

Abstract
CD8+ T cells specific for cancer cells are detected within tumours. However, despite their presence, tumours progress. The clinical success of immune checkpoint blockade and adoptive T cell therapy demonstrates the potential of CD8+ T cells to mediate antitumour responses; however, most patients with cancer fail to achieve long-term responses to immunotherapy. Here we review CD8+ T cell differentiation to dysfunctional states during tumorigenesis. We highlight similarities and differences between T cell dysfunction and other hyporesponsive T cell states and discuss the spatio-temporal factors contributing to T cell state heterogeneity in tumours. An important challenge is predicting which patients will respond to immunotherapeutic interventions and understanding which T cell subsets mediate the clinical response. We explore our current understanding of what determines T cell responsiveness and resistance to immunotherapy and point out the outstanding research questions.
CD8+ T cells have the ability to selectively detect and eradicate cancer cells. Tumours express antigens, which include tumour-specific (mutant and viral) neoantigens1–6 and self-antigens (also known as tumour-associated or shared antigens)7,8, and CD8+ T cells reactive against such antigens are found in patients with cancer (reviewed in REFS9,10). However, despite their presence, even tumours expressing highly immunogenic neoantigens often progress. The coexistence of growing tumours and T cells, described by Ingegerd and Karl Erik Hellstrom in 1968 (REF.11) and now referred to as the ‘Hellstrom paradox’12, suggests that tumour-reactive CD8+ T cells become dysfunctional over the course of tumorigenesis.
Much of our knowledge of CD8+ T cell dysfunction has come from studying T cells isolated from established, progressing tumours: tumour-infiltrating lymphocytes (TILs) reactive against tumour antigens express multiple inhibitory receptors (such as PD1, LAG3, CTLA4 and TIM3) and fail to produce effector cytokines (interferon-γ (IFNγ) and TNF) or cytotoxic molecules (such as granzymes and perforin). We refer to hyporesponsive T cells in cancer as ‘dysfunctional’, but several other terms are also widely used, including ‘anergy’, ‘tolerance’ and ‘exhaustion’13–15. These last terms were initially used to describe hyporesponsive T cell states in non-tumour settings, with ‘anergy’ referring to priming or activation of naive T cells in the absence of co-stimulation in vitro and in vivo (reviewed in REF.16), ‘tolerance’ referring to peripheral T cells with specificity for self-antigens (reviewed in REFS13,17), whereby loss of tolerance leads to autoimmunity, and ‘exhaustion’ referring to pathogen-specific T cells during chronic infections, in which attenuated responses lead to a host–pathogen stalemate without causing detrimental immunopathology (reviewed in REF.18). Although there are phenotypic, functional and molecular similarities between dysfunctional TILs and anergic, tolerant and/or exhausted T cells, there are important distinctions between these states, which are discussed in this Review.
In this Review, we summarize our current understanding of tumour-reactive CD8+ T cell differentiation during tumorigenesis, the factors determining TIL dysfunction and heterogeneity, molecular programmes and key transcription factors that are critical for TIL function, including T cell factor 1 (TCF1; encoded by TCF7) and thymocyte selection-associated high mobility group box protein TOX, and potential mechanisms of responsiveness or resistance to cancer immunotherapy, such as immune checkpoint blockade (ICB) and cancer vaccines. For topics related to T cell dysfunction but not extensively discussed here, including inhibitory receptor–ligand interactions, metabolic programmes and the immunosuppressive tumour microenvironment (TME), we refer readers to other, excellent reviews19–21.
Hyporesponsive CD8+ T cell states
CD8+ T cell differentiation is a tightly regulated process, and changes in the nature, context and duration of antigen encounter determine the trajectory of CD8+ T cell differentiation, resulting in T cell states ranging from functional effector T cell states (for example, in acute infection, autoimmunity or graft-versus-host disease) to hyporesponsive T cell states (for example, in tumours, chronic infection or self-tolerance). When naive antigen-specific CD8+ T cells encounter antigens in an acute inflammatory context (for example, viral or bacterial infection), T cells undergo clonal expansion and differentiate into cytolytic effector T cells. After pathogen or antigen clearance, most effector T cells die (contraction), but a small fraction survive and form long-lasting memory T cells. As T cells differentiate from naive to effector and memory states, distinct transcriptional and epigenetic programmes define state-specific phenotypic and functional properties22,23.
Tolerance.
‘Self-tolerance’ refers to the hyporesponsive state of self-antigen-reactive T cells, and is necessary to prevent autoimmunity. Tolerance arises through both central tolerance and peripheral tolerance mechanisms. Naive, self-reactive T cells that encounter a self-antigen in normal tissue in a non-stimulatory context on non-activated and/or non-professional antigen-presenting cells (APCs)24,25 fail to receive full priming and activation signals, which can result in apoptosis26–28 or the establishment of a cell-intrinsic self-tolerance programme29,30 (reviewed in REFS13,17). Peripheral self-tolerant CD8+ T cells appear to be antigen experienced and fail to produce effector cytokines and/or proliferate in response to T cell receptor (TCR) stimulation.
Over the last decades, self-tumour antigens (including gene-amplified oncogenes) have been the major targets of immunotherapy, including adoptive T cell therapies (engineered TCR-expressing T cells and chimeric antigen receptor T cells). Although impressive results have been observed in haematological malignancies31 and melanoma32, successes in targeting self-tumour antigens in other solid tumour types have been limited. In contrast to self-reactive T cells, neoantigen-specific T cells are not hampered by central or peripheral self-tolerance mechanisms, and thus, theoretically, should exhibit superior antitumour responses. However, as discussed later, during the early stages of tumour development, neoantigens may be presented in a non-inflammatory context, inducing a hyporesponsive T cell state similar to peripheral self-tolerance induction. T cells specific for self-tumour antigens within progressing tumours display the same hallmarks of dysfunction as neoantigen-specific T cells33. Whether self-tumour antigen-specific or neoantigen-specific T cells show functional differences in response to immunotherapeutic interventions and/or whether a clinical response is driven mainly by self-tumour antigen-specific or neoantigen-specific T cells remains largely unknown34.
Ignorance.
In the case of self-antigens that are expressed at a low level or in anatomical sites sequestered from immune recognition (immune privileged sites), T cells can be ‘ignorant’ of their cognate antigen and remain in a phenotypically antigen-inexperienced naive state35–39. This ignorance can be overcome if these antigens are overexpressed by cancer cells during tumorigenesis. For example, healthy individuals have been found to harbour phenotypically and functionally naive (ignorant) CD8+ T cells specific for Melan-A (also known as MART1)35. However, in patients with melanoma, Melan-A antigen is presented in tumour-draining lymph nodes (TDLNs), activating Melan-A-specific T cells, which migrate into the tumour40. Immunological ignorance is not restricted to self-antigens but can also impact tumour-specific neoantigens. For example, during early tumorigenesis, transformed cells are embedded within normal tissues or organs and can be undetected (FIG. 1). Thus, T cells specific for tumour neoantigens may remain ignorant until tumours progress to the point that tumour antigens are presented in TDLNs and activate antigen-specific T cells41.
Fig. 1 |. Carcinogenesis and tumour-specific CD8+ T cell differentiation: a two-phase differentiation programme.

An initiating oncogenic mutation occurs within a single cell in an otherwise normal tissue. Neoantigen-specific (naive) CD8+ T cells do not efficiently infiltrate and survey normal peripheral tissues; thus, during early tumorigenesis, tumour-specific CD8+ T cells may remain ignorant of antigen-expressing cancer cells. When tumours progress and sufficient tumour antigen is presented in draining lymph nodes, tumour-specific T cells can be activated in the draining lymph node or within tumour tissue in a largely non-inflammatory, non-stimulatory context, inducing an anergic, early dysfunctional T cell state (phase 1). The tumour continues to progress, inducing an immunosuppressive microenvironment. Persistent tumour antigen and microenvironmental signals drive tumour-specific T cells into a late dysfunctional state (phase 2).
Anergy.
‘Anergy’ describes the hyporesponsive state of T cells activated in the absence of inflammation and/or co-stimulatory signals16,42,43. Proper T cell activation generally requires the simultaneous stimulation of TCR (signal 1) and co-stimulatory receptors (for example, CD28; signal 2) leading to the activation of several critical signalling pathways and the induction of activation-associated genes16. TCR engagement in the absence of co-stimulation does not effectively activate these pathways, resulting in anergy. Naive tumour-specific T cells can be suboptimally primed when they encounter an antigen on cancer cells or APCs that lack expression of co-stimulatory ligands (for example, CD80 or CD86), and thus ‘anergy’ has also been used to describe T cell differentiation and dysfunction in the context of tumours44. T cell anergy and T cell exhaustion share some key molecular features45,46; as anergy and exhaustion are the hyporesponsive T cell states that are most associated with tumour-specific T cell differentiation and dysfunction, we focus on these transcriptional circuits in this Review.
Exhaustion.
The term ‘exhaustion’ was initially used to describe hyporesponsive T cell states during chronic infections (reviewed in REF.18). During chronic infections, naive pathogen-specific T cells expand and initially differentiate into functional effector T cells47. In contrast to acute infections, in which pathogens are quickly eliminated, allowing memory T cell formation, during chronic infections sterilizing immunity is not achieved, pathogens persist and antigen-specific T cells are chronically stimulated by the cognate antigen. Chronic antigen stimulation results in gradual loss of effector functions in a well-described hierarchical manner concomitant with the upregulation of numerous inhibitory receptors, a state described as ‘exhaustion’47,48. T cell exhaustion should be viewed not as a non-responsive T cell state but rather as an adaptive state of hyporesponsiveness: exhausted T cells keep pathogens in check without excessive effector function, which would cause immunopathology49–51. Therefore, T cell exhaustion is an ‘effective’ hyporesponsive state that has evolved to allow a host–pathogen ‘stalemate’49,50,52. The fact that exhausted T cells serve an important immune function is demonstrated by the rapid expansion of pathogens after CD8+ T cell depletion or the emergence of immune escape mutants52–54. T cells in late-stage, progressing tumours become hyporesponsive owing to continuous encounters with tumour antigens and share many key features with T cells in chronic infection, and thus ‘exhaustion’ has been used to describe T cell dysfunction in established, progressing tumours.
