

Summary
Invasive microbial pathogens often disrupt epithelial barriers, yet the mechanisms used to dismantle tight junctions are poorly understood. Here, we show that the obligate pathogen Chlamydia trachomatis uses the effector protein TepP to transiently disassemble tight junctions early during infection. TepP alters the tyrosine phosphorylation status of host proteins involved in cytoskeletal regulation, including the filamentous actin-binding protein EPS8. We determined that TepP-and EPS8 are necessary and sufficient to remodel tight junctions and that the ensuing disruption of epithelial barrier function promotes secondary invasion events. Genetic deletion of EPS8 renders epithelial cells and endometrial organoids resistant to TepP-mediated tight junction remodeling. Finally, TepP and EPS8 promote infection in murine models of infections, with TepP mutants displaying defects in ascension to the upper genital tract. These findings reveal a non-canonical function of EPS8 in the disassembly of epithelial junctions and an important role for Chlamydia pathogenesis.
eTOC Blurb
Dolat et al. show the obligate intracellular pathogen, Chlamydia trachomatis, disrupts epithelial tight junctions to promote infection. The authors identify a role for the C. trachomatis secreted effector TepP in repurposing the host actin-binding protein EPS8 to dismantle tight junctions and promote invasion of polarized epithelia.
Graphical Abstract
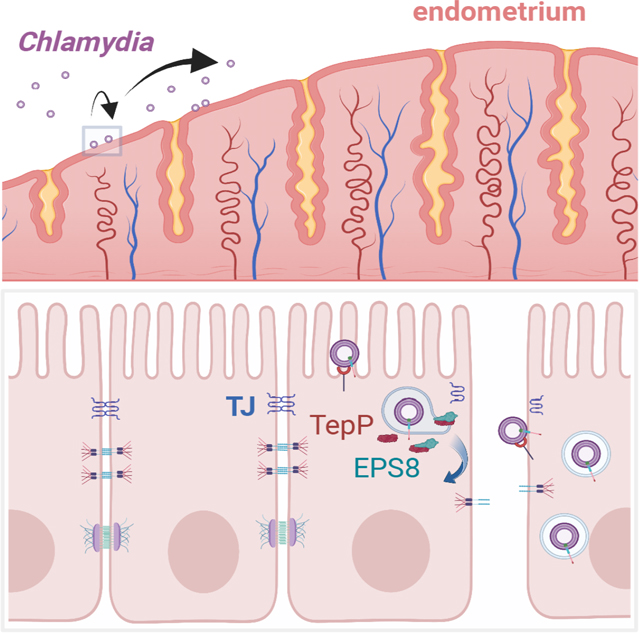
Introduction
Chlamydia trachomatis is an obligate intracellular pathogen responsible for the majority of sexually transmitted bacterial infections and remains the leading cause of preventable infectious blindness (WHO, 2018). In urogenital C. trachomatis infection, the ensuing inflammatory response drives the manifestation of disease including pelvic inflammatory diseases, tubal scarring and infertility (Haggerty et al., 2010). C. trachomatis targets polarized columnar epithelial cells in the cervical transition zones and upper female reproductive tract. Epithelial polarity is spatially coupled to cell-cell contacts, including tight junctions, adherens junctions and desmosomes (Garcia et al., 2018). Apically tight junctions maintain epithelial polarity while gating the movement of solutes and ions through the paracellular space (Zihni et al., 2016). Bacterial and viral pathogens can disrupt epithelial organization and polarity by directly and indirectly targeting tight junctions to promote microbial invasion or transit to the underlying tissue (Guttman and Finlay, 2009; Paradis et al., 2021). In polarized epithelial infection models, C. trachomatis disrupts epithelial cell-cell junctions (Dolat and Valdivia, 2021; Kessler et al., 2012), but the mechanism mediating this disruption is unknown.
Chlamydiae exhibit a biphasic lifecycle – the extracellular and infectious elementary body (EB) and the intracellular, replication competent reticulate body (RB) (Abdelrahman and Belland, 2005). The EB form binds epithelial plasma membrane proteoglycans (e.g. heparan sulfate) and receptors, such as integrin B1, ephrin receptor A2, EGFR and PDGFR2 (Elwell et al., 2008; Patel et al., 2014; Rosmarin et al., 2012; Stallmann and Hegemann, 2016; Subbarayal et al., 2015). Effector proteins are translocated into target cells via a Type III secretion (T3S) system to stimulate actin assembly and the formation of a membrane-bound pathogen-containing vacuole termed an “inclusion” (reviewed in (Elwell et al., 2016)). Early inclusions migrate to the peri-nuclear region where the bacteria differentiate into the replicative RB form (Al-Zeer et al., 2014; Mital et al., 2015). Eventually RBs asynchronously differentiate back into the EB form. At the end of the infectious cycle the inclusion is extruded extracellularly or EBs are released after lysis of the host cell (Hybiske and Stephens, 2007).
A subset of T3S effectors is pre-engaged with the T3S chaperone Slc1, to regulate their secretion during the invasion of epithelial cells (Brinkworth et al., 2011; Chen et al., 2014). Two effectors directly rearrange the actin cytoskeleton to promote EB entry and inclusion formation – translocated actin phosphorprotein (TarP) and translocated membrane-associated effector A (TmeA) (Jewett et al., 2006; Keb et al., 2018). These effectors converge on the host Arp2/3 actin nucleation complex, and mutant strains in either effector exhibit a reduced invasion of epithelial cells (Faris et al., 2020; Keb et al., 2021). In addition TarP can activate the Arp2/3 complex by promoting interactions with the guanine nucleotide-exchange factor Rac1 (Lane et al., 2008). Following the secretion of TarP, the effector TepP (translocated early phosphorprotein) is delivered and phosphorylated by Src kinases (Chen et al., 2014). TepP is the most abundant effector in the EB form and interacts with the Crk adaptor proteins (Crk I/II, CrkL) and the PI3K complex, which stimulates the formation of PI(3,4,5)P3 on the inclusion membrane (Carpenter et al., 2017; Saka et al., 2011). TepP also regulates the expression of immunity-related genes during early infection (Chen et al., 2014) and dampens the recruitment of neutrophils in an ex vivo endometrial organoid infection model (Dolat and Valdivia, 2021).
The phosphorylation of effectors can impact function and activation of signaling events that promote infection and evade innate immune responses (Backert and Selbach, 2005). For example, Tyr phosphorylation of the Enteropathogenic Escherichia coli effector Tir is required for its interaction with the host adaptor protein Nck, actin assembly, and pedestal formation (Gruenheid et al., 2001). Similarly, Tyr phosphorylation of the Helicobacter pylori effector CagA regulates its interaction with the SHP2 phosphatase and promotes the elongation of gastric epithelial cells (Higashi et al., 2002). To modulate the inflammatory state of infected cells, Tyr phosphorylation of the Bartonella henselae effector BepD stimulate STAT3 activation and the production of anti-inflammatory cytokines (Sorg et al., 2020). The effect of TepP phosphorylation, however, is less clear. While TepP phosphorylation promotes interactions with Crk in vitro (Chen et al., 2014), Src-mediated phosphorylation is not required for the recruitment of Crk or PI3K to nascent inclusions (Carpenter et al., 2017). Nonetheless, the Tyr phosphorylation of multiple epithelial proteins is TepP-dependent, but their identity is unknown (Chen et al., 2014).
Here, we find that TepP directly engages the host epidermal growth factor receptor pathway substrate 8 (EPS8) to alter epithelial cell shape and disrupt tight junctions. EPS8, an actin-capping and bundling protein, normally regulates the assembly and stability of actin-based structures such as stereocilia, filopodia and microvilli (Postema et al., 2018; Vaggi et al., 2011; Zampini et al., 2011). TepP recruits EPS8 to nascent inclusions, triggering the disassembly of epithelial cell-cell junctions, loss of transepithelial electrical resistance (TEER), and cell dispersion. Chlamydia then exploits TepP-dependent cell-cell junction breakdown to promote the entry of additional infectious units, presumably by enabling bacterial access to basolateral receptors. The TepP and EPS8-interaction-dependent processes are required for optimal infection of the upper genital tract. These results uncovered an alternative mechanism for Chlamydia invasion of cells and details how a bacterial effector can repurpose the function of a cytoskeletal regulatory protein to perform non-canonical activities that enhance infection.
Results
The C. trachomatis effector TepP disrupts epithelial tight junctions and promotes cell dispersion
We observed that polarized human endocervical epithelial cells (A2ENs) infected with Chlamydia trachomatis strain LGV-L2 (CTL2) transiently dispersed (Fig 1A). The organization of tight junction proteins showed that the nascent inclusion recruits the scaffolding protein, ZO-1 (Fig 1B), which correlated with a transient (< 6h) loss of epithelial barrier function as measured by transepithelial electrical resistance (TEER) (Fig 1C). Live imaging of A2EN cells infected with C. trachomatis indicated a dynamic reorganization of the epithelial monolayer and disruption to cell-cell contacts (Fig 1D, Movie 1). This dispersion phenotype was conserved among C. trachomatis serovars as infection with urogenital C. trachomatis serovars also disrupted barrier function (Fig 1E).
Figure 1. The Chlamydia effector TepP modulates epithelial tight junctions and promotes cell dispersion.

(A-E) C. trachomatis disrupts tight junctions and promotes epithelial cell dispersion. (A) Phase contrast images of polarized A2EN cells on collagen-coated coverslips infected with CTL2 for 1 h. (B) Confocal images of polarized A2EN cells on collagen-coated filters infected with CTL2 for 1 h and stained for ZO-1 (green), F-actin (magenta), MOMP (orange), and DNA (blue). Arrows denote ZO-1-positive inclusions near disassembling tight junctions. Mock represents uninfected cells. Inset scale bar, 1 μm. (C) Transepithelial electrical resistance (TEER) was measured every 30 min for 7 h in polarized A2EN cells infected with CTL2 (n=3). Data are represented as the mean +/− SD. (D) Still frames from time-lapse microscopy of A2EN cells infected with CTL2. Arrows denote gaps forming between cells. Time display, hours: minutes (E) TEER was measured every 30 min for 7 h in polarized A2EN cells mock-infected or infected with C. trachomatis serovar D (CTD) (n=3). Data are represented as the mean +/− SD.
(F-H) TepP is required for tight junction disruption and epithelial cell dispersion. (F) Fluorescence images of polarized A2EN cells on collagen-coated coverslips infected with the indicated strain for 1 h and stained for F-actin (white) and DNA (blue). (G) Quantification of the percent of cell-free area (n = 30 fields of view; unpaired, two-tailed t-test). (H) Maximum projections and orthogonal views from confocal images of A2EN cells polarized on collagen-coated filters infected with CTL2 or tepP mutant bacteria for 1 h and stained for MOMP (green), F-actin (magenta), and DNA (blue). (I) TEER was measured every 30 min for 7 h in polarized A2EN cells infected with a tepP mutant or its complemented counterpart (n = 3). Data are represented as the mean +/−SD.
(J-K) TepP does not regulate synchronized host cell entry. Non-confluent (J) or polarized (K) A2EN cells were infected with the indicated strain for 30 min and processed for inside-outside staining as a measure of cell invasion. Quantification shows the percent of EBs inside the cell (n = 60 cells; Welch’s t-test). (L) TepP-deficient C. trachomatis CTL2 are cleared faster from the mouse genital tract. Quantification of normalized bacterial genome counts at the indicated time (n = 8–9 mice per time point, Mann-Whitney U test). Color indicates values from two independent experiments.
(G, J, K) Box and Whisker plots represent the median and interquartile range (IQR) +/− either 1.5*IQR or min and max values.
*p < 0.05, **p<0.01, ***p<0.001, ****p<0.0001
To identify effectors that mediate this early disruption of tight junctions, we infected polarized A2EN cells with C. trachomatis mutants defective for Slc1-chaperoned early effectors and measured the degree of epithelial cell dispersion after one hour. A strain lacking the effector TepP completely abrogated cell dispersion (Fig 1F–H), which was rescued upon complementation with TepP expression from a plasmid vector (Fig 1I). This effect is not the indirect result of a lower invasion efficiency as the tepP mutant did not display a defect in synchronized entry in non-confluent or polarized A2EN cells (Fig 1J–K). In contrast, tmeA and tarP mutants had no impact on cell dispersion. These mutants exhibited defects in host cell entry in non-polarized cells as previously reported (Ghosh et al., 2020; Keb et al., 2018); although those defects were markedly milder in polarized cells (Fig 1J–K).
These observations suggest that TepP functions may be most relevant in polarized epithelial cells. Indeed, inactivation of tepP significantly reduced the production of infectious units (IFUs) in A2ENs but not HeLa cells (Carpenter et al., 2017). To assess if TepP was important during animal infections, we challenged mice transcervically with wild-type or tepP mutant CTL2, the strain most tractable for genetic manipulation, and measured bacterial burdens over time in the upper genital tract (UGT). The tepP mutant was cleared at a faster rate than wild-type bacteria from the genital tract (Fig 1L), indicating that TepP an important role in Chlamydia survival in host tissues.
TepP promotes the tyrosine phosphorylation of the host actin-binding protein EPS8
TepP alters the global pattern of tyrosine phosphorylated (p-Tyr) proteins during the first 12 hours of infection (Carpenter et al., 2017; Chen et al., 2014). By immunofluorescence microscopy, we observed a significant TepP-dependent spatial redistribution of p-Tyr proteins in Chlamydia infected cells (Fig 2A). To identify p-Tyr proteins, A2EN cells were mock-infected or infected with either a tepP null mutant complemented with TepP (pTepP) or an empty vector (pVector), subjected to immunoprecipitation with anti-phospho-Tyr antibodies (Fig 2B), and processed for a quantitative proteomic analysis by LC/MS-MS (Fig 2C). A pathway enrichment analysis of the Tyr phosphoproteomes of cells infected with TepP+ or TepP− Chlamydia indicated that components of the Rho/Ras mediated signaling pathways, regulation of cytoskeletal dynamics, and cell motility were significantly overrepresented among p-Tyr proteins (Fig 2D). Similar results were obtained when comparing mock-infected to cells infected with TepP+ Chlamydia, indicating that TepP centrally regulates Tyr-based signaling (Fig S1A–B, Table 1). We also identified TepP-independent p-Tyr proteins involved in cytoskeletal organization, namely focal adhesion kinase (FAK1) (Fig S1C–D, Table 1), a putative interactor of the effector TarP (Thwaites et al., 2014).
Figure 2. TepP alters the tyrosine phosphorylation profile of host proteins and prominently targets the actin-binding protein EPS8.

(A-D) TepP alters the host cell phosphoproteome. (A) Confocal images of HeLa cells infected with the indicated strain for 1 h and stained for pTyr (cyan), F-actin (magenta) and DNA (yellow). Inset shows enhanced DNA signal to show inclusions. (B) Whole cell lysates from A2EN cells infected with tepP mutants transformed with pVector or pTepP and subjected to western blot analysis for pTyr, MOMP, and α-tubulin. (C) Volcano plot representing the change in abundance of Tyr phospho-peptides from A2EN cells infected with a tepP mutants transformed with pTepP vs pVector. (D) Bar graph depicts a GO enrichment analysis of biological processes for differentially phosphorylated proteins.
