

Abstract
The earliest events in the folding of a protein are in general poorly understood. We used NMR R2 relaxation dispersion experiments to study transient local collapse events in the unfolded-state (U) conformational ensemble of apomyoglobin (apoMb). Local residual secondary structure (seen in regions corresponding to the A, D-E and H helices of the folded protein) is largely unchanged over the pH range 2.3–2.75, yet a significant pH-dependent increase in the conformational exchange contribution to the R2 relaxation rate (Rex) indicates that transient intramolecular contacts occur on a μs-ms timescale at pH 2.75. Comparison of 15N and 13CO relaxation dispersion data at pH 2.75 for residues in the A, B, G and H regions, which participate in the earliest folding intermediates, indicates that chain collapse and secondary structure formation are rapid and concomitant. Increasingly stabilizing conditions (lower temperature, higher pH) result in the observation of relaxation dispersion in the C, CD and E regions of the protein, which are known to fold at later stages. Mutation of Trp14 in the A helix region to Ala eliminates conformational exchange throughout the protein; mutation of hydrophobic residues in other regions results in selective inhibition of conformational exchange in the B, G or H regions. The R2 dispersion data for WT apoMb at pH 2.75 and 10 °C are best fit to a 4-state model ABGH ⇆ AGH ⇆ U ⇆ ABCD that includes on-pathway (AGH and ABGH) and off-pathway (ABCD) transiently folded states, both of which are required to explain the behavior of the mutant proteins. The off-pathway intermediate is destabilized at higher temperatures. Our analysis provides insights into the earliest stages of apoMb folding where the collapsing polypeptide chain samples both productive and non-productive states with stabilized secondary structure.
Graphical Abstract

INTRODUCTION
Major advances in understanding protein folding mechanisms have been realized in recent years.1–6 Nevertheless, structural details surrounding the earliest folding events remain scarce, since biophysical techniques rarely yield simultaneous mechanistic and high-resolution structural information about transient (short lifetime) states. A current model for the folding process invokes a funnel-shaped energy landscape7 that is divided by energy barriers into discrete conformational states. Folding is driven towards the native-state energy well, while populating a set of intermediate states, each of which exhibits a mixture of native-like and non-native-like properties. Energy barriers between intermediate states are created either by unfavorable enthalpic interactions or an uncompensated entropic penalty involved in forming increasingly compact states. The existence of early intermediates that form before the rate-limiting step is well established.8 On-pathway intermediates with extensive native-like contacts support productive folding by narrowing the search of conformational space and providing a directed pathway for folding.1 Off-pathway, non-productive intermediates, on the other hand, generally contain significant non-native features that reduce the rate of folding9,10 by requiring their unfolding before the productive pathway can be accessed.
Measurement of transient folding processes occurring within the unfolded ensemble generated under mild denaturing conditions, such as acidic pH for apomyoglobin, is a promising method for identifying elusive, partly-folded states that are not sufficiently populated to be observable under the more stabilizing conditions used in rapid mixing techniques. NMR Carr-Purcell-Meiboom-Gill (CPMG) R2 relaxation dispersion experiments11 can readily be used to give information on transient folding and unfolding of proteins.12–15 Other techniques that give information of comparably high resolution include hydrogen-deuterium exchange coupled with mass spectrometry5 and site-directed spin labeling coupled with NMR16 or EPR.17 R2 relaxation dispersion experiments provide information on conformational exchange processes on the μs-ms time scale, including kinetics and the chemical shift differences between the ground states and higher energy states that have populations as low as 1%. The structural features of the higher-energy conformational states can be obtained at high resolution, as each NMR resonance serves as an independent probe of folding.
The folding pathway of apomyoglobin has been studied extensively.18 In the folded state at neutral pH, apomyoglobin contains helical structure in 7 regions (termed A-E, G, H) that approximate the helices found in crystal structures of the heme-containing holoprotein (Figure 1).19 Acid-unfolded apomyoglobin (apoMb, pH 2.3, hereinafter abbreviated as U) transiently samples a small population of helical structure in the A (residues 7–11), D-E (residues 52–61), and H (residues 127–147) regions of the sperm whale myoglobin amino acid sequence, as evidenced by the deviation of backbone chemical shifts from random coil values and by residual dipolar couplings.20,21 Helical structure is stabilized (populations of 50–70% from 13Cα and 13C’ secondary chemical shifts) in the same general regions of the equilibrium molten globule state (MG, pH 4.1, 50°C) and in residues 101–116, which form the G helix in native apoMb.13,22,23 Site-directed spin label (SDSL) data reveal additional complexity within the acid-unfolded state indicating the presence of a number of transient collapsed states formed by local clusters of hydrophobic residues.16 NMR-detected ultra-fast mixing H/D exchange experiments24 reveal a hierarchical folding mechanism starting with an initial collapse of the polypeptide chain, accompanied by helix formation in the A, G, and H helices and followed by sequential folding of the B helix, the E helix and the C helix.
Figure 1.

Locations of residual helical structure in the myoglobin sequence, calculated from published backbone chemical shifts for apoMb at pH 6.1,19 pH 4.1,22 and pH 2.3,20 and for the holoprotein (containing heme) at pH 6.125 using the program TALOS+.26 Bars represent the extent of helical structure identified by TALOS+ in each case except that of the pH 2.3 state, where the hatched, light-colored bars show residues that sample helical dihedral angles.20 The locations of helices are mapped onto the backbone structure of holomyoglobin (1MBC).27 The F helix of holomyoglobin is dynamically disordered in the apoprotein.19
In the present work we utilize R2 relaxation dispersion of the backbone 15N, 1H and 13CO nuclei to investigate and quantify transient folding events occurring on the μs-ms timescale in acid-unfolded apoMb. The effects of temperature, pH, and mutation of key hydrophobic residues on the localization and extent of conformational exchange can be successfully modeled by a multi-state sequential folding pathway with an off-pathway intermediate. Kinetic exchange rates and chemical shift differences between the unfolded state and transiently populated intermediates, obtained from global fitting of R2 dispersion data, reveal significant similarity between the on-pathway intermediate states and the equilibrium MG state.
METHODS
Mutagenesis, expression, purification, sample preparation.
