

Abstract
Objective
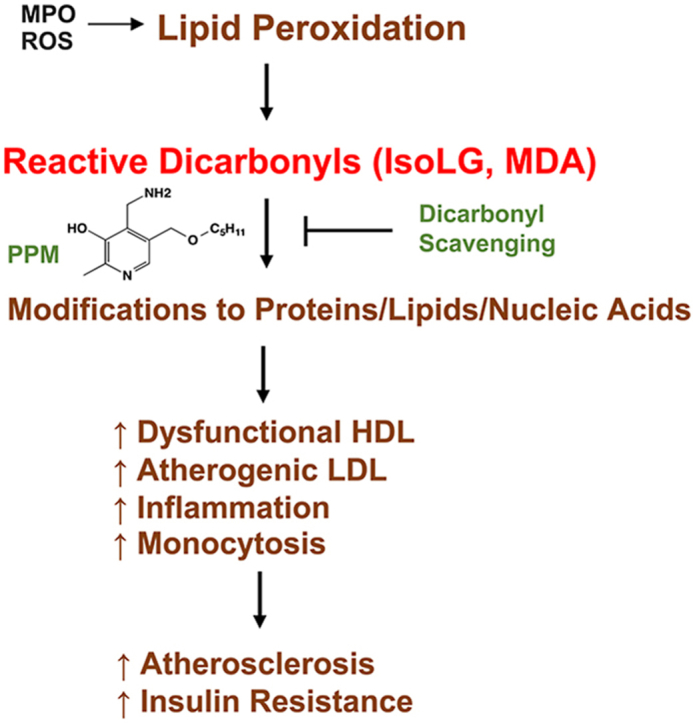
Oxidative stress contributes to the development of insulin resistance (IR) and atherosclerosis. Peroxidation of lipids produces reactive dicarbonyls such as Isolevuglandins (IsoLG) and malondialdehyde (MDA) that covalently bind plasma/cellular proteins, phospholipids, and DNA leading to altered function and toxicity. We examined whether scavenging reactive dicarbonyls with 5′-O-pentyl-pyridoxamine (PPM) protects against the development of IR and atherosclerosis in Ldlr−/− mice.
Methods
Male or female Ldlr−/− mice were fed a western diet (WD) for 16 weeks and treated with PPM versus vehicle alone. Plaque extent, dicarbonyl-lysyl adducts, efferocytosis, apoptosis, macrophage inflammation, and necrotic area were measured. Plasma MDA-LDL adducts and the in vivo and in vitro effects of PPM on the ability of HDL to reduce macrophage cholesterol were measured. Blood Ly6Chi monocytes and ex vivo 5-ethynyl-2′-deoxyuridine (EdU) incorporation into bone marrow CD11b+ monocytes and CD34+ hematopoietic stem and progenitor cells (HSPC) were also examined. IR was examined by measuring fasting glucose/insulin levels and tolerance to insulin/glucose challenge.
Results
PPM reduced the proximal aortic atherosclerosis by 48% and by 46% in female and male Ldlr−/− mice, respectively. PPM also decreased IR and hepatic fat and inflammation in male Ldlr−/− mice. Importantly, PPM decreased plasma MDA-LDL adducts and prevented the accumulation of plaque MDA- and IsoLG-lysyl adducts in Ldlr−/− mice. In addition, PPM increased the net cholesterol efflux capacity of HDL from Ldlr−/− mice and prevented both the in vitro impairment of HDL net cholesterol efflux capacity and apoAI crosslinking by MPO generated hypochlorous acid. Moreover, PPM decreased features of plaque instability including decreased proinflammatory M1-like macrophages, IL-1β expression, myeloperoxidase, apoptosis, and necrotic core. In contrast, PPM increased M2-like macrophages, Tregs, fibrous cap thickness, and efferocytosis. Furthermore, PPM reduced inflammatory monocytosis as evidenced by decreased blood Ly6Chi monocytes and proliferation of bone marrow monocytes and HSPC from Ldlr−/− mice.
Conclusions
PPM has pleotropic atheroprotective effects in a murine model of familial hypercholesterolemia, supporting the therapeutic potential of reactive dicarbonyl scavenging in the treatment of IR and atherosclerotic cardiovascular disease.
Keywords: Cholesterol efflux, High-density lipoprotein (HDL), Malondialdehyde (MDA), Macrophage, Myeloperoxidase (MPO), 5′-O-Pentyl-pyridoxamine (PPM)
Abbreviations: AUC, Area under the curve; Arg1, Arginase1; CCR2, C–C chemokine receptor 2; CD, Chow diet; COX, Cyclooxygenase; EdU, 5-ethynyl-2′-deoxyuridine; FH, Familial hypercholesterolemia; GTT, Glucose tolerance test; HSPC, Hematopoietic stem and progenitor cells; H&E, Hematoxylin and eosin; HDL, High density lipoprotein; HOMA-IR, Homeostatic model assessment for insulin resistance; HOCl, Hypochlorous acid; IR, Insulin resistance; ITT, Insulin tolerance test; IsoLG, Isolevuglandinst; LDL, Low density lipoprotein; MDA, Malondialdehyde; MPO, Myeloperoxidase; NETs, neutrophil extracellular traps; PPM, 5′-O-pentyl-pyridoxamine; ONE, 4-Oxo-nonenal; ROS, Reactive oxygen species; RCT, Reverse cholesterol transport; T2D, Type 2 diabetes; WD, Western diet
Graphical abstract
Highlights
-
•
Reactive dicarbonyl scavenging with PPM prevents atherosclerosis in Ldlr−/− mice.
-
•
PPM improves insulin sensitivity and decreases hepatic fat in male Ldlr−/− mice.
-
•
PPM increases the net cholesterol capacity of HDL from Ldlr−/− mice.
-
•
PPM promotes plaque stability as evidenced by increased efferocytosis, Tregs, and collagen.
-
•
PPM prevents monocytosis by impairing CD11b+ monocyte and CD34+HSPC proliferation.
1. Introduction
Atherosclerosis is a systemic inflammatory disorder that initiates early in life and develops in the arterial wall over decades. Clinical and mechanistic studies have shown that many factors facilitate the development of atherosclerosis, presenting potential targets for therapeutic modification. Although clinical trials of lowering LDL-C have consistently been demonstrated to reduce cardiovascular events, a considerable residual risk of cardiovascular disease still exists, which may be due in part to inflammation [[1], [2], [3]]. This residual inflammatory risk highlights the need to find novel therapeutic approaches for targeting atherosclerosis to achieve more effective reductions in cardiovascular risk.
Pleotropic functions of HDL, including anti-inflammatory function and mediation of reverse cholesterol transport (RCT), are central in protecting against cardiovascular disease [4]. Cholesterol efflux capacity, a measure of HDL function in the first step of RCT, was reported to be inversely associated with measures of cardiovascular events, independent of HDL-C levels [[5], [6], [7]]. Recent observations support the concept that qualitative changes in HDL composition promote its atheroprotective functions [8,9]. HDL is also identified as an important target for treatment of type 2 diabetes (T2D), which is associated with increased risk for atherosclerotic cardiovascular events. HDL promotes the uptake of glucose in skeletal muscle and increases the synthesis and secretion of insulin from pancreatic β cells and thus may have beneficial effects on glycemic control [10,11]. Clinical studies found that reconstituted HDL increased plasma insulin and decreased plasma glucose in patients with T2D by activating AMP-activated protein kinase [11,12]. Thus, mounting evidence demonstrates that HDL function is crucial in chronic inflammatory disorders, such as T2D and atherosclerosis.
Studies have shown that myeloperoxidase (MPO) promotes the formation of dysfunctional HDL and there is abundant dysfunctional HDL in human atherosclerotic lesions [[13], [14], [15]]. Several reactive dicarbonyls including malondialdehyde (MDA), 4-oxo-nonenal (ONE), and isolevuglandins (IsoLG) are endogenously formed during lipid peroxidation. Immunization with MDA epitopes leads to atheroprotection in mouse and rabbit models [16,17], highlighting the importance of MDA epitopes in atherosclerosis. Synthetic MDA-lysine can promote monocyte activation and vascular complications via induction of inflammatory pathways [18]. Modifying apoAI or HDL by MDA or IsoLG blocks cholesterol efflux from macrophages [[19], [20], [21]]. In addition to lipid dicarbonyl modification, myeloperoxidase (MPO) mediated chlorination of tyrosine and oxidation of tryptophan in apoAI impairs cholesterol efflux [14,15]. Furthermore, dysfunctional apoAI is present in human atherosclerotic lesions including MDA modified HDL and chlorinated and oxidized apoAI residues [14,15,21]. Interestingly, our recent studies showed that IsoLG, MDA, and ONE modification of HDL reduces its anti-inflammatory function in macrophages treated with lipopolysaccharide (LPS) [19,20,22]. Importantly, HDL from subjects with familial hypercholesterolemia (FH) versus normocholesterolemic controls has reduced cholesterol efflux capacity and anti-inflammatory function and contains significantly more IsoLG, MDA, and ONE adducts, which likely contribute to the dysfunction of FH-HDL [19,20,22]. In addition, HDL from humans with coronary artery disease versus control subjects have increased MDA adducts, which results in reduced endothelial anti-inflammatory and repairing effects [23]. Taken together, mounting evidence supports the concept that HDL atheroprotective function can be impaired and that reactive dicarbonyls such as MDA, ONE and IsoLG and other oxidative modifications of apoAI contribute to HDL dysfunction, insulin resistance (IR), and the development of atherosclerosis.
In the current studies, we utilized a potent reactive dicarbonyl scavenger, 5′-O-pentyl-pyridoxamine (PPM), to dissect the role of reactive carbonyl species in causing HDL dysfunction in promoting the development of IR and atherosclerosis. Our earlier studies demonstrated that, compared to lysine residues, PPM reacts nearly 2000 times faster with the reactive dicarbonyl, IsoLG, thereby preventing lysine modification [24]. We found that scavenging reactive dicarbonyls with PPM prevents MPO-mediated impairment of HDL cholesterol efflux capacity in vitro, and, importantly, in vivo administration of PPM to hypercholesterolemic mice increased HDL cholesterol efflux capacity without affecting plasma cholesterol levels. Consistent with the effects on HDL function, administration of PPM to Ldlr−/− mice consuming a western diet reduced IR, atherosclerotic lesion extent, lesion pro-inflammatory macrophages, blood Ly6Chi monocytosis, and lesion necrotic area. Furthermore, PPM treatment increased the plaque fibrous cap thickness and efferocytosis, thereby promoting features of plaque stability. Taken together, our results suggest that reactive dicarbonyl scavenging has high therapeutic potential in improving inflammation, preventing HDL dysfunction, IR, and atherosclerosis development.
2. Material and methods
2.1. Mice
Apoe−/−and Ldlr−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were maintained in micro-isolator cages with ad libitum access to a rodent chow diet containing 4.55% fat (PMI 5010, St. Louis, Mo) and water. The Institutional Animal Care and Use Committee of Vanderbilt University Medical Center approved mouse protocols. Experimental procedures and animal care were performed according to the regulations of Vanderbilt University's Institutional Animal Care and Usage Committee.
