

SUMMARY
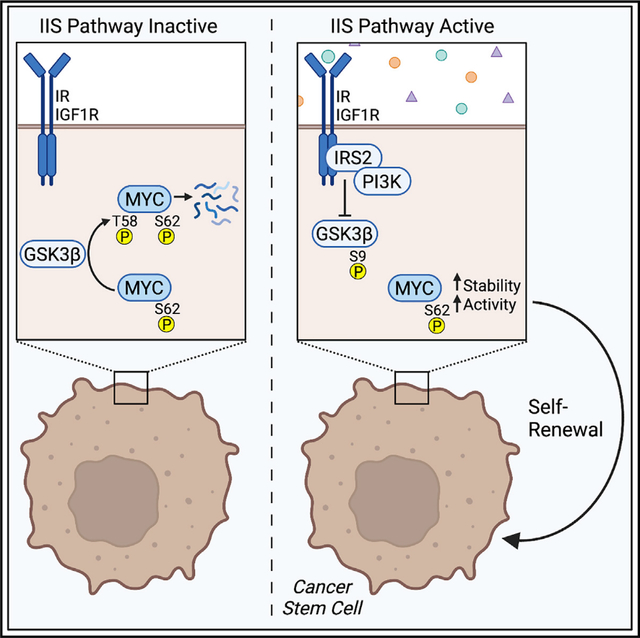
Despite the strong association of the insulin/insulin-like growth factor (IGF) signaling (IIS) pathway with tumor initiation, recurrence, and metastasis, the mechanism by which this pathway regulates cancer progression is not well understood. Here, we report that IIS supports breast cancer stem cell (CSC) self-renewal in an IRS2-phosphatidylinositol 3-kinase (PI3K)-dependent manner that involves the activation and stabilization of MYC. IRS2-PI3K signaling enhances MYC expression through the inhibition of GSK3β activity and suppression of MYC phosphorylation on threonine 58, thus reducing proteasome-mediated degradation of MYC and sustaining active pS62-MYC function. A stable T58A-Myc mutant rescues CSC function in Irs2−/− cells, supporting the role of this MYC stabilization in IRS2-dependent CSC regulation. These findings establish a mechanistic connection between the IIS pathway and MYC and highlight a role for IRS2-dependent signaling in breast cancer progression.
In brief
Lee et al. show that the insulin/IGF signaling pathway regulates breast cancer stem cell (CSC) function in an IRS2- and PI3K-dependent manner. This regulation involves the activation and stabilization of MYC, as shown by the ability of a stable T58A-MYC mutant to restore CSC function in IRS2−/− cells.
Graphical Abstract
INTRODUCTION
Cancer stem cells (CSCs) represent a sub-population of tumor cells that have the properties of self-renewal and pluripotency, i.e., the ability to repopulate the full heterogeneity of the tumor.1,2 These cells are sufficient to initiate primary and recurrent tumor growth after drug treatment as well as contribute to secondary metastatic tumor growth.3 Considerable progress has been made in recent years in defining the nature of CSCs in solid tumors, especially in breast cancer, and understanding how they contribute to tumor behavior.4,5 Despite this progress, much less is known about how the metabolic state of an individual impacts CSCs and, consequently, tumorigenesis and progression. This issue is exemplified by the link between insulin and cancer. Elevated insulin levels, which are a consequence of diseases such as obesity and diabetes, are an independent risk factor for tumor initiation and recurrence across many cancer types.6 For breast cancer, elevated insulin levels are an independent risk factor for cancer development in post-menopausal, non-diabetic women, and high fasting insulin levels are associated with breast cancer recurrence and reduced overall survival.7,8 Hyperinsulinemia also increases the expression and bioavailability of insulin-like growth factor 1 (IGF-1) and IGF-2, further enhancing activation of the insulin/IGF signaling (IIS) pathway. Pre-clinical studies using mouse models support the hypothesis that IIS drives breast cancer progression and demonstrate that reduction of circulating insulin/IGF levels reduces tumor growth and metastasis.9–13 The association of IIS with tumor initiation, recurrence, and metastasis suggests a role for this pathway in the regulation of breast CSCs, but this connection has not been investigated rigorously.
Although there is some evidence that both the insulin receptor (IR) and the related insulin-like growth factor-1 receptor (IGF1R) can influence CSC function, the relative efficacy of their regulation and the mechanism involved have not been determined.14,15 In this study, we sought to dissect the role of IIS ligands, receptors, and adapter proteins in the regulation of breast CSC function. Mechanistically, we focused on the role of the insulin receptor substrate (IRS) proteins because they are critical effectors of IR/IGF1R signaling in both normal physiology and cancer.16 Moreover, these signaling adaptors play important, but distinctive, roles in breast cancer progression.17 The data we report reveal that insulin- and IGF-mediated signaling through IRS2 plays a key role in sustaining the function of breast CSCs because it enables a phosphatidylinositol 3-kinase (PI3K)-dependent signaling pathway that culminates in maintaining the expression and activation of MYC and MYC-dependent CSC functions.
RESULTS
Activation of the IIS pathway supports breast CSC self-renewal
To investigate IIS pathway regulation of breast CSC function, we initially evaluated the requirement of IR/IGF1R signaling for serial passage mammosphere formation in the triple-negative breast cancer (TNBC) cell line SUM-159 (Figure 1A). Only cells with stem-like properties survive in low-attachment conditions, and their ability to self-renew is required for spheroid growth over several passages.18 Assays were performed in the presence of B27 supplement lacking insulin (B27−) or containing insulin (B27+). Cells were plated in low-attachment wells and grown for 5–7 days, at which time mammospheres were counted and then dissociated to re-plate for additional passages (Figure 1A). In the presence of insulin, mammosphere formation was sustained over 3 serial passages. In contrast, mammosphere formation was significantly diminished in the absence of insulin at all passages. Cells grown in the presence of either B27+ or B27− had a comparable adherent growth rate, supporting that the lack of mammosphere formation in B27− media does not result from decreased proliferation (Figure S1A). These results substantiate that IIS supports breast CSC self-renewal.
Figure 1. IIS pathway activation increases breast cancer stemness.

(A) Mammosphere assays were performed with SUM-159 cells in the presence of B27 supplement containing insulin (B27+) or without insulin (B27−). Scale bar, 200 μm.
(B and C) Mammosphere assays (B) and in vitro limiting-dilution assays (C) were performed with SUM-159 cells in B27+, B27−, or B27− medium supplemented with individual ligands (50 ng/mL).
(D–F) SUM-159 cells treated with non-targeting guide RNA (sgNT), IR knockout (KO) cells (IR−/−), or IGF1R KO cells (IGF1R−/−) were assayed for mammospheres in B27+ supplement (D), ALDH activity (E), and tumor initiation by in vivo limiting-dilution assay (F). For (E), the ALDH inhibitor DEAB was included as a negative control. For (F), cells were injected into the mammary fat pads of NCG mice in 10-fold serial dilutions at the concentrations indicated.
For (C) and (F), data are presented as a log-log plot, and the frequency of stem cells is calculated by extreme limiting-dilution analysis. The mammosphere data shown represent the mean ± SD of a representative experiment performed three times independently. The ALDH data shown represent the mean ± SD of six independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S1.