Mutagenesis was performed in the pET17b vector using the Quik-Change mutagenesis kit (Stratagene). Protein expression and purification of non-deuterated samples were carried out as previously described.25,28 Deuterated, 15N-labeled samples were prepared by growing BL21 DE3 E.coli in M9 media with progressively higher concentrations of 2H2O before transformation, followed by large-scale growth in 100% 2H2O using 12C,2H-glucose and (15NH4)2SO4 as the sole carbon and nitrogen sources respectively. 13C-glucose was used as sole carbon source for expression of apoMb uniformly labeled with 13C. Samples were prepared by dissolving lyophilized protein in 5 mM HCl at the approximate protein concentration of 100 μM, followed by desalting on a Hi-Trap (GE Healthcare) column. Desalting is critical for removing residual TFA from HPLC purification and was carried out using an Akta FPLC while monitoring UV absorbance and ionic conductivity to ensure adequate separation of protein and unwanted solute. Following desalting, the sample was concentrated to >200 μM (final concentration 200 μM after pH correction and addition of 2H2O) and pH corrected with NaOH. This method effects complete back-exchange of amide deuterons in deuterated proteins. The pH for samples of non-deuterated proteins was set to 2.75 (unless otherwise noted), and for deuterated protein samples at 2.85 to account for the destabilizing effect of aliphatic deuteration.29 2H2O was added to a final concentration of 10%. The sample was split in half and loaded into separate NMR tubes for simultaneous measurements at two static magnetic fields.
NMR data acquisition.
15N and 1H resonance assignments at pH 2.75–2.85 were transferred from published values at pH 2.320 by following individual cross peaks in a series of high-resolution HSQC spectra across the pH and temperature range 2.3 to 2.75 and 25 to 10°C, respectively. 13CO assignments at pH 2.75 were transferred onto a 3D HNCO30 spectrum from published values at pH 2.3 10°C. Sample temperature calibration and constant time, relaxation compensated R2 relaxation dispersion experiments11 were performed for 15N, 1H, and 13CO as previously described13 on 800 and 500 MHz Bruker spectrometers. In brief, effective R2 relaxation rates (R2eff) were measured as a function of the pulse spacing (τcp) in the CPMG sequence. Full relaxation dispersion profiles were acquired for 15N/2H and non-deuterated 15N/13C labeled wild type protein and for 15N-labeled L32A and L135A mutants. To facilitate comparisons of the patterns of dispersing residues in the wild type and mutant proteins at varying pH values and temperatures, the exchange contributions () to the backbone amide 15N R2 relaxation rates at a single magnetic field strength were determined from two-time-point relaxation dispersion data acquired with CPMG pulsing frequencies (1/τcp) of 100 and 4000 s−1.
Data Fitting.
Full R2 relaxation dispersion profiles were fit globally using the in-house program GLOVE31 by optimizing the exchange rates, intrinsic transverse relaxation rate and chemical shift difference Δω between each of the states. Error bars were defined by spectral signal to noise as well as repeat data points. 2-state, 3-state and 4-state models were used at different stages of the analysis of R2 dispersion data. A closed-form expression for dependence of on 1/τcp was used to model 2-state exchange processes.12 3-site exchange was modeled as previously described13,32 using a semi-analytical expression. A fully numerical matrix formulation was used to model 4-site exchange and parameterized to constrain chemical shift differences for any one of the three conformational transitions. The Akaike Information Criterion33 was used in combination with reduced χ2 to compare fitting models. The uncertainty of the kinetic and chemical shift parameters was determined using an extensive ~10,000-point grid search of the global kinetic parameter space as previously described.13 The fully optimized parameters obtained from the set of highest quality fits were analyzed to yield average parameter values and uncertainties.
Ideally, relaxation dispersion data from different samples and nuclear probes would be fitted simultaneously to extract kinetic parameters. However, the exquisite sensitivity of R2 dispersion to small variations in the populations of excited states associated with slight differences in salt concentration and pH resulted in small but unavoidable variation in the kinetic rates between separate, yet identically prepared apoMb samples. The destabilizing effect of aliphatic deuteration33 (compensated in the present work by increasing the pH by 0.1 unit) adds additional variability. Despite incorporation of a temperature compensation element in the CPMG pulse sequences, further variability arises from differences in RF-induced sample heating for each of the nuclei (15N, 1HN, 13CO). For these reasons, relaxation dispersion data sets for different samples or different nuclei were fit independently.
A core set of the highest quality dispersion curves obtained from resonances that are either fully resolved or significantly more intense than any adjacent partially overlapping peaks were used in the determination of kinetic parameters. Additional dispersion curves obtained from the remaining resonances, excluding those with severe spectral overlap, were added to the core dataset for analysis of structure in the transiently folded states.
RESULTS
pH-induced transient contacts in the unfolded state.
As the pH is raised from 2.3, the lowest pH practicable for apomyoglobin under low-salt conditions, to pH 2.75, the protein populates helical conformations in the same A, D/E and H regions as those seen at pH 2.320 with slightly increased helicity in H, as measured by the 13CO chemical shift deviations from random coil values (termed secondary chemical shifts) (Figure 2A). In contrast to the chemical shifts, the conformational exchange contribution to the backbone amide 15N R2 relaxation rate (termed Rex, the difference in the effective R2 at low and high pulsing frequencies) is highly sensitive to pH (Figure 2B). Many residues in regions corresponding to the A, B, C, G and H helices in folded apoMb exhibit values of 15N Rex greater than 5 s−1 at pH 2.75 and 10 °C (Figure 2B, red), indicating the presence of extensive μs-ms timescale conformational exchange processes. Much smaller Rex values are observed at pH 2.3 (Figure 2B, black) in regions of the sequence where large exchange contributions are observed at pH 2.75. The increased magnitude of Rex under conditions that are slightly more favorable for folding (pH 2.75 versus pH 2.3) strongly suggests that the conformational exchange can be attributed to transient collapse that, under more favorable conditions, could form part of the folding process. Exchange between the unfolded state and elements of isolated helical structure would be expected to occur on a sub-μs timescale,34 too fast to monitor by R2 relaxation dispersion.35 We therefore conclude that dispersion must be associated with long-range hydrophobic interactions that lead to formation of transiently collapsed states. As we will show later, the dispersion data also reflect formation of transient helical secondary structure.
Figure 2.