2.2. Treatment of Ldlr−/− mice with PPM to examine the impact on atherosclerosis
5′-O-pentyl-pyridoxamine (PPM) was solubilized in water and prepared as a 100 mM stock and stored as small aliquots at -80 °C until use. Fresh working solutions were prepared before each assay and diluted in water to the appropriate concentrations. As recommended by the scientific statement on animal atherosclerosis studies from the American Heart Association, studying the atherosclerosis in both male and female mice is a standard procedure to evaluate potential gender specific contributions to atherosclerosis. Therefore, for the atherosclerosis studies, female and male Ldlr−/− mice on a western diet (WD) were treated with water alone or with 1 g/L of PPM in the drinking water for 16 weeks after pretreatment of mice on chow diet with PPM or 2 weeks. The % by weight composition of the WD (Harlan, TD.88,137) used in the experiments was 19.5% Casein, 17.3% protein, 0.3% DL-methionine, 34% sucrose, 21% anhydrous milk fat, 0.2% cholesterol, 5% cellulose (fiber), 3.5% mineral mix (AIN-76, 170,915), 0.4% calcium carbonate, 1% vitamin mix (Teklad, 40,060), 15% corn starch, and 0.004% ethoxyquin.
2.3. Reagents
Fresh human HDL was isolated from normal subjects by sequential density ultracentrifugation as previously described [20]. Maloncarbonyl bis-(dimethylacetal) (cat#: 820,756) and H2O2 (cat#: 216,763) were purchased from Sigma–Aldrich (St. Louis, MO). The chemiluminescent Western Lightning ultra-reagent was obtained from PerkinElmer Inc. (Waltham, MA). Primary antibody to apoAI (cat#: NB600-609) CCR2 (cat#: NBP2-67700) were purchased from Novus Biologicals. Primary anti-MOMA2 (cat#: MCA519G) was purchased from BioRad. Secondary antibody HRP conjugated-rabbit anti-goat IgG (cat#: AD106P) and primary anti-MPO antibody (cat#: AF3667) were purchased from Sigma and R&D systems, respectively. Arg1 antibody (cat#: GTX109242) was purchased from GeneTex. Secondary fluorescent antibodies (FITC goat-anti-rabbit Secondary antibody, cat#: A11034; AF568 Donkey-anti-rabbit Secondary antibody, cat#: A10042; AF488 Donkey-anti-rat Secondary antibody, cat#: A21208) for immunofluorescence staining were obtained from Invitrogen.
2.4. Effect of PPM on modification of HDL with MDA
HDL modification with MDA was done in 50 mM phosphate buffer (pH 7.0) containing 100 μM DTPA. For the PPM inhibition reaction, 1 mM PPM or 5 mM PPM was incubated with 1 mg/mL HDL (cat#: J64903) for 10 min and then 250 μM MDA was added to initiate the reaction. The reactions were carried out at 37 °C for 24 h and stopped by dialysis against PBS overnight.
2.5. Effect of PPM on modification of HDL with MPO
The MPO-mediated HDL modification reactions were done in phosphate buffer (50 mM, pH 7.0) containing 100 μM DTPA, 1 mg/mL of HDL protein (cat#: J64903), 70 nM purified human MPO (Athens Research, cat#: 16-14-130,000, A430/A280 ratio of 0.79), and 100 mM NaCl [25]. For the PPM inhibition reaction, 1 mM PPM was incubated with 1 mg/mL HDL for 10 min and then 70 nM MPO, 160 μM H2O2, and 100 mM NaCl were added to initiate the reaction. The reactions were incubated at 37 °C for 1 h. In all reactions, working solutions of H2O2 were made fresh daily by diluting 30% H2O2 according to the extinction coefficient for H2O2 at 240 nm, 39.4 M−1cm−1 [26].
2.6. Effect of PPM on crosslinking of lipid-free ApoAI with hypochlorous acid
The hypochlorous acid (HOCl)-mediated ApoAI modification reactions were performed in 50 mM phosphate buffer (pH 7.0) containing 100 μM diethylenetriaminepentaacetic acid. For the PPM inhibition reaction, PPM was incubated with 0.2 mg/mL ApoAI for 10 min and then 50 μM HOCl were added to initiate the reaction. The reactions were carried out at 37 °C for 1 h. The samples were immediately used for western blot to determine the effect of PPM on crosslinking of lipid-free ApoAI.
2.7. Isolation of peritoneal macrophages
Peritoneal macrophages were isolated 4 days after intraperitoneal injection of 5% of thioglycolate. Peritoneal cells were collected by rinsing with Ca2+ and Mg2+ free PBS and then centrifuged and resuspended in DMEM containing 10% heat-inactivated FBS (Sigma Aldrich, cat#: B207151) and 100 units/ml penicillin/streptomycin. Cells were plated onto 24-well culture plates (1∗106 cells/well) and allowed to adhere for 2 h. Nonadherent cells were removed by washing two times with DPBS, and adherent macrophages were used for experiments.
2.8. Serum lipid measurements, FPLC lipoprotein profile, and ApoB-depleted serum preparation
Serum total cholesterol and triglyceride (TG) levels were measured by the enzyme-based method (Fisher, cat#: 890,339 RC) after overnight fasting. Lipid profiling of FPLC-separated lipoprotein fractions was done using gel filtration analysis with a Superose 6 column on a Waters 600 FPLC system. Mouse serum (100 μl) was injected onto the column and separated with buffer containing 0.15 M NaCl, 0.01 M Na2HPO4, and 0.1 mM EDTA (pH 7.5), at a flow rate of 0.5 mL/min. Cholesterol content of the fractions was determined using an enzymatic assay (Fisher, cat#: 891,069 RA) [27]. ApoB-depleted serum was prepared by adding 200 μL of PEG 8,000 (20% in 200 mM glycine) to 500 μL serum [28,29]. After incubation for 15 min at room temperature, and then centrifugation at 1,900 g for 20 min, the supernatant containing the apoB-depleted serum was collected and used for cholesterol efflux experiments [30].
2.9. Measurement of HDL capacity to reduce macrophage cholesterol
To measure the capacity of HDL to reduce macrophage cholesterol content, an enzymatic cholesterol assay was used as described [31,32]. Briefly, Apoe−/− macrophages were incubated for 48 h with DMEM containing acetylated LDL (Alfa aesar, cat#: J65029, 40 μg/ml). The cells were then washed and incubated for 1 h in DMEM containing 0.1% BSA. The cells were then washed and incubated for 24 h in DMEM supplemented with HEPES in the presence of 50 μg/mL human HDL, modified HDL, or 2.5% apoB-depleted serum from mice. Cellular cholesterol was measured after incubation with DMEM alone or with cholesterol acceptor using an enzymatic cholesterol assay [31,32]. Briefly, isopropanol/NP-40 (vol/vol:9/1) was added to the dried isopropanol lipid extracts and vortexed for 10 s. Then 40 ul of each sample was added to 96-well plates and catalase solutions (Sigma, cat#:C1345) in 50 mM potassium phosphate buffer were added to the wells for both standards and samples to lower the background. Then, 100 ul of the enzyme and fluorescent probe mixture (ADHP, cat#: AnaSpec # 85,500; cholesterol oxidase, Cat#: C8649–100U; cholesterol esterase, Cat#:C9281–100UN; horseradish peroxidase, Cat#: P8375-1kun) were prepared in reagent A buffer (sodium chloride, cholic acid, Triton-100 in 100 mM potassium phosphate buffer) and added into each well. The fluorescence intensity (excitation wavelength 530 nm, emission wavelength 580 nm) was measured immediately after incubation at 37 °C for 25 min s. The cholesterol efflux capacity was calculated as ((cellular cholesterol with DMEM alone - cellular cholesterol with acceptor)/cellular cholesterol with DMEM alone ∗ 100).
2.10. Western blotting
Samples were incubated with β-mercaptoethanol and loading buffer for 10 min at 55 °C and then resolved by NuPAGE Bis-Tris electrophoresis using 4–12% gels for 1.5 h. The proteins were transferred onto PVDF membranes (Amersham Bioscience) at 150 V for 1.5 h. Membranes were blocked with 5% milk at room temperature for 2 h and incubated with primary antibodies specific to human apoAI or MDA overnight at 4 °C. The secondary antibodies conjugated with HRP were incubated with the membranes for 2 h at room temperature. Protein bands were visualized with ECL western blotting detection reagents (GE Healthcare) and quantified using ImageJ software (NIH).
2.11. Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Insulin levels were measured by ELISA (Mercodia, cat#: 10-1113-01). For the GTT, mice were fasted for 4 h, and 10 μL blood was taken from the tail vein for measurement of baseline levels. Mice were then injected i.p. with 1 g/kg of glucose. Blood samples were then taken at 15, 30, 45, 60, 90, 120 min and analyzed for blood glucose with a clinical meter [33]. For ITT, mice were fasted for 4h, and after taking a baseline blood sample, the mice were injected intraperitoneally with insulin (0.5 U/kg). Blood samples were then taken at 15, 30, 60, 90, 120 min and analyzed for blood glucose with a clinical meter [33]. The homeostatic model assessment for insulin resistance (HOMA-IR) was calculated using the equation fasting insulin (μU/ml) × fasting plasma glucose (mmol/liter)/22.5.
2.12. Hematoxylin and eosin (H&E)
Staining of liver tissue and NAFLD severity score H&E staining of liver tissue was performed as previously described [34]. Briefly, livers were immediately embedded in paraffin, sectioned, and mounted on glass microscope slides. The sections were stained with hematoxylin and eosin and examined using light microscopy. Nonalcoholic fatty liver disease severity score, which estimates the severity of fatty liver by the summation of steatosis, lobular inflammation, and hepatocyte balloon degeneration, was evaluated semi-quantitatively: based on the severity of steatosis, lobular inflammation and hepatocellular ballooning.