(E-G) TepP regulates the Tyr phosphorylation of EPS8. EPS8 was immunoprecipitated from A2EN cells infected with CTL2 or tepP mutant bacteria (E) or tepP mutants transformed with pTepP or pVec (F) for 1 h and subjected to western blot analysis for pTyr, EPS8, α-tubulin, and Chlamydia HtrA. (G) Whole cell lysates HeLa, HeLaCas9 and HeLaCas9 EPS8 KO cells uninfected or infected for 2 h were subjected to western blot analysis for pTyr, EPS8, MOMP, and α-tubulin.
(H) EPS8 is not required for synchronized entry into host cells. Quantification of the percent CTL2 EBs inside in HeLaCas9 and HeLaCas9 EPS8 KO cells at 30 min post-infection (n = 57–58 cells; unpaired, two-tailed t-test).
(I-J) EPS8 localizes at sites of CTL2 invasion and early inclusion trafficking. Time-lapse still frames from MEF (I) and HeLa cells (J) expressing GFP-EPS8 (cyan) infected with CTL2-mCh (magenta) and imaged by spinning disk microscopy. Time display, min:sec.
(K-N) EPS8 recruitment to early inclusions is actin-independent. (K) Fluorescence images of HeLa cells infected with CTL2-mCh (green), incubated with DMSO or Latrunculin A for 1 h, and stained for EPS8 (magenta) and DNA (blue). (L) Quantification of EPS8-positive inclusions (n = 54–56 cells; unpaired, two-tailed t-test). (M) Fluorescence images of HeLa cells expressing GFP-tagged EPS8 (green) or indicated deletions were infected with CTL2-mCh (magenta) for 1 h and stained for DNA (blue). (N) Quantification of EPS8-positive inclusions (n = 47–63 cells; unpaired, two-tailed t-test). (H, L, N) Scatter plots represent individual cells and the line denotes the median. ****p<0.0001
The most prominent targets of TepP-dependent phosphorylation were the host actin-binding protein EPS8 and EPS8 Like 2 (EPS8L2) (Fig 2C, Table 1). EPS8 was first identified as a substrate of EGFR signaling and as a regulator of EGF internalization, Rab5-dependent endocytic trafficking, and Rac1-based signaling (Lanzetti et al., 2000; Scita et al., 1999). The C-terminal domain of EPS8 contains an actin capping and bundling domain (Disanza et al., 2004; Hertzog et al., 2010), which promotes the assembly of actin-rich structures, such as stereocilia, filopodia and microvilli (Vaggi et al., 2011; Zampini et al., 2011; Zwaenepoel et al., 2012). To confirm if EPS8 is phosphorylated in a TepP-dependent manner, we immunoprecipitated EPS8 from A2EN cells infected with wild-type Chlamydia, a tepP mutant, or its complemented version, followed by immunoblotting with anti-pTyr antibodies. Phosphorylation of EPS8 was detected in A2EN cells infected with Chlamydia expressing TepP but not uninfected cells or cells infected with tepP mutants (Fig 2E–F). We further confirmed EPS8 as a major target of TepP-dependent Tyr phosphorylation by infecting HeLa cells where EPS8 had been inactivated by CRISPR/Cas9-mediated gene editing (Fig 2G). Although EPS8 was previously reported to localize to sites of C. trachomatis invasion and siRNA-mediated silencing of EPS8 expression reduced entry (Lane et al., 2008), we found no significant difference in invasion efficiencies between HeLaCas9 and HeLaCas9 EPS8 KO cells (Fig 2H). Using time-lapse imaging, we monitored EGFP-EPS8 localization during the initial stage of C. trachomatis invasion and found that EPS8 is transiently recruited to Chlamydia EBs during entry and dissociates quickly before a more stable pool of EPS8 associates with nascent inclusions (Fig 2I, Movie 2). This stable pool exhibits dynamic behavior where tubular-like EPS8 elements emanate from and into early inclusions (Fig 2J, Movie 3).
TepP regulates the recruitment of EPS8 to early inclusions in an actin- and phosphorylation-independent manner
Because C. trachomatis stimulates actin polymerization during invasion, we first assessed if EPS8 recruitment to inclusion was an indirect consequence of its binding to actin filaments. We observed no difference in EPS8 recruitment to early inclusions in cells treated Latrunculin A, which disassembles actin filaments (Fig 2K–L). We next generated EGFP-tagged EPS8 constructs lacking various functional domains and tested their ability to localize to the nascent inclusion. The actin-binding domain (ABD) was dispensable for the recruitment of EPS8 to nascent inclusions (Fig 2M–N). Similarly, the EPS8 C-terminal SH3 and ABD alone did not localize to inclusions (Fig 2M–N), even though the SH3 domain interacts with Abi1, which localizes to Chlamydia entry sites (Lane et al., 2008). Altogether, these observations imply that EPS8 recruitment to early inclusions is uncoupled from its F-actin binding properties.
Given the role of TepP in regulating EPS8 modifications we posited that the recruitment of EPS8 to inclusions would be dependent in TepP. Indeed, TepP and EPS8 colocalize in punctate and tubule-like structures at the nascent inclusion (Fig 3A). EPS8 localization to the inclusion only early in infection, rapidly diminished by 12 h and was absent from inclusions by 20 h (Fig 3B–C), which correlates with the kinetics of TepP secretion and phosphorylation (Chen et al., 2014). We next infected A2EN cells with C. trachomatis mutants lacking effectors that are translocated early in infection. We found that strains lacking TepP abolished EPS8 recruitment; with tarP and tmeA mutants showing only slight reductions in their association with EPS8 (Fig 3D–E) (Fig 1). Moreover, we determined that the recruitment of EPS8 to nascent inclusions is spatially restricted by TepP. Co-infections with tepP mutant and wild-type bacteria indicated that within a single epithelial cell EPS8 was only recruited to wild-type bacteria (Fig 3F–G).
Figure 3. TepP associates with EPS8 on early inclusions.

(A-C)) Morphology and association of EPS8 with inclusions. (A) Confocal images of HeLa cells infected with CTL2 for 1 h and stained for TepP (green), EPS8 (magenta), and DNA (blue). (B) Fluorescence images of HeLa cells infected with CTL2-mCh (magenta) for the indicated time and stained for EPS8 (green) and DNA (blue) and (C) quantification of EPS8-positive inclusions over time (n = 32 cells).
(D-G) TepP is required for EPS8 recruitment to early inclusions. (D) Fluorescence images of A2EN cells infected with the indicated GFP-expressing strain (green) and stained for EPS8 (magenta) and DNA (blue). (E) Quantification of EPS8-positive inclusions (n=30 cells; unpaired, two-tailed t-test). (F) Fluorescence images of HeLa cells co-infected with GFP-expressing CTL2 (top) or tepP mutant (bottom) bacteria (green) and CTL2-mCh (white) for 1 h and immunostained for EPS8 (magenta) and DNA (blue). (G) Quantification of EPS8-positive inclusions within co-infected cells (one strain, n = 69 cells, two strains, n = 55–57 cells; unpaired, two-tailed t-test).
(H-I) Ectopic expression of TepP drives the relocalization of EPS8. (H) Confocal images of HeLa cells transfected with mCh or mCh-TepP and stained for EPS8 (green) and DNA (blue). (I) Coimmunoprecipitation of EPS8 and TepP. Whole cells lysates of 293T cells co-expressing mCh-TepP with EGFP or EGFP-EPS8 were immunoprecipitated using the GFP Trap system and subjected to western blot analysis for GFP, mCh, and α-tubulin.
(C, E, G) Box and Whisker plots represent the median and interquartile range +/− either the min and max values or 1.5*IQR.
**p<0.01, ***p<0.001, ****p<0.0001
We next expressed TepP in HeLa cells and found that EPS8 re-localized to TepP-positive tubulovesicular structures (Fig 3H). The colocalization of TepP and EPS8 in transfected cells was independent of F-actin as it was resistant to actin depolymerization with Latrunculin A (Fig S2A) and did not require the ABD of EPS8 (Fig S2B). Furthermore, TepP and EPS8 co-expressed in 293T cells co-immunoprecipitated, suggesting an association between TepP and EPS8 (Fig 3G). Despite TepP driving the Src-dependent phosphorylation of EPS8 (Carpenter et al., 2017; Maa et al., 1999), we did not observe differences in the recruitment of EPS8 to nascent inclusions in fibroblasts deficient in Src-family kinases (Src/Fyn/Yes) (Fig S2C–D). Furthermore, we mutated the four Tyr residues to Phe in EGFP-EPS8 and determined that this variant still co-immunoprecipitated with TepP with similar efficiency as wild-type EPS8 (Fig 3I), co-localized to nascent inclusions (Fig S2E–F) and associated with ectopically expressed TepP (Fig S2G). Taken together, these data indicate that TepP associates with EPS8 in an actin- and phospho-Tyr independent manner.
TepP and EPS8 are required for Chlamydia-mediated modulation of epithelial morphology and the disruption of tight junctions and barrier function
Although the association between TepP and EPS8 did not require F-actin polymerization we postulated that this interaction could influence cytoskeletal dynamics given the role of EPS8 in F-actin bundling and capping. Indeed, we observed that Chlamydia infection impacted epithelial cell morphology and that HeLa cells infected with wild-type Chlamydia, but not tepP mutants, became elongated with an increased aspect ratio early in infection (Fig 4A–B). TepP was sufficient to stimulate these morphological alterations as ectopic expression of TepP in HeLa cells increased the cell aspect ratio (Fig 4C–D). To test if TepP drives these changes in cell shape through EPS8, we transfected HeLaCas9 and HeLa EPS8 KO cells with vectors expressing mCh or TepP-mCh. EPS8-deficient HeLa cells exhibited elongated cell shape compared to the parental HeLaCas9 cells (Fig 4E–G), but the expression of TepP did not significantly change the cell aspect ratio between the HeLaCas9 and HeLa EPS8 KO cells (Fig 4F–G). Taken together, these data show that TepP acts through EPS8 to modify epithelial cell shape.
Figure 4. TepP and EPS8 modulate cell morphology and promote the disruption of tight junctions.

(A-B) TepP promotes epithelial cell elongation during infection. (A) Fluorescence images of HeLa cells infected with GFP-expressing CTL2 or tepP mutant bacteria (green) for 1 h and stained for EPS8 (white). (B) Quantification of the cell aspect ratio (n = 106–110 cells; unpaired, two-tailed t-test).
(C-G) TepP-dependent elongation of epithelial cell requires EPS8. (C) Fluorescence images of HeLa cells expressing mCh or mCh-TepP (magenta) and stained for F-actin (white). (D) Quantification of the cell aspect ratio (n = 106–112 cells; unpaired, two-tailed t-test). (E) Fluorescence images of HeLaCas9 or HeLaCas9 EPS8 KO cells expressing mCh or mCh-TepP and stained for F-actin (white). (G) Quantification of the cell aspect ratio (n = 24–32 cells; unpaired, two-tailed t-test).
(H-M) EPS8 is required for the dispersion of polarized A2EN cells. (H) Whole cell lysates from uninfected and infected A2ENCas9 and A2ENCas9 EPS8 KO cells were subjected to western blot analysis for EPS8, pTyr and α-tubulin. (I) Confocal images of A2ENCas9 and A2ENCas9 EPS8 KO cells immunostained for EPS8 (green), F-actin (magenta) and DNA (blue). Transmission electron micrographs of the lateral membrane contacts showing apical tight junctions (blue arrows). Scale bars, 0.5 μm. (J) TEER measurements in A2ENCas9 and A2ENCas9 EPS8 KO cells over ten days. Data are represented as the mean +/− SD (n=3). (K) Confocal images of polarized A2ENCas9 or A2ENCas9 EPS8 KO cells mock infected or infected with CTL2 for 1 h and stained for F-actin and quantification of cell-free area (L) (n = 30 fields of view; unpaired, two-tailed t-test). (M) TEER was measured every 30 min for 7 h in polarized A2ENCas9 and A2ENCas9 EPS8 KO cells infected with CTL2. Data are represented as the mean +/− SD (n=3).
(N) EPS8 is not required for synchronized entry into A2EN cells. A2ENCas9 or A2ENCas9 EPS8 KO cells were infected with CTL2 for 30 min and processed for inside-outside staining. Quantification of the percent of EBs inside (n = 30 cells; unpaired, two-tailed t-test).
(O-R) EPS8 and TepP are required for the recruitment of ZO-1 to early inclusions. (O) Confocal images of A2ENCas9 and A2ENCas9 EPS8 KO cells infected with CTL2-GFP (green) and stained for ZO-1 (magenta) and DNA (blue) and quantification of the percent of ZO-1 positive inclusions (n = 66–74 cells, unpaired, two-tailed t-test) (P). (Q) Confocal images of A2ENCas9 infected with GFP-expressing CTL2 or tepP mutant bacteria (green) and stained for ZO-1 (magenta) and DNA (blue) and quantification of the percent ZO-1-positive inclusions (n = 22–23 cells; unpaired, two-tailed t-test) (R). Arrows denote ZO-1 localization to nascent inclusions.
Box and Whisker plots represent the median and interquartile range +/− min and max values (L, P) or 1.5*IQR (B, D, G, N, R).
***p<0.001, ****p<0.0001
To determine if the interaction between TepP and EPS8 is important for the epithelial dispersion phenotype observed in polarized A2EN cells, we generated Cas9-expressing A2EN cells (A2ENCas9) and deleted EPS8 by CRISPR/Cas9 editing (Fig 4H–I). Depletion of EPS8 did not affect the formation of tight and adherens junctions or the establishment of epithelial barrier functions as assessed by TEER over ten days (Fig 4I–J, S3A–B). However, A2EN EPS8 KO cells were resistant to tight junction disruption during early infection (Fig 4K–M) even though they were as permissible for Chlamydia invasion as the A2ENCas9 parental cells (Fig 4N). In endometrial organoids, a model that more closely recapitulates the polarized epithelia of the upper genital tract, nascent inclusions disrupt adherens junctions and recruit the tight junction scaffolding protein ZO-1 (Dolat and Valdivia, 2021), which may underlie the disruption of tight junctions. Indeed, ZO-1 was recruited robustly to nascent inclusions in polarized A2ENCas9 cells but not A2EN EPS8 KO cells (Fig 4O–P). Reciprocally, we found that TepP is required for the recruitment of ZO-1 (Fig 4Q–R). The disruption to epithelial organization was not limited to tight junctions as TepP and EPS8 were also required for the disassembly of adherens junctions (Fig S3C–D). Tight junctions were eventually repaired as ZO-1 localization to the inclusion diminished over time and correlated with the recovery of TEER (Fig S3E–F).
We next assessed the impact of EPS8 in TepP-mediated disruption of tight junction in the endometrial organoid infection model. In endometrial organoids, TepP was essential for remodeling epithelial organization and reducing epithelial barrier function, as assessed by the leakage of fluorescent dextran from the lumen of infected organoids (Fig 5A–D, Movie 4). EPS8 localized to the apical domain of polarized endometrial epithelia (Fig 5E), where it is known to regulate microvilli elongation. As in A2EN cells, TepP was required for the recruitment of EPS8 to nascent inclusions in endometrial organoids (Fig 5F–G). We next disrupted the Eps8 exon 4 in mice by CRISPR/Cas9-mediated gene editing (Fig 5H) and derived organoids from Eps8+/+ and Eps8−/− mice. These organoids were morphologically indistinguishable (Fig S4B–F). Infections of Eps8+/+ organoids, but not Eps8−/−, with C. trachomatis led to the disruption of epithelial organization (Fig 5I). Epithelial cell disruption is likely conserved among TepP-expressing Chlamydia species as organoids infected with Chlamydia muridarum, a mouse adapted pathogen, also significantly impacted epithelial organization (Fig 5J) in Eps8+/+ but not Eps8−/− organoids. Moreover, tight junctions were preserved in Eps8−/− organoids during early infection and maintain their barrier functions in dextran leakage assays (Fig 5K–M, Movie 5). These findings are consistent with a model wherein TepP requires EPS8 to disrupt cell-cell junctions during invasion in cell culture and organoid models.