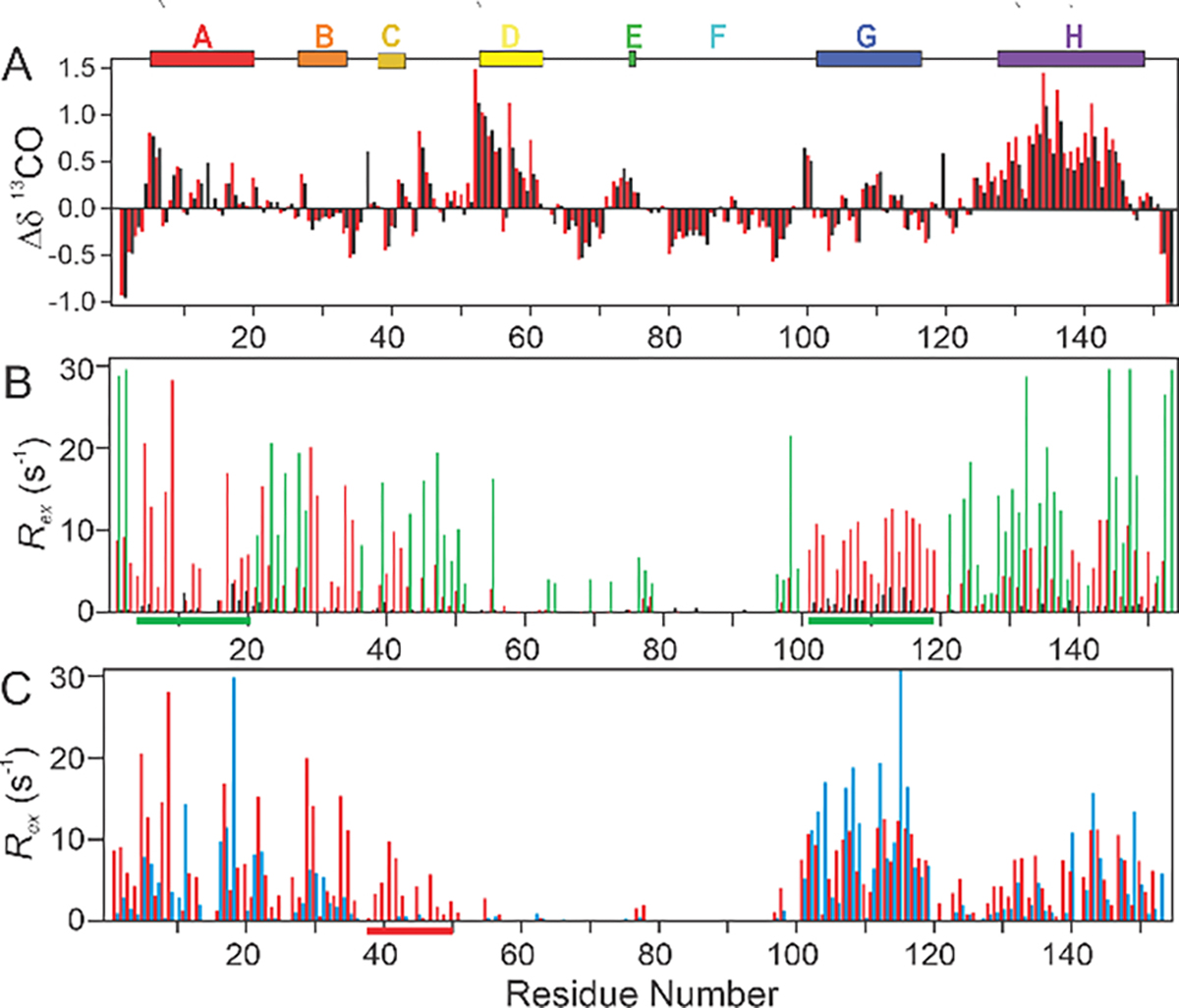
A. Backbone 13CO secondary chemical shifts at 25°C and pH 2.3 (black) or pH 2.75 (red). Secondary shifts were calculated as the difference between the observed chemical shift and sequence-corrected36 random-coil chemical shifts measured at pH 2.3 10°C in 8M urea.37 B. Comparison of 15N Rex at 10 °C and pH 2.3 (black), pH 2.75 (red) and pH 3.2 (green) at a static magnetic field of 800 MHz. Green bars at the bottom of the graph indicate regions where resonances were broadened beyond detection at pH 3.2. C. Comparison of backbone 15N Rex at pH 2.75 and 10°C (red bars) or 25°C (blue bars) at a static magnetic field of 800 MHz (sample: acid-unfolded 15N/12C labeled WT apoMb). Residues in the C helix and the C-D loop, which only display significant Rex at lower temperatures, are highlighted in red along the residue axis. Rex values were determined from two-time-point data acquired at 800 MHz. Colored bars at the top of the figure denote locations of helices in the MG intermediate at pH 4.1.22
As the pH is raised to 3.2 (Figure 2B, green), many of the residues with exchange contributions at pH 2.75 are broadened beyond detection, and the B, CD and H regions show enhanced relaxation dispersion. Several new residues in the DE and E regions also show relaxation dispersion with Rex > 2 s−1 at this pH. This evidence for transient folding of the CD and E residues above pH 3 supports a model of sequential folding in which additional structural elements are recruited under increasingly stabilizing conditions associated with increased pH. Transient structure in the CD and E regions is stabilized only at this higher pH and is not obligatory for collapse in the A, B, G, and H regions.
Increasing the temperature from 10°C to 25°C makes little difference to the 15N Rex values of the G region (Figure 2C), but the exchange observed at 10°C in the C and D regions and the CD loop are melted out at 25°C (indicated by a red bar at the bottom of Figure 2C). Residues in the A, B, and H regions show a decrease in Rex at 25°C, yet still yield average Rex values > 2 s−1. The temperature dependence of Rex clearly indicates that transient contacts involving the C and D regions are not obligatory for transient folding of the A, B, G, and H regions.
Mutagenesis reveals on and off-pathway transiently folded states.
Single point mutations that abrogate hydrophobic contacts at key locations in the myoglobin structure were used to selectively knock out transient interactions in specific regions of the apoMb polypeptide (Figure 3). Substitution of W14, located in the A helix of apoMb, with alanine was previously shown to affect the kinetic folding process.38 The W14A mutation effectively eliminates 15N exchange contributions throughout the protein at pH 2.75 and 25°C (Figure 3A), but raising the pH of the W14A mutant to 3.1 (Figure S1A) results in an increase of Rex to values comparable to those of the WT protein at pH 2.75. These observations reveal the important contribution of W14 to the stability of the collapsed states that are present under the lowest-pH conditions. The strong dependence of Rex in the A, G, and H regions on the presence of the W14 side chain (Figure 3A) indicates that bulky hydrophobic groups in the A region are crucial for collapse events throughout the sequence, including long range interactions between the A and GH regions.
Figure 3.

A-D. 15N Rex for four mutants of apoMb, compared with WT. A. 15N Rex at 25°C and pH 2.75 for WT (gray) and W14A mutant (red). B. 15N Rex at 10°C and pH 2.75 for WT (gray) and at 25°C and pH 2.79 for L32A mutant (orange). C. 15N Rex at 25°C and pH 2.7 for WT (gray) and at pH 3.0 for I111A mutant (blue) D. 15N Rex at 10°C and pH 2.75 for WT (gray) and at 10°C and pH 2.77 for L135A mutant (purple). Vertical arrows show the mutation sites. Colored bars at the top of the figure denote positions of helices at pH 4.1 (top row) and 6.1 (bottom row) (Figure 1). E. Average area buried upon folding (AABUF),39 calculated with a 7-residue moving average along the sequence of apomyoglobin.
