

Abstract
Glyoxalase 2 is the second enzyme of the glyoxalase system, catalyzing the detoxification of methylglyoxal to d-lactate via SD-Lactoylglutathione. Recent in vitro studies have suggested Glo2 as a regulator of glycolysis, but if Glo2 regulates glucose homeostasis and related organ specific functions in vivo has not yet been evaluated. Therefore, a CRISPR-Cas9 knockout of glo2 in zebrafish was created and analyzed. Consistent with its function in methylglyoxal detoxification, SD-Lactoylglutathione, but not methylglyoxal accumulated in glo2−/− larvae, without altering the glutathione metabolism or affecting longevity. Adult glo2−/− livers displayed a reduced hexose concentration and a reduced postprandial P70–S6 kinase activation, but upstream postprandial AKT phosphorylation remained unchanged. In contrast, glo2−/− skeletal muscle remained metabolically intact, possibly compensating for the dysfunctional liver through increased glucose uptake and glycolytic activity. glo2−/− zebrafish maintained euglycemia and showed no damage of the retinal vasculature, kidney, liver and skeletal muscle. In conclusion, the data identified Glo2 as a regulator of cellular energy metabolism in liver and skeletal muscle, but the redox state and reactive metabolite accumulation were not affected by the loss of Glo2.
Keywords: Glyoxalase 2, Methylglyoxal, SD-Lactoylglutathione, P70–S6K, Zebrafish
Graphical abstract
Highlights
-
•
Methylglyoxal-dependent SD-Lactoylglutathione accumulation in glo2−/− zebrafish.
-
•
Glucose metabolism in liver and skeletal muscle are regulated by Glo2.
-
•
Glo2−/− animals show reduced hepatic P70–S6 kinase activation.
-
•
Glo2 function does not significantly influence the redox state.
-
•
Loss of Glo2 does not cause vascular damage in eyes or kidney.
1. Introduction
The glyoxalase system is located in the cytosol of most organisms [1] and comprised of two enzymes, namely Glyoxalase 1 (Glo1) [[2], [3], [4]] and Glyoxalase 2 (Glo2) [4]. Its primary function is the detoxification of methylglyoxal (MG) [2,5]. MG is a reactive carbonyl species (RCS) [6] and a spontaneously formed metabolite in different metabolic pathways, including glycolysis [1,7,8]. It causes the formation of advanced glycation end products (AGEs) [[9], [10], [11]] and directly contributes to the development of end-organ damage in various diseases, including diabetes mellitus [1,12]. Glo1 has intensively been studied and was identified as the rate-limiting enzyme of the glyoxalase system [1]. It catabolizes the first step of MG detoxification by converting MG to SD-Lactoylglutathione (SDL) [[13], [14], [15], [16]], a most likely non-toxic compound [17,18], with the support of the coenzyme glutathione [3,4,19]. Surprisingly, recent glo1 knockout studies in flies [20], zebrafish [21] and mice [22,23] have shown only a 50% increase of MG in the different glo1 knockout animals and mild diabetic organ alterations have been identified in flies [20] and zebrafish only [21]. Glo2 as the second enzyme of the glyoxalase system has sparsely been studied. Glo2 further degrades SDL generated by Glo1 to d-lactate and restores glutathione [16]. It has two isoforms, which are located in the cytosol and mitochondria of most vertebrates [16,24], respectively. While cytosolic Glo2 enables MG detoxification [1], the relevance of the mitochondrial Glo2 isoform is mostly unknown. As Glo2 restores glutathione, it has been suggested that it could be critical for the cellular redox status in general, but no additional data could validate this hypothesis so far [[25], [26], [27], [28]].
Some evidence has recently suggested a protective role for Glo2 in preventing S-acetylation of mitochondrial proteins when acetyl Co-A accumulates [29]. Additional studies suggested that Glo2 could be a novel regulatory factor of glycolysis [30] and protect the cellular mitochondrial energy metabolism. Furthermore, some reports in cancer research have proposed that Glo2 could be an independent driver of prostate cancer development [31], acting through mechanisms involving AKT/mTOR signaling pathways. However, no specific in vivo data on the regulation of AKT/mTOR by Glo2 has been published. Recently, glo2 knockout mice have been generated but, these mice, similar to the glo1 knockout mice [22,23], did not show any obvious organ alterations. The report on the glo2 knockout mice also failed to investigate and identify a Glo2-related metabolic function and if glucose homeostasis and glucose driven metabolic pathways are altered in the mutant [32].
Since zebrafish is more susceptible to the loss of Glo1 than mice [[21], [22], [23]] and therefore provides better mechanistic insights into its function, we have created a zebrafish knockout line for Glo2 to determine the physiological functions of Glo2, its effect on the cellular redox status and to address the question whether Glo2 regulates the cellular energy metabolism and glucose homeostasis in zebrafish. Our data could identify that glo2 knockout zebrafish developed reduced hexose concentrations in the liver accompanied by reduced P70–S6 kinase phosphorylation, and strong increases in glycolytic activity in skeletal muscle as a possible compensation mechanism, while the cellular redox status was unaffected.
2. Results
2.1. Expression of glo2 and generation of glo2−/− knockout zebrafish
A comparison of the amino-acid sequence of the glo2 gene across zebrafish, human and mouse showed high similarities of the sequences across the three species, including in the three domains representing the substrate binding site (Fig. 1A). Except for a change from tyrosine in human and mouse to phenylalanine in zebrafish in position 188 and a change from arginine to lysine in position 293, they are identical, thereby confirming the suitability of zebrafish as an animal model for the study of Glo2′s function. Expression analyses of glo2 using RT-qPCR showed a 4.6-fold increase of glo2 expression compared to b2m over the first 120 h of zebrafish development (Fig. 1B). Complementary to expression studies, activity measurements in adult zebrafish revealed that the activity of Glo2 is strongest in liver, with 4.65 mU/mg of tissue more than twice as much activity as the kidney with 2.15 mU/mg, whereas skeletal muscle showed almost no organ specific Glo2 activity (Fig. 1C), thereby suggesting the liver as the most important organ in the analysis of Glo2′s function. As there is very little in vivo data on the physiological functions of Glo2 as well as its role in the glyoxalase system and its importance for metabolic diseases, a knockout zebrafish line of glo2 using CRISPR-Cas9 was created. The gRNA was designed for a target site in exon 2 of the glo2 gene and injected together with a Cas9-mRNA in Tg(wt1b:EGFP) zebrafish embryos. The resulting F0 positive mosaic mutants were crossed with Tg(wt1b:EGFP) wildtype zebrafish. An animal with a 49 base pair deletion (Δ 49) in the target site, resulting in a frame shift, was identified and chosen to establish the line used in the following experiments (Fig. 2A). To confirm that the selected knockout resulted in a loss of the Glo2 protein, a Western blot analysis of Glo2 was conducted and confirmed the partial loss of Glo2 in glo2+/− mutants and the complete loss of Glo2 in glo2−/− adult zebrafish (Fig. 2B). Glo2 activity measurements further supported these findings, demonstrating a partial and complete loss of the entire Glo2 activity in glo2+/− and glo2−/− animals, respectively (Fig. 2C). The gross morphology of 96 hpf glo2−/− larvae appeared unchanged (Fig. 2D) and adult glo2−/− zebrafish were found in the expected Mendelian distribution (Fig. 2E), they had the same body weight (Fig. 2F), length (Fig. 2G) and gross morphology (data not shown) as their glo2+/+ littermates, indicating no adversity to survival based on the knockout of glo2.
Fig. 1.

Alignment of the glo2 sequence across different species, expression patterns and organ specific activity of Glo2 in zebrafish. A) Alignment of the amino acid sequences of Glo2 in zebrafish (line 1), human (line 2) and mouse (line 3). The 3 domains making up the substrate binding site are highlighted in orange and show great similarities across species. Created with UniProt alignment tool (Access: 12/08/2021). B) Wildtype zebrafish embryos and larvae showed an increasing expression of glo2 mRNA over the first five days of development. Measured using RT-qPCR, compared to b2m. One datapoint represents one clutch of 50 larvae. N = 3. C) Adult wildtype zebrafish showed a very strong activity of Glo2 in the liver, followed by kidney and brain, with little relative activity in skeletal muscle. SD-Lactoylglutathione was used as substrate, activity was measured spectrophotometrically. N = 6. One datapoint represents organ-tissue from one single zebrafish. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 2.