B27 supplement contains supraphysiological levels of insulin (~3 μg/mL), which activate both the IR and IGF1R (Figure S1B). To dissect further the regulation of CSC function by the IIS pathway, mammosphere assays were performed using B27− medium that was supplemented with more physiological levels (50 ng/mL) of IGF-1, IGF-2, or insulin to determine how each individual ligand supports CSC self-renewal (Figure 1B).7 We also assessed stem cell frequency in the presence of individual ligands by in vitro limiting-dilution assay (Figure 1C). All ligands increased mammosphere formation and stem cell frequency when compared with B27− medium alone, with IGF-1 and IGF-2 supporting enhanced CSC activity when compared with insulin. A similar relative ligand dependency was observed in Irs1−/−, Irs2−/− double knockout PyMT mouse mammary tumor cells with restored IRS2 expression (Figure S1C). The enhanced efficacy of IGF-1 and IGF-2 for CSC regulation may relate to the fact that at 50 ng/mL, these ligands stimulated phosphorylation of both IR and IGF1R, whereas insulin stimulated phosphorylation of only the IR (Figure S1D). To directly address the relative ability of the IR and IGF1R to regulate CSC function, IR and IGF1R knockout SUM-159 cells were generated using CRISPR-Cas9 gene editing, and these cells were assayed for serial passage mammosphere formation. Cells expressing both receptors (non-targeting guide RNA [sgNT]) or IGF1R alone (IR−/−) formed a similar number of mammospheres across both passages. However, cells expressing only IR (IGF1R−/−) exhibited a reduced ability to form mammospheres upon serial passage (Figure 1D). Together, our results suggest that the IGF1R may play a more dominant role than the IR in CSC self-renewal, although a role for hybrid receptors comprised of IGF1R and IR heterodimers is also possible.
To examine further the ability of IR and IGF1R to regulate CSC function, stem cell frequency and tumor-initiating potential were evaluated in the receptor knockout SUM-159 cells. The enzymatic activity of aldehyde dehydrogenase (ALDH) is increased in normal mammary epithelial stem cells and breast CSCs, and ALDH activity can be used to determine the frequency of stem cells in a heterogeneous population of cells using the ALDEFLUOR assay.19 As a validation of this assay, in parental SUM-159 cells, ALDH+ cells formed significantly more mammospheres than unsorted or ALDH− cells (Figure S1E). Knockout of either IGF1R or IR decreased the ALDH+ population of cells by approximately 50% when compared with control (sgNT) cells that express both receptors (Figure 1E). To assess tumor initiation, in vivo limiting-dilution assays were performed in which sgNT, IGF1R−/−, and IR−/− SUM-159 cells were injected into the mammary fat pads of NCG mice in 10-fold serial dilutions. A decrease in tumor initiation was observed for both IR−/− and IGF1R−/− cells when compared with sgNT control cells. Extreme limiting-dilution analysis (ELDA) revealed a significant reduction in CSC frequency in the absence of either IGF1R or IR expression (Figure 1F).20 Together with the results from our in vitro ligand studies, our data support that the IGF ligands and the IGF1R are more effective in regulating CSC function; however, insulin and the IR also have CSC regulatory properties, albeit with reduced potential. The concentration of individual ligands in the tumor microenvironment and their ability to co-activate IR and IGF1R, or hybrid receptors, may play the determinant role in CSC regulation.
IRS2 regulates breast CSC self-renewal in a PI3K-pathway-dependent manner
IRS proteins are signaling adaptors for both the IR and IGF1R. Upon ligand stimulation and recruitment to the IR/IGF1R, IRS proteins are phosphorylated on tyrosine residues by their intrinsic receptor tyrosine kinases, generating de novo docking sites for downstream signaling effectors.17 Of the IRS family, IRS1 and IRS2 are ubiquitously expressed, while other members display limited tissue or species expression.17 Although IRS1 and IRS2 share structural homology and functional features, they have been shown to play distinct roles in breast cancer. Specifically, expression of either IRS1 or IRS2 is sufficient for mammary tumorigenesis, whereas IRS2 plays a dominant role in metastatic progression.21,22 IRS1−/−, IRS2−/− double knockout SUM-159 cells were used to assess the independent abilities of IRS1 and IRS2 to support CSC self-renewal.23 Cells expressing either empty vector (EV), IRS1 or IRS2 were assessed for serial passage mammosphere formation (Figure 2A). Cells expressing either EV or IRS1 formed a similar number of mammospheres across all passages, and mammosphere numbers decreased after 3 passages, as was observed in the absence of insulin. In contrast, cells that express IRS2 exhibited increased mammosphere numbers over multiple passages. A similar selective dependency on IRS2 for serial passage mammosphere formation was observed in Irs1−/−, Irs2−/− double knockout PyMT mouse mammary tumor cells with restored IRS1 or IRS2 expression (Figure 2B). This IRS2-driven mammosphere formation was dependent upon upstream IR/IGF1R signaling because cells expressing IRS2 were significantly impaired in their ability to form mammospheres in B27− supplement (Figure 2C). Single knockout of IRS2 in both SUM-159 and PyMT cells decreased serial mammosphere formation (Figures 2D and 2E), further demonstrating the primary role of IRS2 in regulating CSC self-renewal. IR and IGF1R expression were not markedly altered by IRS knockout or re-expression, supporting that the observed differences in CSC function were not due to increases or decreases in upstream receptor signaling (Figure S2A).
Figure 2. IRS2 supports self-renewal of CSC in a PI3K-dependent manner.

(A and B) IRS1−/−, IRS2−/− SUM-159 cells (A) and Irs1−/−, Irs2−/− PyMT cells (B) expressing empty vector (EV), IRS1, or IRS2 were assayed for mammospheres in B27+ supplement.
(C) Irs1−/−, Irs2−/− PyMT cells expressing EV or Irs2 were assayed for mammospheres with B27+ or B27− supplement.
(D and E) IRS2−/− SUM-159 cells (D) or Irs2−/− PyMT cells (E) were assayed for mammospheres in B27+ supplement.
(F and G) ALDH activity was examined in SUM-159 cells (F) and PyMT cells (G). The ALDH inhibitor DEAB was included as a negative control.
(H) Irs2fl/fl PyMT and Irs2−/− PyMT cells were injected into the mammary fat pads of NOD/SCID mice in 10-fold serial dilutions.
(I and J) IRS1−/−, IRS2−/− SUM-159 cells (I) and Irs1−/−, Irs2−/− PyMT cells (J) expressing EV, Irs2-WT (WT), or Irs2-Y5F (Y5F) were assayed for mammospheres in B27+ supplement.
(K and L) IRS1−/−, IRS2−/− SUM-159 cells (K) and Irs1−/−, Irs2−/− PyMT cells (L) expressing EV, Irs2-WT, or Irs2-Y5F were assayed for ALDH activity.
(M) Irs1−/−, Irs2−/− PyMT cells expressing EV, Irs2-WT, or Irs2-Y5F were injected into the mammary fat pads of NOD/SCID mice in 10-fold serial dilutions. For (H) and (M), data are presented as a log-log plot, and the frequency of stem cells is calculated by extreme limiting-dilution analysis. The mammosphere data shown represent the mean ± SD of a representative experiment performed three times independently. The ALDH data shown represent the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figures S2 and S3.
To determine if IRS2 increases CSC frequency, the percentages of ALDH+ cells were determined for SUM-159 and PyMT cells in the presence or absence of IRS2 (Figures 2F, 2G, and S2B). For both cell lines, loss of IRS2 expression resulted in a significant decrease in ALDH+ cells, reflecting a decrease in CSCs in the population. To validate a role for IRS2 in CSC function, in vivo limiting-dilution assays were performed to determine the impact of Irs2 on tumor initiation. PyMT:Irs2fl/fl and PyMT:Irs2−/− tumor cells were injected into the mammary fat pads of NOD/SCID mice in 10-fold serial dilutions and monitored for tumor formation. Fewer mice formed tumors when injected with Irs2−/− cells at each cell concentration, and the frequency of CSCs was significantly lower in the absence of Irs2 expression (1/201,105 versus 1/25,870 cells) (Figure 2H). Together, these results demonstrate divergent impacts of IRS1 and IRS2 on CSC self-renewal and highlight a distinct role for IRS2-dependent signaling in breast CSC regulation by the IIS pathway.
