein et al., 2010).
The potential link between CDK8 and β-catenin is also supported by an independent study with 127 cases of colorectal adenocarcinoma patients (Seo et al., 2010). In this study positive expression of CDK8 was detected in 76% of the cases. Consistent with the study by Firestein et al. (2010), CDK8 expression is closely associated with the high mortality of colorectal cancer patients, gender (more common in women than in men), as well as the expression of β-catenin in the cytoplasm (Firestein et al., 2010; Seo et al., 2010). However, unlike the study by Firestein et al. (2010), this independent study observed significantly positive correlations between the expression of CDK8 and disease stage, or lymph node and distant metastasis (Seo et al., 2010).
Consistent with these reports, recent bioinformatic analyses identified CDK8 as one of the most significant colorectal cancer-associated genes (Nagaraj and Reverter, 2011). Collectively, these studies support an important role of CDK8 in colorectal tumorigenesis. Thus, targeting CDK8 may represent a promising approach in treating a substantial set of colorectal cancers.
In addition to colorectal cancers, CDK8 expression is significantly associated with β-catenin activation in gastric adenocarcinoma (N = 60) (Kim et al., 2011). Positive correlation was observed between CDK8 levels and lymph node metastasis, suggesting that, similar to colorectal cancers, elevated expression of CDK8 predicts poor prognosis in gastric cancers (Kim et al., 2011).
How does CDK8 affect the transcription activities of β-catenin? At least two mechanisms were proposed. First, CDK8 may directly modulate β-catenin-activated transcription. The β-catenin target genes are suppressed by the homologs of CDK8 module subunits MED12 and MED13 in Wnt-regulated cell fusion in Caenorhabditis elegans (Yoda et al., 2005), but activated by homologs of MED12 and MED13 during the development of wing and eye imaginal discs in Drosophila (Carrera et al., 2008). A positive effect of MED12 and MED13 on β-cetenin-activated transcription is also observed in cultured mammalian and Drosophila cells (Kim et al., 2006; Carrera et al., 2008). β-catenin is shown to directly interact with MED12 via its C-terminal domain in mammalian cells, or indirectly interacts with MED12 and MED13 via Pygopus (Kim et al., 2006; Carrera et al., 2008). Pygopus interacts with Armadillo (the β-catenin ortholog in Drosophila) through Legless, and Pygopus and Legless serve as transcription coactivators for β-catenin (Townsley et al., 2004; Hoffmans et al., 2005). The mechanisms underlying these opposite effects are still not known. CDK8 is proposed to phosphorylate β-catenin at several serine residues, but this possibility remains to be rigorously examined (Firestein and Hahn, 2009; Galbraith et al., 2010).
An alternative mechanism is that CDK8 may indirectly activate β-catenin-dependent transcription by inhibiting E2F1, which antagonizes with β-catenin (Morris et al., 2008). In this indirect mechanism, CDK8 directly phosphorylates E2F1, thereby inhibiting transcription activities of E2F1 in Drosophila and mammalian cells (Morris et al., 2008). E2F1 may antagonize with β-catenin by directly competing with β-catenin in binding with the promoter of target genes, such as c-MYC, PPARδ and CD44 (Morris et al., 2008; Ji and Dyson, 2010), or by directly activating the expression of factors that promote the degradation of β-catenin, such as AXIN1, AXIN2 and SIAH1 (Hughes and Brady, 2005; Hallstrom et al., 2008; Morris et al., 2008). Interestingly, E2F1 promotes β-catenin destruction through unknown mechanisms that are independent of GSK3 (Hughes and Brady, 2005; Hallstrom et al., 2008; Morris et al., 2008). Together, these mechanisms may explain how CDK8 positively regulates β-catenin-dependent transcription. It is unclear whether the same regulatory network among CDK8, E2F1, and β-catenin operates during normal development, or in other types of cancer, such as gastric cancers.
In addition to amplification of CDK8 in colorectal cancer, elevated expression of the CDK8 gene is reported to play a major role in promoting the proliferation of melanoma cells (Kapoor et al., 2010). The histone variant macroH2A (mH2A) is lost in over 80% of vertical growth phase and metastatic melanomas (N = 218). Gene expression profiling analyses revealed CDK8 as one of the five genes that are elevated for more than 2 folds in mH2A-deficient melanoma cells. Moreover, knocking down CDK8 suppresses the proliferative advantage induced by mH2A loss in melanoma cells in vitro and in vivo, suggesting that CDK8 is the major effector of melanoma progression caused by mH2A loss (Kapoor et al., 2010). Knockdown of MED12 resulted in a similar effect to knockdown of CDK8 in the proliferation of melanoma cells, suggesting that the effect of CDK8 is dependent on the Mediator complex (Kapoor et al., 2010). It will be important to identify the transcription programs that are dysregulated by increased CDK8 in melanoma cells. Overall, these results suggest that CDK8 functions as an oncoprotein in melanoma like colorectal cancers.
The examples discussed above revealed that CDK8 is aberrantly gained in human cancers. However, loss or reduction of CDK8 is also found in a few types of cancers. For example, the CDK8 gene is deleted in esophageal squamous cell carcinoma (Chattopadhyay et al., 2010). Likewise, the expression of CDK8 is significantly reduced in bladder cancers (Mitra et al., 2006). In addition, a CDK8 point mutation (D189 N) was found in diverse tumor samples (Greenman et al., 2007), but the functional consequence of this point mutation is still unknown. Our bioinformatic analyses suggest that this point mutation is likely to cause a loss of CDK8 kinase activity (see below). This implies that CDK8 may not always behave as an oncoprotein in all human cancers, and its activity needs to be tightly regulated.
CycC is the major regulatory partner of CDK8 (Tassan et al., 1995; Leclerc et al., 1996; Taatjes, 2010; Schneider et al., 2011). Genome-wide gene expression analyses have revealed that the CCNC gene (encoding CycC) is significantly up-regulated in gastric cancer, colorectal cancers, adenocarcinoma, leukemia, lymphoma patients, and a few hepatoma cell lines (Liu et al., 2003a, 2003b; Su et al., 2004; Galamb et al., 2007). However, the CCNC gene is also frequently deleted in a subset of acute lymphoblastic leukemia, osteosarcoma, and gastric cancers (Li et al., 1996; Ohata et al., 2006; Yang et al., 2007; van Delft et al., 2011). The discrepancies of CycC status in gastric cancers are likely due to different patient samples analyzed by these two studies (Galamb et al., 2007; Yang et al., 2007). Nevertheless, these observations suggest that dysregulation of CycC may play important roles in gastric cancers. Because CDK8 is over-expressed in gastric adenocarcinoma (Kim et al., 2011), systematic analyses of CDK8 and CycC in the same set of gastric cancer samples may help to clarify the status of these factors in gastric cancers. It is important to note that the functional consequences of overexpression or deletion of CCNC on the activities of CDK8 kinase or the CDK8 module have not been examined.
Dysregulation of CDK8-CycC causes cancers not only in humans, but also in fish. Walleye dermal sarcoma is a type of common skin tumor caused by walleye dermal sarcoma virus. Recent studies have revealed that the virus encodes a nuclear retro-viral Cyclin, which specifically competes with the endogenous CycC in binding with CDK8 and CDK3, thereby deregulating cell proliferation and differentiation (Rovnak and Quackenbush, 2002; Brewster et al., 2011). This mechanism is proposed to explain how the virus causes the dermal sarcoma in walleye. Interestingly, CDK3 is present only in vertebrates (Malumbres and Barbacid, 2009), and CDK3-CycC is reported to regulate the G0 to G1 transition of the cell cycle in mammalian cells (Ren and Rollins, 2004). However, several commonly used mouse strains harbor a nonsense mutation in the T-loop of CDK3, resulting in loss of CDK3 protein and its activity, suggesting that CDK3 is not an essential gene for normal development of mice (Ye et al., 2001). Together, these observations suggest that deregulation of CDK8-CycC by the retro-viral Cyclin may represent the major cause for walleye sarcoma. This case is similar to the afore-mentioned examples of melanoma and colon cancers: dysregulation of CDK8-CycC “drives” tumorigenesis.
