

Abstract
Background and Purpose:
KATP channels are negative regulators of lymphatic vessel excitability and contractility and are proposed to be targets for immune cell products that inhibit lymph transport. Previous studies in rat and guinea pig mesenteric lymphatics found that nitric oxide (NO)-mediated inhibition of lymphatic contraction was prevented or reversed by the KATP channel inhibitor, glibenclamide. We revisited this hypothesis using mouse lymphatic vessels and KATP channel knock out mice.
Experimental Approach:
Mouse popliteal lymphatics were isolated and contractility was assessed using pressure myography. K+ channel expression was determined by PCR analysis of FACS-purified lymphatic smooth muscle cells.
Key Results:
The NO-producing agonist, ACh, and the NO donor, NONOate, both produced dose-dependent inhibition of spontaneous lymphatic contractions that were blocked by the soluble guanylate cyclase inhibitor, ODQ, or the PKG inhibitor, Rp-8-Br-PET-cGMPS. Surprisingly, the inhibitory effects of both were preserved in Kir6.1−/− vessels, suggesting that KATP channels did not mediate NO-induced responses. We hypothesized a role for BK channels, given their prominence in arterial smooth muscle. Indeed, BK channels were expressed in mouse lymphatic smooth muscle and NS11021 (a BK channel activator) caused dilation and reduced contraction frequency, whereas iberiotoxin and Penitrem A (BK channel inhibitors) produced right-shifts in NONOate concentration-response curves.
Conclusion and Implication:
NO inhibition of mouse lymphatic contractions primarily involves activation of BK channels rather than KATP channels. Thus, BK channels are a potential target for therapeutic reversal of lymph pump inhibition by NO generated by immune cell activation of iNOS in chronic lymphedema.
Keywords: lymphatic muscle, Kir6.1, SUR2, NONOate, acetylcholine, iberiotoxin, apamin
Introduction
Lymphedema develops when the production of interstitial fluid exceeds the capacity of the lymphatic system to absorb that fluid and return it to the central veins. In developed countries, lymphedema is most often seen developing over months or years after removal of lymphatic nodes and/or lymphatic collecting vessels during cancer surgery. Lymphedema typically manifests in dependent extremities, where gravitational loads exacerbate the pressure gradients against which lymph must be transported. Lymph transport normally relies on robust spontaneous contractions of collecting lymphatic vessels that move lymph towards the heart, and one-way valves to prevent reflux (Scallan et al., 2016). Once lymphedema begins to develop, lymphatic pressures become elevated, lymphatic vessels dilate, and lymphatic valves exhibit increasing degrees of incompetence (Olszewski, 2002, Olszewski, 1977); this combination of factors leads to impairment of lymphatic pump function (Scallan et al., 2016).
Stagnant lymph results in reduced immune cell trafficking and/or infiltration (Rockson et al., 2018, Gardenier et al., 2017, Torrisi et al., 2016). Subsequent cytokine release promotes nitric oxide (NO) production through the activation of iNOS, which further impairs lymphatic smooth muscle (LSM) contractility and exacerbates lymphatic pump dysfunction (Liao et al., 2011, Jiang et al., 2019, Norden and Kume, 2020). The overproduction of NO in chronic ileitis, Crohn’s disease and other forms of inflammatory bowel disease also leads to lymphatic pump dysfunction and may contribute to the initiation and/or progression of those diseases by altering immune cell trafficking through lymphatic networks (von der Weid et al., 2011, Von Der Weid and Rehal, 2010).
LMCs express the same KATP channel isoforms—Kir6.1 and SUR2B—as other smooth muscle cells (Mathias and von der Weid, 2013, Davis et al., 2020). NO activates sGC, promoting cGMP production and PKG activation, which may result in phosphorylation and increased activity of Kir6.1 channels (Mathias and von der Weid, 2013, Seino and Miki, 2003, Foster and Coetzee, 2016). KATP channel activation hyperpolarizes the LMC membrane and hence slows or silences the ionic pacemaker that drives spontaneous lymphatic contractions. This has led to the proposal that inhibitory actions of NO on lymphatic contractile strength and pacemaking rate (von der Weid et al., 2001) may be mediated by KATP channel activation in lymphatic muscle cells (LMCs). Consistent with this suggestion, high doses of the KATP channel antagonist glibenclamide attenuates the inhibitory action of NO on the spontaneous contractions of guinea pig mesenteric lymphatics (Mathias and von der Weid, 2013). Likewise, glibenclamide reverses rat mesentery lymphatic contractile dysfunction in the TNBS model of chronic ileitis and in an intestinal inflammation model (Chen et al., 2017b).
Knowing the specific molecular target(s) by which lymphatic pumping is inhibited in inflammatory states, including inflammatory bowel disease and secondary lymphedema (Norden and Kume, 2020, Jiang et al., 2019), is critical for eventual therapeutic correction of lymphatic dysfunction in these diseases. Although the above findings are consistent with a role for KATP channel activation in mediating the inhibitory effects of NO on lymphatic smooth muscle, they hinge on the specificity of glibenclamide, which can have off-target effects on other ion channels, including L-type Ca2+ channels (Lee and Lee, 2005) and other K+ channels (Schaffer et al., 1999). The development of lymphatic contractile function assays in mice (Liao et al., 2011, Scallan and Davis, 2013) and the availability of Kir6.1 null mice (Miki et al., 2002) affords the opportunity to more definitively test the hypothesis that NO acts though KATP channel activation to inhibit lymphatic contractile function.
Methods
Mice
Mice were used as the test species to take advantage of Kir6.1−/−, SUR2-STOP and reporter animals; we have documented previously the reliability and reproducibility of contraction parameter tests on mouse popliteal lymphatic vessels studied ex vivo (Scallan et al., 2013; Castorena et al., 2018; Zawieja et al., 2018). Wild type (WT) male or female mice (20-28 g) on the C57BL/6 background (RRID:IMSR_JAX:000664) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Kir6.1−/− mice were a gift from Susumu Seino (Kobe University). SUR2-STOP mice were generated by the introduction of a premature stop codon (p.Y1148Stop) as described previously (Ran et al., 2013), which prevents transcription of the SUR2 subunit. For FACS analysis of LMCs, SMMHC-CreERT2 mice (RRID:IMSR_JAX:019079) or Prox1-CreERT2 mice (a gift from Taija Makinen, Uppsala University) were crossed to ROSA26mTmG reporter mice (RRID:IMSR_JAX:007676). Genotypes were determined by PCR with Taq DNA Polymerase Premix (Intact Genomics Catalog #3249). Mice were studied at 6-10 weeks of age from either sex, depending upon availability; in the case of SMMHC-CreERT2 mice only male mice were used as the transgene is located on the Y-chromosome. Mice were anesthetized by i.p. injection using a mixture of ketamine/xylazine (100/10 mg kg−1 body weight) and euthanized by intracardiac injection of KCl. All animal protocols were reviewed and approved by the University of Missouri Animal Care and Use Committee and conformed to the National Institutes of Health’s Guide for the care and use of laboratory animals (8th edition, 2011). Mice were housed and bred under pathogen-free conditions in a controlled environment (22 ± 2 °C, 12/12-hr light/dark cycle) of the animal facility of University of Missouri School of Medicine.
Vessel isolation, Pressure Myograph and Data Acquisition
For the isolation of mouse popliteal lymphatic vessels, a proximal-to-distal incision was made in the skin of the lateral thigh to expose the superficial saphenous vein. Afferent popliteal lymphatic vessels on either side of this vein were removed and transferred to a Sylgard dissection dish with Krebs buffer containing albumin. After pinning and cleaning, a vessel was cannulated using two glass micropipettes (40-50 μm, o.d.), pressurized to 3 cmH2O and further cleaned of any remaining tissue in order to track diameter accurately. The vessel was cut to a length that contained only one valve. Polyethylene tubing (PE-190) attached to the back of each micropipette holder was connected to a 2-channel microfluidic device (Elveflow OB1 MK3, Paris) for computer control of pressure on the stage of an inverted microscope. Input and output pressures were transiently set to 10 cmH2O immediately after set up and the vessel was stretched axially to remove slack, which minimized longitudinal bowing and associated diameter-tracking artifacts during subsequent protocols. With input and output pressure held at 3 cmH2O spontaneous contractions typically began within 15-30 min of warm-up; each vessel was allowed to stabilize at 37 °C for 30-60 min before beginning an experimental protocol. A suffusion line connected to a peristaltic pump exchanged the chamber contents with Krebs buffer at a rate of 0.5 ml/min. Custom-written LabVIEW programs (National Instruments; Austin, TX) measured the inner diameter of the vessel from video images obtained at 30 fps using a Basler A641fm firewire camera (Davis et al., 2006).
Assessment of basal contractile function and concentration-response of drugs
Spontaneous contractions were recorded with equal input and output pressures (3 cmH2O) to prevent a pressure gradient for forward flow through the vessel during the experiment. Concentration-response curves to acetylcholine (ACh), which stimulates NO production (Scallan and Davis, 2013), were performed over the range of 1 × 10−9 M to 3 × 10−7 M. Concentration-response curves to the direct NO donor, sodium-NONOate, were performed over the range of 1 × 10−8 M to 3 × 10−6 M. To evaluate the effects of pharmacological blockade of KATP channels, glibenclamide (GLIB) concentration-response curves were performed in WT vessels over the range of 1 × 10−8 M to 1 × 10−5 M. Concentration-response curves to the BK channel activator, NS11021, were tested over the range of 1 × 10−7 M to 1 × 10−5 M. In each case, a small, predetermined volume of the agonist or antagonist was added to the 3 mL bath, followed by thorough mixing while the bath was stopped. Each concentration was given in 2 min intervals in a cumulative manner, and the total concentration-response curve was completed within 20 min to prevent changes in bath osmolality from evaporation. At the end of every experiment, all vessels were perfused with Ca2+-free Krebs buffer containing 3 mM EGTA for 30 min, and passive diameters were recorded at the pressure used in the protocols (typically 3 cmH2O).
Contractile Function parameters
After an experiment, custom-written analysis programs (LabVIEW) were used to detect peak end-diastolic diameter (EDD), end-systolic diameter (ESD) and contraction frequency (FREQ) on a contraction-by-contraction basis. These data were used to calculate several commonly reported parameters that characterize lymphatic vessel contractile function. Each of the contractile parameters represents the average of the respective values from all the recorded contractions at a particular concentration during a 2 min period. From concentration-response protocols, the following parameters were calculated and graphed:
| (1) |
| (2) |
| (3) |
| (4) |
where EDDavg and FREQavg represent the average EDD and frequency during the baseline period before the addition of a drug to the bath. DMAX represents the maximum passive diameter obtained under Ca2+ free Krebs buffer.