Figure 5. TepP promotes EPS8-dependent epithelial barrier disruption in endometrial organoids.

(A-E) TepP is required for organoid barrier disruption. (A) 3D reconstruction of confocal images of endometrial organoids infected with CTL2 or tepP mutant bacteria for 8 h and stained for Chlamydia IncG (magenta), F-actin (white) and DNA (blue). (B) Confocal images of ZO1-GFP (magenta) organoids uninfected or infected with CTL2-mCh (green) for 14 h and stained for DNA (blue). (C) Endometrial organoids were injected with CTL2 or tepP mutant bacteria diluted in fluorescent dextran (white) and imaged live for 14 h. Still frames from the start and end of imaging are shown. (D) Quantification of luminal dextran intensity over time (n = 11 organoids).
(E-G) TepP is required for EPS8 recruitment to early inclusions in endometrial organoids. (E) Confocal image from a medial section of an endometrial organoid stained for EPS8 (green), F-actin (magenta) and DNA (blue). (F) Confocal images of organoids infected with CTL2 or tepP mutant bacteria (white) for 8 h and stained for EPS8 (green), F-actin (magenta) and DNA (blue). (G) Quantification of EPS8-positive inclusions (n = 23 cells; unpaired, two-tailed t-test). Box and Whisker plots represent the median and interquartile range +/− min and max values.
(H-M) EPS8 is required for infection-mediated disruption of epithelial barrier in organoids. (H) Top: Schematic of exon targeting of the mouse Eps8 locus. Middle: PCR analysis of exon 4 deletion in wild-type, heterozygous, and knockout mice. Bottom: Western blot analysis of primary endometrial fibroblasts from heterozygous or knockout mice with anti-EPS8 and β-actin antibodies. (I) Confocal images of Eps8+/+ or Eps8−/− organoids infected with GFP-expressing CTL2 (green) for 4 h and stained for F-actin (magenta) and DNA (blue). (J) Fluorescence images of Eps8 +/+ or Eps8 −/− organoids infected with GFP-expressing C. muridarum (green) for 4 h and stained for F-actin (magenta) and DNA (blue). (K-L) Confocal images of Eps8+/+ or Eps8−/− organoids uninfected (K) or infected with CTL2-GFP for 4 h (L) and immunostained for ZO-1 (white) and DNA (blue). (M) Quantification of luminal dextran intensity over time in Eps8 +/+ or Eps8 −/− organoids infected with CTL2 (n = 9–10 organoids). (D, M) Data are represented as the mean +/− SD.
***p<0.001
The disruption of tight junctions promotes Chlamydia entry into polarized epithelia
Time-lapse microscopy of polarized A2EN cells labeled with fluorescent SiR-actin and infected with GFP-expressing C. trachomatis indicated that EBs often land and enter near sites of junction remodeling (Fig 6A, Movie 6). We hypothesized that the disruption of tight junctions can promote entry of Chlamydia at newly exposed basolateral receptors. Therefore, we developed an assay to assess the impact of an initial infection on subsequent ones (Fig 6B). Polarized A2EN cells were first infected synchronously with a GFP-expressing wild-type or tepP mutant Chlamydia for 1.5 hours followed by an asynchronous infection with non fusogenic mCh-expressing Chlamydia incA mutants. Because the non fusogenic incA mutant makes independent inclusions, the efficiency of secondary invasions can be determined by quantifying the number of mCh-positive inclusions. We observed that epithelial cell dispersion during a primary infection with wild-type Chlamydia enhanced the number of secondary inclusions as compared to primary infections with tepP mutants (Fig 6C). Similarly, polarized A2EN EPS8 KO cells were less permissive for secondary invasion events compared to the parental A2ENCas9 controls (Fig 6D). Importantly, primary infections with Chlamydia tepP mutants showed no further decrease in the number of secondary inclusions formed in A2EN EPS8 KO cells (Fig 6E), providing additional support to the premise that monolayer remodeling by TepP occurs largely through EPS8. Finally, the primary infection did not affect the number of secondary inclusions formed when infections were performed in non-polarized A2EN cells (Fig 6F), further indicating that this effect is unique to polarized cells.
Figure 6. The disruption of tight junctions by Chlamydia promotes secondary invasion events.

(A) Cell invasion at disrupted epithelial cell-cell junctions. Time-lapse still frames of polarized A2EN cells labeled with SiR-actin (magenta) and infected with GFP-expressing CTL2 (green). Time display, min:sec. Arrows denote EBs landing near remodeling cell-cell junctions and moving into cells (orthogonal view).
(B-F) Quantitative assessment of secondary Chlamydia infections upon tight junction disruption. (B) Schematic of experimental workflow to assess secondary infections. Polarized A2EN cells are infected synchronously with GFP-expressing Chlamydia (primary infection) before mCh-expressing non-fusogenic incA mutant bacteria (secondary infection) was added to the media for an additional 16–18 h. (C) Super plots of the number of secondary inclusions in A2EN cells after primary infection with GFP-expressing CTL2 or tepP mutants (n = 90 fields of view, unpaired, two-tailed t-tests). (D) Super plots of the number of secondary inclusions in polarized A2ENCas9 and A2ENCas9 EPS8 KO cells (n = 90 fields of view; unpaired, two-tailed t-test). (E) Scatter plots show the number of secondary inclusions in A2ENCas9or A2ENCas9 EPS8 KO cells after primary infections with tepP mutant bacteria (n = 30 fields of view; unpaired, two-tailed t-test). (F) Scatter plots show the number of secondary inclusions in non-confluent A2ENCas9 and A2ENCas9 EPS8 KO cells after a primary infection with CTL2-GFP (n = 30 fields of view; unpaired, two-tailed t-test).
(G-K) Ectopic expression of TepP is sufficient to promote infection of polarized MDCK cells. (G) Quantification of inclusion per field in polarizing and polarized MDCK cells infected with CTL2 for 20 h (n=20 fields of view, unpaired, two-tailed t-test). (H) MDCK cells expressing mCh or mCh-TepP (not shown) were allowed to polarize and then infected with CTL2-GFP (green) for 20 h. (I) Quantification of inclusions per field in MDCK cells expressing mCh or mCh-TepP and infected with GFP-expressing CTL2 or tepP mutants (n=20 fields of view, unpaired, two-tailed t-test). (J) Fluorescence images of MDCK cells expressing mCh or mCh-TepP (magenta), infected with CTL2-GFP (green) for 20 h, and stained for DNA (blue). (K) Quantification of the percent of infected cells expressing mCh or mCh-TepP (n=20 fields of view, unpaired, two-tailed t-test).
(C-F, K) Scatter plots show inclusion number per field and the bar denotes the median.
(G, I) Box and Whisker plots represent the media and interquartile range +/− min and max values.
****p<0.0001
To test whether TepP was sufficient to promote entry through the disruption of cell junctions, we used Madin-Darby Canine Kidney (MDCK-I) cells, an epithelial model that is recalcitrant to Chlamydia infection when polarized (Fig 6G) (Moore et al., 2008). First, we determined that TepP expression in MDCK-1 cells increased EPS8 localization to the lateral membrane and disrupted ZO-1 positive tight junctions (S5A–D). Moreover, TepP-positive punctate and tubulovesicular structures co-localized with EPS8 at tight junctions and in areas of junction remodeling (Fig S5E). Consistent with these observations, TepP expression delayed monolayer formation and reduced TEER during MDCK polarization (Fig S5F–G). In this model, TepP expression in polarizing MDCK cells significantly enhanced Chlamydia infection (Fig 6H). Importantly, wild-type and tepP mutant Chlamydia invaded TepP-expressing MDCK cells with equivalent efficiency (Fig 6I) and inclusions were preferentially found in cells expressing TepP-mCh (Fig 6J–K).
TepP and EPS8 promotes Chlamydia shedding and survival in the murine genital tract
Our results indicate that TepP and EPS8 regulate epithelial organization during C. trachomatis entry and that TepP prolonged the survival of CTL2 strains following a transcervical challenge. In A2EN EPS8 KO cells, C. trachomatis inclusions were significantly smaller than in the parental lines and there was a marked reduction in the production of IFUs (Fig 7A–C). However, IFU production of CTL2 tepP mutants in A2EN EPS8 KO cells was still lower than wild-type CTL2, indicating that TepP targets other host processes that are important for IFU production (Fig 7D). Because of the limitations of mouse models for human Chlamydia upper genital tract infections (Coers et al., 2009), we tested the role of EPS8 in Chlamydia survival and pathogenesis for the highly-related mouse pathogen C. muridarum. Eps8+/+ and Eps8−/− littermates were infected intravaginally with C.muridarum and bacterial shedding was assessed every three to four days. The number of recoverable IFUs from vaginal swabs was consistently higher in wild-type mice and persisted longer (Fig 7E), further supporting a pro-infection role for EPS8.
Figure 7. TepP and EPS8 promotes infection in polarized endocervical cells and in the murine female genital tract.

(A-D) EPS8 promotes the expansion of CTL2 inclusions and bacterial replication. Confocal images of A2ENCas9 and A2ENCas9 EPS8 KO cells infected with CTL2 (MOI < 1) for 30 h and immunostained for the inclusion membrane protein Cap1 (white) and DNA (blue). (B) Quantification of inclusion size (n=59–65 inclusions; unpaired, two-tailed t-test). Scatter plots represent the range of inclusion size and the bar denotes the median. (C) Normalized IFU production of CTL2 in A2ENCas9 and A2ENCas9 EPS8 KO cells, and (D) CTL2 and tepP mutants in A2ENCas9 cells and A2ENCas9 EPS8 KO cells.
(E) EPS8 promotes C. muridarum infection in vivo. Eps8+/+ (blue), Eps8+/− (yellow) and Eps8−/− (red) mouse littermates were infected intravaginally with C. muridarum. The number of recoverable IFU were quantified and plotted (n = 6–7 mice for 3 −14 dpi, Mann-Whitney U test; n= 3–4 mice for 18, 21 dpi; unpaired, two-tailed t-test).
(F-G) EPS8 and TepP promote C. muridarum replication. (F) Normalized production of C. muridarum IFUs after replication in Eps8−/− fibroblasts stably expressing EPS8 (+EPS8) or an empty vector (−EPS8). (G) Normalized IFU production of C. muridarum wild-type and tepP mutant bacteria in mouse fibroblasts (H-L) TepP promotes C. muridarum colonization and pathology in the upper genital tract. (H) Mice were infected intravaginally with C. muridarum wild-type or tepP mutant bacteria and swabbed at the indicated day. The number of recoverable IFU were quantified (n= 8–16 mice; Mann-Whitney U test). (I) Mice were infected transcervically with C. muridarum wild-type or tepP mutant bacteria and swabbed at the indicated day. The number of recoverable IFU were quantified (n=10 mice; Mann-Whitney U test). (J) Hematoxylin and eosin stained sections of the uterine horns and ovaries from mice infected with C. muridarum wild-type or tepP mutant bacteria at 14 d post-infection. Arrows denote increased levels of cellular infiltrates. (K-L) Scatter plots of acute pathology scores of the uterine horns and ovaries (n=5 mice; unpaired, two-tailed t-test). Data represent scores of individual uterine horns and ovaries and the line denotes the median. (M) Eps8+/+, Eps8+/−, and Eps8−/− mouse littermates were infected transcervically with C. muridarum tepP mutant bacteria and swabbed at the indicated day. The number of recoverable IFU were quantified (n=10 mice; Mann-Whitney U test). (N) Normalized IFU production of C. muridarum wild-type and tepP mutant bacteria in Eps8−/− fibroblasts complemented with EPS8 (+EPS8) or an empty vector (-EPS8).
All bar graphs represent the mean +/− SD (n= 3 independent experiments; unpaired, two-tailed t-test).
*p < 0.05, **p<0.01, ***p<0.001, ****p<0.0001
We next generated a tepP mutant in C. muridarum (Fig S6A), which phenocopies C. trachomatis tepP mutants in that it failed to recruit EPS8 to early inclusions and promote dispersion of polarized A2EN cells (Fig S6B–D). C. muridarum tepP mutants were also significantly impaired in the production of IFUs in mouse fibroblast cells (Fig 7G) and did not ascend to the upper genital tract (UGT) after vaginal infections with complete clearance within six days (Fig 7H). We next bypassed the lower genital tract by infecting mice transcervically and found tepP mutant EBs were shed for up to 12 days, albeit at significantly lower levels than wild-type C. muridarum (Fig 7I). Moreover, the uterine horns and ovaries exhibited significantly reduced acute pathology (Fig 7K–L). To test whether the decrease in bacterial burden in the UGT for C. muridarum tepP mutants was dependent on EPS8-mediated manipulation of host functions, we compared bacterial shedding between infected Eps8+/+ and Eps8−/− littermates. The level of bacterial shedding was lower in Eps8 KO mice and the infection was cleared faster (Fig 7M). Furthermore, IFU production of the tepP mutant is again significantly reduced in the absence of EPS8 (Fig 7N). Collectively, these data indicate that EPS8 promotes infection and that TepP performs additional functions that are also important for successful colonization of the UGT.
Discussion
C. trachomatis disrupts epithelial cell organization and cell-cell junctions in cell culture (Dolat and Valdivia, 2021; Kessler et al., 2012; Prozialeck et al., 2002), but the mechanisms underlying this process or consequences to infection are largely unknown. Here we report that the effector TepP transiently disrupts epithelial tight junction organization to promote host cell entry. We propose based on live-imaging (Fig 6A) and secondary infection assays (Fig 6B) that TepP-mediated disruption of cell junctions and polarity increases the availability of basolateral receptors that enhance the binding and entry of additional bacteria. A similar process has been documented during Pseudomonas aeruginosa infection in polarized MDCK cells, where the bacterium alters epithelial polarity by stimulating the positioning of basolateral components to the apical surface to promote colonization (Kierbel et al., 2007). Similarly, the Coxsackie virus activates Src and Abl kinases and Rho GTPase signaling to reorganize the actin cytoskeleton and bind to the Coxsackie and Adenovirus receptor (CAR), a tight junction component that is necessary for virus entry in intestinal epithelial cells (Coyne and Bergelson, 2006). C. trachomatis binds to heparan sulfate proteoglycans that are largely localized to the basolateral compartment of polarized epithelia in the female reproductive tract (Hayashi et al., 1988; Inki, 1997; Zhang and Stephens, 1992), supporting the notion that altering epithelial polarity can increase EB binding, but further studies are necessary to identify the mechanism of host receptor reorganization.