Besides these types of cancers, whether CDK8 and CycC are also aberrant in other types of cancers is still not explored. Given the fundamental roles of the CDK8 module and the Mediator complex in regulating transcription, we predict that analyzing clinical data from other types of cancers may yield additional evidences linking the dysregulation of CDK8 with tumorigenesis. Nevertheless, the clinical data that we summarized here raise an important question: how does deregulated CDK8-CycC contribute to tumorigenesis? To address this question, it is critical to elucidate the upstream pathways that control CDK8-CycC activity and identify the downstream substrates of CDK8-CycC.
Currently, very little is known about the upstream regulators of CDK8 or CycC in different cellular and developmental contexts (Fig. 2). The only information available is that oxidative stress destabilizes CycC in yeast, and CycC levels are developmentally regulated in the social amoeba Dictyostelium (Krasley et al., 2006; Greene et al., 2010). The downstream consequences of CDK8-dependent phosphorylation in metazoans are also poorly understood (Taatjes, 2010). So far several CDK8 substrates have been identified in metazoans. These include RNA Pol II C-terminal domain; histone H3; general transcription factors, such as Cyclin H of CDK7-CycH, MED13 and CDK8 itself; and several transcription factors, such as p53, E2F1, Notch-intracellular domain, and SMADs (reviewed in Galbraith et al., 2010 and Taatjes, 2010). The effects of phosphorylation can be either positive or negative, or even unknown in some cases (Fig. 2). If the Mediator complex is required for most of the RNA Pol II-dependent transcription, then we expect that CDK8 has additional substrates that remain to be identified.
Fig. 2.
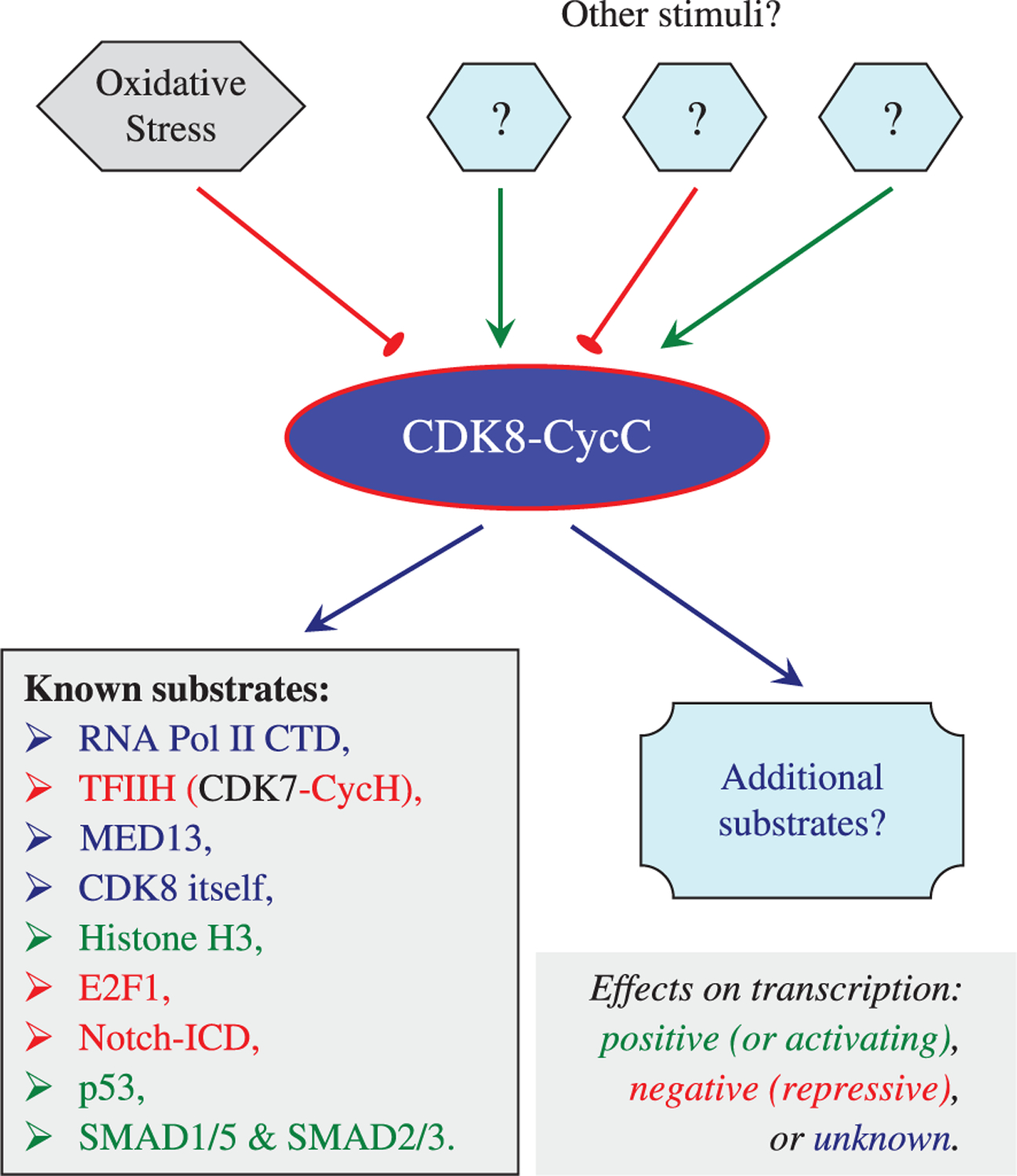
Context-specific functions of CDK8-CycC. Very little is known about the upstream regulators of CDK8-CycC. About 10 downstream effectors of CDK8 have been identified in multi-cellular organisms, and the transcriptional effect of their phosphorylation by CDK8 varies. It is likely that CDK8 has additional substrates that remain to be identified. Characterization of both upstream regulators and downstream effectors of CDK8-CycC is the key to understand how dysregulation of CDK8 contributes to tumorigenesis.
5. Bioinformatic and computational study of CDK8
Although addressing the function and regulation of CDK8 in vivo requires rigorous experimentation, here we sought to gain insights into the function of CDK8 by using a bioinformatics and computational approach. Genome sequences of many organisms are essentially completed. By taking advantage of available DNA and protein sequences and bioinformatics tools, we performed sequence and structural analyses of CDK8 to understand its function. The predictions from this bioinformatics and computational approach may help to guide future experimentation.
5.1. Functional domain analysis of CDK8
Human CDK8 has 464 amino acids (Fig. 3A) with a molecular weight of 53 kDa. The Ser/Thr kinase (STK) domain of human CDK8 includes the overlapped ATP and cyclin binding sites and the activation loop located at its N-terminal region (~19–335 aa). The C-terminal region (336–464 aa) of human CDK8 was searched against InterPro, an integrated database of predictive protein “signatures” used for the classification and automatic annotation of proteins and genomes, but no functional domains were identified. CDK activity is typically regulated through four major mechanisms: (i) binding by activating cyclins, (ii) binding by inhibitory cyclin-dependent kinase inhibitors (CKIs), (iii) inhibitory phosphorylation of the CDK, and (iv) activating phosphorylation of the CDK. The activating phosphorylation is catalyzed by a CDK-activating kinase (CAK) (Liu and Kipreos, 2000). Therefore, we discuss the relationship between sequence and function of CDK8 by focusing on the ATP binding site, the cyclin binding site, and the activating loop of the CDK8 kinase domain.
Fig. 3.

The sequence analyses of CDK8. A: the amino acid sequence of human CDK8. The amino acids of human CDK8 predicted to be involved in ATP binding are represented in red and highlighted in yellow; the amino acids of CDK8 predicted to have interactions with cyclin are in pink and highlighted in green; the predicted CDK8 activation loop is highlighted in blue. B: the sequence alignment of CDK8 in several representative species. The red rectangles highlight the SMSACRE motif and the activation loop.