Mutation of the B helix residue L32 to A40 has a more localized effect, eliminating nearly all exchange contributions in the B, C, and CD regions at pH 2.79, 10°C (Figure 3B) and decreasing Rex for a number of residues in the A region and the N-terminal residues of the G region, while causing only minimal changes in Rex in the H region and the majority of G. The effects of the L32A substitution show that the folding of the A, G, and H regions can occur independently of contributions from region B. The L32A mutation also abrogates Rex in C and CD, suggesting that formation of structure in these regions requires interactions with B. The effects of the L32A mutation provide additional evidence that long range interactions that lead to collapse of the A, G, and H regions are independent of other regions. These observations indicate that the A, G, and H regions dock cooperatively to form a collapsed cluster (U⇆AGH) in the earliest stages of folding, consistent with a spin labeling study that revealed transient interactions between the A and GH regions of unfolded apoMb at pH 2.3.16
Hydrophobic collapse and folding of the G and H regions is affected by the mutants I111A (Figure 3C) and L135A (Figure 3D).28 Nearly all exchange contributions in G and H are eliminated by these mutations. Rex in the A, B, and C regions is decreased to a much smaller extent than in G and H, indicating that A, B, and C can collapse independently of contributions from G and H (U⇆ABC). In strong support of the propensity of the ABC cluster for independent collapse, significant Rex is observed in the A and B regions in a truncation construct of the apoMb sequence spanning helices A through E (Figure S1B). Formation of the ABC cluster is abrogated by the W14A mutation (Figure 3A) suggesting that it is stabilized by non-native interactions between the A and BC regions. The loss of dispersion in the C and CD regions caused by the L32A substitution in region B provides additional evidence that ABC forms a cooperatively collapsed state independent of AGH.
Working Models for the Collapse Mechanism.
It has long been known that the A, G and H helices of apomyoglobin participate in the earliest folding steps.41 Our observations on the effects of temperature, pH and mutagenesis on the Rex values of acid-unfolded apoMb show that additional states are present in the conformational ensemble, with contacts between other regions of the molecule. The simplest model for such a mechanism is a three-state equilibrium involving the unfolded state, the productive AGH species and an unproductive ABCD species (Scheme I). The ABCD state is populated at 10°C but is destabilized and does not contribute to relaxation dispersion at 25°C (Figure 2C) or when the hydrophobic side chain of L32 is replaced by alanine (Figure 3B). The participation of the B region in the productive pathway can be modeled in two ways, either in Scheme II, where B collapses largely concomitantly with A, G and H or in Scheme III, where the initial collapse includes only A, G and H, with B added later. While the B region is clearly involved in the ABCD state, at what stage it is incorporated into the productive state can be evaluated by fitting data to each of the alternate schemes outlined below.
Scheme I.

Scheme II.

Scheme III.

Mapping the transient folding mechanism using 15N, 1H and 13CO R2 dispersion.
In order to elucidate the structural features of the states that participate in the earliest collapse, as well as the sequence of events, full 15N, 1H, and 13CO R2 relaxation dispersion profiles were acquired at two static magnetic fields (11.7 and 18.8 T) (Figure 4; a complete set of dispersion curves is shown in Figure S2). 15N and 1H dispersion data11,12 were acquired at pH 2.85, 10°C using perdeuterated (2H/15N) apoMb. 15N and 13CO R2 dispersion data42 were acquired with non-deuterated 15N, 13C-labeled apoMb at pH 2.75, 10°C. The data were globally fit to each of Schemes I–III.
Figure 4.

Representative dispersion curves for WT apomyoglobin at 800 MHz and 10°C. Data from 15N (pH 2.85, black), 1H (pH 2.85, red) and 13CO (pH 2.75, green) experiments are plotted for the residues indicated. The solid lines are global fits to Scheme III for all nuclei and all residues showing dispersion. The top axis (green) shows 1/τCP for the 13CO measurements, and the bottom axis (black) shows 1/τCP for the 15N and 1H measurements.
To reduce the dimensionality of the fitting models, constraints on Δω were imposed to allow adjustable Δω parameters only for residues expected, on the basis of the results for the mutant proteins (Figure 3, Figure S1), to form transient contacts during a given transition; Δω parameters for all other residues were held constant. In fitting to Scheme III, for instance, Δω for residues in the B region were constrained to zero for the U⇆AGH step (ΔωU-AGH = 0), since B does not fold in this step, but allowed to float for the other two equilibria (U⇆ABCD and AGH⇆ABGH). Since the L32A mutation selectively blocks formation of the ABGH and ABCD states while leaving Rex in the G and H helices largely unperturbed, the parameters ΔωAGH-ABGH and ΔωU-ABCD for residues of the G and H helices were constrained to zero. Similar reasoning shows that chemical shifts of residues in the C and CD regions should be exclusively sensitive to formation of the off-pathway state and are not affected by the formation of the AGH or ABGH states. Therefore, the constraints ΔωU-AGH=0 and ΔωAGH-ABGH=0 were imposed on residues in the C and CD regions. ΔωAGH-ABGH for residues of the A region were also constrained to zero since the change in Rex in the A region upon mutation of L32 in region B can be fully explained by the effect on the off-pathway state ABCD. While all residues are fit to a single set of global exchange rates, the resulting constraints applied to the 4-state model (ABGH⇆AGH⇆U⇆ABCD) reduce the number of adjustable local Δω parameters to that of a 2-state model for residues in regions C, CD, G, and H and that of a 3-state model for residues in regions A and B.
A comparison of statistical parameters (Akaike information criterion, AIC, and reduced χ2) obtained for the global fits to 15N and 13CO R2 dispersion curves (Table 1) shows that data for residues in regions A, C, CD, G, and H are equally well fit by Schemes I–III, but the dispersion data for the B helix are poorly fit by Scheme I. We therefore eliminate this scheme from further consideration.
In Scheme II, ABGH is formed in a single step which seems unlikely in view of the results for the mutant L32A, which show that destabilization of B does not significantly perturb folding in A, G, or H (Figure 3B). Schemes II and III cannot be distinguished on the basis of statistics alone, since the fit to Scheme III does not show a statistically significant improvement over Scheme II (Table S1). This is because only B helix residues can discriminate between the two exchange models; dispersion profiles for residues in helices A, G, and H are fit equally well by both exchange schemes (black data points in Figure 5) since folding of the AGH state occurs on both pathways. However, the two models can be distinguished on the basis of Δω values for residues in the B region (see below).
Figure 5.

Chemical shift changes Δω for 15N and 13C, calculated from the global fits to all nuclei, all residues, plotted against the equilibrium chemical shift difference Δδ between the unfolded state at pH 2.320 and the molten globule state at pH 4.1.22 A. 15N, Scheme II. B. 15N, Scheme III. C. 13C, scheme II. D. 13C, Scheme III. Values for the B helix residues are shown as red squares. A line with slope =1 is shown for reference.
There is a linear correlation between ΔωU-AGH for 15N and 13CO determined from global fits to Scheme III and the equilibrium chemical shift difference (ΔδU-MG) between the unfolded state at pH 2.320 and the pH 4.1 molten globule22 (black data points in Figure 5). This correlation shows that the folding process probed by relaxation dispersion involves exchange between unfolded apoMb and a compact, transiently populated molten globule state.
