

Abstract
The emergence of the coronavirus 2019 (COVID-19) arose the need for rapid, accurate and massive virus detection methods to control the spread of infectious diseases. In this work, a device, deployable in non-medical environments, has been developed for the detection of non-amplified SARS-CoV-2 RNA. A SARS-CoV-2 specific probe was designed and covalently immobilized at the surface of glass slides to fabricate a DNA biosensor. The resulting system was integrated in a microfluidic platform, in which viral RNA was extracted from non-treated human saliva, before hybridizing at the surface of the sensor. The formed DNA/RNA duplex was detected in presence of SYBR Green I using an opto-electronic system, based on a high-power LED and a photo multiplier tube, which convert the emitted fluorescence into an electrical signal that can be processed in less than 10 min. The limit of detection of the resulting microfluidic platform reached six copies of viral RNA per microliter of sample (equal to 10 aM) and satisfied the safety margin. The absence of non-specific adsorption and the selectivity for SARS-CoV-2 RNA were established. In addition, the designed device could be applicable for the detection of a variety of viruses by simple modification of the immobilized probe.
Keywords: DNA-biosensor, Fluorescence, Microfluidic, RNA extraction, SARS-CoV-2 detection, Silica slide
Graphical abstract
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a pathogenic coronavirus which emerged in late 2019 (Zhu et al., 2020) and caused the ongoing major pandemic of the coronavirus disease 2019 (COVID-19). As of October 5, 2022, more that 616 million COVID-19 confirmed cases were reported, with more than 6.5 million deaths (https://covid19.who.int/, accessed Oct 6, 2022). In addition to the vaccines developed against SARS-CoV-2 (Kyriakidis et al., 2021) social distancing, mass screening of the population and quarantine of the infected people have been efficient in mitigating the pandemic (Mercer and Salit, 2021; Pavelka et al., 2021). In late 2020, Slovakia conducted multiple rounds of population-wide rapid antigen testing to reduce SARS-CoV-2 transmission (Pavelka et al., 2021). This massive population scale testing required thousands of health-care workers. After two rounds of mass testing, the prevalence of the disease decreased by 58% after one week (Pavelka et al., 2021). This example highlights the need for novel viral detection techniques which can perform massive, sensitive, reliable and rapid screening, without requiring large numbers of specialized manpower. Population surveillance of the virus load is of particular importance in the situation of highly contagious emerging new viruses for which specific drugs are not available.
While intensive efforts were devoted to the field of COVID-19 diagnostic testing (Kevadiya et al., 2021; Weissleder et al., 2020), several requirements to match the needs for fast and accurate massive testing are still not met. Reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) was the first, and still most used, technique to detect SARS-CoV-2 RNA genome in nasopharyngeal fluid samples (Liu et al., 2020). Despite its high specificity and sensitivity, the RT-qPCR workflow is time-consuming, depends on the availability of reagents and consumables, and requires specialized human and equipment resources which are implemented in hospitals and biomedical laboratories. Another common technique used for the assessment of SARS-CoV-2 infection, relies on the detection of antibodies specific to the Spike (S) and Nucleocapsid (N) proteins. SARS-CoV-2 antibodies are most frequently detected through the ELISA (Enzyme-linked Immunosorbent assay) and LFIA (Lateral Flow Immunoassay) technologies. The ELISA is faster than RT-qPCR but presents the risk of cross-reactivity to antibodies from other coronaviruses (Lv et al., 2020) and does not allow for early detection of the infection (Udugama et al., 2020). These limitations are also associated with LFIA, which, however, is easier to process, faster and cheaper. Alternative methods to assess the presence of the virus are provided by antigen tests which detect specific viral proteins in respiratory tract samples (Nerenz et al., 2021). They mostly rely on lateral flow devices which contain labeled antibodies for complexation of SARS-CoV-2 antigen and immobilized capture antibodies allowing visualization of the antibody-antigen complex. While the LFIA technique used for antigen tests is very fast (less than 20 min), it suffers from low sensitivity and specificity (Feng et al., 2020).
Recent developments for SARS-CoV-2 detection include clustered regularly interspaced short palindromic repeats (CRISPR)-Cas based methods (Bruch et al., 2021; Choi et al., 2021; Ganbaatar and Liu, 2021; Liu et al., 2021) and reverse transcription loop-mediated isothermal amplification (RT-LAMP), as simpler alternatives to the gold-standard RT-qPCR workflow. The RT-LAMP approach, which abolishes the need for thermal cycling, uses four to six primers which target different gene regions, and provides result within 30–40 min from sample collection. However, only one target can be implemented in each assay, thus limiting the accuracy of the test. In addition, optimization of primers remains challenging (Lino et al., 2022). CRISPR-based diagnostics enabled the development of multiplexed and portable nucleic acid detection devices based on several Cas enzymes for the identification of human viruses, and was recently approved by the US Food and Drug Administration (FDA) for COVID-19 screening (Javalkote et al., 2022). Reaching attomolar sensitivity, CRISPR-based technologies hold the potential for being developed as point-of-care diagnostics. However, the need for an amplification step is still a limitation in term of time efficiency. Nanomaterials based technologies, using gold nanoparticles (Gutiérrez-Gálvez et al., 2022) or nanodiamonds (Li et al., 2022) were also investigated for SARS-CoV-2 detection in human serum samples or nasopharyngeal swabs.
Despite the emergence of alternative technologies to the current gold-standard RT-qPCR-based diagnostic kits, the requirements for the development of a portable device for massive and fast COVID-19 screening must still improve (Hwang et al., 2021). In particular, it should be deployable in areas of high population mixing, such as airports, train stations, and ports. The processing should be within reach of non-medical personnel and without the need for a clinical environment. Additional characteristics must be met, including the delivery of robust results within minutes and the minimization of potential human error in both test processing and result interpretation. Also, the diagnostic platform should be low cost for accessibility to third world countries and not vulnerable to reagent shortages. A major step toward time reduction of the test workflow would be the elimination of nucleic acid amplification.