Comparative genomics analysis of all MED subunits, including CDK8 and CycC, has revealed highly conserved motifs in evolution (Bourbon, 2008). Four CDK8 evolutionary conserved motifs, including two CDK8-specific inserts, one CDK8-specific activation segment and one CDK8-specific kinase domain extension, have been identified. We selected CDK8 from the representative species for sequence analysis in order to focus on investigating the ATP, cyclin binding sites and activation loop. The sequence alignment analysis of CDK8 by Vector NTI ClustalW algorithm reveals the conserved amino acids across the species including yeast and metazoans (Fig. 3B). CDK8 catalyzes transfer of the γ-phosphoryl group from ATP to Ser/Thr residues on protein substrates. Eighteen amino acids of human CDK8 are predicted to participate in ATP binding based on the structures of other CDKs with bound ATP or ATP analogs (Russo et al., 1996; Brown et al., 1999; Lolli et al., 2004). Those eighteen residues are highlighted in Fig. 3A and listed in Table 1. The CDK8 alignment study shows that among those predicted residues, G30, V35, A50, K52, I79, D103, A155, N156 and D173, are highly conserved across the representative species. The Lys41 of CDK7 interacts with the triphosphate of ATP in the active site (Lolli et al., 2004). Its corresponding residue Lys52 in human CDK8 is conserved in all the species that we analyzed. It has been experimentally shown that the D173A point mutation completely inactivates the kinase activity (Akoulitchev et al., 2000; Firestein et al., 2008), but this D173A mutation does not affect the transcription repression function of CDK8 for certain genes (Knuesel et al., 2009a). Our sequence alignment study shows that Asp173 is highly conserved from yeast to man. Lys52 and D173 of CDK8 are conserved in all of the eukaryotes (Bourbon, 2008).
Table 1.
The amino acid residues predicted to be located within the activation loop and involved in binding with ATP and cyclin in human CDK8.
| Residues | References | |
|---|---|---|
| ATP binding | 27VGRG, 35V, 50A, 52K, 79I, 97FDYA, 103D, 106H, 155AN, 158L, 173D | Russo et al., 1996; Brown et al., 1999; Lolli et al., 2004 |
| Cyclin binding | 13R, 57TGI, 61M, 64CREIA, 71REL, 84V, 86L, 88H, 91R, 93V, 140H, 143HANW, 178RL, 180FNSP, 185K, 190LD, 321I, 325TSE | Honda et al., 2005; Huang et al., 2007; Baumli et al., 2008, Schneider et al., 2011 |
| Activation loop | 172ADMGFARLFNSPLKPLADLDPVVVTFW | Russo et al., 1996; Martinez et al., 1997; Wu et al., 2003 |
Next, we analyzed the cyclin binding site of CDK8. The thirty-five predicted amino acids of CDK8 for cyclin binding are summarized in Table 1 and Fig. 3A. Our sequence alignment indicates that 15 residues (R13, G57, C64, R65, E66, R71, E72, L73, L86, H88, H143, N145, W146, R178 and D191) out of 35 are conserved as predicted based on literature. Ironically, CDKs are known by their predicted cyclin-association element sequence (PFTAIRE or PCTAIRE). In contrast, CDK8 has a SMSACRE motif (Fig. 3B) in the region corresponding to the PFTAIRE or PCTAIRE motif. Our sequence alignment shows that the SMSACRE motif of CDK8 is only conserved in vertebrates and SACRE in the SMSACRE motif is conserved from yeast to metazoan. The structure of human CDK8 and CycC complex recently solved by X-ray crystallography shows the contacts between Met61 and Arg65 in the SMSACRE motif of human CDK8 and CycC (Schneider et al., 2011). This structure information explains the experimental data that the double-point mutation of R65A/E66A in the SMSACRE motif greatly affects the capacity of CDK8 to bind to CycC (Barette et al., 2001).
For full activation, most protein kinases require the phosphorylation of Thr, Ser, or Tyr residues within the activation loop (or A-loop), also known as the regulatory T-loop. It contains the predicted residues for both ATP and cyclin binding. It has been suggested that the activation loop is flexible. Without cyclin, this loop blocks the ATP binding site because the position of several key amino acid residues is not optimal for ATP binding. In the presence of cyclin, two α-helices change their positions to permit ATP binding (Lolli et al., 2004). Thus, we analyzed the putative activation loop of CDK8, which is shown in both Fig. 3A and Table 1. Our alignment study of CDK8 sequences identified a conserved Thr196 in the activation loop from yeast to human (Fig. 4A). This Thr residue is conserved across a vast spectrum of eukaryotes (Bourbon, 2008). In addition, we found that Thr196 of CDK8 is conserved in both CDK8 and CDK7 of the representative species we analyzed (Fig. 4B). The experiment demonstrated that Thr170 of CDK7, corresponding to Thr196 of CDK8, is phosphorylated and Ser164 of CDK7 is not phosphorylated (Lolli et al., 2004). The phosphorylated Thr170 makes one hydrogen bond through its phosphate group to the main chain nitrogen of Gln22 from the glycine loop between β1/β2 and approaches the γ phosphate of ATP (Lolli et al., 2004), suggesting the importance of Thr phosphorylation in ATP binding and/or its kinase activity.
Fig. 4.
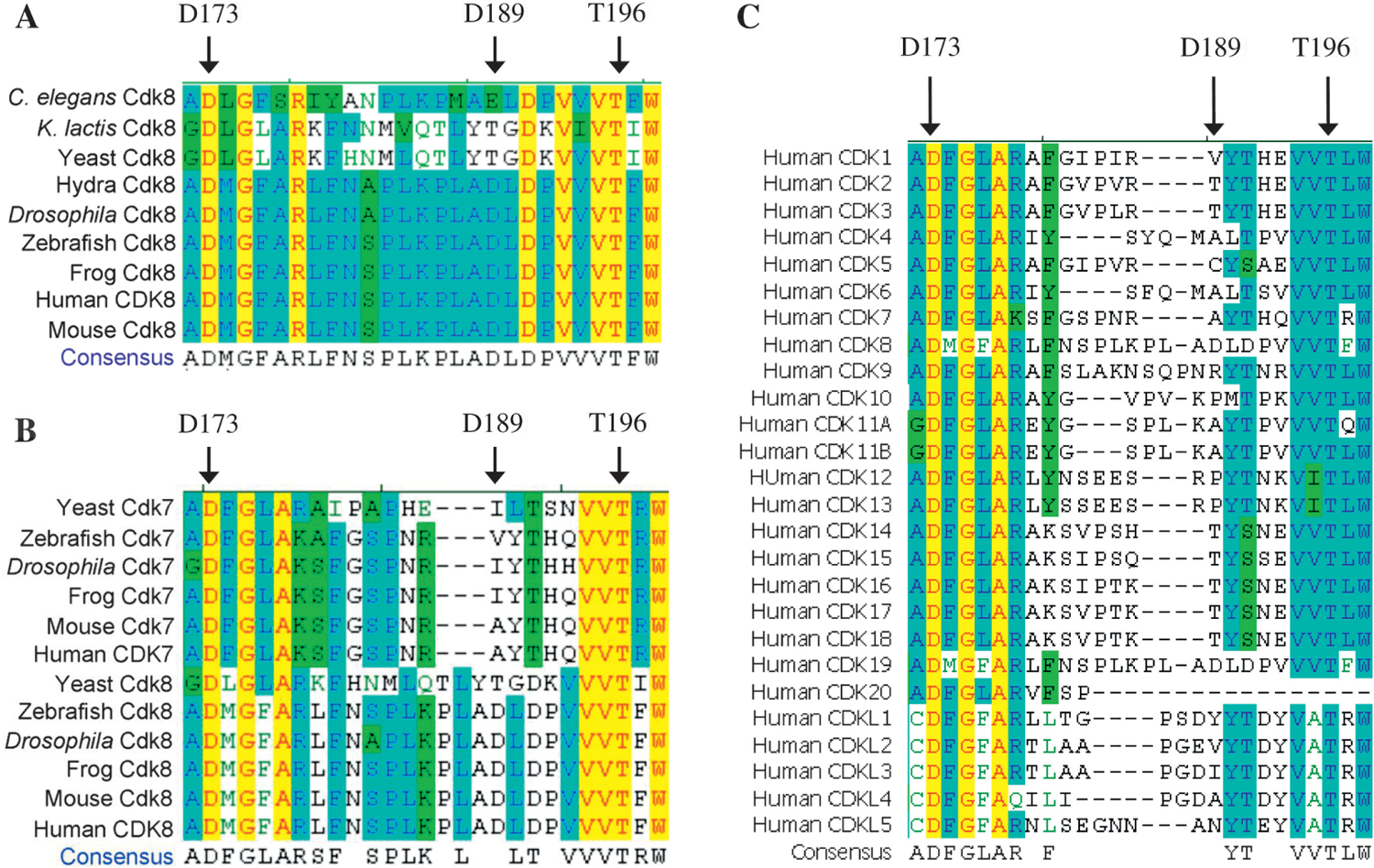
The sequence alignment of the predicted activation loop of CDKs. A: the sequence alignment of the predicted CDK8 activation loops from the representative species. B: the sequence alignment of CDK8 and CDK7 activation loops from the representative species. C: the sequence alignment of human CDK activation loops.