Tumour-induced CD8+ T cell dysfunction
Tumour-induced CD8+ T cell dysfunction was previously thought to be established late during cancer development, induced by the immunosuppressive TME of established solid tumours21 (comprising, for example, myeloid-derived suppressor cells (MDSCs)55, tumour-associated macrophages56,57, FOXP3+CD4+ regulatory T cells58, interleukin-10 (IL-10), transforming growth factor-β and indoleamine 2,3-dioxygenase, inhibitory checkpoint signalling pathways19, and physiological and metabolic changes (such as hypoxia and low nutrient levels)) (reviewed in REFS59,60). However, cancers in humans usually derive from a single transformed cell and develop slowly, sometimes over several years, through clonal evolution61. Between the initial transformation event in a normal cell and the eventual progression to a clinically detectable established tumour, cancers are present as undetectable lesions, which must evade T cell recognition and elimination. In patients, when and how during tumorigenesis tumour-specific T cells differentiate to a hyporesponsive state and how this state evolves over time remain incompletely understood. Evidence from clinically relevant genetic mouse models and human cancers indicates that oncogenic and tumour-suppressor pathways and the tissue of origin can shape innate and adaptive immune cell recruitment, activation and phenotypes, including those of CD8+ T cells62–66. For example, activation of the WNT–β-catenin pathway in cancer cells results in inadequate dendritic cell (DC)-mediated priming of antigen-specific CD8+ T cells in TDLNs and poor tumour infiltration (T cell exclusion)67. T cell exclusion is an important immune evasion mechanism of so-called cold tumours (reviewed in REF.68).
Phase 1: initial tumour antigen emergence associated with ignorance, anergy or early dysfunction.
Carcinogenesis occurs in a sequence of stages — initiation, promotion and progression — and is generally initiated by somatic driver mutations in an otherwise normal cell. After the initiating oncogenic hit, the mutated clone gradually accumulates additional mutations and epigenetic changes that drive clonal expansion and accelerate growth and survival69. Before cancers become clinically detectable, there can be a long latency period. During the early stage of tumorigenesis, also referred to as the ‘preneoplastic phase’ or ‘early malignant phase’, mutations can create neoantigens and these can be presented on transformed cells. Generally, naive CD8+ T cells do not patrol peripheral tissues but recirculate between lymph nodes and the spleen, eventually encountering antigens on DCs presenting antigens picked up from peripheral tissues. During the early malignant phase, when there are only a few transformed cells, antigen release to DCs is likely to be minimal41,70. Thus, the initial sensing of tumour-initiating mutations falls mainly to tissue-resident immune cells, such as macrophages, DCs, natural killer cells and/or innate-like lymphocytes71,72. Studies in mouse models have shown that transformed cells that undergo oncogene-induced senescence induce innate immune responses, which can be tumour promoting or tumour inhibitory, depending on the oncogenes, tissue context and specific model73–75. When and how tissue-resident immune cells respond to tumour-initiating, early events and subsequently engage adaptive immune cells from the periphery is difficult to study in humans. Recent studies of patients with lung cancer revealed that the sensing of cancer cells by the immune system and their escape occurs in pre-invasive stages of lung tumorigenesis76. Productive priming of tumour-specific CD8+ T cell and CD4+ T cell responses during early tumorigenesis would require sufficient cancer cell death and/or antigen release and uptake by APCs for antigen presentation, and appropriate activation of APCs. In acute and even chronic infections, pathogenic antigens appear ‘acutely’ and at a high level, leading to antigen presentation by pathogen-associated molecular pattern (PAMP)-activated APCs and optimal priming of naive pathogen-specific CD8+ T cells and CD4+ T cells. This is in stark contrast to tumorigenesis, in which tumour-reactive CD8+ T cells may remain ignorant during the early malignant phase until a sufficient mass of cancer cells and tumour antigens have formed and/or are presented in TDLNs41,70,77. Even if tumour antigens are picked up and presented by APCs, PAMP-mediated innate immune activation does not occur, and there is a lack of CD4+ T cell help (MHC class II expression is generally restricted to professional APCs), resulting in suboptimal T cell priming, activation and differentiation to a hyporesponsive state akin to anergy and tolerance. Ignorance and early induction of anergy-like T cell hyporesponsiveness may explain why even cancers with highly immunogenic neoepitopes are able to develop and progress78 (FIG. 1).
Phase 2: persistent antigen and TCR stimulation associated with late dysfunction.
Ignorance and early T cell hyporesponsiveness allow early malignant lesions to progress to fully established cancers, with increased antigen presentation in secondary lymphoid tissues and increased immune cell tumour infiltration. As cancers progress and accumulate more mutations, more tumour antigens are expressed. Neoantigens created during the trunk of a cancer’s somatic evolutionary tree (trunk mutations include oncogenic driver mutations or early passenger mutations) are likely expressed on every cancer cell, and neoantigens derived from driver mutations are unlikely to be lost. Mutational antigens that arise later during carcinogenesis may be expressed on only a subset of the cancer cells. Non-essential passenger mutations may be lost, further contributing to antigenic heterogeneity. CD8+ T cells, especially those specific for trunk mutation antigens expressed on all cancer cells, will receive continuous antigen–TCR stimulation in tumours, driving T cells into an exhausted cell state.
Thus, tumour-specific T cell differentiation evolves in two phases: early during tumour development T cells enter an anergy-like early dysfunctional state upon encountering antigens in a non-inflammatory context without CD4+ T cell help (phase 1). This hyporesponsive state allows cancer cells to grow. With cancer progression, T cells are continuously stimulated by tumour antigens and enter a late dysfunctional state (phase 2) (FIG. 1). Since chronic antigen–TCR stimulation is the driver of T cell exhaustion, late dysfunctional T cells specific for highly expressed tumour antigens share many phenotypic, molecular and epigenetic traits with pathogen-specific T cells in chronic infection. However, important differences exist, which are discussed in the next sections.
Programmes of T cell dysfunction in cancer
Technological advances have allowed us to begin dissecting the molecular mechanisms regulating T cells in tumours: these include bulk and single-cell transcriptional and epigenetic profiling of tumour-reactive T cells, and analysis of TCR repertoires and the tumour immune landscape33,46,79–93, as well as perturbation screens (such as using short hairpin RNAs and CRISPR approaches)94–98. In this section we focus on key upstream transcriptional regulators of T cell differentiation and dysfunction, noting that several key regulators of T cell dysfunction also play a critical role during functional effector T cell differentiation.
Dynamic T cell differentiation programmes during tumorigenesis.
Following a clonal CD8+ T cell population over the entire course of tumorigenesis in an autochthonous tumour model revealed that tumour-specific T cells rapidly differentiate to a hyporesponsive state without appearing to pass through a functional effector phase (FIG. 2). With continued exposure to tumour antigens, T cells differentiated to a more profound hyporesponsive state within the progressing tumour79. While both early and late dysfunctional tumour-specific T cells had equally impaired cytotoxic capacity, early dysfunctional T cells could regain effector function upon removal from the tumour (plastic dysfunction), in contrast to late dysfunctional T cells, which could not be rescued (fixed or imprinted dysfunction). Early and late dysfunctional tumour-specific T cells can be distinguished by surface protein expression; whereas PD1 and LAG3 are expressed by both early and late dysfunctional T cells, late dysfunctional T cells express additional inhibitory receptors, such as CD38, CD39, CD101 and TIM3 (REF.99). Thus, T cell differentiation within tumours is progressive, and distinct T cell dysfunction states exist83,84,100. Indeed, distinct chromatin accessibility patterns were associated with early and late dysfunction, both of which were distinct from chromatin accessibility patterns of functional naive, effector and memory CD8+ T cells99. Human PD1+ TILs from melanoma and lung tumours shared chromatin accessibility patterns with late dysfunctional tumour-specific T cells from mouse tumours, suggesting that TILs within tumours may not be reprogrammable by immunotherapeutic approaches such as ICB (see later). Epigenetic programmes and states of T cell dysfunction have been demonstrated in other mouse tumour models46,101,102. Interestingly, two distinct states of CD8+ T cells were identified and associated with tumour regression or progression in a cohort of patients with melanoma treated with ICB84.
Fig. 2 |. Models of CD8+ T cell differentiation and dysfunction in tumours and exhaustion during chronic infections.

a | Linear differentiation model. Naive tumour-specific CD8+ T cells encounter tumour antigens, inducing a programme of early dysfunction. Dysfunction is initially plastic but eventually becomes fixed. T cell factor 1 (TCF1) is progressively lost with time and antigen encounter, while TOX expression increases. Early dysfunction and late dysfunction are epigenetically encoded and can be associated with specific surface marker and transcription factor expression. Early dysfunctional T cells can be reprogrammed; late dysfunctional T cells are resistant to therapeutic reprogramming. b | Branched differentiation model. Tumour-specific T cells infiltrate the tumour. Tertiary lymphoid structures or lymphoid-like structures could represent specialized niches within the tumour microenvironment that facilitate the maintenance of tumour-specific progenitor-like/less dysfunctional CD8+ T cells which self-renew and give rise to more differentiated dysfunctional T cells (terminally differentiated). c | In chronic infection, pathogen-specific T cells differentiate into early effector T cells, which differentiate into progenitor exhausted T cells that reside within secondary lymphoid organs (for example, white pulp of the spleen) and self-renew, and give rise to terminally exhausted T cells found in peripheral tissue and red pulp.
Early dysfunction molecular programmes.
For effective CD8+ T cell activation, co-ligation of TCR and CD28 leads to activation of the MAPK, JNK, PI3K–AKT and IKK signalling pathways and the mobilization of intracellular calcium, which subsequently activates multiple transcription factors, including nuclear factor of activated T cells (NFAT) and FOS–JUN dimers (AP-1)103. Cytosolic NFAT is dephosphorylated by the calmodulin-dependent phosphatase calcineurin and translocates to the nucleus, where it cooperates with AP-1 to induce a transcriptional programme associated with effector function (including effector target genes IL2 and IFNG). TCR engagement in the absence of co-stimulation causes suboptimal MAPK, PI3K–AKT and IKK pathway activation, resulting in insufficient AP-1 activity. Using a mutant constitutively active form of NFAT that is unable to interact with AP-1, Rao and colleagues demonstrated that activated NFAT in the absence of AP-1 cooperation (known as partnerless NFAT) induces a transcriptional programme distinct from that triggered during functional effector differentiation, characterized by repressive transcription factors (EGR2, EGR3, IKZF2, IRF4 and TOX) and other negative regulatory proteins (CBL-B)45. Integrated transcriptional and epigenetic analyses identified that NFAT binding to monomeric binding sites occurred predominantly in promoters and regulatory elements of anergy-associated genes, whereas composite NFAT–AP1 binding elements were found in the promoters and regulatory elements of effector genes. Thus, NFAT partnered with active AP-1 promotes T cell activation and effector differentiation, whereas partnerless NFAT promotes anergy. Indeed, analysis of chromatin accessibility found that sites containing NFAT-binding motifs were significantly more open in dysfunctional tumour-specific CD8+ T cells than in functional effector CD8+ T cells46,99.