A major signaling effector that is recruited to the phosphorylated IRS proteins in response to insulin/IGF stimulation is class I PI3K, the activation of which plays an essential role in the insulin-dependent regulation of normal systemic-glucose metabolism.24 PI3K and its downstream effector AKT have also been implicated in the regulation of CSC function.25,26 A role for PI3K/AKT signaling in the IIS-dependent regulation of CSCs was indicated by our finding that AKT activation is dependent upon the presence of insulin in B27 medium (Figure S1B). To test the requirement of PI3K/AKT pathway activation in IRS2-dependent CSC regulation, cells were pretreated with either an AKT inhibitor (MK2206) or a PI3K inhibitor (BKM120) and analyzed for mammosphere formation (Figures S3A and S3B). Pharmacologic inhibition of both AKT and PI3K decreased mammosphere formation by IRS2-expressing cells while having no effect on EV-expressing cells. Importantly, at the concentrations used, these inhibitors did not reduce cell viability (Figure S3C).
To more directly demonstrate that IRS2-dependent PI3K activation is required for the regulation of CSC function, wild-type murine Irs2 (mIrs2-WT) and a mIrs2 mutant in which the 5 tyrosines located within canonical PI3K binding motifs have been mutated to phenylalanine (mIrs2-Y5F) were expressed in IRS1−/−, IRS2−/− SUM-159 and Irs1−/−, Irs2−/− PyMT cells (Figures 2I and 2J). These five tyrosine residues (Y538, Y649, Y671, Y734, and Y814) are essential for the majority of PI3K association with Irs2 in response to insulin or IGF-1 stimulation (Figure S3D).27 Significantly more mammospheres were formed by cells expressing mIrs2-WT when compared with EV cells over three passages (Figures 2I and 2J), as we had observed for cells expressing human IRS2-WT. In contrast, mIrs2-Y5F cells exhibited similar mammosphere-forming potential as EV cells at each serial passage. Expression of mIrs2-WT, but not mIrs2-Y5F, also increased the percentage of ALDH+ cells (Figures 2K, 2L, and S3E). In in vivo limiting-dilution assays, Irs1−/−, Irs2−/− PyMT cells expressing mIrs2-Y5F formed fewer tumors, and the frequency of CSCs in this population was reduced when compared with mIrs2-WT-expressing cells (Figure 2M). mIrs2-Y5F tumors also exhibited a significantly delayed onset of tumor initiation (Figure S3F). These findings establish that IRS2 signaling promotes CSC self-renewal and supports breast tumor initiation through a PI3K-dependent mechanism.
The IIS pathway sustains MYC activation through an IRS2/PI3K-dependent suppression of GSK3β activity
Gene regulatory programs that control both normal mammary stem cell and CSC function have been identified.2,4,28 To explore the mechanism by which IRS2 regulates CSC self-renewal, we interrogated the METABRIC database of human breast tumors using cBioportal to identify genes that show a high correlation with IRS2 expression.29 The gene that demonstrated the highest positive association with IRS2 was MYC (Figure S4A). The MYC protooncogene is one of the most commonly amplified genes in cancer, and deregulated expression of MYC drives tumorigenesis in many tissues, albeit in concert with additional genetic alterations.30 MYC has been implicated in the regulation of CSCs, and its activation is both necessary and sufficient to sustain the CSC phenotype.31,32 Of relevance for IRS2-dependent regulation of CSC function, MYC expression is upregulated in hyperinsulinemic conditions.9
To validate the positive correlation between IRS2 and MYC in human breast tumors, MYC mRNA levels were evaluated in SUM-159 and PyMT cells. MYC expression was lower in IRS2−/− SUM-159 cells relative to IRS2+/+ (sgNT) cells (Figure S4B) and increased when exogenous IRS2 expression was restored in both IRS2−/− SUM-159 cells (Figure S4C) and Irs1−/−, Irs2−/− PyMT cells (Figure S4D). MYC protein levels exhibited similar IRS2-dependent changes in expression (Figures 3A and 3B). Importantly, MYC and IRS1 did not show a high positive correlation in human breast tumors (Figure S4A), and restoration of exogenous Irs1 expression in Irs1−/−, Irs2−/− PyMT cells did not increase Myc mRNA expression (Figure S4D), highlighting the distinctive correlation between MYC and IRS2. MYC’s activity as a transcription factor is regulated by phosphorylation on serine 62 (pS62-MYC) by ERK and CDK kinases.33 To determine if MYC is also activated in an IRS2-dependent manner, we initially evaluated MYC expression and phosphorylation in the presence of a proteasome inhibitor to prevent MYC degradation in cells either lacking IRS2 or with restored IRS2 expression. Active pS62-MYC levels were significantly associated with IRS2 expression (Figures 3C and 3D).
Figure 3. IRS2/PI3K/ GSK3β axis sustains MYC expression.
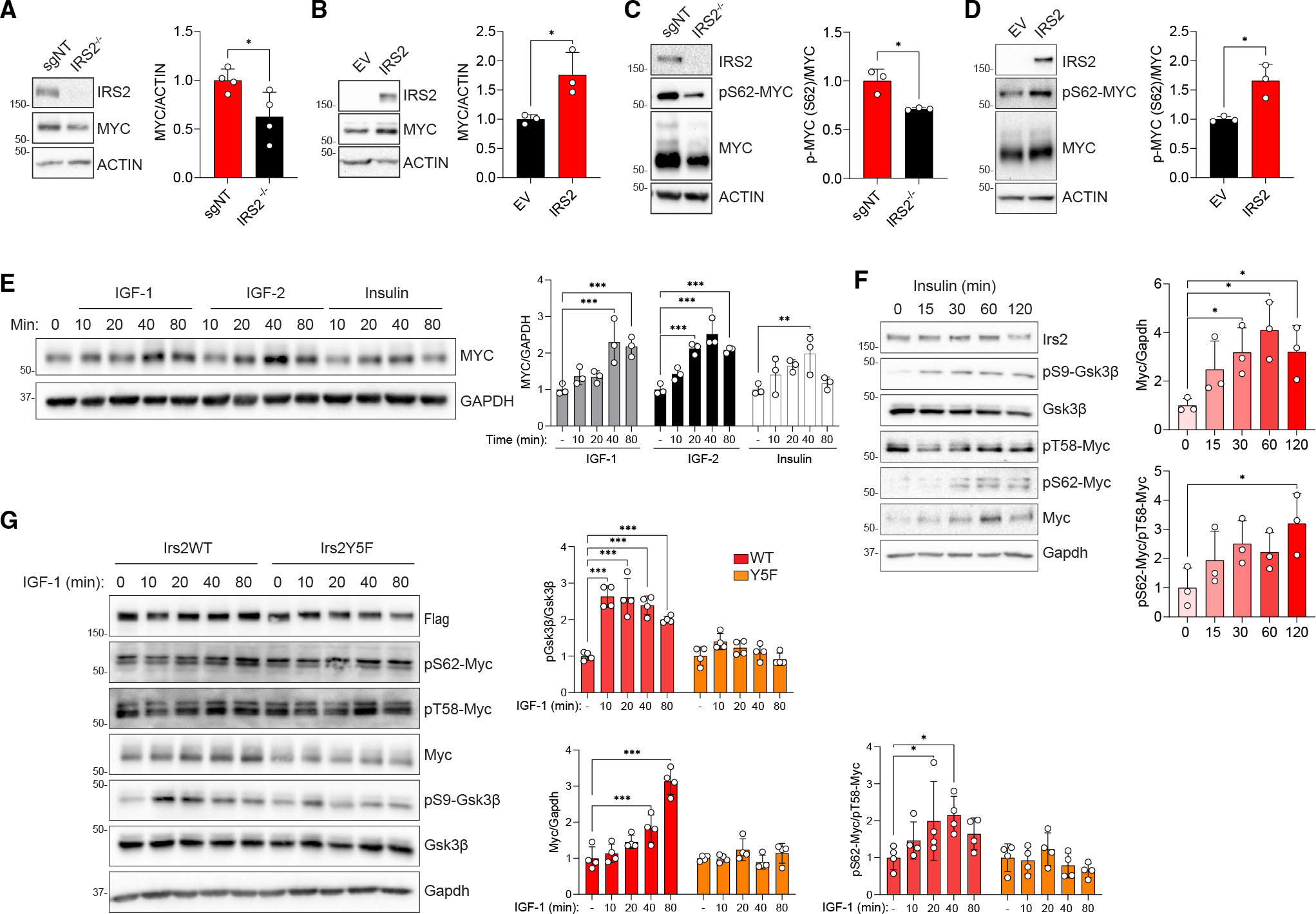
(A and B) Relative expression of MYC protein in IRS2−/− SUM-159 cells (A) or IRS2−/− SUM-159 cells with restored IRS2 expression (B).