The dispersion data also provide insights into folding of the B helix. The 15N and 13CO ΔωU-ABGH values for B helix residues obtained from fits to Scheme III (Figure 5B, D, red data points) are similar to the corresponding equilibrium shift differences (ΔδU-MG) for these residues, consistent with transient folding of secondary structure in B following formation of the AGH core to form the ABGH state. In contrast, the 15N and 13CO Δω values for residues of the B region obtained from fits to Scheme II (ΔωU-ABGH, Figure 5A, C, red data points) are very much smaller than the equilibrium chemical shift differences (ΔδU-MG) between the U and MG (4.1, 50 °C) states, which would be inconsistent with folding of B in the same step as collapse of A, G and H. The small amplitude of ΔωU-ABGH for residues of the B region obtained from Scheme II probably arises from inappropriately forcing data from the B region to fit to a cooperative folding transition U⇆ABGH. We note that Scheme III is fully consistent with ultra-fast H/D exchange pulse labeling measurements which show that the B helix folds more slowly than AGH and that it is stabilized by docking to the preformed AGH core.24
Rate constants (k) and mole fractions (P) derived from the global fits of 15N, 1H and 13CO relaxation dispersion data at two fields to Scheme III are summarized below. These values represent averages of several sets of measurements on protonated and deuterated WT apomyoglobin samples. The fitted parameters from the individual data sets are shown in Table S2.

Rates (s−1) for the forward and reverse processes are shown above and below the equilibrium arrows, and the mole fraction P of each species is shown below it.
Forward and reverse rate constants for the individual processes U⇆AGH and U⇆ABCD were also determined from two-state fits to dispersion data from mutants L32A and L135A respectively. Kinetic parameters derived from two-state fits to 15N dispersion curves acquired at two static magnetic fields (11.7 and 18.8 T) at pH 2.79, 25°C (L32A) and pH 2.77, 10°C (L135A) are in excellent agreement with the rates extracted from Scheme III fits of 15N dispersion profiles of the WT protein at pH 2.75, 10°C (Table S2), providing an independent check on the robustness of the fits.
Evidence for cooperative, single-step formation of ABCD.
For the L111A mutant and the A-E helix truncation fragment (Figure 3, Figure S1), only residues in the A and B regions have significant Rex, suggesting that the ABCD state could potentially fold non-cooperatively in a two-step process. Two-step formation of the ABCD state (U⇆AB⇆ABCD) was tested by systematically removing R2 dispersion curves of the C and CD regions from the 4-state Scheme III global fit to determine whether data from these regions bias the global kinetic parameters. Exclusion of these data from the fits had no effect on the kinetic parameters, suggesting that formation of the ABCD state is likely cooperative. Interestingly, large differences in Rex between WT apoMb and the A-E fragment are observed for residues 5–7, 11, and 16–18 which pack against the H and G helices in the folded apoMb structure.
Secondary structure of the transient states.
The linear correlation between absolute values of ΔωU-ABGH derived from fits to Scheme III and the chemical shift difference ΔδU-MG (Figure 5) shows that the transient state observed in the relaxation dispersion experiments is very similar in structure to the pH 4.1 equilibrium molten globule, which contains well-folded helices in the A, B, G, and H regions.13,22,23 Residues 60–97 exhibit no relaxation dispersion and have 13CO chemical shifts that are close to random coil values. We infer that this region remains largely unstructured and highly dynamic in the transient molten globule state, as in the pH 4.1 equilibrium molten globule.22,23 Chemical shifts of the transient states were calculated from the ground state chemical shifts Δδ and the value of Δω∗sign(ΔδU,MG). Since the absolute sign of Δω cannot be directly obtained from R2 dispersion, the signs of Δω and Δδ were assumed to be identical based on the correlation between the absolute values of Δω and ΔδU,MG. Secondary 13CO chemical shifts were determined by subtracting the random coil chemical shifts at the same pH and temperature predicted using the program POTENCI43 from the chemical shifts of the transient states. The 13CO secondary shifts (Figure 6) were used as a proxy for helical content in the transient states. The 13CO resonances of many residues in the A region are broad and weak at 10°C and reliable values of Δω could not be determined (Figure 6B). These data were therefore supplemented by secondary 13CO chemical shifts calculated from ΔωU-ABGH parameters obtained from a 3-state fit (U⇆AGH⇆ABGH) to 13CO data acquired at two static magnetic fields (11.7 and 18.8 T) at 25°C pH 2.75 (Figure 6A; the 13CO relaxation dispersion curves at 25°C are shown in Figure S3). Exchange broadening is less severe at 25°C and the ABCD state is not populated at this temperature (Figure 2C), thus the 3-state model shown above can be reliably applied.
Figure 6.

13CO secondary shifts calculated for the apoMb folding intermediates derived from the relaxation dispersion analysis.
The 13CO secondary shifts allow us to define the helix boundaries for the transient ABGH state (Figure 6). The helix population, a measure of the extent of structural sampling of helical conformations, was estimated by dividing the average 13CO secondary shift for residues in each helical region by the average 13CO secondary chemical shift (2.1 ppm) of a fully folded helix.20 For the transient ABGH state, helical structure is observed for residues 5–21 (A helix, 95% population at 25°C), residues 102–116 (G helix, 90% population at both 10°C and 25°C), and residues 127–148 (H helix, 70% population at 10°C and 90% population at 25°C). The boundaries of the short B helix are not well defined and it is populated to a lesser extent than the other helices.
The 15N, 1H, and 13CO chemical shifts of the transient ABCD state observed at 10°C, determined from Δω values from the Scheme III fit, differ substantially from random coil values, indicating chain collapse and secondary structure formation. However, the helical content is poorly defined by the 13CO secondary shifts. There appears to be a population of helix (~50%) in the D region and there is likely helix in the A and B regions also, but the sparsity of data due to broadened or overlapped resonances precludes determination of helix boundaries and populations. The ΔωU-ABCD values correlate poorly with the equilibrium chemical shift difference ΔδU-MG (Figure S4), suggesting that the A, B, C and CD regions pack differently in the transient ABCD state than in the equilibrium molten globule. Thus, the packing of the helices in the transient ABCD state appears to be non-native.
DISCUSSION
Transient folding of apoMb occurs by a sequential mechanism.