Late dysfunction molecular programmes.
While early dysfunctional T cells in tumours have little or no effector function, during chronic viral infection CD8+ T cells initially enter a functional effector state. However, with continued tumour exposure, late dysfunctional TILs become phenotypically and transcriptionally similar to terminally differentiated exhausted T cells during chronic viral infection, and a shared critical feature is loss of TCF1 expression and high expression of TOX99,104–107 (FIG. 2). Originally identified as a transcription factor essential for thymocyte development108, TCF1 is now known to be important for peripheral T cells (reviewed in REF.109). The Restifo group demonstrated that TCF1 promotes memory T cell differentiation, and enforced TCF1 expression after T cell activation inhibits effector differentiation110. The Held group showed that Tcf7−/− CD8+ T cells could mount a normal effector response to acute infection but failed to form memory and respond to secondary challenge111. More recent studies from the Reiner and Held groups suggest that early after activation, a small subset of effector-phase CD8+ T cells retain TCF1 expression, possibly as a result of asymmetric division, and do not differentiate to effector cells but rather give rise to central memory T cells112,113.
Several studies in chronic viral infection found two populations of virus-specific CD8+ T cells: TCF1+ (PD1+CXCR5+TIM3−) CD8+ T cells have a memory/stem cell-like phenotype, self-renew and give rise to terminally differentiated TCF1low (PD1+CXCR5−TIM3+) T cells106,107,114–117. These two populations are spatially segregated, with the TCF1+ progenitor population in the T cell zones (white pulp) of secondary lymphoid tissues, while the terminally differentiated, exhausted TCF1low T cell population is predominantly found in peripheral tissues and the splenic red pulp, the reservoir of infected cells and/or antigens. Progenitor exhausted T cells and terminally exhausted T cells represent two distinct T cell states or populations with distinct epigenetic programmes associated with reprogrammability: progenitor exhausted T cells can be reinvigorated by ICB, whereas terminally exhausted T cells cannot104–107.
Although tumour-infiltrating T cells initially express TCF1, they subsequently downregulate TCF1, and most tumour-reactive dysfunctional T cells within tumours are TCF1low cells, as are tissue-infiltrating terminally exhausted CD8+ T cells during chronic viral infection106,107,118. Thus, tissue residency appears to be correlated with loss of TCF1 expression. Given that TCF1 expression in a subset of progenitor-like virus-specific T cells is required for maintenance of the exhausted T cell population during chronic viral infection, an important question for tumour immunologists is how tumour-specific T cell populations are maintained. In chronic viral infection, there is evidence of branched differentiation, where progenitor-like T cells in lymphoid tissues both self-renew and give rise to more terminally differentiated exhausted T cells106,119,120. As has been shown for acute infection, this branched differentiation may be driven by asymmetric cell division121. However, evidence of a stable population of tumour-reactive TCF1+ progenitors within mouse or human tumours is still lacking, and instead longitudinal TIL data may support a linear differentiation model with TCF1+ early dysfunctional TILs differentiating to TCF1low late dysfunctional TILs99. It remains to be determined whether TCF1low tumour-reactive TILs can self-renew, especially those within intratumoural niches and tertiary lymphoid structures (TLSs) (see later), or whether TCF1+ progenitors in secondary lymphoid organs, especially the TDLNs, maintain the tumour-specific T cell pool (FIG. 2). Regardless, as discussed in detail later, a shared critical feature is the loss of TCF1 expression in non-reprogrammable T cells.
In the last 2 years, another high mobility group box DNA-binding protein, TOX, has emerged as an important regulator of T cell dysfunction programmes in tumours and chronic infection120,122–124. TOX is thought to bind DNA in a sequence-independent yet structure-dependent manner and facilitate binding of protein cofactors through its transactivation domain125. Previously, TOX was known to be required for thymic development of CD4+ T lineage cells, natural killer cells and innate lymphoid cells126–128. More recently, TOX has been shown to play an important role in peripheral CD8+ T cell differentiation during infection, tumorigenesis and autoimmunity118,120,122–124,129–132. Tumour mouse models demonstrated that TOX, regulated by NFAT, is highly expressed in dysfunctional tumour-specific T cells but not in non-tumour reactive bystander T cells, suggesting that high TOX expression is driven by TCR stimulation46,122. TOX is expressed in human TILs83,122,124,130, and its expression positively correlates with inhibitory receptor expression and negatively correlates with TCF1 expression83,122,130.
In a mouse tumour model, TOX-deficient tumour-specific T cells in tumours no longer upregulated inhibitory receptor expression but still remained completely hyporesponsive122. Thus, despite the association between TOX and inhibitory receptor expression (exhaustion phenotype) and functional hyporesponsiveness, T cell function or dysfunction and T cell phenotype (TOX and inhibitory receptor expression) are not completely co-regulated (FIG. 3). This conclusion is supported by several other observations. First, functional effector T cells (generated through in vitro stimulation or in vivo during an acute infection) transiently express TOX and several inhibitory receptors, with expression levels decreasing once antigen–TCR stimulation ceases122–124. Second, functional tissue-resident memory T cells express TOX and inhibitory receptors133–135. Third, polyfunctional effector memory CD8+ T cells in humans have elevated TOX expression levels (coupled with inhibitory receptor expression), including cytomegalovirus-specific and Epstein–Barr virus-specific T cells132,136. Thus, TOX and/or inhibitory receptors are not exclusively linked to hyporesponsive CD8+ T cell states. Although PD1, LAG3, CTLA4, CD39, TIGIT and other molecules are often regarded as exhaustion markers, they can also be considered activation markers as they are also transiently expressed 24–48 hours after naive T cell activation. In this setting, inhibitory receptors act as a physiological negative-feedback mechanism to prevent overstimulation of activated antigen-specific CD8+ T cells19,137. Indeed, TOX-deficient CD8+ T cells lacking inhibitory receptor expression fail to persist in settings of chronic antigen stimulation such as chronic infection and cancer, suggesting that the TOX-induced exhaustion phenotype protects T cells from overstimulation120,122–124 (FIG. 3). While TOX-deficient tumour-specific CD8+ T cells in autochthonous tumour models did not show increased effector function and remained hyporesponsive122, in chronic infection models, TOX-deficient progenitor exhausted T cells had increased effector functions123,124, and in transplantation tumour models, tumour-reactive effector T cells with reduced TOX expression levels (through heterozygous deletion or short hairpin RNA) had increased antitumour activity124,130,131. Thus, TOX appears to have distinct roles in effector, progenitor exhausted, anergic, terminally exhausted and dysfunctional T cells, and TOX expression levels may be crucial in determining fate and function. More work is required to understand the mechanisms that drive TOX expression (TCR and non-TCR signals) and determine its role and functional consequences in distinct T cell subsets and activation states.
Fig. 3 |. Two modules of tumour-specific T cell dysfunction programming: loss of effector function and exhaustion phenotype.
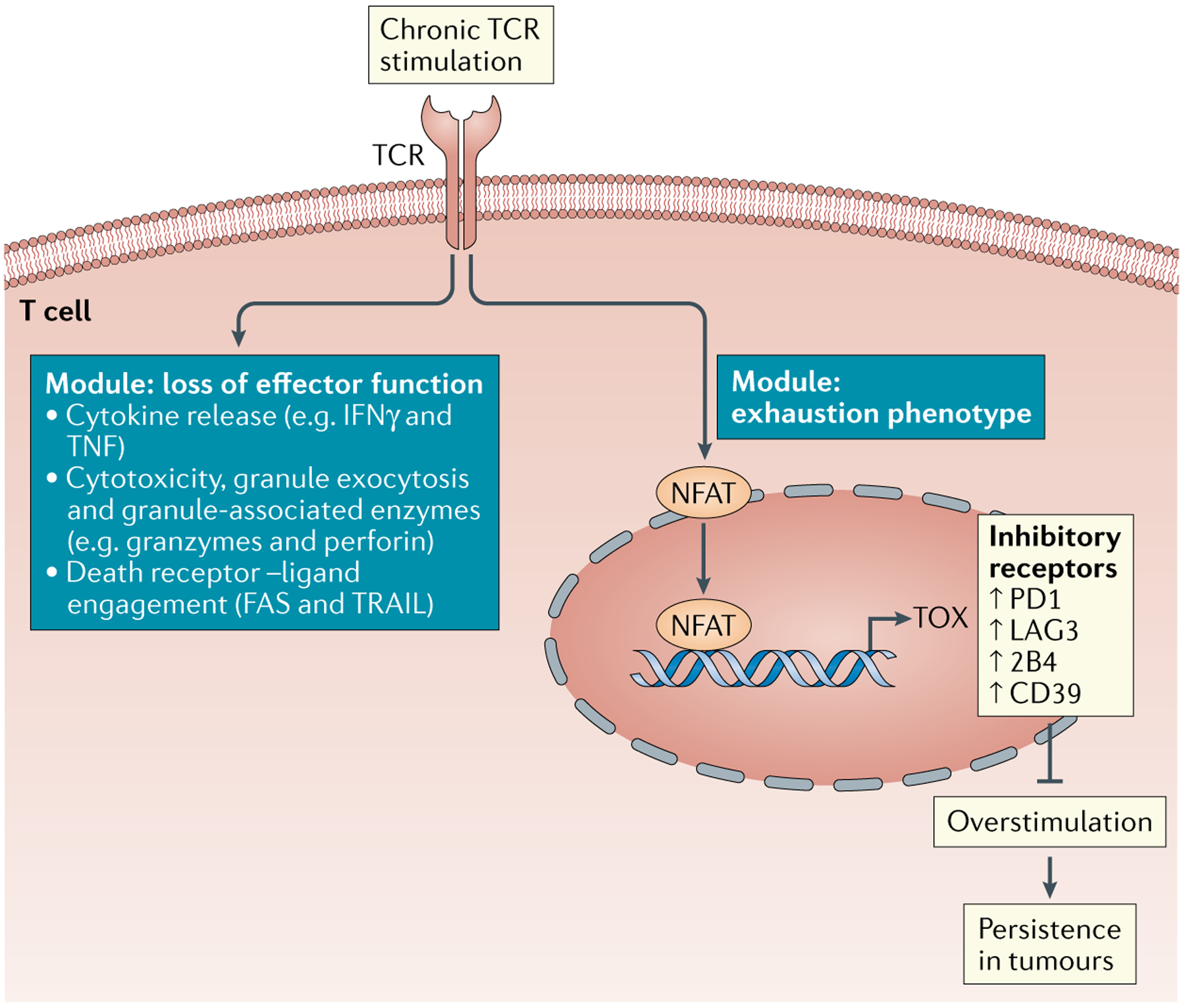
Tumour-specific T cells in solid tumours are continuously triggered by tumour antigens. Chronic T cell receptor (TCR) stimulation leads to nuclear factor of activated T cells (NFAT)-mediated expression of TOX. TOX induces a programme and phenotype associated with exhaustion, including the expression of inhibitory receptors (such as PD1, LAG3, 2B4 and CD39) and downregulation of transcription factors (such as T cell factor 1 (TCF1)). TOX-mediated exhaustion programming and expression of inhibitory receptors prevents T cells from overstimulation and allows T cells to persist in tumours in the face of chronic tumour antigen encounter. TOX-independent mechanisms may regulate the loss of cytotoxic effector function, including cytokines and cytotoxic molecules. IFNγ, interferon-γ.
