

Abstract
A new variant of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), named Omicron (Pango lineage designation B.1.1.529), was first reported to the World Health Organization by South African health authorities on 24 November 2021. The Omicron variant possesses numerous mutations associated with increased transmissibility and immune escape properties. In November 2021, Mexican authorities reported Omicron’s presence in the country. In this study, we infer the first introductory events of Omicron and the impact that human mobility has had on the spread of the virus. We also evaluated the adaptive evolutionary processes in Mexican SARS-CoV-2 genomes during the first month of the circulation of Omicron. We inferred 160 introduction events of Omicron in Mexico since its first detection in South Africa; subsequently, after the first introductions there was an evident increase in the prevalence of SARS-CoV-2 during January. This higher prevalence of the novel variant resulted in a peak of reported cases; on average 6 weeks after, a higher mobility trend was reported. During the peak of cases in the country from January to February 2022, the Omicron BA.1.1 sub-lineage dominated, followed by the BA.1 and BA.15 sub-lineages. Additionally, we identified the presence of diversifying natural selection in the genomes of Omicron and found six non-synonymous mutations in the receptor binding domain of the spike protein, all of them related to evasion of the immune response. In contrast, the other proteins in the genome are highly conserved; however, we identified homoplasic mutations in non-structural proteins, indicating a parallel evolution.
Keywords: Omicron, introductions, natural selection, human mobility
1. Introduction
The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), emerged at the end of 2019 in the province of Wuhan, China, and rapidly spread to other countries, causing more than 5.2 million deaths worldwide as of 18 November 2022 (Platto, Xue, and Carafoli 2020; Zhu et al. 2020). As new variants of SARS-CoV-2 emerged in different regions of the world, the World Health Organization (WHO) designated new variants as variants of interest, variants under monitoring, and variants of concern (VOCs), depending on their epidemiological and clinical behavior (Aleem, Samad, and Slenker 2022). The emergence of VOCs has been of particular interest because they are associated with remarkable increases in the incidence of COVID-19 worldwide (Choi and Smith 2021). For example, the Alpha (B.1.1.7), Gamma (P.1.X), and Delta (B.1.617.2) variants were associated with drastic increases in the number of infections and COVID-19-related deaths in the UK, Brazil, and India, respectively (Tao et al. 2021).
On 24 November 2021, health authorities in Botswana and South Africa reported the circulation of a new SARS-CoV-2 variant that was later designated Omicron and classified as a VOC by the WHO (Viana et al. 2022). This new variant displayed several mutations across the genome, some previously found in other VOCs (Thakur and Ratho 2022). One of the most critical traits of Omicron is the presence of a high number of mutations (thirty-two amino acid substitutions) in the spike protein compared with other VOCs, which might confer this variant an increased affinity toward the angiotensin-converting enzyme 2 (ACE2 receptor) enhancing entry into the host cell and the ability to escape neutralization by antibodies induced by either natural infection or vaccination (Hui et al. 2022; Thakur and Ratho 2022). Moreover, recent in vitro studies reported that the replication rates for Omicron are almost seventy times higher in bronchial tissue than in lung tissue in contrast to the Delta variant, which might explain the high transmissibility of Omicron (Chi-wai et al.).
Omicron has already spread to seventy-seven countries around the world, and the accession numbers of the genomes of these countries come from Global Initiative on Sharing All Influenza Data (GISAID) (Thakur and Ratho 2022). The Omicron variant was first detected in Mexico in November 2021 and has rapidly spread since, probably causing the recent increase in symptomatic COVID-19 cases observed in the country. Due to the high infectivity of Omicron in contrast to other VOCs, the study of the evolution and transmission patterns of this variant in different countries is crucial for the implementation of effective strategies to reduce its impact in a clinical context. In this work, we focused on identifying the early introduction events of Omicron in Mexico and analyzed mobility patterns in the first weeks after the initial introduction events to understand how the virus spread across the country. In addition, we analyzed adaptation signals throughout the genome to search for evidence of natural selection and homoplasic mutations.
2. Materials and methods
2.1. Collection of genomes
A total of 641 genomes obtained in Mexico corresponding to the period between November and March were downloaded from the GISAID platform (Thakur and Ratho 2022) (https://www.gisaid.org/). Additionally, the first twenty genomes of Omicron reported in any other country were downloaded; however, there were fewer genomes available for some countries because there was a lack of active genomic surveillance systems. Finally, 2,148 Omicron SARS-CoV-2 genomes sequenced worldwide were analyzed (including 187 genomes of other, non-Omicron lineages to guide the phylogeny). Additionally, to analyze the spread and diversification of Omicron in Mexico, genomes of this variant sequenced between November 2021 and March 2022 were downloaded from GISAID considering the following inclusion parameters: complete genomes, high coverage, and low coverage exclusion. Additionally, for those countries that reported more than 200 genomes in total (between November 2021 and March 2022), we randomly selected 200 genomes from all the sequences collected during each epidemiological week.
2.2. Phylogenomic and phylogeographic analyses
Genomes were aligned codon-wise using the MAFFT v7 software (Katoh, Rozewicki, and Yamada 2019) trimming the 5ʹ and 3ʹ ends and were further classified according to their viral lineage assignment under the Pango nomenclature system using Pangolin version v3.1.7 (Rambaut et al. 2020).
To estimate the phylogenetic tree and phylogeographic relationships among SARSCOV-2 genomes, a maximum likelihood phylogenetic tree (ML) was constructed using the IQ tree (Nguyen et al. 2015) using a GTR+Γ model of nucleotide substitution with empirical base frequencies and four free site rate categories and statistical support (-alrt 1000). Sequences from lineages other than Omicron were added to this phylogeny in order to establish the monophyly of Omicron clades, while sequences from the original Wuhan outbreak were used to root the tree. The tree was evaluated for a temporal signal using TempEst (Rambaut et al. 2016) (the outliers were eliminated).