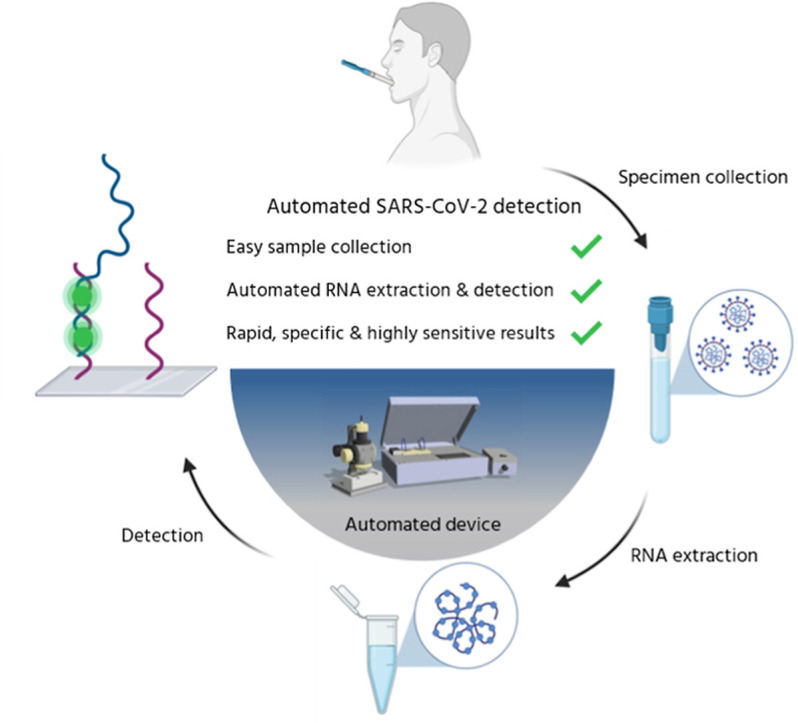
We herein present the development of a microanalysis platform for SARS-CoV-2 viral charge detection in human saliva samples at attomolar concentrations and which does not require transcription and amplification steps (Fig. 1 ). From collection to readout, the sample is processed within a few minutes, including extraction of viral RNA, sensing and fluorescence detection. The system was designed to include four components: a sampling unit for RNA extraction (Fig. 1, ②), a microfluidic unit containing the screening biosensor (Fig. 1, ③), a detection system (fluorescent measurements, Fig. 1, ④), and electronic drivers controlling liquid heating, handling and mixing. The core of the biosensor component consisted of single-strand (ss)DNA coated SiO2 slides integrated within the microfluidic device (Fig. 1, ③) which was conceived as a disposable kit. The ssDNA probes were selected for the specific targeting of SARS-CoV-2 virus sequences, allowing the identification of viral charge by the detection of RNA/DNA hybridization events at the surface of the sensing slides. The diagnostic system was designed to process fluorescence signals from the hybridization events. In addition, the choice of saliva samples (Fig. 1, ①) rather than nasopharyngeal swabs was dictated by the ease of (self)-harvesting, the reduction of variability due to higher sample volume, and the reduced invasiveness which promotes higher acceptability rates (Tobik et al., 2022). This is also supported by a recent study pointing toward the good level of agreement between salivary RT-PCR and standard nasal/oropharyngeal swab RT-PCR within early symptom onset in the context of a large-scale surveillance testing among staff working in the hotel quarantine system in Australia (Jenney et al., 2022).
Fig. 1.

Test workflow for the identification of SARS-CoV-2 viral charge in human saliva samples based on the direct detection of viral RNA at the surface of ssDNA-coated sensing SiO2 slides. From collection to readout, the sample is processed through extraction of the viral genetic material (step ②), sensing on DNA-functionalized silica slides (step ③) and fluorescence detection (step ④).
2. Material and methods
2.1. Commercial materials and instrumentation
2.1.1. Materials for slides functionalization and characterization
Borosilicate glass slides (10 mm × 10 mm × 0.5 mm) were purchased from SCHOTT AG. AziGrip4 amine coating was performed by SuSoS, Switzerland. CaCl2, EDC.HCl, HOBt and MES hydrate were bought from Sigma Aldrich. Succinic anhydride was purchased from TCI. Sodium acetate was purchased from Acros Organic. MgCl2 was bought from Roth, NaCl from Fisher Scientific and KCl from Alfa Aesar. PBS buffer was purchased from Gibco. SSC 20X buffer was purchased from Alfa Aesar. Linear oligonucleotides were purchased from GenScript with HPLC purification. Molecular beacons were bought from Integrated DNA technologies. Coatings were performed with a MSC-100 Cooling Thermoshaker Incubator form Labgene Scientific, Switzerland. All coatings were performed with Milli-Q ultrapure water. Fluorescence measurements of Cy3-modified oligos were carried out with a Synergy H1 multiplate reader from BioTek, at λexc = 532 nm, λem = 568 nm. XPS: X-ray Photoelectron Spectroscopy measurements were carried out on an Axis Supra (Kratos Analytical) using the monochromated Ka X-ray line of an Aluminium anode at the X-Ray Diffraction and Surface Analytics Platform (EPFL–ISIC–XRDSAP, Sion, Switzerland). The pass energy was set to 40eV with a step size of 0.15eV. Charge neutralization was done using a low energy electron gun and the spectra were referenced at 284.8eV using the aliphatic component of the C 1s orbital.
2.1.2. Materials and reagents for probe selection, RNA extraction and evaluation of the sensing slides
Primers were purchased from SIGMA. Amplirun total SARS-CoV-2 Control and Amplirun MERS Coronavirus RNA Control were purchased from Vircell (Granada, Spain). Lucigen QuickExtract DNA(USA) kit was bought from Biosearch Technologies (Teddington, UK). COVID-19 negative saliva was purchased from Lee Biosolutions, Inc. (Maryland Heights, MO, USA).
2.1.3. Components of the test bench
The H10722-20 photomultiplier tube was purchased from Hamamatsu Photonics. Transmission optical filter #86–354, reception optical filter #86–333 and dichroic filter #86–333 were purchased from Edmund Optics; the InGaN High Power LED SMB1N–490H-02 was purchased from Roithner LaserTechnik. The two microcontrollers STM32F7691-DISCO and STM32F407VGT and the different voltage converters were purchased from Mouser Electronics. Capacitors, inductors, resistors, diodes, mosfets and the various connectors were purchased from Digi-Key Electronics. The optical cage system and lens mounts were bought from Edmund Optics. Custom-made mechanical parts and the microfluidic circuit were produced by Chiser SA, Switzerland. Printed circuit boards for transmission and reception were manufactured by PCBway.