2.13. Measurement of atherosclerotic lesions and immunofluorescence analyses
We followed the guidelines for development and execution of experimental design and interpretation of atherosclerosis in animal studies [35]. The mean lesion area from 15 serial sections was used for the aortic root atherosclerotic lesion size per mouse as described [36]. Immunofluorescence staining was performed using sections that were 40–60 μm distal of the aortic sinus. Briefly, aortas were flushed with PBS through the left ventricle, and the entire aorta was dissected and en face was stained for the neutral lipid using Sudan IV as described [36]. The extent of atherosclerosis was examined by Oil-Red-O staining of neutral lipids in cross-sections of the proximal aorta (15 alternate 10 μm cryosections for each mouse aorta). Images of the proximal and distal aorta lesions were analyzed using the KS300 imaging system (Kontron Electronik GmbH) [37,38]. MDA was detected by immunofluorescence staining using anti-MDA primary antibody (FITC, Abcam#: ab27615). Immunofluorescence staining was used to examine the levels of MOMA2, MPO, arginase 1 (Arg1), and C–C chemokine receptor 2 (CCR2) in atherosclerotic lesions [37]. For each mouse, two sections were stained, and quantitation was performed on the entire cross section of all stained sections. Briefly, 5 μm cross-sections of the proximal aorta were fixed in cold acetone (Sigma), blocked with 5% goat serum dilution (GSD), and incubated overnight at 4 °C with primary antibody to MOMA2, MPO, Arg1, or CCR2. After incubation for 1h at 37 °C with the fluorescent labeled secondary antibody, the nuclei were stained with Vectashield Mounting medium with DAPI (Vector, cat#: H1200). The images were captured using an Olympus IX81 fluorescence microscope with SlideBook 6 (Intelligent-Image) software and quantified using ImageJ software (NIH). The apoptotic cells and efferocytosis in atherosclerotic lesions were measured by TUNEL and macrophage staining of cross-sections of the proximal aorta. Briefly, 5 μm sections were fixed in 2% paraformaldehyde, treated with 3% citric acid, and then stained using the in situ TMR red cell death detection kit (Roche Applied Science, cat#: 12156792910) [39]. Macrophages were stained with anti-MOMA2 antibody and nuclei were counterstained using Hoechst. TUNEL-positive (TUNEL+) cells and efferocytosis were then quantitated in proximal aortic sections as previously described [39].
2.14. Masson's trichrome staining
Masson's Trichrome staining procedure (Sigma, cat#: HT15-1 KT) was used for measurement of lesion necrotic core size and fibrous cap thickness as previously described [36,39] Briefly, 5 μm sections of the proximal aorta were fixed for 30 min with Bouin's solution and stained with hematoxylin for nuclei (black). Biebrich scarlet solution and phosphotungstic/phosphomolybdic acid were added to stain cytoplasm (red), and Aniline blue solution was applied to stain collagen (blue). Images were obtained and analyzed for fibrous cap thickness and % necrotic area using ImageJ software (NIH).
2.15. Measurement of prostaglandin (PG) metabolites
Male Ldlr−/− mice were given vehicle (n = 8) or 1 mg/mL PPM (n = 8) for 8 weeks. Urine was collected for 6 h in metabolic cages with 2 mice per cage after 8 weeks of treatment with PPM and analyzed by LC/MS for 2,3-dinor-6-ketoPGF1 (2,3DN) and 11-dehydro TxB2 (11dTxB2) by the Eicosanoid Analysis Core at Vanderbilt University. The creatinine levels are measured using a kit (Enzo Life Sciences; cat # ADI-907-030A). The urinary metabolite levels in each sample were normalized using the urinary creatinine level of the sample and expressed as ng/mg creatinine.
2.16. Measurements of aortic ONE-lysyl adducts and IsoLG-lysyl-lactam adducts
The aortic tissues were isolated, homogenized and then the ONE-Lysyl and IsoLG-lysyl adducts were measured by LC/MS/MS using a Waters Xevo-TQ-Smicro triple quadrupole mass spectrometer as previously described [36].
2.17. RNA isolation and quantitative RT-PCR
Total tissue and cell RNA were extracted and purified using PureLink RNA Mini kit (Invitrogen 12183018A) as described in the manufacturer's protocol. Complementary DNA was synthesized with iScript reverse transcriptase (Bio-Rad). Relative quantitation of the target mRNA was performed using specific primers, SYBR probe (Bio-Rad) and iTaqDNA polymerase (Bio-Rad) on IQ5 Thermocylcer (Bio-Rad). The data were analyzed using the 2-ΔΔCt method, and the results of are presented as the fold change of target gene expression in a sample relative to a reference sample, normalized to a reference gene (18S) [36,40].
2.18. Flow analysis of white blood cells
To measure the numbers of blood neutrophils, Ly6Chi monocytes, and Ly6Clow monocytes in Ldlr−/− mice, blood cells were collected and stained with a cocktail of antibodies targeting these cells [37]. Briefly, 100 μL of blood was collected using Natelson Blood collection tubes, and then incubated with Fc inhibitor CD16/32 for 10 min at RT. Lysis buffer (3 mL) was pre-warmed at 37 °C, added to the blood samples, and incubated at room temperature for 30 min to lyse red blood cells. Samples were centrifuged at 1200 rpm for 10 min, and the cell pellets were re-suspended with 300 ul of FACS buffer (2% FBS in PBS). Antibodies Pacific blue Ly6G (1A8, Biolegend), PE-Cy7 CD11b (M1/70, Biolegend), PE anti-mouse CD3 Antibody (17A2, Biolegend), FITC Anti-mouse CD19 for B cells (6D5, Biolegend) and APC/Cy7 anti-mouse Ly6C (HK1.4, Biolegend) were added, and then incubated with cells on ice for 45 min. The blood T regulatory (Treg) and T helper 2 (Th2) cells were detected using antibodies, where CD3+FoxP3+ positive cells were determined as Treg cells and CD4+GATA-3+ positive populations were Th2 cells. For all flow cytometry assays where propidium iodide or 7-AAD dye were not used, we used gates based on the forward and side scatter to remove dead cells and debris for cell viability from the analysis. Cells were washed with FACS buffer twice and analyzed using FACS DiVa v6.1 software (BD Biosciences) in the Research Flow Cytometry Core Laboratory, Veterans Administration Medical Center.
2.19. Bone marrow cell analyses and proliferation of monocytes and HSPC
The bone marrow cells were treated with 5 μm EdU for 24 h to detect proliferation. The cells were then washed, blocked with Fc inhibitor CD16/32, and stained with FITC anti-mouse CD11b (monocyte marker) or CD34 (HSPC marker, eBioscience, Cat#: 2356471), and EdU positive cells were detected using Click-iT EdU Flow Cytometry Assay Kit (Invitrogen, C10646). Briefly, cells were fixed and permeabilized, and stained with Alexa Fluor 594 picolyl azide in Click-iT EdU buffer following the manufacturer's protocol. For the fluorescence microscopy assay, the stained cells were placed on glass slides, the images captured, and dual CD11b/EdU positive cells were counted as proliferating HSPC. For the flow cytometry assay, dual CD34/EdU positive cells were considered as proliferating HSPC using FACS DiVa v6.1 software and equipment (BD Biosciences).
2.20. Statistics
The data are presented as mean ± SEM visualized by either box plots or bar charts. The normality of the sample populations was examined by the Kolmogorov–Smirnov test. Between-group differences were assessed with two-sided unpaired t-test (2 groups) and one-way ANOVA (>2 groups, Bonferroni's correction for multiple comparisons). Their nonparametric counterparts, Mann–Whitney test (2 groups) and nonparametric Kruskal–Wallis test (more than 2 groups, Bunn's correction for multiple comparison), were used when assumptions for parametric methods were not satisfied. The statistical analysis was performed using Graphpad Prism 7. Statistical tests were determined to be statistically significant at two-sided significance level of 0.05 after correction for multiple comparisons (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001).
3. Results
3.1. Effects of PPM on ApoAI crosslinking, HDL cholesterol efflux capacity, and MDA-LDL adducts
As MDA-apoAI adducts have been localized to atherosclerotic lesions [21], and oxidation of HDL lipids using a MPO/H2O2/Cl− system promotes formation of apoAI-MDA adducts [19], we examined the in vitro effects of PPM on MDA-mediated apoAI crosslinking and impairment of net cholesterol efflux capacity. Modification of HDL with 250 μM MDA increased the degree of apoAI crosslinking (Figure 1A and B) and decreased the capacity of the HDL to reduce Apoe−/− macrophage cholesterol content by 64% (Figure 1C). Addition of PPM significantly decreased the MDA-mediated crosslinking of apoAI (Figure 1A and B) and improved the impairment of HDL net cholesterol efflux capacity (Figure 1C). Besides lipid dicarbonyl modification of apoAI, the MPO generated oxidant, hypochlorous acid (HOCl), can also directly modify apoAI resulting in chlorinated tyrosine or oxidized tryptophan residues, which promote crosslinking and impair cholesterol efflux [[13], [14], [15]]. As PPM is an electron rich molecule that may readily react with HOCl [41], we investigated whether PPM could also inhibit HOCl mediated crosslinking of lipid-free apoAI (Figure 1D). Similar to other studies [[13], [14], [15]], when lipid-free apoAI was incubated with HOCl, there was extensive crosslinking, and consistent with PPM neutralization of HOCl [41], PPM prevented HOCl mediated crosslinking of lipid-free apoAI (Figure 1D). In addition, MPO-mediated HDL modification reduced the net cholesterol efflux capacity by 54.5% and PPM prevented any impairment in HDL cholesterol efflux capacity in Apoe−/− macrophages (Figure 1E). More importantly, administration of PPM to Ldlr−/− mice on a western diet increased the net cholesterol efflux capacity of HDL (Figure 1F) showing that PPM scavenging in vivo preserves HDL function. As apoB adducts increase foam cell formation and are localized to plaques [[42], [43], [44]], we examined the effects of PPM on LDL MDA content. Compared to treatment with water alone, PPM reduced the MDA-LDL adducts by 40% in Ldlr−/− mice on a WD for 16 weeks (Figure 1G) [9,19].
Figure 1.

PPM prevents MPO- and MDA-mediated HDL modification and preserves HDL cholesterol efflux capacity. A-B. PPM prevents the crosslinking of apoAI on HDL caused by MDA modification. C. PPM prevents MDA mediated HDL cholesterol efflux dysfunction in Apoe−/− macrophages. Cholesterol-enriched Apoe−/− macrophages were incubated for 24h with DMEM alone or containing 50 μg HDL protein/ml, and the % decrease in cellular cholesterol was then measured as described in the Methods. D. PPM prevents the HOCl mediated crosslinking of lipid-free apoAI. E. PPM prevents MPO-mediated HDL modification and preserves HDL cholesterol efflux capacity in Apoe−/− macrophages. Cholesterol-enriched Apoe−/− macrophages were incubated for 24h with DMEM alone or containing HDL (50 μg protein/ml) and the % decrease in cellular cholesterol was measured. F. PPM treatment improves the cholesterol efflux capacity of HDL in Ldlr−/− mice. Cholesterol-enriched Apoe−/− macrophages were incubated for 24h with DMEM alone or containing 2.5% (vol/vol) apoB-depleted serum, and the cellular cholesterol was then measured. G. PPM reduces the plasma levels of MDA-LDL in Ldlr−/− mice on WD for 16 weeks. Graph data are expressed as mean ± SEM (n = 3 in each group); The in vitro studies are 3 independent experiments, ∗P < 0.05, ∗∗P < 0.01 by t-test or one-way ANOVA with Bonferroni's post hoc test.