A time-scaled phylogenetic tree was built using TreeTime v0.7.4 (Sagulenko, Puller, and Neher 2018), which specified a clock rate of 8 × 10−4 substitutions per site per year according to the Nextstrain workflow (Hadfield et al. 2018).
The phylogeographic analysis determined and quantified the introduction events of the Omicron lineage in Mexico. This analysis was reconstructed from the temporally scaled tree previously created using TreeTime v0.7.4. We used two different approaches based on several models and inference methods. To identify independent introduction events of Omicron in Mexico, we used a discrete diffusion model implemented in the software package BEAST v1.10.4 (Drummond et al. 2012). The Bayesian analysis through Markov chain Monte Carlo (MCMC) was run on 100 million steps and combined (after discarding 10 per cent of each run as burn-in) to produce a posterior tree distribution.
The time-scaled phylogeny previously built was a fixed empirical tree and considered two possible ancestral locations: ‘Mexico’ and ‘other location’, according to the described pipeline from Dellicour et al. (2021a).
The MCMC convergence and mixing properties were inspected using Tracer v1.72 (Rambaut et al. 2014), and the effective sample size values above 200 were achieved for all parameters. The phylogenies were annotated with a 10 per cent burn-in using TreeAnnotator. Subsequently, the ‘Seraphim’ package (Dellicour et al. 2016) extracted the spatio-temporal information from the data and visualized the phylogeographic reconstructions.
Second, we used PastML (Ishikawa et al. 2019). Briefly, PastML reconstructed the ancestral character states and their changes along with the trees, with ML marginal posterior probabilities approximation (MPPA) and Felsenstein 1981 model options. For the parameters in PastML, we used MPPA as a prediction method (standard settings). We added the character predicted by the joint reconstruction, even if it was not selected by the Brier score (option forced_joint).
On the other hand, the Omicron sequences from Mexico that were downloaded from November to March were built in to reconstruct an ML phylogeny using IQ-TREE v.2.1.1, with the GTR+Γ substitution model and statistical support (-alrt 1000), and the phylogenetic tree was visualized in iTOL v5 (Letunic and Bork 2021).
2.3. Daily mobility, cases report, and government stringency
The mobility data used in this study were obtained from the Google Mobility Reports service (https://www.google.com/covid19/mobility/) for all Mexican states. The data were grouped into the following mobility categories: (1) grocery and pharmacy, (2) retail and recreation, (3) parks, (4) workplaces, (5) residential, and (6) transit stations. The service has been updated daily since 26 February 2020. The last date of collection was 17 April 2022. The data were treated in weekly variations. Finally, we summed all six categories of mobility in order to have a single unitary value representing the mobility of weekly periods.
The country was divided into seven regions to analyze the mobility as follows: northeast (NE; comprising the states of Coahuila, Nuevo León, and Tamaulipas), northwest (NW; Baja California, Baja California Sur, Chihuahua, Durango, Sonora, and Sinaloa), central north (CN; Aguascalientes, Guanajuato, Querétaro, San Luis Potosí, and Zacatecas), central south (CS; Mexico City, State of Mexico, Morelos, Hidalgo, Puebla, and Tlaxcala), west (W; Colima, Jalisco, Michoacán, and Nayarit), southeast (SE; Guerrero, Oaxaca, Chiapas, Veracruz, and Tabasco), and south (S; Campeche, Yucatán, and Quintana Roo).
Moreover, daily cases of the COVID-19 transmission in Mexico were retrieved from the service https://datos.covid-19.conacyt.mx/. The regionalized (as mentioned earlier) daily cases were analyzed in a 7-day window to eliminate data granularity. Next, the data were converted into cases/100,000 inhabitants. Finally, we also obtained data regarding the Oxford Stringency Index (Hale et al. 2020), which measures how governments implemented social restriction measures to contain the COVID-19 pandemic. The data are publicly available as obtained from the Our World in Data COVID-19 repository (https://github.com/owid/covid-19-data). All data analyses were conducted through in-house scripts written in R (R Core Team 2022).
2.4. Natural selection analysis
Natural selection can manifest in directional, diversifying, purifying, and episodic selection. We evaluated these types of natural selection in Omicron genomes from Mexico. First, the Omicron SARS-CoV-2 genome was annotated using RCoV19 (https://ngdc.cncb.ac.cn/ncov/?lang=en) (Gong et al. 2020). The search for directional selection signals was performed with fixed effects likelihood (FEL), single likelihood ancestor counting (SLAC) (Delport et al. 2010), and Fast Unconstrained Bayesian AppRoximation (FUBAR), assuming that the selection pressure is constant across the phylogeny for each site (Murrell et al. 2013). Furthermore, the episodic diversifying selection was evaluated with the mixed-effects model (Murrell et al. 2012).
2.5. Homoplasy test
We evaluated the sequences of Omicron (from Mexico) with mutations that have arisen independently several times (homoplasies). Homoplasies are likely indicative of adaptation due to convergent evolution but can also originate in recombination or in errors during sequence data processing. Homoplasies were detected on phylogeny using HomoplasyFinder (Crispell, Balaz, and Gordon 2019). We collected all the Omicron sequences from Mexico and used two sequences from Botswana (NICD-N22418 and NICD-N22397) as reference genomes. Additionally, we used Snippy (https://github.com/tseemann/snippy) to detect single-nucleotide variations using the Omicron genome NICD-N22418 as reference genomes.