(C and D) Relative levels of MYC expression and pS62-MYC phosphorylation in MG132-treated IRS2−/− SUM-159 cells (C) or IRS1−/−, IRS2−/− SUM-159 cells with restored IRS2 expression (D).
(E) IRS1−/−, IRS2−/− SUM-159 cells expressing IRS2 were serum starved and stimulated with each ligand (50 ng/mL) for the time periods indicated.
(F) Irs1−/−, Irs2−/− PyMT cells expressing Irs2-WT were stimulated with insulin (500 ng/mL) for the time periods indicated.
(G) Irs1−/−, Irs2−/− PyMT cells expressing Irs2-WT or Irs2-Y5F were stimulated with IGF1 (50 ng/mL) for the time periods indicated.
The data shown in all graphs represent the mean ± SD of three or more independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S4.
To investigate further the mechanism by which IRS2 signaling regulates MYC, the ability of IIS pathway ligands to regulate expression was assessed. MYC protein expression increased in response to stimulation with all ligands, although expression increased to a greater extent in response to IGF-1 and IGF-2 stimulation (Figure 3E), in keeping with the enhanced ability of these ligands to support CSC function at 50 ng/mL (Figure 1). However, this short-term stimulation with IIS ligands did not result in a corresponding increase in MYC mRNA expression (Figure S4E). This differential regulation of protein and mRNA supports an additional post-transcriptional mechanism of MYC regulation by the IIS pathway. This post-transcriptional mechanism of regulation was also indicated by the finding that MYC mRNA levels were not significantly different in cells expressing mIrs2-WT and mIrs2-Y5F (Figure S4D). MYC activity and protein stability are regulated by multistep phosphorylation and ubiquitination, and this post-translational modification plays an important role in proper MYC function.33 MYC is activated and stabilized by phosphorylation on pS62-MYC, which primes phosphorylation of threonine 58 (pT58-MYC) by GSK3β, creating a binding site for PP2A. PP2A dephosphorylates S62, and MYC phosphorylated only on T58 is then ubiquitinated and targeted for proteasomal degradation. Mutation of T58 in human tumors correlates with elevated MYC protein expression and activity33,34 and a T58A-Myc mutant is transforming in the mammary gland.35,36 These findings support the importance of regulating T58 phosphorylation for the control of normal MYC expression and function and prevention of its oncogenic activity.
In previous studies, we identified pS9-GSK3β as a preferential downstream target of IRS2 signaling in response to insulin and IGF-1 stimulation.27 Given that phosphorylation of GSK3β inhibits its kinase activity,37 we hypothesized that IRS2 signaling regulates MYC function through the suppression of T58-MYC phosphorylation, resulting in stabilization of pS62-MYC and sustained activity. To investigate this potential mechanism of MYC regulation by IRS2, cells were stimulated over a time course with insulin at a concentration that activates both the IR and IGF1R (Figure S1D). The insulin-dependent increase in MYC expression, which was maximal after 60 min of stimulation, was preceded by an increase in pS9-GSK3β and a decrease in pT58-Myc at 15 min (Figure 3F). Enhanced S62-Myc phosphorylation was observed at 30 min, subsequent to the decrease in pT58-Myc, reflecting the stabilization of active Myc. This regulation requires recruitment and activation of PI3K since cells expressing mIrs2-Y5F did not exhibit changes in expression of total Myc or active Myc (pS62-Myc/pT58-Myc) in response to IGF-1 stimulation (Figure 3G). Myc levels were sustained by insulin stimulation in cells expressing mIrs2-WT, but not in EV- or mIrs2-Y5F-expressing cells, when evaluated in the presence of cyclohexamide (Figure S4F). In contrast, in cells expressing only mIrs1, Myc activation was not sustained in response to IGF-1 stimulation (Figure S4G). These signaling studies identify an essential role for IRS2-dependent PI3K activation in the regulation of MYC expression and function by the IIS pathway.
IRS2 promotes CSC self-renewal through the regulation of MYC expression and activation
The MYC protein contains a basic helix-loop-helix-leucine zipper domain through which it dimerizes with its essential partner MAX and binds DNA to regulate gene transcription.38 The gene regulatory function of MYC is dependent upon this heterodimerization and compounds that stabilize the MYC monomer and prevent MYC-MAX heterodimerization inhibit its transcriptional activity.39 To establish a role for MYC in the IRS2-dependent regulation of breast CSCs, cells were treated with one such compound, 10074-G5, to disrupt MYC function.40 Inhibition of MYC activity resulted in a significant reduction in the ALDH+ CSC population in cells expressing WT-IRS2 (Figures 4A and S4H). In addition, reduced numbers of mammospheres formed upon serial passage of IRS2-WT cells when 10074-G5 was included in the mammosphere culture medium, identifying MYC as an essential downstream effector of IRS2-dependent CSC selfrenewal (Figure 4B).
Figure 4. MYC-mediated IRS2 regulation of breast cancer stemness.

(A) ALDH activity of IRS1−/−, IRS2−/− SUM-159 cells expressing EV or IRS2 after treatment with the Myc inhibitor 10074-G5. The ALDH inhibitor DEAB was included as a negative control.
(B) IRS1−/−, IRS2−/− SUM-159 cells expressing EV or IRS2 were assayed for mammospheres in the absence or presence of 10074-G5 in B27+ supplement.
(C) Irs2−/− PyMT cells expressing EV, Myc-WT, and Myc-T58A were analyzed for mammospheres in B27+ supplement.
(D) In vitro limiting-dilution assay of Irs2−/− PyMT cells expressing EV, Myc-WT, and Myc-T58A.
(E) Model for IRS2/PI3K-dependent regulation of breast cancer stemness. IRS2/PI3K mediates IIS-induced inactivation of GSK3β through phosphorylation on serine 9. Inactive pS9-GSK3β lacks the ability to phosphorylate threonine 58 of MYC, thus blocking proteasome-mediated degradation of MYC and allowing active pS62-MYC to accumulate. Created with BioRender.com.
The mammosphere data shown in all graphs represent the mean ± SD of a representative experiment performed three times independently. The ALDH data shown represent the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
See also Figure S4.
To validate the importance of IRS2-dependent regulation of MYC expression and function in the mechanism by which the IIS pathway regulates CSC function, Irs2−/− PyMT cells were generated with modest overexpression of either WT-Myc or a stableT58A-Myc mutant (Figures 4C and S4I).35 Expression of T58A-Myc significantly enhanced serial passage mammosphere formation and increased the stem cell frequency of Irs2−/− cells when compared with EV-expressing cells (Figures 4C and 4D). WT-Myc did not significantly increase CSC frequency or self-renewal, demonstrating that sustaining Myc stability is necessary in the absence of Irs2 expression.
DISCUSSION
In this study, we establish a role for IRS2-PI3K signaling in the control of MYC function and the regulation of breast CSCs. All ligands of the IIS pathway support CSC self-renewal in cells that express IRS2, and this requires the ability of IRS2 to recruit and activate PI3K. Signaling through IRS2-PI3K increases the ALDH+ population of CSCs and promotes CSC self-renewal in an MYC-dependent manner. IRS2 stabilizes MYC expression through the suppression of GSK3β and inhibition of phosphorylation on T58-MYC, thus reducing proteasome-mediated degradation of MYC and sustaining active pS62-MYC. This sustained activation of MYC is not observed in response to signaling through IRS1. CSC function is rescued in Irs2−/− cells by a stable T58A-Myc mutant, but not WT-Myc, highlighting the importance of IRS2-dependent MYC stabilization in its mechanism of CSC regulation. Together our findings establish a mechanistic connection between IIS and MYC and highlight a role for IRS2-dependent signaling in breast cancer progression.