FACS analysis
Inguinal-axillary lymphatic vessels from tamoxifen-treated SMMHCCreERT2;Rosa26mTmG mice were dissected as described previously (Zawieja et al., 2018). Cleaned vessel segments were transferred to a 1-ml tube of low-Ca2+ PSS containing (in mM): 137 NaCl, 5.0 KCl, 0.1 CaCl2, 1.0 MgCl2, 10 HEPES, 10 Glucose, and 1 mg ml−1 BSA at 37 °C for 10 min. The solution was decanted and replaced with a similar solution containing 1 mg ml−1 papain (Sigma, St. Louis, MO) and 1 mg ml−1 dithioerythritol. The vessels were incubated for 30 min at 37 °C with occasional agitation, then transferred to a new tube containing low-Ca2+ PSS containing 25 mg ml−1 collagenase H (FALGPA U ml−1, Sigma), 0.7 mg ml−1 collagenase F (Sigma), 20 mg ml−1 trypsin inhibitor (Sigma), 1mg ml−1 elastase (Worthington), and incubated for 5-7 min at 37 °C. The resultant dispersed cells from digested vessels were sedimented by centrifugation (300 g, 4 min), resuspended in 0.6 ml PSS containing 1 mM Ca solution, and filtered through a nylon filter with 35-μm mesh size to obtain single cell suspension. Smooth muscle cells expressing GFP were sorted by fluorescence-activated cell sorting (FACS) with a Beckman-Coulter MoFlo XDP instrument using an excitation laser (488 nm) and emission filter (530/40 nm), with 70 μm nozzle at a sheath pressure of 45 psi and sort rate of 100 events per second. Sorting was performed at the Cell and Immunobiology Core facility at the University of Missouri
End-Point RT-PCR
End-point RT-PCR was used to detect message for KATP, SK and BK channels as well as canonical endothelial or smooth muscle cell markers in FACS-purified lymphatic endothelial or smooth muscle cells from mouse. Total RNA was extracted from sorted cells using the Arcturus PicoPure RNA isolation kit (Thermo Fisher Scientific, Waltham, MA) with on-column DNase I treatment (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA then was synthesized using the High Capacity cDNA Reverse Transcription Kit Applied Biosystems™ (Thermo Fisher Scientific, Waltham, MA). PCR was performed in a reaction mixture containing first-strand cDNA as the template, 2 mM MgCl2, 0.25 μM primers, 0.2 mM deoxynucleotide triphosphates; and GoTaq® Flexi DNA polymerase (Promega, Madison, WI). The PCR program comprised an initial denaturation step at 95°C for four minutes; followed by 35 repetitions of the following cycle: denaturation (94°C, 30s), annealing (57°C, 30s) and extension (72°C, 30s). This was followed by a final elongation step for 5 min at 72°C. The Amplified PCR products were loaded on a 1.5 % agarose gel by electrophoresis, stained with SYBR-Safe (Thermo Fisher Scientific, Waltham, MA), and visualized by UV transillumination. All primers were designed to amplify intron-spanning DNA regions; primer sequences are listed in supplementary Table. 1.
Solutions and chemicals
Krebs buffer contained (in mM) 146.9 NaCl, 4.7 KCl, 2 CaCl2·2H2O, 1.2 MgSO4, 1.2 NaH2PO4·H2O, 3 NaHCO3, 1.5 NaHEPES, and 5 D-glucose (pH = 7.4). Krebs-BSA buffer was prepared with the addition of 0.5 % bovine serum albumin. During cannulation, Krebs-BSA buffer was present both luminally and abluminally, but during the experiment, the bath solution was constantly exchanged with Krebs solution without albumin. All chemicals and drugs were purchased from Sigma-Aldrich (St. Louis, MO), with exception of BSA (United States Biochemicals; Cleveland, OH), MgSO4, Na-HEPES (ThermoFisher Scientific; Pittsburgh, PA), iberiotoxin, ODQ, NS11021 (Tocris Bioscience, Bristol, UK) and apamin (Alomone Labs, Jerusalem, Israel). Sodium NONOate, acetylcholine, Rp-8-Br-PET-cGMPS, iberiotoxin and apamin were dissolved in distilled water. Glibenclamide, ODQ and NS11021 were dissolved in DMSO and the total amount of DMSO was set below 0.4 %, which was determined in separate protocols to be the threshold vasoactive dose of DMSO. Penitrem A was dissolved in methanol, which by itself had no effect on contraction at the concentrations used.
Statistical Procedures
The data collection and statistical analyses comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). A priori power calculations were performed for the initial protocols (Figs. 1-6), but additional animals were subsequently added to several protocols to balance the numbers of male and female mice. The number N refers to the number of different animals used in each protocol; the number n refers to the total number of vessels included per group. Values are means ± SEM. Statistical analysis was undertaken only for studies where each group size ≥ 5. Randomization was not a feature of study design. Blinding of the operator was not feasible in most cases because vessel responses observed by the operator to manage the experiment permitted inferences about the treatment. However, data analyses for most protocols were performed semi-blinded by an independent analyst.
Figure 1.
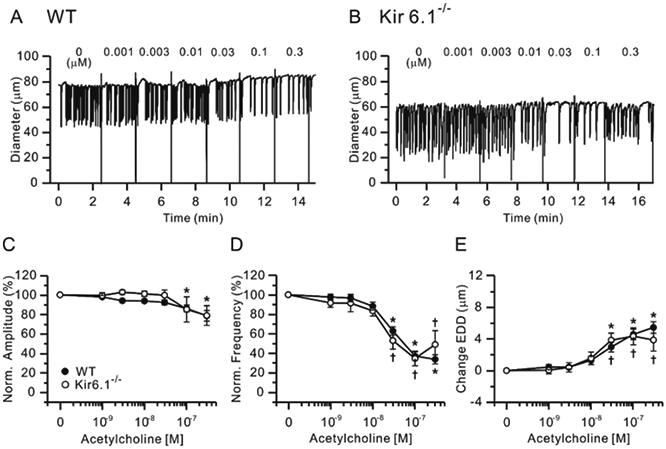
Effects of ACh. Representative traces showing concentration-dependent effects of the NO-producing agonist ACh on the spontaneous contractions of WT (A) and Kir6.1−/− (B) mouse popliteal lymphatics held at a constant pressure of 3 cmH2O. Normalized amplitude (C), normalized frequency (D) and change in EDD (E) plotted as a function of NONOate concentration, from 10 nM to 3 μM. Filled circles indicate WT vessels (n=25) and open circles indicate Kir6.1−/− vessels (n=9). All data are means ± SEM. There were no significant differences between WT and Kir6.1−/− data points at any NONOate concentration. * Significant difference in WT data from control (absence of ACh). † Significant difference in Kir6.1−/− data from control.
Figure 6.
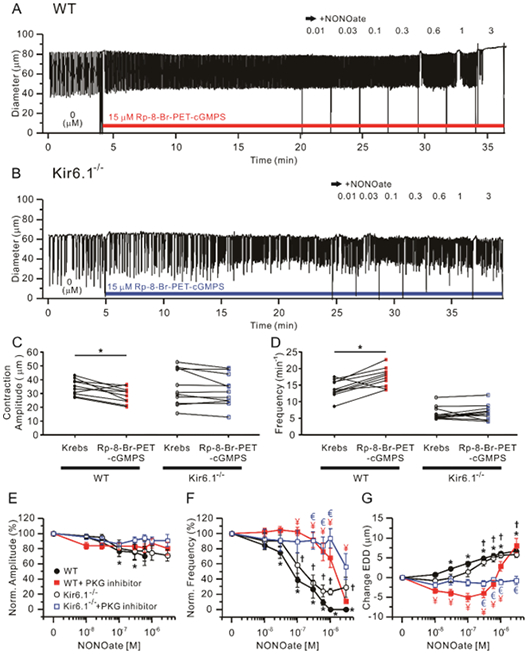
The PKG inhibitor, 15 μM Rp-8-Br-PET-cGMPS blocked contractions and dilations of WT and Kir6.1−/− vessels to increasing concentrations of NONOate. Representative traces of NONOate concentration-dependent responses after pre-incubation with 15 μM Rp-8-Br-PET-cGMPS in WT (A) and Kir6.1−/− (B) popliteal lymphatics. Comparisons of contraction amplitude (C) and frequency (D) before (“Krebs”) and after treatment of Rp-8-Br-PET-cGMPS, * Significant using paired Student’s t-test). Lymphatic contraction parameters were normalized to their respective values before Rp-8-Br-PET-cGMPS treatment (E-G). The effect of NONOate on normalized amplitude (E), normalized frequency (F) and change in EDD (G) in the presence of 15 μM Rp-8-Br-PET-cGMPS. Filled red rectangles indicate NONOate responses of WT vessels pretreated with Rp-8-Br-PET-cGMPS (n=10) and open blue rectangles represent NONOate responses of Kir6.1−/− pretreated with Rp-8-Br-PET-cGMPS (n=11). For comparison, the responses to NONOate only in each group of WT (n=24) and Kir6.1−/− (n=9) are represented by filled black circles and open black rectangles respectively. All data are means ± SEM. * Significant difference in WT data from control (absence of NONOate). † Significant difference in Kir6.1−/− data from control (absence of NONOate). ¥ (red) Significant difference between filled black circle and filled red rectangle data points. € (blue) Significant difference between open black circle and open blue rectangle data points (P < 0.05).
Because baseline values of spontaneous contraction frequency are highly variable among lymphatic vessels (Scallan et al., 2016, Zawieja et al., 2018), we used normalized values of amplitude, frequency and change in EDD to compare IC50 values; this is a common practice for analyses of concentration-response relationships. In these cases, Kruskal-Wallis non-parametric ANOVAs, followed by Bonferroni’s post hoc tests, were used to make multiple comparisons between data sets. Post-hoc tests were conducted only if F achieved P < 0.05 and there was no significant variance homogeneity. Dunnett’s post-hoc tests were used to assess differences from the control data point (i.e., in the absence of drug). Differences between absolute amplitude or frequency in the presence or absence of drug were assessed using paired Student’s t-tests. To calculate IC50 values the concentration-response curves were fit using the Hill equation (IGOR, Wavemetrics, Portland OR). All other statistical analyses were performed using Prism5 (GraphPad Software Inc., CA, USA), with significance for all tests set at P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org. the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
Results
NO is not a major mechanism of KATP channel activation in mouse lymphatic muscle
We previously showed that mouse lymphatic smooth muscle cells expressed mRNA for Kir6.1 and SUR2B KATP channel subunits and that popliteal lymphatic vessels from Kir6.1−/− and SUR2[STOP] mice (both of which lack functional KATP channels) were ~100-fold less sensitive than WT vessels to the KATP channel activator, pinacidil (Davis et al., 2020). Those observations support the conclusion that mouse lymphatic vessels normally express functional KATP channels. However, the intrinsic contractile function of Kir6.1−/− and SUR2[STOP] lymphatic vessels was not significantly different from that of WT vessels over a wide range of pressures, suggesting that KATP channels do not significantly influence contractile behavior under normal conditions, although it is conceivable that Kir6.1−/− mice might chronically compensate for the lack of functional Kir6.1 channels with up- or down-regulation of other conductances (Davis et al 2020). Nevertheless, KATP channels are likely to play roles in other aspects of lymphatic contractile function. Hence, in the present study, we sought to determine whether the inhibitory effects of NO on lymphatic contractile strength, pacemaking rate and tone were mediated by KATP channels, as suggested by previous studies (Mathias and von der Weid, 2013, von der Weid, 1998, Davis et al., 2020).