3. Results
3.1. Phylogenetic evidence for multiple introductions of Omicron and human mobility facilitating its spread
On 3 December 2021, the first Whole genome sequencing-confirmed case of symptomatic COVID-19 caused by Omicron (B.1.1.519) was reported by the Instituto Nacional de Diagnóstico y Referencia Epidemiológica, corresponding to a patient from South Africa that arrived in Mexico City on 16 November 2021. Since then and up to 27 December 2021, 254 genomes were sequenced in Mexico, corresponding to the states of Baja California (n = 1), Chiapas (n = 1), Guerrero (n = 1), Hidalgo (n = 1), Mexico City (n =159), Oaxaca (n = 1), Puebla (n = 4), Quintana Roo (n =18), Sinaloa (n = 2), State of Mexico (n = 35), Tabasco (n = 12), Tamaulipas (n = 5), Veracruz (n = 1), and Yucatán (n = 13). Of note, most of the Omicron sequences come from Mexico City (27.13 per cent) and the State of Mexico (9.88 per cent), likely since most international flights to Mexico arrive via the country’s capital. However, the proportion of Omicron sequences reported is consistent with the average number of SARS-CoV-2 sequences reported (by state) in Mexico since the beginning of the pandemic. In addition, other states with international airports, as well as major destinations such as Baja California Sur, Quintana Roo, Yucatán, and Sinaloa, also reported an increase in reported Omicron sequences during the first days of December (Supplementary Fig. S1). This suggests that for the cases in Mexico City and the State of Mexico, the higher number of sequences is due to a more intense genomic surveillance in these states.
To determine Omicron introductions in Mexico, we constructed a phylogenetic tree using 641 Omicron genome sequences from Mexico, and 3,690 foreign sequences deposited in GISAID from the month of November 2021 to the end of March 2022. First, we inferred the phylogeography of the sequences to trace the movement of Omicron in Mexico through time and space. We noted that the sequences from the Mexican genomes do not form a single clade but rather are scattered throughout the tree, consistent with multiple introductions in the country. In Fig. 1A, the green branches correspond to the Mexican sequences, while the gray branches correspond to international sequences.
Figure 1.
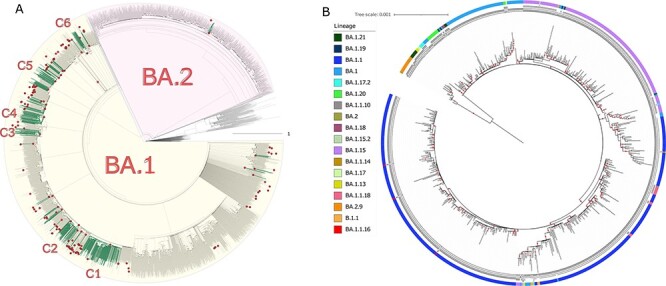
Omicron phylogeography and phylogenomic analysis. (A) The analysis on the temporal scale shows the 160 introduction events in Mexico. The nodes shown as outline circles correspond to the most recent common ancestor (MRCA) of clades resulting from an introduction event. The green branches correspond to the Mexican sequences. The gray branches, on the other hand, correspond to samples from other countries. (B) Maximum likelihood phylogeny of the Mexican lineages, each indicated in the legend. Only bootstrap values ≥80 are indicated with red dots, and the scale bar represents the number of substitutions per site.
We evaluated introductions through a discrete phylogeographic analysis following the Dellicour pipeline (Dellicour et al. 2021a). For this purpose, we used a time-scaled phylogenetic tree, and subsequently, introduction events were identified in the maximum clade credibility tree with an associated highest posterior density (HPD) interval by reporting the number of introduction events. The analysis detected a minimum number of 160 introduction events of the Omicron lineage (95 per cent HPD interval = 142.1–170) identified from the phylogenetic analysis of 641 samples collected in Mexico (Fig. 1A; Supplementary Fig. S2).
Two major clades were identified in a discrete phylogeographic analysis, corresponding to the BA.1 and BA.2 sub-lineages. The clade of the BA.1 sub-lineage is the largest and includes six clades (C1–C6) with more than twenty sequences from Mexico (supported nodes with a posterior probability >80) representing local transmission chains (Fig. 1A). The largest clade consists of 168 sequences corresponding to the BA.1.1 that prevailed in Mexico during the Omicron wave; the oldest sequence within the clade is from Mexico City, 12 September 2021, and the presence of this clade extends to March. This clade includes sequences from across the country, but mainly from Mexico City, Michoacán, Veracruz, and the State of Mexico. Clade 2 corresponded to the BA.1.1 sequence from across the country. Clade 3 corresponded mainly to the BA.1.1, which consisted of sequences from the north of the country. Clade 4 consisted of sequences of the BA.1.15 from across the country, and it was the largest clade representing this sub-lineage. Clades 5 and 6 were mainly composed of sequences from the BA.1.15, although it also contains sequences corresponding to the BA.1.17. In general, the sequences of these clades correspond to the period from November 2021 to March 2022, when the BA.1.1 and BA.1.15 predominated, both co-circulating in the country. There were introductions of BA.2 as early as February 2022, and the oldest sequence corresponds to 10 February 2022, but it was not in the predominant lineage during the month of its appearance.
The PastML analysis predicted Botswana as the location of the tree root of Omicron, suggesting that the origin of the Omicron variant might have been in South Africa. Four main clusters emerged from the Botswana cluster; the oldest clusters were located in Hong Kong, India, Australia, and South Africa (Supplementary Fig. S2), which could indicate that the first transmission events and further spread of the Omicron variant introduced in Mexico probably occurred in those locations. After the Botswana cluster, there is the cluster of South African sequences, which is linked to another one from India, from which a small cluster of Mexican sequences emerges; this result suggests that the first independent introductions of Omicron in Mexico (four sequences) could have come from South Africa or India.
On the other hand, another small cluster from Mexico is connected to the Singapore cluster, which comes from a large group of sequences belonging to Hong Kong (Supplementary Fig. S3). Altogether, these results suggest that the introduction of the Omicron variant in Mexico could have occurred through importation from different locations in Africa, India, and Southeast Asia. Moreover, this analysis showed that the sequence similarities shared between these sequences and the first sequences from Mexico probably delineate the transmission route of Omicron from these regions toward Mexico.
As mentioned earlier, the peak of Omicron was reached in mid-January 2022; thus an ML phylogenetic tree was built to understand the distribution of Omicron during this peak in the context of the first genomes circulating in Mexico. We found multiple introductions of different Omicron sub-lineages.