Generation and validation of glo2−/−zebrafish using CRISPR-Cas9. A) The target site (highlighted in yellow) and PAM site (highlighted in green) for CRISPR-Cas9 were selected in exon 2 of the glo2 gene. The gene-segment containing the target site was amplified with PCR and analyzed with Sanger sequencing. Subsequent genotyping was conducted using PCR, followed by electrophoresis of the PCR product in an agarose gel. Zebrafish with a 49 base pair deletion were selected for the generation of the glo2−/− line. B) The Western blot analysis using a Glo2-specific antibody showed a complete loss of Glo2 in adult glo2−/− zebrafish and a partial loss of Glo2 in glo2+/− zebrafish. Samples were derived from homogenized adult zebrafish tissue, β-Actin was used as loading control. Original images have been cropped. C) Glo2 activity measurements showed a 92 % loss of activity from glo2+/+ to glo2−/− samples and a 47 % loss from glo2+/+ to glo2+/− zebrafish. Tissue was derived from homogenized adult zebrafish. SD-Lactoylglutathione was used as substrate, activity was measured spectrophotometrically. One datapoint represents tissue from one zebrafish. N = 2/2/1. D) The gross morphology of glo2−/− larvae appeared unchanged at 96 hpf upon analysis with a light microscope. Scale bar = 500 μm. E) The distribution of genotypes in heterozygous matings showed the expected distribution and no significant loss of glo2−/− zebrafish, indicating normal survival. F) Adult glo2−/− zebrafish showed a normal body weight compared to glo2+/+ zebrafish. One datapoint represents one zebrafish. N = 10. G) Adult glo2−/− zebrafish showed a normal body length compared to glo2+/+ zebrafish. One datapoint represents one zebrafish. N = 10. For statistical analysis chi-square test, (E) and normality tests (F, G) were used, followed by unpaired t-test (F), or Mann-Whitney test (G). Data presented as mean ± SD. ns = p > 0.05, UTR: untranslated region, E: exon, WT: wildtype, kD: kilodalton. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.2. glo2−/− zebrafish showed an increase in SD-Lactoylglutathione after methylglyoxal incubation
As the second enzyme in the glyoxalase system, Glo2 degrades SDL to d-lactate and glutathione [14,16]. To determine whether a similar function can be observed in zebrafish in vivo, we have measured SDL at 120 hpf and found a significant increase of SDL in glo2−/− larvae after methylglyoxal incubation, compared to glo2+/+ larvae (Fig. 3A). The endogenous accumulation and uptake of methylglyoxal were unchanged in glo2−/− zebrafish compared to glo2+/+, as was evident in the baseline measurements of methylglyoxal before and after incubation with methylglyoxal (Fig. 3B). Measurements of glyoxal and 3-deoxyglucosone, both as baseline and after incubation with methylglyoxal, were also unchanged compared to wildtype animals, thereby confirming that the glo2−/− zebrafish have a specific increase of SDL, but not an increase of reactive carbonyl species in general (Fig. 3C and D). Thus, the data demonstrate for the first time that Glo2 contributes to the degradation of SDL in vivo and that the degradation capacity is dependent on the tissue concentration of methylglyoxal. As the increase in SDL did not result in any measurable change in survival rates and development of glo2−/− larvae as shown in Fig. 2D and E, these data furthermore suggest that SDL is most likely non-toxic in physiological concentrations even upon long-term exposure, as had long been postulated [17,18].
Fig. 3.

glo2−/−zebrafish showed an increase in SD-Lactoylglutathione after incubation in methylglyoxal. A) Incubation of the larvae in 500 μM MG caused a tenfold increase of SDL, with a more pronounced increase of SDL in glo2−/− larvae compared to glo2+/+ larvae, (B) but showed no increase in baseline MG. Incubation with 500 μM MG caused an equal increase of MG in glo2−/− and glo2+/+ larvae. C) The levels of glyoxal were unchanged in glo2−/− larvae and were uninfluenced by incubation in 500 μM MG. D) The levels of 3-deoxyglucosone were unchanged in glo2−/− larvae and were uninfluenced by incubation in 500 μM MG. Measurements were conducted using LC-MS/MS. One datapoint represents one clutch of 46 larvae, collected at 120 hpf. For statistical analysis normality tests were used for all samples, followed by unpaired t-test (A, B, C, D), or Mann-Whitney test (A, C, D). Data presented as mean ± SD. N = 5. ns = p > 0.05, ** = p < 0.01, **** = p < 0.0001. SDL: SD-Lactoylglutathione, MG: methylglyoxal, 3-DG: 3-deoxyglucosone, LC-MS: liquid chromatography/mass spectrometry.
2.3. The redox status of larvae and the adult liver were unaffected by the loss of Glo2
Glo2 restores glutathione as a part of its substrate SD-Lactoylglutathione and has thus been suggested to affect the redox capabilities of cells. Therefore, we have measured glutathione and glutathione-disulfide with and without addition of external methylglyoxal to determine if the increase of SDL, as observed in Fig. 3A, transferred to other glutathione-species. Both glutathione (Fig. 4A) and glutathione-disulfide (Fig. 4B) were unchanged in glo2−/− larvae. Therefore, neither the loss of Glo2 nor the incubation with methylglyoxal affected the glutathione metabolism significantly. Additionally, the NADH/NAD (Fig. 4C) and NADPH/NADP (Fig. 4D) ratios in the adult liver of glo2−/− zebrafish, as well as the determination of various compounds related to cellular redox activity, including FAD (Fig. 4E), nicotinamide (Fig. 4F), ADPR (Fig. 4G), AMP (Fig. 4H) and GMP (Fig. 4I), were unaffected by the loss of Glo2 function. Thus, the loss of Glo2 activity does not significantly affect cellular glutathione concentrations or the redox capabilities of glo2−/− animals, both in larval and adult stages.
Fig. 4.

The redox status of larvae and the adult liver was unaffected by the loss of Glo2. A) The levels of glutathione remained unchanged in glo2−/− zebrafish under normal conditions as well as after incubation in 500 μM MG. Measurements were conducted using LC-MS/MS. One datapoint represents one clutch of 46 larvae, collected at 120 hpf. B) The levels of glutathione-disulfide remained unchanged in glo2−/− zebrafish under normal conditions as well as after incubation in 500 μM of MG. Measurements were conducted using LC-MS/MS. One datapoint represents one clutch of 46 larvae, collected at 120 hpf. C) NADH/NAD ratios and NADPH/NADP (D) ratios were normal in glo2−/− animal livers, indicating no increased redox stress. Liver FAD (E), nicotinamide (F), ADPR (G), AMP (H) and GMP (I) were unchanged in glo2−/− animals compared to glo2+/+ animals. Measured via GC-MS. One datapoint represents the liver of one adult zebrafish. N = 5. For statistical analysis normality tests were used for all samples, followed by unpaired t-test (A, B, C, D, E, F, G), or Mann-Whitney test (A, H, I). Data presented as mean ± SD. ns = p > 0.05. FAD: Flavin adenine dinucleotide, GSH: Glutathione, GSSG: Glutathione-disulfide, ADPR: adenosine diphosphate ribose, AMP: adenosine monophosphate, GMP: guanosine monophosphate, LC-MS: liquid chromatography/mass spectrometry, GC-MS: gas chromatography/mass spectrometry.
2.4. Adult glo2−/− livers showed reduced hexose concentrations
Since the liver showed the highest activity of Glo2 (Fig. 1C) and in vitro studies had suggested a link between the loss of glo2 and changes in glycolytic activity [30], we have investigated the glucose metabolism in the livers of adult glo2−/− zebrafish next. Thus, we have performed a semi-targeted metabolome analysis as a screening tool to determine whether the loss of Glo2 alters the liver's metabolism. It showed that adult glo2−/− livers exhibit a 41 % reduced hexose concentration compared to glo2+/+ zebrafish (Fig. 5A). The hexose pool of the liver is primarily comprised of glucose, indicating that the liver glucose content is significantly reduced in glo2−/− zebrafish. Interestingly, the expression of the glucose transporters of the glut family remained unchanged, except for a downregulation of the fructose transporter glut5 (Suppl. Fig. 1), as did the expression of the three rate limiting enzymes of glycolysis (Suppl. Fig. 2 A–C). Yet, the reduced concentration of hexoses in the glo2−/− liver was accompanied by an increase of glucokinase activity from 0.08 mU/mg to 0.13 mU/mg (Fig. 5B) when measured 2 h postprandially, but not by changes in both phosphofructokinase (Fig. 5C) and pyruvate kinase activity (Fig. 5D). To determine whether the reduced hexose content of the liver was the result of diminished gluconeogenesis, cPEPCK (Suppl. Fig. 2D) and g6pase (Suppl. Fig. 2E) expression was measured and found unaltered in glo2−/− livers. Beyond the effect of the loss of Glo2 on glucose metabolism, analysis of RNA-sequencing data, based on the KEGG (Kyoto encyclopedia of genes and genomes) pathways, revealed that fatty acid degradation and metabolism were significantly downregulated in glo2−/− samples, with NES scores of −2.12 and −1.92, respectively (Suppl. Fig. 3), suggesting that the regulatory mechanisms proposed for Glo2 might not only affect glucose metabolism but extend to fatty acid metabolism as well. These data further suggest the reduced fatty acid metabolism as a possible influencing factor resulting in the reduced hexose content of the liver. In conclusion, the data show a strong reduction of hexoses in glo2−/− liver, but key enzyme expression and activity measurements of both glycolysis and gluconeogenesis revealed only slight changes in glycolytic and no changes in gluconeogenetic processes as a direct result of Glo2 loss.
Fig. 5.

Adult glo2−/−liver showed a reduced concentration of hexoses. A) GC-MS revealed a reduced hexose content of the glo2−/− liver. N = 4. B) Glucokinase showed increased postprandial activity in glo2−/− liver tissue. N = 6. C) The activity of phosphofructokinase remained unaltered in the postprandial state in glo2−/− zebrafish. N = 6. D) The activity of pyruvate kinase remained unaltered in the postprandial state in glo2−/− zebrafish. N = 6. One datapoint represents the liver of one zebrafish. For statistical analysis normality tests were used for all samples, followed by unpaired t-test (A, C), or Mann-Whitney test (B, D). Data presented as mean ± SD. ns = p > 0.05, * = p < 0.05, ** = p < 0.01. GC-MS: gas chromatography/mass spectrometry, GCK: glucokinase, PFK: phosphofructokinase, PK: pyruvate kinase.