2.2. Identification of probes for SARS-CoV-2
To design 24- bp-long probes complementary to regions of the SARS-CoV-2 genome not subjected to reported mutations, sequencing data of SARS-CoV-2 were first collected from NCBI Nucleotide GenBank (https://www.ncbi.nlm.nih.gov/nuccore/1798174254, accessed Oct 6, 2022). A sliding windows approach was used to identify 1000 probes targeting highly polymorphic regions of the SARS-CoV-2 genome with length between 20- and 50 bp (home-brew R script). The binding capacity of each probe was assessed, and those with higher specificity against the virus were selected. A R script was implemented to calculate the GC content of each probe as the fraction of C or G nucleotide over the length of the probe. The self- and heterodimerization tendencies of each probe were also investigated. The Gibbs free energy change (ΔG) was calculated using mathFISH tool (http://mathfish.cee.wisc.edu). The larger negative values the higher propensity for identical primers to hybridize to each other rather than to the template. Probes with ΔG = ≥ −20 kcal/mol were excluded, and this step reduced the probes to 200. Then, all the probes with possible cross reactivity with other coronavirus (SARS-CoV-1 and MERS) were discarded and the number of probes was reduced to 50. In-house script blasted the probes’ sequences to the other coronavirus genomes, and probes with more than 75% identity were excluded. The candidates were ranked using the E-value of the probe-target duplex (blastn). A final list of five probes was provided.
To test the probes, Amplirun total SARS-CoV-2 was resuspended in 200 μL of QuickExtract RNA Extraction kit to obtain a concentration of 50 copies/μL of the virus. The RNA extraction was performed in 5 min at 80 °C. Total RNA extracts were reverse-transcribed using the SuperScript III First-strand Synthesis SuperMix System kit (Invitrogen) to generate cDNA. Quantitative Real-Time (qRT)-PCR amplification was performed using the KAPA SYBR FAST qPCR Master Mix (2x) ABI Prism on a StepOnePlus Real-Time PCR system (Applied Biosystems).
2.3. Preparation of the sensing surfaces
The amino-modified slides were immersed in 3 mL of an AcONa 0.3 M solution containing 150 mg of succinic anhydride and shaken overnight at 25 °C, 1000 rpm. The slides were washed three times with MiliQ (30 mL), three times with acetonitrile (30 mL) and three times with dichloromethane (30 mL). The succinic-modified slides were then immersed in 2 mL of a solution of EDC.HCl (50 mM) and HOBt (60 mM) in MES 0.1 M, to which 10 μL of a 100 μM solution of amino-modified DNA was added. The slides were shaken for 12 h at 37 °C, 1000 rpm. After rinsing the slides with MiliQ, they were incubated twice for 10 min at 25 °C, 1000 rpm with 10 mL of Tween 20 (0.1% solution), and twice for 10 min with 10 mL of a saline solution (NaCl 0.1 M, KCl 0.1 M, MgCl2 0.1 M, CaCl2 0.1 M) to remove adsorbed DNA strands (non-covalently conjugated). After being thoroughly rinsed with MiliQ, the slides were kept in MiliQ at 4 °C.
2.4. Characterization of the sensing surfaces
DNA-functionalized slides were immersed in 1.35 mL of SSC 4X buffer to which 150 μL of a solution of the complementary probe (10 μM in PBS 0.1X) tagged with Cy3 were added. The slides were incubated for 3 h at 25 °C, 1000 rpm. After rinsing the slides with MiliQ, they were incubated twice for 10 min at 25 °C, 1000 rpm with 10 mL of Tween 20 (0.1% solution), and twice for 10 min with 10 mL of a saline solution (NaCl 0.1 M, KCl 0.1 M, MgCl2 0.1 M, CaCl2 0.1 M) to remove the adsorbed Cy3-modified DNA strands. Slides were then immersed in 2 mL PBS 0.1X and incubated for 15 min at 85 °C for 10–15 min at 1000 rpm to dissociate the DNA duplex. The Cy3-tagged complementary DNA released in the supernatant was detected and quantified by fluorescence spectrophotometry (λex = 532 nm, λem = 568 nm) at 25 °C.
2.5. Design and engineering of the test bench
The microfluidic circuit was composed of an INPUT well, a PRE-TREATMENT chamber, a SCREENING chamber (loaded with ssDNA probes) and an OUTPUT well. The test bench was composed of two sub-systems, one allowing for the pre-treatment of the saliva samples through heating, mixing and movement of the fluids (sub-system 1), the other one allowing for the capture and analysis of the fluorimetric (see section 2.6) feedback signals (sub-system 2). The sub-systems were connected to individual evaluation boards (Sedra and Kenneth, 2009).
Sub-system 1 was controlled by a STM32F7691-DISCO board, equipped with a touch display for the management of sample pre-treatment by vibration/heating phases and liquid displacements. The vibration was performed using a double drive shaft DC motor DC6-24V RK370 coupled with two eccentric masses attached to the microfluidic platform where the disposable chip is placed. The heating was performed by two different PCB integrated heater 50 Ω resistors (HK5163R157L12A, Minco), connected to the microfluidic platform below the input well and below the screening chamber. The liquid displacement was performed using a piezoelectric micropump (mp6, Bartels Mikrotechnik) coupled with the microfluidic platform and a hermetic lock system.
Sub-system 2 was controlled by a STM32F407VGT board for the management of the functions required for the analysis through a computer terminal graphical interface coupled with a customized fluorescence microscope.
2.6. RNA extraction
RNA extraction was performed using the Lucigen Quick Extract DNA Extraction Solution. To mimic the future use of device kept at RT, experiments were performed with Lucigen Quick Extract DNA Extraction Solution kept at RT for more than three months and, as comparison, following manufacturer's instructions. Anticipating the future use of the devices in areas with no access to fridge and freezer, the storage temperature of the extraction solution was set to RT. Human saliva with and without SARS-CoV2 virus was mixed with the Quick Extract DNA Extraction at a 1:1 ratio. After vortexing, the mixture was incubated for 5 min at 80 °C. No purification step was performed. Results, as measured by the nanodrop 1000 Spectrophotometer, were comparable with those obtained following the recommended manufacturer's protocol, and no significative difference was observed (Supporting Information, Fig. S7).