During this period, the following sub-lineages were identified for the first time in Mexico City: ‘BA.1’ on 16 November 2021 imported from South Africa, ‘BA.1.1’ on 19 November 2021, ‘BA.1.1.10’ on 21 December 2021, ‘BA.1.17’ on 18 December 2021, ‘BA.1.17.2’ on 16 December 2021, and ‘BA.1.18’ on 10 December 2021, whereas ‘BA.1.15’ was first identified on 3 December 2021 in Tamaulipas.
In the tree, the first genomes are distributed within the clades of the Omicron sub-lineages, indicating that these first introductions gave rise to the spread of the variant throughout the country. However, not all the introduced lineages reached a high frequency, such as BA.1.7, BA.18, and BA.1.17.2. Interestingly, the BA.1, BA.1.1, and BA.1.15 sub-lineages were the ones that dominated and increased their frequency after their introduction, mostly during the Omicron peak, when they represent the largest clades in the phylogenetic tree (Fig. 1B).
Interestingly, the BA.1.1 lineage is further subdivided into two large clades (blue navy), followed by the BA.1.5 lineage distributed in three clades (purple) and by BA.1 (blue), the most ancestral lineage in the tree. In contrast, the other lineages are found in a lesser proportion (Fig. 1B).
We also investigated whether the introduction and spread of the Omicron variant could be partially due to trends of increased population mobility (Fig. 2). We obtained human mobility data and divided the country into seven regions NE, NW, CN, CS, W, SE, and S (Supplementary Fig. S4). First, we found a mobility peak late in December 2021 (Fig. 2A), involving all Mexican regions. The mobility increase observed in December 2021 is the highest in the Omicron selection period. Second, Fig. 2B shows the increase in reported cases in January 2022. For that, we identified that the correlation between mobility and cases is spaced out in time (i.e. it takes time for more mobility to result in more cases). On average, it took 6 weeks for the two variables to switch between the negative and positive correlation (for details, see Supplementary Table S2) except for the NW region. The odd behavior with the Northwestern region can explained as the mobility levels of this region were positive in October 2021 while all the other Mexican regions were negative. Thus, it contributed to the correlation between cases and mobility being positive disregarding the 6-week interval reported in the other areas (the breakdowns of mobility levels for NW Mexico are included in Supplementary Fig. S5). We have also included a government stringency analysis in Fig. 2C. From that, we observed that around January 2022, the restrictions imposed by the government of Mexico were not as severe and long as it has previously been implemented in other COVID-19 waves.
Figure 2.
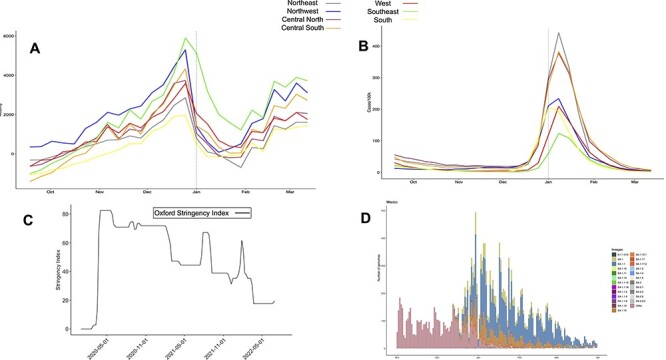
Mobility. (A) Google mobility trends (retail, recreation, grocery, pharmacy, parks, public transport, workplace, and residential) of each Mexican region are computed from 18 September 2021 to 12 March 2022. Mobility levels above 0 indicate a trend above the baseline. The baseline was a period of observation from before the pandemic (the baseline day is the median value from the 5-week period (3 January–6 February 2020). (B) Daily COVID cases are computed in each Mexican region from 18 September 2021 to 12 March 2022. (C) Weekly analysis of the Government Restrictions Index in the pandemic period in Mexico (1 January 2020–10 August 2022). (D) Number of SARS-CoV-2 genomes obtained at the national level. The BA.1.1 lineage was the most abundant in January and February. The BA.2 was the most abundant in March, and its frequency begins to increase.
As can be seen in Fig. 2D, the Omicron lineage peaked in mid-January 2022, where the B.1.1 lineage predominated across the country, followed by the BA.1.15 and BA.1 lineages. Therefore, the increase in cases is mainly associated with the BA.1.1 lineage. In addition, different sub-lineages of Omicron such as BA.1.7, BA.18, and BA.1.17.2 have circulated in Mexico but have not increased in frequency and perhaps may have become extinct.
As mentioned earlier, the peak of mobility increased in December 2021 in all regions of Mexico; therefore, we also evaluated the prevalence and spread of Omicron sub-lineages over time in the seven regions (Fig. 3). Initially, in November, the prevalence of the Delta variant across the country was 99 per cent. However, in December, there was an increase in mobility, and most of the cases in the CS and S regions seem to be related to the BA.1 (∼10.80 per cent), BA.1.1 (∼74.3 per cent), and BA.1.15 (∼11.25 per cent) lineages, whereas some states are located on the US–Mexico border (Baja California, Chihuahua, and Coahuila). In December, NE and NW had a higher prevalence of Delta (70 per cent) than the CS and S states, which had a 60 per cent prevalence of Omicron sub-lineages. By January, Omicron was then already dominant in all regions with three sub-lineages BA.1.1, BA.1.1.15, and BA.1. In addition, there were already a diversity of Omicron sub-lineages such as BA.2 (first detected in Jalisco on 9 January 2022), BA.1.1.10, BA.1.17, BA.1.17.2, BA.1.18, and BA.1.15. By February, BA.1.1 already dominated over the other sub-lineages in all regions, while BA.2 increased in frequency at the end of March, especially in the CS and S regions.
Figure 3.