2.5. glo2−/− zebrafish showed a reduction of liver P70–S6 kinase activation while maintaining euglycemia and unchanged AKT phosphorylation
Since the glo2−/− liver had shown alterations in the glucose metabolism and reduced concentrations of hexoses, we have proceeded to analyze the liver insulin signaling cascade and its effect in glo2−/− animals. First, the phosphorylation of AKT was measured, which acts downstream of insulin receptor, but upstream of P70–S6 kinase [[33], [34], [35]]. The data revealed that AKT phosphorylation was reduced in a fasting state when insulin signaling is low, but strongly upregulated in a postprandial state as a response to increased insulin signaling (Suppl. Fig. 4A–D). Therefore, postprandial AKT activation can be considered as a surrogate marker for insulin signaling intensity. Subsequently, adult glo2−/− animals were fed and liver AKT activation analyzed postprandially, but found unchanged as compared to glo2+/+ livers (Fig. 6A, B, C). Similarly, the expression of insra and insrb, the two Insulin receptor variants found in zebrafish [36,37], were unaffected (Fig. 6D). Additionally, the measurements of both fasting as well as 1, 2 and 3 h postprandial blood glucose measurements were unchanged in glo2−/− animals (Fig. 6E). Most surprisingly, postprandial P70–S6 kinase phosphorylation in the liver of adult glo2−/− animals was determined and found reduced by 70 % compared to glo2+/+ zebrafish (Fig. 6F). Additionally, a GC-MS measurement of hepatic myo-inositol, which has previously been linked to the regulation of glucose uptake into skeletal muscle [38], showed an upregulation in glo2−/− livers (Fig. 6G). In summary, glo2−/− zebrafish showed a strong downregulation of postprandial P70–S6 kinase phosphorylation as a consequence of the loss of Glo2. Remarkably, the insulin signaling cascade upstream of P70–S6 kinase remained unaffected and glo2−/− fish maintained euglycemia. Therefore, Glo2 does not act as an independent regulator of insulin signaling, or AKT activity as in vitro studies had previously suggested [31]. Yet, Glo2 was identified as a novel regulator of liver glucose metabolism and P70–S6 kinase activity, which can be regulated independently of AKT.
Fig. 6.

glo2−/−zebrafish showed a reduction of liver P70–S6 kinase activation while maintaining euglycemia and unchanged AKT phosphorylation.A) The quantification of the pAKT Western blot in glo2−/− livers showed no change in phosphorylation in the postprandial liver compared to the liver of wildtype zebrafish, indicating intact insulin signaling in glo2−/− livers. B) The total amount of AKT was unchanged in glo2−/− and glo2+/+ zebrafish liver. C) The quantification of the housekeeping gene β-actin showed no significant difference in signal volume, indicating equal loading in all samples. D) The expression of insulin receptor A/B mRNA remained unaltered in glo2−/− zebrafish. Measured using RT-qPCR, normalized to b2m. N = 4. E)glo2−/− and glo2+/+ zebrafish showed similar blood glucose kinetics from fasting to 3 h postprandial state. N = 3-5 adult zebrafish. F)glo2−/− zebrafish showed a reduction of the phosphorylation of P70–S6 kinase. N = 3/4. G) The relative amount of myo-inositol was increased in glo2−/− zebrafish. Determined through GC/MS. N = 4. One datapoint represents one zebrafish. N = 3/4. For statistical analysis normality tests were used (A, B, C, D, E, F, G), followed by unpaired t-tests (A, B, C, D, E, F, G). Data presented as mean ± SD. ns = p > 0.05, * = p < 0.05. insr: insulin receptor, pAKT: phosphorylated AKT, tAKT: total AKT, GC-MS: gas chromatography/mass spectrometry.
2.6. Adult glo2−/− skeletal muscle showed an increase in uptake of hexoses and activity of glycolysis and TCA cycle
In contrast to the glo2−/− livers, which had revealed reduced hexose content and only a slight increase in glycolytic activity, glo2−/− skeletal muscle showed an opposite regulation. Not only hexoses but various other metabolites of glycolysis were found to be strongly upregulated in the glo2−/− skeletal muscle, including G6P, F6P and lactate (Fig. 7A). The increase was not limited to glycolysis but also extended to various metabolites of the TCA cycle, namely succinic, fumaric, and malic acid (Fig. 7B), which were also strongly upregulated, as determined via GC-MS. The increase of glycolytic activity was further confirmed by activity measurements of hexokinase (Fig. 7C), phosphofructokinase (Fig. 7D) and pyruvate kinase (Fig. 7E), where pyruvate kinase activity was increased by 26 % in glo2−/− zebrafish indicating an increase in glycolytic activity. Gluconeogenesis activation, as determined by expression analyses of mPECK [39] (Fig. 7F), revealed a reduction in glo2−/− skeletal muscle, proving that the increased hexose concentration is most likely the result of increased uptake of blood glucose, and not the result of gluconeogenesis. Despite the strong increase in hexose uptake and glycolytic activity, the expression patterns of glucose transporters were unchanged (Suppl. Fig. 5). In similar fashion, the expression patterns of the rate limiting enzymes of glycolysis hexokinase (Suppl. Fig. 6A), phosphofructokinase (Suppl. Fig. 6B) and pyruvate kinase (Suppl. Fig. 6C) were unaffected by the loss of glo2−/− in skeletal muscle, as was g6pase (Suppl. Fig. 6D), the remaining rate limiting enzyme of gluconeogenesis. Additionally, the expression of the insulin receptors in the skeletal muscle of glo2−/− animals (Fig. 7G) revealed no apparent changes, similar to the unchanged expression of insulin receptors observed in the liver (Fig. 6D). In summary, glo2−/− skeletal muscle shows strong alterations to its energy metabolism, apparent in its increase in uptake of hexoses as well as glycolytic and TCA cycle activity.
Fig. 7.

Adult glo2−/−skeletal muscle showed an increased concentration of hexoses and activity of glycolysis and TCA cycle. A) GC/MS analysis of the metabolome of glo2−/− skeletal muscle showed an increase in hexoses, G6P, F6P and lactate. N = 3–6. B) The GC/MS analysis of various metabolites of the TCA cycle also showed an increase. N = 6. C) Activity measurements of hexokinase were unchanged in glo2−/− zebrafish skeletal muscle. N = 6. D) Activity measurements of phosphofructokinase were unchanged in glo2−/− zebrafish. N = 6. E) Pyruvate kinase activity was increased, indicating increased glycolysis. N = 6. F)mPEPCK expression was reduced in glo2−/− zebrafish. Measured via RT-qPCR. N = 4. One datapoint represents skeletal muscle tissue from one zebrafish. G) Skeletal muscle insulin receptor expression was unchanged in glo2−/− animals. Measured via qPCR, normalized to b2m. N = 4. For statistical analysis normality tests were used for all samples, followed by unpaired t-test (A, B, C, D, E, F, G). Data presented as mean ± SD. ns = p > 0.05, * = p < 0.05, ** = p < 0.01. insra: insulin receptor A, insrb: insulin receptor B, Hex: hexoses, G6P: glucose-6-phosphate, F6P: fructose-6-phosphate, Lac: lactate, TCA cycle: tricarboxylic acid cycle, SA: succinic acid, FA: fumaric acid, MA: malic acid, OA: oxalic acid, HK: hexokinase, PFK: phosphofructokinase, PK: pyruvate kinase, mPEPCK: phosphoenolpyruvate carboxykinase 2 (mitochondrial).
2.7. The loss of Glo2 did not lead to end organ damage in eye and kidney
To determine whether the measured changes in glucose metabolism coincided with complications commonly found in patients with metabolic disorders such as diabetes, we examined the eyes and kidney in glo2−/− animals. A trypsin digest and microscopic analysis of adult retinae, conducted as previously reported [40], revealed that vascular mural cell counts (Fig. 8A and B), endothelial cell counts (Fig. 8A, C) and vessel diameter (Fig. 8A, D) were unaffected by the loss of Glo2. To complement these datasets, confocal microscopy of Tg(fli1:EGFP) glo2−/− mutants was used to quantify angiogenetic processes and proved that retinal blood vessel sprouts (Suppl. Fig. 7A and B), branching (Suppl. Fig. 7A, C) and blood vessel density (Suppl. Fig. 7A, D) were equally unaltered [41]. In conclusion, neither the structure of individual blood vessels nor angiogenesis in adult retinae were affected by the knockout of Glo2. The kidney was analyzed first using light microscopy (Fig. 9A), showing no alterations in proximal or distal tubules and glomeruli. Measurements of glomerular basement membrane thickness, an early pathological alteration in diabetic kidney disease [42], via transmission electron microscopy (Suppl. Fig. 8A and B), also revealed no pathological thickening. Other organs, such as the glo2−/− liver (Fig. 9B) and glo2−/− skeletal muscle (Fig. 9C) remained normal, despite their observed metabolic alterations. In conclusion, the data have identified unaltered eyes and kidneys in glo2−/− zebrafish, indicating that Glo2 is no independent risk factor for the development of vascular organ damage in metabolic disorders.
Fig. 8.

Adult retinae displayed normal cell distribution and vessel morphology. A) Light-microscopical analysis of retinal digests showed unchanged vasculature. Black arrow: vascular mural cell, white arrow: endothelial cell, red arrow: erythrocyte. Scale bar = 100 μm. B) Vascular mural cell count remained unchanged in glo2−/− retinae. C) Endothelial cell count remained equally unchanged in glo2−/− retinae. D) Vessel diameter also remained unchanged in glo2−/− retinae. One datapoint represents the measurements in one 100 μm grid square. N = 3/4. For statistical analysis normality tests were used for all samples, followed by unpaired t-test (B, C, D). Data presented as mean ± SD. ns = p > 0.05. vMC: vascular mural cells, EC: endothelial cells. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Fig. 9.

Histological analysis of adult liver, skeletal muscle and kidney HE stains and PAS reaction showed normal tissue morphology in glo2−/−zebrafish. A)The analysis of glomeruli, proximal tubules and distal tubules of the kidney did not reveal any significant structural changes.B) Analysis of the liver did not reveal any significant alterations of the liver parenchyma.C)The structure and appearance of skeletal muscle fibers remained unchanged in glo2−/− zebrafish. Scale bar = 100 μm for A, B, C. N = 6.