2.7. Optical fluorescence measurements
The device for digital fluorescence microscopy measurements was composed of: i) an excitation light source (InGaN High Power LED emitter, SMB1N–490H-02) (Steranka et al., 2002) with a peak emission wavelength at 490 nm (Bryant, 2014); ii) five bandpass OD6 optical filters (Lakowicz, 2006); and iii) a photomultiplier tube (PMT) (Hamamatsu, H10722-20) with an anode radiant sensitivity of 8V/nW (PMT gain of 105) as detector (Polyakov, 2013). The bandpass optical filters were integrated as follows. Three bandpass optical filters with a central wavelength at 525 nm (15 nm bandwidth) (Edmund Optics, #86–354) were integrated in the receiver stage. Two bandpass optical filters with a central wavelength at 497 nm (20 nm bandwidth) (Edmund Optics, #86–333) were integrated in the transmission stage. An additional dichroic filter (Edmund Optics, #86–333) with a cut-on wavelength at 516 nm was present in the optical path. The fluorophore SYBR Green I was associated with cDNA probes coated on the sensing slides, allowing the detection of viral RNA/DNA duplexes through the formation of a complex absorbing at 497 nm and emitting at 520 nm (Valeur, 2001).
3. Results and discussion
3.1. Identification and selection of probes for SARS-CoV-2
Five probes targeting highly conserved regions of SARS-CoV-2 were selected based on their specificity, GC content, secondary structure and stability of the probe-target duplex estimated by the thermodynamic model (Table 1 ). The algorithm selected 24-bp-long probes as an optimum length to maximize their specificity (shorter sequences would have high similarity with other genomes) while ensuring high accessibility to their target and fast hybridization (longer sequences would increase the hybridization time and risk of self-folding). Previous reports highlighted the impact of the probe length on the efficiency and kinetics of hybridization events, indicating 18 to 24 bp capture probes as optimal systems (Hua et al., 2022; Xu et al., 2017). Due to its higher stability as compared to the other candidates, the putative probe (PP) 1 was used to develop the conjugation protocols on SiO2-based slides and evaluate the sensing capacities of the microfluidic device. The specificity of PP1 was controlled via qPCR using saliva samples containing SARS-CoV-2 viral RNA, MERS viral RNA virus and mammalian RNA (Supporting Information, Fig. S1).
Table 1.
Selected probes for SARS-CoV-2 detection. Top five probes in silico identified with their main features. The highlighted PP1 was then used for further work.
| Primer ID (PP) | Sequence (5’ → 3′) | Size (bp) | GC (%) | Tm (°G) | DG (kcal/mol) | Start | Stop | Gene |
|---|---|---|---|---|---|---|---|---|
| PP1 | AACAGCAAGAAGTG CAACGCCAAC | 24 | 50 | 65.80 | −27.47 | 25512 | 255535 | ORF3a |
| PP2 | CCATTCTTCGTAAGG GTGGTCGCA | 24 | 54.17 | 65.52 | −27.11 | 1427 | 1450 | ORF1ab |
| PP3 | TCAATAGCCGCCAC TAGAGGAGCT | 24 | 54.17 | 65.68 | −26.91 | 15150 | 15173 | ORF1ab |
| PP4 | CCAAAATTACAATC TAGTCAAGCGTG | 26 | 38.46 | 59.78 | −25.01 | 20625 | 20650 | ORF1ab |
| PP5 | GAAATCTAAAACAA CACGAACGTCAT | 26 | 34.62 | 59.67 | −24.96 | 28231 | 28206 | ORF8 |
3.2. Immobilization of DNA probes on silica substrates
Our strategy for the preparation of the sensing core of the microfluidic device relied on the covalent conjugation of the selected DNA probe to the surface of SiO2 slides, previously modified with the amino-containing AziGrip4 Amine coating (SuSoS). Several requirements must be met for the efficient detection of viral RNA on sensing surfaces. The probes must be stable and accessible to its complementary analytes. The lateral spacing between the DNA strands, their orientation toward the surface and their density are additional key parameters which impact the hybridization performance (Nimse et al., 2014; Teles and Fonseca, 2008). The most frequently reported methods rely either on the immobilization of pre-synthetized DNA probes or on the stepwise synthesis of the oligonucleotide on the surface. The immobilization of pre-synthetized DNA strands is generally preferred as it is easier to perform and control, allows uniform or spotted deposition, and ensures the sequence integrity of the probe (Nimse et al., 2014). Among the various technologies developed so far, three main strategies were highlighted for ssDNA immobilization on solid surfaces, including physical adsorption led by electrostatic or hydrophobic interactions, immobilization through streptavidin-biotin interactions and covalent conjugation (Nimse et al., 2014). In order to benefit from higher stability and durability of the resulting functionalized materials, we herein focused on covalent conjugation strategies for the immobilization of PP1 on SiO2 slides.
Commercial amino-functionalized slides were first reacted in presence of succinic anhydride to obtain carboxylic groups at the surface (Scheme 1 a). PP1, modified at its 5′-end with an amino group was further conjugated to the slides in the presence of EDC/HOBt as coupling agent. Thorough washing cycles with Tween 20 and saline solutions were used to remove any adsorbed probes from the surface.
Scheme 1.

a: Schematic illustration of PP1 immobilization at the surface of amino-modified slides. b: Schematic illustration of the procedure for PP1 quantification at the surface of the sensors. A Cy3-labeled complementary reverse probe (Supporting Information, Table S2) was added to the functionalized slides, followed by removal of non-hybridized sequences through washing cycles. The resulting DNA duplexes were denaturated by thermal treatment for quantification of the Cy3-labeled sequences released in the supernatant.
The functionalization of the glass slides was assessed by X-ray photoelectron spectroscopy (XPS) (Supporting Information, Table S3). The presence of the phosphate backbone of ssDNA probes at the surface of S-PP1 slides was identified by the acquisition of a signal at the P 2p region (Supporting Information, Fig. S3). A negative control was performed by incubation of S-Suc slides with 5′-amino modified PP1, in the absence of coupling agent. XPS analysis of the resulting materials did not show the presence of a phosphorous signal, which gave evidence for the covalent attachment of the DNA probe on S-PP1 slides (for XPS spectra of the C 1s and N 1s region, see Supporting information, Figs. S4 and S5).