Chlamydia infection modulates Tyr phosphorylation-mediated signaling (Birkelund et al., 1994; Fawaz et al., 1997). In addition, TarP and TepP are major effector proteins that are also targets of Tyr phosphorylation by Src kinases (Carpenter et al., 2017; Chen et al., 2014; Clifton et al., 2004). We compared the impact of Chlamydia infection, and TepP in particular, on the global Tyr phosphoproteome in A2EN epithelial cells. Infection with wild-type Chlamydia or tepP mutants resulted in higher level of Tyr phosphorylation of receptor tyrosine kinase signaling pathways (e.g. EGFR), regulators of the actin cytoskeleton, and proteins involved in cell motility and morphology as compared to uninfected cells, but the targets did not significantly overlap. For instance, EPS8 and the related EPS8L2, host actin-binding proteins that regulate actin-based structures (Offenhäuser et al., 2004), emerged as major targets of TepP-dependent phosphorylation (Fig 2C). We determined that EPS8 is recruited to bacterial entry sites and remained strongly associated with nascent inclusions in a manner that is strictly dependent on TepP (Fig 2–3). While EPS8 phosphorylation was not required for its recruitment to nascent inclusion, these phosphorylation sites are in residues that mediate interactions with different host partners involved in cell signaling, endocytosis, vesicle trafficking, cytoskeleton organization (Cunningham et al., 2013) and promote tumor cell growth (Shahoumi et al., 2020). It is also possible that TepP-dependent phosphorylation may stabilize EPS8 given its abundance is linked to Src-dependent phosphorylation (Fig S2) as previously reported (Maa et al., 1999) and EPS8 levels were reduced during infection in the presence of Src inhibitors (Fig S3F).
TepP expression was sufficient to redistribute EPS8 to internal puncta and induce a change in cellular morphology in HeLa cells (Fig 3, S2) which parallels what is seen in cells infected with TepP-expressing Chlamydia (Fig 1). In transfected MDCK cells, TepP re-localized EPS8 to ZO-1 positive tight junctions and disrupted barrier function (Fig S5). EPS8 is not a canonical regulator of epithelial cell junctions and Eps8−/− mice do not exhibit any major phenotypes except for deafness due to defects in auditory cilia (Zampini et al., 2011). However, EPS8 can interact with tight junction proteins, such as Claudin-4, based on proximity-labeling proteomic approaches (Fredriksson et al., 2015). In murine endothelial cells, EPS8 is transiently recruited to adherens junctions upon stimulation to promote VE-cadherin ubiquitination, internalization and turnover (Giampietro, 2016). This latter finding suggests a potential role for EPS8 in the regulated disassembly of junctional complexes. Chlamydia appears to have co-opted a similar process to disassemble epithelial cell junctions since ESP8-deficient A2EN cells, which exhibit normal polarization and maintain barrier functions (Fig 4), and endometrial organoids derived from Eps8−/− mice were resistant to TepP-mediated disruption of tight junctions.
Our initial observation that C. trachomatis CTL2 tepP mutants are cleared faster in the murine upper genital tract (Fig 1) gave us impetus to pursue a molecular characterization of TepP. Because this mouse model of human Chlamydia infections exhibits a short kinetic window of bacterial residence in the UGT, we opted to use C. muridarum, a mouse adapted Chlamydia that infects the vaginal vault and ascends to the UGT where it stimulates pathologies similar to that observed during human infections (Shah et al., 2005). Eps8−/− mice exhibited lower levels of infection and faster clearance (Fig 6), indicating that EPS8 promotes infection and survival in the UGT. The phenotype of C. muridarum tepP mutants, however, was more severe with these mutants being unable to ascend to the UGT after vaginal inoculation and displaying rapid clearance and low pathology if directly inoculated in the UGT. These results suggests that in addition to the manipulation of EPS8, other TepP functions are important for colonization of the reproductive tract. TepP activates Type I interferon responses (Chen et al., 2014), which promotes C. muridarum infection and pathology in mice (Nagarajan et al., 2008), and functions to limit neutrophil recruitment in an endometrial co-culture model with neutrophils (Dolat and Valdivia, 2021). In addition, TepP binds to PI3K and modules the levels of phosphoinositides in infected cells (Carpenter et al., 2017). We postulate that a combination of these additional activities together with the manipulation of TJs contribute to the severe virulence defects of tepP mutants.
STAR METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Raphael Valdivia (raphael.valdivia@duke.edu)
Materials availability
Bacterial strains, plasmids and cell lines will be made available on request.
Data and code availability
Phospho-proteomic datasets were deposited to MassIVE and will be available on the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request
Experimental model and subject details
Cell lines and conditioned medium
Vero (CCL-81), HeLa (CCL-2), SYF (CRL-2459), SYF + c-Src (CRL-2498), MEF (CF-1), 293T (CRL-3216), MDCK-I (CCL-34) cells were purchased from ATCC and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) containing 10% fetal bovine serum (FBS; Sigma-Aldrich). A2EN endocervical epithelial cells were a gift from Dr. Alison Quayle (Louisiana State University Health Sciences Center) and cultured in keratinocyte serum-free media (GIBCO) containing 10% FBS and growth factors according to the manufacturer. Eps8−/− fibroblasts stably expressing EPS8 or an empty vector were a gift from Dr. Giorgio Scita (The Firc Institute of Molecular Oncology, Milan, Italy). L-WRN cells were purchased from ATCC (CRL-3276) and cultured in DMEM containing 0.5 mg/mL geneticin (Gibco) and 0.5 mg/mL hygromycin B (Thermo Fisher Scientific) at 37°C with 5% CO2. Conditioned medium was generated as previously described (Miyoshi and Stappenbeck, 2013).
Chlamydia strains and propagation
C. trachomatis L2/434/Bu (CTL2; ATCC VR-902B), Serovar D (Gift from Dr. Michael Starnbach), the genetically modified C. trachomatis L2 tarp (Gift from Dr. Ken Fields), tepP (Dolat et al., 2021), tmeA, tmeB, M923 (Sixt et al., 2017), and the C. trachomatis L2 strains expressing p2TK2-GFP or p2TK2m-Cherry were propagated in Vero cells, harvested by water lysis at 44–48 h post-infection, sonicated, diluted in SPG (sucrose-phosphate-glutamate) buffer to 1x concentration (75 g/L sucrose, 0.5 g/L KH4PO4, 1.2 g/L Na2HPO4, 0.72 g/L glutamic acid, pH 7.5), and stored as single use aliquots at −80°C. The C. muridarum MoPn and CM001 (Poston et al., 2018) and CM006-GFP strains were propagated in Vero cells and harvested by water lysis and sonication at 36 – 40 h post-infection.
Murine Infection Models
All animal experiments were approved and performed in accordance with the Duke University Institutional Animal Care and Use Committee. Animals were housed in a specific pathogen free environment with unrestricted access to food and water. Six-week-old female C57BL/6J mice (Jackson Laboratory, No. 000664) or 6–8 week old female Eps8 transgenic age-matched littermates (derivation described below) were injected subcutaneously with 2.5 mg medroxyprogesterone (TEVA Pharmaceuticals) to synchronize their estrous cycles one week prior to infections. Intravaginal infections were performed with 1 × 106 – 1 × 107 mCherry-expressing C. muridarum (CM001), MoPn, or tepP mutant EBs. Transcervical infections were performed with 1 × 107 CTL2 or tepP mutant EBs or 5 × 105 C. muridarum MoPn or tepP mutant EBs per mouse using an NSET Embryo Transfer device (ParaTechs).
Organoid and stromal cell isolation from the mouse endometrium
Epithelial organoids and primary stromal fibroblasts were cultured as previously described (Dolat and Valdivia, 2021). Uterine horns from 6 – 12 week old female C57BL/6J (Jackson Labs, No. 000664) or Eps8 littermates were isolated, washed in cold phosphate buffered saline (PBS; Gibco), cleared of adipose tissue, cut into ~ 2 mm pieces, and incubated in DMEM (Gibco) containing 0.2% collagenase A (Thermo Fisher Scientific), 10% FBS and 1 U/mL penicillin/streptomycin (Gibco) for 2.5 h at 37°C. The tissue pieces were subsequently rinsed three times with PBS, shaken vigorously with 12 mL of PBS containing 0.1% bovine serum albumin (BSA; Equitech Bio), and passed through a 70 μm strainer (Falcon); the flow through contained the stromal cell population. The previous step was repeated prior to inverting the filter over a 50 mL tube and the epithelial glands were recovered by passing 20 mL PBS containing 0.1% BSA. Fractions containing stromal cells and epithelial glands were centrifuged at 600 g for 5 min at 10°C. The stromal cells were resuspended in 2–3 mL of 50% L-WRN conditioned media containing 50 μg/mL gentamicin (Gibco) and plated in a 24 well plate (0.5 mL per well). Epithelial glands were resuspended ice cold growth factor reduced Matrigel (Corning) before adding DMEM/F12 (Gibco) at a 1:1 final ratio. Cell suspensions were plated as follows - 35 μL drops per well in a 24 well plate or 125 μL drop in a 35 mm dish (Cellvis) - and incubated at 37°C at 5% CO2 for 40 min before overlaying 50% L-WRN conditioned media in DMEM/F12 containing 50 μg/mL gentamicin (Gibco) and 50 ng/mL EGF (StemCell Technologies).
Generation of Eps8 knockout mice
Targeted Eps8 (ENSMUST00000058210.12) editing in C57BL/6 (Jackson Labs, No. 000664) mice was performed as follows: guide RNAs were designed using the CRISPOR algorithm (Concordet and Haeussler, 2018). High quality guides flanking Eps8 exon 4 (sgRNA3: 5’ CTGAGTTTCGTTACCTACTATGG 3’ and sgRNA4: 5’ TTACTGTTCAAGCGCTTAACTGG 3’) were synthesized by in vitro transcription and screened via Guide-It screening (Clonetech). The CRISPR/Cas9 ribonucleotide complex containing 16 μM Cas9 (IDT Cat No. 1081059) and 0.4 μg/μL of each sgRNA was generated in RNP buffer containing 100 mM HEPES pH 7.5, 750 mM KCl, 5 mM MgCl2, 20 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP; Sigma-Aldrich, cat. no. C4706), and 50% glycerol (Modzelewski et al., 2018). The complex was incubated at 37°C for 15 min prior to direct embryo electroporation using the Nepagene NEPA21 Type II electroporator with the CUY501P1–1.5 electrode with the following settings: Poring Pulse 40V; Length 3.5 ms; Interval 50 ms, No 4; D.Rate% 10; Polarity + and a Transfer Pulse 7V; Length 50ms; Interval 50 ms; No. 5; D.Rate% 40; Polarity +/−. The Eps8 mutant founders were backcrossed twice to the C57BL/6 (Jackson Laboratory; No.00064). Genotyping was performed from mouse tail cuts digested in ~150 μL buffer containing 50 mM Tris pH 8.0, 100 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, and proteinase K at 55°C for 4 h, heat-inactivated at 95°C for 10 min, and centrifuged at 12,000 g for 2 min. PCR analysis of the Eps8 locus was performed with 1 μL of the tail digestion using the primers: 5’-ATTTATTCTACTATAGCTGACGTC-3’ and 5’-TAGAGGGAGGAGGATTCATAGTTC-3’. Founder mutants were backcrossed twice with C57BL/6J (Jackson Labs, No. 000664) and disruption of EPS8 expression was determined by western blot as detailed below.
Method details
Insertional mutagenesis of CTL0063 (tmeA), CTL0064 (tmeB), and C. muridarum TC0268 (tepP)
The CTL0063 and CTL0064 and C. muridarum TC0268 sequences were analyzed for target insertion sites using the TargeTron™ algorithm (Sigma-Aldrich). The primers for CTL0063: IBS(5’-AAAAAAGCTTATAATTATCCTTACTCAACCTATTGGTGCGCCCAGATAGGGTG-3’), EBS1d (5’-CAGATTGTACAAATGTGGTGATAACAGATAAGTCCTATTGTCTAACTTACCTTTCTTTGT-3’), EBS2 (TGAACGCAAGTTTCTAATTTCGGTTTTGAGTCGATAGAGGAAAGTGTCT), and the primers for CTL0064: IBS(5’-AAAAAAGCTTATAATTATCCTTACTTCACTGACCCGTGCGCCCA GATAGGGTG-3’), EBS1d (5’-CAGATTGTACAAATGTGGTGATAACAGATAAGTCTGACCCCTT AACTTACCTTTCTTTGT-3’), EBS2 (5’-TGAACGCAAGTTTCTAATTTCGGTTTGAAGTCGATAG AGGAAAGTGTCT-3’) were used with the EBS Universal Primer (Sigma-Aldrich) to retarget the pDFTT3::aadA plasmid (Lowden et al., 2015) according to the TargeTron™ manual. The primers for TC0268: IBS (5’-AAAAAAGCTTATAATTATCCTTAGCTACCGCCTACGTGCGCCCAGATAGG GTG-3’), EBS1d (5’- CAGATTGTACAAATGTGGTGATAACAGATAAGTCGCCTACTCTAACTT ACCTTTCTTTGT-3’), and EBS2 (5’- TGAACGCAAGTTTCTAATTTCGATTGTAGCTCGATAG AGGAAAGTGTCT-3’) were used with the EBS Universal Primer to retarget the pDFTT3::bla plasmid as above. The vectors were transformed into E. coli DH5α (Invitrogen) and clones were isolated and sequenced (Eton Bioscience) using the T7-promoter primer. CTL2 was transformed with each plasmid as follows: 1 × 108 IFU were incubated with 10 μg DNA in buffer containing 0.9 mM calcium chloride for 30 min at 25°C and added to a confluent monolayer of Vero cells in a six well plate, centrifuged at 1,500 g for 30 min at 10°C. At 12 h post-infection, 150 μg/mL spectinomycin (Millipore) was added. The infections were passaged every 44 – 48 h. Insertions were verified by PCR analysis of the spectinomycin cassette using the primers: aadA forward (5’-GTAACGCGTCCCGGGCCTGATAGTTTGGCTGTGAG-3’) and aadA reverse (5’-TCTACGCGTTGCCTGACGATGCGTGGAG-3’) and the junction spanning the Chlamydia DNA and the intronic insertion using the primers: CTL0063 sense (5’- ATGAGTATTCGACCTAC −3’) and CTL0064 antisense (5’- ACTTCGAACACGCAATGCATC-3’) with the TargeTron universal primer (Sigma-Aldrich). C. muridarum MoPn transformations were performed as above except that 1 × 107 IFU were used in the transformation reaction and EBs were harvested and passaged every 36 h post-infection. Transformants were verified by PCR analysis using primers targeting full-length tepP (5’- GGTACCCGTCGTGTTGTCTAAGTCTC-3’) and (5’- GCGGCCGCTTATTGATTATCTAGTTCC-), the junction spanning Chlamydia DNA (5’- ACTACCTGTATCAACCTCTGATAG) and the universal primer above, and the bla cassette used above. C. muridarum MoPn and tepP mutant was confirmed by whole-genome sequencing.