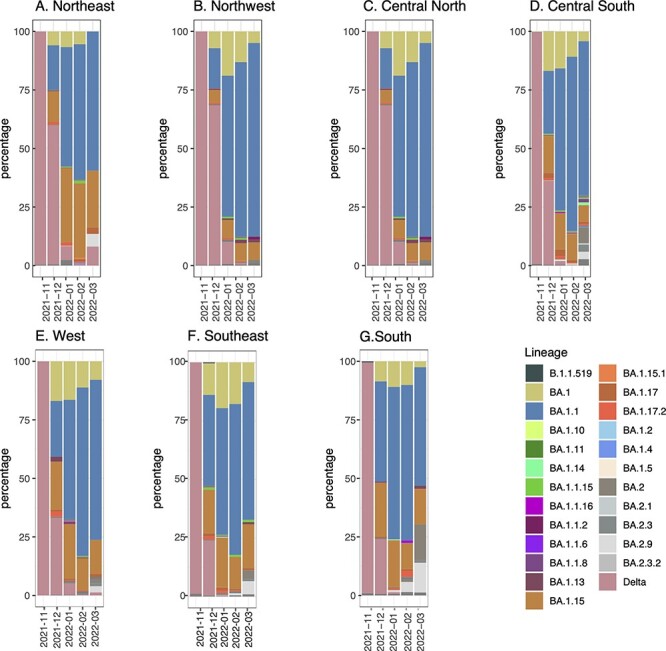
Distribution of the Omicron lineages in Mexico between November 2021 and March 2022. (A) Northeast, (B) Northwest, (C) Central North, (D) Central South, (E) West, (F) Southeast, and (G) South. The B.1.1, B.1, and B.1.15 lineages were predominant in all regions in January.
3.2. Analysis of natural selection of Omicron
We then searched for evidence of positive selection in Omicron genomes isolated in Mexico with different algorithms focused on a codon-based phylogenetic framework by calculating the ratio of non-synonymous (dN) to synonymous (dS) substitutions per coding sequence site (dN/dS). The evaluation of selection throughout the genome revealed that sites are highly conserved and show no evidence of episodic and directional selection. Few non-synonymous mutations were found across the genome (dN/dS > 1); only the gene that codes for the spike protein was found six sites under positive selection. These six sites were detected by the FUBAR method, which assumes that selection pressure is constant for each site throughout the entire phylogeny, using a Bayesian algorithm to infer rates (dN/dS) (Ben et al. 2013). Thus, this gene was subjected to a distinct natural selection strength.
The sites under positive natural selection found in the spike protein were L371S, R346K, N417K, E484A, S496G, and N1098D (Fig. 4). These mutations are located within the receptor binding domain (RBD, residues 319–541). The R346K mutation, originally described in the Mu variant, can alter interactions with monoclonal antibodies (mAbs) (Fratev 2022). The E484A mutation has been shown to confer up to tenfold higher resistance to vaccine sera (Wang et al. 2021: 1). Mutations L371S and S496G by a neutral polar amino acid are unlikely to cause a difference in the binding affinity of the receptor (Rath, Padhi, and Mandal 2022). Finally, the N1098D mutation occurs in the S2 subunit of the spike protein (amino acid residues 686–1,273). These changes in the spike protein are substitutions from nonpolar to polar amino acids that could have significant effects on the infectivity of the virus by promoting better interactions between the spike protein and the cellular receptor (ACE2). Interestingly, these mutations could be local adaptations of the virus to the Mexican population.
Figure 4.
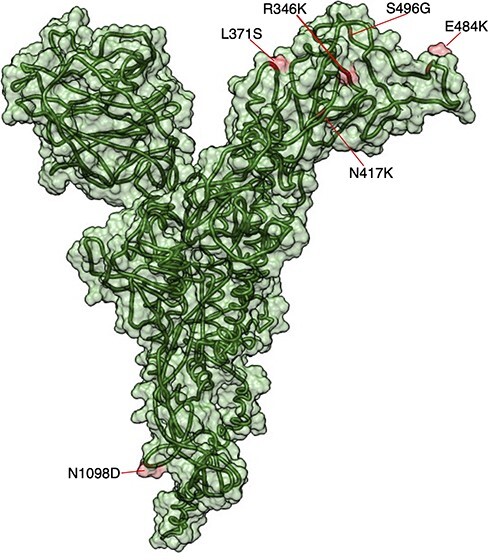
Omicron SARS-COV Spike protein model. Sites that will develop under positive selection using the FUBAR method are shown in red on the protein.
3.3. Detection of homoplasies
Homoplasies can be produced by base-pair substitutions or recombination and can evolve naturally under neutral evolution or positive natural selection. To detect homoplasies in the Omicron sequences in Mexico, we used HomoplasyFinder (Crispell, Balaz, and Gordon 2019). We used 244 Omicron (Mexico) sequences to construct a phylogeny and added two South African genomes, NICD-N22418 and NICD-N22397, as reference genomes. We identified homoplasies at 25 different positions (Supplementary Fig. S6), representing 24.5 per cent of the 102 polymorphic sites identified. Of these, eighteen were located within ORF1, specifically within the region coding for non-structural protein nsp11. In addition, the homoplasy at Position 15854 was located in the RNA polymerase, and the remaining homoplasies (at Positions 1694–9870) were located in the non-structural proteins (NSPs). Homoplasy changes were most common in the genome from thymine to cytokine (Supplementary Table S1).
On the other hand, using SNIPPY (NICD-N22418 and NICD-N22397 as reference genomes), we found that the homoplasies at Positions 1694, 2205, 5250, and 5465 can be directly observed from the Botswana genomes, suggesting that the rest of the homoplasies might be generated by the descendants of the internal nodes.