Collective analysis of the effects of mutagenesis, pH, and temperature and the agreement of the data with different global fitting models shows that the initial stages of apoMb folding likely include a productive sequential series of transient collapse events (U⇆AGH⇆ABGH) that lead to the obligate44 molten globule intermediate ABGH, and a non-productive process U⇆ABCD that competes under some conditions. Both pathways result in formation of collapsed states that contain helical structure. Nevertheless, available evidence suggests that the ABCD state is a non-productive off-pathway intermediate that must unfold prior to forming the productive ABGH molten globule. This assessment is consistent with results of NMR-detected ultra-fast mixing H/D exchange experiments.24 Within 0.4 ms after initiation of refolding at pH 5.8, core segments of the A, G, and H helices are folded and fully protected from H/D exchange,24 while a majority of amides in the B and C helices are only marginally protected. The structure formed at 0.4 ms corresponds to the Ia state, initially described by Jamin and Baldwin,45 which exhibits only partial folding of the B, C, and E helices. Subsequent collapse and folding in these regions form the intermediate state termed Ib, as H/D exchange protection in the B helix significantly increases between 0.4 and 6 ms. The H/D exchange experiments clearly show that the B helix folds after the AGH cluster, consistent with the sequential formation of the AGH and ABGH states in Scheme III. The kinetic sequence of events, in which the A, G, and H helices fold prior to the B helix,24 strongly refutes any suggestion that the ABCD state could be part of an alternate, productive folding pathway where, for example, G and H might fold onto the ABCD core. It should be noted that the rate of structure formation in the AGH core during refolding at pH 5.8 (> 40,000 s−1)24 is, as expected46 very much faster than under the denaturing conditions used in the present relaxation dispersion experiments (pH 2.75 – 2.85; kU-AGH = 30 s−1).
The A, G, and H helices in the transient ABGH state are more than 80% populated at pH 2.75, 25°C, as estimated from 13CO secondary chemical shifts (Figure 6), which is somewhat higher than the populations of the same helices in the equilibrium MG (pH 4.1 50°C).22 The stabilizing effect of the lower temperature (25 versus 50°C) likely outweighs the destabilizing effect of the lower pH (pH 2.75 versus 4.1). The matching populations for the A, G, and H helices (80%) support the conclusion that transient folding from U to AGH is cooperative. The population of the B helix (40–50%) is substantially lower than that of the other helices in the transient ABGH state, reflecting the sequential two-step folding of ABGH. The relatively low population of helical structure in the B region indicates that the ABGH state formed here likely corresponds to either the Ia or a mixture of Ia and Ib states described by Jamin and Baldwin45 and Uzawa et al.24 The R2 dispersion data suggest that conversion between Ia and Ib is consistent with an increasing population of docked/folded B region rather than an increase in the length of the helix in the B region. The most significant difference between the transient ABGH state observed by relaxation dispersion at pH 2.75 and the pH 4.1 equilibrium MG is the lack of structure in the ABGH state in the C and D regions, which have substantial helical propensity in the equilibrium MG.22 Extrapolating from this information, we can conclude that partial folding and docking of the C and D helices in the pH 4.1 MG state does not induce significant rearrangement of the folded ABGH core.
In contrast, the helical structure in the CD region in the ABCD state is longer (residues 40–46) than the corresponding helix in either the equilibrium MG (39–41) or the N-state (37–41) and extends into the native CD turn. The topological arrangement and hydrophobic packing of the B, C, and D helices in the ABCD state may resemble that of the native state, since these helices interact in folded apoMb. However, the A helix necessarily forms extensive non-native contacts in the ABCD state, given the lack of packing interactions between A and either B, C, or D helices in the native state. This combination of native and non-native interactions provides sufficient driving force to form a non-productive transient state detectable under some solution conditions, yet affords little stability.
Correlation with surface area buried upon folding.
There is a striking correlation (Figure 3E) between regions of WT apoMb that exhibit relaxation dispersion at 10 °C, pH 2.75 and the average area buried upon folding (AABUF), a parameter which measures a combination of residue size and hydrophobicity and is calculated from the amino acid sequence.39 In previous studies we have shown that the earliest chain collapse and folding events are driven by hydrophobic interactions between regions where there are peaks in the AABUF profile for the Mb sequence.16,47,48 A more recent sequence-based method (EFoldMine)49 also predicts early folding sites in the A, G, H and C-terminal region of the B helix, in close agreement with the 13CO secondary chemical shift profile at 25 °C (Figure 2A). While EFoldMine correctly identifies early, on-pathway folding sites, it fails to identify the C-D regions that fold in the off-pathway ABCD intermediate. Formation of this off-pathway intermediate is likely driven by interactions between clusters of residues in the A, B, and CD regions that are associated with maxima in the AABUF profile (Figure 3).
The U-state ensemble consists of a diverse array of transient structures formed on multiple timescales.
Three biophysical techniques have been brought to bear on the issue of the composition and dynamics of transient collapsed states in apoMb at pH 2.3. The secondary chemical shifts measured at equilibrium at pH 2.320 give a measure of the population of helical structure under these conditions: the percentages calculated from 13Cα, 1Hα, and 13CO secondary chemical shifts indicate 15% helix for A, 22% non-native helix for the D/E region and 20% helix in H, with no discernible helix in the G region. Similar populations of helix were observed in peptides corresponding to the D/E and H regions, showing that a propensity for local helix formation is an intrinsic property of these sequences.20,50 Increasing the pH to 2.75 causes very little change in the 13CO secondary chemical shifts (Figure 2) showing that the population of helix in the ground state is unchanged. However, this small increase in pH creates more favorable conditions for folding and leads to transient formation of a small population (< 5%) of helical “excited” states that can be observed by relaxation dispersion. The greatly increased helicity of these states (> 80% helix in the A, G, and H regions) reflects hydrophobic collapse and stabilization of helical structure through long-range packing interactions. Although relaxation dispersion is not observed at pH 2.3, site-directed spin labeling (SDSL) measurements16 reveal both local and long-range hydrophobic interactions between clusters of residues with high values of the AABUF parameter, leading to a small population of collapsed states formed through native-like interactions between the A/B and G/H regions and through non-native interactions between the A, B, and C regions.16 The long-range interactions between the A/B and G/H regions are abrogated in 8M urea and only local interactions persist between A and B or G and H. The SDSL analysis thus complements the current relaxation dispersion measurements by showing that under even more strongly denaturing conditions at pH 2.3, transient native-like contacts between the A, B, G, and H regions occur, as do non-native interactions between A, B, and C. The populations of the collapsed states observed by SDSL at pH 2.3 are comparable to those observed by relaxation dispersion at higher pH; it is therefore likely that the absence of measurable relaxation dispersion at pH 2.3 reflects a shift to a faster exchange timescale, not detectable by relaxation dispersion, under the more strongly denaturing conditions.
The apomyoglobin folding pathway.
Relaxation dispersion experiments are unique in their ability to characterize elusive, partly folded states that are present at very low population and are not observable by conventional rapid mixing methods. The current relaxation dispersion measurements provide new insights into the earliest events that likely initiate apoMb folding and, in conjunction with the wealth of prior work, extend our understanding of the folding pathway (summarized in Figure 7).
Figure 7.