The IIS pathway is comprised of multiple ligands (IGF-1, IGF-2, insulin) and receptors (IR-A, IR-B, IGF1R, IR/IGF1R hybrids).41 Adding to this complexity, each ligand stimulates a sub-set of receptors with varying affinities. In particular, IGF-1 and IGF-2 bind hybrid receptors with higher affinity than insulin.42,43 In our current study, we determined that insulin is a poor regulator of CSC function at concentrations that stimulate only the IR. However, at elevated concentrations, insulin activates both the IR and IGF1R, and under these conditions, CSC function is enhanced. IGF-1 and IGF-2 expression and bioavailability are enhanced systemically when insulin levels are elevated, and our data support that these ligands have enhanced CSC regulatory activity, potentially due to their ability to co-activate the IR and IGF1R or hybrid receptors. These findings may explain epidemiological studies that have reported a positive correlation between high insulin levels and breast cancer risk.44,45 IGF-1 and IGF-2 are also independently associated with increased risk and poor outcomes in breast cancer, and these ligands have been linked to breast cancer disparities in African American populations.46–50 While previous studies have focused on the mitogenic role of the IIS ligands for these breast cancer associations, we now suggest that the ability of each ligand to regulate breast CSCs also contributes to breast cancer development and progression.
Although high insulin, IGF-1, and IGF-2 levels have been known for some time to be independent risk factors for breast cancer risk and poor outcomes, the mechanism underlying this association has remained an open question.6 Tumor growth and progression to metastasis are enhanced in hyperinsulinemic mouse models.10 A potential role for MYC in this insulin-dependent tumor promotion was initially suggested by the finding that mammary tumor cells isolated from hyperinsulinemic mice express elevated MYC protein expression.9 However, the mechanism by which MYC expression is regulated in hyperinsulinemic environments and the functional consequences of elevated MYC expression were not explored in this study. Our data now provide insight into these questions by revealing that IIS enhances MYC expression and regulates MYC function through an IRS2-PI3K-GSK3β signaling axis that stabilizes active MYC and that is necessary to promote breast CSC self-renewal. It is worth noting that MYC expression in tumors is deregulated more frequently at the protein level than by genetic alterations, which highlights the significance of the IIS-dependent regulation of MYC phosphorylation and enhancement of protein expression for breast cancer outcomes.51
The selective ability of IRS2 to mediate the IIS-dependent regulation of CSC function identified in this study provides a mechanistic explanation for the differential roles of IRS1 and IRS2 in breast cancer progression. Mammary tumor metastasis is significantly diminished in the absence of Irs2 expression, and Irs1 does not compensate for this loss.21,22 In fact, tumors lacking Irs1 have elevated expression and phosphorylation of Irs2, and these tumors are more metastatic when compared with WT tumors. In previous studies, we and others identified a role for IRS2, but not IRS1, in the regulation of invasion, a property of tumor cells that facilitates the metastatic spread of cells to distant sites in the body.21,52 CSCs have also been implicated in metastatic colonization by supporting the initiation of tumor growth in distant organs.3 The combined ability of IRS2 to mediate signaling that supports both invasion and CSC function may explain the dominant role of IRS2 in metastatic progression. From a clinical perspective, identifying an IRS2-specific signaling pathway that promotes breast cancer progression may allow for selective targeting of aggressive tumor cells and avoid the common adverse metabolic side effects of current anti-IIS strategies aimed at inhibiting receptor activation or function. The identification of MYC as an effector of IRS2-dependent regulation of breast CSC self-renewal reveals one potential approach to achieve this goal.
Limitations of the study
There are some limitations to our analysis of IIS pathway involvement in breast CSC regulation. We performed in vivo limitingdilution assays to demonstrate that IIS signaling promotes tumor initiation. CSCs have also been implicated in chemoresistance and metastasis, and it will be important in future studies to determine whether the IIS pathway also contributes to these essential CSC functions. Our data support the conclusion that dual activation of the IR and IGF1R promotes optimal CSC function. However, we were limited in our ability to determine whether hybrid receptors contribute to this function.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Leslie M. Shaw (leslie.shaw@umassmed.edu).
Materials availability
Plasmids and cell lines generated in this study will be freely available upon request.
Data and code availability
All data reported in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells, antibodies, and reagents
SUM-159 cells that were authenticated by STR profiling at the University of Arizona Genetics Core in February 2022 were a kind gift from Art Mercurio (UMass Chan Medical School, Worcester, MA) and were grown in F12 media (Gibco) containing 5% FBS (Sigma), 5 μg/mL insulin (Sigma) and 1 μg/mL hydrocortisone (Sigma). IRS1/IRS2 double knockout (KO) (IRS1−/−, IRS2−/−), IRS1 KO (IRS1−/−) and IRS2 KO (IRS2−/−) SUM-159 cells were generated by CRISPR/Cas9-mediated gene editing.23 IR (IR−/−) and IGF1R (IGF1R−/−) knockout SUM-159 cells were generated by CRISPR/Cas9-mediated gene editing by electroporation of a ribonucleoprotein (RNP) complex of CRISPR-gRNA and Cas9 protein prepared according to manufacturer’s protocol (IDT). Guide RNAs used were as follows: sgIR-1, GAGAACTGCACGGTGATCGA; sgIR-2, TCGGTAATGACCGTGAGCTT; sgIGF1R-1, GATACGGGACCAGTCGATAG; sgIGF1R-2, GTTGTTCCGGATATCCATGC. For the IR and IGF1R KOs, the two guides were electroporated simultaneously. The control non-targeting guide RNA (sgNT) was purchased from IDT. Murine mammary tumor cells were isolated from female FVB MMTV-PyMT:Irs1fl/fl, Irs2fl/fl or PyMT:Irs2fl/fl mice and null cells were generated by infection with adenoviral Cre-recombinase.27 PyMT mouse mammary tumor cells were grown in low-glucose (1 g/L) DMEM (Corning) containing 10% FBS. All cells tested negative for mycoplasma by PCR (Abm, May 2022). Human IRS1 and IRS2 were kindly provided by Adrian Lee (University of Pittsburgh, Pittsburgh, PA). Murine Irs1 and Irs2 were a kind gift from Morris White (Children’s Hospital, Boston, MA) and the mIrs2-Y5F mutantwas generated previously.27 cDNAs were subcloned into the pCDH-CMV-MCS-EF1-puro lentiviral vector (System Bioscience) with a C-terminal FLAG tag and cells were infected and selected in 2 mg/mL puromycin (Gold Biotechnology). pD40-His/V5-c-MycWT and c-MycT58A were a gift from Rosalie Sears (Addgene plasmids # 45597 and #45598).35 For mammosphere assays with AKT or PI3K inhibitors, cells were pretreated with MK2206 (Selleckchem) or BKM120 (Selleckchem) for 24 hrs. For proteasome inhibition, cells were pre-treated with 10 uM MG132 (Sigma) for 3 hrs prior to extraction. For MYC inhibition, cells were pretreated overnight with 10074-G5 (Selleckchem) (ALDH assays) or 10074-G5 was included in the culture media (mammosphere assays). Primary antibodies used in this study: rabbit anti-HA Tag (Cell Signaling Technology, Cat#3724; RRID: AB_1549585), rabbit anti-IRS1 (Bethyl Laboratories, Cat# A301-158A; RRID: AB_2125761), rabbit anti-IRS2 (Cell Signaling Technology, Cat# 4502; RRID: AB_2125774), rabbit anti-phospho-c-Myc (Thr58) (Cell Signaling Technology, Cat# 46650), rabbit anti-phospho-c-Myc (Ser62) (Cell Signaling Technology, Cat# 13748; RRID: AB_2687518), rabbit anti- GSK-3β (Cell Signaling Technology, Cat# 9315 RRID: AB_490890), rabbit anti-phospho-GSK-3β (Ser9) (Cell Signaling Technology, Cat# 9336; RRID: AB_331405), rabbit anti-insulin receptor β (Cell Signaling Technology, Cat# 3025; RRID: AB_2280448), rabbit anti-IGF-1 receptor β (Cell Signaling Technology, Cat# 3027, RRID: AB_2122378), rabbit anti-PI3 kinase p85 (Cell Signaling Technology, Cat# 4292; RRID: AB_329869), mouse anti-actin (Thermo Fisher Scientific, Cat# MA5-11869; RRID: AB_11004139), mouse anti-GAPDH (Santa Cruz Biotechnology, Cat# sc-32233; RRID: AB_627679), mouse anti-p-Tyr (Santa Cruz Biotechnology, Cat# sc-7020; RRID: AB_628123), mouse anti-α-tubulin (Sigma-Aldrich, Cat# T5168; RRID: AB_477579), mouse anti-Flag (Sigma-Aldrich, Cat# F3165; RRID: AB_259529).