3.2. Effect of PPM on IR and hepatic fat in Ldlr−/− mice
As oxidative stress and HDL dysfunction impact IR, we next examined whether PPM impacts insulin sensitivity in male Ldlr−/− mice, which develop IR and increased fat deposition when consuming a WD versus a chow diet [45,46]. After 16 weeks on a WD, male Ldlr−/− mice had significantly increased fasting blood glucose and insulin levels versus chow diet (CD) fed controls (Figure 2A and B). Administration of PPM to Ldlr−/− mice consuming a WD reduced the fasting glucose and insulin levels (Figure 2A and B). We also examined HOMA-IR levels as an indicator of IR and found that treatment with PPM reduced the HOMA-IR in Ldlr−/− mice on a WD to HOMA-IR values that were similar to those observed in Ldlr−/− mice consuming a chow diet (Figure 2C). To determine whether PPM improved insulin or glucose tolerance after 4 h s fasting, GTT and ITT were done after 12 weeks of male Ldlr−/− mice consuming a WD and being treated with PPM (Figure 2D–G). Treatment of Ldlr−/− mice consuming a WD with PPM nearly normalized the dynamic change of glucose levels in response to glucose challenge and significantly decreased the glucose tolerance test area under the curve (AUC) (Figure 2D and E). Consistent with the GTT results, PPM reduced the dynamic changes of glucose levels in response to insulin challenge in male Ldlr−/− mice and significantly reduced the insulin tolerance test AUC (Figure 2F and G). We next examined the effects of PPM on hepatic fat accumulation in male Ldlr−/− mice. Compared to male Ldlr−/− mice on a chow diet, the hepatic fat levels were increased by 3.6-fold in Ldlr−/− mice consuming a western diet (Figure 2H and I). Consistent with oxidative stress impacting fat deposition, PPM prevented the hepatic fat accumulation in Ldlr−/− mice on a western diet (Figure 2H and I). Similar to other reports [46,47], administration of a western versus chow diet to female Ldlr−/− mice did not affect fasting glucose levels or the response to glucose challenge, and PPM had no impact on fasting glucose levels and GTT (Figs. S1A–S1C). Taken together, these results demonstrate that scavenging reactive dicarbonyls with PPM protects against IR and hepatic fat accumulation in male Ldlr−/− mice.
Figure 2.

PPM reduces fasting glucose and insulin levels and improves insulin sensitivity in male Ldlr−/−mice. A-B. PPM reduces fasting glucose and insulin levels in male Ldlr−/− mice. C. PPM normalizes HOMA-IR values in male Ldlr−/− mice (n = 9 in group on a chow diet, n = 10 in group on a western diet, n = 9 in group on a western diet and treated with PPM). D-E. Effect of PPM on glucose levels in male Ldlr−/− mice in response to injection of glucose using IPGTT as described in Methods (n = 9–10 in each group). F-G. Effect of PPM on glucose levels in male Ldlr−/− mice in response to injection of insulin using IPITT as detailed in Methods. AUC, area under the curve. H–I. PPM prevents hepatic fat deposition in Ldlr−/− mice (n = 6), as determined by H&E staining (H) and measured using Image J (I). Scale bar = 100 μm. A.U., arbitrary units. For each experiment, graph data are expressed as mean ± SEM; ∗P < 0.05 by one-way ANOVA with Bonferroni's post hoc test.
3.3. Effects of PPM on atherosclerosis extent and plaque dicarbonyl-lysyl adducts
To examine the effect of scavenging reactive dicarbonyls on atherosclerosis, male Ldlr−/− mice were fed a WD for 16 weeks and treated with water alone or containing PPM. As shown in Figure 3A and B and S2A-S2C, PPM did not change the plasma cholesterol, TG levels, body weight, water intake, or ALT activities. Importantly, administration of PPM versus water alone reduced the extent of proximal aortic atherosclerosis by 46% (Figure 3C–D). In addition, PPM treatment reduced the en face aortic lesion content by 52% in male Ldlr−/− mice (Figure 3E–F). We next examined the effects of PPM on atherosclerosis in female Ldlr−/− mice, which studies suggest are more susceptible to atherosclerosis than male Ldlr−/− mice [48]. Similar to male Ldlr−/− mice, PPM treatment of female mice on a western diet for 16 weeks reduced the extent of proximal aortic atherosclerosis by 48% without changing plasma cholesterol, triglyceride levels, or lipoprotein FPLC profile (Figs. S3A–S3E). As with male Ldlr−/− mice, PPM did not change body weight, water intake, and ALT activities in female Ldlr−/− mice (Figs. S2D–S2F). Consistent with PPM being an effective scavenger of reactive dicarbonyls, immunohistochemistry analysis of MDA-lysyl adducts in plaques revealed that PPM significantly reduced the MDA adduct-positive area in the proximal aortic lesions of female Ldlr−/− mice (Figs. S4A–S4B). In addition, compared to water alone, treatment with PPM significantly decreased the levels of ONE-lysyl adduct in aortic lesions of male Ldlr−/− mice (Fig. S4C). PPM also impacted the levels of IsoLG-lysyl adduct in the aorta of male Ldlr−/− mice, with a trend towards reduced levels of adducts (p = 0.058, Fig. S4D). These results demonstrate that PPM treatment effectively decreases atherosclerosis development in experimental mouse models without changing plasma cholesterol and suggests that improved HDL function as well as reduced apoB modification and decreased plaque cellular adducts contribute to the impact on atherosclerosis.
Figure 3.

PPM reduces aortic root and en face atherosclerotic lesions in male Ldlr−/−mice. A-B. PPM does not affect plasma cholesterol levels or triglyceride levels in male Ldlr−/− mice. C-D. PPM reduces the aortic root atherosclerotic lesions in male Ldlr−/− mice. E&F. PPM reduces en face atherosclerotic lesion area in male Ldlr−/− mice. Male Ldlr−/− mice were pretreated with 1 g/L of PPM for 2 weeks on chow diet. Then mice were treated with 1 g/L of PPM for 16 weeks on WD. Oil-Red-O staining of atherosclerosis lesion in male Ldlr−/− mice with vehicle (n = 8) or PPM (n = 10) is shown. Scale bar = 200 μm. Data are expressed as mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 by two-sided unpaired t-test. G&H. Immunohistochemistry staining of Arg1+ macrophages was performed as described in Methods. H. PPM increases the number of Arg1+ macrophages in lesions of Ldlr−/− mice. I&J. Immunohistochemistry staining of CCR2+ macrophages was performed as described in Methods. J. PPM reduces the number of CCR2+ macrophages in lesions of Ldlr−/− mice. K&L. Pro-inflammatory enzyme MPO staining of atherosclerotic lesions in Ldlr−/− mice is shown. L. PPM reduces MPO expression in atherosclerotic lesions of Ldlr−/− mice. G, I, & K. Scale bar = 40 μm n = 6 in each group, for each experiment, graph data are expressed as mean ± SEM; ∗P < 0.05 by two-sided unpaired t-test.
3.4. In vivo and in vitro effects of PPM on macrophage inflammation
As macrophage inflammation is modulated by HDL and oxidative stress, we next examined the effects of scavenging reactive dicarbonyls on atherosclerotic lesion macrophage inflammatory markers. Immunohistochemical staining of the proximal aortic sections from male Ldlr−/− mice demonstrates that compared to water alone, PPM increased the number of anti-inflammatory Arg1 positive M2 macrophages by 120% (Figure 3G and H) and decreased the numbers of pro-inflammatory CCR2 positive macrophages by 58% (Figure 3I and J). In addition, PPM treatment reduced the expression of MPO, which is a marker of neutrophil extracellular traps (NETs) that promote the conversion of anti-inflammatory macrophages to a proinflammatory phenotype [49,50], in the proximal aortic lesions of male Ldlr−/− mice (Figures 3K and 3L). In addition, similar effects of PPM on macrophage Arg1 levels were seen in proximal aortas of female Ldlr−/− mice (Figs. S3F–S3G). Examination of the serum proinflammatory cytokines revealed that scavenging reactive dicarbonyls with PPM decreased the levels of IL-1β and TNF-α by 34% and 62% in male Ldlr−/− mice (Figures 5SA-5SB). We also examined the effects of PPM versus water alone on hepatic inflammatory markers in male Ldlr−/− mice consuming a western diet for 16 weeks. The mRNA levels of proinflammatory CCL2 and CCL4 were decreased by 20%, and 23% in PPM compared to water alone treated Ldlr−/− mice (Fig. S5C and S5D). In contrast, PPM increased the hepatic mRNA levels of anti-inflammatory Arg1 by 2.2-fold (Fig. S5E). PPM did not significantly impact the hepatic mRNA levels of CCL3 and CXCL2 in male Ldlr−/− mice (Data not shown). Importantly, PPM treatment of male Ldlr−/− mice reduced the aortic levels of IL-1β and IL-6 mRNA (Fig. S6A and S6B). Next, we examined the ex vivo effects of PPM on macrophage inflammatory markers. Thioglycolate elicited peritoneal macrophages were isolated from PPM and water alone treated male Ldlr−/− mice consuming a WD for 16 weeks and then stimulated with oxidized LDL or LPS. Macrophages from PPM versus vehicle treated Ldlr−/− mice had 28% and 33% reductions in mRNA levels of IL-1β and IL-6 in response to oxidized LDL (Figure 4A and B). In addition, there was a significant reduction in expression of IL-1β and IL-6 in LPS stimulated macrophages from PPM versus vehicle treated Ldlr−/− mice (Figure 4C and D). In contrast, PPM versus water alone administration increased Arg1 expression by 3.6-fold in LPS stimulated macrophages (Figure 4E). Importantly, in vitro incubation of macrophages with PPM and oxidized LDL compared to oxidized LDL alone markedly reduced the mRNA levels of IL-1β and TNF-α (Figure 4F and G) suggesting that PPM minimizes inflammation by preventing cellular oxidative modifications. Due to the striking impact on inflammatory cytokines and macrophage phenotype in vivo, we also measured urinary prostaglandins to evaluate whether PPM might be inhibiting cyclooxygenase (COX). Urine samples were analyzed for 2,3-dinor-6-ketoPGF1 (2,3DN) and 11-dehydro TxB2 (11dTxB2) by LC/MS/MS. We found that there were no significant differences in levels of these major urinary prostaglandin metabolites of Ldlr−/− mice treated with PPM compared to the vehicle control (Fig. S7), indicating that PPM was not significantly inhibiting COX in vivo in mice.
Figure 5.

PPM reduces the number of apoptotic cells, increases efferocytosis, and stabilizes the atherosclerotic plaque of male Ldlr−/−mice. A-B. Images depict TUNEL staining of nuclei (red) and merged images show TUNEL+, MOMA2+ (green) and Hoechst+ (blue) staining of atherosclerotic lesions in male Ldlr−/− mice. Scale bar = 50 μm. C. PPM reduces the apoptotic cells in atherosclerotic lesions of male Ldlr−/− mice (n = 6). B&D. PPM enhances macrophage efferocytosis of apoptotic cells. Higher magnification images of the TUNEL staining (B) were used to quantitate (D) efferocytosis of dead cells in aortic root sections (n = 6 per group). Efferocytosis was quantitated as the free (white arrows) vs macrophage-associated TUNEL-positive cells (yellow arrows) in the proximal aortic sections (D). E-H. The plaque stability was measured by using Trichrome staining of atherosclerotic lesions of Ldlr−/− mice (n = 6 in each group). The necrotic area (F), collagen content (G), and fibrous cap thickness (H) and necrotic area were quantitated in atherosclerotic lesions of Ldlr−/− mice (n = 6 in each group). Scale bar = 40 μm. For each experiment, graph data are expressed as mean ± SEM; ∗P < 0.05 by two-sided unpaired t-test.