Chlamydia transformations with shuttle plasmid vectors
The C. muridarum CM001 strain was transformed with p2TK2spec-Nigg-mCherry (Cortina et al., 2019) as follows: Approximately 107 IFU were incubated with 15 μg DNA in buffer containing 0.9 mM calcium chloride for 40 min, added to confluent Vero cells in a six well plate, and centrifuged at 1,500 g for 30 min at 10°C. At 15 h post-infection, 500 ug/mL spectinomycin was added. The infections were passaged every 36–40 hours until inclusions were present and fluorescent. Transformants were subsequently plaque-purified to obtain a clonal strain. The CTL0063 (tmeA) and the CTL0064 (tmeB) insertional mutants described above were transformed with the GFP-expressing E.coli-Chlamydia shuttle vector p2TK2 (Agaisse and Derré, 2013) as previously described (Dolat and Valdivia, 2021). In brief, 1 × 108 IFU were incubated with 10 μg DNA in buffer containing 0.9 mM calcium chloride for 30 min at 25°C and added to a confluent monolayer of Vero cells in a six well plate, centrifuged at 1,500 g for 30 min at 10°C. At 12 h post-infection, 1 U/mL penicillin was added. The infections were passaged every 44 – 48 h until GFP-positive inclusions were present when the penicillin concentration was raised to 5 U/mL prior to plaque purification. Clonal isolates were verified using GFP expression and primers targeting the bla cassette (5’-CGATCTGTCTATTTCGTTCA-3’) and (5’-CGGTATTATCCCGTATTGAC-3’).
Chlamydia infections in HeLa cells and fibroblasts
HeLa and MEF cells were cultured No.1 glass coverslips (Bellco Glass) coated with 30 μg/mL Type I Collagen (Invitrogen) in 10 mM glacial acetic acid (Sigma-Aldrich) for 10 min at 25°C. Coverslips and filters were rinsed once with media to remove excess acetic acid. The indicated Chlamydia strain (MOI=20) was diluted in the cold medium, vortexed for 15 s, and centrifuged onto cells at 1,500 g for 10–20 min at 10°C. The cells were replenished with warm media and incubated at 37°C for the indicated time. To depolymerize actin filaments after invasion, cells were infected for one hour prior to the addition of 1 μM Latrunculin A (Sigma-Aldrich) for 1 h.
A2EN polarization, infections, transepithelial electrical resistance (TEER), and live-cell imaging of cell dispersion
A2EN cells were polarized on either No. 1 glass coverslips (Bellco Glass) or polycarbonate or polyester transwell filters (0.4 μm pore; Corning) coated with Type I Collagen as above. A2EN cells (250–350k per coverslips or transwell filter) were cultured for 4–5 days in media containing 2 mM CaCl2 prior to infections, infected (MOI=50) by centrifugation at 500 g for 5 min at 25°C, and replenished with warm media. TEER measurements were performed at the indicated timepoints using an Epithelial Volt/Ohm Meter (World Precision Instruments). TEER was calculated by subtracting TEER measurement from a blank filter in media and multiplying the resistance (Ohm) measurement by the filter area.
For the dual invasion assay in Figure 6, A2EN cells were infected as above with GFP-expressing CTL2 or tepP mutants (polarized MOI = 50; nonpolarized = 10). After 1.5 h, the mCherry-expressing M923 (incA) mutant (polarized MOI = 10; nonpolarized = 10) was added directly to the media. The infection proceeded for an additional 16–18 h. Cells were fixed with 3% paraformaldehyde for 20 min, rinsed five times with PBS, mounted on coverslides with FluorSave (EMD Millipore) mounting medium, and imaged on the Nikon widefield microscopy described below. For live cell imaging, 35 mm glass bottom dishes (Mat-Tek) were coated with collagen as above prior to plating 1.0 × 106 – 1.5 × 106 A2EN cells. Brightfield microscopy was performed on an inverted Zeiss AxioObserver Z.1 microscope equipped with a motorized stage, XCite 120XL metal halide light source, and a Pecon XL S1 incubator. Images were acquired using a Plan Apo DICII 20x/0.80 NA objective (Olympus) and an EM-CCD camera (Photometrics) every three minutes for 14 h at 37°C.
Endometrial organoid infections
Endometrial organoids were microinjected using an Eppendorf FemtoJet 4x coupled with a Stereo Microscope (Nikon). C. trachomatis CTL2 was diluted in PBS to a final concentration of 5 × 105 – 5 × 106 IFU/mL, vortexed for 30 s, and ~ 5 μL was pipetted into a glass capillary (Sutter Instrument Company) using a microloader tip (Eppendorf). The glass capillary tip was broken off and the organoids were punctured once using a steep vertical angle and injected with equal volumes. When co-injected with fluorescent dextran, C. trachomatis CTL2 was diluted in PBS containing 0.01 mg/mL 3kD Texas-Red dextran (Invitrogen). Live imaging was performed on the Nikon Ti2 described below using a humidified Okolab environmental chamber set to 37°C.
MDCK infections, transfections, and TEER
MDCK cells were plated on collagen-coated coverslips as above. Non-polarized and polarized cells – either 4 × 104 or 1.2 × 105 per coverslip – were cultured for two days, infected with CTL2-GFP (MOI = 10) by centrifugation at 500 g for 10 min at 25°C, and refed with warm media. Note that non-polarized cells achieved polarity in two days. At 18 – 20 h post-infection, cells were rinsed twice with warm PBS, fixed with 3% PFA in PBS for 20 min, and quenched with 0.25% ammonium chloride for 5 min. TEER measurements were performed as described above in 7.5 × 104 cells cultured on collagen-coated polycarbonate filters. Transfections were performed using jetPRIME reagents (PolyPlus) with 1,500 ng of the indicated vector, and cells were cultured for two days prior to infections (MOI=10) and/or fixation.
CRISPR design, targeting and generation of knockout cell lines
Gene edits in HeLa and A2EN cells using the CRISPR/Cas9 system in cells stably expressing Cas9 as generated by transduction with the lentiCas9-Blast (Addgene, #52962) using 100 μg/mL Blasticidin (Sigma-Aldrich) selection. Virus was produced in HEK293T cells by co-transfecting using the jetPRIME system (Polyplus) containing a single guide RNA (sgRNA) in a lentiGuide-Puro vector (Addgene, #52962), and a psPAX2 (Addgene; #12260) and pMD2.G (Addgene; #12259). The following sgRNA was used to target EPS8: TCATCTCTTCCAGTGTGATG. Cells were infected with virus particles in media containing 8 ug/mL polybrene for two days prior to selection with 1 μg/mL puromycin (Sigma-Aldrich) for two days and subsequent passaging in 10 μg/mL puromycin for two weeks. Single clones were isolated by limiting dilution in 96 well plates. Knockout efficiency was assessed by western blot analysis as described below.
Plasmids, primers, mutagenesis, and transfections
To generate C1-mCherry-TepP, the TepP ORF was PCR amplified from genomic DNA using primers 5’-TCTCGAGCTCAAATGAGCATCGGGGGAGTACG-3’ and 5’- GGTGGATCCTTATTCCCTATCGA CTTCTCTATC-3’ and the Sac1 and BamH1 restriction sites. EGFP-EPS8 was a gift from Dr. Matthew Tyska (Vanderbilt University). The four tyrosine residues in EPS8 were sequentially mutated to phenylalanine using the NEB Q5 site-direct mutagenesis kit (New England Biolabs) according to the manufacturer’s protocol using the following primers: Y525 5’-TGTTTTGAGTGGTTCAAAATTTCTA TCTATATGGCGATTAGAAGTTGG-3’and 5’-CCAACTTCTAATCGCCATATAGATAGAAA TTTTGAACCACTCAAAACA-3’; Y540 5’-CCTTGCTACAAAGTCAAACTTGGATTTGGCATATT TCTTGG-3’ and 5’-CCAAGAAATATGCCAAATCCAAGTTTGACTTTGTAGCAAGG-3’; Y602 5’-TCTGTATAGTATGAGTAAAAGGTGGATCAGCACGCC-3’ and 5’-GGCGTGCTGATC CACCTTTTACTCATACTATACAGA-3’; Y774 5’-GTACAGTGATTTGGCTAAAGACTCTC GCCCCTTCA-3’ and 5’-TGAAGGGGCGAGAGTCTTTAGCCAAATCACTGTAC-3’. Clonal isolates were sequenced to verify each mutation. The EGFP-EPS8 truncation mutants were generated using the In-Fusion Snap Assembly Kit (Takara Bio) according to the manufacturer’s protocol. The EGFP-EPS8-ΔABD (nt 1–1944) used primers: 5’-TCTCGAGCTCAAGCTATGAATGGTCATATTTCTAATCATCCC-3’ and 5’-CCGCGGTACCGTCGATTATGACACAGGAACAGGTGCTG-3’, and the EGFP-EPS8-SH3/ABD (1593–2469) use primers: 5’-TCTCGAGCTCAAGCTATGCAACCCAAGAAATATGCCAAATC-3’ and 5’-CCGCGGTACCGTCGATTAGTGACTGCTTCCTTCATCA-3’. The reactions were transformed into E. coli Stellar cells and clonal isolates were sequenced. Transfections were performed using jetPRIME (PolyPlus) reagents according to the manufacturer. HeLa and MEFs cells (5 × 104) were cultured on coverslips or 35 mm glass bottom dishes coated with collagen as above and transfected with 500 ng of the indicated plasmid for 24 – 48 h. In Fig S2, MEFs were incubated 1 μM Latrunculin A (Sigma-Aldrich) for 1 h.
Time-lapse spinning disk microscopy
All cell lines were cultured on collagen-coated 35 mm glass bottom dishes (Cellvis) as described above. HeLa cells expressing EGFP-EPS8 or Eps8 −/− MEFs complemented with EGFP-EPS8 were infected with CTL2-mCherry (MOI = 50) for 1 h prior to imaging on an inverted Zeiss AxioObserver Z.1 microscope equipped with a motorized stage, XLIGHT V2 spinning disk unit (Crest Optics), and an Insert P environmental chamber (Zeiss). Images were acquired using a 60x/NA 1.4 objective (Olympus), an LDI multiline laser (89 North), a sCMOS ORCA Flash V3 camera (Hamamatsu), and the Metamorph software (Molecular Devices) every 15 s at 37°C. Live imaging of actin dynamics during infection was performed by first incubating polarized A2EN cells with 200 nM Sir-Actin (Cytoskeleton) for 1.5 h and subsequently infecting with GFP-expressing CTL2 (MOI=50) for 1 h prior to imaging on the same microscope using 0.5 μm slices every 30 s.
Determination of infectious progeny
Infectious units (IFU) were quantified using two 96 well plates of confluent cell lines indicated. The input IFU was assessed by counting inclusions at 24 h from infections of serial dilutions where the MOI < 1. In the second plate, the infection proceeded for 48 h (C. trachomatis CTL2) or 36 h (C. muridarum) and infectious units were recovered by rinsing cells once with water and incubating cells with water for 20 min. Cell lysates were serially diluted and confluent Vero cells were infected by centrifugation 1,500 g for 20 min at 10°C. The number of inclusions were counted in dilutions where the MOI < 1. The number of infectious units from the full cycle was compared to the input IFU to determine the output IFU. Three independent replicates in three biological replicates were performed and IFU output to input was normalized to control cell line.
Phosphotyrosine profiling in Chlamydia-infected A2EN cells
A2EN cells (1.5 × 107) were cultured in 150 mm dish and, in triplicate, mock-infected or infected (MOI=50) with tepP mutants complemented with TepP (pTepP) or an empty vector (pVector) (Carpenter et al., 2017) in HBSS at 4°C for 1 h prior to adding warm media for 4 h. The cells were washed once with cold PBS and collected in cold PBS containing cOmplete protease inhibitors (Millipore), scraped and transferred to a 50 mL conical tube, centrifuged at 2,000 g for 5 min at 4°C. The supernatant was discarded and the pellet was resuspended in 1 mL cold PBS containing protease inhibitors and phosphatase inhibitors and centrifuged at 2,000 g for 5 min at 4°C to isolate unlysed cells. The supernatant was discarded and the pellet was frozen at −80C. Samples were thawed and incubated with 100 mM ammonium bicarbonate and 8M urea, probe sonicated three times for 5 s, and centrifuged at 12,000 g for 5 min at 4°C. 6.6 mg of each sample was removed and normalized to 8.31 mg/mL with 50 mM ammonium bicarbonate containing 8M urea. All samples were then reduced for 20 min at 80°C with 10 mM dithiolthreitol and alkylated for 40 min at room temperature with 22 mM iodoacetamide. Samples were then diluted 1.6M urea with 50 mM ammonium bicarbonate. Trypsin was added to a 1:50 ratio (enzyme to total protein) and allowed to proceed for 18 h at 37°C. Samples were then acidified with 0.2% TFA (pH 2.5) and subjected to C18 SPE cleanup (Sep-Pak, 50 mg bed). Following elution, all samples were then frozen and then lyophilized to dryness. 6.6 mg digested peptide for each sample was resuspended in 1.0 mL IAP Buffer (Cell Signaling Technology) by vortexing and brief bath sonication, and transferred to an aliquot of pY-1000 PTMScan enrichment beads (Cell Signaling Technology). IP was performed for two hours at 4°C using end-over-end mixing. After spinning to settle the beads and removing the supernatant, pTyr enrichment beads were washed with three aliquots of PBS, one aliquot of water, and then eluted with two 50 μl aliquots of 0.15% TFA in water, for approximately 10 minutes at room temperature. Eluates were combined and taken through a C18 STAGE tip desalting cleanup, and resulting peptides were dried via speedvac. Samples were then spiked with 100 fmol pre-digested Bovine alpha casein. Quantitative LC/MS/MS was performed on 4 μL of each phosphopeptide enriched sample, using a nanoAcquity UPLC system (Waters Corp) coupled to a Thermo QExactive Plus high-resolution accurate mass tandem mass spectrometer (Thermo) via a nanoelectrospray ionization source. Briefly, the sample was first trapped on a Symmetry C18 300 mm 180 mm trapping column (5 l/min at 99.9/0.1 v/v water/acetonitrile), after which the analytical separation was performed using a 1.7 um Acquity BEH130 C18 75 mm 250 mm column (Waters Corp.) using a 5-min hold at 3% acetonitrile with 0.1% formic acid and then a 90-min gradient of 3 to 30% acetonitrile with 0.1% formic acid at a flow rate of 400 nanoliters/minute (nL/min) with a column temperature of 55°C. Data collection on the QExactive Plus mass spectrometer was performed in a data-dependent acquisition (DDA) mode of acquisition with a r = 70,000 (@ m/z 200) full MS scan from m/z 375 – 1600 with a target AGC value of 1e6 ions followed by 10 MS/MS scans at r-17,500 (@ m/z 200) at a target AGC value of 5e4 ions. A 20s dynamic exclusion was employed to increase depth of coverage. The total analysis cycle time for each sample injection was approximately 2 h. Sample order of data collection was interwoven between conditions to minimize temporal bias. Following the 9 LC-MS/MS analyses, data was imported into Rosetta Elucidator v3.3 (Rosetta Biosoftware, Inc), and all LC-MS/MS runs were aligned based on the accurate mass and retention time of detected ions (“features”) which contained MS/MS spectra using PeakTeller algorithm (Elucidator) and intensity-scaled based on a robust mean (10%) normalization of the identified pY phosphopeptide features. The relative peptide abundance was calculated based on area-under-the-curve (AUC) of aligned features across all runs. The overall dataset had 23,849 quantified isotope (peptide) groups. Additionally, 124,764 MS/MS spectra were acquired for peptide sequencing by database searching. This MS/MS data was searched against a SwissProt_Human database and Chlamydia database which also contained a reversed-sequence “decoy” database for false positive rate determination as well as Casein_Bovine as a surrogate internal standard. Database searching was performed within Mascot Server (Matrix Science) and annotated at a Mascot ion tolerance of 20.0 which resulted in a peptide false discovery rate of 0.70%. Searching allowed variable M (oxidation, +16 Da), and STY (phosphorylation, +80 Da). Differential expression of phosphopeptides were analyzed in Excel (Microsoft) using a heteroscedastic unpaired, two-tailed t-test and Volcano plots were generated in R Studio. Gene ontology enrichment analysis was performed in gProfiler (Raudvere et al., 2019) to identify the over-represented biological processes within dataset comparisons. Pathways were plotted if they reached significant p < 0.01.