4. Discussion
In this study, we performed a phylogenetic and phylogeographic analysis to infer Omicron introductions in Mexico and how mobility might affect virus spread following these introductions. Subsequently, we analyzed the adaptive evolutionary processes in local genomes during the first month of the circulation of Omicron in Mexico. According to the data obtained from the genomes collected, the earliest detected genomes come mainly from Mexico City, the State of Mexico, Quintana Roo, and Yucatán, with the first of the reported cases being imported from South Africa. However, most of the introduction cases in Mexico were singletons, which indicates multiple independent introductions (160 independent introductions). These results are like those reported during the first epidemic wave in New York City by Dellicour et al. (2021b); discrete phylogeographic analysis found 116 independent introductions in the city and relatively small clades. These results show the heterogeneity of the virus required to establish a successful local transmission (Dellicour et al. 2021b). The multiple introductions of variants in Mexico have already been explored in other lineages, such as B.1.1.519 (Taboada et al. 2021) and Alpha (Zárate et al. 2022). For example, in the Alpha lineage, the introductions probably came from the USA, contributing to the spread of the Alpha lineage from the north to the south of the country (Zárate et al. 2022).
In the case of Omicron, the first increase in Omicron-related cases was observed in Mexico City, likely due to the arrival of international travelers harboring the virus. As indicated by the PastML phylogeographic analysis, the Mexican clusters likely emerged from variants previously circulating in South Africa and India. The other introductions correspond to viruses that circulated in Hong Kong or Singapore, and the BA.1 lineage arose from viruses circulating in South Africa. Variants can also spread in the world prior to COVID-19 through increased travel between countries (Hollingsworth, Ferguson, and Anderson 2007). An example of this could be South Africa. This country has the continent’s main airport hub (Newfarmer, Page, and Tarp 2018), with mobility data showing that by November 2021, there had been approximately 93,000 trips from South Africa to other 30 non-African regions. Moreover, at the time the WHO declared Omicron as VOCs, the likelihood of importation of the new variant in North America, Europe, the Middle East, and Oceania was over 50 per cent (Bai et al. 2021). Therefore, international human mobility is likely the critical event for the introduction of new variants of the virus.
From the moment a novel variant arrives in a country, internal mobility is among the factors that facilitate its local transmission (Fakir and Bharati 2021; Oka, Wei, and Zhu 2021). We demonstrated the latter by observing an increase in the number of cases that positively correlated with elevated mobility trends, spaced by an average time span of 6 weeks. In addition, in moments of higher mobility and, consequently, higher cases, we noticed that the stringency levels were not on pair as they were in less transmissible VOCs. Added to the fact that the mobility trends of a population are likely to follow a seasonal pattern (i.e. end of year celebrations) as reported by Hoogeveen, Kroes, and Hoogeveen (2022), we argue that a suitable scenario for mass transmission could have been created during the holidays. Next, we showed that the increase in cases reported in early January 2022 was due to the BA.1.1, BA.1, and BA.15 sub-lineages, and there does not appear to be any regionalization of these sub-lineages. In addition, the local transmission chains may reflect the higher internal mobility observed in the SW and CS parts of the country; this has been previously observed for the Delta variant in Mexico (Castelán-Sánchez et al. 2022).
Once we know the introduction of Omicron in Mexico, we assessed the adaptive changes in the genomes of Mexico. Because natural selection is one of the most important evolutionary forces that generate diversity and show signs of adaptation in an organism, we assessed natural selection. We used the FUBAR analysis, which shows better performance than SLAC or FEL (Delport et al. 2010; Ben et al. 2013). Results showed six sites of positive selection on spike protein (dN/dS > 1). The residues subject to positive selection are located in the RBD of the spike protein surrounding the receptor binding site (R346K, L371S, K417N, E484A, S496G, and N1098D) and are commonly associated with antibody evasion. For example, the amino acid change R346K, previously discovered in the Mu lineage (B.1.621) (Laiton-Donato et al. 2021), is associated with increased resistance to convalescent plasma therapy and decreased neutralization of antibodies (Liu et al. 2022). The R346K substitution was previously detected in genomes from Mexico City with a local prevalence of 40 per cent in the capital city, and phylogenetic analyses indicate that the Mexico City sequences form a monophyletic clone (Cedro-Tanda et al. 2022). Genomes with the R346K mutation are now designated as the BA.1.1 sub-lineage (Mohandas et al. 2022), which predominated in Mexico and spread throughout the country.
Amino acid substitution L371S was associated with a significant increase in the binding affinity of spike protein to human ACE2 (Kimura et al. 2022). Amino acid substitution K417N has previously been noted in the Alpha and Gamma lineages and has been associated with escape from mAbs (bamlanivimab/LY-CoV555) (Starr et al. 2021) and escape from neutralization by hyperimmune sera from individuals vaccinated with the BNT162b2 vaccine (Hoffmann et al. 2021). The amino acid substitution E484A is under selection in genomes in Mexico; at the same site, previous lineages such as Beta, Gamma, and Mu had the E484K substitution, which has been associated with escape from neutralization by vaccines. Currently, the E484A mutation is present in Omicron, which weakens mAb binding to the RBD and promotes escape from natural and vaccine-induced neutralizing antibodies (Shah and Woo 2022). Amino acid modification S496G is associated with bypassing mAb binding but shows no significant difference in binding to the RBD (Kannan et al. 2022).
In general, the sites under selection in the spike protein of the Omicron variants in Mexico are associated with the evasion of humoral immune responses. In addition, several recent studies have proven that natural selection is evident in the spike of a glycoprotein (Chaw et al. 2020; Tang et al. 2020). Changes in the amino acid sequence of the RBD can drastically affect the binding affinity of spike for the ACE2 receptor and also lead to an increase in the infectivity and transmission of SARS-CoV-2.
The methods used to detect natural selection determined sites under positive selection in the genes encoding the spike protein but not in other genomic regions of Omicron. However, the ORF1ab, ORF3a, and ORF8 proteins have been reported to have sites that evolve under positive natural selection (Velazquez-Salinas et al. 2020). Many of the mutations that occur repeatedly and independently multiple times in the genome (homoplasies) result in non-synonymous changes at the protein level and are candidates to present possible adaptive processes due to positive natural selection. Therefore, we investigated the presence of homoplasies, i.e. recurrent mutations, in Mexican genomes and found that most of the changes were related to the ORF1a nsp11 gene, RNA polymerase, and NSPs. The recurrent mutational signal in ORF1a, which encodes the NSPs nsp11 and nsp13, has been reported previously (van Dorp et al. 2020). In our analysis, nsp11 was the recurrent gene with homoplasies.