CDK7 associates with Cyclin H and MAT1 to form a complex called CDK-activating kinase (CAK), which has essential roles in both the cell-division cycle and transcription (Fisher, 2005). CDK7 or CAK has been reported to phosphorylate CDK1, CDK2, CDK4 and CDK6 (Fisher, 2005) including CDK7 itself at its activating residue Thr170 (Larochelle et al., 2001). In order to answer whether this Thr is conserved in other CDKs or not, we performed sequence alignments of all human CDKs and CDK likes. We found that this Thr is conserved in all human CDKs and CDK likes except CDK20, which does not have a corresponding amino acid at position of Thr170 of CDK7 in the alignment (Fig. 4C). Our sequence studies along with the literature demonstrate that this Thr residue in the activation loop is highly conserved. No study has shown that Thr196 of CDK8 can be phosphorylated in vivo or in vitro, thus it will be interesting to test this hypothesis. In addition, we have noticed that both Asp residues (D173 and D189) of the kinase-dead point mutation and human cancer mutation (Akoulitchev et al., 2000; Greenman et al., 2007; Firestein et al., 2008) are located in this putative activation loop (Fig. 4).
Finally, our sequence alignment analysis shows a short and conserved polyglutamine (polyQ) stretch at the C-terminal regions in the majority of species compared in Fig. 3B. Kluyveromyces lactis and yeast cells lack of an obvious polyQ motif. The polyQ motif has been found in transactivation domains of transcription factors or regulators. Drosophila has ~5 times longer polyQ motif than other species (Fig. 3B). The longer polyQ of CDK8 appears to be the unique feature of insect species (Bourbon, 2008). It is unclear whether transactivation function of Drosophila and yeast CDK8 differ from other species. Collectively, the bioinformatics analysis based on literature and our CDK8 sequence alignment has identified putative ATP and cyclin binding and phosphorylation sites in CDK8.
5.2. Phylogenetic study of CDK8
Phylogenetic studies of all CDKs have been reported (Doonan and Kitsios, 2009; Malumbres and Barbacid, 2009). To explore more details of CDK8 evolution, we built phylogenetic trees of CDK8 using MEGA 5. Our study shows four distinct CDK8 groups: vertebrate, insect, worm and yeast (Fig. 5A). Yeast CDK8 is distinct from CDK8 of multi-cellular organisms. Drosophila CDK8 is closely related to vertebrates (Fig. 5A).
Fig. 5.
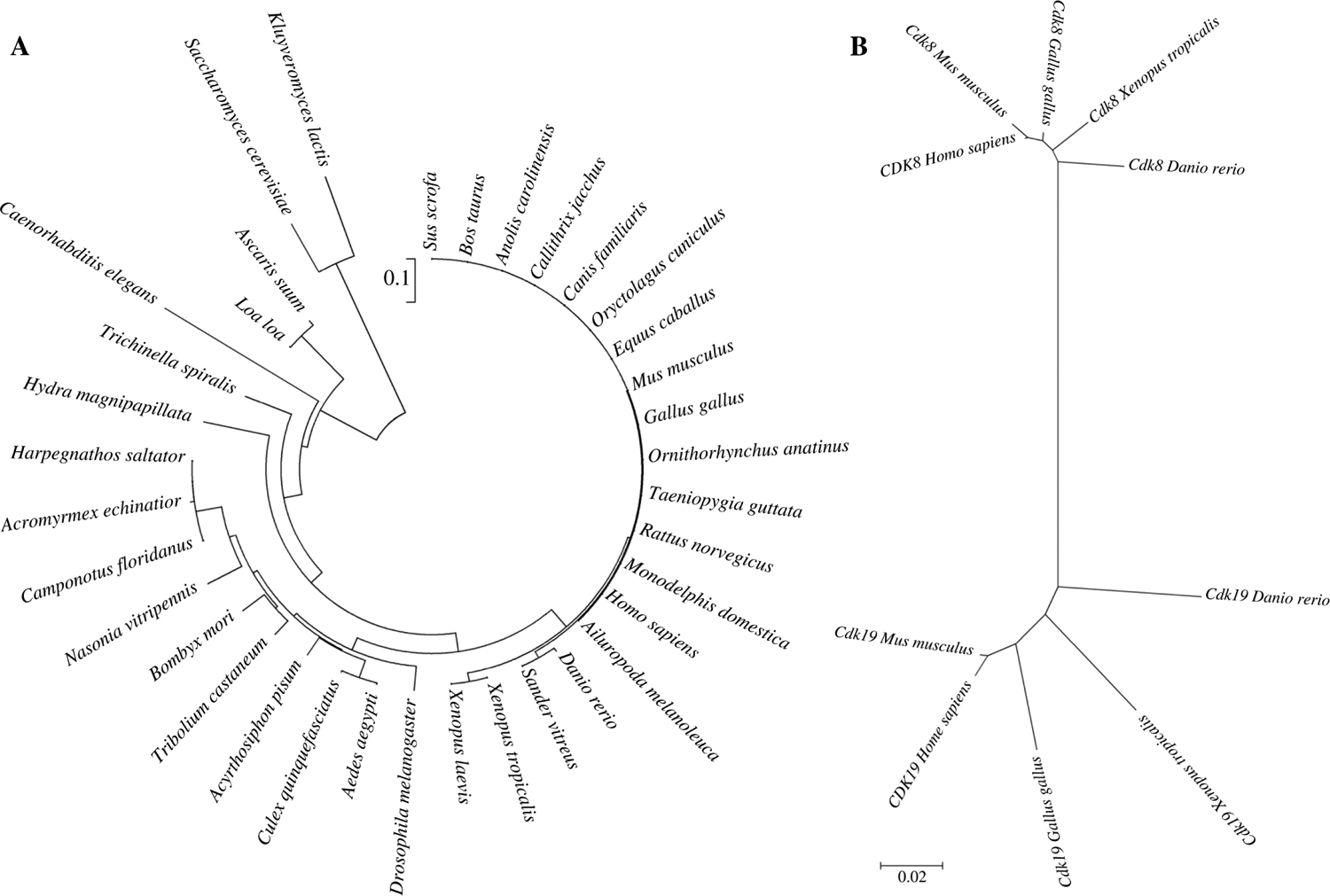
The phylogenetic analyses of CDK8. A: the phylogenetic tree of CDK8 from yeast to humans. B: the phylogenetic tree of CDK8 and CDK19 in the representative species.
The duplication of genes and their subsequent functional divergence is a fundamental process of adaptive evolution. CDK19 (also known as CDC2L6, CDK8-like or CDK8L) is the paralog of CDK8 with a highly conserved amino acid sequence in the kinase and cyclin binding domains at the N-terminal regions. Disruption of CDK19 has been linked to microcephaly and mild mental retardation in human (Mukhopadhyay et al., 2010). Both CDK8 and CDK19 contain an identical SMSACRE motif (Sato et al., 2004). Additionally, CDK8 and CDK19 associate with seemingly identical Mediator complexes (Galbraith et al., 2010; Taatjes, 2010), suggesting not only sequence homology, but also functional similarities. While CDK8 and CDK19 share sequence and functional similarity, they differ in their C-terminal regions (Sato et al., 2004), which raise the possibility that differential interactions mediated through their C-terminal regions might provide them with substrate specificity (Galbraith et al., 2010). CDK19 has been found only in vertebrates, and interestingly, some vertebrates do not have CDK19.
We performed phylogenetic study of CDK8 and CDK19 to explore when gene duplication of CDK8 and CDK19 occurred. Not surprisingly, we have observed two distinct groups: CDK8 and CDK19, suggesting the ancestor of CDK8 and CDK19 was duplicated during the early vertebrate evolution (Fig. 5B). Intriguingly, MED12 and MED13 have also undergone gene duplication to generate MED12L and MED13L (Sato et al., 2004; Bourbon, 2008). It is unknown whether different combinations of CDK8, CDK19, MED12, MED12L, MED13 and MED13L are assembled in vivo depending on different physiological conditions (Galbraith et al., 2010). Taken together, our phylogenetic analysis may be helpful to guide future experimentation to understand the specific function of CDK8 and CDK19.