Immunoprecipitations
In Fig 2E–F, A2EN cells were mock-infected or infected with the indicated strain (MOI=100) for 1 h, rinsed twice with PBS, and transferred to an Eppendorf tube by scraping in buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM PMSF with fresh addition of Halt alkaline phosphatase inhibitor (Thermo Fisher Scientific) and cOmplete cocktail protease inhibitors (Millipore) according to the manufacturer. Lysates were rotated at 4°C for 20 min, sonicated for 10s, rotated for an additional 10 min at 4°C, and cleared by centrifugation at 9,600 g for 10 min at 4°C. Input samples were collected before the lysate was incubated with 5 μL anti-EPS8 (Abcam; 96144) overnight at 4°C with rotation. The next day, 300 μL magnetic Protein A beads (Sigma-Aldrich) was rinsed five times with cold lysis buffer, resuspend in 150 μL lysis buffer, and 50 μL was added to each sample. The tubes were incubated with rotation at 4°C for 2.5 h, gently washed five times with lysis buffer, resuspended in Lamaelli buffer, and boiled for 10 min. In Fig 3G, EGFP-EPS8 was co-immunoprecipitated with TepP-mCherry using the GFP Trap system (ChromoTek). Six well plates were coated with 30 μg/mL Type I Collagen (Invitrogen) in 10 mM acetic acid for 10 min and rinsed once with media. 293T cells (6 × 105 per well) were co-transfected with the indicated plasmid (1 μg total DNA per well) using the jetPRIME transfection protocol (Polyplus) for 24 h. Cells were gently rinsed twice with PBS and collected in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Igepal, 1 mM PMSF containing fresh Halt alkaline phosphatase (Thermo Fisher Scientific) and Complete Midi cocktail protease inhibitor (Millipore). Samples were sonicated for 10 s, incubated with rotation at 4°C for 30 min, and cleared by centrifugation at 9,600 g for 5 min at 4°C. The supernatant was collected and incubated with GFP Trap beads (25 μL slurry per sample), which were previously washed five times with 0.5 mL lysis buffer. The samples were incubated with rotation at 4°C for 1.5 h, washed three times with lysis buffer, resuspended in Lamaelli buffer, vortexed, and boiled for 10 min. Equal volumes were analyzed by SDS-PAGE and western blot as below, and probed using anti-GFP (Invitrogen, A11122; 1:2000), anti-DsRed (Clonetech, 632496; 1:2000) and anti-tubulin as below.
Western Blots
In Fig 2E–F and Fig 4A, cells were cultured in a six well plate and mock-infected or infected with CTL2 (MOI=50) for 1 – 2 h, rinsed twice with PBS, transferred to an Eppendorf tube by scraping in buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 5 mM EDTA, 1 mM PMSF, Halt alkaline phosphatase inhibitor (Thermo Fisher Scientific) and Complete midi-cocktail protease inhibitors (Millipore). The cell solution was subsequently incubated on ice for 30 min after brief (10 s) sonication. The lysates were cleared by centrifugation at 9,600 g for 5 min at 4°C, diluted in Lamaelli buffer and boiled for 5 min. Equal volumes were resolved using a 10% SDS-PAGE gel, transferred to a nitrocellulose membrane for 1 h at 4°C, blocked with PBS containing 5% milk and 0.1% Tween-20 for 1 h at 25°C, and sequentially probed with anti-EPS8 (Abcam; 1:2,000), anti-phospho-Tyr (Cell Signaling Technologies; 1:2,000), anti-MOMP (Gift from Dr. Ken Fields; 1:1000), and anti-alpha-tubulin (Sigma-Aldrich; 1:5,000) diluted in 5% milk and 0.1% Tween-20. In Fig 5J, primary stromal cells were rinsed twice with PBS and transferred to an Eppendorf tube by scraping in buffer containing 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 0.1% SDS, 1 mM PMSF and 1x cOmplete protease inhibitors (Millipore), sonicated for 10 s and incubate on ice for 30 min. The lysates were cleared by centrifugation at 9,600 g for 5 min at 4°C, diluted in Lamaelli buffer and boiled for 5 min. Equal volumes were resolved using a 10% SDS-PAGE gel, transferred to a nitrocellulose membrane for 1 h at 4°C, blocked with PBS containing 5% milk and 0.1% Tween-20 for 1 h at 25°C, and sequentially probed with anti-EPS8 (BD Biosciences; 1:2,500) and anti-actin (Sigma-Aldrich; 1:5,000). In Fig S3, HeLa cells were cultured in six well plates and treated with DMSO or 10 μM PP2 (Abcam) for 2 h and infected with CTL2 (MOI=50) for 1 h in the presence of DMSO or 10 μM PP2. Cells were washed twice with PBS and collected in lysis buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 1 mM PMSF, 1x cOmplete protease inhibitor (Millipore) and Halt alkaline phosphatase inhibitors (Thermo Fisher Scientific), briefly sonicated, and incubated on ice for 30 min. Lysates were cleared by centrifugation at 9,600 g for 5 min at 4°C, diluted in Lamaelli buffer and boiled for 5 min. Equal volumes were analyzed by SDS-PAGE as above. The nitrocellulose membrane was blocked with PBS containing 5% milk and 0.1% Tween-20 for 1 h at 25°C, and sequentially probed with anti-phospho-tyrosine (Cell Signaling Technologies; 1:2,500), anti-EPS8 (Abcam; 1:2,000), anti-tubulin (Sigma; 1:5,000) and anti-HtrA (Gift from Dr. Wilhelmina Huston; 1:1,000). All membranes were probed with anti-rabbit or anti-mouse LI-COR secondary antibodies (1:10,000) conjugated to near-infrared dyes prior to scanning on an Odyssey LI-COR.
Antibodies
For immunofluorescence microscopy, the following antibodies were used: mouse anti-EPS8 (BD Bioscience 610413; 1:200), anti-phopsho-Tyr (Cell Signaling Technologies 9411S; 1:200), anti-MOMP (Santa Cruz 57678; 1:500), anti-C.muridarum LPS (Gift from D. Rockey; 1:100), and rabbit anti-ZO-1 (Cell Signaling Technologies D7D12; 1:200), anti-E-cadherin (Cell Signaling Technologies 24E10; 1:500), anti-Cap1 (Gift from A. Subtil; 1:250), anti-IncG (Gift from T. Hackstadt; 1:250), anti-TepP and anti-Slc1 (both generated and validated in Chen et al. 2014; 1:100 and 1:500, respectively). The following secondary antibodies were used: cross-adsorbed goat-anti-mouse (H+L) and goat-anti-rabbit (H+L) conjugated to Alexa-488 (A-11029; A-11008), Alexa-555 (A-21428; A21422), or Alexa-647 (A-21244; A-21236.) All secondary antibodies were diluted 1:1000, vortexed gently and centrifuged at 8,000 g for 1 min.
Immunofluorescence microscopy and inside-outside staining
Cells on coverslips were washed twice with warm PBS and incubated with warm PBS containing 3% formaldehyde (Electron Microscopy Services) for 20 minutes at 25°C. The samples were quenched with 0.25% ammonium chloride for 5 min, rinsed three times with PBS, permeabilized with PBS containing 0.1% tx-100 for 10 min, and incubated with 2% BSA for 20 min at 25°C. Primary antibodies were diluted in 2% BSA and incubated on cells for either 1.5 h at 25°C or overnight at 4°C, washed three times for 3 min with 2% BSA, and incubated with secondary antibodies diluted in 2% BSA for 1 h at 25°C. In Fig 3A, HeLa cells were permeabilized with 2% BSA containing 0.1% saponin for 30 min at 25°C, and antibodies were subsequently diluted in the same buffer and incubations were performed as described above. Organoids were fixed and stained as previously performed (Dolat and Valdivia, 2021). To assess bacterial entry (inside-outside), cells were infected with GFP-expressing CTL2 and mutant strains (MOI = 20) and fixed as above at 30 min post-infection but not permeabilized. Cells were blocked with 2% BSA for 20 min and labeled with anti-MOMP as above. In images where actin was visualized, cells were incubated with phalloidin (Acti-Stain 488 or 555, 1:500; Cytoskeleton) for the final 20 min of secondary incubation. Cells nuclei were labeled with Hoechst for 10 min with 2 ng/mL Hoechst (Thermo Fisher Scientific). Cells were mounted on glass coverslides using Vectashield (Vector Laboratories; H-1000) or FluorSave Reagent (Millipore).
Airyscan confocal imaging was performed on a laser scanning microscope (LSM 880; Zeiss) equipped with a motorized stage, Airyscan detector (Hamamatsu), and diode (405 nm), argon ion (488 nm), double solid-state (561 nm), and helium-neon (633 nm) lasers. Images were acquired using a C-Apochromatic 40x/1.2NA water (Zeiss) or 60x/1.4 NA oil objective (Zeiss), 0.3 μm sections and deconvolved using automatic Airyscan Processing in the Zen Software (Zeiss). Widefield deconvolution imaging was performed on an inverted microscopy (Nikon Ti2) equipped with a motorized stage, ORCA-Flash 4.0 V2 sCMOS camera (Hamamatsu), LED light source (Sola Light Engine). Images were acquired using a Plan Apo 10x/0.45 NA (Nikon) or Plan Apo 60x/1.4 NA oil objective (Nikon) and deconvolved using in the Nikon Elements Software. In Fig S5E, MDCK cells were imaged using an EVOS FL Cell Imaging System (Thermo Fisher Scientific) equipped with 10x/0.3 NA objective and a CCD camera. All images were exported to ImageJ and Adobe Photoshop where only linear adjustments to intensity were done.
Transmission Electron Microscopy
A2ENCas9 and A2EN EPS8 KO cells were cultured in collagen-coated six well plates for four days. Cells were rinsed twice with Hank’s Buffered Saline Solution and fixed with buffer containing 0.05% malachite green, 2.5% glutaraldehyde, 100 mM sodium cacodylate (pH 6.7) for two hours at 25C. Cells were washed four times for five min with 100 mM sodium cacodylate and stained with 0.8% K3Fe(CN)6 with 1% osmium tetroxide. The cells were rinsed four times for 5 min with 100 mM sodium cacodylate, stained with 1% tannic acid for 20 min, and washed once for 5 min with 100 mM sodium cacodylate and twice for 5 min with molecular grade distilled water. The cells were stained with 0.5% uranyl acetate overnight at 4C, washed five times for 5 min with molecular grade distilled water, dehydrated with a series of ethanol incubations, and embedded in resin and vertically cut. Ultrathin sections were post-stained with uranyl acetate and lead citrate. Images were acquired on a FEI Tecnai G2 Twin electron microscope at 6500x magnification.
Image analysis
All analyses were performed in ImageJ (Schindelin et al., 2012). Cell dispersion was quantified by fluorescent intensity segmentation of the F-actin signal. Binary masks were generated using the Minimum Error algorithm and the percent of area covered in each field of view was measured. The cell free area was determined by subtracting the area coverage from 100. Host cell entry (inside-outside) was quantified from maximum projections by encircling individual cells, identifying the number of GFP-positive EBs (total) and the MOMP-positive EBs (outside) using the find maxima algorithm in ImageJ. The number inside (MOMP-negative) was defined by subtracting the number outside (MOMP-positive) from the total number of EBs, which was divided by the number of total EBs to determine the percent inside. Recruitment of the host proteins (EPS8 and ZO-1) to nascent inclusions was performed in either maximum projections or 3D reconstructions by encircling individual cells and identifying the number of total EBs using the find maximum algorithm. Inclusions positive for either host protein was marked manually. To measure the impact of Chlamydia infection on individual cell morphology, cell aspect ratio was manually derived by using the line tool to measure the distance through the center of the cell and through the nucleus to define the length and the width.
Organoid size was determined by subtracting the background from brightfield images to reduce out of focus signal, generating a binary image and extracting the perimeter of each organoid. Epithelial cell height in the organoid was measured manually using the ImageJ line tool. In the dextran leakage assays, the fluorescent dextran intensity was segmented and the mean intensity profile over time was derived using z-axis profile function. To measure EPS8 and ZO-1 intensity in MDCK cells, the lateral membrane was sampled 2–3 times using a 5 micron line. The total intensity was measured and divided by the length of the line to derive intensity per micron. In the dual invasion assay, the number of mCherry-positive, secondary inclusions was quantified in background-subtracted images from ten fields of view using the find maxima algorithm. In Fig 7, inclusion size was measured by fluorescence intensity segmentation of the inclusion membrane protein using binary masks of Cap1. Each inclusion was highlighted, and the area was exported. The number of inclusions per mouse swab were imaged using the same EVOS FL Cell Imaging System and objective described above. Background-subtracted images were processed with the denoise algorithm to remove small outliers using the default settings and inclusion number was enumerated using the find maxima algorithm. All plots were generated in either Microsoft Excel or R Studio.
Quantification of bacterial burden in mice
To measure C. muridarum burden, mice were swabbed with 30 turns of a sterile polyester tipped applicator (Puritan), which was dipped in 0.5 mL SPG buffer and turned 30 times. Confluent Vero cells were infected with 25 μL or serial dilutions by centrifugation at 1,500 g for 10 min, cultured for 20 – 24 h and fixed with either ice cold methanol for 10 min or 3% formaldehyde (Electron Microscopy Services) in PBS for 20 min. Mice infected transcervically with C. trachomatis CTL2 were sacrificed at 2, 4 and 6 days post-infection, and the upper genital tracts were excised and trimmed of adipose tissue and immediately homogenized in 1.5 mL PBS. DNA was extracted using a DNeasy kit (Qiagen) from 100 μL of homogenate following procedures recommended by the manufacturers.
Histological analysis of the female reproductive tract
Semiquantitative histopathology grading was performed by a board-certified veterinary anatomic pathologist masked to the treatment group on the upper genital tract from tissue excised at 14 dpi, cleared of adipose tissue and fixed in 10% formalin for two days at room temperature. The tissue was paraffin embedded, cut at 5 μm sections, and stained with hematoxylin and eosin. Sections were imaged on an Olympus BX51 microscope equipped with a DP70 digital camera (Olympus). Tissue was scored using the following numeric criteria: 0 = normal, 1 = rare foci, 2 = scattered aggregates (1–4) or mild diffuse, 3 = numerous aggregates (> 4) or moderate diffuse, 4 = severe diffuse infiltration or confluence. The genital tract from mice infected for 45 d was excised and imaged using an iPhone XR. Gross pathology, such as hydrometra and hydrosalpinx, which are characterized by fluid-filled sacs or enlargements, were counted.