5. Conclusions
The analyses presented here show multiple introductions of Omicron that took place in different regions of CS Mexico and then spread to the rest of the country during the first months of its circulation. We have also gathered evidence to show that higher levels of mobility may have facilitated the spread of the variant to the outer regions of the country. Here, we show that the increased mobility at the end of the year resulted in a peak of Omicron cases; on average, the peak was observed 6 weeks after an increase in mobility. The identification of introductions is essential to determine the spread of the virus in a country.
We also highlight the presence of diversifying natural selection in the spike protein and the presence of homoplasy mutations in NSPs, which may be possible indicators of adaptation of the virus to the Mexican population. It is crucial to identify possible adaptation signatures in SARS-CoV-2 for the continued development of vaccines and treatments.
Supplementary Material
Acknowledgements
We thank the Consorcio Mexicano de Vigilancia Genómica (CoviGen). We thank the submitting laboratories for generating the genetic sequence and metadata and sharing them through the GISAID initiative on which this research is based. We thank Valeria Flores Almaraz for assisting with the construction of the maps.
Contributor Information
Hugo G Castelán-Sánchez, Programa de Investigadoras e Investigadores por México, Grupo de Genómica y Dinámica Evolutiva de Microorganismos Emergentes, Consejo Nacional de Ciencia y Tecnología, Av. Insurgentes Sur 1582, Crédito Constructor, Benito Juárez, Ciudad de México C.P. 03940, México.
León P Martínez-Castilla, Programa de Investigadoras e Investigadores por México, Grupo de Genómica y Dinámica Evolutiva de Microorganismos Emergentes, Consejo Nacional de Ciencia y Tecnología, Av. Insurgentes Sur 1582, Crédito Constructor, Benito Juárez, Ciudad de México C.P. 03940, México.
Gustavo Sganzerla-Martínez, Laboratory of Immunity, Shantou University Medical College, Shantou, People’s Republic of China, No. 22 Xinling Road Shantou, Guangdong Province 515041, China; Department of Microbiology and Immunology, Dalhousie University, Halifax, 5850 College street, Halifax, NS B3H 4R2, Canada.
Jesús Torres-Flores, Programa de Investigadoras e Investigadores por México, Grupo de Genómica y Dinámica Evolutiva de Microorganismos Emergentes, Consejo Nacional de Ciencia y Tecnología, Av. Insurgentes Sur 1582, Crédito Constructor, Benito Juárez, Ciudad de México C.P. 03940, México; Laboratorio Nacional de Vacunología y Virus Tropicales, Escuela Nacional de Ciencias Biológicas-IPN, Prolongación de Carpio y Plan de Ayala s/n, Col. Santo Tómas, Alcaldía Miguel Hidalgo CDMX C.P. 11340, México.
Gamaliel López-Leal, Programa de Investigadoras e Investigadores por México, Grupo de Genómica y Dinámica Evolutiva de Microorganismos Emergentes, Consejo Nacional de Ciencia y Tecnología, Av. Insurgentes Sur 1582, Crédito Constructor, Benito Juárez, Ciudad de México C.P. 03940, México.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
None declared.
References
- Aleem A., Samad A. B. A., and Slenker A. K. (2022) ‘Emerging Variants of SARS-CoV-2 and Novel Therapeutics against Coronavirus (COVID-19)’, in StatPearls, 1–10. Treasure Island, FL: StatPearls. [Google Scholar]
- Bai Y. et al. (2021) ‘International Risk of SARS-CoV-2 Omicron Variant Importations Originating in South Africa’, medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelán-Sánchez H. G. et al. (2022) ‘Comparing the Evolutionary Dynamics of Predominant SARS-CoV-2 Virus Lineages Co-circulating in Mexico’, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedro-Tanda A. et al. (2022) ‘Early Genomic, Epidemiological, and Clinical Description of the SARS-CoV-2 Omicron Variant in Mexico City’, Viruses, 14: 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw S.-M. et al. (2020) ‘The Origin and Underlying Driving Forces of the SARS-CoV-2 Outbreak’, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi-wai M. C. et al. HKUMed Finds Omicron SARS-CoV-2 Can Infect Faster and Better Than Delta in Human Bronchus But with Less Severe Infection in Lung. Consulting <https://www.med.hku.hk/en/news/press/20211215-Omicron-sars-cov-2-infection> accessed 12 Apr 2022.
- Choi J. Y., and Smith D. M. (2021) ‘SARS-CoV-2 Variants of Concern’, Yonsei Medical Journal, 62: 961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispell J., Balaz D., and Gordon S. V. (2019) ‘HomoplasyFinder: A Simple Tool to Identify Homoplasies on a Phylogeny’, Microbial Genomics, 5: e000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellicour S. et al. (2016) ‘SERAPHIM: Studying Environmental Rasters and Phylogenetically Informed Movements’, Bioinformatics, 32: 3204–6. [DOI] [PubMed] [Google Scholar]
- Dellicour S. et al. (2021a) ‘A Phylodynamic Workflow to Rapidly Gain Insights into the Dispersal History and Dynamics of SARS-CoV-2 Lineages’, Molecular Biology and Evolution, 38: 1608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellicour S. et al. (2021b) ‘Dispersal Dynamics of SARS-CoV-2 Lineages during the First Epidemic Wave in New York City’, PLoS Pathogens, 17: e1009571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W. et al. (2010) ‘Datamonkey 2010: A Suite of Phylogenetic Analysis Tools for Evolutionary Biology’, Bioinformatics, 26: 2455–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2012) ‘Bayesian Phylogenetics with BEAUti and the BEAST 1.7’, Molecular Biology and Evolution, 29: 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakir A. M. S., and Bharati T. (2021) ‘Pandemic Catch-22: The Role of Mobility Restrictions and Institutional Inequalities in Halting the Spread of COVID-19’, PLoS One, 16: e0253348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratev F. (2022) ‘R346K Mutation in the Mu Variant of SARS-CoV-2 Alters the Interactions with Monoclonal Antibodies from Class 2: A Free Energy Perturbation Study’, Journal of Chemical Information and Modeling, 62: 627–31. [DOI] [PubMed] [Google Scholar]
- Gong Z. et al. (2020) ‘An Online Coronavirus Analysis Platform from the National Genomics Data Center’, Zoological Research, 41: 705–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-Time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale T. et al. (2020) ‘Variation in Government Responses to COVID-19’, Blavatnik School of Government Working Paper, p. 31 <https://www.bsg.ox.ac.uk/sites/default/files/2022-08/BSG-WP-2020-032-v14.1.pdf>.