5.3. Computational study of CDK8
During the course of our computational study of CDK8, its structure was not experimentally determined, but human CDK4 structure has been examined by X-ray crystallography (Takaki et al., 2009). When our manuscript was under review, the structure of human CDK8-CycC complex with sorafenib, an anti-cancer drug, was solved (Schneider et al., 2011). Human CDK4 and CDK8 sequences share 30.8% identity and 44.2% similarity. As a step toward a more complete understanding of CDK8 function, we sought to develop a hypothetical structure of human CDK8 by using human CDK4 (PDB ID: 3G33) as the template. Human CDK8 has 464 amino acids and only the region corresponding to human CDK4 structure solved by experiment was included in our molecular modeling (Modeller 9.9) and molecular dynamics (MD) simulations (Amber 11). The alignment of human CDK4 and CDK8 by ClustalW was imported into Modeller version 9.9 (Sali and Blundell, 1993) to generate the initial structure of the human CDK8, which was then optimized and analyzed by MD simulations.
Thanks to increased software efficiency and computational power, molecular dynamics simulation is a growing computational and theoretical field to study protein structure and function. The molecular dynamics simulations were based on the procedure described previously (Simmerling et al., 2002). This hypothetical structure of human CDK8 with the lowest energy during our 50-ns MD simulation was generated, which can be used as a starting point for understanding the detailed function of the protein and for future modification and optimization by more sophisticated theoretical methods and/or for comparison with actual structures solved by experiment (Schneider et al., 2011). The hypothetical CDK8 structure is shown in Fig. 6A. The residue D173 was predicted to be involved in ATP binding and the D173A mutant completely lost its kinase activity. In our hypothetical model, the side chain of D173 is hydrogen-bonded to the side chains of Lys134 and Asn137. The point mutation D189 N was found in human cancers (Greenman et al., 2007). The side chain of Asp189 participates in three hydrogen bonds, two with amino group of Phe20 and one with the main chain of Leu190. Thr196 is predicted to be phosphorylated based on the sequence similarity of this site with CDK7 Thr170 (Lolli et al., 2004). Thr196 is hydrogen bonded to the main chain of Pro232. The predicted activation loop of human CDK8 contains 27 amino acids (Fig. 4). The CDK8 structure experimentally solved contains a 16-amino acid deletion in the activation loop (Schneider et al., 2011). The activation loop of human CDK4, the template of our hypothetical CDK8 structure, has 23 amino acids with four-amino acid shorter than human CDK8 (Fig. 4C). Fig. 6B shows two representative conformations of CDK8 activation loop during the MD simulation.
Fig. 6.
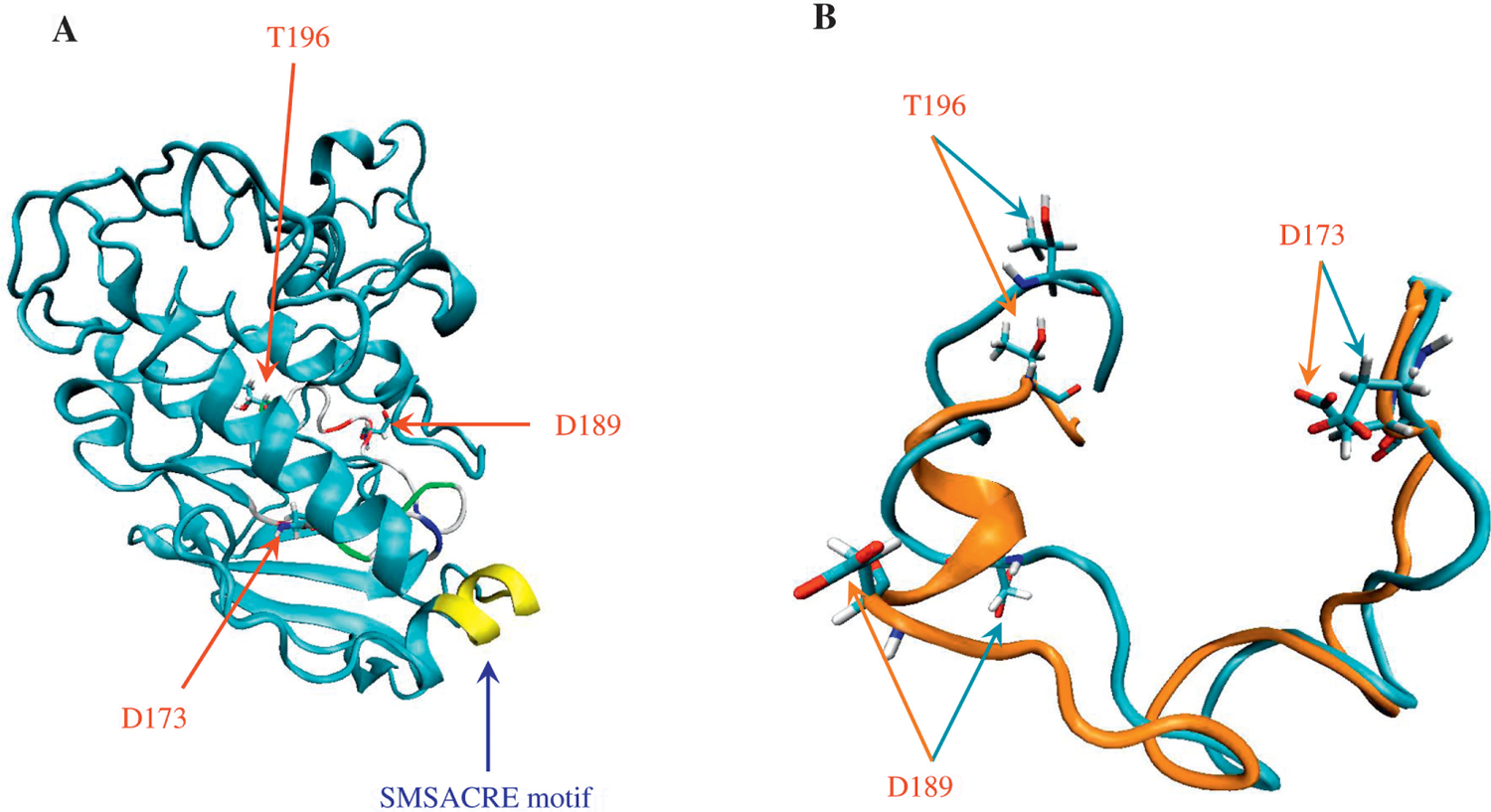
The hypothetical structure and the predicted activation loop of CDK8. A: the hypothetical structure of human CDK8 with the lowest energy from the molecular dynamics simulations. The molecular dynamics simulations revealed the heating phase to show a gradual increase in temperature with a few abrupt elevations; temperature at the equilibration phase was approximately 325 K, as expected. The energy profile (kinetic energy, potential energy, and total energy) was calculated. The kinetic energy was stable, as predicted by the stability of the temperature plot, which is directly proportional to the kinetic energy. The total and potential energy plots showed a gradual decrease, implying that the system had achieved an energy state more stable than the starting structure. The potential energy plot, which showed the structure with the lowest energy, was of greatest interest; its coordinates will be provided upon request. The backbone root-mean-square deviation (RMSD) plot, generated by using the lowest-energy structure as the reference, showed a decrease in RMSD during the simulation trajectory, indicating the equilibration steps are gradually approaching to the structure with the lowest energy. B: the models of human CDK8 activation loop. Cyan color represents the activation loop from the lowest potential energy structure (−7313 kcal/mol) during 50-ns simulation. Orange color represents the activation loop from the highest energy structure (−6677 kcal/mol) during 50-ns simulation. All the molecular dynamics simulation input, output and trajectory files are available upon request.
6. Important implications in therapeutic approaches
As summarized earlier, both CDK8 and CycC are either amplified or deleted in a variety of human cancers. It is important to clarify the functional consequence of these dosage variations: amplifying one of two subunits of the CDK8 module may generate partial complexes with MED12 and MED13, which may lead to a gain of CDK8 kinase activity but at the same time generate dominant negative effects on the function of the CDK8 module. By contrast, the CDK8 point mutation (D189 N) found in cancer patients (Greenman et al., 2007) likely causes reduced or loss of CDK8 kinase activity. If this point mutation does not affect CDK8 protein stability and its ability to interact with other proteins, then we expect that the structure of the CDK8 module will be intact and such a mutant CDK8 module may be still capable of inducing the conformational changes of the small Mediator complex. Therefore, to understand the molecular mechanisms of amplification, deletion, and mutation of CDK8 and/or CycC on tumorigenesis, it will be important to understand the consequence of such perturbations on the activities of the CDK8 kinase or the transcription function of the CDK8 module. Distinguishing these differences will help to define the role of CDK8 as an oncoprotein or a tumor suppressor in different cancers, which will guide the design of more effective therapeutic strategies.