Schematic diagram of the folding pathway of apomyoglobin inferred from equilibrium and relaxation dispersion experiments of acid-unfolded apoMb and stopped-flow/quench flow experiments at higher pHs. The productive and non-productive pathways are shown with green and red arrows, respectively. The time scales for formation of the AGH, ABGH, and AB(E)GH intermediates and native apoMb are from hydrogen exchange pulse labeling experiments.24,51 Secondary structure for the transient states are mapped onto the backbone structure of myoglobin.27 The secondary structure predictions were made using TALOS+26 from excited state 15N, 1H, and 13CO, chemical shifts derived from the Δω parameters obtained from relaxation dispersion experiments at pH 2.85 (15N, 1H) and 2.75 (13CO). ΔωU,ABGH parameters for 13CO determined from a 3-state (ABGH⇆AGH⇆U) fit to data at 25°C were used to supplement predictions of helical structure in ABGH by assigning residues with helical population greater than 50% as helical. The figure representing the transient state AB(E)GH is reproduced from reference 13 with permission. Colors show regions predicted to be helix (red), coil (green) and dynamic residues (blue). Thin white tubes indicate regions that have U-state chemical shifts (Δω≈0, no R2 relaxation dispersion) and are unfolded. 13CO secondary chemical shifts at 10°C obtained from fits of Scheme III were similarly used to identify regions of helix in the ABCD state. Tube radii are scaled proportional to the helix population in A and B as determined by the 13CO secondary chemical shifts at 25°C (ABGH) or 10°C (ABCD). The cylinder schematic provides a conceptual model for packing of the helices in the transient non-productive ABCD structure. The structure figures were prepared using MolMol52 using the Protein Data Bank coordinate file 1MBC.27
Based on SDSL measurements16 and the current relaxation dispersion measurements, we infer that folding is initiated by transient coalescence of clusters of residues with high AABUF scores. At these earliest stages, chain collapse and folding can proceed along competing pathways to form either native-like (AGH and ABGH) or non-native (ABCD) intermediates. The ABCD intermediate is relatively unstable and must be unfolded before productive collapse and secondary structure formation can occur. While these processes occur on a millisecond time scale under the denaturing conditions used in the relaxation dispersion experiments (Table S2), collapse and secondary structure formation is much more rapid at higher pH and the AGH intermediate is fully formed in 400 μs or less.24,53,54 Folding then proceeds by docking of the B helix (5 ms) to form an energetically frustrated ABGH intermediate that slowly progresses to the native state. The apoMb folding landscape is highly rugged and folding is impeded by energetic bottlenecks at the interfaces between the A/G/H, the B/E/G, and the E/G/H helices, which must be relieved before the native structure can be reached.28,55,56
Supplementary Material
ACKNOWLEDGMENT
We thank Gerard Kroon for assistance with NMR experiments. This work was supported by the National Institutes of Health Grants GM131693 (HJD), and DK34909 and the Skaggs Institute for Chemical Biology (PEW). DJF and DWM were the recipients of Ruth L. Kirschstein National Research Service Awards GM075713 and GM84594 respectively.
ABBREVIATIONS
- apoMb
apomyoglobin
Footnotes
ASSOCIATED CONTENT
Supporting Information.
A table of kinetic parameters, and figures showing complete sets of relaxation data.
The following files are available free of charge. Supplementary.pdf
Any additional relevant notes should be placed here.
REFERENCES
- (1).Roder H; Maki K; Cheng H Early events in protein folding explored by rapid mixing methods. Chem. Rev. 2006, 106, 1836–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Neudecker P; Lundström P; Kay LE Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys. J. 2009, 96, 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dill KA; MacCallum JL The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- (4).Schuler B; Hofmann H Single-molecule spectroscopy of protein folding dynamics--expanding scope and timescales. Curr. Opin. Struct. Biol. 2013, 23, 36–47. [DOI] [PubMed] [Google Scholar]
- (5).Hu W; Walters BT; Kan ZY; Mayne L; Rosen LE; Marqusee S; Englander SW Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7684–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gelman H; Gruebele M Fast protein folding kinetics. Quart. Rev. Biophys. 2014, 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dill KA; Chan HS From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997, 4, 10–18. [DOI] [PubMed] [Google Scholar]
- (8).Matthews CR Pathways of protein folding. Annu. Rev. Biochem. 1993, 62, 653–683. [DOI] [PubMed] [Google Scholar]
- (9).Sosnick TR; Mayne L; Hiller R; Englander SW The barriers in protein folding. Nat. Struct. Biol. 1994, 1, 149–156. [DOI] [PubMed] [Google Scholar]
- (10).Krishna MM; Lin Y; Englander SW Protein misfolding: optional barriers, misfolded intermediates, and pathway heterogeneity. J. Mol. Biol. 2004, 343, 1095–1109. [DOI] [PubMed] [Google Scholar]
- (11).Loria JP; Rance M; Palmer AG III. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999, 121, 2331–2332. [Google Scholar]
- (12).Tollinger M; Skrynnikov NR; Mulder FA; Forman-Kay JD; Kay LE Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001, 123, 11341–11352. [DOI] [PubMed] [Google Scholar]
- (13).Meinhold DW; Wright PE Measurement of protein unfolding/refolding kinetics and structural characterization of hidden intermediates by NMR relaxation dispersion. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9078–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Korzhnev DM; Religa TL; Banachewicz W; Fersht AR; Kay LE A transient and low-populated protein-folding intermediate at atomic resolution. Science 2010, 329, 1312–1316. [DOI] [PubMed] [Google Scholar]
- (15).Korzhnev DM; Vernon RM; Religa TL; Hansen AL; Baker D; Fersht AR; Kay LE Nonnative interactions in the FF domain folding pathway from an atomic resolution structure of a sparsely populated intermediate: an NMR relaxation dispersion study. J. Am. Chem. Soc. 2011, 133, 10974–10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Felitsky DJ; Lietzow MA; Dyson HJ; Wright PE Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6278–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lopez CJ; Oga S; Hubbell WL Mapping molecular flexibility of proteins with site-directed spin labeling: A case study of myoglobin. Biochemistry 2012, 51, 6568–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dyson HJ; Wright PE How does your protein fold? Elucidating the apomyoglobin folding pathway. Acc. Chem. Res. 2017, 50, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Eliezer D; Wright PE Is apomyoglobin a molten globule? Structural characterization by NMR. J. Mol. Biol. 1996, 263, 531–538. [DOI] [PubMed] [Google Scholar]