First, we tested the involvement of KATP channels in the response of popliteal lymphatics to the endothelium-dependent vasodilator, acetylcholine (ACh). ACh inhibits lymphatic pumping via production of NO from lymphatic endothelium (Scallan and Davis, 2013), similar to how it induces relaxation of arteries and arterioles (Chataigneau et al., 1999). Increasing concentrations of ACh (from 1 nM to 0.3 μM) were applied to spontaneously contracting popliteal lymphatics isolated from WT and Kir6.1−/− mice. Representative traces of WT and Kir6.1−/− lymphatic vessel responses to ACh are shown in Fig. 1A-B, respectively. Progressively higher concentrations of ACh reduced the frequency of contraction and dilated vessels from WT mice (Fig. 1D, E), with modest but significant reductions in contraction amplitude at 0.1 and 0.3 μM (Fig. 1C). Surprisingly, and contrary to predictions from the literature (von der Weid, 1998, Mathias and von der Weid, 2013), similar effects of ACh were observed in popliteal lymphatics from Kir6.1−/− mice. Indeed, the IC50 values for frequency and EDD to ACh were not significantly different between in Kir6.1−/− and WT vessels (Table 1).
Table 1.
Summary of IC50 values
| Drug | WT | Kir6.1−/− | |
|---|---|---|---|
| ACh | FREQ | 2.70 × 10−8 M | 2.14 × 10−8 M |
| EDD | 3.14 × 10−8 M | 1.22 × 10−8 M | |
| NONOate | FREQ | 8.63 × 10−8 M | 9.51 − 10−8 M |
| EDD | 9.50 × 10−8 M | 1.85 × 10−7 M | |
| GLIB+NONOate | FREQ | 7.27 × 10−8 M | - |
| EDD | 1.45 × 10−7 M | - | |
| ODQ+NONOate | FREQ | # | # |
| EDD | # | # | |
| Rp-8-Br-PET-GMPS+NONOate | FREQ | 1.36 × 10−6 M | # |
| EDD | 1.12 × 10−6 M | # | |
| NS11021 | FREQ | 2.53 × 10−6 M | 1.08 × 10−6 M |
| EDD | # | # | |
| IbTX+NONOate | FREQ | # | - |
| EDD | # | - | |
| Penitrem A+NONOate | FREQ | # | - |
| EDD | # | - | |
| IbTX+ACh | FREQ | 1.25 × 10−7 M | - |
| EDD | 1.53 × 10−8 M | - | |
| Penitrem A+ACh | FREQ | 2.14 × 10−7 M | - |
| EDD | 2.05 × 10−8 M | - | |
| Apamin+NONOate | FREQ | 2.69 × 10−7 M | - |
| EDD | 2.42 × 10−7 M | - |
Could not be fit to the Hill equation.
Because ACh is an indirect source of NO, we also tested the responses of WT and Kir6.1−/− popliteal lymphatics to the specific NO donor, NONOate (at concentrations from 10 nM to 3 μM), Representative recordings showing the response of WT and Kir6.1−/− lymphatics to NONOate are shown in Fig. 2A and B, respectively. In light of previous studies using NONOate concentrations as high as 100 μM (Mathias and von der Weid, 2013), the high sensitivity of WT and Kir6.1−/− vessels was surprising, with NONOate almost completely stopping contraction in vessels of both genotypes below 1 μM, and higher doses more completely inhibiting contractions than ACh. The summary data for NONOate effects (Fig. 2C-E) are similar to those for ACh in that there was no significant difference between the response of WT and Kir6.1−/− vessels. There was a trend for EDD and frequency to be less sensitive to NONOate in WT than Kir6.1−/− vessels (~3-5-fold, Fig. 2D, E), and some Kir6.1−/− lymphatics exhibited a few persistent contractions at high concentrations of NONOate (Fig. 2B, D); this was not significant, and IC50 values for NONOate on normalized frequency and EDD were similar between WT and Kir6.1−/− vessels, as summarized in Table 1. Because contractions stopped completely at concentrations above 1 μM NONOate, the full dose-dependence of contraction amplitude could not be determined. Collectively, the observation that the inhibitory effects of both ACh and NONOate were comparable between WT and Kir6.1−/− lymphatics suggests that KATP channels are not the major transduction mechanism through which NO inhibits mouse popliteal lymphatic contractions.
Figure 2.
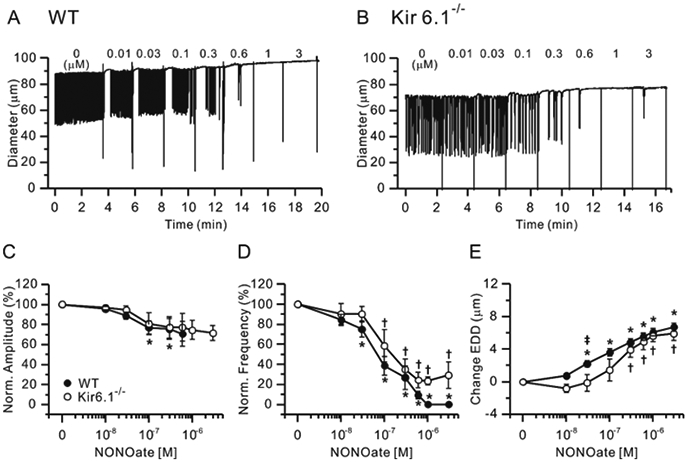
Effects of NONOate. Representative traces showing concentration-dependent effects of the NO-donor NONOate on the spontaneous contractions of WT (A) and Kir6.1−/− (B) mouse popliteal lymphatics. Normalized amplitude (C), normalized frequency (D) and change in EDD (E) plotted as a function of NONOate concentration, from 10 nM to 3 μM. Filled circles indicate WT vessels (n=24) and open circles indicate Kir6.1−/− vessels (n=9). All data are means ± SEM. There were no significant differences between WT and Kir6.1−/− data points at any NONOate concentration. * Significant difference in WT data from control (absence of NONOate). † Significant difference in Kir6.1−/− data from control. ǂ Significant difference between WT and Kir6.1−/− data.
Glibenclamide has off-target effects on mouse lymphatic vessel contractions
Previous studies implicating a role for KATP channels in NO-induced inhibition of lymphatic contractile function used glibenclamide (GLIB, 10 μM) to inhibit KATP channels (von der Weid, 1998). We assessed possible off-target non-KATP effects of GLIB in mouse lymphatic vessels by performing concentration-response curves to GLIB. Because GLIB acts by binding to the SUR subunit (Seino and Miki, 2003), we tested the responses of vessels from both Kir6.1−/− and SUR2[STOP] mice. Representative responses to GLIB are shown in Fig. 3 for WT (A) and Kir6.1−/− (B) vessels. Increasing concentrations of GLIB caused slight but progressive reductions in EDD and amplitude and increases in frequency, starting at 30 nM for the WT vessel (A). In the Kir6.1−/− vessel, increasing concentrations of GLIB led to only slight reductions in EDD with little effect on amplitude or frequency. Summary data are shown in Fig. 3C-E. WT vessels showed significant reductions in contraction amplitude at GLIB concentrations ≥ 300 nM; amplitude was slightly, but not significantly, reduced in Kir6.1−/− or SUR[STOP] vessels. There were significant differences between WT and Kir6.1−/− vessels at 1 and 10 μM and between WT and SUR2[STOP] vessels at 3 and 10 μM. GLIB produced biphasic changes in contraction frequency of WT vessels, with concentrations ≤ 300 nM progressively increasing frequency, and concentrations ≥1 μM decreasing frequency back toward control values (Fig. 3D). The contraction frequencies of Kir6.1−/− or SUR2[STOP] vessels were unaffected by lower concentrations of GLIB, but concentrations ≥ 1μM again led to decreases in frequency in both cases. At 10 μM GLIB, frequency was inhibited by 32% in Kir6.1−/− vessels and by 15% in SUR2[STOP] vessels (Fig. 3D). GLIB tended to produce slight constrictions in all three types of vessels, with the greatest effect in Kir6.1−/− (Fig. 3E). Decreased frequency at high concentrations of GLIB in Kir6.1−/− and SUR2[STOP] vessels is not consistent with an action on Kir6.1/SUR2 channels and, together with significant GLIB-induced decrease of amplitude and EDD in SUR2[STOP] vessels at concentrations ≥1 μM, suggests that off-target effects could be prominent at the dose of 10 μM used in previous studies.
Figure 3.
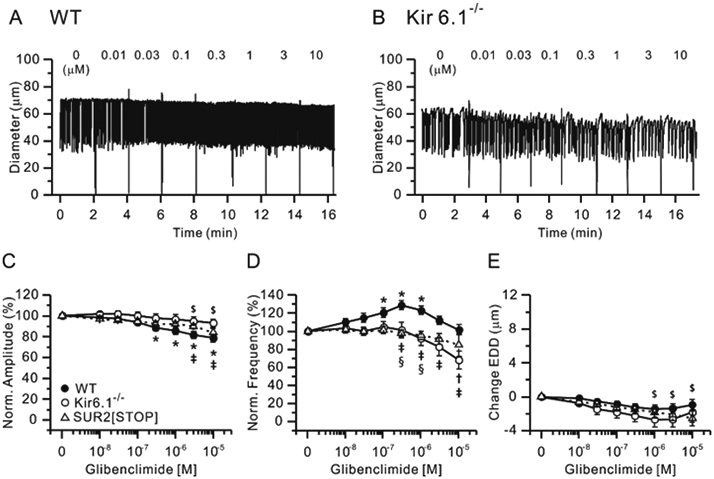
Effects of glibenclamide (GLIB). Representative recordings showing concentration-dependent effects of the KATP channel antagonist GLIB on the spontaneous contractions of WT (A) and Kir6.1−/− (B) mouse popliteal lymphatics. Normalized amplitude (C), normalized frequency (D) and change in EDD (E) plotted as a function of GLIB concentration, from 10 nM to 10 μM. Filled circles indicate WT vessels (n=22), open circles indicate Kir6.1−/− vessels (n=10) and open triangles indicate SUR2[STOP] vessels (n=14). All data are means ± SEM. * Significant difference in WT data from control (in the absence of GLIB). $ Significant difference in SUR2[STOP] data from control. † Significant difference in Kir6.1−/− data from control (none). ǂ Significant difference between WT and Kir6.1−/− data. § Significant difference between WT and SUR2[STOP] data.