Given the potent effect of CDK8 in cancer cell proliferation and the high frequency of the gain of CDK8 in melanoma and colorectal cancers, it is of great interest to target CDK8 for cancer therapy. There are two straightforward approaches for this: first, one can screen for small molecule inhibitors of CDK8 kinase activity. This approach has attracted considerable interest of pharmaceutical companies (Osherovich, 2008). While the effort to identify such CDK8-sepcific inhibitors is certainly worth trying, it may be challenging to achieve because it is difficult to ensure that such inhibitors do not affect other CDKs considering their close similarities. Another major obstacle for this approach is to develop sophisticated readouts for CDK8-specific activities in multi-cellular organisms (Osherovich, 2008). Furthermore, irreversibly inhibiting CDK8 kinase over an extended period may also generate adverse effects on cell viability. An alternative approach is to design methods to reduce the protein levels of CDK8 or CycC, such as RNA interference (RNAi) in cancer cells with amplified CDK8 or CycC. Compared to the small molecular inhibitor approach, this method is easier in specifically targeting CDK8. However, the major technical challenge for this alternative approach is to deliver such CDK8-specific shRNAs that target CDK8 or CycC in cancer cells.
If successful, these approaches may specifically benefit patients with increased levels of CDK8 or CycC. This idea is exciting and supported by recent reports showing that silibinin, an active ingredient of the health supplement milk thistle extract, can effectively block the proliferation of colon cancer cells by down-regulating CDK8 and CycC (Kaur et al., 2010; Velmurugan et al., 2010). It will be important to determine whether silibinin inhibits the activities of both CDK8 kinase and CDK8 module and if so, how.
7. Drosophila as a model system to study the function and regulation of CDK8
The study of the fundamental roles of the Mediator complex and the CDK8 module in fine-tuning transcription has progressed rapidly since its discovery in yeast two decades ago. As we summarized here, the accumulating evidence in recent years clearly links dysregulation of CDK8 and CycC to a variety of human cancers. Several challenges remain for future studies. In particular, it will be important to understand how dysregulation of CDK8 contributes to tumorigenesis. To address this question, it is essential to identify both upstream regulators and downstream effectors of CDK8. An important task will be to identify such CDK8 targets and then elucidate how these interactions are regulated in vivo. Furthermore, because CDK8 is a kinase and functions as an oncoprotein in a number of cancers, it will be important to develop CDK8 specific inhibitors and evaluate the specificity and efficacy of such inhibitors in treating cancers.
Because of many advantages, Drosophila is an ideal experimental system to elucidate the function and regulation of CDK8 in vivo. CDK8 is highly conserved during evolution and Drosophila CDK8 is closely related to human CDK8 (Fig. 5). The sequence alignment identifies the conservation of amino acid residues and domains (Fig. 3), and this information has important implications in its structure, function, and regulation. Furthermore, functions and regulations of many proteins are conserved, but are less complex in Drosophila than in mammals. For examples, paralogs for CDK8, MED12 and MED13, known as CDK8L (or CDK19), MED12L, and MED13L respectively, exist in mammals, which can also form very similar CDK8 modules (Taatjes, 2010). No paralog is found for all of the Mediator subunits in Drosophila (Bourbon, 2008), making it easier to identify the exact functions of these subunits compared to mammalian cells. Drosophila also provides a plethora of sophisticated genetic tools and reagents, and molecular markers for in vivo analyses. The rich literature of Drosophila developmental biology provides an ideal system to study the specific functions of CDK8 in different cellular and developmental contexts. Finally, this system allows one to perform un-biased large-scale genetic screens at relatively low cost. These unique features enable to address more challenging questions using other experimental systems.
Taken together, we expect that combined with other model systems and experimental approaches, Drosophila will continue to provide insights into the fundamental mechanisms of cancer biology. These complementary approaches will facilitate the efforts to develop more efficient treatment of human cancers and other diseases.
Acknowledgements
This work was supported by a sub-award of the NIH LEQSF-INBRE grant (P20RR016456) to WX, and a grant from the American Heart Association (11SDG7590123) to JYJ. We thank Christine Hermann and Fajun Yang for thorough reading and critical comments on the manuscript. We are grateful to two anonymous reviewers for their very careful and constructive comments, which help us to improve the paper substantially. The bioinformatics and computational analyses were performed at the Louisiana Optical Network Initiative (LONI) and Louisiana Immersive Technologies Enterprise (LITE).
References
- Akoulitchev S, Chuikov S, Reinberg D, 2000. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 407, 102–106. [DOI] [PubMed] [Google Scholar]
- Barette C, Jariel-Encontre I, Piechaczyk M, Piette J, 2001. Human cyclin C protein is stabilized by its associated kinase cdk8, independently of its catalytic activity. Oncogene 20, 551–562. [DOI] [PubMed] [Google Scholar]
- Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN, 2008. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 27, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernecky C, Grob P, Ebmeier CC, Nogales E, Taatjes DJ, 2011. Molecular architecture of the human Mediator-RNA polymerase II-TFIIF assembly. PLoS Biol. 9, e1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund S, Gustafsson CM, 2005. The yeast Mediator complex and its regulation. Trends Biochem. Sci 30, 240–244. [DOI] [PubMed] [Google Scholar]
- Bourbon HM, 2008. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 36, 3993–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon HM, Aguilera A, Ansari AZ, Asturias FJ, Berk AJ, Bjorklund S, Blackwell TK, Borggrefe T, Carey M, Carlson M, Conaway JW, Conaway RC, Emmons SW, Fondell JD, Freedman LP, Fukasawa T, Gustafsson CM, Han M, He X, Herman PK, Hinnebusch AG, Holmberg S, Holstege FC, Jaehning JA, Kim YJ, Kuras L, Leutz A, Lis JT, Meisterernest M, Näär AM, Nasmyth K, Parvin JD, Ptashne M, Reinberg D, Ronne H, Sadowski I, Sakurai H, Sipiczki M, Sternberg PW, Stillman DJ, Strich R, Struhl K, Svejstrup JQ, Tuck S, Winston F, Roeder RG, Kornberg RD, 2004. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol. Cell 14, 553–557. [DOI] [PubMed] [Google Scholar]
- Brewster CD, Birkenheuer CH, Vogt MB, Quackenbush SL, Rovnak J, 2011. The retroviral cyclin of walleye dermal sarcoma virus binds cyclin-dependent kinases 3 and 8. Virology 409, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NR, Noble ME, Endicott JA, Johnson LN, 1999. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Carlson M, 1997. Genetics of transcriptional regulation in yeast: connections to the RNA polymerase II CTD. Annu. Rev. Cell Dev. Biol 13, 1–23. [DOI] [PubMed] [Google Scholar]
- Carrera I, Janody F, Leeds N, Duveau F, Treisman JE, 2008. Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc. Natl. Acad. Sci. USA 105, 6644–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamassimi A, Napoli C, 2007. Mediator complexes and eukaryotic transcription regulation: an overview. Biochimie 89, 1439–1446. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay I, Singh A, Phukan R, Purkayastha J, Kataki A, Mahanta J, Saxena S, Kapur S, 2010. Genome-wide analysis of chromosomal alterations in patients with esophageal squamous cell carcinoma exposed to tobacco and betel quid from high-risk area in India. Mutat. Res 696, 130–138. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW, 2011. Function and regulation of the Mediator complex. Curr. Opin. Genet. Dev 21, 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway RC, Sato S, Tomomori-Sato C, Yao T, Conaway JW, 2005. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem. Sci 30, 250–255. [DOI] [PubMed] [Google Scholar]
- Ding N, Tomomori-Sato C, Sato S, Conaway RC, Conaway JW, Boyer TG, 2009. MED19 and MED26 are synergistic functional targets of the RE1 silencing transcription factor in epigenetic silencing of neuronal gene expression. J. Biol. Chem 284, 2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding N, Zhou H, Esteve PO, Chin HG, Kim S, Xu X, Joseph SM, Friez MJ, Schwartz CE, Pradhan S, Boyer TG, 2008. Mediator links epigenetic silencing of neuronal gene expression with x-linked mental retardation. Mol. Cell 31, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan JH, Kitsios G, 2009. Functional evolution of cyclin-dependent kinases. Mol. Biotechnol 42, 14–29. [DOI] [PubMed] [Google Scholar]