- (20).Yao J; Chung J; Eliezer D; Wright PE; Dyson HJ NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry 2001, 40, 3561–3571. [DOI] [PubMed] [Google Scholar]
- (21).Mohana-Borges R; Goto NK; Kroon GJA; Dyson HJ; Wright PE Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J. Mol. Biol. 2004, 340, 1131–1142. [DOI] [PubMed] [Google Scholar]
- (22).Eliezer D; Chung J; Dyson HJ; Wright PE Native and non-native secondary structure and dynamics in the pH 4 intermediate of apomyoglobin. Biochemistry 2000, 39, 2894–2901. [DOI] [PubMed] [Google Scholar]
- (23).Eliezer D; Yao J; Dyson HJ; Wright PE Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 1998, 5, 148–155. [DOI] [PubMed] [Google Scholar]
- (24).Uzawa T; Nishimura C; Akiyama S; Ishimori K; Takahashi S; Dyson HJ; Wright PE Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13859–13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jennings PA; Stone MJ; Wright PE Overexpression of myoglobin and assignment of the amide, Cα and Cβ resonances. J. Biomol. NMR 1995, 6, 271–276. [DOI] [PubMed] [Google Scholar]
- (26).Shen Y; Delaglio F; Cornilescu G; Bax A TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kuriyan J; Wilz S; Karplus M; Petsko GA X-ray structure and refinement of carbon-monoxy (Fe II)-myoglobin at 1.5 Å resolution. J. Mol. Biol. 1986, 192, 133–154. [DOI] [PubMed] [Google Scholar]
- (28).Nishimura C; Dyson HJ; Wright PE Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J. Mol. Biol. 2006, 355, 139–156. [DOI] [PubMed] [Google Scholar]
- (29).Hattori A; Crespi HL; Katz JJ Effect of side-chain deuteration on protein stability. Biochemistry 1965, 4, 1213–1225. [DOI] [PubMed] [Google Scholar]
- (30).Grzesiek S; Bax A Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 1992, 96, 432–440. [Google Scholar]
- (31).Sugase K; Konuma T; Lansing JC; Wright PE Fast and accurate fitting of relaxation dispersion data using the flexible software package GLOVE. J. Biomol. NMR 2013, 56, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sugase K; Dyson HJ; Wright PE Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [DOI] [PubMed] [Google Scholar]
- (33).Motulsky H; Christopoulos A Fitting models to biological data using linear and nonlinear regression: a practical guide to curve fitting; Oxford University Press, Oxford, 2004. [Google Scholar]
- (34).Werner JH; Dyer RB; Fesinmeyer RM; Andersen NH Dynamics of the primary processes of protein folding: Helix nucleation. J. Phys. Chem. B 2002, 106, 487–494. [Google Scholar]
- (35).Arai M; Sugase K; Dyson HJ; Wright PE Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schwarzinger S; Kroon GJA; Foss TR; Chung J; Wright PE; Dyson HJ Sequence dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001, 123, 2970–2978. [DOI] [PubMed] [Google Scholar]
- (37).Schwarzinger S; Kroon GJA; Foss TR; Wright PE; Dyson HJ Random coil chemical shifts in acidic 8 M urea: implementation of random coil chemical shift data in NMRView. J. Biomol. NMR 2000, 18, 43–48. [DOI] [PubMed] [Google Scholar]
- (38).Nishimura C; Lietzow MA; Dyson HJ; Wright PE Sequence determinants of a protein folding pathway. J. Mol. Biol. 2005, 351, 383–392. [DOI] [PubMed] [Google Scholar]
- (39).Rose GD; Geselowitz AR; Lesser GJ; Lee RH; Zehfus MH Hydrophobicity of amino acid residues in globular proteins. Science 1985, 229, 834–838. [DOI] [PubMed] [Google Scholar]
- (40).Nishimura C; Wright PE; Dyson HJ Role of the B helix in early folding events in apomyoglobin: evidence from site-directed mutagenesis for native-like long range interactions. J. Mol. Biol. 2003, 334, 293–307. [DOI] [PubMed] [Google Scholar]
- (41).Jennings PA; Wright PE Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 1993, 262, 892–896. [DOI] [PubMed] [Google Scholar]
- (42).Lundström P; Hansen DF; Kay LE Measurement of carbonyl chemical shifts of excited protein states by relaxation dispersion NMR spectroscopy: comparison between uniformly and selectively 13C labeled samples. J. Biomol. NMR 2008, 42, 35–47. [DOI] [PubMed] [Google Scholar]
- (43).Nielsen JT; Mulder FAA POTENCI: prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins. J. Biomol. NMR 2018, 70, 141–165. [DOI] [PubMed] [Google Scholar]
- (44).Tsui V; Garcia C; Cavagnero S; Siuzdak G; Dyson HJ; Wright PE Quench-flow experiments combined with mass spectrometry show apomyoglobin folds through an obligatory intermediate. Protein Sci. 1999, 8, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jamin M; Baldwin RL Two forms of the pH 4 folding intermediate of apomyoglobin. J. Mol. Biol. 1998, 276, 491–504. [DOI] [PubMed] [Google Scholar]
- (46).Jackson SE; Fersht AR Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435. [DOI] [PubMed] [Google Scholar]
- (47).Nishimura C; Dyson HJ; Wright PE Enhanced picture of protein-folding intermediates using organic solvents in H/D exchange and quench-flow experiments. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 4765–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dyson HJ; Wright PE; Scheraga HA The role of hydrophobic interactions in initiation and propagation of protein folding. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13057–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Verona G; Mangione PP; Raimondi S; Giorgetti S; Faravelli G; Porcari R; Corazza A; Gillmore JD; Hawkins PN; Pepys MB; et al. Inhibition of the mechano-enzymatic amyloidogenesis of transthyretin: role of ligand affinity, binding cooperativity and occupancy of the inner channel. Sci. Rep. 2017, 7, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Waltho JP; Feher VA; Merutka G; Dyson HJ; Wright PE Peptide models of protein folding initiation sites. 1. Secondary structure formation by peptides corresponding to the G- and H-helices of myoglobin. Biochemistry 1993, 32, 6337–6347. [DOI] [PubMed] [Google Scholar]
- (51).Nishimura C; Dyson HJ; Wright PE The apomyoglobin folding pathway revisited: structural heterogeneity in the kinetic burst phase intermediate. J. Mol. Biol. 2002, 322, 483–489. [DOI] [PubMed] [Google Scholar]
- (52).Koradi R; Billeter M; Wüthrich K MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 1996, 14, 51–55. [DOI] [PubMed] [Google Scholar]
- (53).Uzawa T; Akiyama S; Kimura T; Takahashi S; Ishimori K; Morishima I; Fujisawa T Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of α-helical content and compactness. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Xu M; Beresneva O; Rosario R; Roder H Microsecond folding dynamics of apomyoglobin at acidic pH. J. Phys. Chem. B 2012, 116, 7014–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Nishimura C; Dyson HJ; Wright PE Energetic frustration of apomyoglobin folding: role of the B helix. J. Mol. Biol. 2010, 396, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Aoto PC; Nishimura C; Dyson HJ; Wright PE Probing the non-native H helix translocation in apomyoglobin folding intermediates. Biochemistry 2014, 53, 3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.