We then tested the effect of pretreating WT vessels with 10 μM GLIB prior to performing a concentration-response curve to NONOate. Representative recordings of WT vessels to NONOate with and without GLIB are shown in Fig. 4A-B and the results from 10 vessels are summarized in Fig. 4E-G. In response to 10 μM GLIB alone, absolute contraction amplitude declined and contraction frequency increased significantly (Fig. 4C, D; paired t-tests), consistent with the results in Fig. 3. Concentration-response relationships for normalized amplitude and frequency to NONOate, in the presence or absence of 10 μM GLIB, are shown in Fig. 4E-G. In the presence of 10 μM GLIB, the spontaneous contraction amplitude and frequency decreased by ~40% and ~80%, respectively, at high concentrations of NONOate (Fig. 4E, F). However, a few spontaneous contractions typically persisted in presence of 10 μM GLIB, in contrast to their complete abolishment in NONOate alone, and normalized frequency plots suggest that NONOate sensitivity of WT vessels was slightly reduced in the presence of 10 μM GLIB (see IC50 values in Table 1). In contrast, NONOate-induced increases in EDD of WT vessels were greater in the presence of 10 μM GLIB (Fig. 4G). These findings are consistent with the persistence of NONOate-induced inhibition of contractions in Kir6.1−/− lymphatics (Fig. 3), further supporting the conclusion that KATP channels are not the major transduction mechanism through which NO inhibits spontaneous contractions of mouse lymphatic vessels.
Figure 4.
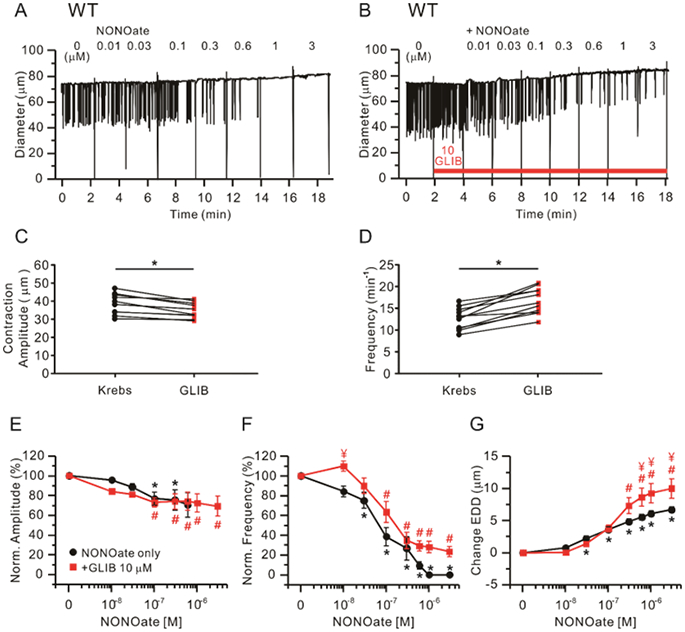
Effect of 10 μM glibenclamide (GLIB) on NONOate-induced responses in WT vessels. Raw traces showing NONOate-induced responses in the absence (A) or presence of 10 μM GLIB (B). Comparisons of contraction amplitude (C) or frequency (D) before (“Krebs”) and after treatment of 10 μM GLIB alone. * Significant using paired Student’s t-test. Lymphatic contraction parameters were normalized to their respective values prior to GLIB treatment (E-G). Comparisons of NONOate-induced effects on normalized amplitude (E), normalized frequency (F) and change in EDD (G) in the absence (n=12; black closed circles) or presence of 10 μM GLIB (n=11; red closed squares). All data are means ± SEM. * Significant difference in data from control for NONOate in the absence of GLIB. # Significant difference in data from control for NONOate in the presence of GLIB. ¥ Significant difference in data between NONOate alone and NONOate + GLIB.
KATP channels are not activated via the NO-sGC-cGMP-PKG pathway
Previous studies showed that NO induces hyperpolarization of guinea pig lymphatic smooth muscle through stimulation of cyclic GMP synthesis, because the responses to NO donors were blocked by the soluble guanylyl cyclase (sGC) inhibitor ODQ at 10 μM (von der Weid, 1998). We tested the role of the NO-sGC-cGMP-PKG pathway in mouse lymphatics, first by using ODQ and second by using a selective PKG inhibitor, Rp-8-Br-PET-cGMPS. NONOate concentration-response relationships were measured after 20 min of ODQ (10 μM) pretreatment in both WT and Kir6.1−/− mouse popliteal lymphatics (Fig. 5A, B). Pretreatment of WT but not Kir6.1−/−vessels with ODQ led to a significant decrease in absolute contraction amplitude and increase in frequency (Fig. 5C, D; paired t-tests). In the continued presence of ODQ, contraction amplitude was not significantly changed in response to increasing concentrations of NONOate (Fig. 5E), for either WT or Kir6.1−/− vessels. However, ODQ essentially completely inhibited frequency decreases and EDD increases at NONOate concentrations <10−5 M in WT and Kir6.1−/− vessels, which both showed persistent contractions at even the highest NONOate concentrations (Fig. 5F, G). Residual effects on frequency and EDD at higher concentrations could not be fit by the Hill equation (Table. 1) in WT and Kir6.1−/− with ODQ treatment.
Figure 5.
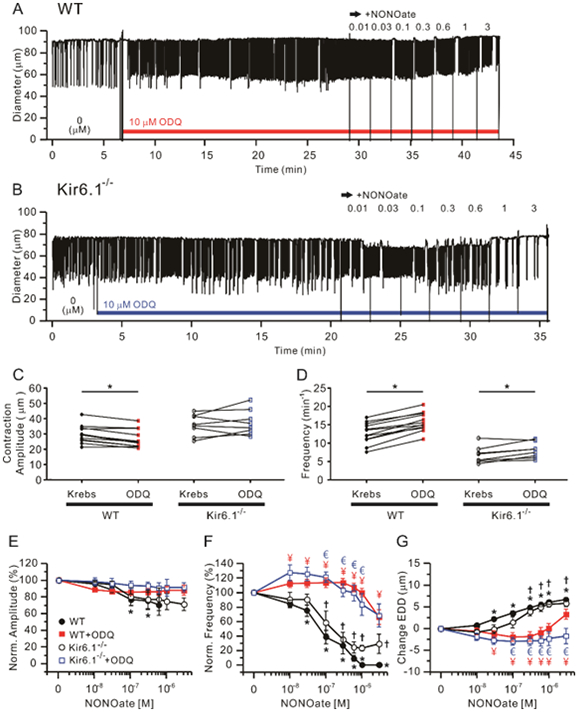
Effects of the soluble guanylate cyclase inhibitor, ODQ on NONOate-induced responses. 10 μM ODQ prevented the responses of both WT and Kir6.1−/− vessels to NONOate. Representative traces of concentration-dependent NONOate responses after pre-incubation with 10 μM ODQ for 20 min in WT (A) and Kir6.1−/− (B) popliteal lymphatics. Summary of absolute contraction amplitude (C) and frequency (D) compared before (“Krebs”) and after the treatment of ODQ. * Significant using paired Student’s t-test). Lymphatic contraction parameters were normalized to their respective values prior to ODQ treatment (E-G). Analysis of the effect of NONOate compared to the presence of 10 μM ODQ on normalized amplitude (E), normalized frequency (F) and change in EDD (G). Filled red rectangles indicate NONOate responses of WT vessels pretreated with ODQ (n=12) and open blue rectangles represent NONOate responses of Kir6.1−/− pretreated with ODQ (n=8). In comparison, the response to NONOate only in each group of WT (n=24) and Kir6.1−/− (n=8) is represented by filled black circles and open black circles respectively. All data are means ± SEM. * Significant difference in WT data from control (absence of NONOate). † Significant difference in Kir6.1−/− data from control (absence of NONOate). ¥ (red) Significant difference between filled black circles and filled red rectangle data points. € (blue) Significant difference between open black circles and open blue rectangle data points.
A similar protocol was performed after pretreatment with Rp-8-Br-PET-cGMPS treatment to test the contribution of PKG to NONOate-induced responses in WT and Kir6.1−/− mouse popliteal lymphatics (Fig. 6A, B). WT, but not Kir6.1−/−, lymphatics showed a significant decrease in contraction amplitude and increase in frequency after pretreatment with 15 μM Rp-8-Br-PET-cGMPS for 20 min (Fig. 6C, D). Again, in the presence of Rp-8-Br-PET-cGMPS, NONOate did not significantly change contraction amplitude of either WT or Kir6.1−/− vessels (Fig. 6E), but again, the IC50 for normalized frequency was right shifted >100-fold in WT vessels and even more so in Kir6.1−/− vessels (although the latter could not be fit by the Hill equation (Fig. 6F, Table 1). Similarly, Rp-8-Br-PET-cGMPS completely prevented NONOate-induced increases in EDD in Kir6.1−/− vessels and in WT vessels below 10−6M (Fig. 6G, Table 1). These significant right-shifts in the NONOate concentration response curves for frequency and EDD in the presence of ODQ or Rp-8-Br-PET-cGMPS in both WT and Kir6.1−/− vessels indicate that the NONOate responses of mouse lymphatics are mediated by the sGC-cGMP-PKG signaling pathway but do not require KATP channels.
mRNA for BK channels is expressed in mouse lymphatic muscle cells
Cyclic GMP-dependent PKG-mediated phosphorylation results in the modification of three serine residues in the c-terminus of the BK channel α-subunit, enhancing BK channel activity (Kyle and Braun, 2014), and many studies of vascular smooth muscle have shown that KCa channels are regulated by the NO-cGMP-PKG signaling axis (Kyle and Braun, 2014, Dopico et al., 2018, Archer et al., 1994b). We hypothesized that the inhibitory effects of NO on lymphatic smooth muscle might also be mediated by KCa channels rather than KATP channels. Functional BK channels were previously demonstrated in sheep mesenteric lymphatic smooth muscle cells by the presence of Ca2+-activated outward K+ currents that were inhibited by the selective BK channel antagonist, penitrem A (Cotton et al., 1997). We first tested whether BK channels were expressed in mouse lymphatic muscle cells (LMCs) in cDNA transcribed from purified LMC mRNA for endpoint RT-PCR with primers designed to amplify Kcnma1 encoding the pore-forming KCa1.1 α-subunit of the tetrameric BK channel complex. Two types of collecting lymphatic vessels were dissected from SMMHCCre;Rosa26mTmG mice, selectively expressing a GFP reporter in smooth muscle. Multiple vessels were dissected, cleaned and enzymatically dissociated and subjected to FACS analysis; RNA was then extracted from the GFP+ cell population (Davis et al., 2020). This procedure was repeated three times on three separate FACS-sorted lymphatic vessel preparations, LMC1, LMC2 and LMC3 from both inguinal-axillary lymphatics (IA) and popliteal lymphatics (POP). Inguinal-axillary vessels are longer and have a higher LMC density, thus providing more GFP+ cells than popliteal vessels. PCR analysis showed mRNA expression for Kcnma1 in all three preparations from both types of vessels as well as in samples of brain tissue used as positive controls (Fig. 7A-B). The preparations also showed clear signals for both smooth muscle markers, α-actin (α-Act) and myosin heavy chain (Myh11), but no message for Prox1 or eNOS, indicating no contamination by lymphatic endothelial cells (LECs). These results suggest that message for BK channels is expressed in LMCs of mouse lymphatic vessels. We performed a similar analysis on inguinal-axillary lymphatics dissected from Prox1Cre;Rosa26mTmG mice, which expressed LEC but not LMC markers (Suppl. Fig. 1). These vessels showed expression for Kcnma1 message in brain but not in LECs.