Mechanisms of effector function loss.
There are several mechanisms for the failure of CD8+ T cells to eliminate cancers. These include T cell-extrinsic mechanisms: cancer cell-mediated mechanisms (such as loss of MHC expression, antigen loss, loss or defects in antigen presentation, or expression of inhibitory receptor ligands) and TME-mediated mechanisms (such as TGFβ, IL-10, nitrogen metabolites, regulatory T cells or MDSCs)21. For example, the Gabrilovich group demonstrated that T cell dysfunction can be caused via MDSC-mediated nitration of tyrosines within the TCR complex, inhibiting TIL binding to peptide–MHC complexes138. Lack of antitumour T cell responses can also result from T cell-intrinsic impairment of cytotoxicity (T cell dysfunction). Cytotoxic CD8+ T cells eliminate target cells through several pathways, including cytokine (IFNγ and TNF) release, granule exocytosis and granule-associated enzymes (perforin and granzymes), or death receptor–ligand engagement (FAS and TRAIL). In addition to these direct cytotoxic mechanisms, tumour-reactive T cells can induce cancer cell death indirectly by targeting tumour stromal cells or the tumour vasculature139–143. Tumour-reactive T cells do not necessarily engage or require all modes of effector functions. While in some mouse models deficiency of one specific effector or cytotoxic pathway results in impaired tumour control, in others the same deficiency has no impact144. Thus, the mode of tumour killing by tumour-reactive T cells is tumour, context and model dependent. We know little about which effector mechanisms operate in human tumours or after ICB therapy, and improved understanding of the critical T cell effector mechanism or mechanisms in tumours will be important to design strategies to improve T cell antitumour responses.
Loss of effector molecule expression is transcriptionally and epigenetically regulated in tumour-reactive TILs99,145, but other mechanisms have been identified. Expression of cytokines and cytotoxic molecules, including IFNγ, can be regulated at the post-transcriptional level146,147. Post-transcriptional silencing was demonstrated in hyporesponsive CD8+ T cells, including tumour-reactive TILs, and blocked mRNA translation was mediated by conserved (A+U)-rich elements within the cytokine 3′ untranslated regions148,149. Similarly, granzyme and perforin expression is regulated transcriptionally and post-transcriptionally, with blocks at both levels associated with T cell dysfunction150. Moreover, cytotoxic molecules can be synthesized but not released from TILs151; since a MAPK-mediated activation signal is required for perforin mobilization or degranulation152,153, altered MAPK signalling in TILs can impair lytic granule release154.
Cytotoxicity of and cytokine release by TILs (or their lack thereof) is difficult to assess in situ, and thus cytotoxic gene mRNA level has been used as a surrogate for T cell functional or dysfunctional status within tumours. However, given the importance of post-transcriptional regulatory mechanisms and the observation that exhausted or dysfunctional CD8+ T cells can have high levels of cytokine or cytotoxic mRNA with little protein or cytotoxic function47,79, care must be taken in using transcriptional data to infer functional status. Ex vivo or in vitro restimulation assays can be used to assess TIL functional status. However, even these functional assays must be interpreted carefully as, first, phorbol myristate acetate and ionomycin, which are commonly used to stimulate TILs ex vivo, directly activate protein kinase C and Ca2+ mobilization independently of TCR engagement and overcome proximal TCR signalling defects154,155, and, second, TIL exposure to exogenous IL-2 in vitro and/or several rounds of proliferation can reverse TIL dysfunction. Thus, ex vivo assays do not necessarily capture the functional status of TILs in vivo. One challenge and urgent need is the development of tools to capture functional TIL states in situ.
Tissue residency programmes.
T cells in tumours harbour molecular programmes associated with tissue residency. Tissue-resident memory T cells develop and seed tissues during infection156 (reviewed in REFS157,158) and are characterized by downregulation of sphingosine 1-phosphate receptor 1, expression of the tissue retention markers CD69, CD49a and CD103, various inhibitory receptors (PD1, TIGIT and TIM3) and transcription factors such as HOBIT, BLIMP1 and RUNX3 (REFS157,159–161). Aside from haematological cancers, most cancers arise in non-lymphoid peripheral tissues. T cells migrate to and infiltrate the tumour tissue, and TILs (both bystander and tumour-reactive TILs) display characteristics of tissue residency, including expression of the tissue retention marker CD103 and inhibitory receptors (reviewed in REF.157). The presence of CD103+CD8+ TILs has been associated with increased survival, and CD103+CD8+ double-positive TILs was identified as a better prognostic marker than total CD8+ TILs157, and were associated with increased survival of patients with melanoma treated with ICB162. With the current data remaining largely correlative in nature, further studies are needed to determine the specificity and potential functional roles of tissue-resident TILs in human tumours. Given the experimental evidence from animal models regarding the potency of tissue-resident memory T cells in cancer elimination and cancer–immune equilibrium160,163,164, promoting tissue-resident memory T cell-associated programmes in tumour-reactive T cells could be an attractive therapeutic approach for prevention of local recurrence.
TIL heterogeneity
Although we have made tremendous advances in identifying TIL phenotypes and transcriptional and epigenetic programmes, especially at single-cell resolution, the major challenge remains to link distinct phenotypes to tumour antigen specificity and functional states, and to determine which antigen reactivities and states contribute to a clinical response to immunotherapeutic interventions.
Tumour-reactive versus bystander TILs.
The TIL population is composed of both tumour-reactive T cells and non-tumour-reactive bystander T cells. T cells can be recruited to the tumour by antigen-dependent and antigen-independent mechanisms, including proinflammatory signals and cytokines (IFNγ, TNF and IL-1), which increase T cell migration to inflammatory sites through the induction of integrins (ICAM1 and VCAM1)165. Bystander T cells can be naive or antigen experienced, including effector and memory T cells (specific for influenza virus, cytomegalovirus or Epstein–Barr virus). The fraction of tumour-reactive T cells is highly variable among cancer types and patients. While increased immune infiltration is thought to correlate with better outcomes and improved response to ICB166, high tumour immune infiltration does not necessarily correlate with increased tumour reactivity. The Schumacher group assessed the intratumoural TCR repertoire of human ovarian and colorectal tumours and demonstrated that only a small fraction of intratumoural CD8+ T cells (approximately 10% or less) had the capacity to recognize cancer cells; in some patients, despite significant T cell infiltration into tumour, no tumour reactivity was detected. Thus, tumours which are highly infiltrated by T cells (‘quantitatively hot’) can represent ‘qualitatively cold’ tumours if the tumour-recognition potential of their TCRs is low or even absent90. Similar observations were made by the Newell group in human colorectal and lung cancers85.
Bystander TILs are phenotypically distinct from tumour-reactive TILs; bystander T cells may express inhibitory receptors, including PD1 or TIGIT, but they generally lack hallmarks of chronic antigen stimulation, expressing low (or lower) levels of exhaustion-associated surface markers and transcription factors and higher levels of quiescence or memory-associated transcription factors, including TCF1. Although PD1 expression on CD8+ TILs has been used for the enrichment of tumour-reactive T cells167–170, PD1 expression is not specific for tumour-reactive T cells. Two recent studies identified CD39 as an additional biomarker to demarcate tumour-reactive TILs from non-tumour-reactive bystander TILs85,171; within PD1+ TILs, CD39+ or CD103+CD39+ TILs were enriched for tumour reactivity in human solid tumours. Increased frequency of CD103+CD39+ TILs correlated with increased survival of patients with head and neck cancer171. Bystander T cells in tumours retain effector function and transcriptional and epigenetic programmes generally associated with memory or effector-like T cell phenotypes79,122 although bystander and tumour-reactive T cells are exposed to the same immunosuppressive factors within the TME46,79,122,172. Thus, continuous tumour antigen encounter is required to induce T cell dysfunction: chronic antigen stimulation leads to high inhibitory receptor expression on tumour-reactive T cells, making chronically stimulated tumour-reactive T cells susceptible to the inhibitory signals present in the TME (for example, by binding to inhibitory ligands, such as PDL1). By contrast, bystander T cells, which are not chronically stimulated by the cognate antigen, do not express high levels of inhibitory receptors and thus are not susceptible to inhibitory signalling from the TME.
Role of T cell-intrinsic factors.
The tumour-reactive T cell repertoire contains T cells specific for different tumour antigens (self-antigens or neoantigens), as well as T cells with distinct TCR clonotypes and affinities for the same tumour antigen. TCR affinity for peptide–MHC determines the kinetics and magnitude of CD8+ T cell responses, including antitumour CD8+ T cell responses173–179. TCRs of self-antigen-specific T cells generally have lower affinity than neoantigen-specific T cells180–182. Both self-antigen-specific and neoantigen-specific TILs differentiate to hyporesponsive states within tumours, sharing many phenotypic and functional characteristics of exhaustion and dysfunction. However, whether differences exist in their molecular or epigenetic programmes and how they contribute to clinical responses to immunotherapy remain largely unknown.
Role of spatio-temporal factors.