RT-qPCR
Quantitative PCR was performed on an a StepOne Plus Real Time PCR Systems (Applied Biosystems) using Power UP SYBR Green (Thermo Fisher Scientific). Quantification of C. trachomatis 16S rRNA and mouse GAPDH were performed in triplicate and based on standard curves from dilutions of purified C. trachomatis L2 and mouse DNA. Mouse PCR targets and primers used were: GAPDH (5’-ACTGAGCAAGAGAGGCCCTA-3’, 5’-TATGGGGGTCTGGGATGGAA-3’), and C. trachomatis PCR targets and primers used were: 16S rRNA (5’-GGAGGCTGCAGTCGAGAATCT-3’, 5’-TTACAACCCTAGAGCCTTCATCACA-3’) (Sixt et al., 2017)
Quantification and statistical analysis
All datasets contain three independent replicates and were analyzed in R Studio for equal variance and normality using a Levene Test or Shapiro test, respectively. Normally distributed datasets were analyzed using an unpaired, two-tailed t-test. Datasets with unequal variance or distributions were analyzed using a Welch’s t-test or a Mann Whitney U test. The indicated statistical test for each experiment can be found in the figure legends and was performed in either R Studio or Microsoft Excel.
Supplementary Material
Table S1, Related to Figure 2. List of differentially Tyr phosphorylated host proteins in mock-infected and infected epithelial cells. Comparison of Tyr phosphorylation of proteins in uninfected A2EN cells or A2EN cells infected with TepP+ Chlamydia (pTepP) and TepP− Chlamydia (pVector). Tab1, pTepP vs pVector; Tab2, pTepP vs Mock; Tab3, pVector vs Mock.
Movie 1, Related to Figure 1. Time-lapse brightfield microscopy of polarized A2EN cells infected with CTL2. Images were acquired every 3 min for ~ 3 h. Time display, hour:min.
Movie 2, Related to Figure 2. Time-lapse spinning disk confocal microscopy of a HeLa cells expressing EGFP-EPS8 and infected with CTL2-mCherry. Images were acquired every 15 s for ~ 26 min. Time display, min:sec.
Movie 3, Related to Figure 5. Time-lapse fluorescence microscopy of endometrial organoids co-injected with CTL2 (left) or tepP mutant (right) bacteria with Texas-Red Dextran. Images were acquired every 10 min for ~ 14 h. Time display, hour:min.
Movie 4, Related to Figure 5. Time-lapse fluorescence microscopy of Eps8+/+ (left) and Eps8−/− (right) endometrial organoids were co-injected with CTL2 and Texas-Red Dextran. Images were acquired every 10 min for ~ 14 h. Time display, hour:min.
Movie 5, Related to Figure 6. Time-lapse spinning disk confocal microscopy of polarized A2EN cells labeled with SiR-actin and infected with CTL2-GFP. Images were acquired every 30s for ~ 26 min. Time display, min:sec.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-EPS8 | Abcam | Cat# 96144; RRID:AB_10678966 |
| Mouse anti-EPS8 | BD Biosciences | Cat# 610143; RRID:AB_397544 |
| Mouse anti-phospho-Tyr | Cell Signaling Technology | Cat# 9411S;RRID:AB_331228 |
| Mouse anti-MOMP | Santa Cruz Biotechnology | Cat# Sc-57678;RRID:AB_1119779 |
| Mouse anti-beta-catenin | BD Biosciences | Cat# 610154;RRID:AB_397555 |
| Rabbit anti-ZO-1 | Cell Signaling Technology | Cat# 8193S;RRID:AB_10898025 |
| Rabbit anti-E-cadherin | Cell Signaling Technology | Cat# 3195; RRID:AB_2291471 |
| Rabbit anti-Slc1 | (Chen et al. 2014) | N/A |
| Rabbit anti-TepP | (Chen et al. 2014) | N/A |
| Rabbit anti-IncG | (Scidmore-Carlson et al. 1999) | N/A |
| Rabbit anti-Cap1 | (Delevoye et al. 2008) | N/A |
| Rabbit anti-HtrA | (Huston et al., 2008) | N/A |
| Mouse anti-LPS | Dan Rockey, Oregon State University | N/A |
| Rabbit anti-GFP | Thermo Fisher Scientific | Cat# A11122;RRID:AB_221569 |
| Rabbit anti-DsRed | Takara Bio | Cat# 632496;RRID:AB_10013483 |
| Mouse anti-alpha-tubulin | Sigma-Aldrich | Cat# T5168;RRID:AB_477579 |
| Mouse anti-beta-actin | Sigma-Aldrich | Cat# A2228;RRID:AB_476697 |
| Rabbit PTMScan phospho-Tyr | Cell Signaling Technology | Cat# 8803 |
| Goat anti-Mouse IgG 680 | LI-COR Biosciences | Cat# 926-68020;RRID:AB_10706161 |
| Goat anti-Rabbit IgG 680 | LI-COR Biosciences | Cat# 926-68021;RRID:AB_10706309 |
| Goat anti-Mouse IgG 800 | LI-COR Bioscences | Cat# 926-32210;RRID:AB_621842 |
| Goat anti-Rabbit IgG 800 | LI-COR Biosciences | Cat# 926-32211;RRID:AB_621843 |
| Goat anti-mouse 488 | Thermo Fisher Scientific | Cat# A-11029;RRID:AB_2534088 |
| Goat anti-rabbit 488 | Thermo Fisher Scientific | Cat# A-11008;RRID:AB_143165 |
| Goat anti-mouse 555 | Thermo Fisher Scientific | Cat# A-21422;RRID:AB_2535844 |
| Goat anti-rabbit 555 | Thermo Fisher Scientific | Cat# A-21428;RRID:AB_2535849 |
| Goat anti-rabbit 647 | Thermo Fisher Scientific | Cat# A-21236;RRID:AB_2535805 |
| GFP-Trap | ChromoTek | Cat# GTMA-20;RRID:AB_2631358 |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Sigma-Aldrich | Cat# D2438 |
| Latrunculin A | Sigma-Aldrich | Cat# I5163 |
| PP2 | Sigma-Aldrich | Cat# P0042 |
| Acti-stain 488 | Cytoskeleton | Cat# PHDG1-A |
| Acti-stain 555 | Cytoskeleton | Cat# PHDH1-A |
| SiR-Actin | Cytoskeleton | Cat# CY-SC001 |
| 16% paraformaldehyde | Electron Microscopy Services | Cat# 15710 |
| Triton X-100 | EMD Millipore | Cat# TX1568-1 |
| Hoechst | Thermo Fisher Scientific | Cat# 62249 |
| Vectashield | Vector Labs | Cat# H-1000 |
| FluorSave Reagent | EMD Millipore | Cat# 345789 |
| Tannic Acid | Electron Microscopy Services | Cat# 21700 |
| Sodium cacodylate trihydrate | Electron Microscopy Services | Cat# 12300 |
| Osmium Tetroxide | Electron Microscopy Services | Cat# 19152 |
| Uranyl acetate | Electron Microscopy Services | Cat# 22400 |
| K3Fe(CN)6 | Electron Microscopy Services | Cat# 20150 |
| Malachite green | Electron Microscopy Services | Cat# 18100 |
| Ammonium chloride | Sigma-Aldrich | Cat# A-5666 |
| jetPRIME Transfection Reagent | PolyPlus | Cat# 89129-924 |
| Texas-Red Dextran (3 kDa) | Thermo Fisher Scientific | Cat# D3329 |
| Spectinomycin | Millipore | Cat# 567570 |
| Penicillin | Sigma-Aldrich | Cat# P3032-10MU |
| Gentamicin | Gibco | Cat# 15750060 |
| Type I collagen | Invitrogen | Cat# A1048301 |
| cOmplete, EDTA_free protease inhibitor | Millipore Sigma | Cat# 11836170001 |
| Halt phosphatase inhibitor | Thermo Fisher Scientific | Cat# 78428 |
| BSA (Fraction V) | Equitech-Bio | Cat# BAC61-1000 |
| Tween-20 | Sigma-Aldrich | Cat# P9416 |
| Collagenase A | Sigma-Alddrich | Cat# 10103578001 |
| Matrigel | Corning | Cat# 356231 |
| EGF | STEMCELL Technologies | Cat# 78016.1 |
| Critical commercial assays | ||
| Deposited data | ||
| Experimental models: Cell lines | ||
| HeLa | ATCC | CCL-2;RRID:CVCL_0030 |
| Vero | ATCC | CCL-81;RRID:CVCL_0059 |
| A2EN | (Buckner et al. 2013) | N/A |
| A2EN Cas9 | This paper | N/A |
| A2EN EPS8 KO | This paper | N/A |
| SYF | ATCC | CRL-2459 |
| SYF + c-Src | ATCC | CRL-2498 |
| MEF | ATCC | CF-1;RRID:CVCL_5251 |
| 293T | ATCC | CRL-3216 |
| MDCK-1 | ATCC | CCL-34;RRID:CVCL_0422 |
| L-WRN | ATCC | CRL-3276 |
| Eps8 −/− pBABE EPS8-GFP | (Werner et al. 2013) | N/A |
| Eps8 −/− pBABE | (Werner et al. 2013) | N/A |
| Experimental models: Mice | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| Eps8 −/− | This paper | N/A |
| ZO-1 GFP | (Foote et al. 2013) | N/A |
| Experimental models: Bacterial strains | ||
| CTL2 434/Bu | ATCC | VR-902B |
| CTD UW-3/Cx | ATCC | VR-885 |
| CTL2-p2TK2 GFP | (Agaisse and Derré 2013) | N/A |
| CTL2-p2TK2 mCherry | (Agaisse and Derré 2013) | N/A |
| CTL2 tepP::GII aadA | (Dolat and Valdivia 2021) | N/A |
| CTL2 tepP::GII aadA + pTepP | (Dolat and Valdivia 2021) | N/A |
| CTL2 tepP::GII aadA + pVector | (Dolat and Valdivia 2021) | N/A |
| CTL2 tmeA::GII aadA | This paper | N/A |
| CTL2 tmeB::GII aadA | This paper | N/A |
| CTL2 tarP::bla | (Ghosh et al. 2020) | N/A |
| CTL2 M923-p2TK2 mCherry | (Sixt et al. 2017) | N/A |
| CTL2-M062G1 [tepPQ103*] + pTepP | (Carpenter et al. 2017) | N/A |
| CTL2-M062G1 [tepPQ103*] + pVector | (Carpenter et al. 2017) | N/A |
| CTL2 tepP::GII aadA p2TK2-GFP | (Dolat and Valdivia 2021) | N/A |
| CTL2 tmeA::GII aadA p2TK2-GFP | This paper | N/A |
| CTL2 tmeB::GII aadA p2TK2-GFP | This paper | N/A |
| C. muridarum CM006-GFP | (Dolat and Valdivia 2021) | N/A |
| C. muridarum CM001-mCherry | This paper | N/A |
| C. muridarum MoPn | Catherine O’Connell (UNC-Chapel Hill) | N/A |
| C. muridarum MoPn tepP :: GII bla | This paper | N/A |
| Oligonucleotides | ||
| See Table 2 | ||
| Recombinant DNA | ||
| C1-mCherry-TepP | This paper | N/A |
| EGFP-EPS8 | (Postema et al. 2018) | N/A |
| EGFP-EPS8–4xYF | This paper | N/A |
| EGFP-EPS8-ΔABD | This paper | N/A |
| EGFP-EPS8-SH3+ABD | This paper | N/A |
| p2TK2-SW2-GFP | (Agaisse and Derré 2013) | N/A |
| pDFTT3::aadA | (Lowden et al. 2015) | N/A |
| pDFTT3::bla | (Johnson and Fisher 2013) | N/A |
| pDFTT3::aadA-CTL0063–280 | This paper | N/A |
| pDFTT3::aadA-CTL0064–187 | This paper | N/A |
| pDFTT3::bla-TC0268–438 | This paper | N/A |
| p2TK2-spec-Nigg-mCherry | (Cortina et al. 2019) | N/A |
| pSW2NiggCDS2 | (Wang et al. 2014) | N/A |
| psPAX2 | Dr. Didier Trono | Addgene #12260;RRID:Addgene_12260 |
| pMD2.G | Dr. Dider Trono | Addgene #12259;RRID_Addgene_12259 |
| Software and algorithms | ||
| NIS Elements | Nikon Instruments | N/A |
| R Studio | N/A | N/A |
| ImageJ | (Schindelin et al. 2012) | N/A |
| Excel | Microsoft | N/A |
| Other | ||
Highlights.
C. trachomatis transiently disrupts epithelial tight junctions
The effector TepP interacts with and recruits EPS8 to nascent inclusions
TepP repurposes EPS8 to disrupt tight junctions and promote Chlamydia entry
TepP and EPS8 promote infection in mouse models of Chlamydia infection
Acknowledgements
We thank Dr. Kenneth Fields (University of Kentucky), Dr. Isabelle Derre (University of Virginia) and Dr. Catherine O’Connell (UNC-Chapel Hill) for Chlamydia strains and plasmids, Dr. Matthew Tyska (Vanderbilt University) for plasmids, Dr. Lisa Cameron at the Duke Light Microscopy Core Facility for assistance with microscopy, Dr. Terry Lechler for the ZO-1 GFP mouse line, Dr. Erik Soderblom and Dr. Arthur Moseley at the Duke Proteomic and Metabolomics Core Facility, Gary Kucera and Cheryl Bock at the Duke Rodent Genetic Engineering Molecular Services Team, Dr. So Young Kim at the Duke RNAi Screening Facility, Dr. Michelle Plue at Duke Shared Materials Instrumentation Facility for assistance with electron microscopy, and Dr. Jeffrey Everitt for assistance with pathological analysis. We also thank Dr. David Tobin and Dr. Carolyn Coyne for critical comments on the manuscript and members of the Valdivia lab for feedback on this project. This work was funded by NIH grants AI134891 (R.H.V.) and AI138372 (L.D.)
Footnotes
Declaration of interests
R.H. Valdivia is a founder of Bloom Sciences (San Diego, CA), which is a microbiome therapeutics company. The current manuscript is unrelated to the work performed with Bloom Sciences.