- Hoffmann M. et al. (2021) ‘SARS-CoV-2 Variants B.1.351 and P.1 Escape from Neutralizing Antibodies’, Cell, 184: 2384–93.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth T. D., Ferguson N. M., and Anderson R. M. (2007) ‘Frequent Travelers and Rate of Spread of Epidemics’, Emerging Infectious Diseases, 13: 1288–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogeveen M. J., Kroes A. C. M., and Hoogeveen E. K. (2022) ‘Environmental Factors and Mobility Predict COVID-19 Seasonality in the Netherlands’, Environmental Research, 211: e113030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K. P. Y. et al. (2022) ‘SARS-CoV-2 Omicron Variant Replication in Human Bronchus and Lung Ex Vivo’, Nature, 603: 715–20. [DOI] [PubMed] [Google Scholar]
- Ishikawa S. A. et al. (2019) ‘A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios’, Molecular Biology and Evolution, 36: 2069–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S. R. et al. (2022) ‘Complex Mutation Pattern of Omicron BA.2: Evading Antibodies without Losing Receptor Interactions’ International Journal of Molecular Sciences, 23: 5534. doi: 10.3390/ijms23105534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Rozewicki J., and Yamada K. D. (2019) ‘MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization’, Briefings in Bioinformatics, 20: 1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I. et al. (2022) ‘SARS-CoV-2 Spike S375F Mutation Characterizes the Omicron BA.1 Variant’, Microbiology <http://biorxiv.org/lookup/doi/10.1101/2022.04.03.486864>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiton-Donato K. et al. (2021) ‘Characterization of the Emerging B.1.621 Variant of Interest of SARS-CoV-2’, Infection, Genetics and Evolution, 95: e05038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., and Bork P. (2021) ‘Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation’, Nucleic Acids Research, 49: W293–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2022) ‘SARS-CoV-2 Variants of Concern and Variants of Interest Receptor Binding Domain Mutations and Virus Infectivity’, Frontiers in Immunology, 13: 825256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas S. et al. (2022) ‘Pathogenicity of SARS-CoV-2 Omicron (R346K) Variant in Syrian Hamsters and Its Cross-Neutralization with Different Variants of Concern’, EBioMedicine, 79: 103997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B. et al. (2012) ‘Detecting Individual Sites Subject to Episodic Diversifying Selection’, PLoS Genetics, 8: e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B. et al. (2013) ‘FUBAR: A Fast, Unconstrained Bayesian Approximation for Inferring Selection’, Molecular Biology and Evolution, 30: 1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newfarmer, R., Page, J., and Tarp, F. eds (2018) Industries without Smokestacks: Industrialization in Africa Reconsidered, WIDER Studies in Development Economics, Oxford: Oxford University Press; <http://fdslive.oup.com/www.oup.com/academic/pdf/openaccess/9780198821885.pdf>. [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T., Wei W., and Zhu D. (2021) ‘The Effect of Human Mobility Restrictions on the COVID-19 Transmission Network in China’, PLoS ONE, 16: e0254403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platto S., Xue T., and Carafoli E. (2020) ‘COVID19: An Announced Pandemic’, Cell Death & Disease, 11: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2014) ‘Tracer V1.6’ <http://tree.bio.ed.ac.uk/software/tracer/>. [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath S. L., Padhi A. K., and Mandal N. (2022) ‘Scanning the RBD-ACE2 Molecular Interactions in Omicron Variant’, Biochemical and Biophysical Research Communications, 592: 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2022) R: A Language and Environment for Statistical Computing (Vienna: R Foundation for Statistical Computing; ) <https://www.R-project.org/> accessed 15 May 2022. [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-Likelihood Phylodynamic Analysis’, Virus Evolution, 4: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M., and Woo H. G. (2022) ‘Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies’, Frontiers in Immunology, 12: 830527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. N. et al. (2021) ‘Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016’, Cell Reports Medicine, 2: 100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taboada B. et al. (2021) ‘Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic’, Viruses, 13: 2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X. et al. (2020) ‘On the Origin and Continuing Evolution of SARS-CoV-2’, National Science Review, 7: 1012–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K. et al. (2021) ‘The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants’, Nature Reviews Genetics, 22: 757–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur V., and Ratho R. K. (2022) ‘OMICRON (B.1.1.529): A New SARS-CoV-2 Variant of Concern Mounting Worldwide Fear’, Journal of Medical Virology, 94: 1821–4. [DOI] [PubMed] [Google Scholar]
- van Dorp L. et al. (2020) ‘Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2’, Infection, Genetics and Evolution, 83: 104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Salinas L. et al. (2020) ‘Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 during the 2020 COVID-19 Pandemic’, Frontiers in Microbiology, 11: 550674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana R. et al. (2022) ‘Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa’, Nature, 603: 679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. (2021) ‘Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7’, Nature, 593: 130–5. [DOI] [PubMed] [Google Scholar]
- Zárate S. et al. (2022) ‘The Alpha Variant (B.1.1.7) of SARS-CoV-2 Failed to Become Dominant in Mexico’, Microbiology Spectrum, 10: e0224021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N. et al. (2020) ‘A Novel Coronavirus from Patients with Pneumonia in China, 2019’, New England Journal of Medicine, 382: 727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.