METHOD DETAILS
Mammosphere assays
Single-cell suspensions were plated in ultra-low-attachment 24-well plates (Corning) at 5,000 cells/well in mammosphere media (DMEM/F12 media containing B27 supplement, 20 ng/mL hEGF (Sigma) and 20 ng/mL bFGF (Gibco)). After 5 days, wells were imaged using a Celigo imaging cytometer (Nexcelom). For additional passages, mammospheres were dissociated using trypsin and Soybean Trypsin inhibitor (Gibco) was added to stop the reaction. Single-cell suspensions were re-plated and cultured for 7 days. Mammospheres of >50 μm in diameter were counted using Image J.
In vitro limiting dilution assays
Single-cell suspensions were plated in ultra-low-attachment 96–2 well plates (Corning) in serial dilutions (5, 10, 25, 50, 100 cells/well) in B27 mammosphere media. After 5 days, wells were imaged using a Celigo imaging cytometer and the number of wells containing ≥1 mammosphere were determined. Cancer stem cell frequency was determined using ELDA.20
ALDEFLUOR assays and sorting
ALDH activity was examined using the ALDEFLUOR™ kit (Stem Cell Technologies) according to the manufacturer’s protocol. Briefly, single-cell suspensions were incubated with either the ALDEFLUOR™ reagent alone or the ALDEFLUOR™ reagent and the ALDH inhibitor diethylaminobenzaldehyde (DEAB) for 40 minutes at 37°C. Cells were centrifuged, washed and stained with 7-AAD (Biolegend) for dead cell exclusion. The fluorescence data were collected using either an LSR II flow cytometer (BD Bioscience) or a ZE5 cytometer (Biorad). To separate ALDH+ and ALDH− population, stained SUM-159 cells were sorted using a BD FACS Melody (BD Bioscience). The sorting gates were established using as negative controls the ALDEFLUOR-stained cells treated with DEAB. Data was processed and analyzed using FCS Express (De Novo Software).
In vivo limiting dilution assays
Cells were injected into the 3rd mammary fat pad of 7-week-old female NOD.CB17-Prkdcscid/NCrCrl (NOD/SCID, Charles River Laboratories, Figures 2H and 2M) or 7-week-old female NOD-Prkdcem26Cd52Il2rgem26Cd22/NjuCrl (NCG, Charles River Laboratories, Figure 1F) mice at the numbers indicated in the Figures. Mice were palpated twice weekly to detect tumor initiation. Cancer stem cell frequency was determined using ELDA.20 All studies were performed according to protocols approved by the UMass Medical School Institutional Animal Care and Use Committee.
Immunoprecipitation and immunoblotting
Cells were serum starved for 4 hrs (PyMT) or overnight (SUM-159) in serum-free medium and then stimulated with ligands at the concentrations and time periods indicated in Figure Legends prior to extraction. For whole-cell extract immunoblots, cells were solubilized at 4°C in lysis buffer (20 mM Tris buffer, pH 7.4, 1% Nonidet P-40, 0.137 M NaCl, 10% glycerol) containing phosphatase inhibitors (Roche) and protease inhibitors (Roche). Cell extracts containing equivalent amounts of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked for 1 hr with 50 mM Tris buffer, pH 7.5, containing 0.15 M NaCl, 0.01% Tween 20, and 5% (wt/vol) dry milk or 5% bovine serum albumin (BSA); incubated overnight at 4°C in the same buffer containing primary antibodies; and then incubated for 1 hr in 5% blocking buffer with milk containing peroxidase-conjugated secondary antibodies. Bands were detected by chemiluminescence using a ChemiDoc XRS+ system (Bio-Rad Laboratories) and band intensities were quantified by densitometry using Image J. Only signals within a linear range were used for quantitation and signals were normalized to total protein and/or housekeeping genes. For immunoprecipitations, cells were extracted in the same lysis buffer containing phosphatase inhibitors (Roche) and protease inhibitors (Roche). Aliquots of cell extracts containing equivalent amounts of protein were precleared for 1 hr with IgG and protein G-Sepharose beads (GE Helathcare) and then incubated for 3 hrs or overnight at 4°C with specific antibodies and protein G-Sepharose beads (GE Healthcare) or anti-Flag M2 affinity gel with constant agitation. The beads were washed three times in extraction buffer. Laemmli sample buffer was added to the samples, and immune complexes were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted as described above.
Quantitative polymerase chain reaction (qPCR)
RNA was extracted using the RNA-easy kit (Qiagen). cDNA was synthesized using an AzuraQuant cDNA synthesis kit (Azura Genomics). qPCR was performed in an Applied Biosystems QuantStudio 6 Flex apparatus using AzuraView GreenFast qPCR Blue Mix (Azura Genomics). The delta –delta Ct method was used to determine relative mRNA expression. Primers used were as follows: hMYC-Fwd 5′-GGCTCCTGGCAAAAGGTCA-3′; hMYC-Rev 5′-CTGCGTAGTTGTGCTGATGT-3′; hACTIN-Fwd 5′-TGAGCGCGGCTACAGCTT-3′; hACTIN-Rev 5′-TCCTTAATGTCACGCACGATTT-3′; mMyc-Fwd 5′-ATGCCCCTCAACGTGAACTTC-3′; mMyc-Rev 5′-GTCGCAGATGAAATAGGGCTG-3′; mActin-Fwd 5′-AGGTGACAGCATTGCTTCTG-3′; mActin-Rev 5′-GCTGCCTCAACACCTCAAC-3′. To detect exogenous Myc expression in cells expressing pD40-His/V5-c-MycWT or c-MycT58A, primers used were: Fwd 5′- TAAGCCTATCCCTAACCCTCTC-3′; Rev 5′-CAGATGGCTGGCAACTAGAA-3′.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analysis was performed using Prism7, Graphpad. Student’s t-test (two-sided) was applied and p-value of <0.05 was considered to indicate statistical significance. For comparison of more than 2 groups, two-way ANOVA was applied.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-HA Tag (C29F4) | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Rabbit polyclonal anti-IRS1 | Bethyl Laboratories | Cat# A301-158A; RRID: AB_2125761 |
| Rabbit polyclonal anti-IRS2 | Cell Signaling Technology | Cat# 4502; RRID: AB_2125774 |
| Rabbit monoclonal anti-c-Myc (E5Q6W) | Cell Signaling Technology | Cat# 18583; RRID: AB_2895543 |