Tumours are spatially heterogeneous and evolve over time, with changes in cancer cell genetic and epigenetic make-up, immune and non-immune (stromal) cell composition, vascular networks, extracellular matrix remodelling and microenvironmental gradients of nutrients, oxygen, pH, ions and other soluble factors. T cells and other immune cells adapt to the TME, and in turn cancer cells and the TME change in response to immune activity: CD8+ T cells can eliminate antigen-expressing cancer cells in distinct tumour regions (cancer elimination) (BOX 1). Cancer cells can change their antigenic landscape, losing antigens or creating new antigens through genetic or epigenetic changes in genes encoding antigens or more globally through altered expression of antigen processing and presentation machinery (cancer immunoediting), ultimately allowing cancer variant outgrowth (cancer escape)183. Thus, antigen density can vary within progressing tumours, and T cells in spatially distinct regions can experience quantitative and qualitative differences in antigen and TCR stimulation. Moreover, TIL phenotypes and functional states evolve with time, because the duration of tumour antigen exposure is the critical determinant of T cell dysfunction and T cell state transitions. Thus, the longer TILs reside in the tumour and are exposed to antigens and immunosuppressive signals, the more exhausted their phenotype and the severer their dysfunctional state. Since T cells infiltrate tumours at different stages of tumorigenesis, the exposure duration to exhaustion-driving or dysfunction-driving factors will vary, contributing to the intratumoural hetero geneity of T cell states. Moreover, inhibitory ligand expression and other microenvironmental factors can vary substantially within the tumour and over time.
Box 1 |. CD8+ T cells in cancer immunosurveillance.
For many decades, it has been vigorously debated as to whether the immune system can detect and eliminate malignant cells, preventing cancer, a hypothesis originally proposed by Burnet and Thomas and referred to as ‘cancer immunosurveillance’239. Immunosuppressed persons have a higher incidence of viral-associated cancers but there is less evidence of increased incidence of non-viral cancers240. Addressing this question using animal models requires experimentally recapitulating tumour initiation and development as it occurs in humans241,242. Autochthonous tumours, which include spontaneous tumours, carcinogen-induced (UV-induced or chemically induced) tumours and genetically induced tumours, are initiated through the de novo transformation of normal cells within intact tissues and organs, and these tumours frequently express tumour antigens. Given that autochthonous tumours can develop and progress in immunocompetent mice as well as in humans despite the expression of highly immunogenic tumour antigens34,243–247, the question remains whether CD8+ T cell-mediated surveillance mechanisms are operative during tumorigenesis. in genetically engineered mouse models, tumour-specific CD8+ T cell hyporesponsiveness is a major mechanism of tumour evasion79,248,249. in some mouse models, including methylcholanthrene-induced cancers, increased tumour incidence was observed in immunodeficient mice compared with immunocompetent mice, with CD8+ T cells playing an important role in immunosurveillance250,251. By contrast, other studies using mouse models with T cell or effector molecule deficiencies failed to reveal significant differences in tumour development252. If T cell-mediated immunosurveillance mechanisms exist in humans, the prediction is that patient cancer genomes will evolve to escape immune recognition and elimination. recent data from the ongoing Tracking Cancer evolution through Therapy (TrACerx) study of non-small-cell lung cancers suggest that the antigen landscape might be shaped by immunoediting mechanisms253; however, other studies failed to find evidence of neoantigen depletion in patient genomes254. Thus, whether the immune system and especially CD8+ T cells survey and/or edit developing tumours remains an open question.
Role of TLSs and intratumoural niches.
Several cancer types (for example, lung, colorectal, pancreatic, hepatocellular and breast cancers) have been shown to develop intratumoural TLSs, which form through chronic inflammatory signals mediated by chemokines and cytokines (including LTα1β2, CXCL13, CXCL19, CXCL21, IL-22 and IL-23) (reviewed in REF.184). The presence of TLSs in tumours has been linked to higher CD8+ T cell infiltration, clinical benefit (with some exceptions) and improved responsiveness to immunotherapy, suggesting that TLSs promote local antitumour immune responses. Several recent studies identified a crucial role for B cells within cancer-associated TLSs from patients treated with ICB185–187: the presence of CD20+ B cells and TLSs before and/or during ICB predicted improved clinical response. Interestingly, B cell-rich and TLS-rich metastatic melanoma tumours had increased numbers of TCF1+ T cells, in contrast to tumours without TLSs, in which T cells had a more differentiated, dysfunctional molecular phenotype187. In a study with a cohort of patients with kidney cancer, TCF1+ T cells were shown to reside in dense APC niches, whereas clonally related, more differentiated CD8+ T cells were found outside these intratumoural niches188. Although at present the mechanism by which B cells and TLSs and intratumoural niches favour-ably affect antitumour immunity and immunotherapy responsiveness is unknown, it is tempting to speculate that these spatially distinct lymphoid-like structures are privileged sites within the TME that facilitate the maintenance of less differentiated and dysfunctional CD8+ T cells, which may be more amenable to immunotherapeutic reprogramming. Additionally, these niches may facilitate tumour antigen presentation by DCs or B cells, which can activate CD4+ T helper cells and CD8+ T cells together for the generation of a more effective antitumour response. However, it is currently not clear whether T cells close to these structures are tumour-reactive or bystander T cells. As most of the current data from human patients regarding tumour specificity and functional states of T cells within and outside TLSs are correlative, further studies are needed to understand the role of TLSs in tumour-specific CD8+ T cell differentiation and their contribution to antitumour immunity and immunotherapy responses.
Which T cells mediate ICB responses?
Although ICB has the potential to induce long-term remissions, even in patients with metastatic solid tumours, most patients fail to achieve durable clinical responses189–191. This has led to much effort to identify predictors or biomarkers of ICB response. At the population level, increased infiltration of CD8+ T cells in tumours correlates with better outcomes, yet the predictive value for individual patients is low166,192,193. Cancer cell-intrinsic factors, such as mutational load and expression of inhibitory receptor ligands (such as PDL1), have been explored as prognostic markers, with mixed results194–198. This has turned the focus to profiling TILs and/or circulating T cells in patients with cancer, both to identify biomarkers for response and to address a hotly debated question: which T cells (tumour resident, peripheral or lymphoid resident) mediate ICB responses, and what are the antigen specificities (low or high affinity, self-antigens or neoantigens) and differentiation states (naive, functional effector, memory or dysfunctional) of responding T cells?
Initially, ICB was thought to act by reinvigorating exhausted or dysfunctional tumour-infiltrating CD8+ T cells (FIG. 4). This idea came from observations in chronic viral infection in which progenitor exhausted (TCF1+ and/or T-bethiEOMESlow) CD8+ T cells (which reside in secondary lymphoid organs) proliferated and had increased effector function in response to ICB106,119. However, there is no evidence to date showing that dysfunctional tumour-reactive CD8+ T cells within tumours in human patients regain functional capacity in response to ICB. Initial studies found that increased proliferation of circulating lymphocytes in patients predicted better ICB responses199, and clonotypic expansion of T cells in the peripheral blood of patients with cancer predicted clinical responsiveness to anti-PDL1 (REF.200). Moreover, the presence of TCF1+ TILs was shown to correlate with improved survival responses to ICB in patients with melanoma84. Several studies have identified TCF1+ TILs in human tumours84,115,201–203; however, the tumour reactivity of TCF1+ TILs in these studies was unknown. Studies in mouse models with tumour-specific CD8+ T cells of known tumour specificity and in defined dysfunctional states have shown that TCF1+ tumour-specific CD8+ T cells mediate antitumour responses in response to ICB therapy and/or vaccination101,202, but it is not known whether these TCF1+ T cells represent a stable, self-renewing stem cell-like progenitor T cell population, or are ‘earlier’, less differentiated dysfunctional TCF1+ T cells99. Li et al. conducted single-cell RNA sequencing and TCR sequencing and assessed tumour reactivity of TILs from patients with melanoma83. They found that TCF1+ TILs include bystander non-tumour-reactive cytotoxic T cell populations, whereas TCF1low TILs had dysfunctional hallmarks, including expression of PD1, LAG3, CD39 and TOX. Additional studies have demonstrated that the pre-existing TIL repertoire in patients with cancer does not expand following ICB, but rather that exhausted intratumoural CD8+ T cells are replaced with non-exhausted T cells recruited from sites outside the tumour and exert antitumour effects. Yost et al. conducted paired single-cell RNA and TCR sequencing on TILs from patients with basal or squamous cell carcinoma and demonstrated that after anti-PD1 therapy, tumour-infiltrating T cell clones did not derive from the pre-existing TIL repertoire, but instead included novel clonotypes not previously observed in the tumour (clonal replacement)204. Dammeijer et al. showed that PDL1 blockade induces CD8+ T cell-mediated antitumour immunity by seeding the tumour with progenitor exhausted PD1+ T cells from the TDLNs, where they associate with PDL1+ DCs; PD1–PDL1 interactions in TDLNs but not in the tumour correlated with a better prognosis in patients with melanoma205. Collectively, these studies suggest that before ICB, tumour-reactive TILs are largely refractory to ICB-mediated reinvigoration, consistent with the epigenetic inflexibility demonstrated in mouse models for dysfunctional tumour-specific TILs99 and exhausted T cells in chronic viral infection104.
Fig. 4 |. Tumour-specific CD8+ T cell subsets and states mediating immunotherapy responses.

a | In the tumour, most tumour-reactive T cells are in a late dysfunctional state that is resistant to immunotherapeutic reprogramming (such as with immune checkpoint blockade or vaccination). b,c | Early dysfunctional T cells which recently entered the tumour and/or progenitor/early dysfunctional tumour-specific T cells residing in tertiary lymphoid structures or intratumoural niches can be functionally rescued and reprogrammed into effector T cells by immunotherapy. d | In the periphery, functional or progenitor-like/early dysfunctional tumour-reactive T cells in secondary lymphoid organs or peripheral blood proliferate and differentiate into cytotoxic effector T cells in the presence of immune checkpoint blockade. Reprogrammed functional T cells infiltrate the tumour and mediate tumour elimination.
However, there may be some tumour-reactive TILs that are amenable to ICB-mediated reprogramming: first, T cells that have recently entered tumours and have not yet undergone terminal differentiation to a TCF1low phenotype and, second, T cells resident in distinct niches within the tumour, such as TLSs185–187 or intratumoural niches188, antigen-loss regions or specific metabolic niches206 (FIG. 4). For example, Tumeh et al. demonstrated that the number of PD1+CD8+ TILs localized to the invasive tumour margin in association with PDL1-expressing cells predicted response to ICB therapy207.
The association of TCF1+ T cells in tumours with better prognosis could reflect an underlying property of these tumours — that they are hot and facilitate T cell infiltration, independently of tumour antigen specificity and due to tumour-intrinsic factors. Such tumours, when treated with ICB, would more likely facilitate the infiltration of tumour-reactive T cells from the periphery, including blood and secondary lymphoid organs (FIG. 4). Indeed, the Rosenberg group and others have shown that tumour-reactive T cells, including neoantigen-specific T cells, can be found in the peripheral blood of patients with cancer and are enriched within the PD1+CD8+ T cell pool40,168,208–210. Interestingly, tumour-reactive PD1+ peripheral blood lymphocytes have a ‘less exhausted’ phenotype, with lower expression of the inhibitory receptors PD1, TIM3 and LAG3 than their tumour-resident counterparts. Thus, circulating, tumour-reactive PD1+ T cells may be targeted by PD1-blocking antibodies, expand, infiltrate the tumour and contribute to the antitumour efficacy of anti-PD1 therapy (FIG. 4).