The amount of DNA probe immobilized at the surface of the sensing slides was further quantified through a protocol adapted from Miyahara et al. (Miyahara et al., 2019) (Scheme 1b)., S-PP1 slides were immersed in an aqueous solution of the 5′-Cy3-labeled complementary DNA strand (Supporting Information, Table S2) to allow hybridization at the surface. Following washing cycles to remove non-hybridized DNA reagent, the slides were heated at 85 °C and the Cy3-labeled DNA released from the duplexes was quantified by fluorescence spectrophotometry (for quantification standard curve, see Supporting Information, Fig. S2) and corresponded to the amount of immobilized PP1. Over repetitive conjugation experiments (n = 4), the quantity of covalently conjugated probe (PP1) was measured at 2.7 ± 0.7 pmol. As a control, S-Suc slides were treated according to the same protocol and led to the detection of only traces of Cy3-labeled DNA (0.1 pmol) (Supporting Information, Table S1).
The stability of the obtained sensors under different storage conditions was evaluated. S-PP1 slides were stored for 10 days at 4 °C, in the absence of light, in various buffers (SSC 4X, Tris.HCl 1 M, PBS 1X), MilliQ water or dried. The amount of non-altered probes at the surface was quantified as previously described (for quantification values, see Supporting Information Table S4). While drying S-PP1 slides with organic solvents led to a drastic alteration of the immobilized probes, storage in both MilliQ water and buffers maintained the integrity of the DNA strands conjugated to the sensing surface. Drying S-PP1 slides without organic solvent treatment did not show detrimental effect to the functionalized surface; however, it required extensive drying time. For further studies, we selected MilliQ water as storage medium to avoid the presence of salts in subsequent experiments and to remove the need of an extra drying step. The stability of S-PP1 slides, stored in MiliQ at 4 °C, was studied over longer durations (Table S4). While no degradation was observed after 15 and 20 days of storage, the amount of non-altered immobilized probes decrease by 50% after 30 days (data not shown). We therefore used S-PP1 slides for sensing experiments within a two weeks range after their production.
3.3. Design, fabrication and characterization of the sensing device test bench
The portable platform for SARS-CoV-2 diagnostics was engineered to allow for an automatic and fully integrated screening workflow, including the RNA extraction from the saliva sample (PRE-TREATMENT chamber), the hybridization of potential viral RNA sequences at the surface of sensing slides incorporated in the SCREENING chamber and the readout of hybridization events through optical fluorescence microscopy (OUTPUT chamber) (Fig. 2 a and b). All these steps were included in the design of a multi-component microfluidic device composed of untreated hydrophilic polycarbonate micro-channels and hosting the sensing slides (S-PP1) (Fig. 2c and d). The hydrophilic properties of the microfluidic chip surfaces were identified as crucial to promote passive transport of the liquid samples by capillary flow (600 μm internal capillary diameter) and reduce surface tension of the aqueous fluids during the transfers from the INLET area to the PRE-TREATMENT and SCREENING chambers. The overall testing workflow (for conceptual design of the device, see Supporting Information, Fig. S6) after saliva sample harvesting started with the extraction of RNA components in the PRE-TREATMENT chamber loaded with a DNA extraction kit (Lucigen), shaken with a miniature vibrating disk motor for mixing enhancement and heated at 80 °C for 5 min. The resulting extract was then flowed to the SCREENING chamber containing the sensing slide for direct viral RNA detection, in the presence of SYBR Green I dye. The device was equipped with a second heating unit (50 °C) to allow annealing of SARS-CoV-2 RNA/DNA probe duplexes, intercalated with the fluorescent dye. A micro volumetric pump was incorporated in the system to flow the supernatant to the OUTPUT well. Redirection to the PRE-TREATMENT well for cyclic passages though the SCREENING chamber was also designed in the test workflow. After a pre-determined number of cycles, the sensing slide could be analyzed by optical digital fluorescence microscopy (Fig. 2e and f). Detection of hybridization events on the sensing slide would provide evidence for the presence of SARS-CoV-2 viral charge in the analyzed saliva samples.
Fig. 2.

a: Concept of the SARS-CoV-2 diagnostics sensing device. b: Manufactured test bench. c: Concept of the microfluidic diagnostic circuit. d: Produced disposable microfluidic kit. e: Concept of the opto-mechanical microscopy setup of the SARS-CoV-2 diagnostics sensing device. f: Manufactured fluorimetric detection system. For the main block diagram of the automated SARS-CoV-2 diagnostics sensing device, see Supporting Information, Fig. S6.
3.4. Validation of S-PP1 sensing slides for SARS-CoV-2 RNA detection at attomolar concentrations
The detection of hybridization events as the surface of S-PP1 slides was first investigated with the customized fluorescence microscope (Fig. 2f) connected to the microfluidic chip (Fig. 2d). Saliva samples (20 μL) containing decreasing concentrations of SARS-CoV-2 virus (50, 25, 12, 6, 3, 1 copies per μL) were added to the surface of S-PP1 slides, with SYBR Green I (1:500 dilution in water). After 10 min at 50 °C, the slides were analyzed on the digital fluorescence microscope. A baseline experiment was performed on S-PP1 slides, in the presence of the fluorescent dye only (no viral charge). Control samples included the use of S-Suc slides (no DNA probe immobilized) with SARS-CoV-2 containing saliva and S-PP1 slides with MERS containing saliva. Fluorescence emission was converted by the PMT for voltage readout of the measurements, with subtraction of the baseline value from the non-intercalated dye (Fig. 3 a). A two-parameter sweep was implemented based on the LED current (proportional to the light emission) and the voltage controlling the PMT gain (exponentially proportional to its output). A 30-point grid search of these parameters was used to identify the optimal conditions for the detection system. To maximize the operating range of the detection system, the parameters were set at 650 mV for the PMT voltage and 250 mA for the LED current. All samples and controls were analyzed and compared under these conditions (Fig. 3b), and the results were normalized to the maximum PMT output value (4.66 V).
Fig. 3.

SARS-CoV-2 RNA detection with S-PP1 slides. a: Light intensities emitted by SYBR Green I upon hybridization of SARS-CoV-2 RNA with PP1 probes at the surface of S-PP1 sensing slides, using decreasing viral loads expressed in copies per μL of analyzed saliva samples. Results are expressed as the mean ± SD of two independent experiments b: Difference between S-PP1 samples and baseline at decreasing viral loads represented as logarithmic colorbars of normalized intensities over LED current intensity (mA) and PMT gain (mV).