3. Discussion
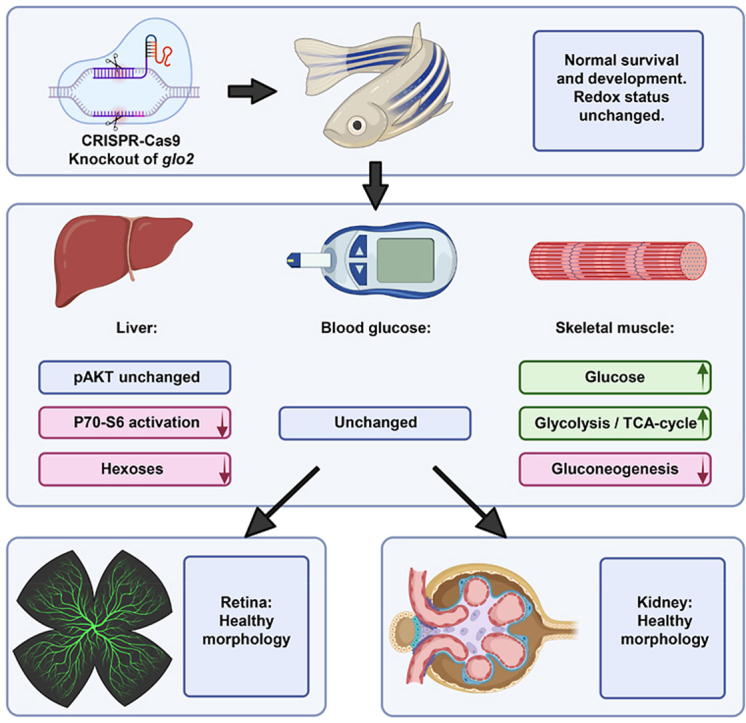
In this study, we have created the first knockout of glo2 in zebrafish and conducted the most thorough in vivo analysis of glo2 to date. glo2−/− animals developed an impaired P70–S6 kinase activation in the liver and showed strong regulation of glucose metabolism in both liver and skeletal muscle while maintaining euglycemia and avoiding end organ damage. Thus, the data have identified Glo2 as a novel regulator of the glucose metabolism (Fig. 10).
Fig. 10.

Loss of Glo2 leads to reduced P70–S6 kinase phosphorylation and hexose content in the liver. The knockout of glyoxalase 2 via CRISPR-Cas9 did not affect the redox status or impair the survival and general development of glo2−/− zebrafish. The liver, the organ with the highest glo2 expression, showed reduced P70–S6 kinase phosphorylation and reduced glucose concentrations, while AKT phosphorylation remained unaffected. Blood glucose remained normal. Skeletal muscle showed a strong increase of glucose concentrations, glycolysis and TCA cycle activity. Gluconeogenesis in contrast was downregulated. The retinal vasculature and kidneys did not show altered morphology and architecture.
The first important finding of this study is that glo2−/− animals accumulate SDL in presence of methylglyoxal. This confirms the basic feature of Glo2 for the first time in an in vivo model, showing that Glo2 uses SDL generated from methylglyoxal for its function. While previous data had postulated that the absence of Glo2 might lead to increased redox stress in the cell, as Glo2 is relevant for glutathione restoration [[25], [26], [27], [28]], our data showed that a global knockout of the enzyme does not influence cellular glutathione concentrations. Additionally, the survival and general development of glo2−/− zebrafish was not limited compared to glo2+/+ animals, indicating that SDL is non-toxic at physiological concentrations, even upon long-term exposure.
The second important finding of this study is that even though glo2−/− did not show a regulation of glycolysis in the liver as had been proposed by in vitro studies [28], the glo2−/− animals revealed various alterations to their glucose metabolism. The liver did show a reduced uptake of hexoses while the skeletal muscle showed an opposite regulation, strong increases in glucose uptake as well as glycolytic and TCA cycle activity. This is particularly interesting as skeletal muscle shows very little intrinsic Glo2 activity. Therefore, it is most likely the result of either reduced uptake of glucose into the liver, prompting skeletal muscle to compensate for the liver, or the result of a liver-skeletal muscle cross talk regulated by Glo2. Previous studies have already identified myo-inositol as a metabolite capable of serving as a messenger influencing skeletal muscle glucose absorption, even independent of insulin [43], and also suggested SD-Lactoylglutathione to be capable of fulfilling regulatory functions in cooperation with Glo2 [44,45]. As both substances were increased in glo2−/− mutants, a possible liver-muscle cross talk mediated by Glo2 needs to be explored in further studies.
The most significant finding of this study is that postprandial liver P70–S6 kinase was found to be strongly regulated through Glo2. Yet, insulin signaling, upstream of P70–S6 kinase, measured through AKT phosphorylation was found intact. This is of particular interest, as previous in vitro analyses had linked Glo2 to AKT regulation, which could not be confirmed in our Glo2 knockout animal model. The second pathway acting upstream of P70–S6 kinase is the AMPK pathway, which primarily serves as a sensor of cellular energy metabolism [46]. Therefore, it seems likely that the reduced liver hexoses could trigger AMPK activation, which in turn suppresses P70–S6 kinase phosphorylation in glo2−/− animals [47]. Thus, Glo2 is most likely a novel regulator of the cellular energy metabolism, contributing to glucose homeostasis by influencing AMPK and P70–S6 kinase activity. The mechanism warrants further research in additional animal and in vitro models.
Lastly, the glo2−/− zebrafish neither developed vascular end organ damage in eyes, kidney or liver, nor hyperglycemia, increased RCS or changes to their redox status, which would normally be associated with vascular damage. Thus, it can be concluded that even though Glo2 creates alterations to the cellular energy metabolism, it cannot be considered as an independent driver of vascular damage in metabolic disease.
In summary, the data revealed Glo2 as a novel regulator of cellular energy metabolism and P70–S6 kinase activity, showing distinct metabolic phenotypes in liver and skeletal muscle. Yet, a direct function of Glo2 in the glutathione metabolism, redox state, regulation of glycolytic activity, accumulation of reactive carbonyl species or AKT activation, as had been previously suggested, could not be identified.
4. Material and methods
4.1. Study approval
All experiments and procedures on animals were approved by the local government authority, the Regierungspräsidium Karlsruhe (license no.: G-98/15), and by Heidelberg University (I-19/02 and I-21/04). All experiments were carried out in accordance with the approved guidelines, including EU Directive 2010/63/EU for animal experiments.
4.2. Protein sequence alignment
The UniProt database and the UniProt alignment tool (www.uniprot.org, access: 12/08/2021) were used to check the alignment of the zebrafish (Q6P963), human (Q16775), and mouse (Q99KB8) amino acid sequences of the Glo2 protein.
4.3. Zebrafish husbandry and zebrafish lines
The zebrafish lines Tg(fli1:EGFP) and Tg(wt1b:EGFP) were raised and staged according to established protocols. Embryos were staged in hours post fertilization (i.e., hpf) and described as larvae after 72 hpf and loss of the chorion. Embryos and larvae were kept in E3 medium at 28.5 °C for the first 6 days, then transferred to adult boxes in the Zebrafish Core Facility of the Medical Faculty Mannheim. For imaging studies 0.003% PTU (N-Phenylthiourea, CAS 103-85-5) was added to suppress pigmentation. Zebrafish were considered as adults after 90 days. Adult zebrafish were kept under a 13 h light/11 h dark cycle. Adult zebrafish were fed twice a day with freshly hatched Artemia salina and flake food, respectively. All shown experiments except for the Glo2 activity and glo2 expression studies were done exclusively on male zebrafish.
4.4. Mutant generation
The CRISPR guide-RNA was designed for a target site in exon 2 of glo2, using ZiFit-targeter Version 3.3. It was cloned into a pT7-gRNA promotor expression vector (Addgene). glo2-CIRSPR-for: 5′-TAGGTGGAACAGTCCGACATGA, glo2-CRISPR-rev: 5′-AAACTCATGTCGGACTGTTCCA. Cas9 mRNA was obtained by transcribing a pT3TS-nCas9n vector by Addgene. The T7 mMessage mMachine Kit and T3 MEGAshortscript Kit by Invitrogen were used according to the manufacturers' protocol to synthesize the required mRNA. To create mutants, Tg(wt1b:EGFP) embryos in the 1 cell stage were injected with 1 nl of a mixture containing 200 pg/nl guide RNA and 200 pg/nl Cas9-mRNA. F0 zebrafish were analyzed to identify germline transmission and were crossed with Tg(wt1b:EGFP) zebrafish and raised. For genotyping, PCR-products were created using glo2 specific primers (glo2-genotyping-for1: 5′-TCTTAGATGGHGATGGTGGGC; glo2-genotyping-rev1: 5′-ACAATGCATGACCAGCTGAC; glo2-genotyping-for2: 5′-GCGGGCTCTATCTCTGGTTT; glo2-genotyping-rev2: 5′-GCATGACCAGCTGACCTTCAA) binding to the flanking regions next to the CRISPR target site in exon 2 of glo2, genotyped via Sanger sequencing (Eurofins genomics). The sequencing data were analyzed with PolyPeakParser (http://yosttools.genetics.utah.edu/PolyPeakParser) and benchling (https://www.benchling.com, last access: 10/23/2022). To create the glo2−/− Tg(fli1:EGFP) line, glo2+/− Tg(wt1b:EGFP) zebrafish were crossed with glo2+/+ Tg(fli1:EGFP) zebrafish. The zebrafish with strong fli1:EGFP fluorescence were selected and crossed with glo2+/+ Tg(fli1:EGFP) zebrafish twice more.