Figure 7.
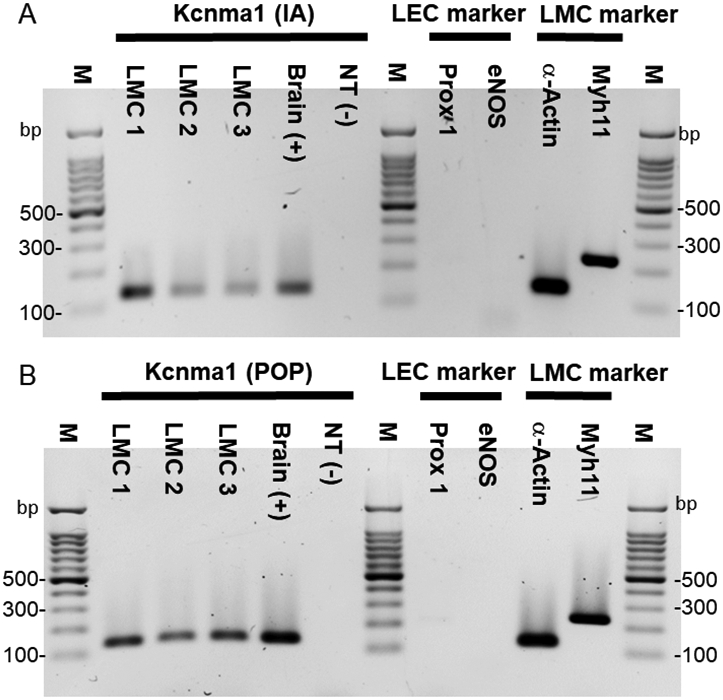
PCR to detect Kcnma1 (KCa1.1 α-subunit) in mouse lymphatic muscle cells (LMCs) isolated from inguinal-axillary (IA) lymphatic vessels (A) and popliteal (POP) lymphatic vessels (B). LMCs were collected by FACS analysis after isolation and digestion of lymphatic vessels from SMMHCCre;Rosa26mTmG mice. Kcnma1 showed strong bands in both LMC1 (168 cells), 2 (144 cells) and 3 (88 cells) from IA samples and LMC1 (76 cells), 2 (79 cells) and 3 (80 cells) from POP samples. A small amount of brain cDNA was used as a positive control (Lane 5). A representative panel of cell markers from LMC3 is shown. α-actin and Myh11 were detected but not Prox1 or eNOS, indicating no contamination by lymphatic endothelial cells (LEC) (Lane 8-11). Kcnma1 (135 bp), Prox1 (218 bp), eNOS (143 bp), α-actin (129 bp) and Myh11 (myosin-11; 238 bp). M = Marker; NT = negative; bp = base pairs.
Activation of BK channels in mouse lymphatic muscle through the NO-sGC-cGMP-PKG signaling axis
We then tested the effects of BK channel activation in popliteal lymphatics from WT and Kir6.1−/− mice using increasing concentrations (100 nM to 10 μM) of the selective BK channel activator, NS11021. Representative traces are shown in Fig. 8A-B for WT and Kir6.1−/− vessels, respectively. Contraction amplitudes of either WT or Kir6.1−/− lymphatics were not significantly affected by NS11021 (Fig. 8C). Normalized frequency declined, while EDD increased slightly, with increasing NS11021 concentration in both, with NS11021 sensitivity being slightly higher for Kir6.1−/− (Fig. 8D, E, Table 1). These results support the conclusion that BK channel activation can inhibit the ionic pacemaker of mouse popliteal lymphatics without significant effects on contraction amplitude or tone.
Figure 8.
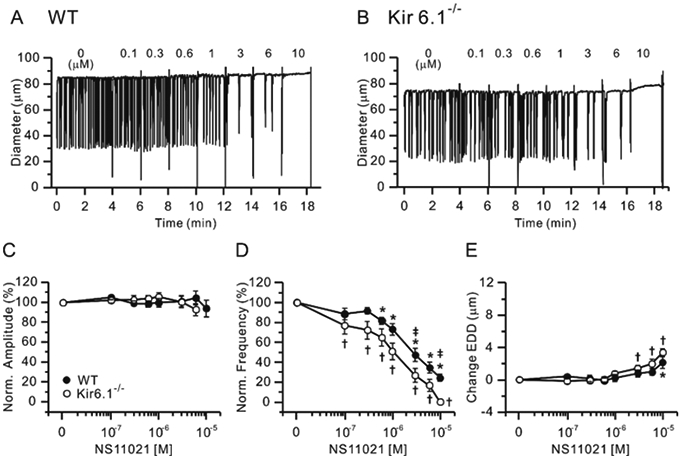
Concentration-response curves for NS11021, a BK channel activator, in WT and Kir6.1−/− lymphatic vessels at pressure = 3 cmH2O. Sample recordings for WT (A) and Kir6.1−/− (B) lymphatics. Normalized amplitude (C), normalized frequency (D) and change in EDD (E) are plotted as a function of NS11021 concentration (from 0.1 μM to 10 μM) and. Filled circles represent WT vessels (n=11) and open circles represent Kir6.1−/− vessels (n=13). All data are means ± SEM. * Significant difference in filled data points (WT) from control (0 M). † Significant difference in open data points (Kir6.1−/−) from control (0 M). ‡ Significant difference in filled (WT) versus open (Kir6.1−/−) data points (P < 0.05).
We then tested the hypothesis that lymphatic smooth muscle BK channels mediate contraction inhibition to NONOate. WT popliteal lymphatic vessels were pretreated with a BK channel inhibitor (Iberiotoxin;IbTX or penitrem A) for 5 min, then exposed to increasing concentrations of NONOate (Fig. 9A, B). Treatment with either 300nM IbTX or 100 nM penitrem A, did not significantly change contraction amplitude (Fig. 9C), but both inhibitors significantly increased contraction frequency (Fig. 9D). Neither IbTX nor penitrem A had any significant effect on NONOate-induced reduction in contraction amplitude, except at the highest concentration of NONOate, at which contractions ceased (Fig. 9E). However, the concentration-frequency curves for NONOate were shifted significantly to the right by both BK inhibitors (Fig. 9F), although spontaneous contractions persisted at the highest concentrations of NONOate. Likewise, both IbTX and penitrem A prevented significant increases in EDD from baseline over the entire concentration range of NONOate (Fig. 9G). The IC50 values for frequency and EDD in WT with IbTX or penitrem A treatment could not be fit to the Hill equation, but the estimated IC50 values appear to be ~10-fold higher for frequency and 50-fold higher for EDD. These data strongly suggest that BK channels are major mediators of the inhibitory effects of NO on spontaneous contractions of mouse lymphatic vessels.
Figure 9.
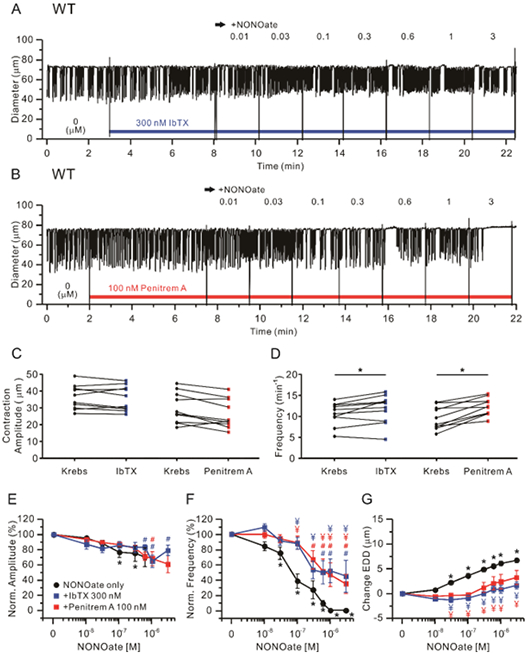
BK channel inhibitors attenuate NONOate-induced inhibition of lymphatic contractions in WT vessels. Representative traces of NONOate concentration-dependent response after pretreatment with 300 nM IbTX (A) or 100 nM penitrem A (B) in WT popliteal lymphatics held at pressure = 3 cmH2O. Summary of absolute contraction amplitude (C) and frequency (D) before (“Krebs”) and after treatment of IbTx or penitrem A. * Significant using paired Student’s t-test). Analysis of the effect of NONOate on normalized amplitude (E), normalized frequency (F) and change in EDD (G) in the presence of IbTX (n=10) or penitrem A (n=10). Filled red rectangles indicate NONOate responses of lymphatics with penitrem A present and filled blue rectangles indicate NONOate responses with IbTX present, compared to the response to NONOate only (n=24), marked as a filled black circles. All data are means ± SEM. * Significant difference in WT data from control (absence of NONOate). # (Blue, IbTX; Red, penitrem A) Only filled rectangle data points differ significantly from the first data points (0 M). ¥ (red) Significant difference between NONOate only (filled black circles) versus NONOate + penitrem A (filled red rectangles), (P < 0.05). ¥ (blue) Significant difference between NONOate only (filled black circles) versus NONOate + IbTX (filled blue rectangles).
We also tested the effects of IbTX and penitrem A on ACh-mediated inhibition of WT lymphatic vessel contractions. As above, neither IbTX nor penitrem A had significant effects on contraction amplitude (Suppl. Fig. 2A), but both significantly increased contraction frequency (Suppl. Fig. 2B). Both blunted the modest decrease in amplitude in response to ACh (Suppl. Fig. 2C) and both shifted the ACh-frequency curve slightly to the right (Suppl. Fig. 2D); however, these rightward shifts were modest compared to the effects of IbTX and penitrem A on the NONOate-frequency curve (Fig. 9F). IbTX, but not penitrem A, blunted the increase in EDD produced by ACh (Suppl. Fig. 3E). These results suggest that BK channels are only modestly involved in the ACh-mediated inhibitory effects on lymphatic contraction.