Outstanding questions and challenges
Cancer immunotherapy, including ICB211,212, vaccines213–215, adoptive T cell therapy216,217 and chimeric antigen receptor T cell therapy31, has generated considerable excitement, inducing durable remissions in a subset of patients and cancer types, yet most patients still fail to achieve long-term responses189–191,218. Despite much effort, we cannot currently predict which patients will respond, nor do we understand the mechanisms that determine success or failure. Recent technological advances have provided important insights into the heterogeneity of CD8+ TILs, demonstrating that distinct T cell subsets exist with different transcriptional and epigenetic programmes, and functional states, and different responses to therapeutic reprogramming. However, important questions and challenges remain.
CD8+ T cells have multiple ways to eliminate tumours, and they can directly target cancer cells or indirectly target tumour stromal cells. We know little about which mechanism or mechanisms are most relevant in human cancers, whether distinct tumours and T cells use different mechanisms, and how these mechanisms are subverted in hyporesponsive TILs. Cytokines or cytotoxic effector molecules are highly regulated at the post-transcriptional level; thus, care must be taken in using human TIL transcriptional data to infer the functional status of T cells in situ. Identifying tumour-reactive T cell populations and their functional states in patients, and understanding how these T cells execute antitumour effector function, will be crucial to understand and predict the efficacy of immunotherapeutic strategies. Does the response come from within the tumour or from outside? Is there an intratumoural niche for stable, self-renewing progenitor tumour-reactive T cell populations or do peripheral blood or secondary lymphoid organs, especially TDLNs, represent the reservoir for these T cell populations? Patient studies and mouse cancer models have provided valuable insights into cancer immunity that have translated into therapeutic advances for patients. While patient studies provide much evidence, they remain largely correlative, and mechanistic studies are necessary to confirm causative relations. Thus, we must continue to develop and use clinically relevant animal cancer models as well as human samples to obtain mechanistic insights to move the field forward. Exciting advances in single-cell resolution multiplexed protein imaging219,220, spatial metabolomics221 and spatially mapped single-cell chromatin accessibility222,223, together with transcriptomics224 and single-cell TCR sequencing, are allowing us to now gain these mechanistic insights, including the impact of spatio-temporal factors on T cells.
One important lesson we have learnt from using ICB is that tumour-reactive T cells present in some patients are capable of proliferating, infiltrating tumours and eliminating cancer cells. This raises the question of whether in patients who fail to achieve durable responses to ICB, tumour-reactive CD8+ T cell responses are not being optimally activated or amplified. Since cancer, in contrast to infection, does not provide sufficient inflammatory or innate stimulation for optimal T cell priming and expansion, there are growing efforts to harness innate immunity and proinflammatory signals to improve anti-cancer responses225, for example, oncolytic viruses226, cGAS–STING agonists227, cytokines228 and vaccines213–215. Clinical trials are under way to test possible synergy between innate-stimulating therapies and ICB229–231.
Furthermore, given the growing evidence that TILs may be in an epigenetically ‘inflexible’ state resistant to ICB and other immunotherapies99,104, in patients without sufficient immune functional reserve to mount new anti-tumour responses, we need to reinvigorate dysfunctional TILs, a difficult task that may require targeted strategies to remodel the CD8+ T cell epigenome. Efforts are under way to combine drugs targeting the epigenome with ICB232,233; understanding when and how these therapies work is complicated because epigenetic remodellers have widespread pleiotropic effects on cancer cells, stromal cells and T cells. An alternative strategy is to target key transcription factors that drive chromatin remodelling in response to suboptimal priming or chronic stimulation. In the non-adoptive therapy setting, pharmacological strategies to target transcription factors could be used. For example, FK506, a calcineurin inhibitor, has been used to modulate NFAT and/or TOX levels in preclinical studies99,122,124. In adoptive T cell settings, effective T cell reprogramming and antitumour responses could be demonstrated by targeting NR4A1 (REFS234,235), TOX131 and JUN236, and CRISPR-mediated reprogramming could emerge as a powerful approach for future immunotherapeutic applications237,238.
The recent rapid growth in the number of successful cancer immunotherapies brought to the clinic and saving lives has been the result of many years of concerted effort by clinicians and basic scientists from many different fields. With continued advances in single-cell profiling technologies and high-throughput data analysis222, genome and epigenome editing, imaging and cellular therapies, we can integrate basic immunology insights and clinical observations and data to bring the benefit of cancer immunotherapy to all patients with cancer.
Acknowledgements
The authors are grateful for support from the V Foundation Scholar Award (M.P.), Vanderbilt Digestive Disease Research Center Young Investigator and Pilot Award (P30DK058404) (M.P.), the Vanderbilt-Ingram Cancer Center Breast Cancer SPORE Career Enhancement Program (P50CA098131) (M.P.), NIH NCI grants DP2CA225212 (A.S.) and U54CA209975, the Lloyd Old Star Awards Program from the Cancer Research Institute (A.S.), the Pershing Square Award (A.S.), and the Josie Robertson Young Investigator Award (A.S.).
Glossary
- Immune checkpoint blockade (ICB)
Therapy for cancer using specific antibodies that block interactions between inhibitory ‘checkpoint’ receptors on immune cells and their ligands on cancer and stromal cells.
- Central tolerance
A process occurring during thymic T cell development in which thymocytes bearing rearranged T cell receptors with too high affinity for self-antigen–MHC complexes are eliminated (negative selection).
- Peripheral tolerance
Process by which self-reactive T cells that escaped central tolerance and entered the periphery are inactivated by induction of apoptosis (peripheral deletion), suppression by CD4+ regulatory T cells or induction of cell-intrinsic hyporesponsive programmes (tolerance).
- Cold tumours
Non-T-cell-inflamed tumours as opposed to hot tumours, which are T cell-inflamed tumours that have high levels of inflammatory cytokines and T cell infiltration and are associated with a better response to immune checkpoint blockade.
- Tertiary lymphoid structures
Organized immune cell aggregates within parenchymal tissues similar to secondary follicles in lymph nodes, comprising a T cell zone of mainly helper and follicular helper CD4+ T cells and mature dendritic cells, as well as a follicular zone with B cells and follicular dendritic cells.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.De Plaen E et al. Immunogenic (tum-) variants of mouse tumor P815: cloning of the gene of tum-antigen P91A and identification of the tum- mutation. Proc. Natl Acad. Sci. USA 85, 2274–2278 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study to identify a somatic mutation in a mouse cancer cell line variant creating a cytolytic T lymphocyte-recognized epitope using a cloning-based approach.
- 2.Mandelboim O et al. CTL induction by a tumour-associated antigen octapeptide derived from a murine lung carcinoma. Nature 369, 67–71 (1994). [DOI] [PubMed] [Google Scholar]; This study uses protein extraction and fractionation to identify a peptide epitope in a mouse lung cancer cell line containing an amino acid-altering mutation recognized by cytolytic T lymphocytes.
- 3.Monach PA, Meredith SC, Siegel CT & Schreiber H A unique tumor antigen produced by a single amino acid substitution. Immunity 2, 45–59 (1995). [DOI] [PubMed] [Google Scholar]; This study is the first to identify a mutation in a mouse cancer cell creating a tumour-specific CD4+ T cell-recognized epitope.
- 4.Coulie PG et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl Acad. Sci. USA 92, 7976–7980 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfel T et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281–1284 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Lennerz V et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl Acad. Sci. USA 102, 16013–16018 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Bruggen P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 (1991). [DOI] [PubMed] [Google Scholar]; This study is the first to show that human cancer cells express antigens recognizable by human cytolytic T lymphocytes.
- 8.Jager E et al. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc. Natl Acad. Sci. USA 97, 4760–4765 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher TN, Scheper W & Kvistborg P Cancer neoantigens. Annu. Rev. Immunol 37, 173–200 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Renkvist N, Castelli C, Robbins PF & Parmiani G A listing of human tumor antigens recognized by T cells. Cancer Immunol. Immunother 50, 3–15 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellstrom I, Hellstrom KE, Pierce GE & Yang JP Cellular and humoral immunity to different types of human neoplasms. Nature 220, 1352–1354 (1968). [DOI] [PubMed] [Google Scholar]; This study describes the paradoxical finding that tumour-reactive immune cells are found in tumours from human patients although the tumours continue to progress, now known as the Hellstrom paradox.
- 12.Hellstrom KE & Hellstrom I From the Hellstrom paradox toward cancer cure. Prog. Mol. Biol. Transl. Sci 164, 1–24 (2019). [DOI] [PubMed] [Google Scholar]
- 13.ElTanbouly MA & Noelle RJ Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat. Rev. Immunol 21, 257–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schietinger A & Greenberg PD Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 35, 51–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speiser DE, Ho PC & Verdeil G Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol 16, 599–611 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Wells AD New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J. Immunol 182, 7331–7341 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Mueller DL Mechanisms maintaining peripheral tolerance. Nat. Immunol 11, 21–27 (2010). [DOI] [PubMed] [Google Scholar]
- 18.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Baumeister SH, Freeman GJ, Dranoff G & Sharpe AH Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol 34, 539–573 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Bader JE, Voss K & Rathmell JC Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 78, 1019–1033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson KG, Stromnes IM & Greenberg PD Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henning AN, Roychoudhuri R & Restifo NP Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol 18, 340–356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Zander R, Khatun A, Schauder DM & Cui W Transcriptional and epigenetic regulation of effector and memory CD8 T cell differentiation. Front. Immunol 9, 2826 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinman RM, Hawiger D & Nussenzweig MC Tolerogenic dendritic cells. Annu. Rev. Immunol 21, 685–711 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Heath WR & Carbone FR Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol 19, 47–64 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Hernandez J, Aung S, Redmond WL & Sherman LA Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med 194, 707–717 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redmond WL, Marincek BC & Sherman LA Distinct requirements for deletion versus anergy during CD8 T cell peripheral tolerance in vivo. J. Immunol 174, 2046–2053 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Kurts C, Kosaka H, Carbone FR, Miller JF & Heath WR Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med 186, 239–245 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redmond WL & Sherman LA Peripheral tolerance of CD8 T lymphocytes. Immunity 22, 275–284 (2005). [DOI] [PubMed] [Google Scholar]; This study shows that naive T cells activated by a self-antigen undergo different fates depending on antigen dose and duration.