4.5. MG, 3-DG and glyoxal measurements
Methylglyoxal, glyoxal and 3-deoxyglucosone (3-DG) were measured via isotope dilution, tandem mass spectroscopy, following derivatization with 1,2-diaminobenzene (CAS 95-54-5) [48]. Briefly, about 5–10 mg of zebrafish tissue or about 50 zebrafish larvae were collected, flash frozen in liquid nitrogen and pelleted. The pellets were homogenized in ice-cold 20 % (wt/vol) trichloroacetic acid (CAS 76-03-9) in 0.9 % (wt/vol) sodium chloride (CAS 7647-14-5) (20 μl) and water (80 μl). An aliquot (5 μl) of the internal standard (13C3-MG; 400 nM) was then added and the samples vortexed. Following centrifugation (14,000 rpm; 5 min at 4 °C), 35 μl of the supernatant was transferred to an HPLC vial containing a 200 μl glass insert. An aliquot (5 μl) of 3 % sodium azide (CAS 26628-22-8) (wt/vol) was then added to each sample followed by 10 μl of 0.5 mM DB in 200 mM HCl (CAS 7647-01-0) containing 0.5 mM diethylenetriaminepentaacetic acid (CAS 67-43-6) (DETAPAC) in water. The samples were then incubated for 4 h at room temperature, protected from the light. Samples were then analyzed by LC-MS/MS using an ACQUITY™ ultra-high-performance liquid chromatography system with a Xevo-TQS LC-MS/MS mass spectrometer (Waters, Manchester, UK). The column was a Waters BEH C18 (100 × 2.1 mm) and guard column (5 × 2.1 mm). The mobile phase was 0.1 % formic acid (CAS 64-18-6) in water with a linear gradient of 0 – 100 % 0.1 % formic acid in 50 % acetonitrile:water over 0 – 10 min; the flow rate was 0.2 ml/min and column temperature was 5 °C. The capillary voltage was 0.5 kV, the cone voltage 20 V, the interscan delay time 100 ms, the source and desolvation gas temperatures 150 and 350 °C, respectively, and the cone gas and desolvation gas flows were 150 and 800 l/h, respectively. Mass transitions (retention time, parent ion > fragment ion; collision energy) were as follows: 5.93 min, 145.0 > 77.1, 24 eV. Acquisition and quantification were completed with MassLynx 4.1 and TargetLynx 2.7 (Waters®). Calibration curves were constructed by plotting peak area ratio of analyte/isotopic standard against analyte concentration (0–20 pmol).
4.6. Glutathione, glutathione disulfide and SD-Lactoylglutathione measurements
Reduced glutathione (GSH), oxidized glutathione (GSSG) and SD-Lactoylglutathione (SDL) were measured by stable dilution, liquid chromatography tandem mass spectrometry. Briefly, about 5 – 10 mg of zebrafish tissue or about 50 zebrafish larvae were collected, flash frozen in liquid nitrogen and pelleted. The pellets were homogenized in ice-cold 3 % (wt/vol) trichloroacetic acid, 0.25 % sodium azide (wt/vol) and 0.5 mM diethylenetriaminepentaacetic acid (DETAPAC) in 0.9 % (wt/vol) sodium chloride (100 μl). Following centrifugation (14.000 rpm; 5 min at 4 °C), 20 μl of the supernatant was transferred to an HPLC vial containing 20 μl of the isotopic standards (20 pmol [glycine-13C2,15N]-GSH and 10 pmol [13C4, 15N2-GSSG]. Samples were then analyzed by LC-MS/MS using an ACQUITY™ ultra-high-performance liquid chromatography system with a Xevo-TQS LC-MS/MS mass spectrometer (Waters, Manchester, UK). The column was a Waters BEH C18 (100 × 2.1 mm) and guard column (5 × 2.1 mm). The mobile phase was 0.1 % formic acid in water with a linear gradient of 0 – 100 % 0.1 % formic acid in 50 % acetonitrile:water over 0 – 10 min; the flow rate was 0.2 ml/min, column temperature was 5 °C and injection volume was 10 μl. The capillary voltage was 2.5 kV, the cone voltage 18 V, the interscan delay time 3 ms, the source and desolvation gas temperatures 150 and 250 °C, respectively, and the cone gas and desolvation gas flows were 150 and 600 l/h, respectively. Mass transitions (retention time, parent ion > fragment ion, collision energy) and retention times were as follows: GSH, 2.33 min, 370.9 > 178.7, 12 eV; GSSG, 5.02 min, 613.1 > 483.9, 18 eV; SDL, 5.19 min 380.1 > 75.5, 36 eV. Acquisition and quantification were completed with MassLynx 4.1 and TargetLynx 2.7 (Waters®). Calibration curves were constructed by plotting peak area ratio of analyte/isotopic standard against analyte concentration (GSH: 0 – 100 pmol; GSSG: 0 – 50 pmol; SDL: 0 – 10 pmol).
4.7. Incubation with methylglyoxal
Approximately 80 fertilized eggs were identified and transferred to a 10 cm Petri dish containing 30 ml of E3 medium. Methylglyoxal (CAS 78-98-8) was added to a 500 mM solution. For imaging studies 0.003 % PTU was added to avoid pigmentation. For the collection of samples, the larvae were anesthetized by adding tricaine (ethyl 3-aminobenzoate methanesulfonate) stock to an end concentration of 160 mg/l. The anesthetized larvae and embryos were flash frozen in liquid nitrogen.
4.8. Enzyme activity assays
For the Glo2 activity assay, flash frozen zebrafish tissue was pelleted. The pellets were resuspended in 100 mM sodium phosphate buffer (pH 7.4) containing 0.1 % Triton X100 and protease inhibitor cocktail (300 μl) and transferred to a bioruptor microtube (1.5 ml) containing 100 mg of protein extraction beads. The samples were sealed and then placed into a pre-cooled Bioruptor® Pico and sonicated for 30 cycles of ON/OFF (run time 30 min). The samples were then centrifuged (14.000 rpm; 5 min; 4 °C) and the resulting supernatant was used for protein determination by Bradford assay [49]. Glyoxalase 2 (Glo2) activity was determined in total extract (10 μg) from the rate of formation of reduced glutathione (GSH) from SD-Lactoylglutathione via the reaction with 5′5-dithiobis (2-nitrobenzoic) acid (DNTB) at 415 nm [50], with 1 U defined as 1 μmol of GSH from SD-Lactoylglutathione per minute at pH 7.4 at 25 °C.
The activity of phosphofructokinase was measured spectrophotometrically. The assay measured the oxidation of NADH using the absorbance at 340 nm. NADH was oxidized in a coupled reaction with aldolase, triosephosphate isomerase and glyceraldehyde-3-phosphate dehydrogenase. 1 U of activity was defined as 1 μm of fructose-6-phosphate and ATP to fructose 1,6-diphosphate and ADP per minute at pH 8.2 at 30 °C. The following reaction buffer was used: 50 mM HEPES, 10 mM KCl, 6.5 mM MgCl2, 1 mM NH4Cl, 5 mM KH2PO4, 0.1 mM AMP, 0.3 mM NADH, 0.5 U/ml aldolase, 0.5 U/ml glyceraldehyde-3-phosphate dehydrogenase, 5 U/ml triosephosphate isomerase, 0.1 mM fructose-6-phosphate, 0.3 mM glucose-6-phosphate, 1.5 mM ATP.
The activity of pyruvate kinase was measured spectrophotometrically utilizing the oxidation of NADH through a coupled reaction with L-lactatedehydrogenase. Absorbance was measured at 340 nm. 1 U was defined as the conversion of 1 μm of phosphoenolpyruvate to pyruvate per minute at pH 7.6 at 37 °C. The following reaction buffer was used: 39 mM potassium phosphate, 0.58 mM phospho (enol)pyruvate, 0.11 mM NADH, 6.8 mM magnesium sulphate, 1.5 mM ADP, 3.4 U/ml L-lactate dehydrogenase.
The activities of hexokinase and glucokinase were measured spectrophotometrically, as described previously [51]. The assay is based on the oxidation of glucose-6-phosphate in the presence of glucose-6-phosphate dehydrogenase and NADP+. The generated NADPH reduces WST-1 and the change in absorbance can be measured at 440 nm. The following buffers were used: For hexokinase: 28 mM Tris at a pH of 7.5, 1.25 mM MgCl2, 0.625 mM of ATP, 1.25 mM of glucose, 0.0125 mM of NADP+, 0.0125 mM of ImPMS, G6PDH 0.875 U/ml and WST at 0.63 mM/100 Ml per well were used. For the glucokinase assay 28 mM Tris at a pH of 7.5, 1.25 mM MgCl2, 5 mM of ATP, 25 mM of glucose, 0.0125 mM of NADP+, 0.0125 mM of ImPMS, G6PDH 0.875 U/ml and WST at 0.63 mM/100 Ml per well were used.
4.9. P70–S6-kinase phosphorylation assay
To determine the phosphorylation of the P70–S6 kinase, a PathScan P70–S6-kinase (1389) ELISA Kit (Cell Signaling) was used according to the manufacturer's instructions. The tissue was collected as described under “preparation of tissues” and flash frozen in liquid nitrogen. The tissue was homogenized using a TissueLyser II (QIAGEN) in the kits own Cell Lysis buffer with added PMSF. The protein concentration was measured using a Pierce BCA protein assay kit (ThermoScientific). A concentration of 0.1 mg/ml of protein was used for the assay. The results were measured spectrophotometrically at 450 nm.
4.10. Blood glucose measurements
To measure the blood glucose values of the zebrafish, they were placed in single boxes after the last feeding of the day and fasted overnight. After 16 – 18 h, zebrafish either remained fasting or were fed with 0.5 g of flake food. The food was added to the fish tank and the zebrafish were given 1 h for food uptake. Afterwards, the zebrafish were transferred to fresh water to stop feeding and the zebrafish were euthanized using ice water after 1, 2 or 3 h to determine the respective 1 h, 2 h and 3 h postprandial blood glucose values. Blood glucose was measured according to the method published previously [52] by inserting a heparinized fine glass needle into the caudal vessel through the side of the zebrafish approximately above the anal fin and transferring blood into the capillary using gentle suction. Blood glucose was measured with a hand-held glucometer (Freestyle, Freedom lite) after transfer of the blood-droplet onto a sheet of parafilm.