Finally, we explored whether IK or SK channels might also be involved in mediating NONOate responses. Previously, we showed that although whole lymphatic vessels from mouse did express one SK channel isoform, Kcnn3 (Behringer et al., 2017), there was no detectable message for SK or IK channels in mouse lymphatic endothelium (Behringer et al., 2017), and a lack of electrical coupling between lymphatic endothelium and smooth muscle (Hald et al., 2018) that could mediate conduction of polarizing signals between those layers. To detect SK channel or IK channel message in a more sensitive and cell-specific manner, we performed RT-PCR analysis on FACS-purified LMCs from SMMHC-Cre;Rosa26MtmG mice. We were unable to detect message for Kcnn1 (encoding SK1 channels), Kcnn3 (encoding SK3 channels) or Kcnn4 (encoding IK channels) in LMCs, but we did detect a weak band for Kcnn2 (encoding the SK2 channel) (Suppl. Fig.3A) and confirmed expression of Kcnj8 (Kir6.1 channels); the same primer sequences detected all 5 K+ channel isoforms in brain (Suppl. Fig. 3B). To further test whether SK channels might mediate part of NO-mediated inhibition of lymphatic contraction, NONOate responses were assessed in WT popliteal lymphatics after pretreatment with the well-established SK channel inhibitor (Castle, 1999), apamin. Apamin (1 μM) was without pattern (Suppl. Fig. 4A-B), and the sensitivity of contraction amplitude to NONOate was not significantly different in the presence or absence of apamin (Suppl. Fig. 4C). Apamin produced a slight right-shift in the NONOate-frequency response curve (Suppl. Fig. 4D) and blunted the NONOate-induced increase in EDD at low but not higher NONOate concentrations (Suppl. Fig. 4E). We did not to evaluate any possible role for functional IK channels because the commonly-used IK channel inhibitor, tram-34 (1 μM) caused significant changes in baseline amplitude and frequency, complicating assessment of any possible effect on NONOate-induced responses. It should be noted that Tram-34 is reported to block non-selective cation channels at nanomolar concentrations (Schilling and Eder, 2007) as well as other types of ion channels (McNeish et al., 2010), which might explain its effects on baseline lymphatic contractility. Collectively, the lack of evidence for expression of SK and IK channels and the very modest effects of the SK inhibitor Apamin suggests that neither SK nor IK channels are involved in the inhibition of spontaneous contractions by the NO-sGC-cGMP-PKG signaling axis in mouse lymphatic vessels.
Discussion and Conclusions
KATP independence of NO effect in mouse lymphatics
In both chronic peripheral lymphedema and inflammatory bowel disease, iNOS activation and subsequent NO production result in lymphatic contractile dysfunction (Chen et al., 2017a). The prevailing view is that KATP channels in lymphatic smooth muscle mediate this response (von der Weid, 1998, Mathias and von der Weid, 2013). This view arises from the finding that in guinea pig mesenteric lymphatics, NO-mediated inhibition of spontaneous contractions was largely prevented by 10 μM GLIB, (von der Weid, 1998, Mathias and von der Weid, 2013), and reversed much of the tonic inhibitory actions of NO produced as a consequence of iNOS activation in guinea pig and rat models of TNBS-induced ileitis (Mathias and von der Weid, 2013, Chen et al., 2017b). The inference from the latter studies is that KATP channels in lymphatic muscle can be activated by immune cell products to inhibit lymph transport, a concept that could apply more widely to include lymphatic dysfunction in other diseases (Jiang et al., 2019, Norden and Kume, 2020).The first aim of this study was to directly test this putative role for KATP channels using mouse models with genetic deletion of KATP channel subunits. In agreement with previous work, we found that the inhibitory effects of NO on the contractile function of mouse popliteal lymphatics are mediated through the sGC-cGMP-PKG signaling axis (Figs. 5-6), but the inhibitory effects of ACh and NONOate on lymphatic contractions are not significantly impaired in popliteal lymphatics from Kir6.1−/− or SUR2[STOP] mice, which lack KATP channels (Figs. 1-2). This leads us to conclude that KATP channels are not the major transduction mechanism through which nitric oxide inhibits lymphatic contractions in the mouse. GLIB does partially attenuate the response to NONOate in WT and KATP knockout mouse lymphatic vessels at 10 μM (as used in previous studies), but at this concentration it already affects spontaneous contraction amplitude, frequency and tone in KATP knockout vessels (Figs. 3-4).
The disparate results of our study and those in guinea pig (Mathias and von der Weid, 2013) and rat (Chen et al., 2017b) might be explained by the different species used but, given our findings that GLIB (at the concentration used in these other studies) alters spontaneous contractility in mice lacking functional Kir6.1 or SUR2 subunits, an alternative explanation is that off-target effects of GLIB on other ion channels may lead to an incorrect conclusion. Indeed, multiple patch clamp studies have found that concentrations of GLIB >1 μM have direct inhibitory effects on L-type Ca2+ channels (Lee and Lee, 2005, Bian and Hermsmeyer, 1994, Sadraei and Beech, 1995), other Kir channels (Song et al., 1996) and Cl− channels (Schultz et al., 1996, Faivre et al., 1998). However, it should be noted that in our studies 10 μM GLIB did not produce the same degree of right shift in the NONOate concentration-response curve observed with sGC or PKG blockade (Table 1), implying that off-target effects of GLIB cannot completely explain its prevention of responses to NO donors in other lymphatic studies (von der Weid, 1998, Mathias and von der Weid, 2013). These discrepancies remain to be fully explained.
Ca2+-activated BK channels as mediators of NO action
Based on precedents from the arterial literature (Archer et al., 1994a, Kyle and Braun, 2014), we investigated whether calcium-activated K+ channels might alternately provide a major target of NO signaling in lymphatic smooth muscle. Message for the BK channel α-subunit, but not for SK or IK isoforms, was detected in mouse lymphatic muscle (Fig. 7 and Suppl. Fig. 3). The SK channel inhibitor, apamin, had almost no effect of NONOate-induced responses (Suppl. Fig. 4). However, a BK channel activator mimicked many of the effects of NONOate (Fig. 8) and two BK channel inhibitors significantly attenuated the inhibitory effects of NONOate on both WT and Kir6.1−/− popliteal lymphatics (Fig. 9). Collectively, these results suggest that functional BK channels are present in mouse lymphatic smooth muscle, and that NO-induced inhibition of spontaneous lymphatic contractions is largely independent of KATP, SK and IK channels but is mediated to a substantial degree by BK channels in lymphatic muscle.
Although we were unable to test this directly in BK channel α-subunit KO mice, we did test two well-characterized pharmacological inhibitors of BK channels, iberiotoxin (IbTX) (Candia et al., 1992) and penitrem A (Knaus et al., 1994) on NONOate and ACh-induced responses of mouse lymphatic vessels. Both agents produced marked right-shifts in the NONOate concentration-response curves for contraction frequency (Fig. 9). However, these rightward shifts were only ~10 to 50-fold compared to the ~100-fold shift produced by ODQ and PKG inhibitors (Figs. 5-6). Assuming complete inhibition of BK channels, this difference suggests that BK channel activation is only part of the mechanism of lymphatic contraction inhibition through the NO-cGMP-PKG pathway.
Other potential mediators of NO action
The more potent inhibition of spontaneous contractions produced by sGC and PKG inhibitors than by BK channel inhibitors suggests that other, unidentified target(s) downstream of the NO-cGMP-PKG signaling axis could be involved. We explored whether IK or SK channels might be involved in NONOate responses. However, the lack of evidence for expression of SK and IK channels and the lack of substantial effect of the SK channel inhibitor apamin suggests that neither are likely to be involved in the inhibition of spontaneous contractions by the NO-sGC-cGMP-PKG signaling axis. Alternate potential components include other ion channels and contractile proteins. PKG is a known regulator of L-type Ca2+ channels, and the isoforms expressed in cardiac (Cav1.2b) and smooth muscle (Cav1.2c) share a conserved phosphorylation site on residue 533 of their α-subunits (Keef et al., 2001, Sperelakis et al., 1994). Thus, PKG-mediated phosphorylation downstream from NO-sGC could be an explanation for the fraction of the sGC/PKG inhibition not explained by BK channels, but we would be unable to test this experimentally because L-type Ca2+ channels are essential for spontaneous lymphatic contractions (Lee et al., 2014) and blocking them would eliminate all contractions (To et al., 2020). However, the fact that BK channel inhibition more potently affected NONOate-induced changes in frequency and tone rather than amplitude, perhaps points to target(s) other than ion channel(s). Thus, another possibility is that PKG induces the activation of Rho kinase and myosin phosphatase targeting subunit (MYPT), which induces smooth muscle relaxation by dephosphorylation of myosin regulatory light-chain subunits (Francis et al., 2010).
Developmental consequences of KATP channels in lymphatic physiology redux
This study revealed several differences between WT and Kir6.1−/− vessels that were not noted previously (Davis et al 2020). As evident in the recordings shown in Figs. 1-3 the EDDs of Kir6.1−/− popliteal lymphatics were often considerably smaller than those of WT vessels. An analysis of the average EDDs for WT and Kir6.1−/− vessels in the ACh, NONOate and GLIB protocols reveals that the EDDs for untreated WT vessels averaged 78.1±2.2 μm, 76.4±2.2 μm and 79.0±2.1 μm, respectively, compared to average EDDs for untreated Kir6.1−/− vessels of 60.7±5.2 μm, 61.7±4.1 μm and 67.1±3.2 μm; all three sets of comparisons were statistically significant. This observation points to potential long-term developmental consequences of KATP channel knockout on lymphatic vessel architecture. Recent publications document a marked increase in arterial vessel diameter and length in Cantú syndrome humans (Leon Guerrero et al., 2016) and mice (Huang et al., 2018, McClenaghan et al., 2020), and these differences are evident even in isolated unpressurized arteries (Huang et al., 2018). Thus, it is possible that mice which are deficient in KATP channels might exhibit opposite effects, i.e., reduced vessel diameters and/or lengths. Compensatory changes in ion channels, e.g., other K+ channels, are also possible in Kir6.1−/− mice, although to our knowledge such changes have not been systematically studied. It is also intriguing, however, that the frequency of Kir6.1−/− vessels was more sensitive to NS11021 than the frequency of WT vessels, raising the possibility that expression or trafficking of BK channels might be upregulated in lymphatic smooth muscle of Kir6.1−/− mice.
Conclusions
In summary, the sGC-cGMP-PKG pathway mediates NO-induced contraction inhibition in mouse lymphatic vessels, as reported in other species. However, our studies in gene knockout animals reveal that KATP channels are not involved in this response; rather, BK channels are primary mediators of the NO-mediated inhibition, but also suggest that other as-yet-to-be-identified target or targets is/are involved. Lymphatic contractile dysfunction is a newly appreciated component of multiple diseases (Norden and Kume, 2020, Jiang et al., 2019), triggered in part by excessive NO production, and treatment with appropriate inhibitors could be therapeutically beneficial.
Supplementary Material
Supplemental Figure 2. Effects of BK channel inhibitors (IbTX and penitrem A) on baseline lymphatic contractions and ACh concentration-response curves in WT lymphatic vessels at pressure = 3 cmH2O. Summary of absolute contraction amplitude (A) and frequency (B) before (Krebs) and after treatment with IbTX (300 nM) or penitrem A (100 nM). * Significant using paired Student’s t-test). Analysis of the effect of ACh on normalized contraction amplitude (C), normalized contraction frequency (D) and change in EDD (E) in the presence of IbTX 300 nM (n=10) or penitrem A 100 nM (n=10). Filled red rectangles indicate ACh responses of lymphatics with penitrem A present and filled blue rectangles indicate ACh responses with IbTX present, compared to the response to ACh alone (n=25), indicated by filled black circles. All data are means ± SEM. # (Blue, IbTX; Red, penitrem A) Only filled rectangle data points differ significantly from the first data points (0 M). # (red) Significant difference between ACh only (filled black circles) versus ACh + penitrem A (filled red rectangles). # (blue) Significant difference between ACh only (filled black circles) versus ACh + IbTX (filled blue rectangles).