Figure 4.

PPM reduces the macrophage inflammatory response to oxidized LDL and LPS. A-E. The ex vivo effect of PPM on the expression of inflammatory genes in macrophages incubated with either oxidized LDL (A–B) or LPS (C–E). A-E. Macrophages were isolated from male Ldlr−/− mice that were fed a western diet for 16 weeks and were treated with vehicle alone or with 1 g/L of PPM. The cells were then incubated for 24h with 40 μg/mL of oxidized LDL (A–B) or 4h with 100 ng/ml of LPS (C–E). A-B. In vivo treatment with PPM reduces the mRNA levels of IL-1β and IL-6 in Ldlr−/− macrophages stimulated with oxidized LDL. C-E. PPM reduces the ex vivo expression of IL-1β and IL-6 and increases Arg1 mRNA levels in Ldlr−/− macrophages in response to LPS. A-E. The mRNA levels were measured by real-time PCR as described in Methods. n = 7 in each group, for each experiment, graph data are expressed as mean ± SEM; ∗P < 0.05 by two-sided unpaired t-test. F-G. PPM reduces the mRNA expression levels of IL-1β and TNF-α in Apoe−/− macrophages in response to 24h with 40 μg/mL ox-LDL. n = 3 independent experiments. Data are expressed as mean ± SEM; ∗P < 0.05, ∗∗P < 0.01, by two-sided unpaired t-test.
3.5. Effects of PPM on plaque cell death, efferocytosis, and features of stability
As plaque inflammation and oxidative stress induces cellular death, we next investigated the effects of PPM on lesion cell death. PPM versus vehicle treatment decreased the number of dead macrophages as evidenced by TUNEL staining in the proximal aortic lesions in both male and female Ldlr−/− mice (Figure 5A–C and Fig. S8A and S8B). From the costaining of TUNEL and live macrophages, the effect of PPM on efferocytosis of apoptotic cells in male and female Ldlr−/− mouse plaques was also examined (Figure 5A,B, and 5D and Fig. S8A and S8C). The number of TUNEL-positive cells not associated with macrophages was decreased by 40% in lesions of male mice treated with PPM versus vehicle (Figure 5D). Similar results on the effects of PPM on plaque efferocytosis were observed in female Ldlr−/− mice (Fig. S8A and S8C). Consistent with the decreased inflammatory cell death and enhanced efferocytosis, PPM versus vehicle treatment decreased the necrotic area by 51.6% and increased the collagen content and fibrous cap thickness by 107.3% and 41.9% in male Ldlr−/− mice, respectively (Figure 5E–H).
3.6. Effects of PPM on plaque T and B cells
As macrophage inflammation and efferocytosis are impacted by T cell populations, we next examined the effects of PPM on plaque T cells. There were similar total numbers of T cells (CD3+) in plaques of PPM versus control treated female Ldlr−/− mice consuming a western diet for 14 weeks (Figure 6A and B). Interestingly, the number of Th2 cells (GATA-3+) were increased by 2.2–fold in plaques of Ldlr−/− mice (Figure 6C and D). In addition, the number of Tregs (Fox3P+) which promote efferocytosis and reduce inflammation were increased by 2.1–fold in PPM versus control Ldlr−/− mice (Figure 6E and F). PPM did not affect the number of B cells (B220+) in plaques of Ldlr−/− mice (Fig. S9A and S9B). Taken together, our data show that PPM suppresses the features of vulnerable plaques in a hypercholesterolemic murine model.
Figure 6.

PPM increases the number of Treg and Th2 lymphocytes in plaques of Ldlr−/−mice. A-B. Immunohistochemistry staining and quantitation of CD3+ (green) T lymphocytes in atherosclerotic lesions of female Ldlr−/− mice. Scale bar = 50 μm. C-D. Immunohistochemistry staining and quantitation of GATA-3+ (green) Th2 lymphocytes in atherosclerotic lesions of female Ldlr−/− mice. Scale bar = 50 μm. E-F. Immunohistochemistry staining of FoxP3+ (green) Treg cells. F. PPM increases the number of Treg cells in plaques of Ldlr−/− mice. Scale bar = 50 μm. For each experiment (n = 6 or 7 in each group), graph data are expressed as mean ± SEM; ∗P < 0.05 by two-sided unpaired t-test.
3.7. Effects of PPM on white blood cell populations in Ldlr−/− mice
Studies have demonstrated that Ly6Chi monocytes preferentially infiltrate the arterial wall and convert to inflammatory M1-like macrophages during inflammation thereby promoting progression of the atheroma [37,51,52]. As inflammatory Ly6Chi monocytosis is reduced by HDL cholesterol efflux and enhanced by oxidative stress, we next examined the effects of reactive dicarbonyl scavenging on subsets of white blood cells in female Ldlr−/− mice consuming a western diet for 14 weeks (Figure 7A and B). Interestingly, we found that compared to water alone, PPM treatment of female Ldlr−/− mice decreased the total number of blood monocytes and markedly reduced the number of Ly6Chi monocytes (Figure 7A–D). In addition, PPM did not affect the number of blood Ly6Clow cells but decreased the ratio of Ly6Chi to Ly6Clow monocytes (Figure 7E and F) in female Ldlr−/− mice. A similar impact of PPM treatment on total monocytes, Ly6Chi cells and Ly6Clow cells was observed in male Ldlr−/− mice (Fig. S10A–S10D). In both female and male Ldlr−/− mice, PPM treatment did not impact the number of circulating neutrophils, T cells, and B cells (Fig. S11A–S11F). Similar to the atherosclerotic lesions, the number of anti-inflammatory Th2 (GATA-3+) and Treg (Fox3P+) cells were increased in blood from PPM versus water alone treated female Ldlr−/− mice (Fig. S12A–S12C).
Figure 7.

PPM decreases the number of blood Ly6Chimonocytes and the ratio of Ly6Chito Ly6cLowmonocytes in female Ldlr−/−mice. A-B. The representative flow cytometry gating strategies for monocyte subsets, neutrophils, T cells, and B cells in peripheral blood of vehicle alone or PPM treated female Ldlr−/− mice fed a western diet for 14 weeks are shown. C. There was a reduction in total blood monocytes in PPM versus water treated Ldlr−/− mice. D. PPM reduced the number of Ly6Chi monocytes E. PPM did not impact the number of Ly6C low monocytes. F. PPM decreased the ratio of Ly6Chi to Ly6C low monocytes. n = 9 in each group. Graph data are expressed as mean ± SEM; ∗P < 0.05 by two-sided unpaired t-test.
3.8. Effects of PPM on monocyte and HSPC proliferation in Ldlr−/− mice
We next examined the impact of PPM on bone marrow Ly6Chi monocytes. Compared to male and female Ldlr−/− mice on a chow diet, the numbers of bone marrow Ly6Chi monocytes increased in male and female Ldlr−/− mice fed a western diet for 16 weeks (Figure 8A and B). Consistent with the decreased numbers of blood Ly6Chi monocytes in PPM treated Ldlr−/− mice, the number of bone marrow Ly6Chi monocytes trended higher in water alone versus PPM treated male and female Ldlr−/− mice fed a western diet (Figure 8A and B). When the Ly6Chi monocyte data for male and female Ldlr−/− mice were grouped together, the number of bone marrow Ly6Chi monocytes was significantly reduced in PPM versus water alone treated Ldlr−/− mice (Figure 8C). Consistent with studies demonstrating that oxidative stress enhances inflammatory monocytosis [53], the number of Ly6Chi monocytes were markedly increased in white blood cells incubated with H2O2 compared with medium alone (Figure 8D). PPM prevented the H2O2 induced increase in Ly6Chi monocytes (Figure 8D). Examination of the proliferation of monocytes, by Edu incorporation and fluorescence microscopy, in bone marrow cells isolated from male Ldlr−/− mice fed a western versus chow diet revealed increased proliferation of CD11b + monocytes after administration of a western diet (Figure 8E and F). Importantly, PPM treatment of male Ldlr−/− mice decreased the western diet induced increase in proliferation of bone marrow monocytes (Figure 8E and F). Similar results were observed using fluorescence microscopy to examine the effects of PPM on monocyte proliferation in bone marrow cells isolated from female Ldlr−/− mice fed a western diet (Fig. S13A and S13B). In addition, measurement of monocyte proliferation as measured by Edu incorporation into CD11b + cells and flow cytometry revealed similar results with bone marrow isolated from female Ldlr−/− mice (Fig. S13C and S13D). Next, we examined the effects of PPM on proliferation of CD34+ hematopoietic stem and progenitor cells. Compared to the proliferation of CD34+ HSPC in bone marrow isolated from chow fed male Ldlr−/− mice, HSPC proliferation was enhanced in bone marrow from mice consuming a western diet (Figure 8G and H). Administration of PPM to the male Ldlr−/− mice prevented the increase in proliferation of bone marrow CD34+ HSPC (Figure 8G and H). Similar effects with PPM treatment on the proliferation of CD34+ HSPC were seen in bone marrow isolated from female Ldlr−/− mice (Fig. S13E and S13F). Collectively, we demonstrate that scavenging reactive dicarbonyls improves HDL function, modulates macrophage polarization, represses Ly6Chi monocytosis, and decreases lesion macrophage inflammation and death resulting in increased plaque stability.
Figure 8.

PPM decreases bone marrow Ly6Chimonocytes, oxidative stress induced accumulation of blood Ly6Chimonocytes, and the proliferation of bone marrow monocytes and HSPC. A-C. PPM decreases the number of bone marrow Ly6Chi monocytes. Male (A) or female (B) Ldlr−/− mice were fed a chow or a western diet for 16 weeks and treated with vehicle alone or PPM. The bone marrow was isolated and the number of Ly6Chi monocytes was measured by flow cytometry as described in the Methods. C. The data for male (n = 4 or 5 per group) and female (n = 9 per group) bone marrow Ly6Chi monocytes were grouped. Data are expressed as mean ± SEM; ∗P < 0.05, ∗∗P < 0.01 by one-way ANOVA with Bonferroni's post hoc test. D. PPM decreases the accumulation of Ly6Chi monocytes in white blood cells subjected to oxidative stress. White blood cells isolated from Ldlr−/− mice fed a chow diet were incubated for 24h in medium alone or with either H2O2 (100 μM) or H2O2 and PPM (100 μM). The number of Ly6Chi monocytes was then measured by flow cytometry as described in the Methods. n = 6 per group. Data are expressed as mean ± SEM; ∗P < 0.05, ∗∗P < 0.01 by one-way ANOVA with Bonferroni's post hoc test. E-H. PPM decreases the proliferation of bone marrow monocytes (E–F) and HSPC (G–H). Bone marrow was isolated from male Ldlr−/− mice fed a chow or western diet for 16 weeks and treated with water alone or with PPM. E-F. Bone marrow cells were incubated for 24h with 5 μM EdU, and then stained with FITC-labeled CD11b antibody (green). EdU (red) was detected as described in the Methods. Dual EdU + CD11b + cells were detected by fluorescence microscopy (E) and quantitated (F). n = 8 per group. Bar = 200 μM. Data are expressed as mean ± SEM; ∗∗∗∗P < 0.0001 by one-way ANOVA with Bonferroni's post hoc test. G-H. Bone marrow cells were incubated for 24h with 5 μM EdU. EdU was detected with Alexa Fluor 594 picolyl azide as described in the Methods. HSPC were stained with FITC-labeled CD34 antibody. Data are expressed as mean ± SEM, N = 5 each group, ∗∗∗∗P < 0.0001 by one-way ANOVA with Bonferroni's post hoc test.