- Ebmeier CC, Taatjes DJ, 2010. Activator-Mediator binding regulates Mediator-cofactor interactions. Proc. Natl. Acad. Sci. USA 107, 11283–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmlund H, Baraznenok V, Lindahl M, Samuelsen CO, Koeck PJ, Holmberg S, Hebert H, Gustafsson CM, 2006. The cyclin-dependent kinase 8 module sterically blocks Mediator interactions with RNA polymerase II. Proc. Natl. Acad. Sci. USA 103, 15788–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B, 2004. The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153. [DOI] [PubMed] [Google Scholar]
- Firestein R, Hahn WC, 2009. Revving the Throttle on an oncogene: CDK8 takes the driver seat. Cancer Res. 69, 7899–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Shima K, Nosho K, Irahara N, Baba Y, Bojarski E, Giovannucci EL, Hahn WC, Fuchs CS, Ogino S, 2010. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 126, 2863–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, Chheda MG, Tamayo P, Finn S, Shrestha Y, Boehm JS, Jain S, Bojarski E, Mermel C, Barretina J, Chan JA, Baselga J, Tabernero J, Root DE, Fuchs CS, Loda M, Shivdasani RA, Meyerson M, Hahn WC, 2008. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP, 2005. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci 118, 5171–5180. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, White JB, Jones KA, 2004. Mastermind recruits CycC: CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16, 509–520. [DOI] [PubMed] [Google Scholar]
- Galamb O, Sipos F, Molnar B, Szoke D, Spisak S, Tulassay Z, 2007. Evaluation of malignant and benign gastric biopsy specimens by mRNA expression profile and multivariate statistical methods. Cytometry B Clin. Cytom 72, 299–309. [DOI] [PubMed] [Google Scholar]
- Galbraith MD, Donner AJ, Espinosa JM, 2010. CDK8: a positive regulator of transcription. Transcription 1, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert V, Osman D, Bras S, Auge B, Boube M, Bourbon HM, Horn T, Boutros M, Haenlin M, Waltzer L, 2010. A genome-wide RNA interference screen identifies a differential role of the mediator CDK8 module subunits for GATA/RUNX-activated transcription in Drosophila. Mol. Cell. Biol 30, 2837–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene DM, Hsu DW, Pears CJ, 2010. Control of cyclin C levels during development of Dictyostelium. PLoS One 5, e10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR, 2007. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, 2004. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol 11, 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg M, Polozkov GV, Hu GZ, Beve J, Gustafsson CM, Ronne H, Bjorklund S, 2004. Site-specific Srb10-dependent phosphorylation of the yeast Mediator subunit Med2 regulates gene expression from the 2-microm plasmid. Proc. Natl. Acad. Sci. USA 101, 3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Mori S, Nevins JR, 2008. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 13, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2000. The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hengartner CJ, Myer VE, Liao SM, Wilson CJ, Koh SS, Young RA, 1998. Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol. Cell 2, 43–53. [DOI] [PubMed] [Google Scholar]
- Hoffmans R, Stadeli R, Basler K, 2005. Pygopus and legless provide essential transcriptional coactivator functions to armadillo/beta-catenin. Curr. Biol 15, 1207–1211. [DOI] [PubMed] [Google Scholar]
- Honda R, Lowe ED, Dubinina E, Skamnaki V, Cook A, Brown NR, Johnson LN, 2005. The structure of cyclin E1/CDK2: implications for CDK2 activation and CDK2-independent roles. EMBO J. 24, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Ferrin-O’Connell I, Zhang W, Leonard GA, O’Shea EK, Quiocho FA, 2007. Structure of the Pho85-Pho80 CDK-cyclin complex of the phosphate-responsive signal transduction pathway. Mol. Cell 28, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TA, Brady HJ, 2005. E2F1 up-regulates the expression of the tumour suppressor axin2 both by activation of transcription and by mRNA stabilisation. Biochem. Biophys. Res. Commun 329, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Janody F, Martirosyan Z, Benlali A, Treisman JE, 2003. Two subunits of the Drosophila mediator complex act together to control cell affinity. Development 130, 3691–3701. [DOI] [PubMed] [Google Scholar]
- Ji JY, Dyson NJ, 2010. Interplay between Cyclin-dependent Kinases and E2F-dependent Transcription. In: Enders GH (Ed.), Cell Cycle Deregulation in Cancer. Springer Science, pp. 23–41. [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E, 2010. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Velmurugan B, Tyagi A, Agarwal C, Singh RP, Agarwal R, 2010. Silibinin suppresses growth of human colorectal carcinoma SW480 cells in culture and xenograft through down-regulation of beta-catenin-dependent signaling. Neoplasia 12, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Han SI, Lim SC, 2011. Roles of cyclin-dependent kinase 8 and beta-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int. J. Oncol 38, 1375–1383. [DOI] [PubMed] [Google Scholar]
- Kim S, Xu X, Hecht A, Boyer TG, 2006. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem 281, 14066–14075. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Lis JT, 2005. Interactions between subunits of Drosophila Mediator and activator proteins. Trends Biochem. Sci 30, 245–249. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B, 1997. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 386, 761–763. [DOI] [PubMed] [Google Scholar]
- Knuesel MT, Taatjes DJ, 2011. Mediator and post-recruitment regulation of RNA polymerase II. Transcription 2, 28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ, 2009a. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 23, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ, 2009b. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol 29, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, 2005. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci 30, 235–239. [DOI] [PubMed] [Google Scholar]
- Krasley E, Cooper KF, Mallory MJ, Dunbrack R, Strich R, 2006. Regulation of the oxidative stress response through Slt2 p-dependent destruction of cyclin C in Saccharomyces cerevisiae. Genetics 172, 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Chen J, Knights R, Pandur J, Morcillo P, Erdjument-Bromage H, Tempst P, Suter B, Fisher RP, 2001. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 20, 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc V, Tassan JP, O’Farrell PH, Nigg EA, Leopold P, 1996. Drosophila Cdk8, a kinase partner of cyclin C that interacts with the large subunit of RNA polymerase II. Mol. Biol. Cell 7, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lahti JM, Kidd VJ, 1996. Alternatively spliced cyclin C mRNA is widely expressed, cell cycle regulated, and encodes a truncated cyclin box. Oncogene 13, 705–712. [PubMed] [Google Scholar]
- Liu J, Kipreos ET, 2000. Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol. Biol. Evol 17, 1061–1074. [DOI] [PubMed] [Google Scholar]
- Liu LX, Liu ZH, Jiang HC, Zhang WH, Qi SY, Hu J, Wang XQ, Wu M, 2003a. Gene expression profiles of hepatoma cell line HLE. World J. Gastroenterol 9, 683–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LX, Jiang HC, Liu ZH, Zhu AL, Zhou J, Zhang WH, Wang XQ, Wu M, 2003b. Gene expression profiles of hepatoma cell line BEL-7402. Hepatogastroenterology 50, 1496–1501. [PubMed] [Google Scholar]
- Liu Y, Kung C, Fishburn J, Ansari AZ, Shokat KM, Hahn S, 2004. Two cyclin-dependent kinases promote RNA polymerase II transcription and formation of the scaffold complex. Mol. Cell. Biol 24, 1721–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolli G, Lowe ED, Brown NR, Johnson LN, 2004. The crystal structure of human CDK7 and its protein recognition properties. Structure 12, 2067–2079. [DOI] [PubMed] [Google Scholar]
- Loncle N, Boube M, Joulia L, Boschiero C, Werner M, Cribbs DL, Bourbon HM, 2007. Distinct roles for Mediator Cdk8 module subunits in Drosophila development. EMBO J. 26, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Roeder RG, 2005. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem. Sci 30, 256–263. [DOI] [PubMed] [Google Scholar]