Supplemental Figure 1. PCR to detect Kcnma1 (KCa1.1 α-subunit) in mouse lymphatic endothelial cells (LECs) isolated from lingual-axillary (IA) lymphatic vessels. LECs were collected by FACS analysis after isolation and digestion of lymphatic vessels from Prox1GFP mice. Kcnma1 showed strong bands in LEC samples 1 (36 cells), 2 (84 cells) and 3 (66 cells). Brain was used as a positive control (Lane 5). A representative panel of cell markers from LEC3 is shown. Prox1 and eNOS were detected but not α-actin or Myh11, indicating no contamination by lymphatic muscle cells (LMC) (Lane 8-11). Kcnma1 (135 bp), Prox1 (218 bp), eNOS (143 bp), α-actin (129 bp) and Myh11 (myosin-11; 238 bp). M = Marker; NT = negative; bp = base pairs.
Supplemental Figure 3. PCR analysis to detect expression of Kcnn1 (SK1 channel), Kcnn2 (SK2 channel), Kcnn3 (SK3 channel), Kcnn4 (IK channel) and Kcnj8 (Kir6.1 subunit) in mouse lymphatic muscle cells (LMCs) isolated from lingual-axillary lymphatic vessels. LMCs were collected by FACS analysis after isolation and digestion of lymphatic vessels from SMMHCCre;Rosa26mTmG mice. No expression of Kcnn1, Kcnn3, and Kcnn4 is detected, but there is faint signal for Kcnn2 in LMCs (A). In contrast, a strong Kcnj8 band is observed, verifying the quality of the cDNA. Brain homogenate was used as a positive control (B). Kcnn1 (352 bp), Kcnn2 (242 bp), Kcnn3 (294 bp), Kcnn4 (290 bp) and Kcnj8 (249 bp). M = Marker; bp = base pairs.
Supplemental Figure 4. Effects of the SK channel inhibitor, Apamin (1 μM), on baseline lymphatic contractions and NONOate concentration-response curves in WT popliteal lymphatics at pressure = 3 cmH2O. Summary of contraction amplitude (A) and frequency (B) compared between before (Krebs) and after treatment of apamin (paired Student’s t-test). When analyzed and normalized lymphatic contractile parameter the values before apamin treatment were used as a standard (C-E). Analysis of the effect of NONOate compared to the presence of 1 μM apamin on normalized amplitude (C), normalized frequency (D) and change in EDD (E). Filled circles indicate NONOate responses of WT vessels (n=24) and open red rectangles represent NONOate responses of WT pretreated with apamin (n=11). All data are means ± SEM. † (red) Only open red rectangle data points (apamin) differ significantly from the first data points (0 M).
Bullet Point Summary:
What is already known
High concentrations of NO donors and KATP channel inhibitors block nitric oxide inhibition of lymphatic pumping.
This has led to the conclusion that KATP channel activation mediates nitric oxide inhibition of lymphatic pumping.
What this study adds
Kir6.1−/− mice lack KATP channels, but NO-inhibition of lymphatic pumping is still present.
Rather, BK channels in lymphatic smooth muscle mediate much of these inhibitory effects.
Clinical significance
BK channels are a potential therapeutic target to counteract NO-mediated inhibition of lymph pumping.
Acknowledgments
This research was supported by National Institutes of Health grants R01 HL-122608 and HL-122578 to M.J.D, and by R35 HL-140024 to CGN. H.J.K was supported in part by National Research Foundation of Korea grant NRF-2020R1A6A3A03037151.
Abbreviations
- NO
nitric oxide
- LSM
lymphatic smooth muscle
- LMCs
lymphatic muscle cells
- cGMP
cyclic guanosine-3’,5’-monophosphate
- WT
wild type
- SUR2
sulfonylurea receptor2
- EDD
end-diastolic diameter
- FREQ
frequency
- GLIB
glibenclamide
- ACh
acetylcholine
- NONOate
diethylamine NONOate sodium salt hydrate
- sGC
soluble guanylyl cyclase
- ODQ
1H-[1,2,4]-oxadiazolo-[4,3-a]-quinoxalin-1-one
- BK channel
large-conductance Ca2+-activated potassium
- IbTX
Iberiotoxin
- NS11021
N'-[3,5-Bis(trifluoromethyl)phenyl]-N-[4-bromo-2-(2H-tetrazol-5-yl-phenyl]thiourea
- Rp-8-Br-PET-cGMPS
2-Bromo-3,4-dihydro-3-[3,5-O-[(R)-mercaptophosphinylidene]-β-D-ribofuranosyl]-6-phenyl-9H-Imidazo[1,2-a]purin-9-one sodium salt
Footnotes
Conflict of interest
The authors have no competing interests to declare or conflicts of interest to disclose. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declaration of transparency and scientific rigour
This work adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Data available on request from the authors: The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
- ALEXANDER SPH, MATHIE A, PETERS JA, VEALE EM, STRIESSNIG J, KELLY E, et al. 2019.The Coincise Guide to PHARMACOLOGY 2019/20: Ion channels. Br J Pharmacol, 176, S142–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARCHER SL, HUANG JM, HAMPL V, NELSON DP, SHULTZ PJ & WEIR EK 1994a. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A, 91, 7583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEHRINGER EJ, SCALLAN JP, JAFARNEJAD M, CASTORENA-GONZALEZ JA, ZAWIEJA SD, MOORE JE JR., DAVIS MJ & SEGAL SS 2017. Calcium and electrical dynamics in lymphatic endothelium. J Physiol, 595, 7347–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIAN K & HERMSMEYER K 1994. Glyburide actions on the dihydropyridine-sensitive Ca2+ channel in rat vascular muscle. J Vasc Res, 31, 256–64. [DOI] [PubMed] [Google Scholar]
- CANDIA S, GARCIA ML & LATORRE R 1992. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca(2+)-activated K+ channel. Biophys J, 63, 583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASTLE NA 1999. Recent advances in the biology of small conductance calcium-activated potassium channels. Perspectives in Drug Discovery and Design, 15, 131–154. [Google Scholar]
- CASTORENA-GONZALEZ JA, ZAWIEJA SD, LI M, SRINIVASAN AS, SIMON AM, WIT CD, TORRE RDL, MARTINEZ-LEMUS LA, HENNIG GW, DAVIS MJ 2018. Mechanisms of connexin-related lymphedema: A critical role for Cx45, but not Cx43 or Cx47, in the entrainment of spontaneous lymphatic contractions. Circ Res, 123(8):964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATAIGNEAU T, FELETOU M, HUANG PL, FISHMAN MC, DUHAULT J & VANHOUTTE PM 1999. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol, 126, 219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y, REHAL S, ROIZES S, ZHU HL, COLE WC & VON DER WEID PY 2017a. The pro-inflammatory cytokine TNF-alpha inhibits lymphatic pumping via activation of the NF-kappaB-iNOS signaling pathway. Microcirculation, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COTTON KD, HOLLYWOOD MA, MCHALE NG & THORNBURY KD 1997. Outward currents in smooth muscle cells isolated from sheep mesenteric lymphatics. J Physiol, 503 (Pt 1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CURTIS MJ, ALEXANDER S, CIRINO G, DOCHERTY JR, GEORGE CH, GIEMBYCZ MA, … AHLUWALIA A 2018. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br J Pharmacol, 175, 987–993. 10.1111/bph.14153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS MJ, KIM HJ, ZAWIEJA SD, CASTORENA-GONZALEZ JA, GUI P, LI M, SAUNDERS BT, ZINSELMEYER BH, RANDOLPH GJ, REMEDI MS & NICHOLS CG 2020. Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J Physiol. 598(15):3107–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS MJ, ZAWIEJA DC & GASHEV AA 2006. Automated measurement of diameter and contraction waves of cannulated lymphatic microvessels. Lymphat Res Biol, 4, 3–10. [DOI] [PubMed] [Google Scholar]
- DOPICO AM, BUKIYA AN & JAGGAR JH 2018. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflugers Arch, 470, 1271–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAIVRE JF, ROUANET S & BRIL A 1998. Comparative effects of glibenclamide, tedisamil, dofetilide, E-4031, and BRL-32872 on protein kinase A-activated chloride current in guinea pig ventricular myocytes. J Cardiovasc Pharmacol, 31, 551–7. [DOI] [PubMed] [Google Scholar]
- FOSTER MN & COETZEE WA 2016. KATP Channels in the Cardiovascular System. Physiol Rev, 96, 177–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANCIS SH, BUSCH JL, CORBIN JD & SIBLEY D 2010. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev, 62, 525–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARDENIER JC, KATARU RP, HESPE GE, SAVETSKY IL, TORRISI JS, NORES GD, JOWHAR DK, NITTI MD, SCHOFIELD RC, CARLOW DC & MEHRARA BJ 2017. Topical tacrolimus for the treatment of secondary lymphedema. Nat Commun, 8, 14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALD BO, CASTORENA-GONZALEZ JA, ZAWIEJA SD, GUI P & DAVIS MJ 2018. Electrical Communication in Lymphangions. Biophys J, 115, 936–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDING SD, SHARMAN JL, FACCENDA E, SOUTHAN C, PAWSON AJ, IRELAND S, … NC-IUPHAR 2018. Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Res, 46, D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG Y, MCCLENAGHAN C, HARTER TM, HINMAN K, HALABI CM, MATKOVICH SJ, ZHANG H, BROWN GS, MECHAM RP, ENGLAND SK, KOVACS A, REMEDI MS & NICHOLS CG 2018. Cardiovascular consequences of KATP overactivity in Cantu syndrome. JCI Insight, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIANG X, TIAN W, NICOLLS MR & ROCKSON SG 2019. The Lymphatic System in Obesity, Insulin Resistance, and Cardiovascular Diseases. Frontiers in Physiology, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEEF KD, HUME JR & ZHONG J 2001. Regulation of cardiac and smooth muscle Ca(2+) channels (Ca(V)1.2a,b) by protein kinases. Am J Physiol Cell Physiol, 281, C1743–56. [DOI] [PubMed] [Google Scholar]
- KILKENNY C, BROWNE W, CUTHILL IC, EMERSON M, & ALTMAN DG 2010. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNAUS HG, MCMANUS OB, LEE SH, SCHMALHOFER WA, GARCIA-CALVO M, HELMS LM, SANCHEZ M, GIANGIACOMO K, REUBEN JP, SMITH AB 3RD & et al. 1994. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry, 33, 5819–28. [DOI] [PubMed] [Google Scholar]