4.11. Metabolome analysis
The analysis of the samples metabolomes was conducted via semi-targeted gas chromatography-mass spectrometry (GC/MS) in cooperation with the Metabolomics Core Technology Platform (MCTP) of the Center for Organismal Studies (COS) of Heidelberg University. The samples were collected as described belowand flash frozen. The frozen, ground sample material from zebrafish was extracted in 360 μL of 100 % MeOH (CAS 67-56-1) for 15 min at 70 °C with vigorous shaking. As internal standard, 20 μl ribitol (CAS 488-81-3) (0.2 mg/ml) were added to each sample. After the addition of 200 μl chloroform (CAS 67-66-3) samples were shaken at 37 °C for 5 min. To separate polar and organic phases, 400 μl of water were added and samples were centrifuged for 10 min at 11.000 g. For the derivatization, 700 μl of the polar (upper) phase were transferred to a fresh tube and dried in a speed-vac (vacuum concentrator) without heating. Pellets of the aqueous phase after extraction were re-dissolved in 20 μl methoximation reagent containing 20 mg/ml methoxyamine hydrochloride (Sigma 226,904, CAS 593-56-6) in pyridine (Sigma 270,970, CAS 110-86-1) and incubated for 2 h at 37 °C with shaking. For silylation, 32.2 μl N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA; Sigma M7891, CAS 24589-78-4) and 2.8 μl Alkane Standard Mixture (50 mg/ml C10 − C40; Fluka 68,281) were added to each sample. After incubation for 30 min at 37 °C, samples were transferred to glass vials for GC/MS analysis, for which a GC/MS-QP2010 Plus (Shimadzu®) fitted with a Zebron ZB 5MS column (Phenomenex®; 30 m × 0.25 mm x 0.25 μm) was used. The GC was operated with an injection temperature of 230 °C and 1 μl sample was injected in splitless mode. The GC temperature program started with a 1 min hold at 40 °C followed by a 6 °C/min ramp to 210 °C, a 20 °C/min ramp to 330 °C and a bake-out for 5 min at 330 °C using Helium as carrier gas with constant linear velocity. The MS was operated with ion source and interface temperatures of 250 °C, a solvent cut time of 7 min and a scan range (m/z) of 40–700 with an event time of 0.2 s. The “GCMS solution” software (Shimadzu®) was used for data processing.
4.12. Reverse-transcription quantitative polymerase chain reaction analysis
The expression of various genes was determined using reverse-transcription quantitative polymerase chain reaction (RT-qPCR). Total RNA was isolated from flash frozen adult zebrafish tissue and larvae. They were homogenized by either using the TissueLyser II (Qiagen) or by pulling the tissue through a 20 G syringe. RNA from the homogenate was isolated using the RNeasy Mini Kit according to the manufacturer's instructions (QIAGEN). 0.5 μg of RNA was treated with a dsDNase to remove DNA contamination and first-strand cDNA was synthesized using a Maxima First Strand cDNA Synthesis Kit with dsDNase (ThermoFisher). The used primers were designed using NCBI Primer-BLAST and are listed in (Suppl. Table 1). The RT-qPCR was conducted using PowerSYBR Green PCR Master Mix (appliedbiosystems) on a QuantStudio3 cycler (applied biosystems). The expression data was normalized to b2m and standardized to wild-type samples.
4.13. Preparation of tissues
The zebrafish were placed in single boxes overnight. Feeding, anesthesia and blood glucose measurements were conducted as described above. Afterwards, zebrafish were weighed on a microscale, pictures of their body were taken and their body dimensions marked on a Petri dish for later measurement. The zebrafish were decapitated and their abdomen opened with scissors. The zebrafish were placed in ice-cold 1x Phosphate Buffered Saline (PBS) solution. The organs were prepared using #5 micro tweezers (Dumont) under a stereomicroscope. 1.5 ml or 2.5 ml safelock tubes (Eppendorf) were weighed. Afterwards, the organs were transferred to the epi tubes, all excess water was removed using a Hamilton syringe and the organ was weighed on a microscale. After weighing, the safelock tube containing the organ was flash-frozen in liquid nitrogen and transferred to a −80 °C freezer for storage. Organs destined for light-, confocal and electron microscopy were not flash frozen, but transferred to 4 % paraformaldehyde (PFA, CAS 30525-89-4)/PBS for light- and confocal microscopy and 3 % glutaraldehyde (GA, CAS 111-30-8) solution, buffered by 0.1 M cacodylate at pH 7.4 for electron microscopy instead.
4.14. Retinal digest
The retinal digest preparations were performed according to a slightly altered version of an established protocol [40]. First, the zebrafish were prepared as described under “preparation of tissues”. The heads of the zebrafish were transferred into a 4 % PFA solution and fixed for 48 h. The fixed retinas were then prepared according to the established protocol. After dissection, the retinas were transferred to distilled water for overnight incubation at 37 °C. On the next day, they were transferred into a 3 % porcine trypsin (Sigma-Aldrich) solution, containing 0.2 M Tris-HCl, and incubated again, at 37 °C for 1.5 h. After the trypsin-incubation, the retinae were placed on microscope slides and the retinal cells were carefully separated from the vasculature by dropping ddH2O from a syringe directly onto the retina. The superfluous cells and water were removed from the slide through aspiration and the remaining vasculature air-dryed. To stain the remaining vasculature, the slides were briefly placed in distilled water and then in fresh, undiluted Mayers hemalum solution for 7 min. Then, they were placed in lukewarm tap-water for 2 min and dehydrated in 70 %, 80 %, 96 % and finally 99.8 % ethanol for 5 min each. Following dehydration, they were placed in xylene for 2 × 5 min, and covered using DPX mounting media (ThermoFisher) and cover slips. The stained vasculature was imaged at 200 × magnification using an upright microscope (Olympus BX51) equipped with a camera (Olympus XC10). The imaged cells were identified based on their location and morphology and counted in six to eight randomly selected 100 μm grid squares of the middle circular third of the retina, avoiding the entrance of the optic nerve and periphery of the retina. The cells were counted over 200 μm, and the numbers calculated as "number of cells/mm2 of capillary area". The analysis of the cell counts and the vessel diameter was conducted using Cell-F software by Olympus Opticals.
4.15. Western blot analysis
For the Glo2 Western Blot analysis, adult zebrafish were euthanized and cut in halves. The front halves of the zebrafish were homogenized using a Tissue Lyser II (Qiagen) and aliquoted. The protein concentration of the sample was determined using the Pierce BCA Protein Assay Kit (ThermoScientific). The Western Blot was conducted according to standard protocols. Briefly, due to partial overlap of the β-Actin and Glo2 bands, a rabbit anti-HAGH (Glo2) primary antibody (Sigma) was used, and after addition of a goat, HRP linked anti-rabbit secondary antibody (Dako) detected through enhanced chemiluminescence (ECL) and stripped using stripping-solution (0.375 g of glycine in 50 ml of H2O with 500 μl 37 % HCl for 30 min). Then the goat anti-β-actin primary antibody (Santa Cruz Biotechnology) was added and incubated over-night, followed by a rabbit, HRP linked anti-goat secondary antibody (DAKO) and the β-actin band was detected with the help of ECL.
For the AKT Western Blot adult fish were fasted overnight and then fed 2 h before dissection or remained fasting. The adult livers were then dissected and homogenized using a 20 G syringe in NP40 lysis buffer with added PhosSTOP phosphatase inhibitors (Roche). Protein concentration was determined using the Pierce BCA Protein Assay Kit (ThermoScientific). 50 μg protein were diluted with Laemmli buffer and loaded into BIORAD Mini-Protean TGX 10% Gels and electrophoresis conducted using a Bio-Rad Mini Protean Tetra System. Proteins were then blotted onto a Protan 0.2 μM Supported nitrocellulose membrane (Amersham). The membranes were blocked using 5 % BSA solution in PBST. For primary antibodies an anti phospho-AKT antibody 4060P from Cell Signaling was used in a 1:1000 dilution in 5 % BSA in PBST, an anti-total AKT antibody 9272S from Cell Signaling in a 1:1000 dilution in 5 % BSA in PBST and an anti-β-actin sc-47778 from Santa Cruz Biotechnology was used in a 1:1000 dilution in 5 % BSA in PBST. Primary antibodies were incubated over night at 4 °C. For secondary antibodies a goat anti‐rabbit HRP-linked antibody, P0448, DAKO, in a 1:1000 dilution in PBST and a rabbit anti-mouse HRP-linked antibody, P0260, DAKO in a 1:1000 dilution in PBST were used. Protein bands were imaged using Western Lightning Plus ECL reagent (PerkinElmer) on a Vilber Fusion Solo S imaging system.
4.16. Microscopical analysis of larvae
To analyze the morphology of 96 hpf larvae, they were anesthetized using tricaine at a concentration of 160 mg/l, gently placed on their side with micro tweezers (Dumont) and analyzed under a stereo – light microscope (Leica MZ 9.5).
4.17. Histological analysis of adult tissue
The zebrafish were euthanized and prepared as described under “preparation of tissues”. After overnight fixation in 10 % buffered formalin solution (or 4 % PFA), representative specimens of the kidney, liver, and skeletal muscle were routinely dehydrated, embedded in paraffin, and cut into 4 μm-thick sections. HE staining and PAS reaction were done using standard protocols on an integrated workstation (Leica ST5020) using hematoxylin solution modified according to Gill III (Merck), 100 % acetic acid (Rotipuran, CAS 64-19-7) and Eosin Y (Merck, CAS 15086-94-9) for HE staining, and periodic acid (Roth, CAS 10450-60-9), Schiff's reagent (SIGMA) as well as hemalum according to Mayer (Roth) for PAS reaction. Coverslipping was conducted on an automatic coverslipper (Leica CV5030) using Consul Mount Histology Formulation (Thermo Scientific).