In the absence of immobilized DNA probe, the readout values were close to the baseline, giving evidence for the absence of non-specific light emission from the interaction of SARS-CoV-2 RNA with the glass slides. At the baseline measurement, a margin of 50% of the base value was added to set the safety margin (which is the threshold above whose reading values were considered as a positive detection of the viral load). All concentrations above 6 copies per μL led to readout values satisfying the safety margin and were thus reliably detected by the sensing slides coupled to the fluorimetric analyzer. SARS-CoV-2 RNA detection at 3 and 1 copies per μL concentrations was below the significance threshold and thus remained uncertain (Fig. 3b). Noticeably, the specificity of S-PP1 sensing slides for SARS-CoV-2 RNA detection was evidenced by the absence of positive readout after incubation with MERS containing saliva.
Further validation of the S-PP1 sensing slides was performed by immunofluorescence detection of SARS-CoV-2 RNA duplexed with the immobilized PP1 probe (Fig. 4 ), after incubation at 50 °C for 10 min with saliva samples at different viral concentrations. The fluorescent emission was dependent on the viral load concentration, and decreased over the studied range in accordance with the diminution of binding events. All viral loads above 6 copies per μl led to clear detection of the SYBR Green I fluorescent signal (Fig. 4a) Several control samples were investigated (Fig. 4b) and did not show any fluorescent signal, thus confirming the specificity of the selected probe and the absence of non-specific binding to the slide surface.
Fig. 4.

a: Immunofluorescence detection of SARS-CoV-2 RNA with S-PP1 slides in saliva samples containing 50, 12, 6 and 3 copies/μL of the virus. Fluorescence was revealed through SYBR Green emission. b: Control experiments; S-PP1 slides with SYBR Green I only, S-PP1 slides with MERS virus at 50 copies/μL, S-PP1 slides with pure saliva, S-Suc slides with SARS-CoV-2 virus at 50 copies/μL. Scale bar: 10 μm.
In order to avoid the use of an external fluorophore during the detection workflow, replacement of PP1 probes by their analog molecular beacon (MB-PP1) versions was investigated (for the sequence of the molecular beacon, see Supporting information, Table S5). While MBs have been thoroughly reported for biosensing applications in solution (Bidar et al., 2021), their immobilization on sensing solid supports have not been extensively studied yet. Regarding glass materials, MBs were previously grafted on beads (Brown et al., 2000) or slides (Yao and Tan, 2004; Fang et al., 1999). In addition to removing the need for the addition of an external fluorophore to detect hybridization events, MBs generally offer a higher selectivity than their linear counterpart (Situma et al., 2007). The same conjugation procedure as for PP1 probes was applied to the immobilization of MB-PP1 oligonucleotides at the surface of the sensing slides, resulting in a grafting density of 4.5 ± 0.9 pmol (n = 2) (see Supporting Information, Table S6 and Scheme S1a). These slides (S-MB-PP1) were assessed for their detection ability toward SARS-CoV-2 RNA according to the protocol above described, using viral loads of 50, 25, 12, 6, 3 and 1 copies/μL and without the addition of the external fluorescent label SYBR Green I as carboxyfluorescein was integrated in the MB-PP1 probe (the principle for fluorescence detection with S-MB-PP1 slides is provided in Scheme S1b; the evaluation of the sensing properties of S-MB-PP1 slides is presented in Supporting Information, Figs. S8 and S9). While the specificity of the sensing slides for SARS-CoV-2 genome was maintained (MERS virus at 50 copies per μL did not lead to fluorescence emission), the sensitivity of S-MB-PP1 slides allowed for clear detection of viral loads above a concentration of 25 copies per μL of analyzed sample. While it was reported that the interaction of glass surfaces with MBs can lead to their partial opening, resulting in high fluorescence background (Situma et al., 2007), S-MB-PP1 sensing slides did not show such background signal. The decreased sensitivity of the detection workflow using S-MB-PP1 sensing surfaces as compared to S-PP1 slides might be the result of a lower concentration of immobilized fluorophore as compared to the concentration of SYBR Green I intercalated in the hybridized duplexes at the surface of S-PP1 slides. Yet, these MB-based sensors could lead to a more user-friendly method of detection as they do not require the addition of and external fluorescent reagent. Further development and optimization could lead to interesting candidate for point-of care detection.
3.5. Discussion
The access to sensing platforms for mass population viral screening, which do not require sophisticated bioanalytical instrumentation and specialized personnel, is of crucial importance to mitigate the spread of contagious emerging viruses and monitor the infection rates at regular time points. The gold standard RT-qPCR viral detection workflow is poorly adapted to mass screening and generally not accessible to developing countries. We herein disclosed the design and engineering of a prototype of an automated and portable microfluidic platform for the quick detection of SARS-CoV-2 virus from saliva samples. The focus on a microfluidic-based system was motivated by several considerations, including the possibility to handle and process very small volumes resulting in limited consumption and reduced cost of reagents and materials (Gwak et al., 2019), and the high degree of control over diagnostic conditions resulting in improved precision and accuracy of the test results (Sithara et al., 2022). In addition, microfluidic platforms are amenable to integration within compact and portable devices, thus favoring their application in point-of-care diagnostic applications (Chen et al., 2010). The sensing unit of the device was based on functionalized borosilicate slides equipped with a DNA probe targeting highly conserved regions of SARS-CoV-2 and selected in silico. One of the most critical parameters affecting the hybridization efficiency of DNA-based biosensors is the density of the probes immobilized at their surface. A quantity of 5 pmol per cm2 was suggested as an optimal amount to avoid steric hindrance while ensuring sufficient density of the probes at the surface for efficient detection of hybridization events (Peterlinz et al., 1997). The conjugation protocol herein disclosed results in the immobilization of DNA probes to amino-coated glass slides within the same magnitude range as the reported optimal quantities. The integrity of S-PP1 slides was maintained over at least two weeks of storage in MilliQ water at 4 °C. The diagnostics workflow included the RNA extraction step for 5 min, the incubation with S-PP1 slides over 10 min, followed by immediate readout from a customized fluorescence microscope connected to the sensing unit. The overall process, after collection of saliva samples, can thus be completed in less than 20 min, does not require RNA amplification and reached a detection limit of 10 aM corresponding to six virus copies per μL of analyte. In addition, the specificity toward SARS-CoV-2 vs other respiratory coronaviruses such as MERS was guaranteed by careful selection of the DNA probe covalently immobilized at the surface of the sensing slides.