- 30.Schietinger A, Delrow JJ, Basom RS, Blattman JN & Greenberg PD Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335, 723–727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is one of the first to show that T cell self-tolerance is an imprinted differentiation state persisting in the absence of a self-antigen.
- 31.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang JC & Rosenberg SA Adoptive T-cell therapy for cancer. Adv. Immunol 130, 279–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baitsch L et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest 121, 2350–2360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first to perform transcriptional profiling on tumour-reactive T cells from human tumours and show exhaustion-associated programmes.
- 34.Stevanovic S et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356, 200–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pittet MJ et al. High frequencies of naive Melan-A/MART-1-specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med 190, 705–715 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi PS et al. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 65, 305–317 (1991). [DOI] [PubMed] [Google Scholar]; This is one of the first studies to describe how ignorance can be overcome, causing autoimmunity.
- 37.Overwijk WW et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med 198, 569–580 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parish IA & Heath WR Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunol. Cell Biol 86, 146–152 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Kurts C et al. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc. Natl Acad. Sci. USA 96, 12703–12707 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zippelius A et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 64, 2865–2873 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Ochsenbein AF et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl Acad. Sci. USA 96, 2233–2238 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz RH T cell anergy. Annu. Rev. Immunol 21, 305–334 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Macian F, Im SH, Garcia-Cozar FJ & Rao A T-cell anergy. Curr. Opin. Immunol 16, 209–216 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Melero I, Bach N & Chen L Costimulation, tolerance and ignorance of cytolytic T lymphocytes in immune responses to tumor antigens. Life Sci. 60, 2035–2041 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Martinez GJ et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that TCR-driven NFAT activation in the absence of its AP-1 binding partners is a key driver of T cell anergy and exhaustion.
- 46.Mognol GP et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl Acad. Sci. USA 114, E2776–E2785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is one of the first to describe epigenetic and transcriptional programmes of tumour-specific CD8+ T cells in mouse models.
- 47.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]; This study is one of the earliest to map transcriptional programmes in CD8+ T cells differentiating during acute versus chronic infection in mice and identifying hallmarks of CD8+ T cell exhaustion.
- 48.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R & Ahmed R Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol 77, 4911–4927 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frebel H et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med 209, 2485–2499 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zinselmeyer BH et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med 210, 757–774 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cornberg M et al. Clonal exhaustion as a mechanism to protect against severe immunopathology and death from an overwhelming CD8 T cell response. Front. Immunol 4, 475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz JE et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Jin X et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med 189, 991–998 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pircher H et al. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature 346, 629–633 (1990). [DOI] [PubMed] [Google Scholar]
- 55.Gabrilovich DI, Ostrand-Rosenberg S & Bronte V Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol 12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cassetta L & Pollard JW Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov 17, 887–904 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Savage PA, Malchow S & Leventhal DS Basic principles of tumor-associated regulatory T cell biology. Trends Immunol. 34, 33–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gajewski TF, Schreiber H & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spranger S & Gajewski TF Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bezzi M et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med 24, 165–175 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Tamborero D et al. A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin. Cancer Res 24, 3717–3728 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wellenstein MD & de Visser KE Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 48, 399–416 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]; This study shows that specific oncogenic mutations in cancer cells can impact immune cell recruitment and antitumour immune responses.
- 68.Joyce JA & Fearon DT T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Ochsenbein AF et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature 411, 1058–1064 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Bottcher JP et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dadi S et al. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell 164, 365–377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kang TW et al. Senescence surveillance of premalignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Eggert T et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30, 533–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frey N, Venturelli S, Zender L & Bitzer M Cellular senescence in gastrointestinal diseases: from pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol 15, 81–95 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Mascaux C et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 571, 570–575 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Spiotto MT et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity 17, 737–747 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Zinkernagel RM Immunity against solid tumors? Int. J. Cancer 93, 1–5 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Schietinger A et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses a genetic cancer mouse model and shows that CD8+ T cell dysfunction is induced early during tumorigenesis and is progressive, driven primarily by antigen exposure, and shares hallmarks with human TILs from late-stage metastatic tumours.
- 80.Giordano M et al. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 34, 2042–2058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singer M et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell 166, 1500–1511 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Azizi E et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses single-cell transcriptional and TCR profiling and shows that TILs in human melanoma are heterogeneous, with tumour-reactive T cells expressing hallmarks of dysfunction.
- 84.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses transcriptional and epigenetic profiling of melanoma-infiltrating TILs from patients who received ICB and identifies features associated with improved response.
- 85.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]; This study finds that a high number of human TILs are not in fact tumour reactive and identifies surface markers such as CD39 that enrich tumour-reactive CD8+ TILs.
- 86.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng C et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Chevrier S et al. An immune atlas of clear cell renal cell carcinoma. Cell 169, 736–749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lavin Y et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scheper W et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med 25, 89–94 (2019). [DOI] [PubMed] [Google Scholar]; This study demonstrates that the fraction of tumour-reactive T cells within a tumour is highly variable, and thus tumours with high numbers of infiltrating T cells could actually have very few tumour-reactive T cells and be quantitatively ‘hot’ but qualitatively ‘cold’ tumours.
- 91.Guo X et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Zhang L et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Savas P et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med 24, 986–993 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Zhou P et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong MB et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell 178, 1189–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patel SJ et al. Identification of essential genes for cancer immunotherapy. Nature 548, 537–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that dysfunctional tumour-specific T cells undergo epigenetic changes over time within tumours which determine therapeutic reprogramm-ability, and biomarkers can be associated with reprogrammable or non-reprogrammable T cell dysfunction states.
- 100.Thommen DS et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res 3, 1344–1355 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Minnie SA et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 132, 1675–1688 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Muller MR & Rao A NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol 10, 645–656 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the epigenetic inflexibility of the exhausted T cell population during chronic infections, limiting reinvigoration by ICB.
- 105.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with He et al. (2016), this study demonstrates that TCF1+CXCR5+ progenitor T cells in secondary lymphoid organs sustain antiviral T cell responses during chronic viral infection and are targets of PD1 therapy.
- 107.Utzschneider DT et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]; Together with Im et al. (2016), this study demonstrates that TCF1-expressing progenitor T cells give rise to the more terminally differentiated exhausted T cell pool during chronic viral infection.
- 108.Verbeek S et al. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 374, 70–74 (1995). [DOI] [PubMed] [Google Scholar]
- 109.Escobar G, Mangani D & Anderson AC T cell factor 1: a master regulator of the T cell response in disease. Sci. Immunol 5, eabb9726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gattinoni L et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jeannet G et al. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl Acad. Sci. USA 107, 9777–9782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pais Ferreira D et al. Central memory CD8+ T cells derive from stem-like Tcf7hi effector cells in the absence of cytotoxic differentiation. Immunity 53, 985–1000 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Lin WW et al. CD8+ T lymphocyte self-renewal during effector cell determination. Cell Rep. 17, 1773–1782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that in the initial cell divisions following T cell activation, the level of TCF1 expression determines whether CD8+ T cells become memory T cells or adopt a terminally differentiated effector fate.
- 114.He R et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–428 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Wu T et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1, eaai8593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wieland D et al. TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun 8, 15050 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leong YA et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol 17, 1187–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 118.Beltra JC et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first to describe the heterogeneity of the exhausted T cell pool consisting of progenitor and terminally differentiated exhausted T cells.
- 120.Yao C et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol 20, 890–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Borsa M et al. Modulation of asymmetric cell division as a mechanism to boost CD8+ T cell memory. Sci. Immunol 4, eaav1730 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that TOX is a key transcription factor driving dysfunctional programmes in tumour-specific T cells, and that the exhaustion phenotype is uncoupled from the loss of effector function.
- 123.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]; Together with Scott et al. (2019) and Khan et al. (2019), this study shows the importance of TOX in regulating exhaustion programmes during chronic viral infection, although TOX is not required for functional CD8+ T cell differentiation.
- 124.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.O’Flaherty E & Kaye J TOX defines a conserved subfamily of HMG-box proteins. BMC Genomics 4, 13 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aliahmad P & Kaye J Development of all CD4 T lineages requires nuclear factor TOX. J. Exp. Med 205, 245–256 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aliahmad P, Seksenyan A & Kaye J The many roles of TOX in the immune system. Curr. Opin. Immunol 24, 173–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Seehus CR et al. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat. Immunol 16, 599–608 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Page N et al. Expression of the DNA-binding factor TOX promotes the encephalitogenic potential of microbe-induced autoreactive CD8+ T cells. Immunity 48, 937–950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang X et al. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol 71, 731–741 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Seo H et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl Acad. Sci. USA 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heim K et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 10.1136/gutjnl-2020-322404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Z et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol 4, eaaw1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hombrink P et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol 17, 1467–1478 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Kurd NS et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 5, eaaz6894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sekine T et al. TOX is expressed by exhausted and polyfunctional human effector memory CD8+ T cells. Sci. Immunol 5, eaba7918 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Alegre ML et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol 157, 4762–4770 (1996). [PubMed] [Google Scholar]
- 138.Nagaraj S et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med 13, 828–835 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kammertoens T et al. Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature 545, 98–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Listopad JJ et al. Fas expression by tumor stroma is required for cancer eradication. Proc. Natl Acad. Sci. USA 110, 2276–2281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Engels B, Rowley DA & Schreiber H Targeting stroma to treat cancers. Semin. Cancer Biol 22, 41–49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang B et al. Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res. 68, 1563–1571 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Zhang B et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J. Exp. Med 204, 49–55 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Martinez-Lostao L, Anel A & Pardo J How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res 21, 5047–5056 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Abd Hamid M et al. Defective interferon gamma production by tumor-specific CD8+ T cells is associated with 5’methylcytosine-guanine hypermethylation of interferon gamma promoter. Front. Immunol 11, 310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Salerno F, Turner M & Wolkers MC Dynamic post-transcriptional events governing CD8+ T cell homeostasis and effector function. Trends Immunol. 41, 240–254 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Salerno F & Wolkers MC T-cells require post-transcriptional regulation for accurate immune responses. Biochem. Soc. Trans 43, 1201–1207 (2015). [DOI] [PubMed] [Google Scholar]
- 148.Salerno F et al. Critical role of post-transcriptional regulation for IFN-gamma in tumor-infiltrating T cells. Oncoimmunology 8, e1532762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates the importance of post-transcriptional regulation of IFNγ in TILs, explaining why high levels of Ifng mRNA found in exhausted/dysfunctional T cells do not correlate with IFNγ expression.