4.18. Collection of zebrafish embryos and larvae
To collect zebrafish embryos and larvae, the larvae were anesthetized in a Petri dish by adding tricaine stock to achieve an end concentration of 160 mg/l. After anesthesia, the larvae were transferred to a 1.5 ml or 2 ml Eppendorf tube with a large-bore glass pipette and excess water was removed using a Hamilton syringe and then flash frozen in liquid nitrogen. For embryos and larvae younger than 120 hpf, the yolk sack was destroyed by pipetting the collected larvae up and down using a 100 μl pipette tip (nerbe plus) and centrifuged for 5 min at 14.000 rpm at 4 °C, then the excess water was removed and the tubes with the embryos/larvae flash frozen in liquid nitrogen.
4.19. Statistics
GraphPad Prism (Version 9.1.1) was used for all statistical analyses. The samples were tested for normality using the Anderson-Darling test, D'-Agostino-Pearson omnibus normality test, Shapiro-Wilk normality test and the Kolmogorov-Smirnov normality test with Dallal-Wilkinson-Lillie for P value wherever applicable. Afterwards, they were analyzed for statistical significance using the unpaired parametric t-test and the Mann-Whitney test when applicable. The distribution of genotypes in larvae was analyzed using the chi-square test. All data are presented as mean ± standard deviation. A significant p value was defined as < 0.05. * = p < 0.05, ** = p < 0.01, **** = p < 0.0001.
4.20. Material requests
The glo2 zebrafish mutants generated in this study are available from the corresponding author with a completed Materials Transfer Agreement.
Author contributions
C.T.T. designed this study, performed experiments, analyzed data and wrote the manuscript. E.L. generated the mutants, performed experiments and analyzed data. K.B. performed experiments and analyzed data. C.S.M. performed retinal digests and analyzed data. V.E. performed imaging studies and analyzed data. H.O. performed imaging studies. T.P. and I.H. performed microscopical studies and analyzed data. T.F. and J.M. performed metabolite and activity measurements and analyzed data. C.S. performed RNA sequencing data analysis. G.P. performed metabolome studies and analyzed data. J.S. and P.P.N. provided conceptual support. J.K. conceived and designed this study and wrote the manuscript. J.K. is the guarantor of this work, has full access to all data of the study and takes responsibility for the integrity and the accuracy of the data and the data analysis.
Declaration of competing interest
Declarations of interest: none.
Acknowledgements
This study was supported by grants from the Deutsche Forschungsgemeinschaft (IRTG 1874/2 DIAMICOM as well as CRC 1118). The funding providers were not involved in the design of the study, the collection, analysis and interpretation of data, or in the writing or submission of this article. The authors thank Björn Hühn for zebrafish maintenance, Dr. Stefan Hillmer, Steffi Gold, Silas Soleymani, Andrea Döbler, Ulrike Ganserer for performance and analysis of electron microscopy images, Dr. Carolina De La Torre and Angelika Duda for RNA quality control and advice. The authors thank Dr. Michael Büttner, Dr. Katja Machemer-Noonan, Dr. Elena Heidenreich and Dr. Hagen Gegner of the Metabolomics Core Technology Platform of the Excellence cluster “CellNetworks” (Heidelberg University), and the Deutsche Forschungsgemeinschaft (grant ZUK 40/2010-3009262) for support regarding UPLC-based metabolite quantification. We also acknowledge the support of the LIMa Live Cell Imaging Mannheim at Microscopy Core Facility Platform Mannheim (CFPM) and the Zebrafish Core Facility Mannheim.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102576.
List of abbreviations
- 3-DG
3-deoxyglucosone
- ADPR
adenosine diphosphate ribose
- AKT
protein kinase B
- AMP
adenosine monophosphate
- b2m
beta-2-microglobulin
- CoA
coenzyme A
- cPEPCK
phosphoenolpyruvate carboxykinase 1 (cytosolic)
- E
exon
- E3
embryo medium
- EC
endothelial cells
- F6P
fructose-6-phosphate
- FA
fumaric acid
- FAD
flavin adenine dinucleotide
- G6P
glucose-6-phosphate
- g6pase
glucose-6-phosphatase
- GA
glutaraldehyde
- GC/MS
gas chromatography/mass spectrometry
- gck
glucokinase
- glut
facilitated glucose transporter member
- GMP
guanosine monophosphate
- GSH
glutathione
- GSSG
glutathione-disulfide
- Hex
hexoses
- HK
hexokinase
- hpf
hours post fertilization
- insra/insrb
insulin receptor a/b
- kD
kilodalton
- KEGG
Kyoto encyclopedia of genes and genomes
- Lac
lactate
- LC-MS/MS
liquid chromatography-mass spectrometry/mass spectrometry
- MA
malic acid
- MAPK
mitogen activated protein kinases
- MG
methylglyoxal
- mPEPCK
phosphoenolpyruvatecarboxykinase 2 (mitochondrial)
- mTOR
mammalian target of rapamycin
- NES
normalized enrichment score
- OA
oxalic acid
- pAKT
phosphorylated AKT
- PFK
phosphofructokinase
- PK
pyruvate kinase
- PPAR
peroxisome-proliferator-activating receptor
- PTU
1-phenyl 2-thiourea
- RCS
reactive carbonyl species
- RT-qPCR
real-time quantitative polymerase chain reaction
- SA
succinic acid
- SDL
SD-Lactoylglutathione
- tAKT
total AKT
- TCA cycle
tricarboxylic acid cycle
- UTR
untranslated region
- vMC
vascular mural cells
- WT
wildtype
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.He Y., Zhou C., Huang M., Tang C., Liu X., Yue Y., Diao Q., Zheng Z., Liu D. Glyoxalase system: a systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020;131 doi: 10.1016/j.biopha.2020.110663. [DOI] [PubMed] [Google Scholar]
- 2.Dakin H.D., Dudley H.W. AN enzyme concerned with the formation of hydroxy acids from ketonic aldehydes. J. Biol. Chem. 1913;14:155–157. doi: 10.1016/S0021-9258(18)88610-0. [DOI] [Google Scholar]
- 3.Hopkins F.G., Morgan E.J. Studies on glyoxalase: 1. A new factor. Biochem. J. 1948;42:23–27. [PMC free article] [PubMed] [Google Scholar]
- 4.Racker E. The mechanism of action of glyoxalase. J. Biol. Chem. 1951;190:685–696. doi: 10.1016/S0021-9258(18)56017-8. [DOI] [PubMed] [Google Scholar]
- 5.Racker E., Krimsky I. The mechanism of oxidation of aldehydes by GLYCERALDEHYDE-3-PHOSPHATE dehydrogenase. J. Biol. Chem. 1952;198:731–743. doi: 10.1016/S0021-9258(18)55530-7. [DOI] [PubMed] [Google Scholar]
- 6.Bellier J., Nokin M.-J., Lardé E., Karoyan P., Peulen O., Castronovo V., Bellahcène A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019;148:200–211. doi: 10.1016/j.diabres.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Phillips S.A., Thornalley P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993;212:101–105. doi: 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- 8.Thornalley P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988;254:751–755. doi: 10.1042/bj2540751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Herreweghe F., Mao J., Chaplen F.W.R., Grooten J., Gevaert K., Vandekerckhove J., Vancompernolle K. Tumor necrosis factor-induced modulation of glyoxalase I activities through phosphorylation by PKA results in cell death and is accompanied by the formation of a specific methylglyoxal-derived AGE. Proc. Natl. Acad. Sci. U.S.A. 2002;99:949–954. doi: 10.1073/pnas.012432399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheung S.-T., Fonda M.L. Reaction of phenylglyoxal with arginine. The effect of buffers and pH. Biochem. Biophys. Res. Commun. 1979;90:940–947. doi: 10.1016/0006-291X(79)91918-1. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K. The reactions of phenylglyoxal and related reagents with amino acids. J. Biochem. 1977;81:395–402. doi: 10.1093/oxfordjournals.jbchem.a131471. [DOI] [PubMed] [Google Scholar]
- 12.Thornalley P.J., Hooper N.I., Jennings P.E., Florkowski C.M., Jones A.F., Lunec J., Barnett A.H. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res. Clin. Pract. 1989;7:115–120. doi: 10.1016/0168-8227(89)90101-0. [DOI] [PubMed] [Google Scholar]
- 13.Thornalley P.J. The glyoxalase system in health and disease. Mol. Aspect. Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-U. [DOI] [PubMed] [Google Scholar]
- 14.Sousa Silva M., Ferreira A.E.N., Tomás A.M., Cordeiro C., Ponces Freire A. Quantitative assessment of the glyoxalase pathway in Leishmania infantum as a therapeutic target by modelling and computer simulation. FEBS J. 2005;272:2388–2398. doi: 10.1111/j.1742-4658.2005.04632.x. [DOI] [PubMed] [Google Scholar]
- 15.Silva M.S., Barata L., Ferreira A.E.N., Romão S., Tomás A.M., Freire A.P., Cordeiro C. Catalysis and structural properties of Leishmania infantum glyoxalase II: trypanothione specificity and phylogeny. Biochemistry. 2008;47:195–204. doi: 10.1021/bi700989m. [DOI] [PubMed] [Google Scholar]
- 16.Sousa Silva M., Gomes R.A., Ferreira A.E.N., Ponces Freire A., Cordeiro C. The glyoxalase pathway: the first hundred years… and beyond. Biochem. J. 2013;453:1–15. doi: 10.1042/BJ20121743. [DOI] [PubMed] [Google Scholar]
- 17.Edwards L.G., Adesida A., Thornalley P.J. Inhibition of human leukaemia 60 cell growth by S-d-lactoylglutathione in vitro. Mediation by metabolism to N-d-lactoylcysteine and induction of apoptosis. Leuk. Res. 1996;20:17–26. doi: 10.1016/0145-2126(95)00095-X. [DOI] [PubMed] [Google Scholar]