4. Discussion
Oxidative stress plays critical roles in the pathogenesis and progression of atherosclerosis. While some studies administering antioxidants to humans showed decreased atherosclerosis, the majority of clinical trials demonstrated no beneficial effect on cardiovascular events, which may be due to the doses used [54] or from impaired physiologic signaling induced by reactive oxygen species (ROS) that could be atheroprotective. Lipid peroxidation produces reactive dicarbonyls, which covalently bind plasma/cellular proteins, phospholipids, and DNA resulting in altered function and toxicity [55]. We demonstrate that reactive dicarbonyl scavenging with PPM effectively increased HDL cholesterol efflux capacity, (Figure 1, Figure 2), and reduced IR and atherosclerosis in hypercholesterolemic Ldlr−/− mice (Figures 3 and S3) without impacting plasma lipids. In addition, PPM decreased monocytosis, systemic inflammation, and features of plaque instability (Figure 3, Figure 5, Figure 7, Figure 8, S5, S6, S8, S10, and S13). Thus, reactive dicarbonyl scavenging may have therapeutic potential to reduce risk of atherosclerotic clinical events.
PPM reduced atherosclerosis without changing plasma cholesterol (Figures 3 and S3) suggesting that PPM preserves normal molecular functions by preventing oxidative modifications. Consistent with this concept, PPM decreased MDA-, ONE-, and IsoLG-lysyl adducts in plaques of Ldlr−/− mice (Fig. S4). MPO and dicarbonyl modified LDL enhance foam cell formation and are present in human plaques [[42], [43], [44],56], and PPM reduced plasma MDA-LDL implicating that the atheroprotection is in part due to decreased LDL modification (Figure 1G). Indeed, antibodies against MDA-apoB protect against atherosclerosis development in mice [44]. HDL protects against atherosclerosis via a number of functions including promotion of cholesterol efflux. As oxidative modifications of HDL enhance particle internalization via scavenger receptors [57,58], we examined the impact of PPM on the ability of HDL to reduce cholesterol mass in cholesterol-enriched macrophages to control for differences in the influx of cholesterol. The removal of cholesterol mass was reduced with MDA and MPO modification of HDL (Figure 1C and E), and PPM prevented these impairments in net cholesterol efflux (Figure 1A and D). Importantly, in vivo treatment of Ldlr−/− mice with PPM increased the HDL net cholesterol efflux capacity (Figure 1F), which probably results from decreased dicarbonyl-ApoAI adducts as we previously showed that PPM treatment of Ldlr−/− mice decreases plasma MDA-HDL [19]. In addition, IsoLG or MDA modification of HDL reduces cholesterol efflux capacity in vitro, and HDL from subjects with FH has impaired cholesterol efflux capacity and contains more IsoLG- and MDA adducts compared to HDL from normocholesterolemic controls [19,20,22,36]. HOCl modification of ApoAI (i.e. oxidized tryptophan, chlorinated tyrosine) also reduces cholesterol efflux [[13], [14], [15]], and our studies suggest that PPM decreases HOCl modification of apoAI in vivo as PPM prevented the HOCl-mediated crosslinking of lipid-free apoAI and impairment of cholesterol efflux (Figure 1D–E). In agreement, the scavenger, pyridoxamine, neutralizes HOCl in vitro and prevents oxidation and chlorination of collagen tryptophan in vivo [41]. It is worth noting that MDA-apoAI and HOCl modified apoAI are present in human plaques [[13], [14], [15],21]. Furthermore, HDL cholesterol efflux capacity is independently associated with atherosclerosis, and impairments in HDL cholesterol efflux capacity increase the risk of cardiovascular events [[5], [6], [7]]. Thus, the finding that PPM treatment of Ldlr−/− mice enhances HDL net cholesterol efflux capacity highlights the therapeutic potential of in vivo dicarbonyl/oxidant scavenging against atherosclerosis.
Ly6Chi monocytes dominate hypercholesterolemia-associated monocytosis in mice and give rise to inflammatory macrophages in the atheroma [51,59]. In addition, humans with hypercholesterolemia and coronary artery disease have increased proinflammatory CD14++CD16+ (intermediate) and CD14++CD16- (classical) monocytes, which promote plaque instability and are the human counterparts to mouse proinflammatory Ly6Chi monocytes) [[60], [61], [62], [63], [64]]. Our results show that PPM reduced Ly6Chi monocytosis in response to hypercholesterolemia, providing a novel mechanism whereby dicarbonyl and/or oxidant neutralization decreases atherosclerosis and plaque destabilization (Figures 7 and S10). The effects of PPM on monocytosis resulted from decreased proliferation of bone marrow HSPC and monocytes (Figures 8 and S13), which is probably due in part to the prevention of cellular oxidative modifications. In agreement, PPM prevented in vitro the H2O2 induced increase in blood Ly6Chi monocytes (Figure 8D). In addition, studies have shown that hypercholesterolemia induces oxidative stress causing enhanced p38 and Notch1 signaling and HSPC proliferation [53,65]. Furthermore, treatment of hypercholesterolemic Apoe−/− and Scarb1−/− mice with antioxidants decreases monocytosis [53,65]. The prevention of lipoprotein modifications likely contributes to the PPM effects on monocytosis. Oxidized LDL enhances HSPC monocyte potential [66]. In addition, HDL reduces proliferation by decreasing ROS in HSPC [53], and, in contrast, HDL dicarbonyl adducts enhance ROS production [67]. Importantly, HDL cholesterol efflux capacity limits HSPC/monocyte proliferation, and PPM improved the ability of HDL from Ldlr−/− mice to remove macrophage cholesterol (Figure 1F) [68].
Inflammation induces the conversion of infiltrating monocytes into proinflammatory M1-like macrophages, which promote plaque progression and instability [69]. The CANTOS trial supports the importance of inflammation in atherosclerotic cardiovascular disease as treatment of humans with an IL-1β monoclonal antibody lowered recurrent cardiovascular events independent of cholesterol lowering [70]. Our studies show that PPM limits inflammation without impacting plasma cholesterol. Indeed, PPM reduced systemic inflammation as evidenced by reduced serum IL-1β and TNF-α and decreased hepatic inflammatory gene expression in Ldlr−/− mice (Figure 4). In addition, PPM reduced plaque M1-like macrophages as substantiated by decreased CCR2 [71] and IL-1β expression (Figure 3 and Figs. S3 and S5). Importantly, PPM decreased the association of NETs MPO with plaque macrophages, which propagates oxidative stress and conversion to proinflammatory macrophages [49,50]. In addition, PPM increased lesional Treg and Th2 cells (Figure 6), which promote conversion to anti-inflammatory M2-like macrophages [72,73]. In agreement with the increase in M2-like macrophages, the plaques of PPM treated Ldlr−/− mice contained more Arg1+ macrophages (Figures 3 and S3). In addition, some studies have shown that Tregs enhance efferocytosis signaling via stimulation of macrophage IL-10 production, and Arg1 metabolizes apoptotic cell arginine to promote continual efferocytosis [73,74]. Consistent with these studies and the increased Tregs and Arg1+ cells, scavenging with PPM enhanced the efferocytosis of lesion apoptotic cells and decreased necrosis (Figures 5 and S8). These anti-inflammatory effects of PPM are in part due to prevention of cellular oxidative modifications as PPM decreased the ex vivo and in vitro inflammatory response to LPS and/or oxidized LDL (Fig. S6). In addition, we previously showed that the scavenger, 2-hydroxybenzylamine, decreases the inflammatory response to oxidized LDL and H2O2 by forming adducts with macrophage dicarbonyls [36]. The prevention of LDL modifications also likely contributed to the PPM effects on plaque macrophage inflammation [56,75]. Furthermore, functional HDL promotes conversion to anti-inflammatory macrophages by increasing STAT6 mediated expression of Arg1 and Fizz-1 and by limiting inflammatory signaling via ERK1/2 and STAT3 [76,77]. In contrast, MDA, IsoLG, and HOCl mediated modifications convert HDL into proinflammatory particles [13,14,19,20]. It is also worth noting that HDL anti-inflammatory signaling is intimately linked to cholesterol efflux capacity [[78], [79], [80]], which is enhanced by PPM scavenging in vivo (Figure 1F).
Similar to other studies, administration of a western diet to male Ldlr−/− mice caused IR, but had no effect in female Ldlr−/− mice (Figures 2 and S1) [[45], [46], [47]]. We show that reactive dicarbonyl scavenging decreased IR in male Ldlr−/− mice, and, consistent with this observation, hepatic fat and inflammation were also reduced (Figure 2, Figure 4). The effects of PPM occurred without changes in plasma lipids, which substantiates the role of oxidative stress in IR development [81], and is in agreement with studies showing that cellular dicarbonyl adducts impair insulin signaling and glucose uptake in adipocytes, hepatocytes, and muscle cells [[82], [83], [84]]. The preservation of HDL function may also contribute to the decreased IR as the cholesterol transporter, ABCA1, enhances insulin signaling and glucose uptake in adipose and muscle [85,86]. In addition, glucose metabolism is improved with injection of apoAI, apoAI peptides, or HDL in diabetic mice and humans with T2DM [11,12,87]. A weakness of our studies is that Ldlr−/− mice may not be the most stringent model for testing the effects of PPM on IR. However, it is worth noting that our results are substantiated by other studies demonstrating that the parent molecule of PPM, pyridoxamine, improves insulin sensitivity and glucose metabolism in more robust models including obese db/db mice and diabetic rats [38,88].
In summary, dicarbonyl scavenging with PPM decreased atherosclerosis and IR without changing plasma lipid levels. Importantly, PPM reduced monocytosis and enhanced features of plaque stability. The effects of PPM on atherosclerosis and insulin sensitivity are likely due to prevention of oxidative damage to cellular and plasma constituents. In particular, reduced modification of apoB containing lipoproteins and improved HDL cholesterol efflux capacity are atheroprotective. Thus, PPM offers therapeutic potential for addressing the residual cardiovascular risk that persists in patients treated with current LDL-C lowering medications.