4. Conclusions
A diagnostic system based on direct detection of SARS-CoV-2 RNA in saliva samples using a customized microfluidic platform was developed for future integration within a fully automated portable device designed for sequential RNA extraction, hybridization at the surface of DNA biosensors and detection by fluorescence microscopy. We herein presented the engineering and validation of the individual components of the diagnostic platform. A probe sequence of 24 bp (PP1) was identified to target specifically the ORF3a gene of the virus. The specificity of the probe toward SARS-CoV-2 was confirmed by qPCR. Then, the selected probe was covalently conjugated to glass surfaces at an average density of 2.7 ± 0.7 pmol (measured on 1 cm2 slides) to provide the biosensing unit of the screening platform. After extraction of SARS-CoV-2 RNA from infected human saliva samples, hybridization to the complementary PP1 probe was achieved by simple incubation with S-PP1 slides for 10 min at 50 °C. Detection of the resulting DNA/RNA duplexes was performed in the presence of SYBR Green I via fluorescence microscopy. This workflow resulted in the detection of SARS-CoV-2 virus from unamplified samples with a limit of detection of 6 copies per μL (10 aM), in 15 min and with high specificity. The approach used and the platform design hold the potential to be easily adapted to the detection of other viruses by simple modification of the immobilized probe sequence. In addition, the absence of amplification step, expensive reagents and sophisticated analytical instrumentation offer promising perspective for various point-of-care infection diagnostics. Future investigations on SARS-CoV-2 and other viral DNA sensors will focus on chemical functionalization strategies allowing fine tuning of the orientation and lateral spacing of the capture probes on the sensing surface.
Funding source
This work was funded by the Swiss National Science Foundation (NRP78, Grant n° 4078P0_198265) and Innosuisse (Innovation project DeMoViS, Grant n° 38934.1 IP-LS).
Data statement
The raw data associated to the manuscript:
Perrine Robin, Laura Barnabei, Stefano Marocco, Jacopo Pagnoncelli, Daniele Nicolis, Chiara Tarantelli, Agatino Christian Tavilla, Roberto Robortella, Luciano Cascione, Lucas Mayoraz, Céline M. A. Journot, Mounir Mensi, Francesco Bertoni, Igor Stefanini Sandrine Gerber-Lemaire. A DNA biosensors-based microfluidic platform for attomolar real-time detection of unamplified SARS-CoV-2 virus.
CRediT authorship contribution statement
Perrine Robin: Conceptualization, Investigation, Methodology, Validation, Data curation, Writing – original draft, Writing – review & editing. Laura Barnabei: Investigation, Methodology, Validation, Data curation. Stefano Marocco: Conceptualization, Investigation, Methodology, Test bench realization, Validation, Data curation, Writing – original draft, Writing – review & editing. Jacopo Pagnoncelli: Conceptualization, Investigation, Methodology, Test bench realization, Validation, Data curation, Writing – original draft, Writing – review & editing. Daniele Nicolis: Conceptualization, Investigation, Methodology. Chiara Tarantelli: Investigation, Methodology, Validation, Data curation. Agatino Christian Tavilla: Conceptualization, Investigation, Methodology. Roberto Robortella: Conceptualization, Investigation, Methodology. Luciano Cascione: Conceptualization, Investigation, Methodology, Test bench realization, Validation, Data curation, Writing – original draft, Writing – review & editing. Lucas Mayoraz: Investigation, Methodology. Céline M.A. Journot: Conceptualization, Investigation, Methodology, Validation. Mounir Mensi: Methodology. Francesco Bertoni: Funding acquisition, Supervision, Project administration, Writing – original draft, Writing – review & editing. Igor Stefanini: Funding acquisition, Supervision, Project administration, Writing – original draft, Writing – review & editing. Sandrine Gerber-Lemaire: Funding acquisition, Supervision, Project administration, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
P.R. and S.G-L. thanks Dr Davide Staedler (University of Lausanne) for helpful scientific discussions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biosx.2022.100302.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
Data availability
The raw data associated to this study are available in open access at: https://doi.org/10.5281/zenodo.7230549
References
- Bidar N., Amini M., Oroojalian F., Baradaran B., Hosseini S.S., Shahbazi M.-A., Hashemzaei M., Mokhtarzadeh A., Hamblin M.R., de la Guardia M. Trends Anal. Chem. 2021;134 [Google Scholar]
- Brown L.J., Cummins J., Hamilton A., Brown T. Chem. Commun. 2000;7:621–622. [Google Scholar]
- Bruch R., Johnston M., Kling A., Mattmüller T., Baaske J., Partel S., Madlener S., Weber W., Urban G.A., Dincer C. Biosens. Bioelectron. 2021;177 doi: 10.1016/j.bios.2020.112887. [DOI] [PubMed] [Google Scholar]
- Bryant J. Analog. Dialogue. 2014;48:1–3. [Google Scholar]
- Chen D., Mauk M., Qiu X., Liu C., Kim J., Ramprasad S., Ongagna S., Abrams W.R., Malamud D., Corstjens P.L.A.M., Bau H.H. Biomed. Microdevices. 2010;12:705–719. doi: 10.1007/s10544-010-9423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-H., Lim J., Shin M., Paek S.-H., Choi J.-W. Nano Lett. 2021;21:693–699. doi: 10.1021/acs.nanolett.0c04303. [DOI] [PubMed] [Google Scholar]
- Fang X., Liu X., Schuster S., Tan W. J. Am. Chem. Soc. 1999;121:2921–2922. [Google Scholar]
- Feng W., Newbigging A.M., Le C., Pang B., Peng H., Cao Y., Wu J., Abbas G., Song J., Wang D.B. Anal. Chem. 2020;92:10196–10209. doi: 10.1021/acs.analchem.0c02060. 2020. [DOI] [PubMed] [Google Scholar]