- KYLE BD & BRAUN AP 2014. The regulation of BK channel activity by pre- and post-translational modifications. Front Physiol, 5, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE S, ROIZES S & VON DER WEID PY 2014. Distinct roles of L- and T-type voltage-dependent Ca2+ channels in regulation of lymphatic vessel contractile activity. J Physiol, 592, 5409–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE SY & LEE CO 2005. Inhibition of Na+-K+ pump and L-type Ca2+ channel by glibenclamide in Guinea pig ventricular myocytes. J Pharmacol Exp Ther, 312, 61–8. [DOI] [PubMed] [Google Scholar]
- LEON GUERRERO CR, PATHAK S, GRANGE DK, SINGH GK, NICHOLS CG, LEE JM & VO KD 2016. Neurologic and neuroimaging manifestations of Cantu syndrome: A case series. Neurology, 87, 270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO S, CHENG G, CONNER DA, HUANG Y, KUCHERLAPATI RS, MUNN LL, RUDDLE NH, JAIN RK, FUKUMURA D & PADERA TP 2011. Impaired lymphatic contraction associated with immunosuppression. Proc Natl Acad Sci U S A, 108, 18784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHIAS R & VON DER WEID PY 2013. Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am J Physiol Gastrointest Liver Physiol, 304, G623–34. [DOI] [PubMed] [Google Scholar]
- MCCLENAGHAN C, HUANG Y, MATKOVICH SJ, KOVACS A, WEINHEIMER CJ, PEREZ R, BROEKELMANN TJ, HARTER TM, LEE JM, REMEDI MS & NICHOLS CG 2020. The Mechanism of High-Output Cardiac Hypertrophy Arising From Potassium Channel Gain-of-Function in Cantu Syndrome. Function (Oxf), 1, zqaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNEISH AJ, JIMENEZ ALTAYO F & GARLAND CJ 2010. Evidence both L-type and non-L-type voltage-dependent calcium channels contribute to cerebral artery vasospasm following loss of NO in the rat. Vascul Pharmacol, 53, 151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIKI T, SUZUKI M, SHIBASAKI T, UEMURA H, SATO T, YAMAGUCHI K, KOSEKI H, IWANAGA T, NAKAYA H & SEINO S 2002. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med, 8, 466–72. [DOI] [PubMed] [Google Scholar]
- NORDEN PR & KUME T 2020. The Role of Lymphatic Vascular Function in Metabolic Disorders. Frontiers in Physiology, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSZEWSKI WL 1977. Pathophysiological and clinical observations of obstructive lymphedema of the limbs. In: CLODIUS L (ed.) Lymphedema. Stuttgart: Georg Thieme Publishers. [Google Scholar]
- OLSZEWSKI WL 2002. Contractility patterns of normal and pathologically changed human lymphatics. Annals of the New York Academy of Science, 979, 52–63. [DOI] [PubMed] [Google Scholar]
- RAN FA, HSU PD, WRIGHT J, AGARWALA V, SCOTT DA & ZHANG F 2013. Genome engineering using the CRISPR-Cas9 system. Nature Protocols, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCKSON SG, TIAN W, JIANG X, KUZNETSOVA T, HADDAD F, ZAMPELL J, MEHRARA B, SAMPSON JP, ROCHE L, KIM J & NICOLLS MR 2018. Pilot studies demonstrate the potential benefits of antiinflammatory therapy in human lymphedema. JCI Insight, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SADRAEI H & BEECH DJ 1995. Ionic currents and inhibitory effects of glibenclamide in seminal vesicle smooth muscle cells. Br J Pharmacol, 115, 1447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCALLAN JP & DAVIS MJ 2013. Genetic removal of basal nitric oxide enhances contractile activity in isolated murine collecting lymphatic vessels. J Physiol, 591, 2139–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCALLAN JP, ZAWIEJA SD, CASTORENA-GONZALEZ JA & DAVIS MJ 2016. Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol, 594, 5749–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFFER P, PELZMANN B, BERNHART E, LANG P, MÄCHLER H, RIGLER B & KOIDL B 1999. The sulphonylurea glibenclamide inhibits voltage dependent potassium currents in human atrial and ventricular myocytes. British Journal of Pharmacology, 128, 1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHILLING T & EDER C 2007. TRAM-34 inhibits nonselective cation channels. Pflugers Arch, 454, 559–63. [DOI] [PubMed] [Google Scholar]
- SCHULTZ BD, DEROOS AD, VENGLARIK CJ, SINGH AK, FRIZZELL RA & BRIDGES RJ 1996. Glibenclamide blockade of CFTR chloride channels. Am J Physiol, 271, L192–200. [DOI] [PubMed] [Google Scholar]
- SEINO S & MIKI T 2003. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol, 81, 133–76. [DOI] [PubMed] [Google Scholar]
- SONG Y, SRINIVAS M & BELARDINELLI L 1996. Nonspecific inhibition of adenosine-activated K+ current by glibenclamide in guinea pig atrial myocytes. Am J Physiol, 271, H2430–7. [DOI] [PubMed] [Google Scholar]
- SPERELAKIS N, XIONG Z, HADDAD G & MASUDA H 1994. Regulation of slow calcium channels of myocardial cells and vascular smooth muscle cells by cyclic nucleotides and phosphorylation. Mol Cell Biochem, 140, 103–17. [DOI] [PubMed] [Google Scholar]
- TO KHT, GUI P, LI M, ZAWIEJA SD, CASTORENA-GONZALEZ JA & DAVIS MJ 2020. T-type, but not L-type, voltage-gated calcium channels are dispensable for lymphatic pacemaking and spontaneous contractions. Sci Rep, 10, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORRISI JS, HESPE GE, CUZZONE DA, SAVETSKY IL, NITTI MD, GARDENIER JC, GARCIA NORES GD, JOWHAR D, KATARU RP & MEHRARA BJ 2016. Inhibition of Inflammation and iNOS Improves Lymphatic Function in Obesity. Sci Rep, 6, 19817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VON DER WEID PY 1998. ATP-sensitive K+ channels in smooth muscle cells of guinea-pig mesenteric lymphatics: role in nitric oxide and beta-adrenoceptor agonist-induced hyperpolarizations. Br J Pharmacol, 125, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VON DER WEID PY & REHAL S 2010. Lymphatic pump function in the inflamed gut. Ann N Y Acad Sci, 1207 Suppl 1, E69–74. [DOI] [PubMed] [Google Scholar]
- VON DER WEID PY, REHAL S & FERRAZ JG 2011. Role of the lymphatic system in the pathogenesis of Crohn's disease. Curr Opin Gastroenterol, 27, 335–41. [DOI] [PubMed] [Google Scholar]
- VON DER WEID PY, ZHAO J & VAN HELDEN DF 2001. Nitric oxide decreases pacemaker activity in lymphatic vessels of guinea pig mesentery. Am J Physiol Heart Circ Physiol, 280, H2707–16. [DOI] [PubMed] [Google Scholar]
- ZAWIEJA SD, CASTORENA-GONZALEZ JA, SCALLAN JP & DAVIS MJ 2018. Differences in L-type Ca(2+) channel activity partially underlie the regional dichotomy in pumping behavior by murine peripheral and visceral lymphatic vessels. Am J Physiol Heart Circ Physiol, 314, H991–H1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 2. Effects of BK channel inhibitors (IbTX and penitrem A) on baseline lymphatic contractions and ACh concentration-response curves in WT lymphatic vessels at pressure = 3 cmH2O. Summary of absolute contraction amplitude (A) and frequency (B) before (Krebs) and after treatment with IbTX (300 nM) or penitrem A (100 nM). * Significant using paired Student’s t-test). Analysis of the effect of ACh on normalized contraction amplitude (C), normalized contraction frequency (D) and change in EDD (E) in the presence of IbTX 300 nM (n=10) or penitrem A 100 nM (n=10). Filled red rectangles indicate ACh responses of lymphatics with penitrem A present and filled blue rectangles indicate ACh responses with IbTX present, compared to the response to ACh alone (n=25), indicated by filled black circles. All data are means ± SEM. # (Blue, IbTX; Red, penitrem A) Only filled rectangle data points differ significantly from the first data points (0 M). # (red) Significant difference between ACh only (filled black circles) versus ACh + penitrem A (filled red rectangles). # (blue) Significant difference between ACh only (filled black circles) versus ACh + IbTX (filled blue rectangles).
Supplemental Figure 1. PCR to detect Kcnma1 (KCa1.1 α-subunit) in mouse lymphatic endothelial cells (LECs) isolated from lingual-axillary (IA) lymphatic vessels. LECs were collected by FACS analysis after isolation and digestion of lymphatic vessels from Prox1GFP mice. Kcnma1 showed strong bands in LEC samples 1 (36 cells), 2 (84 cells) and 3 (66 cells). Brain was used as a positive control (Lane 5). A representative panel of cell markers from LEC3 is shown. Prox1 and eNOS were detected but not α-actin or Myh11, indicating no contamination by lymphatic muscle cells (LMC) (Lane 8-11). Kcnma1 (135 bp), Prox1 (218 bp), eNOS (143 bp), α-actin (129 bp) and Myh11 (myosin-11; 238 bp). M = Marker; NT = negative; bp = base pairs.
Supplemental Figure 3. PCR analysis to detect expression of Kcnn1 (SK1 channel), Kcnn2 (SK2 channel), Kcnn3 (SK3 channel), Kcnn4 (IK channel) and Kcnj8 (Kir6.1 subunit) in mouse lymphatic muscle cells (LMCs) isolated from lingual-axillary lymphatic vessels. LMCs were collected by FACS analysis after isolation and digestion of lymphatic vessels from SMMHCCre;Rosa26mTmG mice. No expression of Kcnn1, Kcnn3, and Kcnn4 is detected, but there is faint signal for Kcnn2 in LMCs (A). In contrast, a strong Kcnj8 band is observed, verifying the quality of the cDNA. Brain homogenate was used as a positive control (B). Kcnn1 (352 bp), Kcnn2 (242 bp), Kcnn3 (294 bp), Kcnn4 (290 bp) and Kcnj8 (249 bp). M = Marker; bp = base pairs.
Supplemental Figure 4. Effects of the SK channel inhibitor, Apamin (1 μM), on baseline lymphatic contractions and NONOate concentration-response curves in WT popliteal lymphatics at pressure = 3 cmH2O. Summary of contraction amplitude (A) and frequency (B) compared between before (Krebs) and after treatment of apamin (paired Student’s t-test). When analyzed and normalized lymphatic contractile parameter the values before apamin treatment were used as a standard (C-E). Analysis of the effect of NONOate compared to the presence of 1 μM apamin on normalized amplitude (C), normalized frequency (D) and change in EDD (E). Filled circles indicate NONOate responses of WT vessels (n=24) and open red rectangles represent NONOate responses of WT pretreated with apamin (n=11). All data are means ± SEM. † (red) Only open red rectangle data points (apamin) differ significantly from the first data points (0 M).