- 149.Villarino AV et al. Posttranscriptional silencing of effector cytokine mRNA underlies the anergic phenotype of self-reactive T cells. Immunity 34, 50–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an early study showing cytokine mRNA translation regulation by 3′ untranslated region (A+U)-rich elements, suggesting a mechanism for loss of effector function in anergic T cells subsequently also shown by Salerno et al. (2019) in TILs.
- 150.Zelinskyy G et al. CD8+ T-cell dysfunction due to cytolytic granule deficiency in persistent Friend retrovirus infection. J. Virol 79, 10619–10626 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Radoja S et al. CD8+ tumor-infiltrating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J. Immunol 167, 5042–5051 (2001). [DOI] [PubMed] [Google Scholar]
- 152.Berg NN, Puente LG, Dawicki W & Ostergaard HL Sustained TCR signaling is required for mitogen-activated protein kinase activation and degranulation by cytotoxic T lymphocytes. J. Immunol 161, 2919–2924 (1998). [PubMed] [Google Scholar]
- 153.Wei S et al. Control of lytic function by mitogen-activated protein kinase/extracellular regulatory kinase 2 (ERK2) in a human natural killer cell line: identification of perforin and granzyme B mobilization by functional ERK2. J. Exp. Med 187, 1753–1765 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Radoja S & Frey AB Cancer-induced defective cytotoxic T lymphocyte effector function: another mechanism how antigenic tumors escape immune-mediated killing. Mol. Med 6, 465–479 (2000). [PMC free article] [PubMed] [Google Scholar]
- 155.Macian F et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731 (2002). [DOI] [PubMed] [Google Scholar]
- 156.Schenkel JM et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Molodtsov A & Turk MJ Tissue resident CD8 memory T cell responses in cancer and autoimmunity. Front. Immunol 9, 2810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gebhardt T, Palendira U, Tscharke DC & Bedoui S Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev 283, 54–76 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 160.Milner JJ et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Milner JJ et al. Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity 52, 808–824 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Edwards J et al. CD103+ tumor-resident CD8+T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin. Cancer Res 24, 3036–3045 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Park SL et al. Tissue-resident memory CD8+ T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371 (2019). [DOI] [PubMed] [Google Scholar]
- 164.Malik BT et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci. Immunol 2, eaam6346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Harjunpaa H, Llort Asens M, Guenther C & Fagerholm SC Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol 10, 1078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fridman WH, Zitvogel L, Sautes-Fridman C & Kroemer G The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol 14, 717–734 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Gros A et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest 124, 2246–2259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pasetto A et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol. Res 4, 734–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Inozume T et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother 33, 956–964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Thommen DS et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Duhen T et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun 9, 2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Simoni et al. (2018), this study shows that CD39 can be used to enrich tumour-reactive CD8+ T cells in human tumours.
- 172.Erkes DA et al. Virus-specific CD8+ T cells infiltrate melanoma lesions and retain function independently of PD-1 expression. J. Immunol 198, 2979–2988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bos R, Marquardt KL, Cheung J & Sherman LA Functional differences between low- and high-affinity CD8+ T cells in the tumor environment. Oncoimmunology 1, 1239–1247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lyman MA et al. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J. Immunol 174, 2563–2572 (2005). [DOI] [PubMed] [Google Scholar]
- 175.Schmid DA et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J. Immunol 184, 4936–4946 (2010). [DOI] [PubMed] [Google Scholar]
- 176.Sette A et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol 153, 5586–5592 (1994). [PubMed] [Google Scholar]
- 177.Zhong S et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl Acad. Sci. USA 110, 6973–6978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Engels B et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 23, 516–526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fritsch EF et al. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res 2, 522–529 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hoffmann MM & Slansky JE T-cell receptor affinity in the age of cancer immunotherapy. Mol. Carcinog 59, 862–870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stone JD, Harris DT & Kranz DM TCR affinity for p/MHC formed by tumor antigens that are self-proteins: impact on efficacy and toxicity. Curr. Opin. Immunol 33, 16–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Blankenstein T, Leisegang M, Uckert W & Schreiber H Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol 33, 112–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dunn GP, Old LJ & Schreiber RD The three Es of cancer immunoediting. Annu. Rev. Immunol 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 184.Sautes-Fridman C, Petitprez F, Calderaro J & Fridman WH Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019). [DOI] [PubMed] [Google Scholar]
- 185.Petitprez F et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020). [DOI] [PubMed] [Google Scholar]
- 186.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cabrita R et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 188.Jansen CS et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that in human kidney cancers TCF1+CD8+ T cells reside in dense APC niches, whereas clonally related, more differentiated CD8+ T cells are found outside these intratumoural niches.
- 189.Haslam A & Prasad V Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2, e192535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kalbasi A & Ribas A Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol 20, 25–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fares CM, Van Allen EM, Drake CG, Allison JP & Hu-Lieskovan S Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am. Soc. Clin. Oncol. Educ. Book 39, 147–164 (2019). [DOI] [PubMed] [Google Scholar]
- 192.Fridman WH, Pages F, Sautes-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 193.Galon J et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006). [DOI] [PubMed] [Google Scholar]; This is one of the earliest studies to show the correlation between immune cell infiltration in human tumours and clinical outcomes.
- 194.Yarchoan M, Hopkins A & Jaffee EM Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med 377, 2500–2501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Herbst RS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schoenfeld AJ et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann. Oncol 31, 599–608 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Havel JJ, Chowell D & Chan TA The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wu TD et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020). [DOI] [PubMed] [Google Scholar]
- 201.Brummelman J et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med 215, 2520–2535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Siddiqui I et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211 (2019). [DOI] [PubMed] [Google Scholar]
- 203.Kurtulus S et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1-CD8+ tumor-infiltrating T cells. Immunity 50, 181–194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses paired single-cell transcriptomic and TCR analysis to show that rather than intratumoural T cells, novel clonotypes from the periphery preferentially expand and infiltrate into tumours in response to immune checkpoint therapy.
- 205.Dammeijer F et al. The PD-1/PD-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell 38, 685–700.e8 (2020). [DOI] [PubMed] [Google Scholar]; This study shows that in a mouse tumour model, PD1 or PDL1 immune checkpoint therapy expanded progenitor T cells in TDLNs rather than TILs, and that these progenitor T cells then infiltrated tumours.
- 206.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gros A et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med 22, 433–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cohen CJ et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest 125, 3981–3991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gros A et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest 129, 4992–5004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sharma P & Allison JP Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol 20, 75–76 (2020). [DOI] [PubMed] [Google Scholar]
- 213.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 214.Ott PA et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Romero P et al. The human vaccines project: a roadmap for cancer vaccine development. Sci. Transl. Med 8, 334ps9 (2016). [DOI] [PubMed] [Google Scholar]
- 216.Leko V & Rosenberg SA Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 38, 454–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR & Greenberg PD Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol. Rev 257, 145–164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schoenfeld AJ & Hellmann MD Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schurch CM et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Allam M, Cai S & Coskun AF Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precis. Oncol 4, 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sun C et al. Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl Acad. Sci. USA 116, 52–57 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rozenblatt-Rosen O et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Thornton CA et al. Spatially mapped single-cell chromatin accessibility. Nat. Comms 12, 1274 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gonzalez-Gugel E, Saxena M & Bhardwaj N Modulation of innate immunity in the tumor microenvironment. Cancer Immunol. Immunother 65, 1261–1268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Russell SJ & Barber GN Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell 33, 599–605 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Flood BA, Higgs EF, Li S, Luke JJ & Gajewski TF STING pathway agonism as a cancer therapeutic. Immunol. Rev 290, 24–38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhou T et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ochoa de Olza M, Navarro Rodrigo B, Zimmermann S & Coukos G Turning up the heat on non-immunoreactive tumours: opportunities for clinical development. Lancet Oncol. 21, e419–e430 (2020). [DOI] [PubMed] [Google Scholar]
- 230.Galluzzi L, Humeau J, Buque A, Zitvogel L & Kroemer G Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol 17, 725–741 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Zou W, Wolchok JD & Chen L PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med 8, 328rv324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Topper MJ, Vaz M, Marrone KA, Brahmer JR & Baylin SB The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol 17, 75–90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chen J et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Liu X et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lynn RC et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Roth TL et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell 181, 728–744 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wei J et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Smyth MJ, Dunn GP & Schreiber RD Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol 90, 1–50 (2006). [DOI] [PubMed] [Google Scholar]
- 240.Engels EA Epidemiologic perspectives on immuno-suppressed populations and the immunosurveillance and immunocontainment of cancer. Am. J. Transpl 19, 3223–3232 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dranoff G Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat. Rev. Immunol 12, 61–66 (2011). [DOI] [PubMed] [Google Scholar]
- 242.Zitvogel L, Pitt JM, Daillere R, Smyth MJ & Kroemer G Mouse models in oncoimmunology. Nat. Rev. Cancer 16, 759–773 (2016). [DOI] [PubMed] [Google Scholar]
- 243.Yang W et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med 25, 767–775 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Tabachnick-Cherny S, Pulliam T, Church C, Koelle DM & Nghiem P Polyomavirus-driven Merkel cell carcinoma: Prospects for therapeutic vaccine development. Mol. Carcinog 59, 807–821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Heemskerk B, Kvistborg P & Schumacher TN The cancer antigenome. EMBO J. 32, 194–203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 247.Blankenstein T Do autochthonous tumors interfere with effector T cell responses? Semin. Cancer Biol 17, 267–274 (2007). [DOI] [PubMed] [Google Scholar]
- 248.Willimsky G & Blankenstein T Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 437, 141–146 (2005). [DOI] [PubMed] [Google Scholar]; This study demonstrates in an autochthonous sporadic cancer mouse model that tumour-specific CD8+ T cells become hyporesponsive early during tumorigenesis.
- 249.Willimsky G et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J. Exp. Med 205, 1687–1700 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shankaran V et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111 (2001). [DOI] [PubMed] [Google Scholar]
- 251.Koebel CM et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]; Together with Shankaran et al. (2001), this study demonstrates that CD8+ T cells contribute to cancer immunosurveillance in methylcholanthrene-induced tumour mouse models.
- 252.Kammertoens T, Qin Z, Briesemeister D, Bendelac A & Blankenstein T B-cells and IL-4 promote methylcholanthrene-induced carcinogenesis but there is no evidence for a role of T/NKT-cells and their effector molecules (Fas-ligand, TNF-alpha, perforin). Int. J. Cancer 131, 1499–1508 (2012). [DOI] [PubMed] [Google Scholar]
- 253.Rosenthal R et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Van den Eynden J, Jimenez-Sanchez A, Miller ML & Larsson E Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet 51, 1741–1748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]