Inclusion and diversity
This work was performed by a team of individuals with diverse background and life experiences. We support inclusive and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelrahman YM, and Belland RJ (2005). The chlamydial developmental cycle. FEMS Microbiol. Rev 29, 949–959. [DOI] [PubMed] [Google Scholar]
- Agaisse H, and Derré I. (2013). A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8, e57090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zeer MA, Al-Younes HM, Kerr M, Abu-Lubad M, Gonzalez E, Brinkmann V, and Meyer TF (2014). Chlamydia trachomatis remodels stable microtubules to coordinate Golgi stack recruitment to the chlamydial inclusion surface. Mol. Microbiol 94, 1285–1297. [DOI] [PubMed] [Google Scholar]
- Backert S, and Selbach M. (2005). Tyrosine-phosphorylated bacterial effector proteins: the enemies within. Trends Microbiol. 13, 476–484. [DOI] [PubMed] [Google Scholar]
- Birkelund S, Johnsen H, and Christiansen G. (1994). Chlamydia trachomatis serovar L2 induces protein tyrosine phosphorylation during uptake by HeLa cells. Infect. Immun 62, 4900–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkworth AJ, Malcolm DS, Pedrosa AT, Roguska K, Shahbazian S, Graham JE, Hayward RD, and Carabeo RA (2011). Chlamydia trachomatis Slc1 is a type III secretion chaperone that enhances the translocation of its invasion effector substrate TARP. Mol. Microbiol 82, 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner LR, Lewis ME, Greene SJ, Foster TP, and Quayle AJ (2013). Chlamydia trachomatis infection results in a modest pro-inflammatory cytokine response and a decrease in T cell chemokine secretion in human polarized endocervical epithelial cells. Cytokine 63, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter V, Chen Y-S, Dolat L, and Valdivia RH (2017). The Effector TepP Mediates Recruitment and Activation of Phosphoinositide 3-Kinase on Early Chlamydia trachomatis Vacuoles. MSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-S, Bastidas RJ, Saka HA, Carpenter VK, Richards KL, Plano GV, and Valdivia RH (2014). The Chlamydia trachomatis type III secretion chaperone Slc1 engages multiple early effectors, including TepP, a tyrosine-phosphorylated protein required for the recruitment of CrkI-II to nascent inclusions and innate immune signaling. PLoS Pathog. 10, e1003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, and Hackstadt T. (2004). A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. USA 101, 10166–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J, Starnbach MN, and Howard JC (2009). Modeling infectious disease in mice: coadaptation and the role of host-specific IFNgamma responses. PLoS Pathog. 5, e1000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet J-P, and Haeussler M. (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortina ME, Ende RJ, Bishop RC, Bayne C, and Derré I. (2019). Chlamydia trachomatis and Chlamydia muridarum spectinomycin resistant vectors and a transcriptional fluorescent reporter to monitor conversion from replicative to infectious bacteria. PLoS One 14, e0217753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, and Bergelson JM (2006). Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124, 119–131. [DOI] [PubMed] [Google Scholar]
- Cunningham DL, Creese AJ, Auciello G, Sweet SMM, Tatar T, Rappoport JZ, Grant MM, and Heath JK (2013). Novel binding partners and differentially regulated phosphorylation sites clarify Eps8 as a multi-functional adaptor. PLoS One 8, e61513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, and Subtil A. (2008). SNARE protein mimicry by an intracellular bacterium. PLoS Pathog. 4, e1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanza A, Carlier M-F, Stradal TEB, Didry D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP, and Scita G. (2004). Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat. Cell Biol 6, 1180–1188. [DOI] [PubMed] [Google Scholar]
- Dolat L, and Valdivia RH (2021). An endometrial organoid model of interactions between Chlamydia and epithelial and immune cells. J. Cell Sci 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C, Mirrashidi K, and Engel J. (2016). Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol 14, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Ceesay A, Kim JH, Kalman D, and Engel JN (2008). RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 4, e1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faris R, McCullough A, Andersen SE, Moninger TO, and Weber MM (2020). The Chlamydia trachomatis secreted effector TmeA hijacks the N-WASP-ARP2/3 actin remodeling axis to facilitate cellular invasion. PLoS Pathog. 16, e1008878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawaz FS, van Ooij C, Homola E, Mutka SC, and Engel JN (1997). Infection with Chlamydia trachomatis alters the tyrosine phosphorylation and/or localization of several host cell proteins including cortactin. Infect. Immun 65, 5301–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote HP, Sumigray KD, and Lechler T. (2013). FRAP analysis reveals stabilization of adhesion structures in the epidermis compared to cultured keratinocytes. PLoS One 8, e71491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson K, Van Itallie CM, Aponte A, Gucek M, Tietgens AJ, and Anderson JM (2015). Proteomic analysis of proteins surrounding occludin and claudin-4 reveals their proximity to signaling and trafficking networks. PLoS One 10, e0117074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Nelson WJ, and Chavez N. (2018). Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb. Perspect. Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Ruelke EA, Ferrell JC, Bodero MD, Fields KA, and Jewett TJ (2020). Fluorescence-Reported Allelic Exchange Mutagenesis-Mediated Gene Deletion Indicates a Requirement for Chlamydia trachomatis Tarp during In Vivo Infectivity and Reveals a Specific Role for the C Terminus during Cellular Invasion. Infect. Immun 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giampietro C. (2016). VE-cadherin complex plasticity: EPS8 and YAP play relay at adherens junctions. Tissue Barriers 4, e1232024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, and Finlay BB (2001). Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol 3, 856–859. [DOI] [PubMed] [Google Scholar]
- Guttman JA, and Finlay BB (2009). Tight junctions as targets of infectious agents. Biochim. Biophys. Acta 1788, 832–841. [DOI] [PubMed] [Google Scholar]
- Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, and Ness RB (2010). Risk of sequelae after Chlamydia trachomatis genital infection in women. J. Infect. Dis 201 Suppl 2, S134–55. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Hayashi M, Boutin E, Cunha GR, Bernfield M, and Trelstad RL (1988). Hormonal modification of epithelial differentiation and expression of cell surface heparan sulfate proteoglycan in the mouse vaginal epithelium. An immunohistochemical and electron microscopic study. Lab. Invest 58, 68–76. [PubMed] [Google Scholar]
- Hertzog M, Milanesi F, Hazelwood L, Disanza A, Liu H, Perlade E, Malabarba MG, Pasqualato S, Maiolica A, Confalonieri S, et al. (2010). Molecular basis for the dual function of Eps8 on actin dynamics: bundling and capping. PLoS Biol. 8, e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, and Hatakeyama M. (2002). SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295, 683–686. [DOI] [PubMed] [Google Scholar]
- Hybiske K, and Stephens RS (2007). Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 104, 11430–11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inki P. (1997). Expression of syndecan-1 in female reproductive tract tissues and cultured keratinocytes. Mol. Hum. Reprod 3, 299–305. [DOI] [PubMed] [Google Scholar]
- Jewett TJ, Fischer ER, Mead DJ, and Hackstadt T. (2006). Chlamydial TARP is a bacterial nucleator of actin. Proc. Natl. Acad. Sci. USA 103, 15599–15604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keb G, Hayman R, and Fields KA (2018). Floxed-Cassette Allelic Exchange Mutagenesis Enables Markerless Gene Deletion in Chlamydia trachomatis and Can Reverse Cassette-Induced Polar Effects. J. Bacteriol 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keb G, Ferrell J, Scanlon KR, Jewett TJ, and Fields KA (2021). Chlamydia trachomatis TmeA Directly Activates N-WASP To Promote Actin Polymerization and Functions Synergistically with TarP during Invasion. MBio 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M, Zielecki J, Thieck O, Mollenkopf H-J, Fotopoulou C, and Meyer TF (2012). Chlamydia trachomatis disturbs epithelial tissue homeostasis in fallopian tubes via paracrine Wnt signaling. Am. J. Pathol 180, 186–198. [DOI] [PubMed] [Google Scholar]
- Kierbel A, Gassama-Diagne A, Rocha C, Radoshevich L, Olson J, Mostov K, and Engel J. (2007). Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J. Cell Biol 177, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, and Carabeo RA (2008). Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 4, e1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti L, Rybin V, Malabarba MG, Christoforidis S, Scita G, Zerial M, and Di Fiore PP (2000). The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature 408, 374–377. [DOI] [PubMed] [Google Scholar]
- Lowden NM, Yeruva L, Johnson CM, Bowlin AK, and Fisher DJ (2015). Use of aminoglycoside 3’ adenyltransferase as a selection marker for Chlamydia trachomatis intronmutagenesis and in vivo intron stability. BMC Res. Notes 8, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maa M-C, Lai J-R, Lin R-W, and Leu T-H (1999). Enhancement of tyrosyl phosphorylation and protein expression of eps8 by v-Src. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1450, 341–351. [DOI] [PubMed] [Google Scholar]
- Mital J, Lutter EI, Barger AC, Dooley CA, and Hackstadt T. (2015). Chlamydia trachomatis inclusion membrane protein CT850 interacts with the dynein light chain DYNLT1 (Tctex1). Biochem. Biophys. Res. Commun 462, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modzelewski AJ, Chen S, Willis BJ, Lloyd KCK, Wood JA, and He L. (2018). Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc 13, 1253–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore ER, Fischer ER, Mead DJ, and Hackstadt T. (2008). The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin-containing exocytic vacuoles. Traffic 9, 2130–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan UM, Prantner D, Sikes JD, Andrews CW, Goodwin AM, Nagarajan S, and Darville T. (2008). Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect. Immun 76, 4642–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offenhäuser N, Borgonovo A, Disanza A, Romano P, Ponzanelli I, Iannolo G, Di Fiore PP, and Scita G. (2004). The eps8 family of proteins links growth factor stimulation to actin reorganization generating functional redundancy in the Ras/Rac pathway. Mol. Biol. Cell 15, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis T, Bègue H, Basmaciyan L, Dalle F, and Bon F. (2021). Tight junctions as a key for pathogens invasion in intestinal epithelial cells. Int. J. Mol. Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AL, Chen X, Wood ST, Stuart ES, Arcaro KF, Molina DP, Petrovic S, Furdui CM, and Tsang AW (2014). Activation of epidermal growth factor receptor is required for Chlamydia trachomatis development. BMC Microbiol. 14, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postema MM, Grega-Larson NE, Neininger AC, and Tyska MJ (2018). IRTKS (BAIAP2L1) Elongates Epithelial Microvilli Using EPS8-Dependent and Independent Mechanisms. Curr. Biol 28, 2876–2888.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston TB, O’Connell CM, Girardi J, Sullivan JE, Nagarajan UM, Marinov A, Scurlock AM, and Darville T. (2018). T Cell-Independent Gamma Interferon and B Cells Cooperate To Prevent Mortality Associated with Disseminated Chlamydia muridarum Genital Tract Infection. Infect. Immun 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozialeck WC, Fay MJ, Lamar PC, Pearson CA, Sigar I, and Ramsey KH (2002). Chlamydia trachomatis disrupts N-cadherin-dependent cell-cell junctions and sequesters betacatenin in human cervical epithelial cells. Infect. Immun 70, 2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J. (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmarin DM, Carette JE, Olive AJ, Starnbach MN, Brummelkamp TR, and Ploegh HL (2012). Attachment of Chlamydia trachomatis L2 to host cells requires sulfation. Proc. Natl. Acad. Sci. USA 109, 10059–10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Thompson JW, Chen Y-S, Kumar Y, Dubois LG, Moseley MA, and Valdivia RH (2011). Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol. Microbiol 82, 1185–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore-Carlson MA, Shaw EI, Dooley CA, Fischer ER, and Hackstadt T. (1999). Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol. Microbiol 33, 753–765. [DOI] [PubMed] [Google Scholar]
- Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegård M, Betsholtz C, and Di Fiore PP (1999). EPS8 and E3B1 transduce signals from Ras to Rac. Nature 401, 290–293. [DOI] [PubMed] [Google Scholar]
- Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, and Ramsey KH (2005). Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex. Transm. Dis 32, 49–56. [DOI] [PubMed] [Google Scholar]
- Shahoumi LA, Khodadadi H, Bensreti H, Baban B, and Yeudall WA (2020). EPS8 phosphorylation by Src modulates its oncogenic functions. Br. J. Cancer 123, 1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, Coers J, and Valdivia RH (2017). The Chlamydia trachomatis Inclusion Membrane Protein CpoS Counteracts STING-Mediated Cellular Surveillance and Suicide Programs. Cell Host Microbe 21, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg I, Schmutz C, Lu Y-Y, Fromm K, Siewert LK, Bögli A, Strack K, Harms A, and Dehio C. (2020). A Bartonella Effector Acts as Signaling Hub for Intrinsic STAT3 Activation to Trigger Anti-inflammatory Responses. Cell Host Microbe 27, 476–485.e7. [DOI] [PubMed] [Google Scholar]
- Stallmann S, and Hegemann JH (2016). The Chlamydia trachomatis Ctad1 invasin exploits the human integrin β1 receptor for host cell entry. Cell Microbiol. 18, 761–775. [DOI] [PubMed] [Google Scholar]
- Subbarayal P, Karunakaran K, Winkler A-C, Rother M, Gonzalez E, Meyer TF, and Rudel T. (2015). EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 11, e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites T, Nogueira AT, Campeotto I, Silva AP, Grieshaber SS, and Carabeo RA (2014). The Chlamydia effector TarP mimics the mammalian leucine-aspartic acid motif of paxillin to subvert the focal adhesion kinase during invasion. J. Biol. Chem 289, 30426–30442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaggi F, Disanza A, Milanesi F, Di Fiore PP, Menna E, Matteoli M, Gov NS, Scita G, and Ciliberto A. (2011). The Eps8/IRSp53/VASP network differentially controls actin capping and bundling in filopodia formation. PLoS Comput. Biol 7, e1002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cutcliffe LT, Skilton RJ, Ramsey KH, Thomson NR, and Clarke IN (2014). The genetic basis of plasmid tropism between Chlamydia trachomatis and Chlamydia muridarum. Pathog Dis 72, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Disanza A, Reifenberger N, Habeck G, Becker J, Calabrese M, Urlaub H, Lorenz H, Schulman B, Scita G, et al. (2013). SCFFbxw5 mediates transient degradation of actin remodeller Eps8 to allow proper mitotic progression. Nat. Cell Biol 15, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2018). Report on global sexually transmitted infection surveillanc (World Health Organization; ). [Google Scholar]
- Zampini V, Rüttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, Xiong H, Hackney CM, Holley MC, Offenhauser N, et al. (2011). Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol. 9, e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, and Stephens RS (1992). Mechanism of C. trachomatis attachment to eukaryotic host cells. Cell 69, 861–869. [DOI] [PubMed] [Google Scholar]
- Zihni C, Mills C, Matter K, and Balda MS (2016). Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol 17, 564–580. [DOI] [PubMed] [Google Scholar]
- Zwaenepoel I, Naba A, Da Cunha MML, Del Maestro L, Formstecher E, Louvard D, and Arpin M. (2012). Ezrin regulates microvillus morphogenesis by promoting distinct activities of Eps8 proteins. Mol. Biol. Cell 23, 1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 2. List of differentially Tyr phosphorylated host proteins in mock-infected and infected epithelial cells. Comparison of Tyr phosphorylation of proteins in uninfected A2EN cells or A2EN cells infected with TepP+ Chlamydia (pTepP) and TepP− Chlamydia (pVector). Tab1, pTepP vs pVector; Tab2, pTepP vs Mock; Tab3, pVector vs Mock.
Movie 1, Related to Figure 1. Time-lapse brightfield microscopy of polarized A2EN cells infected with CTL2. Images were acquired every 3 min for ~ 3 h. Time display, hour:min.
Movie 2, Related to Figure 2. Time-lapse spinning disk confocal microscopy of a HeLa cells expressing EGFP-EPS8 and infected with CTL2-mCherry. Images were acquired every 15 s for ~ 26 min. Time display, min:sec.
Movie 3, Related to Figure 5. Time-lapse fluorescence microscopy of endometrial organoids co-injected with CTL2 (left) or tepP mutant (right) bacteria with Texas-Red Dextran. Images were acquired every 10 min for ~ 14 h. Time display, hour:min.
Movie 4, Related to Figure 5. Time-lapse fluorescence microscopy of Eps8+/+ (left) and Eps8−/− (right) endometrial organoids were co-injected with CTL2 and Texas-Red Dextran. Images were acquired every 10 min for ~ 14 h. Time display, hour:min.
Movie 5, Related to Figure 6. Time-lapse spinning disk confocal microscopy of polarized A2EN cells labeled with SiR-actin and infected with CTL2-GFP. Images were acquired every 30s for ~ 26 min. Time display, min:sec.
Data Availability Statement
Phospho-proteomic datasets were deposited to MassIVE and will be available on the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request