- Malik S, Guermah M, Yuan CX, Wu W, Yamamura S, Roeder RG, 2004. Structural and functional organization of TRAP220, the TRAP/mediator subunit that is targeted by nuclear receptors. Mol. Cell. Biol 24, 8244–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M, 2009. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Martin ES, Tonon G, Sinha R, Xiao Y, Feng B, Kimmelman AC, Protopopov A, Ivanova E, Brennan C, Montgomery K, Kucherlapati R, Bailey G, Redston M, Chin L, DePinho RA, 2007. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 67, 10736–10743. [DOI] [PubMed] [Google Scholar]
- Martinez AM, Afshar M, Martin F, Cavadore JC, Labbe JC, Doree M, 1997. Dual phosphorylation of the T-loop in cdk7: its role in controlling cyclin H binding and CAK activity. EMBO J. 16, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAloose D, Newton AL, 2009. Wildlife cancer: a conservation perspective. Nat. Rev. Cancer 9, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Donner AJ, Knuesel MT, York AG, Espinosa JM, Taatjes DJ, 2008. Cooperative activity of cdk8 and GCN5L within Mediator directs tandem phosphoacetylation of histone H3. EMBO J. 27, 1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra AP, Almal AA, George B, Fry DW, Lenehan PF, Pagliarulo V, Cote RJ, Datar RH, Worzel WP, 2006. The use of genetic programming in the analysis of quantitative gene expression profiles for identification of nodal status in bladder cancer. BMC Cancer 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittler G, Kremmer E, Timmers HT, Meisterernst M, 2001. Novel critical role of a human Mediator complex for basal RNA polymerase II transcription. EMBO Rep. 2, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris EJ, Ji JY, Yang F, Di Stefano L, Herr A, Moon NS, Kwon EJ, Haigis KM, Näär AM, Dyson NJ, 2008. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature 455, 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Kramer JM, Merkx G, Lugtenberg D, Smeets DF, Oortveld MA, Blokland EA, Agrawal J, Schenck A, van Bokhoven H, Huys E, Schoenmakers EF, van Kessel AG, van Nouhuys CE, Cremers FP, 2010. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum. Genet 128, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LC, Kornberg RD, 2000. Mediator of transcriptional regulation. Annu. Rev. Biochem 69, 729–749. [DOI] [PubMed] [Google Scholar]
- Näär AM, Lemon BD, Tjian R, 2001. Transcriptional coactivator complexes. Annu. Rev. Biochem 70, 475–501. [DOI] [PubMed] [Google Scholar]
- Näär AM, Taatjes DJ, Zhai W, Nogales E, Tjian R, 2002. Human CRSP interacts with RNA polymerase II CTD and adopts a specific CTD-bound conformation. Genes Dev. 16, 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj SH, Reverter A, 2011. A Boolean-based systems biology approach to predict novel genes associated with cancer: application to colorectal cancer. BMC Syst. Biol 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata N, Ito S, Yoshida A, Kunisada T, Numoto K, Jitsumori Y, Kanzaki H, Ozaki T, Shimizu K, Ouchida M, 2006. Highly frequent allelic loss of chromosome 6q16–23 in osteosarcoma: involvement of cyclin C in osteosarcoma. Int. J. Mol. Med 18, 1153–1158. [PubMed] [Google Scholar]
- Osherovich L, 2008. CDK8 is enough in colorectal cancer. SciBX 1, 5–7. [Google Scholar]
- Ren S, Rollins BJ, 2004. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 117, 239–251. [DOI] [PubMed] [Google Scholar]
- Rovnak J, Quackenbush SL, 2002. Walleye dermal sarcoma virus cyclin interacts with components of the mediator complex and the RNA polymerase II holoenzyme. J. Virol 76, 8031–8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Pavletich NP, 1996. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol 3, 696–700. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL, 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Samuelsen CO, Baraznenok V, Khorosjutina O, Spahr H, Kieselbach T, Holmberg S, Gustafsson CM, 2003. TRAP230/ARC240 and TRAP240/ARC250 Mediator subunits are functionally conserved through evolution. Proc. Natl. Acad. Sci. USA 100, 6422–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Tomomori-Sato C, Parmely TJ, Florens L, Zybailov B, Swanson SK, Banks CA, Jin J, Cai Y, Washburn MP, Conaway JW, Conaway RC, 2004. A set of consensus mammalian mediator subunits identified by multidimensional protein identification technology. Mol. Cell 14, 685–691. [DOI] [PubMed] [Google Scholar]
- Schneider EV, Bottcher J, Blaesse M, Neumann L, Huber R, Maskos K, 2011. The structure of CDK8/CycC implicates specificity in the CDK/Cyclin family and reveals interaction with a deep pocket binder. J. Mol. Biol 412, 251–266. [DOI] [PubMed] [Google Scholar]
- Seo JO, Han SI, Lim SC, 2010. Role of CDK8 and beta-catenin in colorectal adenocarcinoma. Oncol. Rep 24, 285–291. [PubMed] [Google Scholar]
- Shahi P, Gulshan K, Näär AM, Moye-Rowley WS, 2010. Differential roles of transcriptional mediator subunits in regulation of multidrug resistance gene expression in Saccharomyces cerevisiae. Mol. Biol. Cell 21, 2469–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA, 2010. Epigenetics in cancer. Carcino-genesis 31, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E, 2009. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 106, 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerling C, Strockbine B, Roitberg AE, 2002. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc 124, 11258–11259. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB, 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes DJ, 2010. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem. Sci 35, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes DJ, Näär AM, Andel F 3rd, Nogales E, Tjian R, 2002. Structure, function, and activator-induced conformations of the CRSP coactivator. Science 295, 1058–1062. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, Florens L, Seidel CW, Lin C, Smith ER, Shilatifard A, Conaway RC, Conaway JW, 2011. Human Mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki T, Echalier A, Brown NR, Hunt T, Endicott JA, Noble MEM, 2009. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc. Natl. Acad. Sci. USA 106, 4171–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey WP, 2001. Transcriptional activation: risky business. Genes Dev. 15, 1045–1050. [DOI] [PubMed] [Google Scholar]
- Tassan JP, Jaquenoud M, Leopold P, Schultz SJ, Nigg EA, 1995. Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc. Natl. Acad. Sci. USA 92, 8871–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley FM, Cliffe A, Bienz M, 2004. Pygopus and Legless target Armadillo/beta-catenin to the nucleus to enable its transcriptional coactivator function. Nat. Cell Biol 6, 626–633. [DOI] [PubMed] [Google Scholar]
- Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, Liu H, Krier C, Stengel RF, Barany F, Gerald WL, Paty PB, Domany E, Notterman DA, 2006. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 66, 2129–2137. [DOI] [PubMed] [Google Scholar]
- van de Peppel J, Kettelarij N, van Bakel H, Kockelkorn TT, van Leenen D, Holstege FC, 2005. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol. Cell 19, 511–522. [DOI] [PubMed] [Google Scholar]
- van Delft FW, Horsley S, Colman S, Anderson K, Bateman C, Kempski H, Zuna J, Eckert C, Saha V, Kearney L, Ford A, Greaves M, 2011. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117, 6247–6254. [DOI] [PubMed] [Google Scholar]
- Velmurugan B, Gangar SC, Kaur M, Tyagi A, Deep G, Agarwal R, 2010. Silibinin exerts sustained growth suppressive effect against human colon carcinoma SW480 xenograft by targeting multiple signaling molecules. Pharm. Res 27, 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerling T, Kuuluvainen E, Makela TP, 2007. Cdk8 is essential for preimplantation mouse development. Mol. Cell. Biol 27, 6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, McNae I, Kontopidis G, McClue SJ, McInnes C, Stewart KJ, Wang S, Zheleva DI, Marriage H, Lane DP, Taylor P, Fischer PM, Walkinshaw MD, 2003. Discovery of a novel family of CDK inhibitors with the program LIDAEUS: structural basis for ligand-induced disordering of the activation loop. Structure 11, 399–410. [DOI] [PubMed] [Google Scholar]
- Yang S, Jeung HC, Jeong HJ, Choi YH, Kim JE, Jung JJ, Rha SY, Yang WI, Chung HC, 2007. Identification of genes with correlated patterns of variations in DNA copy number and gene expression level in gastric cancer. Genomics 89, 451–459. [DOI] [PubMed] [Google Scholar]
- Ye X, Zhu C, Harper JW, 2001. A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc. Natl. Acad. Sci. USA 98, 1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda A, Kouike H, Okano H, Sawa H, 2005. Components of the transcriptional Mediator complex are required for asymmetric cell division in C. elegans. Development 132, 1885–1893. [DOI] [PubMed] [Google Scholar]
- Yudkovsky N, Ranish JA, Hahn S, 2000. A transcription reinitiation intermediate that is stabilized by activator. Nature 408, 225–229. [DOI] [PubMed] [Google Scholar]