Author contributions
Jiansheng Huang: Conceptualization, Investigation, Visualization, Writing - Original Draft, Formal analysis; Huan Tao: Investigation, Visualization, Supervision, Writing - Review & Editing, Patricia G. Yancey: Conceptualization, Supervision, Visualization, Formal analysis; Writing - Original Draft; Zoe Leuthner: Investigation, Writing - Review & Editing, Linda S. May-Zhang: Investigation, Visualization, Writing - Review & Editing, Ju-Yang Jung: Investigation, Writing - Review & Editing, Youmin Zhang: Investigation; Lei Ding: Investigation; Venkataraman Amarnath: Resources, Dianxin Liu; Investigation; Sheila Collins: Supervision, Writing - Review & Editing; Sean S. Davies: Supervision, Methodology, Writing - Review & Editing; MacRae F. Linton: Conceptualization, Supervision, Writing - Review & Editing, Formal analysis, Funding acquisition
Acknowledgments
This work was supported by National Institutes of Health Grants: HL116263, HL148137, and HL146134. Jiansheng Huang was supported by America Heart Association Postdoctoral Fellowship (19POST34450266). The analyses of urinary prostaglandin metabolites were performed in the Vanderbilt University Eicosanoid Core Laboratory.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2022.101651.
Declaration of competing interest
M.F.L., S.S.D., V.A. are inventors on a patent application for the use of 2-HOBA and related dicarbonyl scavengers for the treatment of cardiovascular disease. M.F.L. has received research support from Amgen, Regeneron, Ionis, Merck, REGENXBIO, Sanofi and Novartis and has served as a consultant for Esperion, Alexion Pharmaceuticals and REGENXBIO. All other authors: Declarations of interest: none.
Appendix ASupplementary data
The following is the Supplementary data to this article:
1
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Linton M.R.F., Yancey P.G., Davies S.S., Jerome W.G., Linton E.F., Song W.L., et al. In: Feingold K.R., et al., editors. Endotext; South Dartmouth, MA: 2019. The role of lipids and lipoproteins in atherosclerosis.https://www.ncbi.nlm.nih.gov/pubmed/26844337 Available from: [Google Scholar]
- 2.Sampson U.K., Fazio S., Linton M.F. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14(1):1–10. doi: 10.1007/s11883-011-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ridker P.M. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016;37(22):1720–1722. doi: 10.1093/eurheartj/ehw024. [DOI] [PubMed] [Google Scholar]
- 4.Fisher E.A., Feig J.E., Hewing B., Hazen S.L., Smith J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arteriosclerosis Thrombosis Vasc Biol. 2012;32(12):2813–2820. doi: 10.1161/ATVBAHA.112.300133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rohatgi A., Khera A., Berry J.D., Givens E.G., Ayers C.R., Wedin K.E., et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371(25):2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saleheen D., Scott R., Javad S., Zhao W., Rodrigues A., Picataggi A., et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 2015;3(7):507–513. doi: 10.1016/S2213-8587(15)00126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenson R.S., Brewer H.B., Jr., Ansell B.J., Barter P., Chapman M.J., Heinecke J.W., et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. 2016;13(1):48–60. doi: 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feig J.E., Hewing B., Smith J.D., Hazen S.L., Fisher E.A. High-density lipoprotein and atherosclerosis regression: evidence from preclinical and clinical studies. Circ Res. 2014;114(1):205–213. doi: 10.1161/CIRCRESAHA.114.300760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J., Wang D., Huang L.H., Huang H. Roles of reconstituted high-density lipoprotein nanoparticles in cardiovascular disease: a new paradigm for drug discovery. Int J Mol Sci. 2020;21(3) doi: 10.3390/ijms21030739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siebel A.L., Heywood S.E., Kingwell B.A. HDL and glucose metabolism: current evidence and therapeutic potential. Front Pharmacol. 2015;6:258. doi: 10.3389/fphar.2015.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalla-Riva J., Stenkula K.G., Petrlova J., Lagerstedt J.O. Discoidal HDL and apoA-I-derived peptides improve glucose uptake in skeletal muscle. J Lipid Res. 2013;54(5):1275–1282. doi: 10.1194/jlr.M032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drew B.G., Duffy S.J., Formosa M.F., Natoli A.K., Henstridge D.C., Penfold S.A., et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;119(15):2103–2111. doi: 10.1161/CIRCULATIONAHA.108.843219. [DOI] [PubMed] [Google Scholar]
- 13.Shao B.H., Tang C.R., Heinecke J.W., Oram J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res. 2010;51(7):1849–1858. doi: 10.1194/jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y., DiDonato J.A., Levison B.S., Schmitt D., Li L., Wu Y., et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med. 2014;20(2):193–203. doi: 10.1038/nm.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao B., Pennathur S., Heinecke J.W. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J Biol Chem. 2012;287(9):6375–6386. doi: 10.1074/jbc.M111.337345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonen A., Hansen L.F., Turner W.W., Montano E.N., Que X., Rafia A., et al. Atheroprotective immunization with malondialdehyde-modified LDL is hapten specific and dependent on advanced MDA adducts: implications for development of an atheroprotective vaccine. J Lipid Res. 2014;55(10):2137–2155. doi: 10.1194/jlr.M053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busch C.J., Binder C.J. Malondialdehyde epitopes as mediators of sterile inflammation. Biochim Biophys Acta. 2017;1862(4):398–406. doi: 10.1016/j.bbalip.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 18.Shanmugam N., Figarola J.L., Li Y., Swiderski P.M., Rahbar S., Natarajan R. Proinflammatory effects of advanced lipoxidation end products in monocytes. Diabetes. 2008;57(4):879–888. doi: 10.2337/db07-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J., Yancey P.G., Tao H., Borja M.S., Smith L.E., Kon V., et al. Reactive dicarbonyl scavenging effectively reduces MPO-mediated oxidation of HDL and restores PON1 activity. Nutrients. 2020;12(7) doi: 10.3390/nu12071937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May-Zhang L.S., Yermalitsky V., Huang J., Pleasent T., Borja M.S., Oda M.N., et al. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J Biol Chem. 2018;293(24):9176–9187. doi: 10.1074/jbc.RA117.001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao B., Pennathur S., Pagani I., Oda M.N., Witztum J.L., Oram J.F., et al. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. 2010;285(24):18473–18484. doi: 10.1074/jbc.M110.118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.May-Zhang L.S., Yermalitsky V., Melchior J.T., Morris J., Tallman K.A., Borja M.S., et al. Modified sites and functional consequences of 4-oxo-2-nonenal adducts in HDL that are elevated in familial hypercholesterolemia. J Biol Chem. 2019;294(50):19022–19033. doi: 10.1074/jbc.RA119.009424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Besler C., Heinrich K., Rohrer L., Doerries C., Riwanto M., Shih D.M., et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121(7):2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies S.S., Brantley E.J., Voziyan P.A., Amarnath V., Zagol-Ikapitte I., Boutaud O., et al. Pyridoxamine analogues scavenge lipid-derived gamma-ketoaldehydes and protect against H2O2-mediated cytotoxicity. Biochemistry. 2006;45(51):15756–15767. doi: 10.1021/bi061860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J., Smith F., Panizzi P. Ordered cleavage of myeloperoxidase ester bonds releases active site heme leading to inactivation of myeloperoxidase by benzoic acid hydrazide analogs. Arch Biochem Biophys. 2014;548:74–85. doi: 10.1016/j.abb.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J., Smith F., Panizzi J.R., Goodwin D.C., Panizzi P. Inactivation of myeloperoxidase by benzoic acid hydrazide. Arch Biochem Biophys. 2015;570:14–22. doi: 10.1016/j.abb.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linton M.F., Atkinson J.B., Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267(5200):1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls S.J., Ruotolo G., Brewer H.B., Kane J.P., Wang M.D., Krueger K.A., et al. Cholesterol efflux capacity and pre-beta-1 HDL concentrations are increased in dyslipidemic patients treated with evacetrapib. J Am Coll Cardiol. 2015;66(20):2201–2210. doi: 10.1016/j.jacc.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Khera A.V., Cuchel M., de la Llera-Moya M., Rodrigues A., Burke M.F., Jafri K., et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hung A.M., Tsuchida Y., Nowak K.L., Sarkar S., Chonchol M., Whitfield V., et al. IL-1 inhibition and function of the HDL-containing fraction of plasma in patients with stages 3 to 5 CKD. Clin J Am Soc Nephrol. 2019;14(5):702–711. doi: 10.2215/CJN.04360418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinet P., Wang Z., Hazen S.L., Smith J.D. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res. 2010;51(11):3364–3369. doi: 10.1194/jlr.D007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shea S., Stein J.H., Jorgensen N.W., McClelland R.L., Tascau L., Shrager S., et al. Cholesterol mass efflux capacity, incident cardiovascular disease, and progression of carotid plaque. Arterioscler Thromb Vasc Biol. 2019;39(1):89–96. doi: 10.1161/ATVBAHA.118.311366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furuhashi M., Tuncman G., Gorgun C.Z., Makowski L., Atsumi G., Vaillancourt E., et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447(7147):959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang W., Menke A.L., Driessen A., Koek G.H., Lindeman J.H., Stoop R., et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One. 2014;9(12) doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daugherty A., Tall A.R., Daemen M., Falk E., Fisher E.A., Garcia-Cardena G., et al. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American heart association. Arterioscler Thromb Vasc Biol. 2017;37(9):e131–e157. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 36.Tao H., Huang J., Yancey P.G., Yermalitsky V., Blakemore J.L., Zhang Y., et al. Scavenging of reactive dicarbonyls with 2-hydroxybenzylamine reduces atherosclerosis in hypercholesterolemic Ldlr(-/-) mice. Nat Commun. 2020;11(1):4084. doi: 10.1038/s41467-020-17915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Babaev V.R., Ding L., Zhang Y., May J.M., Ramsey S.A., Vickers K.C., et al. Loss of 2 akt (protein kinase B) isoforms in hematopoietic cells diminished monocyte and macrophage survival and reduces atherosclerosis in ldl receptor-null mice. Arterioscler Thromb Vasc Biol. 2018 doi: 10.1161/ATVBAHA.118.312206. ATVBAHA118312206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maessen D.E., Brouwers O., Gaens K.H., Wouters K., Cleutjens J.P., Janssen B.J., et al. Delayed intervention with pyridoxamine improves metabolic function and prevents adipose tissue inflammation and insulin resistance in high-fat diet-induced obese mice. Diabetes. 2016;65(4):956–966. doi: 10.2337/db15-1390. [DOI] [PubMed] [Google Scholar]
- 39.Tao H., Yancey P.G., Babaev V.R., Blakemore J.L., Zhang Y., Ding L., et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015;56(8):1449–1460. doi: 10.1194/jlr.M056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Y.R., Linton M.F., Fazio S. Rapid quantification of murine ABC mRNAs by real time reverse transcriptase-polymerase chain reaction. J Lipid Res. 2002;43(12):2180–2187. doi: 10.1194/jlr.d200020-jlr200. [DOI] [PubMed] [Google Scholar]