| Rabbit monoclonal anti-phospho-c-Myc (Thr58) (E4Z2K) | Cell Signaling Technology | Cat# 46650 |
| Rabbit monoclonal anti-phospho-c-Myc (Ser62) (E1J4K) | Cell Signaling Technology | Cat# 13748; RRID: AB_2687518 |
| Rabbit monoclonal anti-GSK-3β (27C10) | Cell Signaling Technology | Cat# 9315 RRID: AB_490890 |
| Rabbit monoclonal anti-phospho-GSK-3β(Ser9) | Cell Signaling Technology | Cat# 9336; RRID: AB_331405 |
| Rabbit monoclonal anti-insulin receptor β (4B8) | Cell Signaling Technology | Cat# 3025; RRID: AB_2280448 |
| Rabbit polyclonal anti-IGF-1 receptor β | Cell Signaling Technology | Cat# 3027, RRID: AB_2122378 |
| Rabbit polyclonal anti-PI3 kinase p85 | Cell Signaling Technology | Cat# 4292; RRID: AB_329869 |
| Mouse monoclonal anti-actin (ACTN05 (C4)) | Thermo Fisher Scientific | Cat# MA5-11869; RRID: AB_11004139 |
| Mouse monoclonal anti-GAPDH (6C5) | Santa Cruz Biotechnology | Cat# sc-32233; RRID: AB_627679 |
| Mouse monoclonal anti-p-Tyr (PY99) | Santa Cruz Biotechnology | Cat# sc-7020; RRID: AB_628123 |
| Mouse monoclonal anti-α-tubulin (B512) | Sigma-Aldrich | Cat# T5168; RRID: AB_477579 |
| Mouse monoclonal anti-Flag M2 | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Mouse (G3A1) mAb IgG1 isotype control | Cell Signaling Technology | Cat# 5415; RRID: AB_10829607 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID: AB_1031062 |
| Anti-FLAG M2 affinity gel | Sigma-Aldrich | Cat# A2220; RRID: AB_10063035 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB 5-alpha Competent E.coli | New England Biolabs | Cat# C2987 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Insulin | Sigma-Aldrich | Cat# I6634 |
| recombinant human epidermal growth factor (hEGF) | Sigma-Aldrich | Cat# E9644 |
| recombinant human fibroblast growth factor-basic (bFGF) | Gibco | Cat# PHC0026 |
| B-27 supplement | Gibco | Cat# 17504044 |
| B-27 supplement, minus insulin | Gibco | Cat# A1895601 |
| recombinant human insulin-like growth factor-2 (IGF-2) | R&D Systems | Cat# 292-G2 |
| recombinant human insulin-like growth factor-1 (IGF-1) | Sigma-Aldrich | Cat# SRP3069 |
| MK-2206 | Selleckchem | Cat# S1078 |
| BKM120 (Buparlisib) | Selleckchem | Cat# S2247 |
| 10074-G5 | Selleckchem | Cat# S8426 |
| MG132 | Sigma-Aldrich | Cat# M8699 |
| Cycloheximide | Sigma-Aldrich | Cat# C4859 |
| Alt-R S.p. Cas9 Nuclease V3 | IDT | Cat# 1081058 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ALDEFLUOR Kit | Stem cell technologies | Cat# 01700 |
| 7-AAD Viability staining solution | Biolegend | Cat# 420403 |
| AzuraQuant cDNA synthesis kit | Azura genomics | Cat# AZ-1995 |
| AzuraView greenfast qPCR blue mix | Azura genomics | Cat# AZ-2401 |
|
| ||
| Deposited data | ||
|
| ||
| METABRIC database of human breast tumors | cBioportal | METABRIC, Nature 2012 & Nat Commun 2016 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| SUM-159 | Kindly provided by Dr. Art Mercurio, UMass Chan Medical School, Worcester, MA | RRID:CVCL_5423 |
| PyMT:Irs1fl/fl, Irs2fl/fl | Landis et al., J Biol Chem. 2014 | N/A |
| PyMT:Irs2fl/fl | Landis et al., J Biol Chem. 2014 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: NOD/SCID | Charles River Laboratories | Strain Code: 394 |
| Mouse: NCG | Charles River Laboratories | Strain Code: 572 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Alt-R Cas9 negative control crRNA | IDT | Cat# 288190998 |
| sgIR-1, GAGAACTGCACGGTGATCGA sgIR-2, TCGGTAATGACCGTGAGCTT | IDT | N/A |
| sgIGF1R-1, GATACGGGACCAGTCGATAG sgIGF1R-2, GTTGTTCCGGATATCCATGC | IDT | N/A |
| Alt-R CRISPR-Cas9 tracrRNA, ATTO550 | IDT | Cat# 1075927 |
| hMYC Fwd 5′-GGCTCCTGGCAAAAGGTCA-3′ Rev 5′-CTGCGTAGTTGTGCTGATGT-3′ |
Genewiz | N/A |
| hACTIN Fwd 5′-TGAGCGCGGCTACAGCTT-3′ Rev 5′-TCCTTAATGTCACGCACGATTT-3′ |
Genewiz | N/A |
| mMyc Fwd 5′-ATGCCCCTCAACGTGAACTTC-3′ Rev 5′-GTCGCAGATGAAATAGGGCTG-3′ |
Genewiz | N/A |
| mActin Fwd 5′-AGGTGACAGCATTGCTTCTG-3′ Rev 5′-GCTGCCTCAACACCTCAAC |
Genewiz | N/A |
| exogenous Myc (Figure S4H) Fwd 5′- TAAGCCTATCCCTAACCCTCTC-3′ Rev 5′-CAGATGGCTGGCAACTAGAA-3′ |
IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| HA-tagged human IRS1 | Kindly provided by Dr. Adrian Lee, University of Pittsburgh, Pittsburgh, PA | N/A |
| HA-tagged human IRS2 | Kindly provided by Dr. Adrian Lee, University of Pittsburgh, Pittsburgh, PA | N/A |
| pCDH-CMV-MCS-EF1-puro | System Biosciences | Cat# CD510B-1 |
| pCDH-IRS1-FFH-EF1-puro | This paper | N/A |
| pCDH-IRS2-FFH-EF1-puro | This paper | N/A |
| pCMV-His-mIrs1 | Kindly provided by Dr. Morris White, Children’s Hospital, Boston, MA | N/A |
| pCMV-His-mIrs2 | Kindly provided by Dr. Morris White, Children’s Hospital, Boston, MA | N/A |
| pCDH-mIrs1-FFH-EF1 -puro | This paper | N/A |
| pCDH-mIrs2WT-FFH-EF1-puro | This paper | N/A |
| pCDH-mIrs2Y5F-FFH-EF1-puro | This paper | N/A |
| pD40-His/V5-c-MycWT | Yeh et al., Nat Cell Biol. 200435 | RRID:Addgene_45597 |
| pD40-His/V5-c-MycT58A | Yeh et al., Nat Cell Biol. 200435 | RRID:Addgene_45598 |
|
| ||
| Software and algorithms | ||
|
| ||
| FCS Express v 7 | De Novo Software | https://denovosoftware.com |
| GraphPad Prism v 9.3.1 | GraphPad Software | https://graphpad.com |
| ImageJ | Open Source | https://imagej.nih.gov |
| Biorender | Biorender | https://biorender.com |
| ELDA:Extreme Limiting Dilution Analysis | Walter Eliza Hall institute of medical research | bioinf.wehi.edu.au/software/elda |
Highlights.
Insulin/IGF signaling (IIS) supports breast cancer stem cell (CSC) self-renewal
IIS regulates stemness in an IRS2- and PI3K-dependent manner
IRS2/PI3K signaling activates and stabilizes MYC to enhance CSC function
ACKNOWLEDGMENTS
We thank Art Mercurio and the Mercurio lab for helpful discussion and comments on the manuscript. This work was supported by National Institutes of Health (NIH) grants CA227993 and CA240655 (L.M.S.) and NIH F31 Predoctoral fellowship CA180706 (J.M.-M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111759.