- Ganbaatar U., Liu C. Front. Cell. Infect. Microbiol. 2021;11 doi: 10.3389/fcimb.2021.663949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Gálvez L., del Caño R., Menéndez-Luque I., García-Nieto D., Rodríguez-Peña M., Luna M., Pineda T., Pariente F., García-Mendiola T., Lorenzo E. Talanta. 2022;240 doi: 10.1016/j.talanta.2021.123203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak H., Kim J., Cha S., Cheon Y.-P., Kim S.-I., Kwak B., Hyun K.-A., Jung H.-I. Biomicrofluidics. 2019;13 doi: 10.1063/1.5100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y., Ma J., Li D., Wang R. Biosensors. 2022;12:183. doi: 10.3390/bios12030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C., Park N., Kim E.S., Kim M., Kim S.D., Park S., Kim N.Y., Kim J.H. Biosens. Bioelectron. 2021;185 doi: 10.1016/j.bios.2021.113177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javalkote V.S., Kancharla N., Bhadra B., Shukla M., Soni B., Sapre A., Goodin M., Bandyopadhyay A., Dasgupta S. Methods. 2022;203:594–603. doi: 10.1016/j.ymeth.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenney A., Chibo D., Batty M., Druce J., Melvin R., Stewardson A., Dennison A., Symes S., Kinsella P., Tran T., Mackenzie C., Johnson D., Thevarajan I., McGrath C., Matlock A., Prestedge J., Gooey M., Roney J., Bobbitt J., Yallop S., Catton M., Williamson D.A. Lancet reg. Health west. Pac. 2022;26 doi: 10.1016/j.lanwpc.2022.100533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevadiya B.D., Macchi J., Herskovitz J., Oleynikov M.D., Blomberg W.R., Bajwa N., Soni D., Das S., Hasan M., Patel M., Senan A.M., Gorantla S., McMillan J., Edagwa B., Eisenberg R., Gurumurthy C.B., Reid St P.M., Punyadeera C., Chang L., Gendelman H.E. Nat. Mater. 2021;20:593–605. doi: 10.1038/s41563-020-00906-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakidis N.C., López-Cortés A., Vásconez González E., Barreto Grimaldos A., Ortiz Prado E. npj Vaccines. 2021;6:28. doi: 10.1038/s41541-021-00292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz J.R. Principles of Fluorescence Spectroscopy. third ed. Springer; 2006. Instrumentation for fluorescence spectroscopy; pp. 27–61. [Google Scholar]
- Li C., Soleyman R., Kohandel M., Cappellaro P. Nano Lett. 2022;22:43–49. doi: 10.1021/acs.nanolett.1c02868. [DOI] [PubMed] [Google Scholar]
- Lino A., Cardoso M.A., Gonçalves H.M.R., Martins-Lopes P. Chemosensors. 2022;10:221. doi: 10.1002/rmv.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Han H., Liu F., Lv Z., Wu K., Liu Y., Feng Y., Zhu C. Clin. Chim. Acta. 2020;505:172–175. doi: 10.1016/j.cca.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.-F., Zhao K.-R., Liu Z.-J., Wang L., Ye S.-Y., Liang G.-X. Biosens. Bioelectron. 2021;176 doi: 10.1016/j.bios.2020.112954. [DOI] [PubMed] [Google Scholar]
- Lv H., Wu N.C., Tsang O.T.Y., Yuan M., Perera R.A.P.M., Leung W.S., So R.T.Y., Chan J.M.C., Yip G.K., Chik T.S.H., Yang Y., Choi C.Y.C., Lin Y., Ng W.W., Zhao J., Poon L.L.M., Peiris J.S.M., Wilson I.A., Mok C.K.P. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T.R., Salit M. Nat. Rev. Genet. 2021;22:415–426. doi: 10.1038/s41576-021-00360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara K., Sakai R., Hara M. Colloid Polym. Sci. 2019;297:927–931. [Google Scholar]
- Nerenz R.D., Hubbard J.A., Cervinski M.A. Adv. Mol. Pathol. 2021;4:217–229. [Google Scholar]
- Nimse S.B., Song K., Sonawane M.D., Sayyed D.R., Kim T. Sensors. 2014;14:22208–22229. doi: 10.3390/s141222208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka M., Van-Zandvoort K., Abbott S., Sherratt K., Majdan M., CMMID COVID-19 working group. Jarčuška P., Krajčí M., Flasche S., Funk S. Science. 2021;372:635–641. doi: 10.1126/science.abf9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlinz K., Georgiadis M.R., Herne T.M., Tarlov M.J. J. Am. Chem. Soc. 1997;119:3401–3402. [Google Scholar]
- Polyakov S.V. Exp. Methods Phys. Sci. 2013;45:69–82. [Google Scholar]
- Sedra A.S., Kenneth C.S. sixth ed. Oxford University Press; 2009. Microlelectronic Circuits; pp. 53–106. [Google Scholar]
- Sithara R., Minu M., Chandra sekhar R. Mater. Adv. 2022;3:1874–1904. [Google Scholar]
- Situma, C., Moehring, A. J., Noor, M. A., Soper, S. A. Anal. Biochem.. 363, 35–45. [DOI] [PMC free article] [PubMed]
- Steranka, F.M., Bhat, J., Collins, D., Cook, L., Craford, M.G., Fletcher, R., Gardner, N., Grillot, P., Götz, W., Keuper, M., Khare, R., Kim, A, Krames, M., Harbers, G., Ludowise, M., Martin, P.S., Misra, M., Mueller, G., Mueller-Mach, R., Rudaz, S., Shen, Y.-C., Steigerwald, D., Stockman, S., Subramanya, S., Trottier, T., Wierer, J.,J. Phys. Status Solidi A 194, 380–388.
- Teles F.R.R., Fonseca L.P. Talanta. 2008;77:606–623. [Google Scholar]
- Tobik E.R., Kitfield-Vernon L.B., Thomas R.J., Steel S.A., Tan S.H., Allicock O.M., Choate B.L., Akbarzada S., Wyllie A.L. Expert Rev. Mol. Diagn. 2022;22:519–535. doi: 10.1080/14737159.2022.2094250. [DOI] [PubMed] [Google Scholar]
- Udugama B., Kadhiresan P., Kozlowski H.N., Malekjahani A., Osborne M., Li V.Y.C., Chen H., Mubareka S., Gubbay J.B., Chan W.C.W. ACS Nano. 2020;14:3822–3835. doi: 10.1021/acsnano.0c02624. [DOI] [PubMed] [Google Scholar]
- Valeur . Molecular Fluorescence: Principles and Applications. Wiley-VCH Verla GmbH; Veinheim: 2001. Characteristics of fluorescence emission; pp. 34–71. [Google Scholar]
- Weissleder R., Lee H., Ko J., Pittet M.K. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.abc1931. [DOI] [PubMed] [Google Scholar]
- Xu S., Zhan J., Man B., Jiang S., Yue W., Gao S., Guo C., Liu H., Li Z., Wang J., Zhou Y. Nat. Commun. 2017;8 doi: 10.1038/ncomms14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G., Tan W. Anal. Biochem. 2004;331:216–223. doi: 10.1016/j.ab.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data associated to this study are available in open access at: https://doi.org/10.5281/zenodo.7230549