- 41.Madu H., Avance J., Chetyrkin S., Darris C., Rose K.L., Sanchez O.A., et al. Pyridoxamine protects proteins from damage by hypohalous acids in vitro and in vivo. Free Radic Biol Med. 2015;89:83–90. doi: 10.1016/j.freeradbiomed.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yla-Herttuala S., Palinski W., Rosenfeld M., Parthasarathy S., Carew T., Butler S., et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw P.X., Horkko S., Tsimikas S., Chang M.K., Palinski W., Silverman G.J., et al. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21(8):1333–1339. doi: 10.1161/hq0801.093587. [DOI] [PubMed] [Google Scholar]
- 44.Schiopu A., Frendeus B., Jansson B., Soderberg I., Ljungcrantz I., Araya Z., et al. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(-/-)/low-density lipoprotein receptor(-/-) mice. J Am Coll Cardiol. 2007;50(24):2313–2318. doi: 10.1016/j.jacc.2007.07.081. [DOI] [PubMed] [Google Scholar]
- 45.Merat S., Casanada F., Sutphin M., Palinski W., Reaven P.D. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler Thromb Vasc Biol. 1999;19(5):1223–1230. doi: 10.1161/01.atv.19.5.1223. [DOI] [PubMed] [Google Scholar]
- 46.Hartvigsen K., Binder C.J., Hansen L.F., Rafia A., Juliano J., Horkko S., et al. A diet-induced hypercholesterolemic murine model to study atherogenesis without obesity and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2007;27(4):878–885. doi: 10.1161/01.ATV.0000258790.35810.02. [DOI] [PubMed] [Google Scholar]
- 47.Li A.C., Brown K.K., Silvestre M.J., Willson T.M., Palinski W., Glass C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106(4):523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arnold A.P., Cassis L.A., Eghbali M., Reue K., Sandberg K. Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2017;37(5):746–756. doi: 10.1161/ATVBAHA.116.307301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warnatsch A., Ioannou M., Wang Q., Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316–320. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Odobasic D., Kitching A.R., Holdsworth S.R. Neutrophil-mediated regulation of innate and adaptive immunity: the role of myeloperoxidase. J Immunol Res. 2016;2016:2349817. doi: 10.1155/2016/2349817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swirski F.K., Libby P., Aikawa E., Alcaide P., Luscinskas F.W., Weissleder R., et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babaev V.R., Hebron K.E., Wiese C.B., Toth C.L., Ding L., Zhang Y., et al. Macrophage deficiency of Akt2 reduces atherosclerosis in Ldlr null mice. J Lipid Res. 2014;55(11):2296–2308. doi: 10.1194/jlr.M050633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao M., Zhao D., Schouteden S., Sorci-Thomas M.G., Van Veldhoven P.P., Eggermont K., et al. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression. Arterioscler Thromb Vasc Biol. 2014;34(9):1900–1909. doi: 10.1161/ATVBAHA.114.304006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts L.J., Oates J.A., Linton M.F., Fazio S., Meador B.P., Gross M.D., et al. The relationship between dose of vitamin E and suppression of oxidative stress in humans. Free Radical Biol Med. 2007;43(10):1388–1393. doi: 10.1016/j.freeradbiomed.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.May-Zhang L.S., Kirabo A., Huang J., Linton M.F., Davies S.S., Murray K.T. Scavenging reactive lipids to prevent oxidative injury. Annu Rev Pharmacol Toxicol. 2020 doi: 10.1146/annurev-pharmtox-031620-035348. [DOI] [PubMed] [Google Scholar]
- 56.Delporte C., Van Antwerpen P., Vanhamme L., Roumeguere T., Zouaoui Boudjeltia K. Low-density lipoprotein modified by myeloperoxidase in inflammatory pathways and clinical studies. Mediators Inflamm. 2013;2013:971579. doi: 10.1155/2013/971579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panzenboeck U., Raitmayer S., Reicher H., Lindner H., Glatter O., Malle E., et al. Effects of reagent and enzymatically generated hypochlorite on physicochemical and metabolic properties of high density lipoproteins. J Biol Chem. 1997;272(47):29711–29720. doi: 10.1074/jbc.272.47.29711. [DOI] [PubMed] [Google Scholar]
- 58.Takata K., Horiuchi S., Morino Y. Scavenger receptor-mediated recognition of maleylated albumin and its relation to subsequent endocytic degradation. Biochim Biophys Acta. 1989;984(3):273–280. doi: 10.1016/0005-2736(89)90293-9. [DOI] [PubMed] [Google Scholar]
- 59.Kimball A., Schaller M., Joshi A., Davis F.M., denDekker A., Boniakowski A., et al. Ly6C(Hi) blood monocyte/macrophage drive chronic inflammation and impair wound healing in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2018;38(5):1102–1114. doi: 10.1161/ATVBAHA.118.310703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berg K.E., Ljungcrantz I., Andersson L., Bryngelsson C., Hedblad B., Fredrikson G.N., et al. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5(1):122–131. doi: 10.1161/CIRCGENETICS.111.960385. [DOI] [PubMed] [Google Scholar]
- 61.Heine G.H., Ulrich C., Seibert E., Seiler S., Marell J., Reichart B., et al. CD14(++)CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int. 2008;73(5):622–629. doi: 10.1038/sj.ki.5002744. [DOI] [PubMed] [Google Scholar]
- 62.Fadini G.P., Simoni F., Cappellari R., Vitturi N., Galasso S., Vigili de Kreutzenberg S., et al. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis. 2014;237(2):805–808. doi: 10.1016/j.atherosclerosis.2014.10.106. [DOI] [PubMed] [Google Scholar]
- 63.Hjuler Nielsen M., Irvine H., Vedel S., Raungaard B., Beck-Nielsen H., Handberg A. Elevated atherosclerosis-related gene expression, monocyte activation and microparticle-release are related to increased lipoprotein-associated oxidative stress in familial hypercholesterolemia. PLoS One. 2015;10(4) doi: 10.1371/journal.pone.0121516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rogacev K.S., Cremers B., Zawada A.M., Seiler S., Binder N., Ege P., et al. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. 2012;60(16):1512–1520. doi: 10.1016/j.jacc.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 65.Tie G., Messina K.E., Yan J., Messina J.A., Messina L.M. Hypercholesterolemia induces oxidant stress that accelerates the ageing of hematopoietic stem cells. J Am Heart Assoc. 2014;3(1) doi: 10.1161/JAHA.113.000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van der Valk F.M., Kuijk C., Verweij S.L., Stiekema L.C.A., Kaiser Y., Zeerleder S., et al. Increased haematopoietic activity in patients with atherosclerosis. Eur Heart J. 2017;38(6):425–432. doi: 10.1093/eurheartj/ehw246. [DOI] [PubMed] [Google Scholar]
- 67.Schill R.L., Knaack D.A., Powers H.R., Chen Y., Yang M., Schill D.J., et al. Modification of HDL by reactive aldehydes alters select cardioprotective functions of HDL in macrophages. FEBS J. 2020;287(4):695–707. doi: 10.1111/febs.15034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tall A.R., Yvan-Charvet L., Westerterp M., Murphy A.J. Cholesterol efflux: a novel regulator of myelopoiesis and atherogenesis. Arterioscler Thromb Vasc Biol. 2012;32(11):2547–2552. doi: 10.1161/ATVBAHA.112.300134. [DOI] [PubMed] [Google Scholar]
- 69.Nahrendorf M., Swirski F.K. Abandoning M1/M2 for a network model of macrophage function. Circ Res. 2016;119(3):414–417. doi: 10.1161/CIRCRESAHA.116.309194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 71.Boring L., Gosling J., Cleary M., Charo I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394(6696):894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 72.Bartlett B., Ludewick H.P., Misra A., Lee S., Dwivedi G. Macrophages and T cells in atherosclerosis: a translational perspective. Am J Physiol Heart Circ Physiol. 2019;317(2):H375–H386. doi: 10.1152/ajpheart.00206.2019. [DOI] [PubMed] [Google Scholar]
- 73.Doran A.C. Inflammation resolution: implications for atherosclerosis. Circ Res. 2022;130(1):130–148. doi: 10.1161/CIRCRESAHA.121.319822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yurdagul A., Jr., Subramanian M., Wang X., Crown S.B., Ilkayeva O.R., Darville L., et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 2020;31(3):518–533. doi: 10.1016/j.cmet.2020.01.001. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Papac-Milicevic N., Busch C.J., Binder C.J. Malondialdehyde epitopes as targets of immunity and the implications for atherosclerosis. Adv Immunol. 2016;131:1–59. doi: 10.1016/bs.ai.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee M.K., Moore X.L., Fu Y., Al-Sharea A., Dragoljevic D., Fernandez-Rojo M.A., et al. High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br J Pharmacol. 2016;173(4):741–751. doi: 10.1111/bph.13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanson M., Distel E., Fisher E.A. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One. 2013;8(8) doi: 10.1371/journal.pone.0074676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yvan-Charvet L., Welch C., Pagler T.A., Ranalletta M., Lamkanfi M., Han S., et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118(18):1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu X., Lee J.Y., Timmins J.M., Brown J.M., Boudyguina E., Mulya A., et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283(34):22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Groenen A.G., Halmos B., Tall A.R., Westerterp M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit Rev Biochem Mol Biol. 2021;56(4):426–439. doi: 10.1080/10409238.2021.1925217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henriksen E.J., Diamond-Stanic M.K., Marchionne E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51(5):993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vidal N., Cavaille J.P., Poggi M., Peiretti F., Stocker P. A nonradioisotope chemiluminescent assay for evaluation of 2-deoxyglucose uptake in 3T3-L1 adipocytes. Effect of various carbonyls species on insulin action. Biochimie. 2012;94(12):2569–2576. doi: 10.1016/j.biochi.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 83.Pillon N.J., Croze M.L., Vella R.E., Soulere L., Lagarde M., Soulage C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology. 2012;153(5):2099–2111. doi: 10.1210/en.2011-1957. [DOI] [PubMed] [Google Scholar]
- 84.Shearn C.T., Fritz K.S., Reigan P., Petersen D.R. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50(19):3984–3996. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- 85.de Haan W., Bhattacharjee A., Ruddle P., Kang M.H., Hayden M.R. ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity. J Lipid Res. 2014;55(3):516–523. doi: 10.1194/jlr.M045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanchez-Aguilera P., Diaz-Vegas A., Campos C., Quinteros-Waltemath O., Cerda-Kohler H., Barrientos G., et al. Role of ABCA1 on membrane cholesterol content, insulin-dependent Akt phosphorylation and glucose uptake in adult skeletal muscle fibers from mice. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(12):1469–1477. doi: 10.1016/j.bbalip.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 87.Edmunds S.J., Liebana-Garcia R., Nilsson O., Domingo-Espin J., Gronberg C., Stenkula K.G., et al. ApoAI-derived peptide increases glucose tolerance and prevents formation of atherosclerosis in mice. Diabetologia. 2019;62(7):1257–1267. doi: 10.1007/s00125-019-4877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abdullah K.M., Abul Qais F., Hasan H., Naseem I. Anti-diabetic study of vitamin B6 on hyperglycaemia induced protein carbonylation, DNA damage and ROS production in alloxan induced diabetic rats. Toxicol Res (Camb) 2019;8(4):568–579. doi: 10.1039/c9tx00089e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.