- 18.Thornalley P.J. Advances in glyoxalase research. Glyoxalase expression in malignancy, anti-proliferative effects of methylglyoxal, glyoxalase I inhibitor diesters and S-d-lactoylglutathione, and methylglyoxal-modified protein binding and endocytosis by the advanced glycation endproduct receptor. Crit. Rev. Oncol. Hematol. 1995;20:99–128. doi: 10.1016/1040-8428(94)00149-N. [DOI] [PubMed] [Google Scholar]
- 19.Kermack W.O., Matheson N.A. The effects of some analogues of glutathione on the glyoxalase system. Biochem. J. 1957;65:48–58. doi: 10.1042/bj0650048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moraru A., Wiederstein J., Pfaff D., Fleming T., Miller A.K., Nawroth P., Teleman A.A. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metabol. 2018;27:926–934. doi: 10.1016/j.cmet.2018.02.003. e8. [DOI] [PubMed] [Google Scholar]
- 21.Lodd E., Wiggenhauser L.M., Morgenstern J., Fleming T.H., Poschet G., Büttner M., Tabler C.T., Wohlfart D.P., Nawroth P.P., Kroll J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight. 2019;4 doi: 10.1172/jci.insight.126154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schumacher D., Morgenstern J., Oguchi Y., Volk N., Kopf S., Groener J.B., Nawroth P.P., Fleming T., Freichel M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metabol. 2018;18:143–152. doi: 10.1016/j.molmet.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang S., Kwon D.M., Kwon K., Park C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017;490:460–465. doi: 10.1016/j.bbrc.2017.06.063. [DOI] [PubMed] [Google Scholar]
- 24.Talesa V., Uotila L., Koivusalo M., Principato G., Giovannini E., Rosi G. Demonstration of glyoxalase II in rat liver mitochondria. Partial purification and occurrence in multiple forms. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1988;955:103–110. doi: 10.1016/0167-4838(88)90183-5. [DOI] [PubMed] [Google Scholar]
- 25.Thornalley P.J. Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003;31:1343–1348. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 26.Jain M., Munoz-Bodnar A., Gabriel D.W. Concomitant loss of the glyoxalase system and glycolysis makes the uncultured pathogen "candidatus liberibacter asiaticus" an energy scavenger. Appl. Environ. Microbiol. 2017;83 doi: 10.1128/AEM.01670-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armeni T., Cianfruglia L., Piva F., Urbanelli L., Luisa Caniglia M., Pugnaloni A., Principato G. S-D-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014;67:451–459. doi: 10.1016/j.freeradbiomed.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Cianfruglia L., Morresi C., Bacchetti T., Armeni T., Ferretti G. Protection of polyphenols against glyco-oxidative stress: involvement of glyoxalase pathway. Antioxidants. 2020;9 doi: 10.3390/antiox9101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James Am, Hoogewijs K., Logan A., Hall A.R., Ding S., Im Fearnley, Murphy M.P. Non-enzymatic N-acetylation of lysine residues by AcetylCoA often occurs via a proximal S-acetylated thiol intermediate sensitive to glyoxalase II. Cell Rep. 2017;18 doi: 10.1016/j.celrep.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaffney D.O., Jennings E.Q., Anderson C.C., Marentette J.O., Shi T., Schou Oxvig A.-M., Streeter M.D., Johannsen M., Spiegel D.A., Chapman E., Roede J.R., Galligan J.J. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem. Biol. 2020;27:206–213. doi: 10.1016/j.chembiol.2019.11.005. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talesa V.N., Ferri I., Bellezza G., Love H.D., Sidoni A., Antognelli C. Glyoxalase 2 is involved in human prostate cancer progression as part of a mechanism driven by PTEN/PI3K/AKT/mTOR signaling with involvement of PKM2 and ERα. Prostate. 2017;77:196–210. doi: 10.1002/pros.23261. [DOI] [PubMed] [Google Scholar]
- 32.Li X., Fargue S., Challa A.K., Poore W., Knight J., Wood K.D. Generation of a GLO-2 deficient mouse reveals its effects on liver carbonyl and glutathione levels. Biochem. Biophys. Rep. 2021;28 doi: 10.1016/j.bbrep.2021.101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saltiel A.R. Insulin signaling in health and disease. J. Clin. Invest. 2021;131 doi: 10.1172/JCI142241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molinaro A., Becattini B., Mazzoli A., Bleve A., Radici L., Maxvall I., Sopasakis V.R., Molinaro A., Bäckhed F., Solinas G. Insulin-driven PI3K-AKT signaling in the hepatocyte is mediated by redundant PI3Kα and PI3Kβ activities and is promoted by RAS. Cell Metabol. 2019;29:1400–1409. doi: 10.1016/j.cmet.2019.03.010. e5. [DOI] [PubMed] [Google Scholar]
- 35.Gray C.W., Coster A.C.F. From insulin to Akt: time delays and dominant processes. J. Theor. Biol. 2020;507 doi: 10.1016/j.jtbi.2020.110454. [DOI] [PubMed] [Google Scholar]
- 36.Das D., Nath P., Pal S., Hajra S., Ghosh P., Maitra S. Expression of two insulin receptor subtypes, insra and insrb, in zebrafish (Danio rerio) ovary and involvement of insulin action in ovarian function. Gen. Comp. Endocrinol. 2016;239:21–31. doi: 10.1016/j.ygcen.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Gong Y., Zhai G., Su J., Yang B., Jin J., Liu H., Yin Z., Xie S., Han D. Different roles of insulin receptor a and b in maintaining blood glucose homeostasis in zebrafish. Gen. Comp. Endocrinol. 2018;269:33–45. doi: 10.1016/j.ygcen.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Bae E.J., Xu J., Da Oh Y., Bandyopadhyay G., Lagakos W.S., Keshwani M., Olefsky J.M. Liver-specific p70 S6 kinase depletion protects against hepatic steatosis and systemic insulin resistance. J. Biol. Chem. 2012;287:18769–18780. doi: 10.1074/jbc.M112.365544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stark R., Guebre-Egziabher F., Zhao X., Feriod C., Dong J., Alves T.C., Ioja S., Pongratz R.L., Bhanot S., Roden M., Cline G.W., Shulman G.I., Kibbey R.G. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem. 2014;289:7257–7263. doi: 10.1074/jbc.C113.544759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wohlfart D.P., Lou B., Middel C.S., Morgenstern J., Fleming T., Sticht C., Hausser I., Hell R., Hammes H.-P., Szendrödi J., Nawroth P.P., Kroll J. Accumulation of acetaldehyde in aldh2.1-/- zebrafish causes increased retinal angiogenesis and impaired glucose metabolism. Redox Biol. 2022;50 doi: 10.1016/j.redox.2022.102249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiggenhauser L.M., Kohl K., Dietrich N., Hammes H.-P., Kroll J. Studying diabetes through the eyes of a fish: microdissection, visualization, and analysis of the adult tg(fli:EGFP) zebrafish retinal vasculature. JoVE. 2017 doi: 10.3791/56674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papadopoulou-Marketou N., Chrousos G.P., Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes. Metab. Res. Rev. 2017;33 doi: 10.1002/dmrr.2841. [DOI] [PubMed] [Google Scholar]
- 43.Chukwuma C.I., Ibrahim M.A., Islam M.S. Myo-inositol inhibits intestinal glucose absorption and promotes muscle glucose uptake: a dual approach study. J. Physiol. Biochem. 2016;72:791–801. doi: 10.1007/s13105-016-0517-1. [DOI] [PubMed] [Google Scholar]
- 44.Galeazzi R., Laudadio E., Falconi E., Massaccesi L., Ercolani L., Mobbili G., Minnelli C., Scirè A., Cianfruglia L., Armeni T. Protein-protein interactions of human glyoxalase II: findings of a reliable docking protocol. Org. Biomol. Chem. 2018;16:5167–5177. doi: 10.1039/c8ob01194j. [DOI] [PubMed] [Google Scholar]
- 45.Ercolani L., Scirè A., Galeazzi R., Massaccesi L., Cianfruglia L., Amici A., Piva F., Urbanelli L., Emiliani C., Principato G., Armeni T. A possible S-glutathionylation of specific proteins by glyoxalase II: an in vitro and in silico study. Cell Biochem. Funct. 2016;34 doi: 10.1002/cbf.3236. [DOI] [PubMed] [Google Scholar]
- 46.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krause U., Bertrand L., Hue L. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur. J. Biochem. 2002;269:3751–3759. doi: 10.1046/j.1432-1033.2002.03074.x. [DOI] [PubMed] [Google Scholar]
- 48.Rabbani N., Thornalley P.J. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 2014;9:1969–1979. doi: 10.1038/nprot.2014.129. [DOI] [PubMed] [Google Scholar]
- 49.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 50.Allen R.E., Lo T.W., Thornalley P.J. Purification and characterisation of glyoxalase II from human red blood cells. Eur. J. Biochem. 1993;213:1261–1267. doi: 10.1111/j.1432-1033.1993.tb17877.x. [DOI] [PubMed] [Google Scholar]
- 51.Zhu A., Romero R., Petty H.R. An enzymatic colorimetric assay for glucose-6-phosphate. Anal. Biochem. 2011;419:266–270. doi: 10.1016/j.ab.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zang L., Shimada Y., Nishimura Y., Tanaka T., Nishimura N. A novel, reliable method for repeated blood collection from aquarium fish. Zebrafish. 2013;10:425–432. doi: 10.1089/zeb.2012.0862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.