REFERENCES
- 1.Visvader JE, and Lindeman GJ (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 8, 755–768. 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 2.Batlle E, and Clevers H (2017). Cancer stem cells revisited. Nat. Med. 23, 1124–1134. 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 3.Chaffer CL, and Weinberg RA (2011). A perspective on cancer cell metastasis. science 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- 4.Liu S, and Wicha MS (2010). Targeting breast cancer stem cells. J. Clin. Oncol. 28, 4006–4012. 10.1200/jco.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prager BC, Xie Q, Bao S, and Rich JN (2019). Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell 24, 41–53. 10.1016/j.stem.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallagher EJ, and LeRoith D (2020). Hyperinsulinaemia in cancer. Nat. Rev. Cancer 20, 629–644. 10.1038/s41568-020-0295-5. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, Hartwick W, Hoffman B, and Hood N (2002). Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J. Clin. Oncol. 20, 42–51. 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- 8.Lawlor DA, Smith GD, and Ebrahim S (2004). Hyperinsulinaemia and increased risk of breast cancer: findings from the British Women’s Heart and Health Study. Cancer Causes Control 15, 267–275. [DOI] [PubMed] [Google Scholar]
- 9.Ferguson RD, Novosyadlyy R, Fierz Y, Alikhani N, Sun H, Yakar S, and LeRoith D (2012). Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res. 14, R8–R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferguson RD, Gallagher EJ, Cohen D, Tobin-Hess A, Alikhani N, Novosyadlyy R, Haddad N, Yakar S, and LeRoith D (2013). Hyperinsulinemia promotes metastasis to the lung in a mouse model of Her2-mediated breast cancer. Endocr. Relat. Cancer 20, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wise TL, and Pravtcheva DD (2006). Delayed onset of Igf2-induced mammary tumors in Igf2r transgenic mice. Cancer Res. 66, 1327–1336. 10.1158/0008-5472.CAN-05-3107. [DOI] [PubMed] [Google Scholar]
- 12.Yakar S, Pennisi P, Kim CH, Zhao H, Toyoshima Y, Gavrilova O, and LeRoith D (2005). Studies involving the GH-IGF axis: lessons from IGF-I and IGF-I receptor gene targeting mouse models. J. Endocrinol. Invest. 28, 19–22. [PubMed] [Google Scholar]
- 13.de Ostrovich KK, Lambertz I, Colby JKL, Tian J, Rundhaug JE, Johnston D, Conti CJ, DiGiovanni J, and Fuchs-Young R (2008). Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am. J. Pathol. 173, 824–834. 10.2353/ajpath.2008.071005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang W-W, Lin R-J, Yu J, Chang W-Y, Fu C-H, Lai A, Yu J-C, and Yu AL (2013). The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res. 15. R39–R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, Orr ZS, Caspi A, Tzukerman M, and LeRoith D (2015). Highly specific role of the insulin receptor in breast cancer progression. Endocr. Relat. Cancer 22, 145–157. 10.1530/ERC-14-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw LM (2011). The insulin receptor substrate (IRS) proteins: at the intersection of metabolism and cancer. Cell Cycle 10, 1750–1756. 10.4161/cc.10.11.15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mardilovich K, Pankratz SL, and Shaw LM (2009). Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 7, 14. 10.1186/1478-811X-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, and Wicha MS (2003). In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270. 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567. 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Nagle JA, Ma Z, Byrne MA, White MF, and Shaw LM (2004). Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol. Cell Biol. 24, 9726–9735. 10.1128/MCB.24.22.9726-9735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, and Shaw LM (2006). Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol. Cell Biol. 26, 9338–9351. 10.1128/MCB.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S, Ward BM, Yu J, Matthew-Onabanjo AN, Janusis J, Hsieh CC, Tomaszewicz K, Hutchinson L, Zhu LJ, Kandil D, and Shaw LM (2018). IRS2 mutations linked to invasion in pleomorphic invasive lobular carcinoma. JCI Insight 3, 97398. 10.1172/jci.insight.97398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorpe LM, Yuzugullu H, and Zhao JJ (2015). PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 15, 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, García-Echeverría C, Schultz PG, and Reddy VA (2009). The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 106, 268–273. 10.1073/pnas.0810956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O, et al. (2015). PIK3-CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 525, 114–118. 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 27.Landis J, and Shaw LM (2014). Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J. Biol. Chem. 289, 18603–18613. 10.1074/jbc.M114.564070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien CA, Kreso A, and Jamieson CHM (2010). Cancer stem cells and self-renewal. Clin. Cancer Res. 16, 3113–3120. 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida GJ (2018). Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 37, 173. 10.1186/s13046-018-0835-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santoro A, Vlachou T, Luzi L, Melloni G, Mazzarella L, D’Elia E, Aobuli X, Pasi CE, Reavie L, Bonetti P, et al. (2019). p53 loss in breast cancer leads to Myc activation, increased cell plasticity, and expression of a mitotic signature with prognostic value. Cell Rep. 26, 624–638.e8. 10.1016/j.celrep.2018.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sears RC (2004). The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3, 1133–1137. [PubMed] [Google Scholar]
- 34.Cohn GM, Liefwalker DF, Langer EM, and Sears RC (2020). PIN1 provides dynamic control of MYC in response to extrinsic signals. Front. Cell Dev. Biol. 8, 224. 10.3389/fcell.2020.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. (2004). A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6, 308–318. 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M,and Sears RC (2011). Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 71, 925–936. 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutherland C, Leighton IA, and Cohen P (1993). Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296 (Pt 1), 15–19. 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blackwood EM, and Eisenman RN (1991). Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251, 1211–1217. 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 39.Madden SK, de Araujo AD, Gerhardt M, Fairlie DP, and Mason JM (2021). Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 20, 3. 10.1186/s12943-020-01291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yap JL, Wang H, Hu A, Chauhan J, Jung KY, Gharavi RB, Prochownik EV, and Fletcher S (2013). Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 23, 370–374. 10.1016/j.bmcl.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lero MW, and Shaw LM (2021). Diversity of insulin and IGF signaling in breast cancer: implications for therapy. Mol. Cell. Endocrinol. 527, 111213. 10.1016/j.mce.2021.111213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benyoucef S, Surinya KH, Hadaschik D, and Siddle K (2007). Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 403, 603–613. 10.1042/BJ20061709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slaaby R (2015). Specific insulin/IGF1 hybrid receptor activation assay reveals IGF1 as a more potent ligand than insulin. Sci. Rep. 5, 7911. 10.1038/srep07911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, and Pollak M (1998). Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 351, 1393–1396. 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 45.Gunter MJ, Hoover DR, Yu H, Wassertheil-Smoller S, Rohan TE, Manson JE, Li J, Ho GYF, Xue X, Anderson GL, et al. (2009). Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 101, 48–60. 10.1093/jnci/djn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalla Singh S, Tan QW, Brito C, De León M, Garberoglio C, and De León D (2010). Differential insulin-like growth factor II (IGF-II) expression: a potential role for breast cancer survival disparity. Growth Horm. IGF Res. 20, 162–170. 10.1016/j.ghir.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fowke JH, Matthews CE, Yu H, Cai Q, Cohen S, Buchowski MS, Zheng W, and Blot WJ (2010). Racial differences in the association between body mass index and serum IGF1, IGF2, and IGFBP3. Endocr. Relat. Cancer 17, 51–60. 10.1677/ERC-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laron Z, and Werner H (2021). Laron syndrome - a historical perspective. Rev. Endocr. Metab. Disord. 22, 31–41. 10.1007/s11154-020-09595-0. [DOI] [PubMed] [Google Scholar]
- 49.Murphy N, Knuppel A, Papadimitriou N, Martin RM, Tsilidis KK, Smith-Byrne K, Fensom G, Perez-Cornago A, Travis RC, Key TJ, and Gunter MJ (2020). Insulin-like growth factor-1, insulin-like growth factor-binding protein-3, and breast cancer risk: observational and Mendelian randomization analyses with approximately 430 000 women. Ann. Oncol. 31, 641–649. 10.1016/j.annonc.2020.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian F, and Huo D (2020). Circulating insulin-like growth factor-1 and risk of total and 19 site-specific cancers: cohort study analyses from the UK biobank. Cancer Epidemiol. Biomarkers Prev. 29, 2332–2342. 10.1158/1055-9965.EPI-20-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu J, Chen Y, and Olopade OI (2010). MYC and breast cancer. Genes Cancer 1, 629–640. 10.1177/1947601910378691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mercado-Matos J, Janusis J, Zhu S, Chen SS, and Shaw LM (2018). Identification of a novel invasion-promoting region in insulin receptor substrate 2. Mol. Cell Biol. 38, e005900–17. 10.1128/MCB.00590-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.